

Abstract
The mitochondrion is a double membrane structured organelle involved in a variety of regulatory functions such as calcium signaling, production of adenosine triphosphate, apoptosis, reactive oxygen species generation, cell growth, and cell cycling. Impaired mitochondrial function is evident in various neurological disorders stemming from both acute and chronic neural injury. Herein, we review the role of mitochondrial regulation in maintaining cellular homeostasis, the consequences of their dysfunction in relation to pathophysiology after neurotrauma, approaches being used to promote their bioenergetic integrity for neuroprotection, and multifaceted methods being used to preserve/rescue their function following both traumatic brain and spinal cord injury.
1. Introduction
The mitochondrion lies at the heart of cell life and cell death, referred to as the “power house” that provides 95% of cellular energy. The organelle is made up of double membranes that form a sac within a sac configuration, with the smooth outer membrane holding numerous transport proteins shuttling materials in and out of the mitochondria. The inner membrane forms folded structures called cristae housing the electron transport chain and adenosine triphosphate (ATP) synthesis apparatus. The inner compartment is known as the matrix which lodges circular DNA and ribosomes necessary for mitochondrial protein translation, as well as numerous enzymes involved with Kreb’s Cycle and fatty acid oxidation, as well as calcium (Ca2+) buffering. Mitochondria undergo fusion and fission events to maintain balance in their number and morphology to adjust to tissue- or cell-specific physiological needs in order to maintain cellular homeostasis (Fischer et al., 2012; Marzetti et al., 2013; Muller et al., 2015; Van Blerkom, 2009). Even in the same cell types, mitochondria change their number and morphologies in response to different environs. Dynamic association of mitochondria with other organelles such as endoplasmic reticulum (ER) is crucial for cellular processes such as autophagy, mitochondrial motility, lipid and Ca2+ fluxes, as well as mitochondrial division (Chakrabarti et al., 2018; Friedman et al., 2011; Hamasaki et al., 2013; Lewis et al., 2016; Mendes et al., 2005; Rizzuto et al., 1998; Szabadkai et al., 2006; Vance, 1990, 2014).
Mitochondria play key roles in neurogenesis, neuronal development, axonal transport, and synaptic plasticity via regulating neurotransmitter synthesis (Beckervordersandforth et al., 2017; Hollenbeck, 2005; Kim et al., 2011; Levy et al., 2003; Todorova and Blokland, 2017; Vos et al., 2010). It is important to note that neurons are more prone to mitochondrial dysfunction compared to astrocytes and oligodendrocytes as they are mainly dependent on mitochondrial oxidative phosphorylation for their energy demands and have a limited capacity to upregulate glycolysis (Fernandez-Fernandez et al., 2012). Mitochondrial dysfunction underlies neuronal death cascade(s) in neurodegenerative diseases and central nervous system (CNS) injuries (Calkins et al., 2011; Liu et al., 2016; Ludwig et al., 2018; Ottolini et al., 2017; Suarez-Rivero et al., 2016; Witte et al., 2014). It is well documented that their dysfunction is directly related to intracellular Ca2+ overload that is associated with their increased reactive oxygen species (ROS) production and decreased ATP synthesis leading to disruption of cellular hemostasis (Adam-Vizi, 2005; Adam-Vizi and Chinopoulos, 2006; Holmstrom and Finkel, 2014; Starkov et al., 2004). Prior to reviewing evidence for targeting mitochondrial dysfunction as a viable neurotherapeutic, a brief description of the complex interactions that allow them to function under normal and pathophysiological conditions ensues.
2. Mitochondrial regulation
2.1. Dynamic structural alterations
Mitochondria frequently join their membranes together to form one mitochondrion by a process termed fusion, but they also divide into separate mitochondria as they move along cytoskeletal tracks, a process known as fission (Hoppins et al., 2007). These processes allow the mitochondria to regulate their morphology and number within the cell and play an important role in maintaining mitochondrial homeostasis under pathological conditions (Hoppins et al., 2007; Meyer et al., 2017; Youle and van der Bliek, 2012). When mitochondria are compromised due to metabolic or environmental stress, functional mitochondria complement damaged mitochondria by diffusing and sharing of their components via fusion. Fission, on the other hand, is the process of creating new mitochondria by mitochondrial division (Youle and van der Bliek, 2012). Fission also helps mitochondrial quality control by elimination of damaged mitochondria or parts of mitochondria containing damaged protein or DNA (Meyer et al., 2017; Youle and van der Bliek, 2012).
Mitochondrial fusion is mediated by three fusogenic proteins: Mfn1 and Mfn2, also called mitofusins are located on the outer mitochondrial membrane with both the C- and N-terminal regions protruding into the cytosol (Koshiba et al., 2004), while Opa1 (optic atrophy 1) is located on the inner mitochondrial membrane (Olichon et al., 2002). Mfn1 and Mfn2 are responsible for outer mitochondrial membrane fusion, whereas OPA1 mediates fusion of inner mitochondrial membranes (Scorrano, 2013). After fusion, mitochondrial fission often occurs to segregate dysfunctional mitochondrial components that go through autophagic degradation; in this way cells are able to retain healthy mitochondria (Twig et al., 2008). Fission results in two uneven mitochondrial daughter units, one of which exhibits higher capacity for subsequent fusion due to higher membrane potential and fusion protein expression, whereas the other unit has a lower membrane potential with reduced fusion protein expression which, in turn, gets degraded via autophagy (Twig et al., 2008).
Mitochondrial fission is mediated by dynamin-related protein 1 (Drp1) and its receptors, mitochondrial fission factor (MFF) (Otera and Mihara, 2011; Otera et al., 2010), as well as MiD49 and MiD51 (Palmer et al., 2011) located on the outer mitochondrial membrane. Drp1 is present in the cytoplasm and in punctate arrays on mitochondrial tubules, and includes both a GTPase domain and a GTPase effector domain. Drp1 is recruited by MFF, MiD49 and MiD51 to mitochondrial surfaces where it promotes fission by tethering of mitochondria at specific positions known as constriction sites. siRNA mediated ablation of Drp1 results in highly elongated, fused mitochondria suggesting a crucial role of Drp1 in mediating mitochondrial fission (Lee et al., 2004; Smirnova et al., 2001). Another receptor protein thought to play a role in mitochondrial fission is Fis 1 (Frank et al., 2001; Hu et al., 2017; Yu et al., 2019), however its specific mechanism of action in mammalian cells is not yet established. Endophilin B1, a protein with fatty acyl transferase, helps in maintaining mitochondrial morphology, and its genetic knockdown causes the formation of vesicles and tubules of outer mitochondrial membrane; and knockdown of both endophilin B1 and Drp1 leads to a mitochondrial phenotype identical to that of the Drp1 single knockdown, suggesting that endophilin B1 functions upstream of Drp1 in the process of mitochondrial fission (Karbowski et al., 2004). Another mitochondrial membrane protein, MTP18, is crucial for fission, and its overexpression increases mitochondrial fission whereas its depletion increases fusion in mammalian COS-7 cells (Tondera et al., 2005). Notably, overexpression of Fis1 does not induce mitochondrial fission in cells lacking MTP18.
2.2. Mitophagy
Mitophagy is the process by which damaged mitochondria are degraded by autophagy that is critical in maintaining mitochondrial quality control by relieving potential cell-damaging proteins from cells (Pickles et al., 2018; Wu et al., 2015). Mitophagy is governed by two cytosolic proteins, PTEN-induced kinase 1 (Pink1), a serine/threonine kinase and Parkin, an n E3 ubiquitin ligase, that are both localized near healthy mitochondria (Pickles et al., 2018; Wu et al., 2015). The two proteins work together to keep cells healthy by helping to clear any damaged mitochondria from the cell (Zhuang et al., 2016).
Under normal physiological conditions, Pink1 is imported into healthy mitochondrial matrix by their translocases present in the inner mitochondrial membrane where it gets cleaved. On the other hand, metabolically dysfunctional mitochondria are not capable of importing and degrading Pink1, resulting in accumulation and stabilization of Pink1 on outer mitochondrial membranes. Accumulated Pink1 then phosphorylates ubiquitin and Parkin to activate the E3 ligase activity of Parkin; activated Parkin then initiates mitochondrial degradation by the autophagosome that consequently fuses with the lysosomes. Activated Parkin also then ubiquitinates other mitophagy receptors present on the outer mitochondrial membrane such as such as NBR1, Nix, TAxBP1, NDP51, optineurin and FUNDC1 that bind to micro-tubule-associated proteins 1A/1B light chain 3B, also known as LC3, to recruit autophagosomes around the damaged mitochondria (Vives-Bauza et al., 2010).
2.3. Mitochondrial-mediated apoptosis
Mitochondria play a key role in the induction of another key regulatory process, apoptosis, in order to initiate cell death. Under normal conditions, anti-apoptotic proteins Bcl-2 and Bcl-XL are generally integrated within the outer mitochondrial membrane, but there are instances of cytosolic, endoplasmic and nuclear membrane localizations where they regulate apoptosis by blocking cytochrome c release from mitochondria (Carthy et al., 2003; Johnson et al., 2000; Kluck et al., 1997; Shimizu et al., 1999; Yang et al., 1997). Mitochondria-mediated apoptosis initiates in response to exogenous and endogenous stimuli such as DNA damage, ischemia, and oxidative stress. Under pathological conditions, cytochrome c is released from mitochondria through the actions of nuclear encoded pro-apoptotic proteins Bax and Bak (Kuwana et al., 2005; Letai et al., 2002). Once cytochrome c is released it binds with apoptotic protease activating factor–1 (Apaf-1) and ATP, which then bind to pro-caspase-9 to create a protein complex known as an apoptosome that cleaves the pro-caspase to its active form of caspase-9; which in turn activates the effector caspase-3 (Bratton et al., 2001; Zou et al., 1997). Cytochrome c release from mitochondria is a defining event during apoptosis, as cells die due to mitochondrial dysfunction even if downstream caspases are inhibited (Colell et al., 2007; Ekert et al., 2004).
2.4. Mitochondrial oxidative stress
The production of free radicals by mitochondria has been considered to play a central role in the degradation of cellular function that appears to underlie the process of aging and age related neurological disorders and CNS trauma (Bains and Hall, 2012; Fatima et al., 2015; Gandhi and Abramov, 2012; Lewen et al., 2000; Schon and Manfredi, 2003; Singh et al., 2006; Sullivan et al., 2007). Under physiological conditions, free radicals produced in mitochondria play key roles as a signaling mechanism, for example, in the regulation of ion-channel activities and in initiating cytoprotective mechanism in stressed cells. Mitochondria have their own anti-oxidant defense system that sequesters free radicals under normal conditions. Free radical production is a byproduct of ATP generation in mitochondria via the electron transport chain (ETC). Electrons escape from the ETC and reduce O2 to O2−•. Normally cells convert O2‒• to hydrogen peroxide (H2O2) utilizing both manganese superoxide dismutase, which is localized to the mitochondria, and copper-zinc superoxide dismutase found in the cytosol. H2O2 is rapidly converted to H2O via catalase and glutathione peroxidase, but has the potential to be converted to the highly reactive hydroxyl radical (OH•) via the Fenton reaction, underlying ROS neurotoxicity. OH• rapidly attacks unsaturated fatty acids in membranes causing lipid peroxidation and the production of 4-hydroxynonenal that conjugates to membrane proteins, impairing their function (Ayala et al., 2014; Bruce-Keller et al., 1998; Kruman et al., 1997; Lee et al., 2011; Mattson, 2009; McCracken et al., 2000; McGrath et al., 2001; Vaishnav et al., 2010; Yang et al., 2019). Such oxidative injury results in significant alterations in cellular function. Glutathione (GSH) is the most abundant intracellular thiol and is a component of the endogenous antioxidant defense system that protects cells from lipid peroxidation and other forms of free radical-mediated damage (Cooper and Kristal, 1997; Penugonda et al., 2005; Price et al., 2006). Glutathione reductase reduces the oxidized form of glutathione (GSSG) to GSH (Grinberg et al., 2005), which then prevents membrane damage by directly scavenging ROS, and also serves as a co-factor of glutathione peroxidase to eliminate H2O2 from the cell (Penugonda et al., 2005, Price et al., 2006). In addition, GSH prevents initiation and propagation of lipid peroxidation by modulating the regeneration of ascorbic acid (vitamin C) and α-to-copherol (vitamin E), both of which are anti-oxidants (Kurutas, 2016; Niki, 1991; Retsky et al., 1993). Given its key role as an endogenous antioxidant, GSH levels have been evaluated in CNS trauma (Pandya et al., 2014; Patel et al., 2014; Visavadiya et al., 2016).
3. Mitochondrial dysfunction in neurotrauma
3.1. Ca2+ overload and mitochondrial permeability transition
Increased understanding of the molecular and cellular mechanisms of apoptosis has led to the identification of specific therapeutic targets for CNS trauma. Necrotic damage shortly after CNS injuries seems to set up the conditions for longer-term apoptosis in a way that reflects the pattern of axonal loss that can be targeted by either blocking apoptotic triggers or by targeting early pre-mitochondrial alterations such as scavenging of ROS and blocking Ca2+ overload (see Section 4a). It is well documented that mitochondria play a critical role in development of secondary pathology following traumatic brain injury (TBI) and spinal cord injury (SCI) via their involvement in post-traumatic apoptosis, cell signaling, cell cycle regulation, Ca2+ homeostasis, and ROS production that determine the fate cells under stress (Adam-Vizi, 2005; Adam-Vizi and Chinopoulos, 2006; Beal, 1996; Coyle and Puttfarcken, 1993; Crowe et al., 1997; Dugan et al., 1995; Hall and Springer, 2004; Lee et al., 2011; Levy et al., 2003; McEwen et al., 2007; Sullivan et al., 2007; Yonutas et al., 2016). Mitochondrial dysfunction after CNS trauma is associated with glutamate-mediated prolonged Ca2+ overload that drives mitochondria to mitochondrial permeability transition (mPT) state formation of the mitochondrial permeability transition pore (mPTP) allowing substances < 1.5 kDa to pass into the cytoplasm (Halestrap and Brenner, 2003) (see Fig. 1). The mPTP formation uncouples respiration from ATP production causing ATP depletion and ultimately cell death when sufficient numbers of mitochondria undergo mPT state. The mPTP is a presumed proteinaceous entity primarily located in the inner mitochondrial membrane and spanning to outer mitochondrial membrane, essentially allowing for translocation of small molecules between the matrix and cytosol. The precise molecular composition of the mPTP is quite controversial, however several identified candidates include the voltage dependent anion channel (VDAC), the adenine nucleotide translocase (ANT), spastic paraplegia 7 (SPG7), phosphate carrier (PiC), and components of the ATP synthase (Alavian et al., 2014; Bernardi and Di Lisa, 2015; Crompton et al., 1998; Giorgio et al., 2013; Halestrap and Brenner, 2003; Halestrap and Richardson, 2015; Leung and Halestrap, 2008; Shanmughapriya et al., 2015).
Fig. 1.
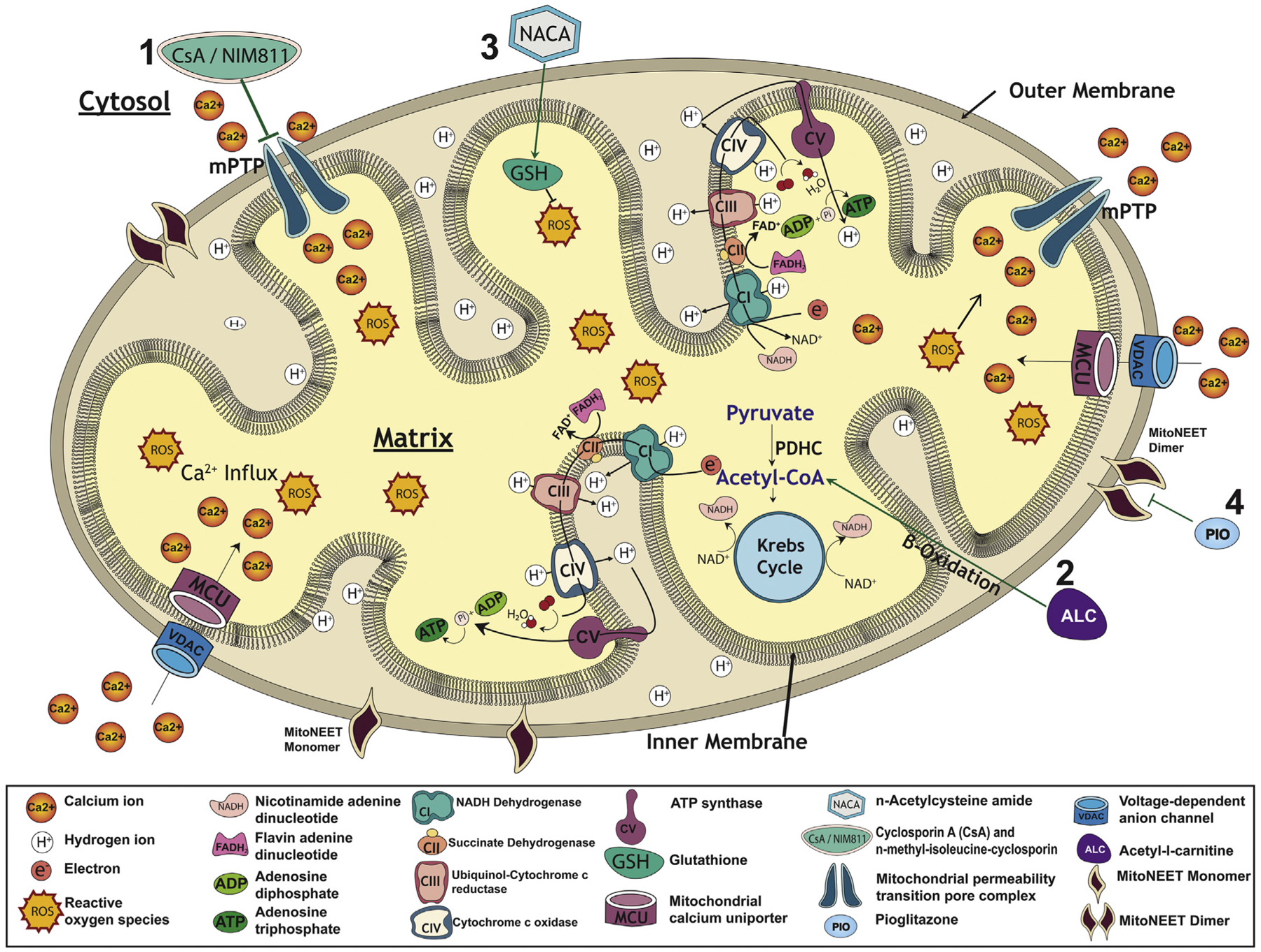
Schematic representation of compromised mitochondrial function after spinal cord injury (SCI) and how various treatment strategies target mitochondrial dysfunction specifically, as discussed in this review. The major functions of mitochondria are to produce adenosine triphosphate (ATP) via electron transport chain which consist of five different enzymatic complexes: NADH dehydrogenase (CI), succinate dehydrogenase (CII), ubiquinol-cytochrome c reductase (CIII), cytochrome c oxidase (CVI) and ATP synthase (CV). They modulate calcium (Ca2+) buffering as it enters the mitochondrial outer membrane via the voltage-dependent anion channel (VDAC) and inner membrane through the mitochondrial calcium uniporter (MCU) to be stored in the mitochondrial matrix. While carrying out their normal functions, mitochondria also produce small amounts of reactive oxygen species (ROS) which are typically eliminated by components of mitochondrial antioxidant system, such as superoxide dismutase (not shown) and glutathione (GSH). Following SCI, N-methyl-d-aspartate (NMDA)-mediated glutamate toxicity results in massive influx of Ca2+ into cells which substantially increases mitochondrial Ca2+ uptake, and beyond a certain concentration threshold it triggers the opening of the mitochondrial permeability transition pore (mPTP). Under pathological conditions, Cyclosporin A (CsA) and N-methyl-isoleucine-cyclosporin (NIM811) maintain mitochondrial function by blocking formation of mPTP (1). SCI-induced ROS affects activities of key mitochondrial enzymes, specifically decreased pyruvate dehydrogenase complex (PDHC) activities reduce the conversion of pyruvate to acetyl-CoA, which in turn leads to decreased ATP production by mitochondrial electron transport system. An alternative biofuel, acetyl-l-carnitine (ALC), can bypass PDHC by providing acetyl-CoA to Kreb’s cycle via β-oxidation of free fatty acids to maintain ATP generation (2). Alternatively, increased mitochondrial oxidative stress can be controlled by mitochondrial-targeted antioxidants such as n-acetylcysteine amide (NACA) which maintains antioxidant glutathione (GSH) levels that has neuroprotective effects (3). mitoNEET is an iron-containing integral protein present as monomer (active form) in the outer mitochondrial membrane that plays an important role in mitochondrial function and metabolism by serving as a potential redox or pH sensor. Following SCI, mitoNEET becomes dimerized and inactive due to increased oxidative stress and Ca2+ overload resulting in formation of mPTP. Pioglitazone (Pio) is thought to bind to mitoNEET and prevent its dimerization, thus maintaining mitochondrial integrity (4).
3.2. b. Targeting the mPTP with cyclosporin A
Cyclophilin D is unequivocally identified as a major component of mPTP complex which is a matrix cyclophilin that regulates pore opening, conferring sensitivity of the pore to inhibition by cyclosporin A (CsA) (Crompton et al., 1988; Halestrap and Davidson, 1990). High-conductance mPTP opening is associated with osmotic swelling, loss of inner mitochondrial membrane potential, uncoupling of oxidative phosphorylation, and metabolic collapse. Given the importance of mitochondria in cellular function, blocking the mPTP formation has been shown to be an effective therapeutic strategy for limiting cell death in CNS trauma (see Fig. 1). It was found that pre-injury treatment with CsA reduced diffuse axonal injury and Ca2+-induced cytoskeletal damage after TBI in rats (Okonkwo and Povlishock, 1999). Post-injury treatment with CsA was then reported to reduce cortical damage significantly after TBI, and the immunosuppressive properties of CsA were not responsible for the neuroprotection since the more potent immunosuppressor FK506 afforded no neuroprotection (Scheff and Sullivan, 1999). In a follow-up study, this same group reported neuroprotective efficacy of CsA via modulation of the mPTP and maintenance of mitochondrial homeostasis after TBI in rats (Sullivan et al., 1999).
4. MitoCeuticals for spinal cord injury
Over the past several decades, there have been numerous pre-clinical evaluations of potential “neuroprotective” pharmacological strategies targeting different aspects of the secondary injury process in animal models of SCI (Hall and Springer, 2004). More importantly, clinical trials have taken place based on such studies (Bracken, 1990; Bracken et al., 1992; Bracken et al., 1997), and the cumulative data indicates that oxidative damage resulting from post-traumatic disruption of Ca2+ homeostasis and the generation of ROS are arguably among the most validated secondary injury targets in SCI clinical trials (Bracken, 2001). However, the modest degree of clinical efficacy for existing pharmacotherapeutic strategies targeting ROS (Hurlbert, 2000) underlies the necessary development of pharmacological agents or alternative approaches that target more directly the origin(s) of oxidative damage in the injured spinal cord; namely the mitochondrion (Semple, 2014). Accordingly, the ensuing sections will review the evidence supporting the use of particular ‘MitoCeuticals’ to promote neuroprotection following acute traumatic SCI.
4.1. Anti-apoptotic strategies
While not directly tied to mitochondrial dysfunction, per se, various studies have used markers of apoptosis as corollaries for tissue sparing and impaired or improved functional recovery. The acute necrosis and secondary pathophysiological cascades after contusion SCI trigger long-term neuroglial apoptosis and neuronal degeneration (Crowe et al., 1997; Pearse et al., 2004), and pharmaceuticals have been used to prevent apoptotic triggering by targeting alterations upstream of mitochondrial damage, such as free radical production and neuronal Ca2+ influx (Matute et al., 2001). For instance, upstream targeting of N-methyl-d-aspartate (NMDA) receptors with the antagonist MK-801 improves motor function following SCI in association with reduced neuronal apoptosis (Wada et al., 1999). More recently, the microRNA-137 has been reported to inhibit cellular apoptosis after SCI via targeting mitogen-activated protein kinase-activated protein kinase 2 (MK2) (Gao et al., 2018), which is elevated after SCI and associated with secondary damage (Ghasemlou et al., 2010). Another intriguing approach is to trigger anti-apoptotic pathways by treadmill exercise via activation of PI3K/Akt after SCI in rats (Jung et al., 2014).
4.2. Cyclosporin A (CsA)
Given the similarities in secondary pathologies of TBI and SCI, members of the same TBI research group at the University of Kentucky cited above used a similar CsA dosage and treatment paradigm following contusion SCI in rats, but demonstrated no protective effects (Rabchevsky et al., 2001). It was later discovered that such differential effects were likely attributed to fundamental differences in brain (cortex) versus spinal cord mitochondria; comparatively the mitochondrial lipid peroxidation, in situ ROS production, mtDNA oxidation, complex I enzyme activity, and NADH-linked respiration were all significantly compromised in spinal cord mitochondria (Sullivan et al., 2004). Mitochondrial bioenergetic dysfunction was then first characterized temporally in a rat contusion model of SCI (Sullivan et al., 2007), reporting significant decline in mitochondrial respiratory capacity beginning 12 h post-injury and persisting to a least 24 h following SCI, which was correlated with increased oxidative stress markers 3-nitrotyrosine, 4-hyroxynonenal and protein carbonyls (Sullivan et al., 2007). Concurrently, colleagues found that pre-treatment with NIM811, a non-immunosuppressive derivative of CsA, before contusion SCI in rats improved the function of synaptic mitochondria isolated 24 h afterwards (McEwen et al., 2007), and that post-injury treatment with CsA increased tissue sparing and reduced cell death (McEwen et al., 2007; Ravikumar et al., 2007).
4.3. Uncouplers and alternative biofuels
The mitochondrial uncoupling agent, 2,4-dinitrophenol significantly maintained normal mitochondrial function in a similar SCI model in terms of respiratory control ratios, respiration rates and enzyme complex activities of the ETC, as well as reduced mitochondrial oxidative stress (Patel et al., 2009). These results suggested that interventions targeting mitochondrial oxidative damage and dysfunction acutely after injury may serve as a beneficial pharmacological treatment for SCI (McEwen et al., 2011). In ensuing studies, the administration of an alternative biofuel acetyl-l-carnitine (ALC) at 1 h post-SCI was found to significantly maintain mitochondrial function at 24 h after injury, and prolonged daily administration significantly increased spinal cord tissue sparing and improved hindlimb locomotor function (Patel et al., 2012; Patel et al., 2010). Importantly, it was the first documentation that SCI results in significant reduction of pyruvate dehydrogenase complex (PDHC) activities which serve to convert pyruvate to acetyl CoA. More importantly, it was found that ALC can bypass PDHC and provide acetyl CoA for the Kreb’s cycle to produce NADH and FADH2 requisite for ATP production through the mitochondrial ETC (Patel et al., 2010) (see Fig. 1). Treatment with another biofuel, β-hydroxy butyrate (BHB), a ketone body, is reported to reduce oxidative stress, improve functional recovery, and reduce neuropathic pain following SCI in mice (Kong et al., 2017; Qian et al., 2017). Moreover, implementation of a ketogenic diet that increases BHB levels attenuated oxidative stress and inflammation, as well as improved forelimb function in rodent models of cervical SCI (Lu et al., 2018; Streijger et al., 2013).
4.4. Boosting endogenous antioxidant systems
As mitochondria are known to produce ROS, the maintenance of redox in mitochondria is essential to maintain the redox-sensitive cellular processes. The sudden change in ROS production or any process affecting the capacity to synthesize reducing equivalent such as GSH or NADPH may impair redox equilibrium of cell. In acute SCI, disrupted redox equilibrium due to GSH depletion results in increased concentration of and free radical leakage which initiates oxidative damage to key proteins and lipids that potentiates the processes of secondary injury (Pisoschi and Pop, 2015). In line with promoting mitochondrial integrity, post-injury treatment with modified GSH precursor, N-acetylcysteine amide (NACA) with enhanced mitochondrial permeability, was found to significantly improve mitochondrial function at 24 h when administered in a dose-dependent manner following contusion SCI, and that continuous delivery of NACA for one week resulted in significantly improved functional neuroprotection at 6 weeks after injury (Patel et al., 2014) (see Fig. 1). It is important to note that the same NACA treatment regimen was also reported to be highly neuroprotective in a rat model of TBI by the same research group (Pandya et al., 2014).
Maintaining GSH levels is crucial for redox equilibrium (the overall balance of oxidation/reduction systems of the cell) which is essential for the maintenance of various cellular processes such as responses to ROS, protection of protein thiols, oxidation–reduction reactions. An agent with both oxidizing and reducing properties that appear on opposite sides of a half-equation constitute a redox couple, for example NAD+ + H+ + 2e− → NADH2. There are many redox couples in a cell that work together to maintain the redox environment. To be an effective buffer, a given redox couple needs to be both highly reducing and present at relatively high concentrations. NAD+/NADH and NADP+/NADPH, which are present at relatively low concentration, do not contribute significantly to the redox environment of the cell (Schafer and Buettner, 2001). On the other hand, the GSSG/2GSH couple is the most abundant redox couple in a cell that plays a major role in determining the cellular redox environment in different cellular compartments such as mitochondria and ER due to its low redox potential and high concentration (Schafer and Buettner, 2001).
Treatment with the antidiabetic drug, pioglitazone, significantly increased functional neuroprotection that was associated with remarkable maintenance of acute mitochondrial bioenergetics after traumatic SCI in mice; but was not associated with immunomodulatory effects (Patel et al., 2017). An alternative theory is that pioglitazone conferred neuroprotection via its interaction with the mitochondrial protein, mitoNEET [referred to as CDGSH iron sulfur domain 1 (CISD1)] (see Fig. 1). The mitoNEET has emerged as the target of thiazolidinedione drugs such as pioglitazone (Geldenhuys et al., 2014). It is critical to note that the mitoNEET regulates the voltage-dependent anion channel (VDAC) in a redox-dependent manner in cells, thus closing the mPTP and likely disrupting VDAC’s flow of metabolites (Lipper et al., 2019). It was recently reported that pharmacological blockade of VDAC1 oligomerization with 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS) reduces apoptosis and oligodendrocyte cell death following SCI correlated with motor functional recovery in rats (Paschon et al., 2019). Collectively, such evidence indicates that mitoNEET can be a target for specific ligands with therapeutic potential to modulate redox and afford neuroprotection after SCI, further reviewed in (Rabchevsky et al., 2017).
4.5. Regulating mitochondrial structural dynamics
Following contusion SCI in rats, mitochondrial fusion is increased in first 8 h and subsequently decreases, whereas mitochondrial fission is increased from 16 to 24 h (Cao et al., 2013; Jia et al., 2016). Both of these processes are interconnected with increased oxidative stress, ATP depletion and apoptosis after neurotrauma. Such changes in fusion/fission lead to altered mitochondrial morphologies. As fusion is reported to increase at 4–8 h post-SCI, electron microscopic assessments confirmed increased mitochondrial volume and disordered cristae, whereas processes of fission (16–24 h post-injury) were correlated with partial mitochondrial membrane rupture and vacuolization, indicating further damage to mitochondria (Jia et al., 2016). Moreover, injury-induced pathological changes such as collapsed mitochondria membrane potential, cytochrome c release, and depletion of ATP and GSH all peaked during fission (16–24 h) compared to levels during fusion (Jia et al., 2016). These results indicate that optimal therapeutic window aiming at maintenance of mitochondrial structural integrity may be less than 8 h after SCI.
Mitophagy is reported to occur within 24 h after contusion SCI in rats (Yu et al., 2013), and its inhibition by knockdown of Bcl-2/E1B-19KD-interacting protein 3 (BNIP3) improved neurological recovery and neuronal survival after injury (Yu et al., 2018). On the contrary, in vivo inhibition of miRNA-124 is neuroprotective, and this was associated with the induction of mitophagy following ischemia–reperfusion injury in rats (Liu et al., 2017). In support of this, a mouse model of spinal ischemia-reperfusion injury showed that rapamycin treatment improved locomotor function that was correlated with promoting mitophagy (Li et al., 2018). Such paradoxical findings illustrate the relatively unexplored avenues to investigate intracellular mitochondrial dynamics in relation to histopathology and functional outcome measures.
Recently, the FDA-approved beta2-adrenoreceptor agonist, formoterol, was shown to promote mitochondrial biogenesis in a severe mid-thoracic mouse contusion SCI model (Scholpa et al., 2019b). PGC-1α is a transcriptional coactivator that is a central inducer of mitochondrial biogenesis (Handschin and Spiegelman, 2006), and formoterol treatment was shown to prevent the SCI-reduced expression of PGC-1α, which was correlated with both significantly improved tissue sparing and hind-limb locomotion following SCI. These investigators also reported a significant increase in skeletal muscle mass using the same injury/treatment paradigm (Scholpa et al., 2019a). Therefore, together with modulating fusion/fission processes, promoting mitochondrial biogenesis is another pharmacological strategy to restore mitochondrial numbers in the injured spinal cord for maintaining muscle function.
5. Mitochondrial transplantation
Along with compelling evidence in the burgeoning field of ‘mitochondrial medicine’ (Armstrong, 2007; Luft, 1994) that promoting mitochondrial integrity in situ using specific pharmaceuticals fosters neuroprotection, attention has shifted to intercellular mitochondria trafficking that affords protection to cells in neurodegenerative states (Birsa et al., 2013). Regulatory mechanisms working to transport, distribute, and clear mitochondria in neurons are compromised after neurotrauma (Vanhauwaert et al., 2019), and this has fostered an emerging concept that after acute CNS injury, mitochondria released into extracellular space may be transferred among cells to support oxidative phosphorylation in recipient cells (Hayakawa et al., 2018; Hayakawa et al., 2016; Nakamura et al., 2019). A logical extension of this concept is the evolution of techniques for mitochondrial transplantation to replace damaged or dysfunctional ones with exogenous healthy mitochondria (see (Gollihue and Rabchevsky, 2017)).
5.1. Premise for mitochondrial transplantation
There is a growing body of evidence that direct or systemic mitochondrial transplantation from autologous sources is beneficial under many experimental conditions such as cardiac ischemia, ischemia-reperfusion of liver and neurodegenerative models (see (Gollihue et al., 2018b). Notably, in a pioneering clinical study, autologous mitochondrial transplantation was performed on five pediatric patients dependent upon extracorporeal membrane oxygenation (ECMO) support due to myocardial dysfunction related to ischemia and reperfusion (Emani et al., 2017). Mitochondria isolated from their own abdominal muscles were injected intracardially and all patients had notable improvement in their myocardial systolic function almost immediately afterwards; and they lived without ECMO. As the first successful clinical trial for mitochondrial transplantation, and based on similar cell death pathways as ischemia-reperfusion injury, such an approach laid the foundation for evaluation in traumatic SCI. In a recent safety and efficacy study, the same group of researchers reported that intracoronary injection of mitochondria provides for the rapid uptake and specific biodistribution of mitochondria throughout the healthy swine heart. It was safe and had no effect on coronary patency. Moreover, they have demonstrated enhanced myocardial function following ischemia-reperfusion injury (Shin et al., 2019). Notably, however, a recent viewpoint article (Bertero et al., 2018) raised several concerns regarding their findings, questioning: 1) how mitochondria could survive the high Ca2+ extracellular environment, 2) whether they remain viable and efficiently produce ATP and 3) if the mitochondria pass through cell membranes to produce sufficient ATP to boost cardiac contractile function. In direct response, the authors (McCully et al., 2020) affirmed that 1) competent cell-free mitochondria were documented in the blood containing physiological Ca2+ and Na+ concentrations (Al Amir Dache et al., 2020); 2) several studies have demonstrated functional integration of exogenous mitochondria into recipient cells in media containing 1.8 mMol Ca2+, representing physiological concentrations (Cowan et al., 2017; Katrangi et al., 2007; Kesner et al., 2016; King and Attardi, 1988; Pacak et al., 2015); 3) mitochondria transplanted by either direct or intracoronary injection increased total tissue ATP content, oxygen uptake, contractile function, and upregulated proteins associated with mitochondrial function (Masuzawa et al., 2013; Shin et al., 2019); 4) transplanted mitochondria produced no systemic inflammatory responses (Emani et al., 2017; Masuzawa et al., 2013; Ramirez-Barbieri et al., 2019; Shin et al., 2019); all cited evidence further supporting the therapeutic potential of mitochondrial transplantation in variety of injuries and diseases.
5.2. Mitochondrial transplantation as an emerging therapeutic for spinal cord injury
Various pharmacological and cell-based therapies are being tested in SCI clinical trials despite scant demonstration of consistent efficacy in animal SCI models. Moreover, if benefits are indeed associated with transplanted cells, it is often uncertain by what mechanism(s) the grafted cells provide protection, notably since they typically do not survive long-term to manifest improvements. Alternatively, it has been posited that transplanted cells may render their effects by “donating” cellular organelles, such as mitochondria, to compromised host cells. Such a novel approach was developed recently to transplant exogenous, well-coupled mitochondria into tissues surrounding contusion SCI sites (Gollihue and Rabchevsky, 2017). The hypothesis was that supplementation of endogenous antioxidant systems via mitochondrial transplantation will increase overall Ca2+ buffering capacity and improve energy production. Cellular incorporation of intraspinal injected transgenic-labeled (tGFP) mitochondria was evident at 24 h, 48 h and 7 days post-injection, by which time the injection boluses were decreased significantly in volume and rostral-caudal spread (Gollihue et al., 2018a). Transplanted mitochondria were co-localized within various cell types including microglia/brain macrophages, endothelial cells, pericytes, astrocytes, and oligodendrocytes, with most abundant internalization in pericytes and brain macrophages and no evidence of up take in neurons (Gollihue et al., 2018a). However, while mitochondrial transplantation into contused rat spinal cords significantly maintained acute bioenergetics, such early preservation did not manifest long-term functional recovery or tissue sparing (Gollihue et al., 2018a). It remains equivocal whether direct focal circumferential injections of mitochondria, alone, around contusion sites is adequate to maintain sufficient bioenergetic integrity in the injured spinal cord to spare compromised tissues.
Many questions still remain before mitochondrial transplantation will garner consideration equal to stem cell therapies for SCI, despite the plethora of negative findings over the past decades, including human clinical trials (Willison et al., 2020). We do not yet understand how transplanted mitochondria maintain OCR, whether they must be internalized into cells in order to improve OCR measures ex vivo, or whether they can function as extracellular mitochondrial vesicles/particles (Hayakawa et al., 2018). Future interrogations to manifest functional integration of transplanted mitochondrial must establish how to maximize their integrity extracellularly upon delivery. This may involve co-administration of ALC/NACA to maintain both host and grafted mitochondria and/or rather delivering mitochondria within specialized supportive hydrogels that controlled their release. As we learn more about the mechanism(s) by which mitochondria enter cells (see Gollihue and Rabchevsky, 2017), it will be intriguing to see whether neurons, in particular, can be selectively targeted, and to determine optimal therapeutic time windows for mitochondrial transplantation after injury.
6. Summary
This review discusses the pivotal roles that mitochondria play in maintaining bioenergetic integrity under normal and injured states of the CNS, and that mitochondrial dysfunction is a validated target to maintain cellular homeostasis and consequent functional neuroprotection after both TBI and SCI. Importantly, further development of compounds that promote bioenergetic integrity for extended periods to modulate mitochondria/redox states may be combined with injectable hydrogel systems for intrathecal delivery of mitochondria for broader distribution at sites of injury in a more clinically relevant manner. Such novel complementary therapeutic approaches may have wide applications for CNS trauma and/or neurodegenerative disorders in which mitochondrial dysfunction is unequivocal.
Acknowledgement
Financial Support: An endowment from the Spinal Cord & Brain Injury Research Center, University of Kentucky (AGR). This sponsor did not contribute to the preparation of this manuscript.
References
- Adam-Vizi V, 2005. Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non-electron transport chain sources. Antioxid. Redox Signal 7, 1140–1149. [DOI] [PubMed] [Google Scholar]
- Adam-Vizi V, Chinopoulos C, 2006. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci 27, 639–645. [DOI] [PubMed] [Google Scholar]
- Al Amir Dache Z, et al. , 2020. Blood contains circulating cell-free respiratory competent mitochondria. FASEB J. 34, 3616–3630. [DOI] [PubMed] [Google Scholar]
- Alavian KN, et al. , 2014. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. U. S. A 111, 10580–10585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong JS, 2007. Mitochondrial medicine: pharmacological targeting of mitochondria in disease. Br. J. Pharmacol 151, 1154–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala A, Munoz MF, Arguelles S, 2014. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med. Cell. Longev 2014, 360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bains M, Hall ED, 2012. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim. Biophys. Acta 1822, 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF, 1996. Mitochondria, free radicals, and neurodegeneration. Curr. Opin. Neurobiol 6, 661–666. [DOI] [PubMed] [Google Scholar]
- Beckervordersandforth R, et al. , 2017. Role of mitochondrial metabolism in the control of early lineage progression and aging phenotypes in adult hippocampal neurogenesis. Neuron 93, 1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, Di Lisa F, 2015. The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol 78, 100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertero E, Maack C, O’Rourke B, 2018. Mitochondrial transplantation in humans: “magical” cure or cause for concern? J. Clin. Invest 128, 5191–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsa N, Norkett R, Higgs N, Lopez-Domenech G, Kittler JT, 2013. Mitochondrial trafficking in neurons and the role of the Miro family of GTPase proteins. Biochem. Soc. Trans 41, 1525–1531. [DOI] [PubMed] [Google Scholar]
- Bracken MB, 1990. Methylprednisolone in the management of acute spinal cord injuries. Med. J. Aust 153, 368. [PubMed] [Google Scholar]
- Bracken MB, 2001. Methylprednisolone and acute spinal cord injury: an update of the randomized evidence. Spine 26, S47–S54. [DOI] [PubMed] [Google Scholar]
- Bracken MB, et al. , 1992. Methylprednisolone or naloxone treatment after acute spinal cord injury: 1-year follow-up data. Results of the second National Acute Spinal Cord Injury Study. J. Neurosurg 76, 23–31. [DOI] [PubMed] [Google Scholar]
- Bracken MB, et al. , 1997. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury. Results of the Third National Acute Spinal Cord Injury Randomized Controlled Trial. National Acute Spinal Cord Injury Study. Jama 277, 1597–1604. [PubMed] [Google Scholar]
- Bratton SB, Walker G, Srinivasula SM, Sun XM, Butterworth M, Alnemri ES, Cohen GM, 2001. Recruitment, activation and retention of caspases-9 and −3 by Apaf-1 apoptosome and associated XIAP complexes. EMBO J. 20, 998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Li YJ, Lovell MA, Kraemer PJ, Gary DS, Brown RR, Markesbery WR, Mattson MP, 1998. 4-Hydroxynonenal, a product of lipid peroxidation, damages cholinergic neurons and impairs visuospatial memory in rats. J. Neuropathol. Exp. Neurol 57, 257–267. [DOI] [PubMed] [Google Scholar]
- Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH, 2011. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet 20, 4515–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Lv G, Wang YS, Fan ZK, Bi YL, Zhao L, Guo ZP, 2013. Mitochondrial fusion and fission after spinal sacord injury in rats. Brain Res. 1522, 59–66. [DOI] [PubMed] [Google Scholar]
- Carthy CM, et al. , 2003. Bcl-2 and Bcl-xL overexpression inhibits cytochrome c release, activation of multiple caspases, and virus release following coxsackievirus B3 infection. Virology 313, 147–157. [DOI] [PubMed] [Google Scholar]
- Chakrabarti R, Ji WK, Stan RV, de Juan Sanz J., Ryan TA, Higgs HN, 2018. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J. Cell Biol 217, 251–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colell A, et al. , 2007. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell 129, 983–997. [DOI] [PubMed] [Google Scholar]
- Cooper AJ, Kristal BS, 1997. Multiple roles of glutathione in the central nervous system. Biol. Chem 378, 793–802. [PubMed] [Google Scholar]
- Cowan DB, Yao R, Thedsanamoorthy JK, Zurakowski D, Del Nido PJ, McCully JD, 2017. Transit and integration of extracellular mitochondria in human heart cells. Sci. Rep 7, 17450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P, 1993. Oxidative stress, glutamate, and neurodegenerative disorders. Science 262, 689–695. [DOI] [PubMed] [Google Scholar]
- Crompton M, Ellinger H, Costi A, 1988. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J 255, 357–360. [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Virji S, Ward JM, 1998. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem 258, 729–735. [DOI] [PubMed] [Google Scholar]
- Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS, 1997. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat. Med 3, 73–76. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Sensi SL, Canzoniero LM, Handran SD, Rothman SM, Lin TS, Goldberg MP, Choi DW, 1995. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-d-aspartate. J. Neurosci 15, 6377–6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekert PG, Read SH, Silke J, Marsden VS, Kaufmann H, Hawkins CJ, Gerl R, Kumar S, Vaux DL, 2004. Apaf-1 and caspase-9 accelerate apoptosis, but do not determine whether factor-deprived or drug-treated cells die. J. Cell Biol 165, 835–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emani SM, Piekarski BL, Harrild D, Del Nido PJ, McCully JD, 2017. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg 154, 286–289. [DOI] [PubMed] [Google Scholar]
- Fatima G, Sharma VP, Das SK, Mahdi AA, 2015. Oxidative stress and antioxidative parameters in patients with spinal cord injury: implications in the pathogenesis of disease. Spinal Cord 53, 3–6. [DOI] [PubMed] [Google Scholar]
- Fernandez-Fernandez S, Almeida A, Bolanos JP, 2012. Antioxidant and bioenergetic coupling between neurons and astrocytes. Biochem. J 443, 3–11. [DOI] [PubMed] [Google Scholar]
- Fischer MT, Sharma R, Lim JL, Haider L, Frischer JM, Drexhage J, Mahad D, Bradl M, van Horssen J, Lassmann H, 2012. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 135, 886–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ, 2001. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 1, 515–525. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK, 2011. ER tubules mark sites of mitochondrial division. Science 334, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S, Abramov AY, 2012. Mechanism of oxidative stress in neurodegeneration. Oxidative Med. Cell. Longev 2012, 428010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Dai C, Feng Z, Zhang L, Zhang Z, 2018. MiR-137 inhibited inflammatory response and apoptosis after spinal cord injury via targeting of MK2. J. Cell. Biochem 119, 3280–3292. [DOI] [PubMed] [Google Scholar]
- Geldenhuys WJ, Leeper TC, Carroll RT, 2014. mitoNEET as a novel drug target for mitochondrial dysfunction. Drug Discov. Today 19, 1601–1606. [DOI] [PubMed] [Google Scholar]
- Ghasemlou N, Lopez-Vales R, Lachance C, Thuraisingam T, Gaestel M, Radzioch D, David S, 2010. Mitogen-activated protein kinase-activated protein kinase 2 (MK2) contributes to secondary damage after spinal cord injury. J. Neurosci 30, 13750–13759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio V, et al. , 2013. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. U. S. A 110, 5887–5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollihue JL, Rabchevsky AG, 2017. Prospects for therapeutic mitochondrial transplantation. Mitochondrion 35, 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollihue JL, Patel SP, Eldahan KC, Cox DH, Donahue RR, Taylor BK, Sullivan PG, Rabchevsky AG, 2018a. Effects of mitochondrial transplantation on bioenergetics, cellular incorporation, and functional recovery after spinal cord injury. J. Neurotrauma 35, 1800–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollihue JL, Patel SP, Rabchevsky AG, 2018b. Mitochondrial transplantation strategies as potential therapeutics for central nervous system trauma. Neural Regen. Res 13, 194–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg L, Fibach E, Amer J, Atlas D, 2005. N-acetylcysteine amide, a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free Radic. Biol. Med 38, 136–145. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Brenner C, 2003. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem 10, 1507–1525. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Davidson AM, 1990. Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem. J 268, 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Richardson AP, 2015. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell. Cardiol 78, 129–141. [DOI] [PubMed] [Google Scholar]
- Hall ED, Springer JE, 2004. Neuroprotection and acute spinal cord injury: a reappraisal. NeuroRx 1, 80–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M, et al. , 2013. Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393. [DOI] [PubMed] [Google Scholar]
- Handschin C, Spiegelman BM, 2006. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev 27, 728–735. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH, 2016. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535, 551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Bruzzese M, Chou SH, Ning M, Ji X, Lo EH, 2018. Extracellular mitochondria for therapy and diagnosis in acute central nervous system injury. JAMA Neurol. 75, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ, 2005. Mitochondria and neurotransmission: evacuating the synapse. Neuron 47, 331–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstrom KM, Finkel T, 2014. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol 15, 411–421. [DOI] [PubMed] [Google Scholar]
- Hoppins S, Lackner L, Nunnari J, 2007. The machines that divide and fuse mitochondria. Annu. Rev. Biochem 76, 751–780. [DOI] [PubMed] [Google Scholar]
- Hu C, Huang Y, Li L, 2017. Drp1-dependent mitochondrial fission plays critical roles in physiological and pathological progresses in mammals. Int. J. Mol. Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlbert RJ, 2000. Methylprednisolone for acute spinal cord injury: an inappropriate standard of care. J. Neurosurg 93, 1–7. [DOI] [PubMed] [Google Scholar]
- Jia ZQ, Li G, Zhang ZY, Li HT, Wang JQ, Fan ZK, Lv G, 2016. Time representation of mitochondrial morphology and function after acute spinal cord injury. Neural Regen. Res 11, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BW, Cepero E, Boise LH, 2000. Bcl-xL inhibits cytochrome c release but not mitochondrial depolarization during the activation of multiple death pathways by tumor necrosis factor-alpha. J. Biol. Chem 275, 31546–31553. [DOI] [PubMed] [Google Scholar]
- Jung SY, Kim DY, Yune TY, Shin DH, Baek SB, Kim CJ, 2014. Treadmill exercise reduces spinal cord injury-induced apoptosis by activating the PI3K/Akt pathway in rats. Exp. Ther. Med 7, 587–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Jeong SY, Youle RJ, 2004. Endophilin B1 is required for the maintenance of mitochondrial morphology. J. Cell Biol 166, 1027–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katrangi E, D’Souza G, Boddapati SV, Kulawiec M, Singh KK, Bigger B, Weissig V, 2007. Xenogenic transfer of isolated murine mitochondria into human rho0 cells can improve respiratory function. Rejuvenation Res. 10, 561–570. [DOI] [PubMed] [Google Scholar]
- Kesner EE, Saada-Reich A, Lorberboum-Galski H, 2016. Characteristics of mitochondrial transformation into human cells. Sci. Rep 6, 26057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, Lee KY, Lu Y, Wang J, Cui L, Kim SJ, Chung JM, Chung K, 2011. Mitochondrial Ca(2+) uptake is essential for synaptic plasticity in pain. J. Neurosci 31, 12982–12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MP, Attardi G, 1988. Injection of mitochondria into human cells leads to a rapid replacement of the endogenous mitochondrial DNA. Cell 52, 811–819. [DOI] [PubMed] [Google Scholar]
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD, 1997. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 275, 1132–1136. [DOI] [PubMed] [Google Scholar]
- Kong G, Huang Z, Ji W, Wang X, Liu J, Wu X, Huang Z, Li R, Zhu Q, 2017. The ketone metabolite beta-hydroxybutyrate attenuates oxidative stress in spinal cord injury by suppression of class I histone deacetylases. J. Neurotrauma 34, 2645–2655. [DOI] [PubMed] [Google Scholar]
- Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC, 2004. Structural basis of mitochondrial tethering by mitofusin complexes. Science 305, 858–862. [DOI] [PubMed] [Google Scholar]
- Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP, 1997. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J. Neurosci 17, 5089–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurutas EB, 2016. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: current state. Nutr. J 15, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD, 2005. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 17, 525–535. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ, 2004. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell 15, 5001–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kosaras B, Del Signore SJ, Cormier K, McKee A, Ratan RR, Kowall NW, Ryu H, 2011. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol. 121, 487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ, 2002. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2, 183–192. [DOI] [PubMed] [Google Scholar]
- Leung AW, Halestrap AP, 2008. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim. Biophys. Acta 1777, 946–952. [DOI] [PubMed] [Google Scholar]
- Levy M, Faas GC, Saggau P, Craigen WJ, Sweatt JD, 2003. Mitochondrial regulation of synaptic plasticity in the hippocampus. J. Biol. Chem 278, 17727–17734. [DOI] [PubMed] [Google Scholar]
- Lewen A, Matz P, Chan PH, 2000. Free radical pathways in CNS injury. J. Neurotrauma 17, 871–890. [DOI] [PubMed] [Google Scholar]
- Lewis SC, Uchiyama LF, Nunnari J, 2016. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 353, aaf5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Gao S, Kang Z, Zhang M, Zhao X, Zhai Y, Huang J, Yang GY, Sun W, Wang J, 2018. Rapamycin enhances mitophagy and attenuates apoptosis after spinal ischemia-reperfusion injury. Front. Neurosci 12, 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipper CH, Stofleth JT, Bai F, Sohn YS, Roy S, Mittler R, Nechushtai R, Onuchic JN, Jennings PA, 2019. Redox-dependent gating of VDAC by mitoNEET. Proc. Natl. Acad. Sci. U. S. A 116, 19924–19929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, et al. , 2016. ATG3-dependent autophagy mediates mitochondrial homeostasis in pluripotency acquirement and maintenance. Autophagy 12, 2000–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Yan L, Jiang X, Yu Y, Liu H, Gu T, Shi E, 2017. Acquired inhibition of microRNA-124 protects against spinal cord ischemia-reperfusion injury partially through a mitophagy-dependent pathway. J. Thorac. Cardiovasc. Surg 154, 1498–1508. [DOI] [PubMed] [Google Scholar]
- Lu Y, Yang YY, Zhou MW, Liu N, Xing HY, Liu XX, Li F, 2018. Ketogenic diet attenuates oxidative stress and inflammation after spinal cord injury by activating Nrf2 and suppressing the NF-kappaB signaling pathways. Neurosci. Lett 683, 13–18. [DOI] [PubMed] [Google Scholar]
- Ludwig PE, Thankam FG, Patil AA, Chamczuk AJ, Agrawal DK, 2018. Brain injury and neural stem cells. Neural Regen. Res 13, 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luft R, 1994. The development of mitochondrial medicine. Proc. Natl. Acad. Sci. U. S. A 91, 8731–8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzetti E, Csiszar A, Dutta D, Balagopal G, Calvani R, Leeuwenburgh C, 2013. Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: from mechanisms to therapeutics. Am. J. Physiol. Heart Circ. Physiol 305, H459–H476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuzawa A, et al. , 2013. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol 304, H966–H982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, 2009. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp. Gerontol 44, 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute C, Alberdi E, Domercq M, Perez-Cerda F, Perez-Samartin A, Sanchez-Gomez MV, 2001. The link between excitotoxic oligodendroglial death and demyelinating diseases. Trends Neurosci. 24, 224–230. [DOI] [PubMed] [Google Scholar]
- McCracken E, Valeriani V, Simpson C, Jover T, McCulloch J, Dewar D, 2000. The lipid peroxidation by-product 4-hydroxynonenal is toxic to axons and oligodendrocytes. J. Cereb. Blood Flow Metab 20, 1529–1536. [DOI] [PubMed] [Google Scholar]
- McCully JD, Emani SM, Del Nido PJ, 2020. Letter by McCully et al regarding article, “Mitochondria do not survive calcium overload”. Circ. Res 126, e56–e57. [DOI] [PubMed] [Google Scholar]
- McEwen ML, Sullivan PG, Springer JE, 2007. Pretreatment with the cyclosporin derivative, NIM811, improves the function of synaptic mitochondria following spinal cord contusion in rats. J. Neurotrauma 24, 613–624. [DOI] [PubMed] [Google Scholar]
- McEwen ML, Sullivan PG, Rabchevsky AG, Springer JE, 2011. Targeting mitochondrial function for the treatment of acute spinal cord injury. Neurotherapeutics 8, 168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath LT, McGleenon BM, Brennan S, McColl D, Mc IS, Passmore AP, 2001. Increased oxidative stress in Alzheimer’s disease as assessed with 4-hydroxynonenal but not malondialdehyde. QJM 94, 485–490. [DOI] [PubMed] [Google Scholar]
- Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, Rodrigues MA, Gomez MV, Nathanson MH, Leite MF, 2005. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J. Biol. Chem 280, 40892–40900. [DOI] [PubMed] [Google Scholar]
- Meyer JN, Leuthner TC, Luz AL, 2017. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 391, 42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Lu K, Reichert AS, 2015. Mitophagy and mitochondrial dynamics in Saccharomyces cerevisiae. Biochim. Biophys. Acta 1853, 2766–2774. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Park JH, Hayakawa K, 2019. Therapeutic use of extracellular mitochondria in CNS injury and disease. Exp. Neurol 324, 113114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niki E, 1991. Action of ascorbic acid as a scavenger of active and stable oxygen radicals. Am. J. Clin. Nutr 54, 1119S–1124S. [DOI] [PubMed] [Google Scholar]
- Okonkwo DO, Povlishock JT, 1999. An intrathecal bolus of cyclosporin A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J. Cereb. Blood Flow Metab 19, 443–451. [DOI] [PubMed] [Google Scholar]
- Olichon A, et al. , 2002. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 523, 171–176. [DOI] [PubMed] [Google Scholar]
- Otera H, Mihara K, 2011. Discovery of the membrane receptor for mitochondrial fission GTPase Drp1. Small GTPases 2, 167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K, 2010. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol 191, 1141–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottolini D, Cali T, Szabo I, Brini M, 2017. Alpha-synuclein at the intracellular and the extracellular side: functional and dysfunctional implications. Biol. Chem 398, 77–100. [DOI] [PubMed] [Google Scholar]
- Pacak CA, Preble JM, Kondo H, Seibel P, Levitsky S, Del Nido PJ, Cowan DB, McCully JD, 2015. Actin-dependent mitochondrial internalization in cardiomyocytes: evidence for rescue of mitochondrial function. Biol. Open 4, 622–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT, 2011. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 12, 565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya JD, Readnower RD, Patel SP, Yonutas HM, Pauly JR, Goldstein GA, Rabchevsky AG, Sullivan PG, 2014. N-acetylcysteine amide confers neuroprotection, improves bioenergetics and behavioral outcome following TBI. Exp. Neurol 257, 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschon V, Morena BC, Correia FF, Beltrame GR, Dos Santos GB, Cristante AF, Kihara AH, 2019. VDAC1 is essential for neurite maintenance and the inhibition of its oligomerization protects spinal cord from demyelination and facilitates locomotor function recovery after spinal cord injury. Sci. Rep 9, 14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Sullivan PG, Pandya JD, Rabchevsky AG, 2009. Differential effects of the mitochondrial uncoupling agent, 2,4-dinitrophenol, or the nitroxide antioxidant, Tempol, on synaptic or nonsynaptic mitochondria after spinal cord injury. J. Neurosci. Res 87, 130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Sullivan PG, Lyttle TS, Rabchevsky AG, 2010. Acetyl-l-carnitine ameliorates mitochondrial dysfunction following contusion spinal cord injury. J. Neurochem 114, 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Sullivan PG, Lyttle TS, Magnuson DS, Rabchevsky AG, 2012. Acetyl-l-carnitine treatment following spinal cord injury improves mitochondrial function correlated with remarkable tissue sparing and functional recovery. Neuroscience 210, 296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Sullivan PG, Pandya JD, Goldstein GA, VanRooyen JL, Yonutas HM, Eldahan KC, Morehouse J, Magnuson DS, Rabchevsky AG, 2014. N-acetylcysteine amide preserves mitochondrial bioenergetics and improves functional recovery following spinal trauma. Exp. Neurol 257, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Cox DH, Gollihue JL, Bailey WM, Geldenhuys WJ, Gensel JC, Sullivan PG, Rabchevsky AG, 2017. Pioglitazone treatment following spinal cord injury maintains acute mitochondrial integrity and increases chronic tissue sparing and functional recovery. Exp. Neurol 293, 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse DD, Pereira FC, Marcillo AE, Bates ML, Berrocal YA, Filbin MT, Bunge MB, 2004. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat. Med 10, 610–616. [DOI] [PubMed] [Google Scholar]
- Penugonda S, Mare S, Goldstein G, Banks WA, Ercal N, 2005. Effects of N-acetylcysteine amide (NACA), a novel thiol antioxidant against glutamate-induced cytotoxicity in neuronal cell line PC12. Brain Res. 1056, 132–138. [DOI] [PubMed] [Google Scholar]
- Pickles S, Vigie P, Youle RJ, 2018. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol 28, R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisoschi AM, Pop A, 2015. The role of antioxidants in the chemistry of oxidative stress: a review. Eur. J. Med. Chem 97, 55–74. [DOI] [PubMed] [Google Scholar]
- Price TO, Uras F, Banks WA, Ercal N, 2006. A novel antioxidant N-acetylcysteine amide prevents gp120- and Tat-induced oxidative stress in brain endothelial cells. Exp. Neurol 201, 193–202. [DOI] [PubMed] [Google Scholar]
- Qian J, Zhu W, Lu M, Ni B, Yang J, 2017. D-beta-hydroxybutyrate promotes functional recovery and relieves pain hypersensitivity in mice with spinal cord injury. Br. J. Pharmacol 174, 1961–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabchevsky AG, Fugaccia I, Sullivan PG, Scheff SW, 2001. Cyclosporin A treatment following spinal cord injury to the rat: behavioral effects and stereological assessment of tissue sparing. J. Neurotrauma 18, 513–522. [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG, Patel SP, Sullivan PG, 2017. Targeting mitoNEET with pioglitazone for therapeutic neuroprotection after spinal cord injury. Neural Regen. Res 12, 1807–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Barbieri G, et al. , 2019. Alloreactivity and allorecognition of syngeneic and allogeneic mitochondria. Mitochondrion 46, 103–115. [DOI] [PubMed] [Google Scholar]
- Ravikumar R, McEwen ML, Springer JE, 2007. Post-treatment with the cyclosporin derivative, NIM811, reduced indices of cell death and increased the volume of spared tissue in the acute period following spinal cord contusion. J. Neurotrauma 24, 1618–1630. [DOI] [PubMed] [Google Scholar]
- Retsky KL, Freeman MW, Frei B, 1993. Ascorbic acid oxidation product(s) protect human low density lipoprotein against atherogenic modification. Anti-rather than prooxidant activity of vitamin C in the presence of transition metal ions. J. Biol. Chem 268, 1304–1309. [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T, 1998. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766. [DOI] [PubMed] [Google Scholar]
- Schafer FQ, Buettner GR, 2001. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med 30, 1191–1212. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Sullivan PG, 1999. Cyclosporin A significantly ameliorates cortical damage following experimental traumatic brain injury in rodents. J. Neurotrauma 16, 783–792. [DOI] [PubMed] [Google Scholar]
- Scholpa NE, Simmons EC, Tilley DG, Schnellmann RG, 2019a. beta2-adrenergic receptor-mediated mitochondrial biogenesis improves skeletal muscle recovery following spinal cord injury. Exp. Neurol 322, 113064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholpa NE, Williams H, Wang W, Corum D, Narang A, Tomlinson S, Sullivan PG, Rabchevsky AG, Schnellmann RG, 2019b. Pharmacological stimulation of mitochondrial biogenesis using the food and drug administration-approved beta2-adrenoreceptor agonist formoterol for the treatment of spinal cord injury. J. Neurotrauma 36, 962–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, Manfredi G, 2003. Neuronal degeneration and mitochondrial dysfunction. J. Clin. Invest 111, 303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorrano L, 2013. Keeping mitochondria in shape: a matter of life and death. Eur. J. Clin. Investig 43, 886–893. [DOI] [PubMed] [Google Scholar]
- Semple BD, 2014. Early preservation of mitochondrial bioenergetics supports both structural and functional recovery after neurotrauma. Exp. Neurol 261, 291–297. [DOI] [PubMed] [Google Scholar]
- Shanmughapriya S, et al. , 2015. SPG7 is an essential and conserved component of the mitochondrial permeability transition pore. Mol. Cell 60, 47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Narita M, Tsujimoto Y, 1999. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399, 483–487. [DOI] [PubMed] [Google Scholar]
- Shin B, et al. , 2019. A novel biological strategy for myocardial protection by intracoronary delivery of mitochondria: safety and efficacy. JACC Basic Transl. Sci 4, 871–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED, 2006. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J. Cereb. Blood Flow Metab 26, 1407–1418. [DOI] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM, 2001. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 12, 2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkov AA, Chinopoulos C, Fiskum G, 2004. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium 36, 257–264. [DOI] [PubMed] [Google Scholar]
- Streijger F, et al. , 2013. Ketogenic diet improves forelimb motor function after spinal cord injury in rodents. PLoS One 8, e78765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Rivero JM, Villanueva-Paz M, de la Cruz-Ojeda P, de la Mata M, Cotan D, Oropesa-Avila M, de Lavera I, Alvarez-Cordoba M, Luzon-Hidalgo R, Sanchez-Alcazar JA, 2016. Mitochondrial dynamics in mitochondrial diseases. Diseases 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Thompson MB, Scheff SW, 1999. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp. Neurol 160, 226–234. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Keller JN, Lovell M, Sodhi A, Hart RP, Scheff SW, 2004. Intrinsic differences in brain and spinal cord mitochondria: implication for therapeutic interventions. J. Comp. Neurol 474, 524–534. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Krishnamurthy S, Patel SP, Pandya JD, Rabchevsky AG, 2007. Temporal characterization of mitochondrial bioenergetics after spinal cord injury. J. Neurotrauma 24, 991–999. [DOI] [PubMed] [Google Scholar]
- Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R, 2006. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol 175, 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorova V, Blokland A, 2017. Mitochondria and synaptic plasticity in the mature and aging nervous system. Curr. Neuropharmacol 15, 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tondera D, Czauderna F, Paulick K, Schwarzer R, Kaufmann J, Santel A, 2005. The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J. Cell Sci 118, 3049–3059. [DOI] [PubMed] [Google Scholar]
- Twig G, et al. , 2008. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnav RA, Singh IN, Miller DM, Hall ED, 2010. Lipid peroxidation-derived reactive aldehydes directly and differentially impair spinal cord and brain mitochondrial function. J. Neurotrauma 27, 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Blerkom J, 2009. Mitochondria in early mammalian development. Semin. Cell Dev. Biol 20, 354–364. [DOI] [PubMed] [Google Scholar]
- Vance JE, 1990. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem 265, 7248–7256. [PubMed] [Google Scholar]
- Vance JE, 2014. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim. Biophys. Acta 1841, 595–609. [DOI] [PubMed] [Google Scholar]
- Vanhauwaert R, Bharat V, Wang X, 2019. Surveillance and transportation of mitochondria in neurons. Curr. Opin. Neurobiol 57, 87–93. [DOI] [PubMed] [Google Scholar]
- Visavadiya NP, Patel SP, VanRooyen JL, Sullivan PG, Rabchevsky AG, 2016. Cellular and subcellular oxidative stress parameters following severe spinal cord injury. Redox Biol. 8, 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, et al. , 2010. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U. S. A 107, 378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos M, Lauwers E, Verstreken P, 2010. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front Synaptic Neurosci. 2, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada S, Yone K, Ishidou Y, Nagamine T, Nakahara S, Niiyama T, Sakou T, 1999. Apoptosis following spinal cord injury in rats and preventative effect of N-methyl-d-aspartate receptor antagonist. J. Neurosurg 91, 98–104. [DOI] [PubMed] [Google Scholar]
- Willison AG, Smith S, Davies BM, Kotter MRN, Barnett SC, 2020. A scoping review of trials for cell-based therapies in human spinal cord injury. Spinal Cord. 10.1038/s41393-020-0455-1. (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- Witte ME, Mahad DJ, Lassmann H, van Horssen J, 2014. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol. Med 20, 179–187. [DOI] [PubMed] [Google Scholar]
- Wu W, Xu H, Wang Z, Mao Y, Yuan L, Luo W, Cui Z, Cui T, Wang XL, Shen YH, 2015. PINK1-Parkin-mediated mitophagy protects mitochondrial integrity and prevents metabolic stress-induced endothelial injury. PLoS One 10, e0132499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X, 1997. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275, 1129–1132. [DOI] [PubMed] [Google Scholar]
- Yang B, Fritsche KL, Beversdorf DQ, Gu Z, Lee JC, Folk WR, Greenlief CM, Sun GY, 2019. Yin-Yang mechanisms regulating lipid peroxidation of docosahexaenoic acid and arachidonic acid in the central nervous system. Front. Neurol 10, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonutas HM, Vekaria HJ, Sullivan PG, 2016. Mitochondrial specific therapeutic targets following brain injury. Brain Res. 1640, 77–93. [DOI] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM, 2012. Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Li M, Ni B, Kong J, Zhang Z, 2013. Induction of neuronal mitophagy in acute spinal cord injury in rats. Neurotox. Res 24, 512–522. [DOI] [PubMed] [Google Scholar]
- Yu D, Li M, Nie P, Ni B, Zhang Z, Zhou Y, 2018. Bcl-2/E1B-19KD-interacting protein 3/light chain 3 interaction induces mitophagy in spinal cord injury in rats both in vivo and in vitro. J. Neurotrauma 35, 2183–2194. [DOI] [PubMed] [Google Scholar]
- Yu R, Jin SB, Lendahl U, Nister M, Zhao J, 2019. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang N, Li L, Chen S, Wang T, 2016. PINK1-dependent phosphorylation of PINK1 and Parkin is essential for mitochondrial quality control. Cell Death Dis. 7, e2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Henzel WJ, Liu X, Lutschg A, Wang X, 1997. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90, 405–413. [DOI] [PubMed] [Google Scholar]