

Abstract
The COVID-19 pandemic has prompted a rapid response in vaccine and drug development. Herein, we modeled a complete membrane-embedded SARS-CoV-2 spike glycoprotein and used molecular dynamics simulations with benzene probes designed to enhance discovery of cryptic pockets. This approach recapitulated lipid and host metabolite binding sites previously characterized by cryo-electron microscopy, revealing likely ligand entry routes, and uncovered a novel cryptic pocket with promising druggable properties located underneath the 617–628 loop. A full representation of glycan moieties was essential to accurately describe pocket dynamics. A multi-conformational behavior of the 617–628 loop in simulations was validated using hydrogen-deuterium exchange mass spectrometry experiments, supportive of opening and closing dynamics. The pocket is the site of multiple mutations associated with increased transmissibility found in SARS-CoV-2 variants of concern including Omicron. Collectively, this work highlights the utility of the benzene mapping approach in uncovering potential druggable sites on the surface of SARS-CoV-2 targets.
Keywords: coronavirus, cryptic pockets, molecular dynamics simulation, benzene mapping, spike protein, hydrogen-deuterium exchange mass spectrometry, COVID-19, omicron, glycans
Graphical abstract
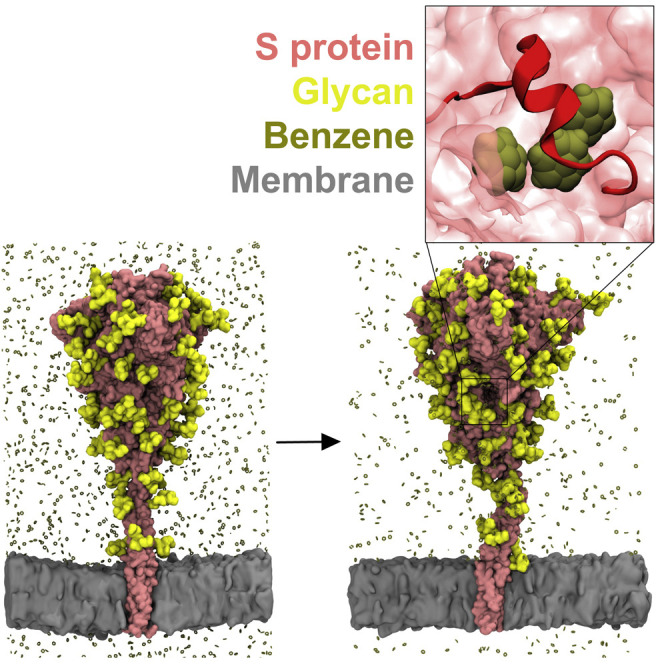
Zuzic et al. build full-length models of the membrane-bound SARS-CoV-2 spike glycoprotein and use a benzene mapping simulation approach to uncover a previously undisclosed experimentally verified cryptic pocket, which represents a potential target for development of drugs against COVID-19.
Introduction
The rapidly spreading outbreak of COVID-19 caused by a novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Gorbalenya et al., 2020), has triggered an unprecedented scale of global socioeconomic meltdown (Wu et al., 2020). SARS-CoV-2 is a large enveloped single-stranded RNA virus with a helical nucleocapsid and with a characteristic crown-like halo of viral envelope proteins. Central to the mechanism of infection is the spike (S) protein on the surface of the virion, which is the primary target for vaccine and therapeutics development. The S protein is a class I viral fusion protein trimer composed of two major subunits: S1, which facilitates host cell recognition by interacting with the human angiotensin converting enzyme 2 (ACE2), and S2, which mediates membrane fusion and entry into the host cell. To date, several structures of the prefusion S protein ectodomain (ECD) and its receptor binding domain (RBD) bound to ACE2 have been resolved using cryo-electron microscopy (cryo-EM) and X-ray crystallography (Cai et al., 2020; Lan et al., 2020; Walls et al., 2020; Wrapp et al., 2020; Wrobel et al., 2020; Yan et al., 2020). The S protein is a large trimeric protein made of various functional domains, densely covered with glycans (Watanabe et al., 2020), and it is structurally divided into two distinctive regions: a club-shaped head and a flexible stalk (Figure 1A). The two dominant conformations of the S protein chains involve the RBD in an “up” state, which renders the S protein competent for receptor binding, and a “down” state, which has inaccessible ACE2-binding surfaces. The S protein is open if any of the three chains are in an “up” conformation; otherwise, the S protein cannot bind to the ACE2 and is therefore considered closed.
Figure 1.

S protein bending dynamics during simulations without benzene
(A) Full-length model of the S glycoprotein with the protein shown in cartoon representation and colored pink (chain A), cyan (chain B), and green (chain C), with the glycans and palmitoylated cysteine residues shown in stick representation and colored yellow and brown, respectively. The different subdomains are labeled as RBD (receptor binding domain), NTD (N-terminal domain), HR1 (heptad repeat 1), CH (central helix), CD (connector domain), HR2 (heptad repeat 2), and TM (transmembrane domain). Enlarged image shows loop 620–640 in red nearby the NTD, CTD1 (C-terminal domain 1), and CTD2 (C-terminal domain 2), with nearby residues D614 and K854 shown in stick representation.
(B) (Left) A snapshot from the end of a simulation of S protein open conformation with dominant glycans. The protein and glycans are colored as in 1A, while the lipid phosphorus atoms are shown in sphere representation and colored gray. (Right) Top, bending angle, Θ, measured as the acute angle between the centers of mass of the ECD (residue 27–1141), the hinge region (residue 1142–1156), and the HR2 domain (residue 1157–1207). Bottom, bending angle, Ψ, measured as the acute angle between the HR2 domain, the hinge region (residue 1208–1212), and the TM domain (residue 1213–1239). Black, dominant glycans; blue, mannose glycans; red, no glycans.
(C) Snapshots of S protein at 10-ns intervals from a simulation, fitted onto the TM domain. The RBD is highlighted in surface representation. Glycan molecules are omitted for clarity. See also Figures S1–S3.
During the course of the pandemic, mutants with a D614G substitution on the S protein rapidly outcompeted other strains. The mutation is now an important feature of all SARS-CoV-2 variants of concern, as it is linked to higher rates of transmission compared with the D614 wild-type (WT) variant (Korber et al., 2020). Disruption of the D614-K854 salt bridge and accompanying structural changes in the S protein lead to better conformational sampling of open states (Yurkovetskiy et al., 2020). In addition, the point mutation stabilizes the S1:S2 complex and thus prevents premature dissociation of the S1 domain, allowing more S proteins to interact with the host receptor (Zhang et al., 2021). The S1:S2 stabilization is linked to ordering of a 620–640 loop, which wedges itself into a gap lined with hydrophobic residues located between an N-terminal domain (NTD) and a C-terminal domain 1 (CTD1) (Figure 1A). In D614 WT, this loop is largely disordered. The same loop is linked to multimerization events, where it extends and inserts into the NTD of a neighboring S protein, effectively forming higher-order structures consisting of two or three S protein components (Bangaru et al., 2020). Structural and biophysical studies, along with molecular dynamics (MD) simulations, have shown that the S protein is highly dynamic (Casalino et al., 2020; Ke et al., 2020; Raghuvamsi et al., 2021; Turoňová et al., 2020). Crucially, several cryo-EM structures have uncovered cryptic pockets in the RBD and NTD, which serve as potential druggable epitopes (Bangaru et al., 2020; Carrique et al., 2020; Toelzer et al., 2020). Linoleic acid (LA) was observed in the cryptic pocket of the RBD from which it also forms stabilizing interactions with the RBDs of neighboring chains (Toelzer et al., 2020). The presence of LA in the binding pocket shifts the S protein equilibrium toward closed conformations, which also has an effect on reduced ACE2 binding in vitro. Rational drug design approaches have been directed toward the LA pocket with the aim of stabilizing receptor binding-incompetent closed conformations (Ellis et al., 2021; Shoemark et al., 2021). It appears that the LA pocket also has long-range effects on allosteric networks that include neighboring chains, NTD, and distant S1/S2 cleavage sites (Oliveira et al., 2022; Tan et al., 2022). The NTD contains a cryptic site that binds a polysorbate (PS) detergent molecule if it is present in vaccine formulation (Bangaru et al., 2020; Ma et al., 2021). The same pocket can accommodate heme metabolites biliverdin and bilirubin, the presence of which change the NTD epitope presentation and modulate the antibody response (Rosa et al., 2021).
Long timescale MD simulations performed on exascale computers have also discovered the existence of numerous cryptic epitopes across the viral proteome (Zimmerman et al., 2021). In recent years, simulations of therapeutically relevant proteins with small organic probes have successfully been used to induce energetically unfavorable opening of hydrophobic cryptic pockets and subsequently identify novel druggable sites (Kuzmanic et al., 2020; Sayyed-Ahmad and Gorfe, 2017; Tan and Verma, 2020). In this work, we thus built a membrane-bound glycosylated model of the S protein and simulated it in the presence of a solution containing benzene probes, to enhance the sampling of novel cryptic pockets that could potentially be targeted by small molecules, peptides, or monoclonal antibodies. Our study not only recapitulates cryptic pockets previously characterized by cryo-EM, but also identifies a novel pocket with promising druggable properties.
Results
Spike model validation
We first built a full-length model of the S protein in the one-RBD-up open conformation using available structural and glycomics data (details in STAR methods). Three different glycosylation patterns were generated: non-glycosylated S protein to represent the original state of the protein, oligomannose-type glycans on all 22 glycosylation sites to represent the unprocessed glycoform, and the most dominant glycans species based on liquid chromatography-mass spectrometry (LC-MS) glycomics data (Watanabe et al., 2020) to represent the final glycoform. To understand the behavior of the S protein in its native environment, the model was then simulated in a model of the endoplasmic reticulum-Golgi intermediate compartment (ERGIC) membrane, where coronaviruses are assembled (Klumperman et al., 1994; Krijnse-Locker et al., 1994). The list of simulations is provided in Table S1.
Interestingly, in all simulations, the S protein did not maintain an upright conformation with respect to the plane of the membrane. Instead, we observed a tilting motion of the ECD of up to 90°, facilitated by the two flexible hinges between the ECD and HR2 domains as well as between the HR2 and transmembrane (TM) domains (Figures 1B and 1C). The flexible bending motion results in orientational freedom of the ECD, presumably yielding a more expansive sampling of the RBD at the host cell surface. This may potentially increase the probability of binding to the ACE2 receptor and hence contribute toward efficient virus-host cell recognition. Such structural dynamics are in good agreement with a range of experimental data including from hydrogen-deuterium exchange mass spectrometry (HDX-MS) (Raghuvamsi et al., 2021), cryo-EM of recombinant S protein ECD (Wrapp et al., 2020), and cryo-electron tomography (cryo-ET) of intact SARS-CoV-2 virions (Ke et al., 2020; Turoňová et al., 2020; Yao et al., 2020), as well as simulation studies of independently built S protein models (Casalino et al., 2020; Choi et al., 2021; Sikora et al., 2020). As expected, in simulations of the S protein modeled with either the predominant glycan species or oligomannose-type glycans, the glycans showed a high degree of mobility resulting in a larger surface area of the S protein being covered by them compared with what is observed in static structures (Figure 2 ). The glycans covered a larger percentage of the stalk surface compared with the ECD, in agreement with previous simulations (Casalino et al., 2020; Sikora et al., 2020). Collectively, the observed protein and glycan dynamics of our S glycoprotein models thus correlate well with other independent experimental and computational studies.
Figure 2.

S protein shielding by glycans during simulations without benzene
(A) Snapshots of S protein ECD (top) and stalk (bottom) colored cyan, pink, and green with glycans in yellow at 10-ns intervals from a simulation of S protein with dominant glycans.
(B) Solvent-accessible surface area (SASA) of the ECD (top) and the stalk (bottom). Black, dominant glycans; blue, mannose glycans; red, no glycans. Percentage of surface area buried by glycans is shown on the right, average from the last 50 ns of the simulations with the error bars showing standard deviations along the trajectories. The radius of solvent probe used for calculation was 0.14 nm.
Benzene mapping recapitulates known cryptic pockets
We next set out to uncover cryptic binding pockets on the surface of the S glycoprotein that could potentially represent targets for rational drug design. Hence, we performed a series of 200 ns simulations of the membrane-embedded, full-length S protein models with a 0.2 M concentration of benzene molecules within the bulk solvent. The benzene parameters have been modified to prevent accumulation within the hydrophobic lipid environment (details in STAR methods) (Zuzic et al., 2020). This was confirmed by the low percentage of benzene found in contact with membrane lipids throughout the simulations, and a similar progression of area per lipid and membrane thickness compared with simulations without benzene, indicating that the presence of benzene did not alter the membrane environment (Figures S1A–S1C). The stable secondary structure of the whole S protein was preserved in simulations with benzene, and the backbone root-mean-square deviation (RMSD) of the ECD was similar to control simulations, irrespective of glycoform, indicating that the overall assembly of the ECD was unaffected by the presence of benzene (Figures S1D–S1E). It should be noted that the trimeric coiled coils forming the HR2 domain partially disintegrated as benzene accumulated at its interface (Figures S2A–S2C), likely because its trimeric interface is primarily composed of an array of hydrophobic residues (Figure S2D). A recent NMR study showed that the HR2 domain adheres transiently to the viral membrane during membrane fusion (Chiliveri et al., 2021). The hydrophobic residues around which benzene accumulated in our simulations, therefore, likely reflect the lipid binding surface. To examine if this structural disintegration affects the ECD, we performed a set of control simulations in the presence of 0.2 M benzene of the isolated ECD (without the HR2 and TM domains present) modeled in the dominant glycans state. The backbone RMSD progression and secondary structural preservation of the ECD from these control simulations were very similar to those from the equivalent full-length S protein simulations (Figures S3A–S3B). This suggests that changes in the structure of the HR2 domain caused by benzene aggregation do not have any detrimental structural impact upon the ECD. Due to the structural deviation of the HR2 domain, as well as the fact that the HR2 domain is more extensively covered by glycans, we therefore only consider cryptic pockets mapped onto the surface of the ECD of the S protein.
The differences between water-only and benzene simulations revealed multiple cryptic pockets on the surface of the S protein—two of which were previously known from structural studies and have hence been used here as positive controls—and one novel pocket located near the functionally interesting loop encompassing residues 617–628 (Figure 3 ). Our control ECD-only simulations revealed very similar apolar surface areas for all three pockets compared with simulations of full-length S protein, further corroborating that HR2 distortion does not impact the ECD (Figure S3C). We also detected pocket densities near a proposed binding site for bacterial lipopolysaccharide (Petruk et al., 2021), but because it is predominantly a surface groove, we have omitted it from further analysis (Figure S4).
Figure 3.

S protein cryptic pockets revealed during simulations in the presence of benzene
LA and PS pockets are named according to their bound ligands (shown in licorice representation in insets), as their binding sites have previously been identified by cryo-EM (Bangaru et al., 2020; Carrique et al., 2020; Toelzer et al., 2020). Multimerization (MM) pocket is a surface groove that extends down the interchain interface and underneath the 617–628 loop (labeled in inset). Pockets are shown as clusters of gray (apolar) and red (polar) spheres. The three chains of S protein are colored as in Figure 1. See also Figure S4.
PS pocket is stabilized in the presence of glycans
A PS detergent molecule has been observed to bind to the NTD when detergent was present in the formulation of the immunogen (Bangaru et al., 2020). The hydrophobic tail was embedded in the hydrophobic groove pointing toward the neighboring chain, with the hydrophilic head more accessible to the protein surface. This site has also recently been shown to bind heme metabolites, which inhibits access to an antibody epitope on the NTD (Rosa et al., 2021). The addition of benzene to the simulation system successfully uncovered the PS-binding pocket, even in the absence of PS (Figure 4A). The outline of the mapped pocket was also in agreement with the shape of the hydrophobic portion of the ligand, as confirmed via structural alignment (Figure 4B).
Figure 4.

PS pocket properties and opening motions
(A) Apolar surface area of the PS pocket across different glycoforms and separated by solvent composition. The dashed line denotes the averaged pocket SASA for cryo-EM structures with bound ligands (PDB: 7JJI [Bangaru et al., 2020], 7NTA, and 7NT9 [Rosa et al., 2021]). p value significance values were calculated using Wilcoxon signed-rank test (∗∗∗∗p ≤ 0.0001; ∗∗∗p ≤ 0.001; ∗∗p ≤ 0.01; ∗p ≤ 0.05; ns > 0.05). “Dominant glycans,” “mannose glycans,” and “no glycans” refers to systems in which the S protein has been modeled with either the most dominant glycan species (Watanabe et al., 2020), oligomannose-type glycans, or without glycans present, respectively. The boxed segment displays the average proportion of polar versus apolar surface area of the PS pocket.
(B) Ligands bound within the PS pocket. PS and biliverdin binding modes are determined from cryo-EM experiments (PDB: 7JJI and 7NT9, respectively), while a simulation snapshot is used to display benzene molecules in the PS pocket. The NTD is shown in blue ribbons and transparent surface representation; Cα atoms of labeled residues are featured as yellow spheres; ligands are shown in licorice and colored by atom types.
(C) Distance measurements between the upper edge (residues 167–180) and lower edge (residues 187, 209–232) segments of the PS pocket. Distance distributions described in violin plots include pooled values from both control (−bnz) and benzene (+bnz) simulations.
(D) The span of motion that describes the opening of the PS pocket. Individual snapshots are distinguished by different-colored Cα spheres. The wide conformations appear only in unglycosylated systems.
The upper edge of the PS pocket (residues 167–180) displayed opening and closing motions that affected the volume of the pocket cavity (Figures 4C and 4D). Notably, pocket opening was greatly affected by the presence of benzene probes and glycans in the simulation system. PS pocket properties for dominant and mannose glycoforms in the presence of benzene closely reflected the pockets with experimentally bound PS and biliverdin ligands (SASAexp = 6.7 ± 0.4 nm2; SASAdom.gly,+bnz = 6.5 ± 1.9 nm2; SASAmann.gly,+bnz = 6.7 ± 2.0 nm2; Figures 4A and 4C). In comparison, water-only simulations failed to generate pocket densities that matched the shape and properties of the ligand in question, with the pockets under those conditions exhibiting lower solvent accessibility (SASAall,-bnz = 4.0 ± 1.2 nm2).
The systems without glycans displayed greater opening motions of the PS pocket upper edge (Figures 4C and 4D), suggesting that the absence of glycans results in an anomalous destabilization of the PS pocket. The fully open pocket conformation was not sampled in glycosylated systems, implying that it is unlikely to be a relevant NTD conformational state. This example highlights the importance of explicit glycan consideration when addressing cryptic pocket properties. Glycan-mediated stabilization may be particularly important in the context of benzene mapping, where the protein conformation is affected by the presence of hydrophobic probes.
LA pocket simulations reveal a common route for ligand entry
A second positive control verifying our method was the LA binding site, which has been shown to exist in the RBD in multiple cryo-EM structures (Bangaru et al., 2020; Carrique et al., 2020; Toelzer et al., 2020), but was not present in the structural templates of our initial S models (Wrapp et al., 2020). We detected increased pocket density in systems with benzene and across all glycoforms. The acyl tail of the LA ligand was accommodated in its entirety in the pocket density, consistent with the nature of the hydrophobic benzene probe, whereas the portion of the pocket outlining the polar carboxylate group was not detected in our simulations. It has been proposed that the presence of LA in the RBD shifts the dynamics of the S protein toward the closed state, whereby all RBDs are in the down configuration (Toelzer et al., 2020), allowing for the interactions of the fatty acid headgroup with the neighboring chain. As our system was modeled using the S protein ECD in the open state, it is conceivable that the arrangement of the RBDs was not able to fully reproduce the complete outline of the LA binding site that also encompasses the fatty acid carboxylate.
The LA pockets exhibited patterns of solvent exposure that were dependent on chain conformation (Figure 5A). Those located on the up-pointing RBD are highly solvent-accessible because they are not shielded by neighboring chains. On the other hand, the pockets located on the down RBDs displayed hydrophobic solvent-accessible surface area (SASA) values that were more similar to the LA pockets in cryo-EM structures with bound ligands (SASAexp = 2.9 ± 0.2 nm2; SASAdown,+bnz = 4.3 ± 1.60 nm2; SASAup,+bnz = 6.4 ± 2.47 nm2). Thus, the down-state RBD is expected to be a better descriptor of an LA-binding mode, even though benzene probes were unable to fully reproduce the LA-mediated stabilization of RBDs. Benzene inside the pocket predominantly interacted with aromatic residues Tyr365, Tyr369, and Phe377 (Figure 5B), whereas Tyr369 also served as a first point of contact for benzene entering the pocket (Figure S5). This entry site (consisting of Tyr369, Ala372, and Phe374) is pertinent to benzene, but is also a likely entry point for LA, as it is located on an exposed flexible loop that allows for ligand ingress. Conversely, the LA pocket on the upward-facing RBD exhibited exaggerated solvent accessibility (Figure 5A) and multiple sites of ligand entry (Figures 5B and S5). Taking into account the fact that the LA ligand induces stabilization of the closed structure, this conformation is unlikely to be reflective of the stable LA-binding state. However, since the simulated “up” pocket was open and accessible for benzene entry, this suggests that the ligand may initially bind to the upward-facing RBD, but stabilize only the down-state conformation.
Figure 5.

LA pocket properties and benzene binding patterns
(A) Apolar surface area of the LA pocket across different glycoforms, solvent compositions and RBD conformations (up versus down-state). The dashed line corresponds to the average apolar SASA for the cryo-EM structure in the presence of bound LA (PDB: 7JJI [Bangaru et al., 2020]). Plot descriptors are the same as in Figure 4A.
(B) Number of per-residue benzene contacts averaged over the course of the simulation, with the error bars showing standard errors along the trajectories. The most prominent contacts for the down-state are highlighted in gray on the plot and labeled on the corresponding structure. The RBD structure is shown in cartoon representation: “up” in purple and “down” in green. LA pocket residues are shown as sticks and colored based on the number of benzene contacts. The down conformation also contains benzene probes bound into the LA pocket, shown as orange spheres. The arrows denote likely routes of ligand entry: solvent-exposed up conformation displays multiple entry pathways, whereas down conformation consistently reveals a single pathway flanked by Tyr369, Ala372, and Phe374 residues. See also Figure S5.
A novel cryptic pocket detected around 617–628 loop
Finally, we also detected a novel pocket with a partial cryptic character located on the side of the S protein, which we term the multimerization (MM) pocket (Figure 6A). The majority of the pocket volume occupies a shallow surface groove in the interchain region of the S protein, while the cryptic component of the pocket is present in the smaller subsection located underneath the 617–628 loop. Although this short loop is missing from our cryo-EM structural template (Wrapp et al., 2020), it was predicted to adopt a predominantly helical structure. In addition, this loop has been shown to be helical in the thermostable disulphide-stabilized S protein construct (Xiong et al., 2020). In cryo-EM structures of S protein with the D614G substitution, this loop forms a helix due to a wider gap between the NTD and CTD1, which correlates with the more open conformation of the D614G S protein (Zhang et al., 2021). The hydrophobic contacts between this loop and a neighboring segment, CTD2, stabilize the cleaved form of the S protein by preventing premature S1 shedding. A cryo-EM structure of an S protein dimer-of-trimer (Bangaru et al., 2020) shows that this loop is involved in the formation of S protein multimers on the viral surface via its insertion into the NTD of the neighboring S protein. The cryo-EM map of the S dimer-of-trimer complex shows one 617–628 loop from each trimer interacting with the neighboring NTD, thus establishing two symmetrical points of contact between the spikes. Interestingly, when involved in multimerization, the loop is extended and exists as a random coil, instead of the predicted helix.
Figure 6.

Properties of the MM pocket
(A) S protein snapshot in surface representation with color-coded chains and an inset image showing a portion of the MM pocket packed underneath the 617–628 loop. Three benzene molecules (in orange) interact with the protein surface and occupy a predominantly hydrophobic binding pocket (spheres representing the pocket are color coded as in Figure 3).
(B) Apolar surface areas of the MM pocket are shown as violin plots and labeled as in Figure 3. Similar to positive controls, the addition of benzene to the system increases overall apolar component of the pocket in question.
(C) MM pocket shown in ribbon representation and colored according to average number of per-residue glycan contacts (calculated across both high-mannose and dominant glycan systems). Glycans predominantly interacted with the 617–628 loop, whereas the pocket interior is not in contact with any glycan component. See also Figures S6 and S7.
Apolar pocket SASA depended on chain conformation (up versus down) and glycosylation content (Figure 6B). In particular, the absence of glycans resulted in a greater solvent exposure, which (similarly to the PS pocket) demonstrated the importance of glycans in pocket stabilization. Glycan contact mapping also revealed contacts predominantly located on the outward-facing portion of the 617–628 loop (Figure 6C). The conformations associated with the MM protein in a “no glycans” system, albeit showing greater apolar SASA content, are less likely to be representative of the S protein in a biological context. Instead, it is apparent that reproducing accurate MM pocket properties in modeling studies demands explicit consideration of glycan moieties, as they interact with and modulate MM pocket exposure.
We then examined the behavior of the loop via HDX-MS experiments carried out at 37°C (details in STAR methods). The peptide (residues 617–632) encompassing the loop exhibited qualitative bimodality, with a major low exchanging deuterium exchange envelope and a minor higher exchanging envelope that reflects either an intrinsic conformational ensemble behavior or intratrimer conformational heterogeneity, with a major lower exchanging population and minor, higher exchanging population in solution (Figure 7 ) (Englander et al., 1996; Narang et al., 2020). This bimodal distribution has been clearly observed in HDX-MS of Spike at 25 °C at a locus region spanning residues 626–636; Multiple peptides displayed bimodal mass spectra, attributed to reversible unwinding of the trimeric S protein (Costello et al., 2022).
Figure 7.

The dynamic properties of the 617–632 peptide
HDX-MS mass spectrum of the peptide at labeled time points (left) shows a bimodal profile with a minor higher exchange population, indicating a conformational ensemble property in solution. In MD simulations, the loop containing this peptide exhibits a similar bimodal characteristic as assessed via solvent-accessible surface area (SASA) and conformational cluster analysis (right).
We compared the loop dynamics observed via HDX-MS with our simulated systems and observed a comparable multimodality in the behavior of the loop, or in this case, its associated peptide. Solvent accessibility of the simulated peptide reflected a similar pattern of behavior, with a large population of states with lower solvent accessibility, and rarer occurrences of states with a more accessible peptide surface. Cluster analysis likewise shows that the peptide is in a helical state for ∼90% of cumulative simulation time, but occasionally rearranges itself into less structured random coil states (Figure S6A).
Discussion
The observed conformational heterogeneity in loop behavior, both in experiment and in simulation, may be of functional importance for the S protein, particularly in the context of controlling the S protein open-closed equilibrium, preventing S1 from premature shedding upon proteolytic cleavage, and higher-order S protein complex formation. Consistently, mutations in the loop have been shown to reduce infectivity and expression of SARS-CoV-2, indicating a potential role for the loop in viral assembly (Bangaru et al., 2020). In our simulations, the MM pocket behind the 617–628 loop interacted with one, two, or rarely three benzene molecules at any given time (Figures 6A and S6B). A multiple sequence alignment indicates that the surface of the MM pocket is well-conserved across different coronaviruses (Figures S7A–S7B). Interestingly, the pocket also contains residue D614, which, when mutated to glycine, results in faster viral transmission, more efficient infection and replication (Hou et al., 2020), and higher S protein density on the viral surface (Zhang et al., 2020). The up-down dynamics of the RBD in D614G S protein correlates with order-disorder conformational changes of the 617–628 loop (Zhang et al., 2021), suggesting that targeting the nearby MM pocket could affect the propensity for RBD opening. Moreover, destabilizing hydrophobic interactions between this loop and CTD2 may promote S1 shedding and therefore reduce the stability of a cleaved S protein trimer. In addition, the pocket is within close proximity to residue A570, which is mutated to aspartate in the SARS-CoV-2 Alpha (B.1.1.7) variant, as well as residue H655, which is mutated to tyrosine in the SARS-CoV-2 Gamma (P.1) and Omicron (B.1.1.529) variants; all three variants of concern are associated with increased transmissibility and immune evasion (Figure S7C) (Davies et al., 2020; Planas et al., 2021). Thus, the predominantly hydrophobic nature of the pocket, its potential to interact with aromatic moieties commonly present in the drug molecules, its well-conserved surfaces, and, most importantly, its proximity to the functionally relevant 617–628 loop and mutated residues in novel SARS-CoV-2 variants with higher transmission rate, are all promising indicators of the potential druggability of the MM pocket.
Our discovery of the MM pocket allows for future exploratory studies including virtual screening of small molecules that may bind to the pocket, coupled to subsequent determination of the effect of binding upon the overall dynamics of the S protein. Due to the strategic location of the pocket vis-à-vis the crucial 617–628 loop described above, we hypothesize that targeting the MM pocket would disrupt the network of hydrophobic interactions between the loop and the CTD2 domain. This could potentially shift the loop dynamics to favor a disordered state, which would destabilize the S1 and S2 contacts upon proteolytic cleavage resulting in premature S1 shedding. Such local structural perturbations would be particularly advantageous against currently circulating SARS-CoV-2 variants of concern including Omicron, as they all carry the D614G mutation, which prevents premature dissociation of the S1 subunit (Zhang et al., 2021). In addition, a recent cryo-EM study suggested that in low pH conditions, as found in intracellular compartments where SARS-CoV-2 is assembled, the region containing the 617–628 loop adopts a fully ordered state resulting in a “locked” conformation of the S protein that prevents premature transition into open and post-fusion conformations during viral assembly (Qu et al., 2021). Disrupting this locked conformation by targeting the MM pocket could therefore reduce the number of functional S proteins in a mature virion.
Although the PS and LA pockets were previously characterized in S protein cryo-EM structures, those provided only a static description of the pockets in their most stable fully ligand-bound state. In this study, we elaborated on pocket dynamic properties, especially with regard to pocket opening and closing motions, the effect of glycans, and the possible route of ligand entry. The LA pocket in particular is an ongoing target for rational drug design and therefore important to address in terms of its dynamic properties. Our observation of a prominent entry route for benzene when RBD is in the down conformation provides an interesting finding that might be relevant for other LA pocket-binding ligands as well. Furthermore, the accessibility of the pocket when in an up-state suggests that ligands might enter the pocket even when the RBD is pointing upward. This is an important point in terms of drug design, as the up-state allows for greater conformational freedom and more flexible entry routes for the ligands. Comparatively, the down-state RBD and its entry site are less accessible and might pose a steric hindrance for effective binding, especially if a given ligand is bulky or branched. Therefore, the upward-pointing RBD might be easier to target with a drug molecule, after which its presence could subsequently stabilize the closed conformation that represents a receptor-binding incompetent state of the S protein.
In summary, we have demonstrated the power of the benzene mapping technique to delineate cryptic hydrophobic pockets that are of interest for drug and monoclonal antibody development targeting the SARS-CoV-2 S glycoprotein. In addition to successfully reproducing two independently identified cryptic pockets, we also uncovered a novel and potentially druggable pocket on the S protein ECD surface. The pocket was present in systems emulating both immature and mature glycosylation states, suggesting its druggability may not be dependent upon the stage of virus maturation. Overall, the predominantly hydrophobic nature of the cryptic pocket, its well-conserved surface, and proximity to regions of functional relevance in viral assembly and fitness, are all promising indicators of its potential for therapeutic targeting. Complementing efforts in antibody-based therapy and vaccine development, rational design of small-molecule drugs targeting S protein pockets may provide an essential therapeutic practice for combatting the COVID-19 pandemic.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Deuterium oxide (chemical) | Cambridge Isotope Laboratories | CAS# 7789-20-0 |
| Tris(2-carboxyethyl) phosphine-hydrochloride (TCEP-HCl) | Sigma-Aldrich | 51805-45-9 |
| GnHCl | Sigma-Aldrich | 50-01-1 |
| Phosphate buffer saline tablets | Sigma-Aldrich | Product No.: P4417-50TAB |
| Deposited data | ||
| SARS-CoV-2 genome | Wu et al. (2020) | GenBank: MN908947 |
| SARS-CoV-2 S ECD open state | Wrapp et al. (2020) | PDB: 6VSB |
| SARS-CoV-2 S RBD bound to ACE2 | Yan et al. (2020) | PDB: 6M17 |
| SARS-CoV-2 S ECD closed state | Cai et al. (2020) | PDB: 6XR8 |
| SARS-CoV HR2 | Hakansson-McReynolds et al. (2006) | PDB: 2FXP |
| HIV-1 gp-41 TM | Dev et al. (2016) | PDB: 5JYN |
| SARS-CoV-2 S model dominant glycans | This work | https://doi.org/10.5281/zenodo.5760159 |
| SARS-CoV-2 S model mannose glycans | This work | https://doi.org/10.5281/zenodo.5760159 |
| SARS-CoV-2 S model no glycans | This work | https://doi.org/10.5281/zenodo.5760159 |
| SARS-CoV-2 S ECD dominant glycans after benzene simulations | This work | https://doi.org/10.5281/zenodo.5760159 |
| SARS-CoV-2 S ECD mannose glycans after benzene simulations | This work | https://doi.org/10.5281/zenodo.5760159 |
| SARS-CoV-2 S ECD no glycans after benzene simulations | This work | https://doi.org/10.5281/zenodo.5760159 |
| HDX data | Raghuvamsi et al. (2021) | ProteomeXchange Consortium: PXD23138 |
| Experimental models: Cell lines | ||
| Human embryonic kidney (HEK293-6E) | NRC, Canada | RRID:CVCL_HF20 |
| Oligonucleotides | ||
| S protein gene of SARS-CoV-2 (1–1208) | Twist Biosciences, Singapore | QHD43416.1 |
| Recombinant DNA | ||
| pTT5 expression vector (plasmid) | Addgene, USA | RRID:Addgene_52367 |
| Software and algorithms | ||
| Modeller v9.21 | Sali and Blundell (1994) | https://salilab.org/modeller/ |
| PSIPRED 4.0 | Jones (1999) | http://bioinf.cs.ucl.ac.uk/psipred/ |
| CHARMM-GUI Membrane Builder | Lee et al. (2019) | https://www.charmm-gui.org |
| GROMACS 2018 | Abraham et al. (2015) | https://www.gromacs.org |
| VMD 1.9 | Humphrey and Dalke (1996) | https://www.ks.uiuc.edu/Research/vmd/ |
| UCSF ChimeraX 1.2.5 | Pettersen et al. (2021) | https://www.rbvi.ucsf.edu/chimerax/ |
| MDPocket | Schmidtke et al. (2011) | http://fpocket.sourceforge.net/ |
| Consurf | Ashkenazy et al. (2016) | https://consurf.tau.ac.il |
| DynamX | Waters Corporation (Milford MA) | DynamX version 3.0 |
| ProteinLynx Global Server | Waters Corporation (Milford MA) | PLGS version 3.0.1 |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Peter J. Bond (peterjb@bii.a-star.edu.sg).
Materials availability
This study did not generate new materials.
Experimental model and subject details
SARS-CoV-2 S glycoprotein models
A full-length model of the wild-type SARS-CoV-2 S protein was built using integrative modelling with a combination of cryo-EM and NMR structures as templates. Three glycoforms of the model were generated: i) with dominant glycans based on mass spectrometric data, ii) with oligomannose-type glycans, and iii) with no glycans. Further details are provided in method details section below.
Methods details
Integrative homology modelling
The S protein RBD defines its predominant functional state, with the open state having one RBD in the “up” conformation, allowing for binding to the host ACE2 receptor (Shang et al., 2020; Yan et al., 2020), with the other two RBDs in the “down” conformation interacting with NTD and other subdomains. We first built a complete model of the SARS-CoV-2 S protein in this open “up-down-down” RBD configuration using Modeller version 9.21 (Sali and Blundell, 1994). The full sequence was obtained from the complete genome of SARS-CoV-2 (GenBank: MN908947) (Wu et al., 2020). The cryo-EM structure of the S protein ECD in the open state (PDB: 6VSB) (Wrapp et al., 2020) was used as the main template for the ECD. Missing loops in the RBD up state were modelled using the cryo-EM structure of SARS-CoV-2 RBD bound to the angiotensin converting enzyme (ACE) 2 receptor (PDB: 6M17) (Yan et al., 2020), while missing loops in the N-terminal domain (NTD) and the C-terminus of the ECD were modelled using the cryo-EM structure of S ECD in the closed state resolved at a higher resolution (PDB: 6XR8) (Cai et al., 2020). The heptad repeat 2 (HR2) domain was modelled based on the NMR structure of SARS-CoV HR2 domain (96% sequence identity) in the prefusion conformation (PDB: 2FXP) (Hakansson-McReynolds et al., 2006). To date, there is no structural information for the transmembrane (TM) domain of the S protein from any coronaviruses. To estimate the position of the TM domain, the PSIPRED secondary structure prediction web server was used (Jones, 1999). The server predicted residue 1213–1237 to be within the TM helix. The presence of a GxxxG motif within this sequence suggests that the three helices of the S protein subunits form an oligomeric assembly in the membrane (Teese and Langosch, 2015). We therefore used the putative TM domain sequence to search for a homotrimeric TM structure with reasonable sequence similarity. The NMR structure of human immunodeficiency virus 1 (HIV-1) gp-41 TM domain (PDB: 5JYN) (Dev et al., 2016), which shares 27% sequence identity, was subsequently used as template. Ten models were built and the three models with the lowest discreet optimised protein energy (DOPE) scores (Shen et al., 2006) were chosen for further stereochemical assessment using Ramachandran analysis (Ramachandran et al., 1963). The model with the lowest number of outlier residues was subsequently selected for further modifications.
Two post-translational modifications were incorporated into the models: palmitoylation and glycosylation. Palmitoylation of two cysteine clusters in the SARS-CoV S protein has been shown to be important in membrane fusion with the host cell (Petit et al., 2007). We therefore added palmitoylation to cysteine residues at position 1236, 1240 and 1243 found in these two membrane proximal clusters. There are 22 N-glycosylation sites on each subunit of the S proteins, which likely play a crucial role in immune evasion by blocking access to protein epitopes. For each model, we built three glycoforms: i) with the most dominant glycan found on each site based on mass spectrometric data (Watanabe et al., 2020); ii) with an oligomannose-type glycan (Man9GlcNAc2) on all sites to represent the unprocessed glycosylated protein; and iii) without glycans. Palmitoylation and glycosylation of the S protein models was performed using the CHARMM-GUI Glycan Reader and Modeller web server (Park et al., 2019).
To simulate the S protein in its native membrane environment (Klumperman et al., 1994; Krijnse-Locker et al., 1994), we embedded our models in an endoplasmic reticulum-Golgi intermediate compartment (ERGIC) membrane. A 25 × 25 nm2 patch representing the ERGIC membrane was built using the CHARMM-GUI Membrane Builder web server (Lee et al., 2019). The membrane is symmetric and the composition is 47% phosphatidylcholine (PC), 20% phosphatidylethanol amine (PE), 11% phosphatidylinositol phosphate (PIP), 7% phosphatidylserine (PS) and 15% cholesterol (Casares et al., 2019; Meer, 1998; van Meer et al., 2008).
Molecular dynamics simulations of benzene-free systems
The CHARMM36 force field was used to parametrize the full-length S glycoprotein models (Huang and MacKerell, 2013). The protein was inserted into the model membrane based on the position of the TM domain. Overlapping lipid residues were removed and the steepest descent method was performed to minimize the system. The system was solvated with TIP3P water molecules and neutralised with 0.15 M NaCl salt. Stepwise minimization and equilibration adapted from CHARMM-GUI standard protocols were performed (Lee et al., 2016). During equilibration, the temperature of the system was maintained at 310 K using the Berendsen thermostat with a time constant of 1 ps, while the pressure was kept at 1 atm by a semi-isotropic pressure coupling using the Berendsen barostat with a time constant of 5 ps (Berendsen et al., 1984). The smooth particle mesh Ewald (PME) method (Essmann et al., 1995) with a real-space cut-off of 1.2 nm was utilised to calculate the electrostatic interactions, whereas the Van der Waals interactions were truncated at 1.2 nm and the force switch smoothing function applied between 1.0 and 1.2 nm. An integration time step of 1 fs and 2 fs were used at the early and late steps of equilibration, respectively, with the LINCS algorithm utilised to constrain all covalent bonds with hydrogen atoms (Hess et al., 1997). After equilibration, 200 ns production simulations were conducted. The Nosé-Hoover thermostat with a time constant of 1 ps was used to maintain the temperature (Hoover, 1985; Nosé, 1984), while the Parrinello-Rahman barostat with a time constant of 5 ps was used to maintain the pressure (Parrinello, 1981). A 2-fs integration time step was employed during this run.
Molecular dynamics simulations of benzene systems
A protocol for setting up benzene probe simulations in membrane systems was adapted from our previous work (Zuzic et al., 2020). Benzene molecule partial charges were adjusted from phenylalanine aromatic ring parameters defined in CHARMM36 force field (Huang and MacKerell, 2013) so that the distribution of charges was uniform across all six carbon atoms. A virtual site was added at a geometric centre of each benzene molecule, and it served as a point of repulsion between the benzene probe and the membrane. As the membrane composition differed from the one used in the original paper, repulsion point positions and the Lennard-Jones σ-value had to be adjusted to be effective for the ERGIC membrane composition. A repulsion point was placed on a carboxyl oxygen (O22/O32), one in each fatty acid tail. Repulsion point atoms were present in all lipid types (including protein-bound palmitoyl group), except for cholesterol. To account for increased gaps between membrane repulsion points due to the presence of cholesterol, the σ-value for the interactions between benzene dummy atoms and membrane repulsion points was increased to 1.4 nm. The setup was tested for the effects of aggregation and probe sequestration on a small ERGIC membrane (7 nm × 7 nm) and during a 100 ns simulation, benzene molecules remained solvated and outside the bilayer.
Benzene probes were added to a simulation box in a 0.2 M concentration. A lower probe concentration allowed for the omission of benzene-benzene repulsions and exclusions, as the probes were less likely to aggregate (Tan et al., 2016). Subsequently, the rest of the system was solvated with TIP3P water and 0.15 M NaCl salt. Minimization, equilibration, and production simulations followed the same protocol as for the benzene-free systems. To determine if the structural distortion caused by benzene on the HR2 domain in full-length S protein simulations has any impact on the formation of cryptic pockets, we repeated our dominant glycans S protein benzene simulations using only the ECD (residue 14–1146). For these simulations, the backbone of the last five residues on the C-terminus was positionally restrained with a force constant of 1000 kJ mol−1 nm−2. All simulations were performed with GROMACS 2018 (Abraham et al., 2015) and the list of simulations is provided in Table S1.
Hydrogen-deuterium exchange mass spectrometry
Deuterium exchange mass spectra of the peptide 617–632 is reported from the data deposited in PreoteomeXchange Consotium via the PRIDE partner repository (Vizca et al., 2016) with dataset identifier: PXD23138 and reported by Palur et al. (Raghuvamsi et al., 2021). Briefly, Deuterium exchange reaction was performed by incubating purified recombinant S protein (PBS, pH 7.4) in PBS buffer containing 90% D2O at 37°C for 1, 10 and 100 min of labelling time. The deuterium exchange reaction was stopped by mixing the reaction mixture with prechilled quenched buffer (1.5 M GnHCl and 0.25 M Tris(2-carboxyethyl) phosphine-hydrochloride (TCEP-HCl)) to lower the pH to 2.4 and incubated in ice for 1 min before online pepsin digestion and mass spectrometry analysis. Quenched samples were injected into nanoUPLC HDX sample manager (Waters, Milford, MA). Immobilised Waters Emzymate BEH pepsin column (2.1 × 30 mm) was used to perform online digestion in 0.1% formic acid in water at 100 μL min−1 flow rate. Pepsin-proteolysed peptides were trapped in a 2.1 × 5 mm C18 trap (ACQUITY BEH C18 VanGuard Pre-column, 1.7 μm, Waters, Milford, MA). Elution of trapped peptides was performed using acetonitrile gradient of 8% to 40% in 0.1% formic acid at flow rate 40 μL min−1 into reverse phase column (ACQUITY UPLC BEH C18 Column, 1.0 × 100 mm, 1.7 μm, Waters) pumped by nanoACQUITY Binary Solvent Manager (Waters, Milford, MA). Electrospray ionisation mode was used to spray ionised peptides and HDMSE mode of detection was implemented on SYNAPT G2-Si mass spectrometer (Waters, Milford, MA). 200 fmol μL−1 of [Glu1]-fibrinopeptide B ([Glu1]-Fib) is injected for lock spray correction at a flow rate of 5 μL min−1. Protein Lynx Global Server (PLGS v3.0, HDMSE mode) was used to identify the mass spectra of undeuterated protein samples on a separate sequence database of each protein sequence. Further, peptides were filtered using minimum intensity cutoffs of 2500 for product and precursor ions, precursor ion mass tolerance of <10 ppm and minimum products per amino acids of 0.2 using DynamX v 3.0 (Waters, Milford, MA). All the experiments were performed in triplicate and not corrected for back exchange.
Analysis
Protein, sugar and membrane structural properties were analysed using GROMACS tools, MDAnalysis (Michaud-agrawal et al., 2011) and VMD (Humphrey and Dalke, 1996). Pockets on the surface of the S protein ectodomain were analysed using MDPocket (Schmidtke et al., 2011). Initial pocket mapping of the entire ectodomain structure was followed by repeated mapping and property characterisation of each individual pocket of interest. Pockets were analysed across all simulated trajectories in 5 ns snapshot intervals. Pocket SASA was determined by calculating apolar contribution of all surrounding residues using GROMACS analysis tools. Benzene and glycan contact maps in pockets were calculated in VMD (Humphrey et al., 1996). Sequence conservation of the S protein was analysed using the ConSurf webserver (Ashkenazy et al., 2016). The HMMER algorithm (Eddy, 1998) was used to pull 40 unique sequences with 35%–95% sequence identity to the SARS-CoV-2 S protein (GenBank: MN908947) (Wu et al., 2020) from the UniProt database (Consortium, 2019). Multiple sequence alignment was built using the MAFFT program (Rozewicki et al., 2019). Structures were visualised using VMD (Humphrey et al., 1996) and USCF ChimeraX (Pettersen et al., 2021).
Quantification and statistical analysis
No statistical analysis was performed.
Additional resources
No additional resources were used.
Acknowledgments
This work was supported by BII of A∗STAR. P.J.B., J.K.M., and F.S. acknowledge funding from grant FY21_CF_HTPO SEED_ID_BII_C211418001 funded by A∗STAR. L.Z. thanks the A∗STAR Graduate Academy (A∗GA) for funding. Simulations were performed on resources of the National Supercomputing Centre, Singapore (https://www.nscc.sg), the A∗STAR Computational Resource Centre (A∗CRC), Iridis 5 supercomputer at the University of Southampton, ARCHER provided by the UK HECBioSim, and the supercomputer Fugaku provided by RIKEN through the HPCI System Research Project (Project ID: hp200303). G.S.A. acknowledges startup funds from The Pennsylvania State University.
Author contributions
Conceptualization, L.Z., F.S., P.J.B.; Methodology, L.Z., F.S., A.T.S., P.V.R.; Investigation, L.Z., F.S., A.T.S., P.V.R., J.K.M., A.B., C.P., N.K.T.; Formal Analysis, L.Z., F.S., P.V.R.; Visualization, L.Z., F.S., P.V.R; Writing – Original Draft, L.Z., F.S., P.V.R., P.J.B; Writing – Review & Editing, L.Z., F.S., P.V.R., J.W., P.M., M.C., S.K., G.S.A, P.J.B; Supervision, J.W., P.M., M.C., S.K., G.S.A, P.J.B; Project Administration, J.W., P.M., M.C., S.K., G.S.A, P.J.B.; Funding Acquisition, J.W., P.M., M.C., S.K., G.S.A, P.J.B.
Declaration of interests
The authors declare no competing interests.
Published: June 3, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.str.2022.05.006.
Supplemental information
Data and code availability
-
•
SARS-CoV-2 models have been deposited at Zenodo and are publicly available as of the date of publication. DOIs are listed in the key resources table. HDX data have been deposited at ProteomeXchange Consortium and is publicly available as of the date of publication. The accession number is listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.
References
- Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Ashkenazy H., Abadi S., Martz E., Chay O., Mayrose I., Pupko T., Ben-Tal N. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44:W344–W350. doi: 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangaru S., Ozorowski G., Turner H.L., Antanasijevic A., Huang D., Wang X., Torres J.L., Diedrich J.K., Tian J.-H., Portnoff A.D., et al. Structural analysis of full-length SARS-CoV-2 spike protein from an advanced vaccine candidate. Science. 2020;370:1089–1094. doi: 10.1126/science.abe1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., DiNola A., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- Cai Y., Zhang J., Xiao T., Peng H., Sterling S.M., Walsh Jr R., Walsh R.M., Rawson S., Rits-Volloch S., Chen B. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020;21:1586–1592. doi: 10.2210/pdb6xr8/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrique L., Duyvesteyn H.M., Malinauskas T., Zhao Y., Ren J., Zhou D., Walter T.S., Radecke J., Huo J., Ruza R.R., et al. The SARS-CoV-2 Spike harbours a lipid binding pocket which modulates stability of the prefusion trimer. BioRxiv. 2020:1–29. Preprint at. [Google Scholar]
- Casalino L., Gaieb Z., Goldsmith J.A., Hjorth C.K., Dommer A.C., Harbison A.M., Fogarty C.A., Barros E.P., Taylor B.C., Mclellan J.S., et al. Beyond shielding: the roles of glycans in the SARS-CoV-2 spike protein. ACS Cent. Sci. 2020;6:1722–1734. doi: 10.1021/acscentsci.0c01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casares D., Escribá P.V., Rosselló C.A. Membrane lipid composition: effect on membrane and organelle structure, function and compartmentalization and therapeutic avenues. Int. J. Mol. Sci. 2019;20:2167–2230. doi: 10.3390/ijms20092167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiliveri S.C., Louis J.M., Ghirlando R., Bax A. Transient lipid-bound states of spike protein heptad repeats provide insights into SARS-CoV-2 membrane fusion. Sci. Adv. 2021;7:1–13. doi: 10.1126/sciadv.abk2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y.K., Cao Y., Frank M., Woo H., Park S.J., Yeom M.S., Croll T.I., Seok C., Im W. Structure, dynamics, receptor binding, and antibody binding of the fully glycosylated full-length SARS-CoV-2 spike protein in a viral membrane. J. Chem. Theor. Comput. 2021;17:2479–2487. doi: 10.1021/acs.jctc.0c01144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium T.U. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello S.M., Shoemaker S.R., Hobbs H.T., Nguyen A.W., Hsieh C.-L., Maynard J.A., McLellan J.S., Pak J.E., Marqusee S. The SARS-CoV-2 spike reversibly samples an open-trimer conformation exposing novel epitopes. Nat. Struct. Mol. Biol. 2022;29:229–238. doi: 10.1038/s41594-022-00735-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies N.G., Barnard R.C., Jarvis C.I., Kucharski A.J., Munday J., Pearson C.A.B., Russell T.W., Tully D.C., Abbott S., Gimma A., et al. Estimated transmissibility and severity of novel SARS-CoV-2 variant of concern 202012/01 in england. MedRxiv. 2020 doi: 10.1126/science.abg3055. Preprint at. [DOI] [Google Scholar]
- Dev J., Park D., Fu Q., Chen J., Ha H.J., Ghantous F., Herrmann T., Chang W., Liu Z., Frey G., et al. Structural basis for membrane anchoring of HIV-1 envelope spike. Science. 2016;353:172–175. doi: 10.1126/science.aaf7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S.R. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Ellis D., Brunette N., Crawford K.H.D., Walls A.C., Pham M.N., Chen C., Herpoldt K.-L., Fiala B., Murphy M., Pettie D., et al. Stabilization of the SARS-CoV-2 spike receptor-binding domain using deep mutational scanning and structure-based design. Front. Immunol. 2021;12:710263–710317. doi: 10.3389/fimmu.2021.710263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander S.W., Sosnick T.R., Englander J.J., Mayne L. Mechanisms and uses of hydrogen exchange. Curr. Opin. Struct. Biol. 1996;6:18–23. doi: 10.1016/s0959-440x(96)80090-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- Gorbalenya A.E., Baker S.C., Baric R.S., de Groot R.J., Drosten C., Gulyaeva A.A., Haagmans B.L., Lauber C., Leontovich A.M., Neuman B.W., et al. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakansson-McReynolds S., Jiang S., Rong L., Caffrey M. Solution structure of the severe acute respiratory syndrome-coronavirus heptad repeat 2 domain in the prefusion state. J. Biol. Chem. 2006;281:11965–11971. doi: 10.1074/jbc.m601174200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comp. Chem. 1997;18:1463–1472. doi: 10.1002/(sici)1096-987x(199709)18:12<1463::aid-jcc4>3.0.co;2-h. [DOI] [Google Scholar]
- Hoover W.G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- Hou Y.J., Chiba S., Halfmann P., Ehre C., Kuroda M., Dinnon K.H., Leist S.R., Schäfer A., Nakajima N., Takahashi K., et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science. 2020;370:1464–1468. doi: 10.1126/science.abe8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., MacKerell A.D. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 2013;34:2135–2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jones D.T. Protein secondary structure prediction based on position-specific scoring matrices 1 1Edited by G. Von Heijne. J. Mol. Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- Ke Z., Oton J., Qu K., Cortese M., Zila V., McKeane L., Nakane T., Zivanov J., Neufeldt C.J., Cerikan B., et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature. 2020;588:498–502. doi: 10.1038/s41586-020-2665-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumperman J., Locker J.K., Meijer A., Horzinek M.C., Geuze H.J., Rottier P.J. Coronavirus M proteins accumulate in the Golgi complex beyond the site of virion budding. J. Virol. 1994;68:6523–6534. doi: 10.1128/jvi.68.10.6523-6534.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E.E., Bhattacharya T., Foley B., et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–827. doi: 10.1016/j.cell.2020.06.043. e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijnse-Locker J., Ericsson M., Rottier P.J.M., Griffiths G. Characterization of the budding compartment of mouse hepatitis virus: evidence that transport from the RER to the Golgi complex requires only one vesicular transport step. J. Cell Biol. 1994;124:55–70. doi: 10.1083/jcb.124.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmanic A., Bowman G.R., Juarez-Jimenez J., Michel J., Gervasio F.L. Investigating cryptic binding sites by molecular dynamics simulations. ACS Appl. Mater. Inter. 2020;53:654–661. doi: 10.1021/acs.accounts.9b00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Lee J., Cheng X., Jo S., MacKerell A.D., Klauda J.B., Im W., Wei S., Buckner J., Jeong J.C., Qi Y., et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theor. Comput. 2016;110:641a–413. doi: 10.1016/j.bpj.2015.11.3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Patel D.S., Ståhle J., Park S.J., Kern N.R., Kim S., Lee J., Cheng X., Valvano M.A., Holst O., et al. CHARMM-GUI membrane builder for complex biological membrane simulations with glycolipids and lipoglycans. J. Chem. Theor. Comput. 2019;15:775–786. doi: 10.1021/acs.jctc.8b01066. [DOI] [PubMed] [Google Scholar]
- Ma J., Su D., Sun Y., Huang X., Liang Y., Fang L., Ma Y., Li W., Liang P., Zheng S. Cryo-electron microscopy structure of S-trimer, a subunit vaccine candidate for COVID-19. J. Virol. 2021;95:e00194-21. doi: 10.1128/jvi.00194-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meer G. Van. Lipids of the golgi membrane. Trends Cell Biol. 1998;8:29–33. doi: 10.1016/s0962-8924(97)01196-3. [DOI] [PubMed] [Google Scholar]
- Michaud-agrawal N., Denning E.J., Woolf T.B., Beckstein O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011;32:2319–2327. doi: 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narang D., Lento C., J Wilson D. HDX-MS: an analytical tool to capture protein motion in action. Biomedicines. 2020;8:224–320. doi: 10.3390/biomedicines8070224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosé S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984;52:255–268. doi: 10.1080/00268978400101201. [DOI] [Google Scholar]
- Oliveira A.S.F., Shoemark D.K., Ibarra A., Davidson D., Berger I., Schaffitzel C., Mulholland A.J. The fatty acid site is coupled to functional motifs in the SARS-CoV-2 spike protein and modulates spike allosteric behaviour. Comput. Struct. Biotechnol J. 2022;20:139–147. doi: 10.1016/j.csbj.2021.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.J., Lee J., Qi Y., Kern N.R., Lee H.S., Jo S., Joung I., Joo K., Lee J., Im W. CHARMM-GUI Glycan Modeler for modeling and simulation of carbohydrates and glycoconjugates. Glycobiology. 2019;29:320–331. doi: 10.1093/glycob/cwz003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. doi: 10.1063/1.328693. [DOI] [Google Scholar]
- Petit C.M., Chouljenko V.N., Iyer A., Colgrove R., Farzan M., Knipe D.M., Kousoulas K.G. Palmitoylation of the cysteine-rich endodomain of the SARS-coronavirus spike glycoprotein is important for spike-mediated cell fusion. Virology. 2007;360:264–274. doi: 10.1016/j.virol.2006.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruk G., Puthia M., Petrlova J., Samsudin F., Strömdahl A.-C., Cerps S., Uller L., Kjellström S., Bond P.J., Schmidtchen A. SARS-CoV-2 Spike protein binds to bacterial lipopolysaccharide and boosts proinflammatory activity. J. Mol. Cell Biol. 2021;12:916–932. doi: 10.1093/jmcb/mjaa067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Meng E.C., Couch G.S., Croll T.I., Morris J.H., Ferrin T.E. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021;30:70–82. doi: 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas D., Saunders N., Maes P., Guivel-Benhassine F., Planchais C., Buchrieser J., Bolland W.H., Porrot F., Staropoli I., Lemoine F., et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature. 2021;602:671–675. doi: 10.1038/d41586-021-03827-2. [DOI] [PubMed] [Google Scholar]
- Qu K., Xiong X., Ciazynska K.A., Carter A.P., Brigss J.A. Structures and function of locked conformations of SARS-CoV-2 spike. BioRxiv. 2021:1–23. Preprint at. [Google Scholar]
- Raghuvamsi P.V., Tulsian N.K., Samsudin F., Qian X., Purushotorman K., Yue G., Kozma M.M., Hwa W.Y., MacAry P., Bond P.J. SARS-CoV-2 S protein:ACE2 interaction reveals novel allosteric targets. Elife. 2021;10:e63646. doi: 10.7554/elife.63646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran G.N., Ramakrishnan C., Sasisekharan V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963;7:95–99. doi: 10.1016/s0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
- Rosa A., Pye V.E., Graham C., Muir L., Seow J., Ng K.W., Cook N.J., Rees-Spear C., Parker E., Dos Santos M.S., et al. SARS-CoV-2 recruits a haem metabolite to evade antibody immunity. Sci. Adv. 2021;7:eabg7607. doi: 10.1101/2021.01.21.21249203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozewicki J., Li S., Amada K.M., Standley D.M., Katoh K. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 2019;47:W5–W10. doi: 10.1093/nar/gkz342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1994;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Sayyed-Ahmad A., Gorfe A.A. Mixed-probe simulation and probe-derived surface topography map analysis for ligand binding site identification. J. Chem. Theor. Comput. 2017;13:1851–1861. doi: 10.1021/acs.jctc.7b00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtke P., Bidon-chanal A., Luque F.J., Barril X. MDpocket: open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics. 2011;27:3276–3285. doi: 10.1093/bioinformatics/btr550. [DOI] [PubMed] [Google Scholar]
- Shang J., Ye G., Shi K., Wan Y., Luo C., Aihara H., Geng Q., Auerbach A., Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–224. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M., Devos D., Melo F., Sali A., Marti-Renom M.A. A composite score for predicting errors in protein structure models. Protein Sci. 2006;15:1653–1666. doi: 10.1110/ps.062095806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemark D.K., Colenso C.K., Toelzer C., Gupta K., Sessions R.B., Davidson A.D., Berger I., Schaffitzel C., Spencer J., Mulholland A.J. Molecular simulations suggest vitamins, retinoids and steroids as ligands of the free fatty acid pocket of the SARS-CoV-2 spike protein. Angew. Chem. 2021;133:7174–7186. doi: 10.1002/ange.202015639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikora M., Gecht M., Covino R., Hummer G. Map of SARS-CoV-2 spike epitopes not shielded by glycans. BioRxiv. 2020 Preprint at. [Google Scholar]
- Tan Y.S., Verma C.S. Straightforward incorporation of multiple ligand types into molecular dynamics simulations for efficient binding site detection and characterization. J. Chem. Theor. Comput. 2020;16:6633–6644. doi: 10.1021/acs.jctc.0c00405. [DOI] [PubMed] [Google Scholar]
- Tan Y.S., Reeks J., Brown C.J., Thean D., Ferrer Gago F.J., Yuen T.Y., Goh E.T.L., Lee X.E.C., Jennings C.E., Joseph T.L., et al. Benzene probes in molecular dynamics simulations reveal novel binding sites for ligand design. J. Phys. Chem. Lett. 2016;7:3452–3457. doi: 10.1021/acs.jpclett.6b01525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z.W., Tee W.-V., Samsudin F., Guarnera E., Bond P.J., Berezovsky I.N. Allosteric perspective on the mutability and druggability of the SARS-CoV-2 Spike protein. Structure. 2022;30:590–607.e4. doi: 10.1016/j.str.2021.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teese M.G., Langosch D. Role of GxxxG motifs in transmembrane domain interactions. Biochemistry. 2015;54:5125–5135. doi: 10.1021/acs.biochem.5b00495. [DOI] [PubMed] [Google Scholar]
- Toelzer C., Gupta K., Yadav S.K.N., Borucu U., Davidson A.D., Kavanagh Williamson M., Shoemark D.K., Garzoni F., Staufer O., Milligan R., et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science. 2020;370:725–730. doi: 10.1126/science.abd3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turoňová B., Sikora M., Schürmann C., Hagen W.J.H., Welsch S., Blanc F.E.C., von Bülow S., Gecht M., Bagola K., Hörner C., et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science. 2020;370:203–208. doi: 10.1126/science.abd5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G., Voelker D.R., Feigenson G.W., Meer G.V. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizca J.A., Csordas A., Griss J., Lavidas I., Mayer G., Perez-riverol Y., Reisinger F., Ternent T., Xu Q., Wang R., et al. Erratum: 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:11033. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Park Y.-J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;183:1735–2292. doi: 10.1016/j.cell.2020.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., Allen J.D., Wrapp D., McLellan J.S., Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science. 2020;369:330–333. doi: 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.-L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel A.G., Benton D.J., Xu P., Roustan C., Martin S.R., Rosenthal P.B., Skehel J.J., Gamblin S.J. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 2020;27:763–767. doi: 10.1038/s41594-020-0468-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G., Hu Y., Tao Z.-W., Tian J.-H., Pei Y.-Y., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X., Qu K., Ciazynska K.A., Hosmillo M., Carter A.P., Ebrahimi S., Ke Z., Scheres S.H.W., Bergamaschi L., Grice G.L., et al. A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020;27:934–941. doi: 10.1038/s41594-020-0478-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H., Song Y., Chen Y., Wu N., Xu J., Sun C., Zhang J., Weng T., Zhang Z., Wu Z., et al. Molecular architecture of the SARS-CoV-2 virus. Cell. 2020;183:730–738.e13. doi: 10.1016/j.cell.2020.09.018. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurkovetskiy L., Wang X., Pascal K.E., Tomkins-Tinch C., Nyalile T.P., Wang Y., Baum A., Diehl W.E., Dauphin A., Carbone C., et al. Structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell. 2020;183:739–751. doi: 10.1016/j.cell.2020.09.032. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Cai Y., Xiao T., Lu J., Peng H., Sterling S.M., Walsh Jr R., Rits-Volloch S., Zhu H., Woosley A.N., et al. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science (80-. ) 2021;372:525–530. doi: 10.2210/pdb7krr/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Jackson C.B., Mou H., Ojha A., Peng H., Quinlan B.D., Rangarajan E.S., Pan A., Vanderheiden A., Suthar M.S., et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020;11:6013. doi: 10.1038/s41467-020-19808-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman M.I., Bowman G., Ward M., Singh S., Vithani N., Meller A., Mallimadugula U., Kuhn C., Borowsky J., Wiewiora R., et al. SARS-CoV-2 simulations go exascale to capture spike opening and reveal cryptic pockets across the proteome. Nat. Chem. 2021;120:299a. doi: 10.1016/j.bpj.2020.11.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuzic L., Marzinek J.K., Warwicker J., Bond P.J. A benzene-mapping approach for uncovering cryptic pockets in membrane-bound proteins. J. Chem. Theor. Comput. 2020;16:5948–5959. doi: 10.1021/acs.jctc.0c00370. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
SARS-CoV-2 models have been deposited at Zenodo and are publicly available as of the date of publication. DOIs are listed in the key resources table. HDX data have been deposited at ProteomeXchange Consortium and is publicly available as of the date of publication. The accession number is listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.