

Abstract
Medulloblastoma (MB) is the most frequent malignant brain tumor in pediatrics. Since the current standard of care for MB consisting of surgery, cranio-spinal irradiation and chemotherapy often leads to a high morbidity rate, a number of patients suffer from long-term sequelae following treatment. Targeted therapies hold the promise of being more effective and less toxic. Therefore, the present study aimed to identify hub genes with an upregulated expression in MB and to search for potential therapeutic targets from these genes. For this purpose, gene expression profile datasets were obtained from the Gene Expression Omnibus database and processed using R 3.6.0 software to screen differentially expressed genes (DEGs) between MB samples and normal brain tissues. A total of 282 upregulated and 436 downregulated DEGs were identified. Functional enrichment analysis revealed that the upregulated DEGs were predominantly enriched in the cell cycle, DNA replication and cell division. The top 10 hub genes were identified from the protein-protein interaction network of upregulated genes, and one identified hub gene [PDZ binding kinase (PBK)] was selected for further investigation due to its possible role in the pathogenesis of MB. The aberrant expression of PBK in MB was verified in additional independent gene expression datasets. Survival analysis demonstrated that a higher expression level of PBK was significantly associated with poorer clinical outcomes in non-Wingless MBs. Furthermore, targeting PBK with its inhibitor, HI-TOPK-032, impaired the proliferation and induced the apoptosis of two MB cell lines, with the diminished phosphorylation of downstream effectors of PBK, including ERK1/2 and Akt, and the activation of caspase-3. Hence, these results suggest that PBK may be a potential prognostic biomarker and a novel candidate of targeted therapy for MB.
Keywords: medulloblastoma, PDZ binding kinase, proliferation, apoptosis, therapeutic target
Introduction
Medulloblastoma (MB) is the most prevalent pediatric central nervous system malignancy (1). The MB classification process has evolved from relying solely on histopathological features to the integration of molecular characteristics (2). The current international consensus recognizes four MB subgroups: Wingless (WNT), Sonic hedgehog (SHH), group 3 and group 4; which are based on distinctive-omic and clinical features (2,3). Somatic CTNNB1 mutations and chromosome 6 loss are common in the WNT subgroup (4), while the amplification of GLI1 or GLI2 and the deletion of Patched 1 (PTCH1) are frequently observed in the SHH subgroup (5). Aberrant MYC amplification can be detected in ~17% of patients with group 3 MB and is considered a defining feature of this subgroup (1). In addition, isochromosome 17q has been found in almost two-thirds of group 4 MB cases, and has been associated with cell division protein kinase (CDK)6 and MYCN amplification (6).
Despite considerable advances being made in the understanding of the molecular characteristics of MB, the current treatments for this disease are limited to maximal safe surgical resection, chemotherapy and cranio-spinal irradiation (1). Molecularly targeted therapy for MB remains in its infancy (7). Furthermore, although the overall survival rates have improved in recent years, a number of survivors suffer from chronic sequelae, such as neurological and neuropsychological disabilities, resulting in a poor quality of life for these children (8). In addition to the effect of the tumor itself, another main cause of these sequelae is the treatments with a high toxicity rate, particularly for patients in the very high-risk group who inevitably receive high-intensity therapeutic regimens (1). Instead of indiscriminately acting on all rapidly dividing cells (not only cancer cells, but also certain normal cells), targeted cancer therapies focusing on interfering with specific molecules involved in oncogenesis hold the promise of being less toxic than traditional chemoradiotherapy (9). Therefore, developing more targeted therapeutic strategies for MB may prove to be instrumental in curtailing the adverse effects of conventional therapies.
In the present study, datasets comprising MB and normal brain samples from the Gene Expression Omnibus database (GEO) were analyzed and a set of hub genes with a significantly upregulated expression in MB was identified. In addition, two MB cell lines, Daoy and D341 belonging to SHH and group 3, respectively (10), were used to investigate the potential of one identified hub gene [PDZ-binding kinase (PBK)], as a therapeutic target for the treatment of MB (please see the flow chart in Fig. 1).
Figure 1.
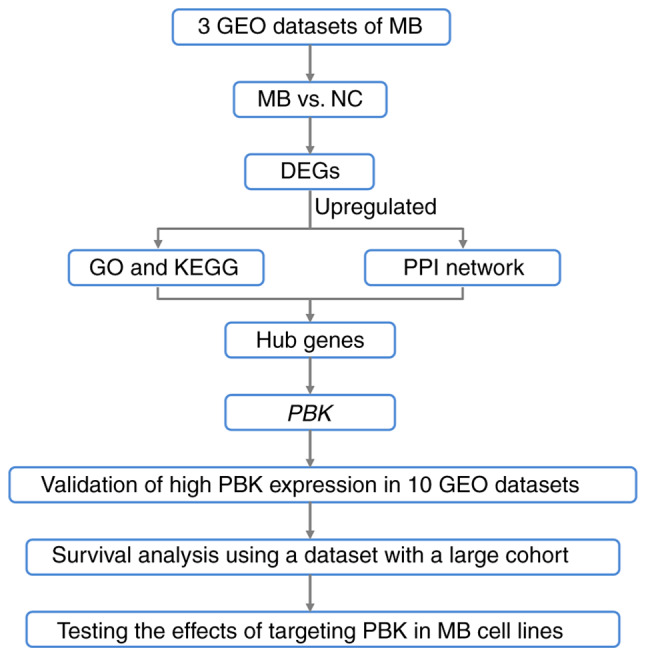
Workflow for identifying the hub genes with an upregulated expression in MB and examining the potential of PBK as a therapeutic target. MB, medulloblastoma; PBK, PDZ binding kinase; GEO, Gene Expression Omnibus; NC, normal control; DEGs, differentially upregulated genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Materials and methods
Identification of differentially expressed genes (DEGs)
RNA-seq and microarray data were retrieved from the GEO database (http://www.ncbi.nlm.nih.gov/geo/). Microarray data comprised GSE35493 (based on Affymetrix Human Genome U133 Plus 2.0 Array, GPL570) (11) and GSE39182 (based on Agilent-014850 Whole Human Genome Microarray 4×44K G4112F, GPL6480) (12). GSE35493 included 21 MB and 9 normal brain samples. GSE39182 included 20 MB and 5 normal samples. In addition, RNA-seq data GSE148389 (based on NextSeq 550, GPL21697) contained 14 normal and 26 tumor tissues (13). All probes were annotated by annotation files. Data processing was performed using R 3.6.0 software (https://www.r-project.org/). DEGs between MB and normal samples in the GSE35493 and GSE39182 microarray datasets were screened using the Limma package (14), and GSE148389 RNA-seq data were analyzed using the edgeR package (15). An adjusted P<0.05 and |log2 fold change (FC)|≥1 were set as thresholds to identify the DEGs. Venn diagrams (http://bioinformatics.psb.ugent.be/webtools/Venn/) were utilized to detect and present the common DEGs among the three datasets.
GO and KEGG pathway analyses of upregulated DEGs
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID, http://davidd.ncifcrf.gov/), which is a database which can be used for processing functional annotation with gene lists (16). The DEG results were entered to obtain the enrichment of the biological process, molecular function and cellular component terms of GO analysis and KEGG pathway terms. A P<0.05 was considered to indicate a statistically significant difference.
Protein-protein interaction (PPI) network construction and identification of hub genes
To assess the functional associations among the upregulated DEGs, the Search Tool for the Retrieval of Interacting Genes (STRING, http://string-db.org/) database was used to construct the PPI network of DEGs (17). Interactions with a combined score >0.4 were considered significant. The PPI network was then visualized using Cytoscape (version 3.8.0; http://cytoscape.org/) (18). Subsequently, hub genes among the upregulated DEGs were identified using the Cytoscape plugin cytoHubba. The top 10 hub genes were calculated according to the maximal clique centrality (MCC) algorithm in cytoHubba (19).
Expression analysis of the hub gene, PBK, and survival analysis
At the beginning of the present study, a number of datasets comprising MB and normal brain samples were downloaded following a search of the GEO database. GSE35493, GSE39182 and GSE148389 were selected to identify DEGs between MB and normal samples as these three datasets had relatively larger sample sizes and were based on different platforms, which was considered beneficial for obtaining more DEGs. The remaining datasets were then used as validation sets to verify the high expression level of PBK in MB, including GSE74195 (20), GSE50161 (21), GSE42656 (22), GSE19360 (23), GSE109401 (24), GSE86574 (25) and GSE62600 (26). PBK expression in the four MB subgroups was also examined in datasets GSE85217 (27), GSE37418 (28) and GSE21140 (29). To date, only a few MB datasets contain survival information, of which GSE85217 is the one with the largest cohort, and the sample size of other datasets is too small to conduct the survival analysis for four subgroups. Therefore, the prognostic values of PBK were tested in GSE85217 with the clinical data of a large cohort of patients with MB (27). Survival curves were drawn using Graphpad Prism 9 (GraphPad Software, Inc.). Survival analysis was completed using the Kaplan-Meier method and overall survival was analyzed using the log-rank test.
MB cell lines and cell culture
The Daoy (HTB-186) and D341 (HTB-187) MB cell lines were obtained from the American Type Culture Collection (ATCC). Daoy cells were cultured in Dulbecco's modified Eagle's medium (DMEM; cat. no. C11995, Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS; cat. no. 10270-106, Gibco; Thermo Fisher Scientific, Inc.), 1% penicillin/streptomycin (P/S; cat. no. 15140-122, Gibco; Thermo Fisher Scientific, Inc.) and 1% non-essential amino acid (NEAA; cat. no. 11140-035, Gibco; Thermo Fisher Scientific, Inc.). D341 cells were cultured in DMEM (cat. no. C11995; Gibco; Thermo Fisher Scientific, Inc.) supplemented with 20% FBS, 1% P/S and 1% NEAA. Both cell lines were maintained under a 95% O2 and 5% CO2 humidified atmosphere in an incubator at 37°C. The PBK inhibitor, HI-TOPK-032, was purchased from MedChemExpress (cat. no. HY-101550).
Western blot analysis
MB cells were plated at a density of 8.5×105 cells in 100-mm cell culture dishes and harvested following treatment with HI-TOPK-032 for 48 h. Daoy cells were treated at 0 (vehicle, DMSO), 1, 2 or 4 µM and D341 cells were treated at 0, 1 or 2 µM. Cells were lysed using RIPA lysis buffer (cat. no. P0013B; Beyotime Institute of Biotechnology), and the protein concentrations were determined using the BCA kit (cat. no. P0012; Beyotime Institute of Biotechnology). Proteins were then separated using SDS-PAGE (10% gel for PBK, ERK1/2, Akt and β-tubulin; 12% gel for cleaved caspase-3 and GAPDH) and transferred to PVDF membranes (Merck Millipore). The membranes were blocked in 5% bovine serum albumin (cat. no. CCS30014.01, MRC, EN MOASAI Biological Technology Co., Ltd.) for 1 h at room temperature, and incubated overnight at 4°C with the following primary antibodies: Anti-PBK (1:1,000; cat. no. 4942S, Cell Signaling Technology, Inc.), anti-phosphorylated (p-)p44/42 MAPK (ERK1/2) (Thr202/Tyr204, 1:1,000; cat. no. AF1015, Affinity Biosciences), anti-p44/42 MAPK (ERK1/2) (1:1,000, cat. no. BF8004, Affinity Biosciences), anti-p-Akt (Ser473, 1:1,000, cat. no. T56569, Abmart Pharmaceutical Technology Co., Ltd.), anti-Akt (1:1,000, cat. no. T55561, Abmart Pharmaceutical Technology Co., Ltd.), anti-cleaved caspase-3 (1:500, cat. no. ab32042, Abcam), anti-β-tubulin (1:5,000, cat. no. AP0064, Bioworld Technology, Inc.) and anti-GAPDH (1:2,000, cat. no. TA-08, OriGene Technologies, Inc.). Subsequently, the membranes were incubated with secondary antibodies (peroxidase-conjugated goat anti-rabbit IgG, 1:5,000; cat. no. ZB-2301, or peroxidase-conjugated goat anti-mouse IgG, 1:5,000, cat. no. ZB-2305; both from OriGene Technologies, Inc.) for 1 h at room temperature. After washing, the membranes were visualized using an enhanced chemiluminescence reagent (Merck Millipore) and a Tanon 5200 Chemiluminescent Imaging System. Gel densities were measured using ImageJ software (Version 1.53m, National Institutes of Health).
Reverse transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted using TRIzol® reagent (cat. no. 15596026, Thermo Fisher Scientific, Inc.) and the RNA concentrations were assayed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc.). cDNA was synthesized using reverse transcription kits (FastKing gDNA Dispelling RT SuperMix; cat. no. KR118, Tiangen Biotech, Co., Ltd.) according to the manufacturer's thermocycler guidelines (the reaction setup comprised of 4 µl of 5X FastKing-RT SuperMix and 1 µg of total RNA, and the final volume was made up to 20 µl with RNase-Free ddH2O; temperature protocol: 15 min at 42°C for gDNA removing and reverse transcription followed by 3 min at 95°C for enzyme inactivation). The sequences of the PCR primer pairs were as follows: PBK forward, CCAAACATTGTTGGTTATCGTGC and reverse, GGCTGGCTTTATATCGTTCTTCT; and actin beta (ACTB) forward, CATGTACGTTGCTATCCAGGC and reverse, CTCCTTAATGTCACGCACGAT. qPCR was then performed according to the manufacturer's instructions [one cycle of initial denaturation at 95°C for 3 min, 40 cycles of PCR (5 sec at 95°C for denaturation and 15 sec at 60°C for annealing/extension), and ended with a melting/dissociation curve stage (15 sec at 95°C, 1 min at 60°C and 1 sec at 95°C)] at a final volume of 20 µl/well using SYBR-Green Talent qPCR Premix (10 µl; cat. no. FP209, Tiangen Biotech, Co., Ltd.), forward and reverse primers (0.6 µl, Sangon Biotech, Co., Ltd.), cDNA (1 µl/50 ng), ROX Reference Dye (0.4 µl, Tiangen Biotech, Co., Ltd.) and RNase-Free ddH2O (7.4 µl, Tiangen Biotech, Co., Ltd.). The reaction was performed on the Applied Biosystems QuantStudio 3 Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific, Inc.). In each run, the expression levels of PBK were normalized to those of ACTB as a housekeeping gene [i.e., the expression level of ACTB was set as 1; the ΔCq value was calculated as follows: ΔCq=Cq(PBK)-Cq(ACTB); the difference in the expression level between PBK and ACTB was 2ΔCq-fold and the expression level of PBK was then normalized as 1/2ΔCq].
Cytotoxicity and cell proliferation assay
The Cell Counting Kit-8 (CCK-8; cat. no. GK10001, Glpbio Technology Inc.) assay was used to detect the viability of the control and HI-TOPK-032-treated cells. The Daoy and D341 cells were seeded in 96-well plates at a density of 2,000 and 10,000 cells per well in 100 µl of culture medium, respectively. For the cytotoxicity assays, the cells were treated with HI-TOPK-032 at 0 (vehicle, DMSO), 0.5, 1, 1.5, 2, 2.5 and 3 µM. Following treatment for 24 h (Daoy cells) or 72 h (D341 cells), 10 µl of CCK-8 solution was added to each well followed by incubation for 3 h. The absorbance values were measured using an ELX808 microplate reader (BioTek Instruments, Inc.) at a wavelength of 450 nm. Cell viability was calculated using the absorbance value of the treated groups divided by the absorbance value of the control groups and multiplied by 100%. Sigmoidal dose-response curves were fitted using non-linear regression in Graphpad Prism 9 (GraphPad Software, Inc.) to determine the IC50 values. For the cell proliferation assay, the absorbance values were measured following treatment with HI-TOPK-032 at 0, 1 or 2 µM for 0, 24, 48, or 72 h.
5-Ethynyl-2′-deoxyuridine (EdU) assays
The cells were plated in 96-well plates at a density of 5,000 (Daoy cells) and 10,000 (D341 cells) cells per well and treated with HI-TOPK-032 at 0, 1 or 2 µM. Following incubation at 37°C for 24 h, EdU assay was performed using a Click EdU cell proliferation kit with Alexa Fluor 594 (cat. no. C0078, Beyotime Institute of Biotechnology) according to the manufacturer's protocol. Images were obtained using an ECLIPSE Ti2-E inverted microscope (Nikon Corporation) and the percentage of EdU-positive cells was quantified using ImageJ software (Version 1.53m; National Institutes of Health).
Apoptosis assay
The MB cells were seeded at a density of 1×105 cells in 60-mm cell culture dishes and treated with HI-TOPK-032 at 0, 2 (D341 cells) or 4 µM (Daoy cells) for 24 h. The cells were then collected and washed twice with PBS followed by staining with Annexin V-APC/7-AAD (Apoptosis Detection kit; cat. no. KGA1017, Nanjing KeyGen Biotech Co., Ltd.) and Hoechst 33342 (cat. no. C1027, Beyotime Institute of Biotechnology) at room temperature for 10 min protected from light. After staining, the cells were transferred to a 24-well plate and fluorescence microscopy images were obtained using an ECLIPSE Ti2-E inverted microscope (Nikon Corporation). Positive cells were quantified using ImageJ software (Version 1.53m; National Institutes of Health).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 9 (GraphPad Software Inc.). All experiments were repeated three times. Quantitative results are presented as the mean ± standard deviation (SD). Statistically significant differences were assessed using an unpaired Student's t-test, one-way or two-way ANOVA with Tukey's HSD post hoc test. A value of P<0.05 was considered to indicate a statistically significant difference.
Results
Identification of upregulated hub genes in MB
The present study first screened for genes differentially expressed between MB and normal brain samples to identify potential therapeutic targets in MB. Following RNA-seq and microarray data analyses, differential mRNA expression between MB and normal samples in three datasets was found (Fig. 2A). In GSE148389, 6,906 DEGs were identified (3,216 upregulated and 3,690 downregulated genes). In GSE35493, 5,568 DEGs were determined (2,793 upregulated and 2,775 downregulated genes). In GSE39182, 2,405 DEGs were detected (818 upregulated and 1,587 downregulated genes). Venn diagrams presented the overlapping DEGs in the three datasets. A total of 282 upregulated and 436 downregulated genes were identified as common between the three datasets (Fig. 2B).
Figure 2.

Identification of upregulated hub genes in MB following enrichment analysis and protein-protein interaction network construction. (A) Volcano plots illustrating upregulated DEGs (red dots) and downregulated DEGs (blue dots) identified in the three datasets with the criteria of adjusted P<0.05 and |log2FC| ≥1 (11–13). (B) Venn diagram of overlapping upregulated or downregulated DEGs among three datasets. (C-E) GO analysis of upregulated DEGs presents the top 10 significant terms of GO analysis in (C) biological process, (D) molecular function, and (E) celluar component. (F) Significant KEGG pathway enrichment terms. (G) The PPI network was constructed with upregulated genes. Red circular nodes represent the top 10 hub genes. MB, medulloblastoma; DEGs, differentially upregulated genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PPI, protein-protein interaction; PBK, PDZ binding kinase; CDC20, cell division cycle 20; KIF2C, kinesin family member 2C; NUSAP1, nucleolar and spindle associated protein 1; TTK, TTK protein kinase; KIF20A, kinesin family member 20A; TOP2A, DNA topoisomerase II alpha; CDK1, cyclin dedendent kinase 1; ASPM, assembly factor for spindle microtubules; AURKA, Aurora kinase A.
Subsequently, the upregulated DEGs were submitted to DAVID to examine their functions and potential roles in the molecular tumorigenesis of MB. Following the enrichment analysis, the top 10 significant terms of GO analysis in three categories (biological process, molecular function and cellular component) and the significant KEGG enrichment terms were screened. In biological process, the upregulated genes were associated predominately with cell division, mitotic nuclear division, DNA replication and the G2/M transition of mitosis (Fig. 2C). In molecular function, the upregulated genes were involved primarily in protein binding, DNA binding and ATP binding (Fig. 2D). In addition, the cytosol, cytoplasm, nucleus and nucleoplasm were significantly associated with upregulated genes in the cellular component (Fig. 2E). The KEGG pathway analysis revealed that upregulated genes were enriched, particularly in DNA replication and the cell cycle (Fig. 2F).
To further investigate the functional associations among the upregulated DEGs, the STRING online database was utilized to analyze the PPI network of these genes. After removing isolated and partially connected nodes, a grid network was constructed using Cytoscape software (Fig. 2G). The Cytoscape plugin cytoHubba was then exploited to determine the hub genes in the PPI network. Finally, the top 10 hub genes [cell division cycle 20 (CDC20), kinesin family member 2C (KIF2C), nucleolar and spindle associated protein 1 (NUSAP1), PBK, TTK protein kinase (TTK), kinesin family member 20A (KIF20A), DNA topoisomerase II alpha (TOP2A), CDK1, assembly factor for spindle microtubules (ASPM) and Aurora kinase A (AURKA)] were determined using the MCC algorithm in cytoHubba (Fig. 2G).
Validation of the high expression and prognostic significance of the hub gene, PBK, in MB
According to the GO and KEGG pathway analysis, the upregulated genes were predominantly enriched in the cell cycle, DNA replication and cell division. Among the top 10 hub genes, PBK encodes PDZ-binding kinase, also known as T-lymphokine-activated killer (T-LAK) cell-originated protein kinase (TOPK), which is a serine/threonine protein kinase belonging to the mitogen-activated protein kinase kinase (MAPKK) family and plays a vital role in mitotic progression (30,31). Furthermore, researchers have previously reported that PBK is highly expressed in cerebellar granule cell precursors of early postnatal mice and functions as a crucial regulator of progenitor proliferation and self-renewal (32). Thus, given that MBs are embryonal tumors originating from stem cells or progenitor cells in the cerebellum or posterior fossa (33), the present study focused on the possible role of PBK in MB. The significant upregulation of PBK was first verified in MB by interrogating and analyzing additional independent gene expression datasets, including data from patients with MB (GSE74195, GSE50161 and GSE42656) and a spontaneous mouse model of MB (GSE19360) (Fig. 3A-D).
Figure 3.

Validation of the aberrant expression of the hub gene, PBK, in MB. (A-D) Significant upregulation of PBK in MB relative to normal brain control examined in expression datasets GSE74195 (27 MB vs. 5 NC) (20), GSE50161 (22 MB vs. 13 NC) (21), GSE42656 (16 MB vs. 9 NC) (22) and GSE19360 (3 MB vs. 3 NC) (23). (E-G) PBK was highly expressed in all MB subgroups compared with normal brain tissue. Sample size: GSE109401 (4 NC, 5 WNT, 5 SHH, 5 G3, 4 G4) (24), GSE86574 (10 NC, 5 SHH, 5 G3, 5 G4) (25) and GSE62600 (1 NC, 2 WNT, 7 SHH, 6 G3, 4 G4) (26). (H-J) PBK expression varied among MB subgroups, with group 4 MBs appearing to have the lowest expression level. Sample size: GSE85217 (70 WNT, 223 SHH, 144 G3, 326 G4) (27), GSE37418 (8 WNT, 10 SHH, 16 G3, 39 G4) (28) and GSE21140 (8 WNT, 33 SHH, 27 G3, 35 G4) (29). Quantitative results are presented as the mean ± SD. Statistical significance was determined using (A-D) an unpaired Student's t test or (E-J) one-way ANOVA with Tukey's HSD post hoc test. *P<0.05. MB, medulloblastoma; PBK, PDZ binding kinase; NC, normal control; WNT, Wingless; SHH, Sonic hedgehog; G3, group 3; G4, group 4.
Since MB is currently divided into four subgroups, the expression level of PBK was also examined in the different subgroups. Of note, PBK was highly expressed in all MB subgroups compared with normal brain samples (Fig. 3E-G), suggesting that PBK may be involved in the tumorigenesis of all subgroups. This is consistent with the function of PBK as a mitotic serine/threonine kinase and the rapid proliferating rate of MB cells. In addition, PBK expression varied among the MB subgroups, with group 4 MBs appearing to have the lowest expression level compared to the other subgroups (Fig. 3H-J). Subsequently, the prognostic significance of PBK in MB was assessed by analyzing the survival information of patients with MB from a large cohort. The overall survival curves revealed that a high PBK expression was significantly associated with poorer clinical outcomes in SHH, group 3 and group 4 MBs, apart from WNT MB (Fig. 4). Taken together, these results indicate that PBK is a crucial upregulated hub gene in MB and is likely to serve as a prognostic marker.
Figure 4.

Prognostic significance of PBK in MB. Survival analysis revealed poorer clinical outcomes in (B) SHH, (C) group 3, and (D) group 4MBs with high expression of PBK. The third quartile was used for defining high or low expression. The log-rank (Mantel-Cox) test was utilized to determine statistically significant differences. MB, medulloblastoma; PBK, PDZ binding kinase; WNT, Wingless; SHH, Sonic hedgehog.
Targeting PBK inhibits the proliferation of MB cells and reduces the phosphorylation of downstream signaling molecules
To further examine the potential of PBK as a therapeutic target for MB, two commonly used MB cell lines, Daoy and D341 belonging to the SHH and group 3 respectively, were employed to perform experiments in vitro. First, PBK expression was detected in Daoy and D341 cells using RT-qPCR and western blot analysis. The results revealed that both cell lines had a robust expression of PBK (Fig. 5A and B). While the D341 cells exhibited a higher PBK mRNA expression, the Daoy cells appeared to have a higher protein expression, suggesting that the translation efficiency of PBK may differ between the two cell lines. However, this inconsistency may also be caused by the different reference genes (housekeeping genes) used in the two assays. The MB cells were then treated with a widely-used PBK inhibitor (HI-TOPK-032) to observe its effects on cell viability and proliferation. In the CCK-8 assay, the viability of the Daoy and D341 cells was effectively inhibited by HI-TOPK-032 in a concentration dependent manner with an IC50 value of 1.241 and 1.335 µM, respectively (Fig. 5C and D). Moreover, HI-TOPK-032 also markedly decreased MB cell growth in a manner dependent on the degree of PBK suppression, as shown in Fig. 5E and F. Consistently, the two MB cell lines treated with HI-TOPK-032 at a concentration of 1 or 2 µM exhibited a lower proliferation rate than the control group in the EdU assay (Fig. 5G). These data demonstrated that targeting PBK with its specific inhibitor significantly impaired the proliferation of MB cells in vitro.
Figure 5.

Targeting PBK inhibits the proliferation of MB cells and reduces the phosphorylation levels of downstream signaling molecules. (A and B) Reverse transcription-quantitative PCR and western blot analysis illustrating the robust expression of PBK in Daoy and D341 cells. (C and D) The viability of Daoy and D341 cells was inhibited by the PBK inhibitor, HI-TOPK-032, with IC50 values of 1.241 and 1.335 µM, respectively. (E and F) Cell proliferation was suppressed by HI-TOPK-032 in a concentration-dependent manner. (G) MB cell lines exhibited a lower proliferation rate in the EdU assay following treatement with HI-TOPK-032. Scale bar, 200 µm. (H) A marked decrease in the levels of p-ERK1/2 and p-Akt was observed in the HI-TOPK-032-treated MB cells. Quantitative results are presented as the mean ± SD. Statistical significance was tested using (E and F) two-way ANOVA or (G) one-way ANOVA with Tukey's HSD post hoc test. *P<0.05. MB, medulloblastoma; PBK, PDZ binding kinase.
The present study then examined the phosphorylation status of two critical downstream targets of PBK to elucidate the mechanisms underlying the effects of HI-TOPK-032 on MB cell proliferation. Western blot analysis revealed that treatment with the PBK inhibitor, HI-TOPK-032, resulted in a slight or moderate reduction in the total levels of downstream signaling molecules, including ERK1/2 and Akt (Fig. 5H). However, a considerable decrement in the phosphorylated form of downstream proteins was observed in the HI-TOPK-032-treated MB cells (Fig. 5H). These two signaling molecules have been reported as essential downstream effectors of PBK in regulating cell proliferation (34,35). Additionally, the level of PBK in the MB cells remained stable following HI-TOPK-032 treatment (Fig. 5H), in accordance with previous studies (36,37). Taken together, these results suggest that HI-TOPK-032 may inhibit the proliferation of MB cells by suppressing the phosphorylation of downstream target proteins of PBK by blocking its kinase activity.
PBK inhibitor HI-TOPK-032 promotes the apoptosis of MB cells with the activation of caspase-3
To examine the effects of PBK inhibitor specifically on cell survival, cell apoptosis was measured by monitoring the externalization of phosphatidylserine (PS) in MB cells treated with HI-TOPK-032. Since HI-TOPK-032 would remain in the cell pellets following centrifugation and its color would interfere with the accuracy of flow cytometry detection, Annexin V/7-AAD-positive cells were observed and quantified under a fluorescence microscope. Annexin V single-positive cells represent early apoptotic cells, while Annexin V and 7-AAD double-positive cells indicate late-stage apoptotic cells and necrotic cells. The percentage of Annexin V single-positive cells was markedly higher in the HI-TOPK-032-treated group as compared with the control group, and the proportion of double-positive cells also exhibited similar results (Figs. 6A and B, and 7A and B). Subsequently, the activation of caspase-3 was examined using western blot analysis to further examine the apoptosis of MB cells. In line with PS externalization, the level of cleaved caspase-3 in the Daoy and D341 cells was substantially increased following treatment with HI-TOPK-032 (Figs. 6C and 7C). Thus, targeting PBK also induces the caspase-dependent apoptosis of MB cells.
Figure 6.

Pharmacological inhibition of PBK promotes the apoptosis of Daoy cells with the activation of caspase-3. (A and B) The percentage of apoptotic cells was significantly higher in the HI-TOPK-032-treated group compared with the control group. Scale bar, 150 µm. Quantitative results are presented as the mean ± SD. Statistical significance according to an unpaired Student's t-test is indicated: *P<0.05. (C) Western blot analysis revealed that the level of cleaved caspase-3 in Daoy cells was substantially increased following treatment with HI-TOPK-032. PBK, PDZ binding kinase.
Figure 7.

Targeting PBK with HI-TOPK-032 induces the apoptosis of D341 cells in vitro. (A and B) The percentage of apoptotic cells was significantly increased in the HI-TOPK-032-treated D341 cells. Scale bar, 100 µm. Quantitative results are presented as the mean ± SD. Statistical significance according to an unpaired Student's t-test is indicated: *P<0.05. (C) Western blot analysis revealed that the level of cleaved caspase-3 in D341 cells was substantially increased following treatment with HI-TOPK-032.
Discussion
As the most frequent malignant brain tumor in pediatrics, MB not only poses a grave threat to the lives of children, but also leads to disabling consequences and a poor quality of life for survivors. As the standard therapeutic regimens for MB (surgical intervention followed by cranio-spinal irradiation and adjuvant chemotherapy) often lead to a high morbidity rate, numerous patients suffer from short- and long-term sequelae following treatment (1). Targeted therapies provide further options for managing this refractory disease and possibilities to reduce the treatment-related toxicity (7). The present study first identified the DEGs between MB and normal brain samples, and then screened 10 hub genes from the significantly upregulated genes in MB. These hub genes have been reported in multiple cancers (e.g., breast, prostate cancer and glioma) by their oncogenic functions (38–47); however, but to the best of our knowledge, PBK has not been previously studied as a therapeutic target for MB. Therefore, PBK was selected as the target gene for further exploration in the present study.
PBK, a mitotic serine/threonine protein kinase belonging to the MAPKK family, has been found to be expressed exclusively in proliferating and multipotent cells, particularly in germinal and fetal cells, as well as cancer cells (35). Additionally, Dougherty et al (32) reported PBK expression in rapidly proliferating central nervous system cells of mice, such as adult subependymal neuronal progenitors and granular cell precursors of the postnatal cerebellum, and its suppression in neurons, mature glia and quiescent cells. MB is a type of embryonal tumor, which is now considered to arise from stem cells or progenitor cells in the cerebellum or posterior fossa (33). Hence, the significant upregulation in PBK expression in MB suggests that this protein may play a critical role in the pathogenesis of MB. The present study then verified the highly expressed level of PBK in MB by examining several independent gene expression datasets. Notably, the expression level of PBK was markedly higher in all MB subgroups than normal brain tissue, and a high PBK expression was associated with a poor clinical outcome in SHH, group 3 and group 4 MBs.
Currently, investigations on targeted therapies for MB are mainly based on distinctive aberrant signaling pathways in a specific subgroup, such as the hedgehog pathway in the SHH subgroup, or more general alterations of certain pathways in MB, such as the PI3K pathway (6,7). Since WNT MBs have a favorable prognosis, major efforts have been made to search for novel therapeutic approaches for non-WNT MBs. Interfering with the PTCH1/SMO/SUFU/GLI axis in the hedgehog pathway with small molecules is considered a promising therapy for SHH MBs. The SMO inhibitor, vismodegib, for instance, is being/has been tested in some clinical trials (NCT01878617) (48). However, resistance has been found in patients with SMO or SUFU mutations, or GLI2 amplifications (7,49). For group 3 and group 4 MBs, fewer targeted treatment options are available due to the insufficient knowledge of their specific tumor drivers (6,7). Accordingly, other novel treatment targets remain to be explored. Of note, PBK is upregulated in all MB subgroups, which also has significant prognostic implications. Therefore, the present study further examined the effects of targeting PBK with its inhibitor, HI-TOPK-032, on MB cell lines [Daoy cells (SHH) and D341 cells (group 3)]. HI-TOPK-032 is the most widely used PBK inhibitor that can occupy the ATP-binding site of PBK and block its kinase activity (37). The pharmacological inhibition of PBK led to a marked decrease in cell proliferation and a notable increase in apoptosis, with the diminished phosphorylation of downstream effectors of PBK, including ERK1/2 and Akt. These results underscore the potential of PBK as a therapeutic target in MB treatment.
Previous studies have also reported the overexpression of PBK in multiple cancer types, such as glioma, leukemia, colorectal, ovarian, skin and lung cancer (31,50). Targeting PBK with HI-TOPK-032 or other novel inhibitors, such as OTS514, OTS964 and ADA-07 in these cancer types has shown promising anticancer efficacy as well (31,35). Notably, a prerequisite for a suitable candidate target for cancer treatment is that its targeting can eliminate cancer cells, while sparing normal tissue. PBK is rarely detectable in neurons and mature glia, but highly expressed in MB cells, rendering it an attractive target for MB treatment. Considering that PBK is also expressed in active stem/progenitor cells in the brain, whether treating MB by PBK inhibition would affect these cells warrants further investigation. Of note, Joel et al (38) found that normal neural stem cells had a better tolerance for HI-TOPK-032 in vitro than glioma initiating cells, suggesting the possibility of identifying an effective dose of PBK inhibitor to destroy cancer cells, with minimal damage to normal stem cells.
It is worth noting that varied responses of MB cells to the PBK inhibitor HI-TOPK-032 were observed in the different assays in the present study. While the IC50 value of HI-TOPK-032 was similar in the Daoy and D341 cells in the CCK-8 assay, the D341 cells appeared to be more sensitive to HI-TOPK-032 than the Daoy cells in the apoptosis assay. The possible reason is that the cell density of Daoy and D341 cells was the same in the apoptosis experiments, and the number of D341 (10,000 cells/well) used in the CCK-8 assay was higher than that of Daoy cells (2,000 cells/well). Although the concentration (or total amount) of HI-TOPK-032 was the same for both cell lines, the absolute quantity of HI-TOPK-032 per cell was different (less for the D341 cells) in the CCK-8 assay. Therefore, it is hypothesized that the cell density may also influence the response of MB cells to HI-TOPK-032. As regards the limitations of the present study, the specific function of PBK in MB cells was not examined by knocking down this protein. Loss-of-function assays are thus required in the future to determine the role of PBK in regulating MB cell proliferation and apoptosis. In addition, it is necessary to evaluate the effects of PBK knockdown in MB cells on tumor growth in vivo by using animal models, particularly orthotopic xenograft models. The preclinical testing of PBK inhibitors in animal models of MB, including patient-derived orthotopic xenograft models, is also an essential step in confirming the efficacy of PBK-targeted therapy.
In conclusion, the present study identified PBK as a hub gene with an upregulated expression in MB. The aberrant expression of PBK was validated in all MB subgroups and higher expression levels of PBK also indicated poorer clinical outcomes in non-WNT MBs. Moreover, targeting PBK with its inhibitor impaired the proliferation and induced the apoptosis of two MB cell lines in vitro. Thus, PBK may prove to be a potential prognostic biomarker and therapeutic target in the management of MB.
Acknowledgements
The authors would like to thank Mr. Enio Barci (Ludwig Maximilian University of Munich) for his comments on the English writing of the manuscript.
Funding Statement
The present study was supported by the 70th China Postdoctoral Science Foundation (grant no. 2021M701618).
Availability of data and materials
The data used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
ML, HW, YD and WZ conceived the study and wrote the manuscript. HW and ML performed the bioinformatics analyses. YD and ML conducted the experiments and analyzed the data. HY, ZZ, QH, YB, PW, MZ and JG made substantial contributions to the design of the study. All authors discussed the results and revised the manuscript. YD, HW, ML and WZ confirm the authenticity of all the raw data. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Northcott PA, Robinson GW, Kratz CP, Mabbott DJ, Pomeroy SL, Clifford SC, Rutkowski S, Ellison DW, Malkin D, Taylor MD, et al. Medulloblastoma. Nat Rev Dis Primers. 2019;5:11. doi: 10.1038/s41572-019-0063-6. [DOI] [PubMed] [Google Scholar]
- 2.Hovestadt V, Ayrault O, Swartling FJ, Robinson GW, Pfister SM, Northcott PA. Medulloblastomics revisited: Biological and clinical insights from thousands of patients. Nat Rev Cancer. 2020;20:42–56. doi: 10.1038/s41568-019-0223-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM, Reifenberger G, et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro Oncol. 2021;23:1231–1251. doi: 10.1093/neuonc/noab106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, Gröbner S, Segura-Wang M, Zichner T, Rudneva VA, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547:311–317. doi: 10.1038/nature22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Juraschka K, Taylor MD. Medulloblastoma in the age of molecular subgroups: A review. J Neurosurg Pediatr. 2019;24:353–363. doi: 10.3171/2019.5.PEDS18381. [DOI] [PubMed] [Google Scholar]
- 6.DeSouza RM, Jones BR, Lowis SP, Kurian KM. Pediatric medulloblastoma-update on molecular classification driving targeted therapies. Front Oncol. 2014;4:176. doi: 10.3389/fonc.2014.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maier H, Dalianis T, Kostopoulou ON. New approaches in targeted therapy for medulloblastoma in children. Anticancer Res. 2021;41:1715–1726. doi: 10.21873/anticanres.14936. [DOI] [PubMed] [Google Scholar]
- 8.Van Ommeren R, Garzia L, Holgado BL, Ramaswamy V, Taylor MD. The molecular biology of medulloblastoma metastasis. Brain Pathol. 2020;30:691–702. doi: 10.1111/bpa.12811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baudino TA. Targeted cancer therapy: The next generation of cancer treatment. Curr Drug Discov Technol. 2015;12:3–20. doi: 10.2174/1570163812666150602144310. [DOI] [PubMed] [Google Scholar]
- 10.Ivanov DP, Coyle B, Walker DA, Grabowska AM. In vitro models of medulloblastoma: Choosing the right tool for the job. J Biotechnol. 2016;236:10–25. doi: 10.1016/j.jbiotec.2016.07.028. [DOI] [PubMed] [Google Scholar]
- 11.Birks DK, Donson AM, Patel PR, Sufit A, Algar EM, Dunham C, Kleinschmidt-DeMasters BK, Handler MH, Vibhakar R, Foreman NK. Pediatric rhabdoid tumors of kidney and brain show many differences in gene expression but share dysregulation of cell cycle and epigenetic effector genes. Pediatr Blood Cancer. 2013;60:1095–1102. doi: 10.1002/pbc.24481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valdora F, Banelli B, Stigliani S, Pfister SM, Moretti S, Kool M, Remke M, Bai AH, Brigati C, Hielscher T, et al. Epigenetic silencing of DKK3 in medulloblastoma. Int J Mol Sci. 2013;14:7492–7505. doi: 10.3390/ijms14047492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanchan RK, Perumal N, Atri P, Chirravuri Venkata R, Thapa I, Klinkebiel DL, Donson AM, Perry D, Punsoni M, Talmon GA, et al. MiR-1253 exerts tumor-suppressive effects in medulloblastoma via inhibition of CDK6 and CD276 (B7-H3) Brain Pathol. 2020;30:732–745. doi: 10.1111/bpa.12829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 17.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8((Suppl 4)):S11. doi: 10.1186/1752-0509-8-S4-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Bont JM, Kros JM, Passier MM, Reddingius RE, Sillevis Smitt PA, Luider TM, den Boer ML, Pieters R. Differential expression and prognostic significance of SOX genes in pediatric medulloblastoma and ependymoma identified by microarray analysis. Neuro Oncol. 2008;10:648–660. doi: 10.1215/15228517-2008-032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griesinger AM, Birks DK, Donson AM, Amani V, Hoffman LM, Waziri A, Wang M, Handler MH, Foreman NK. Characterization of distinct immunophenotypes across pediatric brain tumor types. J Immunol. 2013;191:4880–4888. doi: 10.4049/jimmunol.1301966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henriquez NV, Forshew T, Tatevossian R, Ellis M, Richard-Loendt A, Rogers H, Jacques TS, Reitboeck PG, Pearce K, Sheer D, et al. Comparative expression analysis reveals lineage relationships between human and murine gliomas and a dominance of glial signatures during tumor propagation in vitro. Cancer Res. 2013;73:5834–5844. doi: 10.1158/0008-5472.CAN-13-1299. [DOI] [PubMed] [Google Scholar]
- 23.Ishida Y, Takabatake T, Kakinuma S, Doi K, Yamauchi K, Kaminishi M, Kito S, Ohta Y, Amasaki Y, Moritake H, et al. Genomic and gene expression signatures of radiation in medulloblastomas after low-dose irradiation in Ptch1 heterozygous mice. Carcinogenesis. 2010;31:1694–1701. doi: 10.1093/carcin/bgq145. [DOI] [PubMed] [Google Scholar]
- 24.Rivero-Hinojosa S, Lau LS, Stampar M, Staal J, Zhang H, Gordish-Dressman H, Northcott PA, Pfister SM, Taylor MD, Brown KJ, Rood BR. Proteomic analysis of medulloblastoma reveals functional biology with translational potential. Acta Neuropathol Commun. 2018;6:48. doi: 10.1186/s40478-018-0548-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amani V, Donson AM, Lummus SC, Prince EW, Griesinger AM, Witt DA, Hankinson TC, Handler MH, Dorris K, Vibhakar R, et al. Characterization of 2 novel ependymoma cell lines with chromosome 1q gain derived from posterior fossa tumors of childhood. J Neuropathol Exp Neurol. 2017;76:595–604. doi: 10.1093/jnen/nlx040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hooper CM, Hawes SM, Kees UR, Gottardo NG, Dallas PB. Gene expression analyses of the spatio-temporal relationships of human medulloblastoma subgroups during early human neurogenesis. PLoS One. 2014;9:e112909. doi: 10.1371/journal.pone.0112909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, Garzia L, Torchia J, Nor C, Morrissy AS, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell. 2017;31:737–754.e6. doi: 10.1016/j.ccell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, Phoenix TN, Hedlund E, Wei L, Zhu X, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–48. doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, Bouffet E, Clifford SC, Hawkins CE, French P, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29:1408–1414. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaudet S, Branton D, Lue RA. Characterization of PDZ-binding kinase, a mitotic kinase. Proc Natl Acad Sci USA. 2000;97:5167–5172. doi: 10.1073/pnas.090102397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang H, Lee MH, Liu K, Dong Z, Ryoo Z, Kim MO. PBK/TOPK: An effective drug target with diverse therapeutic potential. Cancers (Basel) 2021;13:2232. doi: 10.3390/cancers13092232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dougherty JD, Garcia AD, Nakano I, Livingstone M, Norris B, Polakiewicz R, Wexler EM, Sofroniew MV, Kornblum HI, Geschwind DH. PBK/TOPK, a proliferating neural progenitor-specific mitogen-activated protein kinase kinase. J Neurosci. 2005;25:10773–10785. doi: 10.1523/JNEUROSCI.3207-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hovestadt V, Smith KS, Bihannic L, Filbin MG, Shaw ML, Baumgartner A, DeWitt JC, Groves A, Mayr L, Weisman HR, et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature. 2019;572:74–79. doi: 10.1038/s41586-019-1434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ayllón V, O'Connor R. PBK/TOPK promotes tumour cell proliferation through p38 MAPK activity and regulation of the DNA damage response. Oncogene. 2007;26:3451–3461. doi: 10.1038/sj.onc.1210142. [DOI] [PubMed] [Google Scholar]
- 35.Herbert KJ, Ashton TM, Prevo R, Pirovano G, Higgins GS. T-LAK cell-originated protein kinase (TOPK): An emerging target for cancer-specific therapeutics. Cell Death Dis. 2018;9:1089. doi: 10.1038/s41419-018-1131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kar A, Zhang Y, Yacob BW, Saeed J, Tompkins KD, Bagby SM, Pitts TM, Somerset H, Leong S, Wierman ME, Kiseljak-Vassiliades K. Targeting PDZ-binding kinase is anti-tumorigenic in novel preclinical models of ACC. Endocr Relat Cancer. 2019;26:765–778. doi: 10.1530/ERC-19-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim DJ, Li Y, Reddy K, Lee MH, Kim MO, Cho YY, Lee SY, Kim JE, Bode AM, Dong Z. Novel TOPK inhibitor HI-TOPK-032 effectively suppresses colon cancer growth. Cancer Res. 2012;72:3060–3068. doi: 10.1158/0008-5472.CAN-11-3851. [DOI] [PubMed] [Google Scholar]
- 38.Joel M, Mughal AA, Grieg Z, Murrell W, Palmero S, Mikkelsen B, Fjerdingstad HB, Sandberg CJ, Behnan J, Glover JC, et al. Targeting PBK/TOPK decreases growth and survival of glioma initiating cells in vitro and attenuates tumor growth in vivo. Mol Cancer. 2015;14:121. doi: 10.1186/s12943-015-0398-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu T, Zhang H, Yi S, Gu L, Zhou M. Mutual regulation of MDM4 and TOP2A in cancer cell proliferation. Mol Oncol. 2019;13:1047–1058. doi: 10.1002/1878-0261.12457. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Qiu R, Wu J, Gudenas B, Northcott PA, Wechsler-Reya RJ, Lu Q. Depletion of kinesin motor KIF20A to target cell fate control suppresses medulloblastoma tumour growth. Commun Biol. 2021;4:552. doi: 10.1038/s42003-021-02075-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chandler BC, Moubadder L, Ritter CL, Liu M, Cameron M, Wilder-Romans K, Zhang A, Pesch AM, Michmerhuizen AR, Hirsh N, et al. TTK inhibition radiosensitizes basal-like breast cancer through impaired homologous recombination. J Clin Invest. 2020;130:958–973. doi: 10.1172/JCI130435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao Y, He J, Li Y, Lv S, Cui H. NUSAP1 potentiates chemoresistance in glioblastoma through its SAP domain to stabilize ATR. Signal Transduct Target Ther. 2020;5:44. doi: 10.1038/s41392-020-0137-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jalalirad M, Haddad TC, Salisbury JL, Radisky D, Zhang M, Schroeder M, Tuma A, Leof E, Carter JM, Degnim AC, et al. Aurora-A kinase oncogenic signaling mediates TGF-β-induced triple-negative breast cancer plasticity and chemoresistance. Oncogene. 2021;40:2509–2523. doi: 10.1038/s41388-021-01711-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pai VC, Hsu CC, Chan TS, Liao WY, Chuu CP, Chen WY, Li CR, Lin CY, Huang SP, Chen LT, Tsai KK. ASPM promotes prostate cancer stemness and progression by augmenting Wnt-Dvl-3-β-catenin signaling. Oncogene. 2019;38:1340–1353. doi: 10.1038/s41388-018-0497-4. [DOI] [PubMed] [Google Scholar]
- 45.Yasukawa M, Ando Y, Yamashita T, Matsuda Y, Shoji S, Morioka MS, Kawaji H, Shiozawa K, Machitani M, Abe T, et al. CDK1 dependent phosphorylation of hTERT contributes to cancer progression. Nat Commun. 2020;11:1557. doi: 10.1038/s41467-020-15289-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng S, Castillo V, Sliva D. CDC20 associated with cancer metastasis and novel mushroom-derived CDC20 inhibitors with antimetastatic activity. Int J Oncol. 2019;54:2250–2256. doi: 10.3892/ijo.2019.4791. [DOI] [PubMed] [Google Scholar]
- 47.Gao Z, Jia H, Yu F, Guo H, Li B. KIF2C promotes the proliferation of hepatocellular carcinoma cells in vitro and in vivo. Exp Ther Med. 2021;22:1094. doi: 10.3892/etm.2021.10528. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Robinson GW, Orr BA, Wu G, Gururangan S, Lin T, Qaddoumi I, Packer RJ, Goldman S, Prados MD, Desjardins A, et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: Results from phase II pediatric brain tumor consortium studies PBTC-025B and PBTC-032. J Clin Oncol. 2015;33:2646–2654. doi: 10.1200/JCO.2014.60.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kool M, Jones DT, Jäger N, Northcott PA, Pugh TJ, Hovestadt V, Piro RM, Esparza LA, Markant SL, Remke M, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25:393–405. doi: 10.1016/j.ccr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wen H, Chen Z, Li M, Huang Q, Deng Y, Zheng J, Xiong M, Wang P, Zhang W. An integrative pan-cancer analysis of PBK in human tumors. Front Mol Biosci. 2021;8:755911. doi: 10.3389/fmolb.2021.755911. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used and/or analyzed during the current study are available from the corresponding author on reasonable request.