

Abstract
Targeting oxidative phosphorylation (OXPHOS) complexes is an emerging strategy to disrupt metabolism of select cancer subtypes and to overcome resistance to targeted therapies. Here, we describe our lead optimization campaign on a series of benzene-1,4-disulfonamides as novel OXPHOS Complex I inhibitors. This effort led to the discovery of compound 23 (DX3–213B) as one of the most potent Complex I inhibitor reported to date. DX3–213B disrupts adenosine triphosphate (ATP) generation, inhibits Complex I function, and results in growth inhibition of pancreatic cancer cells in low nanomolar range. Importantly, the oral administration of DX3–213B resulted in significant in vivo efficacy in a pancreatic cancer syngeneic model without obvious toxicity. Our data clearly demonstrate that OXPHOS inhibition can be a safe and efficacious strategy to treat pancreatic cancer.
Graphical Abstract
INTRODUCTION
Oxidative phosphorylation (OXPHOS) is essential for cell energy metabolism, ATP synthesis, reactive oxygen species (ROS) generation, and biomass production.1 Emerging evidences have demonstrated that OXPHOS inhibition can be beneficial to treat select cancers. For example, cancers with SMARCA4-mutation,2, 3 ENO1-deficiency and PGD-null4, 5 are significantly dependent on OXPHOS. Additionally, OXPHOS is implicated in the resistance mechanism to targeted therapies including tyrosine kinase inhibitors.6, 7 8–10 OXPHOS inhibitors are quite effective in targeting cancer stem cells and immunosuppressive cells that are known to rely on OXPHOS for their energy metabolism.11–14 Therefore, potent inhibition of OXPHOS can provide unique opportunity to treat select cancers, including hard-to-treat leukemia, glioma, and pancreatic cancers.
Pancreatic cancer cells are metabolically dependent on the OXPHOS pathway. It is well known that pancreatic-tumor-initiating cells show strong reliance on OXPHOS.14, 15 Moreover, ablation of KRAS in the KPC mouse model produces a subpopulation of surviving dormant tumor cells with strong reliance on OXPHOS and decreased dependence on glycolysis for cellular energetics. These surviving cells showed high sensitivity to OXPHOS inhibition preventing tumor recurrence, suggesting that OXPHOS inhibitors in combination with KRAS pathway inhibitors can be an effective treatment for pancreatic cancer.16 Additionally, inhibition of mitochondrial oxygen consumption suppresses the progression of pancreatic cancer.17 Importantly, in pancreatic cancer patients, overexpression of OXPHOS genes correlates with poor prognosis.18 In multiple clinical cohorts, treatment with the well-accepted OXPHOS inhibitor metformin, shows positive correlation with survival in pancreatic cancer patients.19 Therefore, inhibition of the OXPHOS pathway represents a novel treatment option for pancreatic cancer patients.
The OXPHOS machinery consists of five complexes (Complex I-V) that transfer electrons from NADH and FADH2 to molecular oxygen, pumping protons from the mitochondria matrix to the intermembrane space to enable complex V to synthesize ATP.20 Complex I is a well-studied complex and its inhibition offers a unique strategy to disrupt cancer cell metabolism.21, 22 Most Complex I inhibitors bind competitively to its substrate binding site, the ubiquinone site, while other inhibitors bind the ND1 subunit hampering the binding of ubiquinone to the complex.4, 23–25 For example, the biguanide anti-diabetic drugs metformin and phenformin reduce the risk of cancer and block cancer cell proliferation by inhibiting Complex I (Figure 1).26–28 Novel Complex I inhibitors with optimized potency and pharmacokinetic properties including BAY 87-224329–31 and its analog IACS-0107592, 4, 32 have also been evaluated in clinical trials against acute myeloid leukemia, lymphoma and advanced solid tumors.33 Although clinical trials of BAY 87-2243 was terminated due to toxicity, IACS-010759 was reported to be well tolerated in Phase I clinical trials.34 We recently reported the discovery of a series of novel benzene-1,4-disulfonamides as a new class of Complex I inhibitors represented by DX3–234 (Figure 1).35
Figure 1.
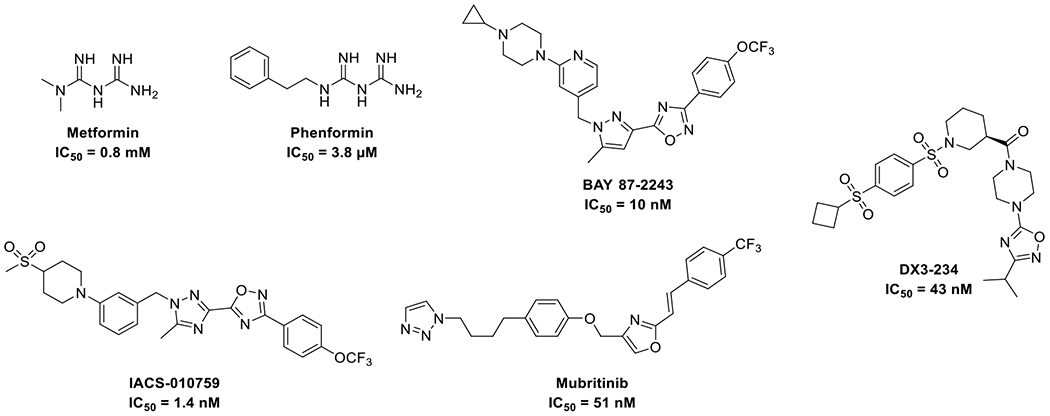
Representative OXPHOS inhibitors.
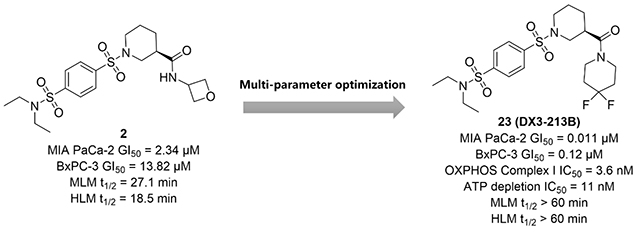
Despite the promising in vivo efficacy of several Complex I inhibitors in various cancers, currently no selective OXPHOS inhibitor has been FDA approved. In our previous work, compound 1 was discovered through a phenotypic screen selective for cytotoxicity in cells grown in galactose-containing media instead of glucose-containing media.35 Our initial optimization campaign produced oxetanyl amide 2 with improved metabolic stability and significant inhibition of MIA PaCa-2 cell proliferation in low micromolar range (Figure 2). Further modifications of the amide moiety to pursue extended interactions led to the discovery of a series of optimized compounds (Figure 2, series I) represented by DX3–234 (Figure 1) with significantly improved potency and metabolic stability. However, series I compounds have high molecular weights (>550), relatively high tPSA (110-130), and high cLogP (>3.5), which could potentially hamper their cellular uptake and penetration to target tissues, limit oral absorption, and other ADMET properties. Herein, we report a robust strategy to keep the overall structure simple by restricting their molecular weights and tPSA while optimizing potency. We designed and synthesized compounds with better overall ligand efficiency (LE) without violating the Lipinski’s “rule of 5”. This effort led to the discovery of series II compounds as represented by optimized compound 23 (DX3–213B, Figure 2). Compared with series I compounds, compound 23 is a highly potent OXPHOS complex I inhibitor, impairs ATP generation, and blocks MIA PaCa-2 cell growth at low nanomolar range, showing significant in vivo efficacy upon oral administration. Therefore, 23 represents an ideal tool compound to better elucidate the role of OXPHOS dependency in select cancer subtypes.
Figure 2.
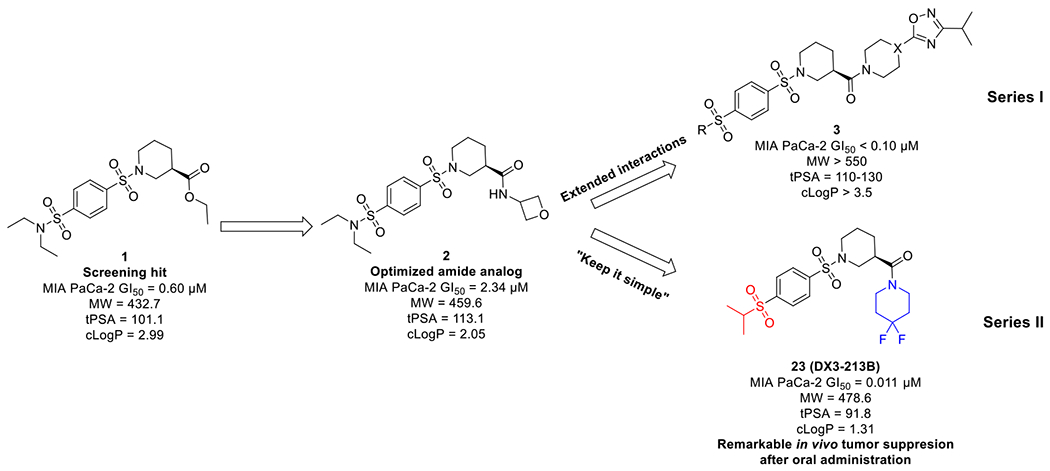
Discovery and optimization of benzene-1,4-disulfonamides as novel OXPHOS inhibitors.
RESULTS AND DISCUSSION
Optimization of lead compound 2
All compounds synthesized during optimization campaign were initially evaluated for their cytotoxicity in a 7-day MTT assay in two pancreatic cancer cell lines, MIA PaCa-2 and BxPC-3. Cancer cells display differences in their metabolic dependency, especially to OXPHOS and glycolysis pathways, therefore, they are expected to respond differently to OXPHOS inhibitors. In our studies, BxPC-3 cells are not as sensitive as MIA PaCa-2 to OXPHOS inhibition, which is probably due to stronger dependency of BxPC-3 cells on the glycolysis pathway. The secondary screening assay was performed in a 3-day MTT assay in glucose- or galactose-containing media to determine the impact of compounds on OXPHOS. Cells grown in high glucose-containing medium utilize the glycolysis pathway for ATP generation, while cells grown in galactose-containing medium exclusively depend on mitochondria for their ATP production. Therefore, cells grown in galactose-containing medium are highly sensitive to OXHPOS inhibition. The optimized compounds were further evaluated for their on-target efficacy by measuring inhibition of Complex I activity and ATP production.
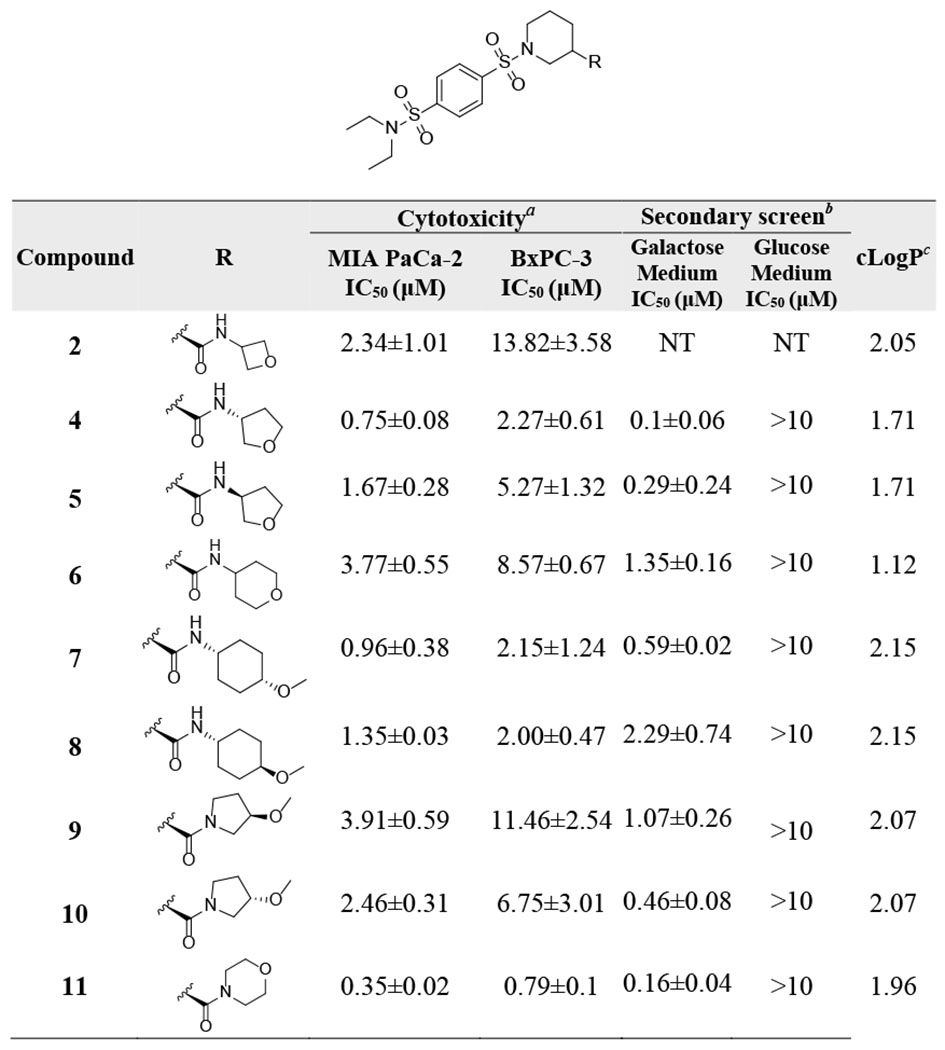
We selected the oxetane containing amide 2 for further optimization because of its reduced cLogP, improved aqueous solubility, and metabolic stability as compared with our original lead 1. Importantly, by addressing the metabolic soft point of the ester group, compound 2 showed improved microsomal stability by more than 30-fold over compound 1 (mouse liver microsome (MLM) t1/2 of 27.1 vs 0.7 min; human liver microsome (HLM) t1/2 of 18.5 vs 0.6 min). However, this amide modification led to a significant loss in cytotoxicity. Notwithstanding, compound 2 with IC50 value of 2.34±1.01 μM and good metabolic stability, is still a promising lead for further optimization.
We hypothesize that the oxetane oxygen in compound 2 might act as a H-bond acceptor or participate in an electrostatic interaction with its target. To improve potency while maintaining drug-like properties, we started optimization by: 1) introducing different oxygen containing cyclic structures to probe optimal distance and orientation of the oxygen to facilitate maximum target engagement; and 2) keeping the cLogP and tPSA at optimal levels to maintain drug-like properties (Table 1). Several efforts including enlarging the four-membered oxetane to five membered tetrahydrofuran (4 and 5) and six membered pyran structure (6) had moderate impact on potency. Importantly, the five membered (R)-3-aminotetrahydrofuran showed improved potency in both cell lines (4, MIA PaCa-2 IC50 = 0.75 ± 0.08 μM, BxPC-3 IC50 = 2.27 ± 0.61 μM). 4-Methoxycyclohexan-1-amines 7 (MIA PaCa-2 IC50 = 0.96 ± 0.38 μM, BxPC-3 IC50 = 2.15 ± 1.24 μM) and 8 (MIA PaCa-2 IC50 = 1.35 ± 0.03 μM, BxPC-3 IC50 = 2.00 ± 0.47 μM) also led to improved potency as compared with 2. The disubstituted cyclic amides 9 and 10 with 3-methoxypyrrolidine exhibited moderate inhibition of cell proliferation, while compound 11 with a morpholine structure showed significantly improved cytotoxicity (MIA PaCa-2 IC50 = 0.35 ± 0.02 μM, BxPC-3 IC50 = 0.79 ± 0.1 μM).
Table 1.
Establishing Structure-activity Relationships Among the Oxygen-containing Amides
|
IC50 values in MIA PaCa-2 and BxPC-3 cells were generated in glucose-containing medium using a 7-day MTT assay.
IC50 values in glucose- and galactose-containing medium were determined using a 3-day MTT assay in MIA PaCa-2 cells.
cLogP values were calculated using ChemDraw Professional 17.0.
NT: not tested.
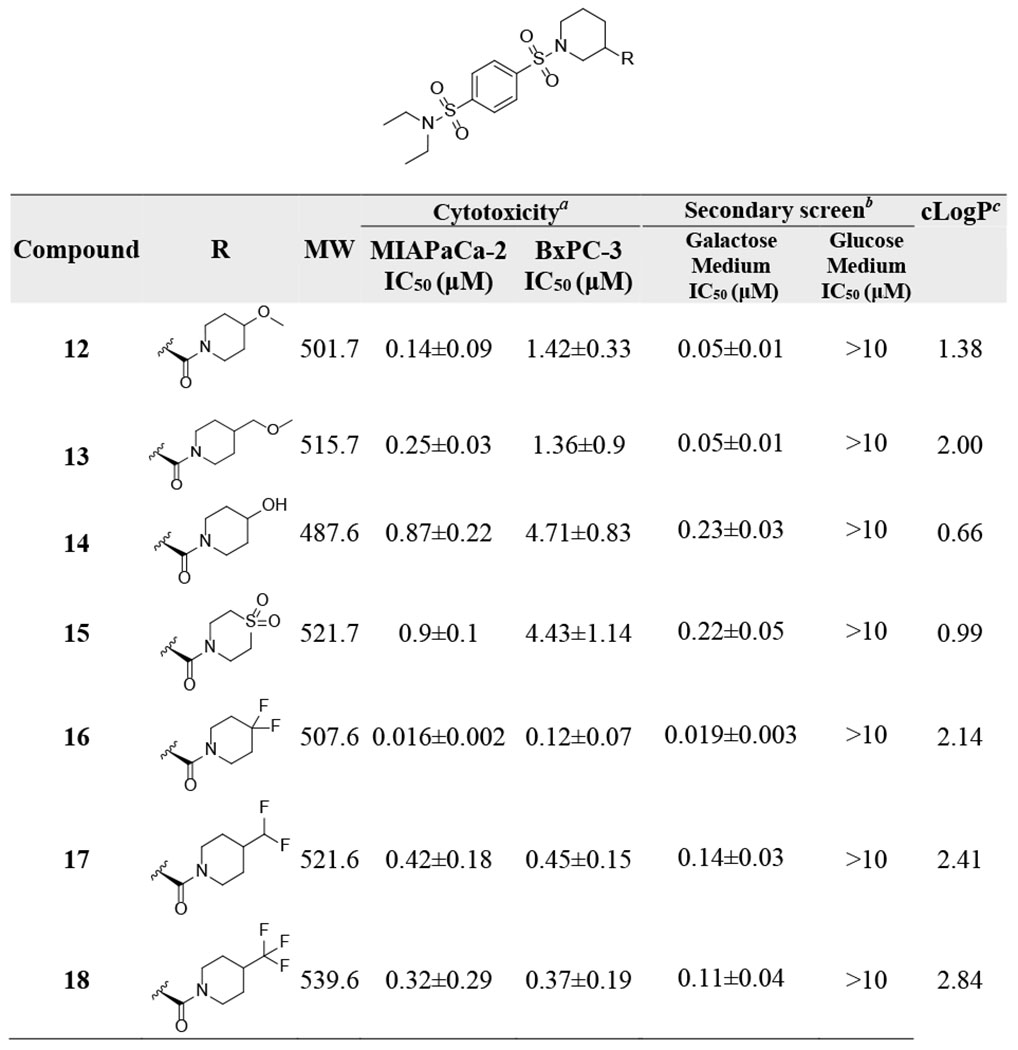
We hypothesized that extending the H-bond acceptor oxygen atom of compound 11 out of the ring might provide optimal distance between the oxygen atom and the amide group. Compounds obtained based on this approach displayed sub-micromolar IC50 values in MIA PaCa-2 cells (Table 2). Compound 12, with a 4-methoxypiperidine structure and lower cLogP than 11, showed a 2-fold improvement in MIA PaCa-2 cell growth inhibition (IC50 = 0.14±0.09 μM). In the secondary screen, compound 12 displayed low IC50 (0.05±0.01 μM) in galactose-containing medium and was inactive in glucose-containing medium, suggesting strong OXPHOS inhibition. Increasing the distance between the oxygen and the amide group by addition of a methylene group (13) was detrimental to activity. Replacement of the methoxy group with a hydroxy group in 14 was not well tolerated (MIA PaCa-2 IC50 = 0.87±0.22 μM, BxPC-3 IC50 = 4.71±0.83 μM). We also investigated the bioisosteric replacement of the morpholine with different H-bond acceptors and probed potential electrostatic interactions by the introduction of fluorine. The replacement of morpholine with a thiomorpholine 1,1-dioxide led to a moderate loss in potency (15: MIA PaCa-2 IC50 = 0.9±0.1 μM, BxPC-3 IC50 = 4.43±1.14 μM) compared to 12. However, the introduction of 4,4-difluoropiperidine (16) led to a significant improvement in inhibition of cell proliferation (MIA PaCa-2 IC50 = 0.016±0.002 μM, BxPC-3 IC50 = 0.12±0.07 μM). In contrast, compounds 17 and 18 with difluoromethane or trifluoromethyl substitutions were less active than 16. Within this series of compounds, 16 showing low nanomolar potency was clearly superior to other derivatives and was selected for further characterization.
Table 2.
Establishing Structure-activity Relationships for Compounds with Bioisosteric Modifications of the Morpholine Moiety
|
IC50 values in MIA PaCa-2 and BxPC-3 cells were generated in glucose-containing medium using 7-day MTT assay.
IC50 values in glucose- and galactose-containing medium were determined using 3-day MTT assay in MIA PaCa-2 cells.
cLogP values were calculated using ChemDraw Professional 17.0.
Evaluation of the metabolic stability of compound 16
Optimization around the amide group of compound 2 led to the discovery of 16 with a 4,4-difluoropiperidine moiety showing low nanomolar potency in MIA PaCa-2 cells (Figure 3). Similar to 2, compound 16 has an optimal cLogP of 2.14. Unfortunately, compound 16 suffers from poor microsomal stability (MLM t1/2 = 2.6 min and HLM t1/2 = 2.7 min). Our previous study suggested that de-ethylation is a possible metabolic pathway of this series of compounds.35 We hypothesized that the newly introduced electro-deficient 4,4-difluoropiperidine would not likely be a metabolic soft point. Therefore, we designed corresponding sulfones or mono substituted sulfonamides to address metabolic liabilities. Reducing the overall cLogP can decrease hydrophobic binding to CYP metabolic enzymes and thus clearance.36 Accordingly, we introduced hydrophilic groups to reduce cLogP. Although those modifications might affect potency, improved metabolic stability would lead to a more balanced profile for late-stage development.
Figure 3.
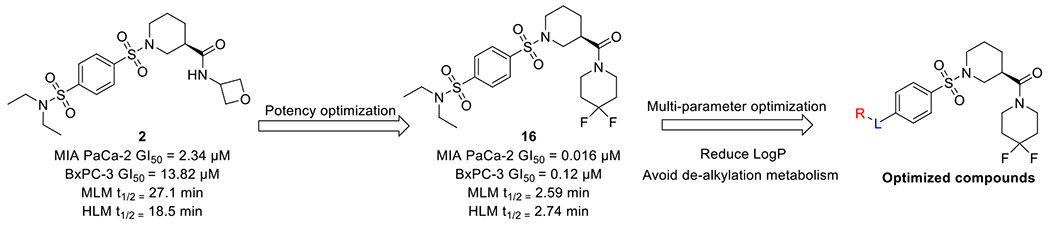
Multi-parameter optimization leading to the discovery of optimized derivatives.
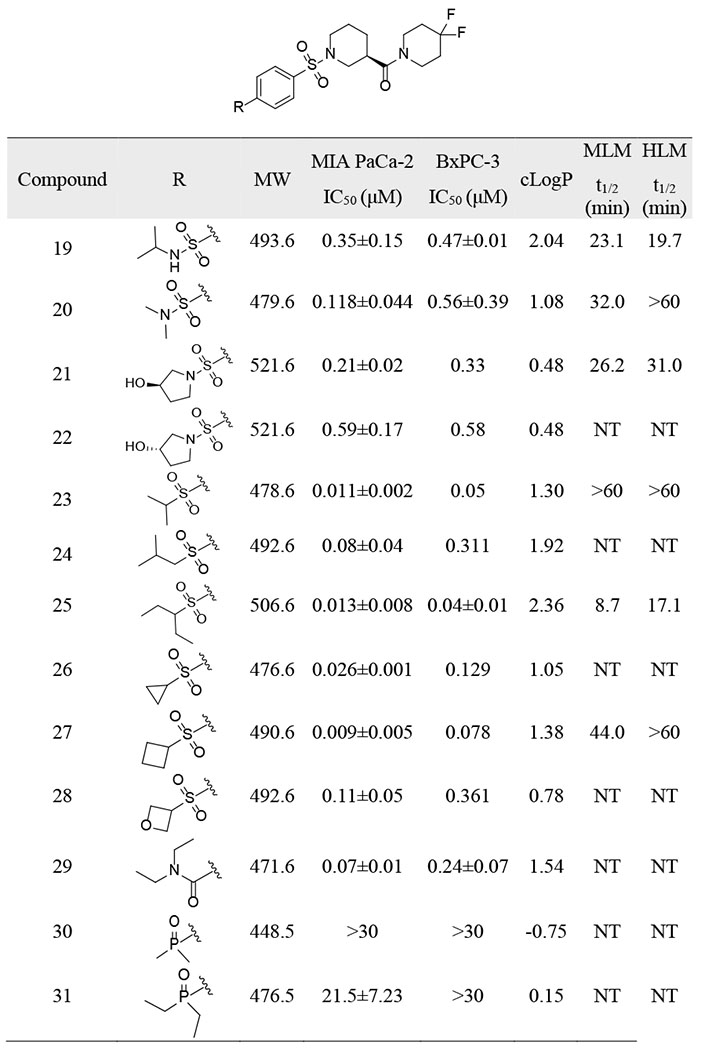
As expected, the mono-substituted sulfonamide 19 resulted in a significant improvement in the metabolic stability (MLM t1/2, 23.1 vs 2.6 min; HLM t1/2, 19.7 vs 2.7 min), indicating that de-alkylation was probably one of the metabolic pathways of 16 (Table 3). Compound 20 with dimethyl sulfonamide exhibited even better stability in microsomes with t ½ > 60 min in HLM, while retaining potent inhibition of pancreatic cancer cell growth (MIA PaCa-2 IC50 = 0.118±0.044 μM, BxPC-3 IC50 = 0.56±0.39 μM). Reduced cLogP as compared with 16 (1.08 v.s. 2.14) was perhaps responsible for its better metabolic stability, despite the presence of a potential de-alkylation metabolic site. The introduction of 3-hydroxypyrrolidine to reduce cLogP (21-22) also led to a robust metabolic profile. Introduction of a sulfone group with different sized alkyl substitutions did not affect potency, and nearly all compounds exhibited IC50 values < 0.1 μM in MIA PaCa-2 cells (23-28). Compounds 23, 25 and 27 were more potent than 16 in both pancreatic cancer cell lines with IC50s of ~10 nM in MIA PaCa-2 cells and < 100 nM in BxPC-3 cells. Compound 25 with a 3-pentyl group suffered from low metabolic stability (MLM t1/2, 8.7 min; HLM t1/2, 17.1 min). Introduction of isopropyl (23) or cyclobutyl (27) led to decreased cLogPs (1.30 and 1.38, respectively) and increased metabolic stability. Compound 23, with t1/2 > 60 min in both MLM and HLM and IC50s of 0.011±0.002 μM and 0.05 μM in MIA PaCa-2 and BxPC-3 cells, showed the most balanced overall profile.
Table 3.
Design of Compounds with Improved Metabolic Stability
|
IC50 values in MIA PaCa-2 and BxPC-3 cells were generated in glucose-containing medium using a 7-day MTT assay.
cLogP values were calculated using ChemDraw Professional 17.0.
MLM t1/2, mouse liver microsome half life.
HLM t1/2, human liver microsome half life.
NT: not tested.
Other modifications to reduce cLogP were also investigated. Compound 29 with an amide structure was less potent (MIA PaCa-2 IC50 = 0.07±0.01 μM, BxPC-3 IC50 = 0.24±0.07 μM). Bioisosteric replacement of sulfones with phosphine oxides resulting in lower cLogP were not tolerated, leading to a complete loss of activity (30 and 31).
Optimized compounds are potent Complex I inhibitors
We further evaluated cellular efficacy for OXPHOS inhibition of representative compounds 16, 20, 21, 23 and 27. All compounds significantly inhibited ATP production of MIA PaCa-2 cells in galactose-containing medium up to low nanomolar range (23, IC50 = 11±1.5 nM) while only partially inhibited ATP production in glucose-containing medium, suggesting that these compounds suppress OXPHOS (Figure 4). In agreement with ATP depletion assay, all the representative compounds showed significant inhibition of cell proliferation only in galactose-containing media (Table 4). We further determined the effect of optimized compounds in reducing NAD+/NADH ratio, which is a direct reflection of cellular OXPHOS Complex I inhibition (Table 4). Results clearly showed that all representative compounds remarkably reduced cellular NAD+/NADH ratios, with the most potent compounds 23 and 27 having IC50 values of 3.6±3.2 nM and 3.2±1.3 nM, respectively. These results demonstrates that our optimization strategy successfully generated OXPHOS inhibitors with IC50 values in low nano-molar range, which was in agreement with their cytotoxicity.
Figure 4.
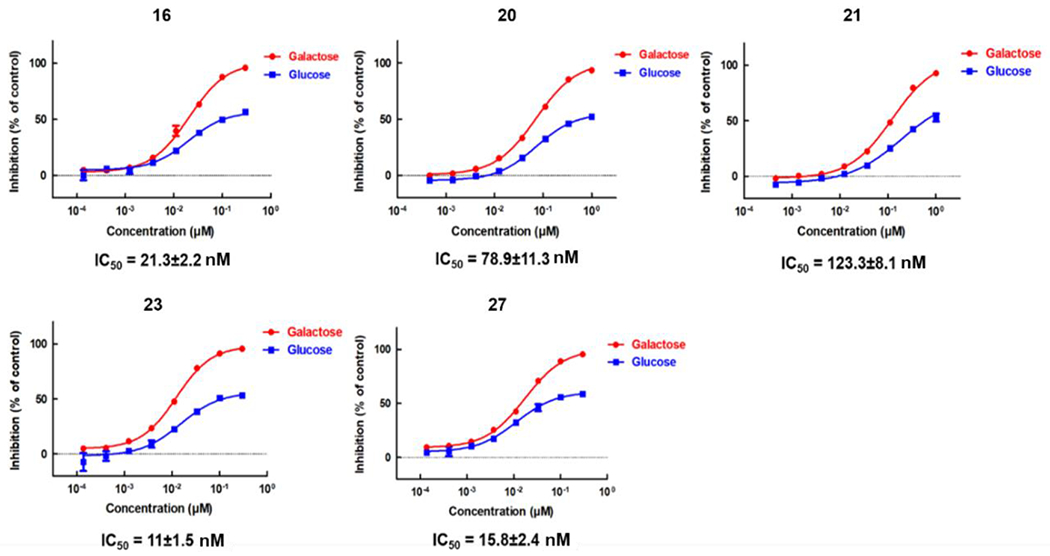
ATP depletion assay of representative compounds in glucose- and galactose-containing medium. The IC50 values shown are for experiments performed in galactose-containing medium.
Table 4.
Potency of Representative Compounds in Multiple Assays
| Cell proliferation (IC50, nM) | ATP Depletion (IC50, nM) | NAD+/NADH ratio (IC50, nM) | |||
|---|---|---|---|---|---|
|
| |||||
| Glucose medium | Galactose medium | Glucose medium | Galactose medium | Glucose medium | |
| 16 | >3000 | 19.4±3.3 | >3000 | 21.3±2.2 | 23±18.1 |
| 20 | >3000 | 72.4±12.6 | >3000 | 78.9±11.3 | 34±15 |
| 21 | >3000 | 135.1±21 | >3000 | 123.3±8.1 | 67.5±18.8 |
| 23 | >3000 | 9.1±5.9 | >3000 | 11±1.5 | 3.6±3.2 |
| 27 | >3000 | 12.6±8.9 | >3000 | 15.8±2.4 | 3.2±1.3 |
IC50 values of proliferation in glucose- and galactose-containing medium were determined using a 3-day MTT assay in MIA PaCa-2 cells.
IC50 values of ATP production in glucose- and galactose-containing medium were determined using CellTiter-Glo after 1 day treatment in MIA PaCa-2 cells.
IC50 values of each compound on the inhibitory effect of NAD+/NADH ratio was determined by NAD/NADH-Glo™ Assay upon 2 days treatment in MIA PaCa-2 cells.
PK study of compound 23 (DX3–213B)
We tested the pharmacokinetic properties of compound 23 in CD-1 mice following i.v. (2 mg/kg) and p.o. (10 mg/kg) administration (Table 5). After i.v. administration, compound 23 showed a high volume of distribution of 5.2 L/kg, a relatively high systemic clearance of 106 mL/min/kg, and an elimination half-life of 1.42 h. Compound 23 was also readily absorbed after oral administration with an AUC (0-inf) of 417 nM.h and oral bioavailability of 11.3%.
Table 5.
PK Parameters of Compound 23 (DX3-213B) in Plasma Following IV and PO Administration
| Route | Dose | C0 | AUC(0-tldc) | AUC(0-inf) | CL | Vss | Bioavailability | t1/2 |
|---|---|---|---|---|---|---|---|---|
| Unit | mg/kg | ng/mL | nM:h | nM:h | mL/min/kg | L/kg | % | h |
| IV | 2 | 354.0 | 650.0 | 657.0 | 106.0 | 5.2 | / | 1.42 |
| PO | 10 | / | 368.1 | 417.1 | 834.8 | / | 11.3 | 3.06 |
PK parameters were estimated using non-compartmental analysis with Phoenix/WINONLIN.
C0 = Initial concentration, AUC (0-tldc) = Area under the concentration-time curve from time zero to time of last detectable concentration, AUC (0-inf) = Area under the concentration-time curve from time zero to infinite, CL = Systemic clearance, Vss: Volume of distribution at steady state, Bioavailability = (AUCT×Doseiv) / (AUCiv×DoseT) ×100%, Terminal elimination half-life (t½) was calculated based on data points (>= 3) in the terminal phase.
Optimized derivatives show single-agent efficacy in a syngeneic model of pancreatic cancer
Compounds 20, 21, 23 and 27 were selected for in vivo efficacy studies using a syngeneic pancreatic cancer model (Figure 5). We performed a comprehensive toxicity study due to potential safety concerns of OXPHOS inhibition. We evaluated different routes of administration and doses in mice and determined that compound 23 was better tolerated when dosed p.o. with an maximum tolerated dose (MTD) of 10 mg/kg. For other compounds, i.p. was the preferred route of administration. The maximum tolerated doses were determined for each compound and were used in the efficacy studies. Briefly, mice were inoculated s.c. with Pan02 cells and treated with compounds 20 (7.5 mg/kg, i.p.), compound 21 (15 mg/kg, i.p.), compound 27 (2.5 mg/kg, i.p.) or compound 23 (10 mg/kg, p.o.) once daily for 28 days. The tumor volume was monitored twice a week. All four compounds were well tolerated for the duration of the study, and none showed obvious signs of toxicity or drop in body weight. The treatment of 23 and 27 resulted in significant tumor growth inhibition (p-value of 0.0008 for 23 and p-value of 0.038 for 27). These results suggest that compounds 23 and 27 are promising tool compounds to better elucidate the role of OXPHOS inhibition in pancreatic cancer.
Figure 5.
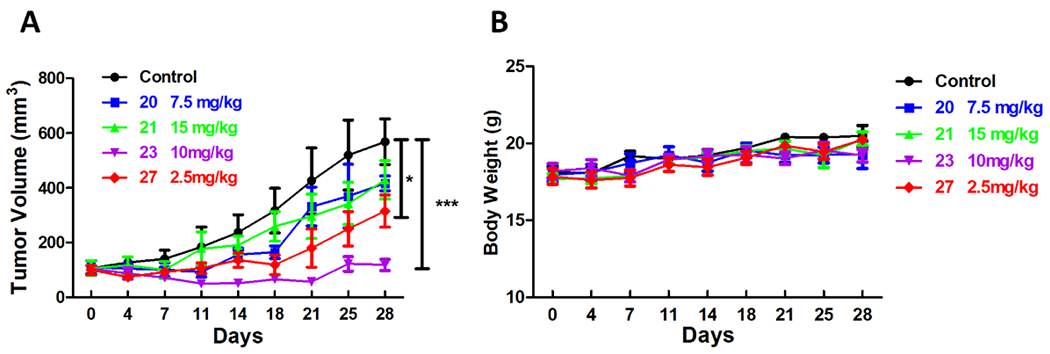
in vivo efficacy study of selected compounds in a Pan02 syngeneic model of pancreatic cancer. (A) Mice (n = 5 per group) were inoculated s.c. with Pan02 cells. When tumors reached approximately 100 mm3 mice were randomized into 5 groups and treated with vehicle or compounds 20 (7.5 mg/kg, i.p.), 21 (15 mg/kg, i.p.), 27 (2.5 mg/kg, i.p.) and 23 (10 mg/kg, po) daily for 28 days. Tumor volume (mean + SEM) was monitored for each group of mice twice a week during 28 days of compound dosing. (B) No significant loss in body weight was observed during treatment.
Chemistry
The general synthetic route for the disulfonamides is shown in Scheme 1. p-bromophenyl sulfonyl chloride (32) was reacted with diethylamine to give sulfonamide intermediate 33. Then a Pd2(dba)3 catalyzed Buchwald-Hartwig type coupling between 33 and benzyl mercaptan, in the presence of XantPhos and DIEA, furnished thioether 34, which was then subjected to oxidative chlorination by 1,3-dichloro-5,5-dimethylimidazolidine-2,4-dione37 to give sulfonyl chloride 35. The reaction between 35 and (R)-piperidine-3-carboxylic acid gave rise to the carboxylic acid 36. A coupling reaction facilitated by HATU and DIEA between 36 and an appropriate amine gave the final products 4-18.
Scheme 1. General Synthetic Route for Disulfonamides.
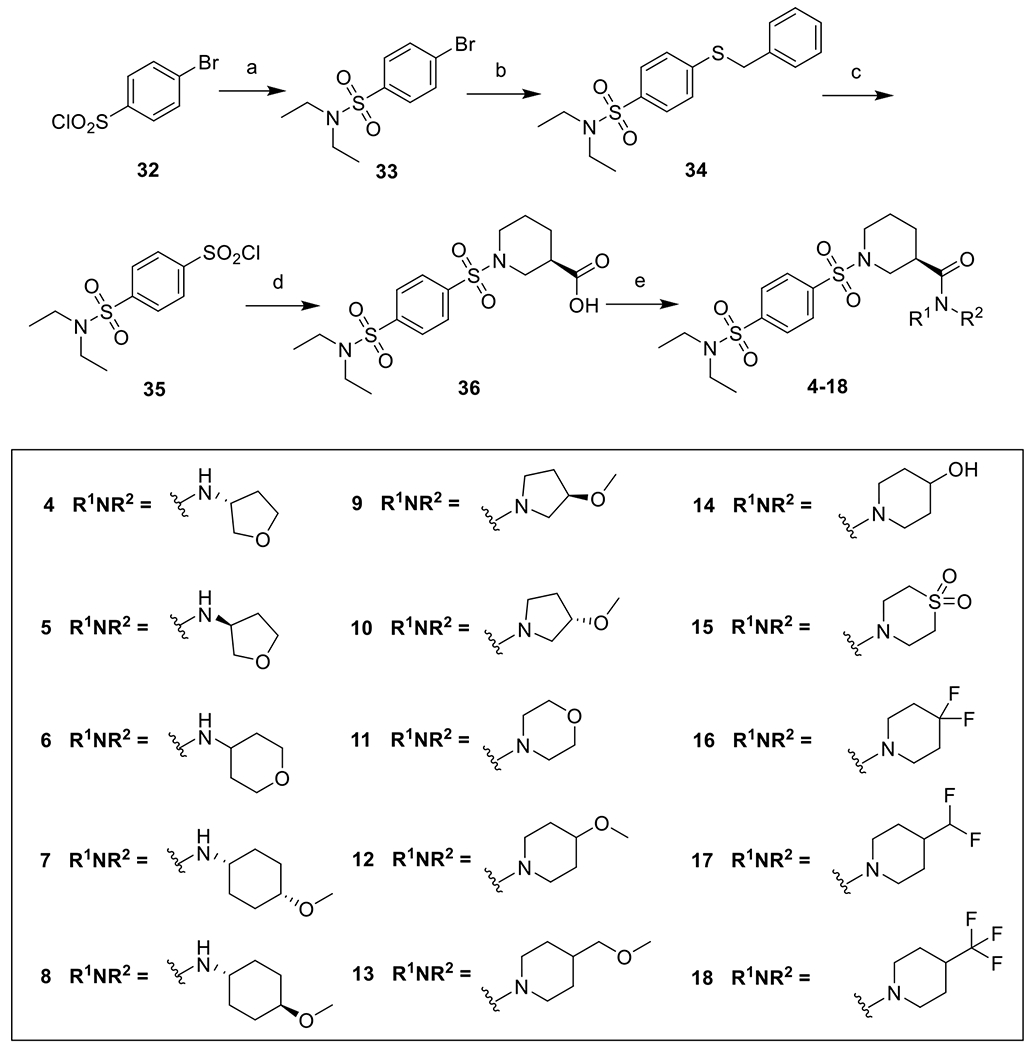
Reagents and conditions: a) diethyl amine, Et3N, DCM, rt, 83%; b) benzyl mercaptan, Pd2(dba)3, XantPhos, DIEA, dioxane, reflux, 91%; c) 1,3-dichloro-5,5-dimethylimidazolidine-2,4-dione, CH3CN, CH3COOH, H2O, 0 °C, 100% crude; d) (R)-piperidine-3-carboxylic acid, Na2CO3, THF, H2O, rt, 73%; e) R1NHR2, HATU, DIEA, DMF, rt, 50-98%.
To facilitate the modification on the left-hand side sulfonamide, an alternative scheme was adopted. 32 was reacted with ethyl (R)-piperidine-3-carboxylate to give 37, which was reacted with benzyl mercaptan as described above to give the thioether 38, which was then hydrolyzed and coupled with 4,4-difluoropiperidine in the presence of HATU and DIEA to give 39. It was then subjected to oxidative chlorination and the corresponding sulfonyl chloride obtained was treated with an appropriate amine to give the final products 19-22 (Scheme 2).
Scheme 2. Alternative Synthetic Route for Disulfonamides.
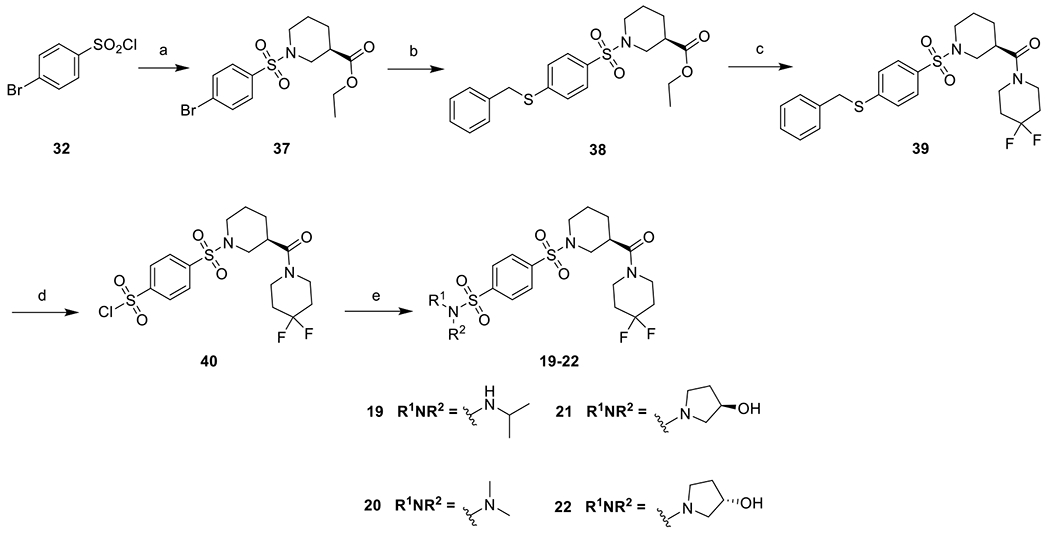
Reagents and conditions: a) ethyl (R)-piperidine-3-carboxylate, Et3N, DCM, rt, 98%; b) benzyl mercaptan, Pd2(dba)3, XantPhos, DIEA, dioxane, reflux, 87%; c) i. LiOH, THF, H2O, rt; ii. 4,4-difluoropiperidine, HATU, DIEA, DMF, rt, 90%; d) 1,3-dichloro-5,5-dimethylimidazolidine-2,4-dione, CH3CN, CH3COOH, H2O, 0 °C, 100% crude; e) R1NHR2, Et3N, DCM, rt, 37-58%.
The sulfones were synthesized in a three-step fashion. Key intermediate 41 was first obtained from 37 using the method described above. It was then coupled with thiols in the presence of catalytic amount of Pd2(dba)3 and XantPhos to give 42a-f, which were then oxidized by mCPBA to give the products 23-28 (Scheme 3).
Scheme 3. Synthetic Route for Sulfones.
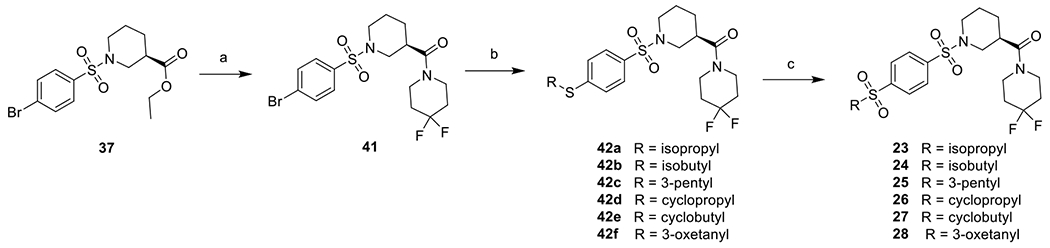
Reagents and conditions: a) i. LiOH, THF, H2O, rt; ii. 4,4-difluoropiperidine, HATU, DIEA, DMF, rt, 91%; b) RSH, Pd2(dba)3, XantPhos, DIEA, dioxane, reflux, 39-90%; c) mCPBA, DCM, rt, 35-68%.
The synthesis of 29 started with a reaction between 43 and (R)-piperidine-3-carboxylic acid to give the carboxylic acid 44. It was then reacted with 4,4-difluoropiperidine in the presence of HATU to give 29. To synthesize the phosphine oxides, 37 was treated with dimethylphosphine oxide or diethylphosphine oxide catalyzed by Pd(OAc)2 at 120 °C to generate intermediates 45a-b. A subsequent hydrolysis and coupling reaction similar to that described above gave rise to the final products 30 and 31.
CONCLUSIONS
Recently, we discovered a series of benene-1,4-disulfonamides as OXPHOS complex I inhibitors showing in vivo efficacy. To improve potency and drug-like properties of this series of compounds, we designed and synthesized a series of derivatives with simple amide group displaying reduced cLogP and tPSA. We discovered that the 4,4-difluoropiperidine moiety significantly improved potency. Multi-parameter optimization led to the discovery of the most potent derivative 23 (DX3–213B) within this series. Compound 23 inhibited the proliferation of MIA PaCa-2 cells with an IC50 value of 9 nM in galactose-containing media and reduced cellular NAD+/NADH ratio by inhibiting OXPHOS complex I with an IC50 of 3.6 nM. ATP depletion was also significantly observed at an IC50 of 11 nM. Oral administration of compound 23 was well tolerated and produced significant tumor growth delay in a syngeneic mouse model of pancreatic cancer. Studies are in progress to evaluate the combination of compound 23 with standard-of-care chemotherapy for pancreatic cancer. Cumulatively, our data suggest that OXPHOS inhibition can be a safe and efficacious approach to treat pancreatic cancer and other cancers that are highly dependent on OXPHOS.
EXPERIMENTAL
General Methods.
All commercial reagents and anhydrous solvents were purchased and used without purification, unless specified. Column chromatography was performed on a Biotage Isolera flash chromatography system on Biotage normal phase silica gel columns. Preparative-HPLC purification was performed on a Shimadzu Semi-Prep LC system. Analytical thin layer chromatography was performed on Merck pre-coated plates (silica gel 60 F254). NMR spectra were recorded on a Bruker Ultrashield 300 MHz or a Bruker Ascend 400 MHz spectrometer using deuterated CDCl3 or CD3OD as solvents. Chemical shifts for proton magnetic resonance spectra (1H NMR) are quoted in parts per million (ppm) referenced to the appropriate solvent peak or 0.0 ppm for tetramethylsilane (TMS). The following abbreviations are used to describe the peak-splitting patterns when appropriate: br, broad; s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; and dd, doublet of doublets. Coupling constants, J, are reported in hertz (Hz). Mass spectra were recorded on a Shimadzu LCMS-2020 system using the electrospray ionization (ESI) ion source. HPLC was used to determine the purity of biologically tested compounds using Shimadzu LC-2030C 3D system equipped with a Kinetex XB-C18 column (2.6 μm, 4.6×75 mm). The following gradient elution conditions were used: acetonitrile/water (10–95 %) or methanol/water (10–95 %), both with 0.1% formic acid as additive, over 15 minutes at a 0.80 mL/min flow rate at room temperature. The purity was established by integration of the areas of major peaks detected at 254 nm, and all final products have >95% purity.
Synthetic procedures for 4–18 (Scheme 1).
4-Bromo-N,N-diethylbenzenesulfonamide (33).
To a solution of diethylamine (730 mg, 10.0 mmol) and triethylamine (2.02 g, 20.0 mmol) was added 4-bromobenzenesulfonyl chloride (2.56 g, 10.0 mmol) portion wise. The mixture was stirred at room temperature overnight. The mixture was concentrated and purified with flash chromatography (10% EtOAc in hexane) to give 33 as a colorless oil. (2.44 g, 83%). 1H NMR (300 MHz, CDCl3) δ 7.73–7.62 (m, 4H), 3.26 (q, J = 7.1 Hz, 4H), 1.15 (t, J = 7.1 Hz, 6H). LC-MS (ESI) m/z 291.9, 293.9 [M + H]+.
4-(Benzylthio)-N,N-diethylbenzenesulfonamide (34).
A solution of 33 (500 mg, 1.71 mmol), DIEA (441 mg, 3.42 mmol) in dioxane (10 mL) was degassed and flushed with argon for three times. Then Pd2(dba)3 (39 mg, 0.043 mmol), XantPhos (50 mg, 0.086 mmol), and benzyl mercaptan (212 mg, 1.71 mmol) was added subsequently. The mixture was degassed and flushed with argon for three times before it was heated under reflux overnight. The mixture was cooled, and the needle-like crystals generated was filtered off. The filtrate was concentrated and purified with flash chromatography (20% EtOAc in hexane) to give 34 as a yellow solid (520 mg, 91%). 1H NMR (300 MHz, CDCl3) δ 7.68 (d, J = 8.1 Hz, 2H), 7.43–7.22 (m, 7H), 4.22 (s, 2H), 3.23 (q, J = 7.2 Hz, 4H), 1.13 (t, J = 7.2 Hz, 6H). LC-MS (ESI) m/z 336.0 [M + H]+.
4-(N,N-Diethylsulfamoyl)benzenesulfonyl chloride (35).
To an ice-cooled solution of 34 (100 mg, 0.30 mmol) in a mixture of CH3CN (2.5 mL), HOAc (0.16 mL) and H2O (0.1 mL) was added 1,3-dichloro-5,5-dimethylimidazolidine-2,4-dione (117 mg, 0.60 mmol) portion wise. The mixture was kept stirring at 0–5 °C for 2h and concentrated. The residue obtained was taken up by DCM, washed with 5% NaHCO3 solution at 0 °C, dried over Na2SO4, filtered and concentrated to give 35 as a white solid (94 mg, 100% crude) which was used in the next step without further purification. 1H NMR (300 MHz, CDCl3) δ 8.22 – 8.04 (m, 4H), 3.32 (q, J = 7.2 Hz, 4H), 1.19 (t, J = 7.1 Hz, 6H).
(R)-1-((4-(N,N-Diethylsulfamoyl)phenyl)sulfonyl)piperidine-3-carboxylic acid (36).
To a solution of 35 (4.0 g, 12.9 mmol) and Na2CO3 (4.1 g, 38.6 mmol) in H2O (50 mL) was added a solution of (R)-piperidine-3-carboxylic acid (1.6 g, 2.66 mmol) in THF (50 mL) dropwise at 0 °C and stirred at room temperature for 3h. THF was removed by evaporation and the pH was adjusted to 3 by 1N HCl solution. It was extracted with EtOAc, and the organic layer was washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated to give 36 as a white solid (3.80 g, 73%). 1H NMR (300 MHz, MeOD) δ 8.07 (d, J = 8.1 Hz, 2H), 7.99 (d, J = 7.9 Hz, 2H), 3.82–3.70 (m, 1H), 3.60–3.48 (m, 1H), 3.36–3.24 (m, 4H), 2.69 (t, J = 10.7 Hz, 1H), 2.65–2.47 (m, 2H), 2.05–1.91 (m, 1H), 1.89–1.76 (m, 1H), 1.71–1.57 (m, 1H), 1.56–1.38 (m, 1H), 1.15 (t, J = 7.1 Hz, 6H). LC-MS (ESI) m/z 405.0 [M + H]+.
(R)-1-((4-(N,N-Diethylsulfamoyl)phenyl)sulfonyl)-N-((R)-tetrahydrofuran-3-yl)piperidine-3-carboxamide (4).
To a solution of 36 (20 mg, 0.05 mmol) and HATU (29 mg, 0.075 mmol) in DMF (1 mL) was added (R)-tetrahydrofuran-3-amine hydrochloride (6.2 mg, 0.05 mmol) and DIEA (19 mg, 0.15 mmol). The mixture was stirred at room temperature overnight. The mixture was then diluted with EtOAc, washed with brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified with flash chromatography (10% MeOH in DCM) to give 4 as a white solid (15 mg, 63%). 1H NMR (300 MHz, CDCl3) δ 7.99 (d, J = 8.5 Hz, 2H), 7.89 (d, J = 8.6 Hz, 2H), 6.01 (d, J = 7.4 Hz, 1H), 4.50 (dt, J = 7.6, 5.8 Hz, 1H), 3.96 (q, J = 7.6 Hz, 1H), 3.83 (qd, J = 9.1, 5.5 Hz, 2H), 3.73–3.54 (m, 3H), 3.31 (q, J = 7.1 Hz, 4H), 2.74 (dd, J = 11.8, 9.6 Hz, 1H), 2.55 (t, J = 10.3 Hz, 1H), 2.44 (dd, J = 10.0, 6.2 Hz, 1H), 2.37–2.20 (m, 1H), 1.91–1.63 (m, 5H), 1.18 (t, J = 7.1 Hz, 6H). LC-MS (ESI) m/z 474.1 [M + H]+. Purity: 96.3%.
(R)-1-((4-(N,N-Diethylsulfamoyl)phenyl)sulfonyl)-N-((S)-tetrahydrofuran-3-yl)piperidine-3-carboxamide (5).
The title compound was synthesized using a similar procedure as described for 4 by employing 36 (20 mg, 0.05 mmol) and (S)-tetrahydrofuran-3-amine hydrochloride (6.2 mg, 0.05 mmol) in DMF (1 mL) to give a white solid (23 mg, 98%). 1H NMR (300 MHz, CDCl3) δ 8.03–7.95 (m, 2H), 7.89 (d, J = 8.7 Hz, 2H), 6.01 (d, J = 7.4 Hz, 1H), 4.57–4.45 (m, 1H), 3.99 (q, J = 7.9, 7.5 Hz, 1H), 3.83 (ddd, J = 9.6, 5.5, 2.4 Hz, 2H), 3.66 (dd, J = 9.6, 2.8 Hz, 2H), 3.57 (d, J = 11.7 Hz, 1H), 3.31 (q, J = 7.2 Hz, 4H), 2.84–2.71 (m, 1H), 2.57 (t, J = 10.0 Hz, 1H), 2.43 (s, 1H), 2.37–2.23 (m, 1H), 1.90–1.63 (m, 5H), 1.18 (t, J = 7.1 Hz, 6H). LC-MS (ESI) m/z 474.0 [M + H]+. Purity: 99.6%.
(R)-1-((4-(N,N-Diethylsulfamoyl)phenyl)sulfonyl)-N-(tetrahydro-2H-pyran-4-yl)piperidine-3-carboxamide (6).
The title compound was synthesized using a similar procedure as described for 4 by employing 36 (20 mg, 0.05 mmol) and tetrahydro-2H-pyran-4-amine (6.9 mg, 0.05 mmol) in DMF (1 mL) to give a white solid (21 mg, 86%). 1H NMR (300 MHz, CDCl3) δ 7.99 (d, J = 8.7 Hz, 2H), 7.89 (d, J = 8.7 Hz, 2H), 5.70 (d, J = 8.0 Hz, 1H), 3.98 (dt, J = 11.2, 3.5 Hz, 3H), 3.66 (d, J = 11.0 Hz, 1H), 3.61–3.42 (m, 3H), 3.31 (q, J = 7.2 Hz, 4H), 2.84–2.73 (m, 1H), 2.60 (d, J = 10.3 Hz, 1H), 2.49–2.36 (m, 1H), 1.97–1.63 (m, 6H), 1.51 (ddd, J = 12.9, 6.5, 4.4 Hz, 2H), 1.18 (t, J = 7.1 Hz, 6H). LC-MS (ESI) m/z 488.2 [M + H]+. Purity: >98%.
(R)-1-((4-(N,N-Diethylsulfamoyl)phenyl)sulfonyl)-N-(cis-4-methoxycyclohexyl)piperidine-3-carboxamide (7).
The title compound was synthesized using a similar procedure as described for 4 by employing 36 (15 mg, 0.037 mmol) and cis-4-methoxycyclohexan-1-amine (6.1 mg, 0.037 mmol) in DMF (1 mL) to give a white solid (12 mg, 63%). 1H NMR (300 MHz, CDCl3) δ 7.98 (d, J = 8.2 Hz, 2H), 7.89 (d, J = 8.2 Hz, 2H), 5.60 (d, J = 8.0 Hz, 1H), 3.89–3.69 (m, 2H), 3.63 (d, J = 11.5 Hz, 1H), 3.44–3.34 (m, 1H), 3.35–3.25 (m, 7H), 2.68 (t, J = 10.8 Hz, 1H), 2.55–2.34 (m, 2H), 1.95–1.77 (m, 4H), 1.77–1.61 (m, 4H), 1.59–1.48 (m, 4H), 1.18 (t, J = 7.1 Hz, 6H). LC-MS (ESI) m/z 538.2 [M + Na]+. Purity: 99.6%.
(R)-1-((4-(N,N-Diethylsulfamoyl)phenyl)sulfonyl)-N-(trans-4-methoxycyclohexyl)piperidine-3-carboxamide (8).
The title compound was synthesized using a similar procedure as described for 4 by employing 36 (15 mg, 0.037 mmol) and trans-4-methoxycyclohexan-1-amine (6.1 mg, 0.037 mmol) in DMF (1 mL) to give a white solid (10 mg, 53%). 1H NMR (300 MHz, CDCl3) δ 7.99 (d, J = 8.2 Hz, 2H), 7.88 (d, J = 8.2 Hz, 2H), 5.62 (d, J = 7.8 Hz, 1H), 3.84–3.70 (m, 1H), 3.67–3.47 (m, 2H), 3.40–3.26 (m, 7H), 3.23–3.11 (m, 1H), 2.79 (t, J = 10.6 Hz, 1H), 2.66–2.55 (m, 1H), 2.47–2.33 (m, 1H), 2.13–1.96 (m, 4H), 1.85–1.63 (m, 4H), 1.44–1.23 (m, 4H), 1.18 (t, J = 7.1 Hz, 6H). LC-MS (ESI) m/z 516.3 [M + H]+. Purity: 98.5%.
N,N-Diethyl-4-(((R)-3-((R)-3-methoxypyrrolidine-1-carbonyl)piperidin-1-yl)sulfonyl)benzenesulfonamide (9).
The title compound was synthesized using a similar procedure as described for 4 by employing 36 (15 mg, 0.037 mmol) and (R)-3-methoxypyrrolidine hydrochloride (5.1 mg, 0.037 mmol) in DMF (1 mL) to give a white solid (14 mg, 78%). 1H NMR (300 MHz, CDCl3) δ 7.97 (d, J = 8.2 Hz, 2H), 7.88 (d, J = 8.3 Hz, 2H), 4.10–3.82 (m, 3H), 3.71–3.41 (m, 4H), 3.39 – 3.22 (m, 7H), 2.80–2.64 (m, 1H), 2.53 (t, J = 11.3 Hz, 1H), 2.34–2.00 (m, 1H), 1.98–1.80 (m, 2H), 1.77–1.66 (m, 1H), 1.56–1.42 (m, 1H), 1.18 (t, J = 7.1 Hz, 6H). LC-MS (ESI) m/z 488.2 [M + H]+. Purity: 96.6%.
N,N-Diethyl-4-(((R)-3-((S)-3-methoxypyrrolidine-1-carbonyl)piperidin-1-yl)sulfonyl)benzenesulfonamide (10).
The title compound was synthesized using a similar procedure as described for 4 by employing 36 (15 mg, 0.037 mmol) and (S)-3-methoxypyrrolidine hydrochloride (5.1 mg, 0.037 mmol) in DMF (1 mL) to give a white solid (14 mg, 78%). 1H NMR (300 MHz, CDCl3) δ 7.97 (d, J = 8.2 Hz, 2H), 7.88 (d, J = 8.3 Hz, 2H), 4.11–3.81 (m, 3H), 3.74–3.39 (m, 4H), 3.38–3.22 (m, 7H), 2.78–2.62 (m, 1H), 2.53 (td, J = 11.3, 4.4 Hz, 1H), 2.35–2.13 (m, 1H), 2.08–1.78 (m, 4H), 1.74–1.57 (m, 1H), 1.56–1.39 (m, 1H), 1.17 (t, J = 7.1 Hz, 6H). LC-MS (ESI) m/z 488.2 [M + H]+. Purity: 97.0%.
(R)-N,N-Diethyl-4-((3-(morpholine-4-carbonyl)piperidin-1-yl)sulfonyl)benzenesulfonamide (11).
The title compound was synthesized using a similar procedure as described for 4 by employing 36 (20 mg, 0.05 mmol) and morpholine (4.4 mg, 0.05 mmol) in DMF (1 mL) to give a white solid (23 mg, 97%). 1H NMR (300 MHz, CDCl3) δ 7.98 (d, J = 8.7 Hz, 2H), 7.89 (d, J = 8.7 Hz, 2H), 3.88 (d, J = 11.6 Hz, 2H), 3.81–3.49 (m, 8H), 3.30 (q, J = 7.1 Hz, 4H), 2.91–2.78 (m, 1H), 2.64–2.53 (m, 1H), 2.36–2.24 (m, 1H), 1.92–1.81 (m, 2H), 1.79–1.64 (m, 1H), 1.55–1.39 (m, 1H), 1.18 (t, J = 7.1 Hz, 6H). LC-MS (ESI) m/z 474.1 [M + H]+. Purity: 99.4%.
(R)-N,N-Diethyl-4-((3-(4-methoxypiperidine-1-carbonyl)piperidin-1-yl)sulfonyl)benzenesulfonamide (12).
The title compound was synthesized using a similar procedure as described for 4 by employing 36 (15 mg, 0.037 mmol) and 4-methoxypiperidine (4.3 mg, 0.037 mmol) in DMF (1 mL) to give a white solid (13 mg, 70%). 1H NMR (300 MHz, CDCl3) δ 7.98 (d, J = 8.2 Hz, 2H), 7.89 (d, J = 8.2 Hz, 2H), 3.95–3.77 (m, 3H), 3.77–3.65 (m, 1H), 3.52–3.43 (m, 1H), 3.42–3.25 (m, 9H), 2.87 (t, J = 11.4 Hz, 1H), 2.57 (dt, J = 15.4, 7.7 Hz, 1H), 2.28 (t, J = 11.8 Hz, 1H), 1.96–1.79 (m, 4H), 1.76–1.60 (m, 3H), 1.52 – 1.39 (m, 1H), 1.18 (t, J = 7.1 Hz, 6H). LC-MS (ESI) m/z 502.3 [M + H]+. Purity: 95.8%.
(R)-N,N-Diethyl-4-((3-(4-(methoxymethyl)piperidine-1-carbonyl)piperidin-1-yl)sulfonyl)benzenesulfonamide (13).
The title compound was synthesized using a similar procedure as described for 4 by employing 36 (15 mg, 0.037 mmol) and 4-(methoxymethyl)piperidine (4.8 mg, 0.037 mmol) in DMF (1 mL) to give a white solid (16 mg, 84%). 1H NMR (300 MHz, CDCl3) δ 7.97 (d, J = 8.0 Hz, 2H), 7.89 (d, J = 8.2 Hz, 2H), 4.59 (d, J = 13.2 Hz, 1H), 3.97–3.81 (m, 3H), 3.41–3.19 (m, 10H), 3.07 (q, J = 13.2 Hz, 1H), 2.93–2.77 (m, 1H), 2.66–2.48 (m, 2H), 2.34–2.21 (m, 1H), 1.94–1.68 (m, 7H), 1.52–1.36 (m, 1H), 1.18 (t, J = 7.1 Hz, 6H). LC-MS (ESI) m/z 516.3 [M + H]+. Purity: 98.8%.
(R)-N,N-Diethyl-4-((3-(4-hydroxypiperidine-1-carbonyl)piperidin-1-yl)sulfonyl)benzenesulfonamide (14).
The title compound was synthesized using a similar procedure as described for 4 by employing 36 (15 mg, 0.037 mmol) and 4-hydroxypiperidine (3.7 mg, 0.037 mmol) in DMF (1 mL) to give a white solid (15 mg, 83%). 1H NMR (300 MHz, CDCl3) δ 7.98 (d, J = 8.2 Hz, 2H), 7.89 (d, J = 8.2 Hz, 2H), 4.09–3.95 (m, 2H), 3.93–3.73 (m, 3H), 3.29 (p, J = 7.2 Hz, 6H), 2.95–2.81 (m, 1H), 2.57 (t, J = 11.3 Hz, 1H), 2.29 (t, J = 11.8 Hz, 1H), 2.05–1.66 (m, 5H), 1.64–1.38 (m, 3H), 1.18 (t, J = 7.1 Hz, 6H). LC-MS (ESI) m/z 488.1 [M + H]+. Purity: >98%.
(R)-4-((3-(1,1-Dioxidothiomorpholine-4-carbonyl)piperidin-1-yl)sulfonyl)-N,N-diethylbenzenesulfonamide (15).
The title compound was synthesized using a similar procedure as described for 4 by employing 36 (15 mg, 0.037 mmol) and thiomorpholine 1,1-dioxide (5.0 mg, 0.037 mmol) in DMF (1 mL) to give a white solid (14 mg, 73%). 1H NMR (300 MHz, CDCl3) δ 7.99 (d, J = 8.2 Hz, 2H), 7.89 (d, J = 8.2 Hz, 2H), 4.26–3.98 (m, 4H), 3.88 (d, J = 11.8 Hz, 2H), 3.31 (q, J = 7.1 Hz, 4H), 3.18–3.01 (m, 4H), 2.89 (t, J = 11.5 Hz, 1H), 2.59 (t, J = 11.4 Hz, 1H), 2.33 (t, J = 11.8 Hz, 1H), 1.89 (d, J = 12.6 Hz, 2H), 1.81–1.68 (m, 1H), 1.51 (dd, J = 14.3, 5.2 Hz, 1H), 1.18 (t, J = 7.1 Hz, 6H). LC-MS (ESI) m/z 544.1 [M + Na]+. Purity: 99.2%.
(R)-4-((3-(4,4-Difluoropiperidine-1-carbonyl)piperidin-1-yl)sulfonyl)-N,N-diethylbenzenesulfonamide (16).
The title compound was synthesized using a similar procedure as described for 4 by employing 36 (15 mg, 0.037 mmol) and 4,4-difluoropiperidine (4.5 mg, 0.037 mmol) in DMF (1 mL) to give a white solid (11 mg, 59%). 1H NMR (300 MHz, CDCl3) δ 7.99 (d, J = 8.2 Hz, 2H), 7.89 (d, J = 8.2 Hz, 2H), 3.89 (d, J = 11.7 Hz, 2H), 3.79–3.58 (m, 4H), 3.30 (q, J = 7.1 Hz, 4H), 2.88 (t, J = 12.3 Hz, 1H), 2.58 (t, J = 11.4 Hz, 1H), 2.30 (t, J = 11.7 Hz, 1H), 2.14–1.94 (m, 4H), 1.93–1.82 (m, 2H), 1.80–1.67 (m, 1H), 1.56–1.42 (m, 1H), 1.18 (t, J = 7.1 Hz, 6H). LC-MS (ESI) m/z 508.2 [M + H]+. Purity: >98%.
(R)-4-((3-(4-(Difluoromethyl)piperidine-1-carbonyl)piperidin-1-yl)sulfonyl)-N,N-diethylbenzenesulfonamide (17).
The title compound was synthesized using a similar procedure as described for 4 by employing 36 (15 mg, 0.037 mmol) and 4-(difluoromethyl)piperidine (6.4 mg, 0.037 mmol) in DMF (1 mL) to give a white solid (22 mg, 63%). 1H NMR (300 MHz, CDCl3) δ 7.98 (d, J = 8.2 Hz, 2H), 7.89 (d, J = 8.2 Hz, 2H), 5.64 (t, J = 56.5 Hz, 2H), 4.69 (d, J = 13.4 Hz, 1H), 3.99 (d, J = 13.7 Hz, 1H), 3.88 (d, J = 11.6 Hz, 2H), 3.30 (q, J = 7.1 Hz, 4H), 3.10 (q, J = 13.5 Hz, 1H), 2.85 (d, J = 12.0 Hz, 1H), 2.58 (q, J = 12.2 Hz, 2H), 2.29 (t, J = 11.7 Hz, 1H), 2.12 – 1.70 (m, 7H), 1.54 – 1.31 (m, 4H), 1.18 (t, J = 7.1 Hz, 6H). LC-MS (ESI) m/z 522.2 [M + H]+. Purity: 99.6%
(R)-N,N-Diethyl-4-((3-(4-(trifluoromethyl)piperidine-1-carbonyl)piperidin-1-yl)sulfonyl)benzenesulfonamide (18).
The title compound was synthesized using a similar procedure as described for 4 by employing 36 (15 mg, 0.037 mmol) and 4-(trifluoromethyl)piperidine (5.7 mg, 0.037 mmol) in DMF (1 mL) to give a white solid (10 mg, 50%). 1H NMR (300 MHz, CDCl3) δ 7.98 (d, J = 8.2 Hz, 2H), 7.89 (d, J = 8.2 Hz, 2H), 4.71 (d, J = 13.6 Hz, 1H), 4.02 (d, J = 13.7 Hz, 1H), 3.88 (d, J = 11.6 Hz, 2H), 3.30 (q, J = 7.1 Hz, 2H), 3.17–3.00 (m, 1H), 2.86 (t, J = 11.8 Hz, 1H), 2.56 (t, J = 12.3 Hz, 2H), 2.29 (t, J = 11.6 Hz, 2H), 2.09–1.83 (m, 4H), 1.74 (d, J = 12.6 Hz, 1H), 1.53–1.41 (m, 2H), 1.18 (t, J = 7.1 Hz, 6H). LC-MS (ESI) m/z 540.2 [M + H]+. Purity: >98%.
Synthetic procedures for 19-22 (Scheme 2).
Ethyl (R)-1-((4-bromophenyl)sulfonyl)piperidine-3-carboxylate (37).
To a solution of ethyl (R)-piperidine-3-carboxylate (0.92 g, 5.86 mmol) and triethylamine (1.18 g, 11.7 mmol) was added 4-bromobenzenesulfonyl chloride (1.50 g, 5.86 mmol) portion wise. The mixture was stirred at room temperature overnight. The mixture was concentrated and purified with flash chromatography (10% EtOAc in hexane) to give 37 as a white solid. (2.16 g, 98%). 1H NMR (300 MHz, CDCl3) δ 7.67 (q, J = 8.6 Hz, 4H), 4.16 (q, J = 7.2 Hz, 2H), 3.85 (d, J = 11.0 Hz, 1H), 3.63 (d, J = 11.8 Hz, 1H), 2.68–2.49 (m, 2H), 2.38 (td, J = 11.3, 3.2 Hz, 1H), 2.02 (d, J = 12.6 Hz, 1H), 1.88–1.77 (m, 1H), 1.77–1.61 (m, 1H), 1.50–1.34 (m, 1H), 1.28 (t, J = 7.1 Hz, 3H).
Ethyl (R)-1-((4-(benzylthio)phenyl)sulfonyl)piperidine-3-carboxylate (38).
The title compound was synthesized using a similar procedure as described for 34 by employing 37 (1.20 g, 3.19 mmol) and benzyl mercaptan (0.40 g, 3.19 mmol) to give a white solid (1.17 g, 87%). 1H NMR (300 MHz, CDCl3) δ 7.63 (d, J = 8.5 Hz, 2H), 7.43–7.26 (m, 7H), 4.24 (s, 2H), 4.15 (q, J = 7.1 Hz, 2H), 3.84 (d, J = 11.3 Hz, 1H), 3.61 (d, J = 11.2 Hz, 1H), 2.69–2.56 (m, 1H), 2.50 (t, J = 10.8 Hz, 1H), 2.34 (td, J = 11.2, 3.0 Hz, 1H), 2.00 (d, J = 13.8 Hz, 1H), 1.87–1.75 (m, 1H), 1.73–1.57 (m, 1H), 1.47–1.34 (m, 1H), 1.28 (t, J = 7.1 Hz, 3H).
(R)-(1-((4-(Benzylthio)phenyl)sulfonyl)piperidin-3-yl)(4,4-difluoropiperidin-1-yl)methanone (39).
To a solution of 38 (139 mg, 0.33 mmol) in THF (2mL) and H2O (2 mL) was added LiOH.H2O (69 mg, 1.65 mmol) at 0 °C and stirred at room temperature for 5h. The mixture was then diluted with H2O, and the pH was adjusted to 3 by 1N HCl solution. The mixture was extracted with EtOAc, and the organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated to give the corresponding carboxylic acid. The carboxylic acid was directly dissolved in DMF (5 mL), then HATU (190 mg, 0.50 mmol), 4,4-difluoropiperidine (40 mg, 0.33 mmol) and DIEA (129 mg, 1.0 mmol) was subsequently added. The mixture was stirred at room temperature overnight. The mixture was then diluted with EtOAc, washed with brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified with flash chromatography (20% EtOAc in hexane) to give 39 as a light-yellow gel (145 mg, 90%). 1H NMR (300 MHz, CDCl3) δ 7.69–7.52 (m, 2H), 7.48–7.27 (m, 7H), 4.23 (s, 2H), 3.92–3.47 (m, 6H), 2.87 (t, J = 11.6 Hz, 1H), 2.46 (t, J = 11.4 Hz, 1H), 2.22 (td, J = 11.9, 2.9 Hz, 1H), 2.12–1.90 (m, 4H), 1.83 (t, J = 7.6 Hz, 2H), 1.69 (d, J = 32.5 Hz, 3H), 1.44 (td, J = 12.9, 9.1 Hz, 1H).
(R)-4-((3-(4,4-Difluoropiperidine-1-carbonyl)piperidin-1-yl)sulfonyl)benzenesulfonyl chloride (40).
The title compound was synthesized using a similar procedure as described for 35 by employing 39 (50 mg, 0.10 mmol) and 1,3-dichloro-5,5-dimethylimidazolidine-2,4-dione (40 mg, 0.20 mmol), colorless gel (47 mg, 100% crude). 1H NMR (300 MHz, CDCl3) δ 8.21 (d, J = 8.6 Hz, 2H), 8.00 (d, J = 8.5 Hz, 2H), 3.88 (d, J = 11.2 Hz, 2H), 3.77–3.55 (m, 4H), 2.95–2.83 (m, 1H), 2.59 (t, J = 11.3 Hz, 1H), 2.33 (td, J = 11.9, 2.8 Hz, 1H), 2.05–1.68 (m, 8H).
(R)-4-((3-(4,4-Difluoropiperidine-1-carbonyl)piperidin-1-yl)sulfonyl)-N-isopropylbenzenesulfonamide (19).
To a solution of isopropylamine (3 mg, 0.05 mmol) and triethylamine (15 mg, 0.15 mmol) was added 40 (24 mg, 0.05 mmol) portion wise. The mixture was stirred at room temperature overnight. The mixture was concentrated and purified with flash chromatography (10% MeOH in DCM) to give 19 as a white solid (10 mg, 37%). 1H NMR (300 MHz, CDCl3) δ 8.05 (d, J = 8.4 Hz, 2H), 7.90 (d, J = 8.3 Hz, 2H), 4.67 (d, J = 7.7 Hz, 1H), 3.88 (d, J = 11.6 Hz, 2H), 3.77–3.49 (m, 5H), 2.96–2.82 (m, 1H), 2.58 (t, J = 11.3 Hz, 1H), 2.37–2.22 (m, 1H), 2.09–1.82 (m, 6H), 1.80–1.65 (m, 1H), 1.56–1.42 (m, 1H), 1.13 (d, J = 6.5 Hz, 6H). LC-MS (ESI) m/z 494.2 [M + H]+. Purity: 99.4%.
(R)-4-((3-(4,4-Difluoropiperidine-1-carbonyl)piperidin-1-yl)sulfonyl)-N,N-dimethylbenzenesulfonamide (20).
The title compound was synthesized using a similar procedure as described for 19 by employing 40 (50 mg, 0.10 mmol) and dimethyl amine (2.3 mg, 0.05 mmol) to give a white solid (14 mg, 58%). 1H NMR (300 MHz, CDCl3) δ 7.95 (s, 4H), 3.90 (d, J = 11.8 Hz, 2H), 3.78–3.58 (m, 4H), 2.89 (t, J = 11.3 Hz, 1H), 2.79 (s, 6H), 2.60 (t, J = 11.3 Hz, 1H), 2.38–2.27 (m, 1H), 2.10–1.83 (m, 6H), 1.80–1.69 (m, 1H), 1.55–1.43 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 171.04, 140.66, 140.34, 128.39, 128.17, 121.17 (t, J = 242.3 Hz), 48.47, 46.26, 42.23, 38.78, 38.64, 37.80, 34.85 (t, J = 23.8 Hz), 33.64 (t, J = 22.9 Hz), 27.29, 24.46. LC-MS (ESI) m/z 502.1 [M + Na]+. Purity: >98%
(4,4-Difluoropiperidin-1-yl)((R)-1-((4-(((R)-3-hydroxypyrrolidin-1-yl)sulfonyl)phenyl)sulfonyl)piperidin-3-yl)methanone (21).
The title compound was synthesized using a similar procedure as described for 19 by employing 40 (24 mg, 0.05 mmol) and (R)-pyrrolidin-3-ol hydrochloride (6.2 mg, 0.05 mmol) to give a white solid (14 mg, 54%). 1H NMR (400 MHz, CDCl3) δ 8.07–8.00 (m, 2H), 7.95–7.88 (m, 2H), 4.38 (s, 1H), 3.90 (d, J = 11.9 Hz, 1H), 3.75–3.61 (m, 4H), 3.57–3.51 (m, 1H), 3.49–3.44 (m, 1H), 3.42–3.35 (m, 1H), 3.26 (dt, J = 11.4, 1.6 Hz, 1H), 2.93–2.84 (m, 1H), 2.39–2.24 (m, 3H), 2.12–1.95 (m, 5H), 1.93–1.81 (m, 3H), 1.77–1.70 (m, 1H), 1.47–1.33 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 171.35, 140.94, 139.72, 128.14, 128.07, 121.08 (t, J = 242.4 Hz), 70.36, 56.10, 48.41, 46.42, 46.22, 42.29, 38.70, 38.42, 34.85 (t, J = 23.7 Hz), 34.46, 33.72 (t, J = 23.8 Hz), 27.21, 24.34. LC-MS (ESI) m/z 522.0 [M + H]+. Purity: 98.4%.
(4,4-Difluoropiperidin-1-yl)((R)-1-((4-(((S)-3-hydroxypyrrolidin-1-yl)sulfonyl)phenyl)sulfonyl)piperidin-3-yl)methanone (22).
The title compound was synthesized using a similar procedure as described for 19 by employing 40 (24 mg, 0.05 mmol) and (S)-pyrrolidin-3-ol hydrochloride (6.2 mg, 0.05 mmol) to give a white solid (15 mg, 58%). 1H NMR (300 MHz, CDCl3) δ 8.13–7.97 (m, 2H), 7.91 (d, J = 8.2 Hz, 2H), 4.52–4.31 (m, 2H), 3.88 (d, J = 11.6 Hz, 1H), 3.67 (dt, J = 19.7, 7.4 Hz, 5H), 3.55–3.39 (m, 2H), 3.31 (td, J = 12.9, 12.0, 7.0 Hz, 2H), 2.88 (t, J = 11.5 Hz, 1H), 2.45 (s, 1H), 2.40–2.13 (m, 3H), 2.13–1.79 (m, 9H), 1.71 (s, 5H), 1.51–1.25 (m, 2H). LC-MS (ESI) m/z 544.0 [M + Na]+. Purity: 96.3%.
Synthetic procedures for 23–28 (Scheme 3).
(R)-(1-((4-Bromophenyl)sulfonyl)piperidin-3-yl)(4,4-difluoropiperidin-1-yl)methanone (41).
The title compound was synthesized using a similar procedure as described for 39 by employing 37 (188 mg, 0.50 mmol) and 4,4-difluropiperidine (67 mg, 0.55 mmol) to give a white solid (205 mg, 91%). 1H NMR (300 MHz, CDCl3) δ 7.70 (d, J = 8.3 Hz, 2H), 7.63 (d, J = 8.4 Hz, 2H), 3.84 (d, J = 11.6 Hz, 2H), 3.77–3.57 (m, 4H), 2.88 (t, J = 11.6 Hz, 1H), 2.50 (t, J = 11.3 Hz, 1H), 2.25 (t, J = 11.8 Hz, 1H), 2.15–1.79 (m, 6H), 1.79–1.66 (m, 1H), 1.54–1.39 (m, 1H).
(R)-(4,4-Difluoropiperidin-1-yl)(1-((4-(isopropylthio)phenyl)sulfonyl)piperidin-3-yl)methanone (42a).
The title compound was synthesized using a similar procedure as described for 34 by employing 41 (200 mg, 0.44 mmol) and propane-2-thiol (34 mg, 0.44 mmol) to give a white solid (177 mg, 90%). 1H NMR (300 MHz, CDCl3) δ 7.65 (d, J = 8.3 Hz, 2H), 7.42 (d, J = 8.3 Hz, 2H), 3.84 (d, J = 11.6 Hz, 2H), 3.77–3.52 (m, 5H), 2.95–2.81 (m, 1H), 2.50 (t, J = 11.3 Hz, 1H), 2.25 (t, J = 11.7 Hz, 1H), 2.14–1.91 (m, 4H), 1.85 (d, J = 12.6 Hz, 2H), 1.76–1.68 (m, 1H), 1.52–1.44 (m, 1H), 1.40 (d, J = 6.7 Hz, 6H).
(R)-(4,4-Difluoropiperidin-1-yl)(1-((4-(isobutylthio)phenyl)sulfonyl)piperidin-3-yl)methanone (42b).
The title compound was synthesized using a similar procedure as described for 34 by employing 41 (50 mg, 0.11 mmol) and 2-methylpropane-1-thiol (10 mg, 0.11 mmol) to give a white solid (43 mg, 85%). 1H NMR (300 MHz, CDCl3) δ 7.63 (d, J = 8.2 Hz, 2H), 7.36 (d, J = 8.2 Hz, 2H), 3.83 (d, J = 11.7 Hz, 2H), 3.78–3.58 (m, 4H), 2.89 (d, J = 6.9 Hz, 3H), 2.48 (t, J = 11.4 Hz, 1H), 2.24 (t, J = 11.7 Hz, 1H), 2.12–1.89 (m, 5H), 1.90–1.66 (m, 3H), 1.53–1.38 (m, 1H), 1.10 (d, J = 6.7 Hz, 6H). LC-MS (ESI) m/z 461.0 [M + H]+.
(R)-(4,4-difluoropiperidin-1-yl)(1-((4-(pentan-3-ylthio)phenyl)sulfonyl)piperidin-3-yl)methanone (42c).
The title compound was synthesized using a similar procedure as described for 34 by employing 41 (90 mg, 0.20 mmol) and pentane-3-thiol (19 mg, 0.18 mmol) to give a white solid (64 mg, 67%). 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 8.6 Hz, 2H), 7.41 (d, J = 8.5 Hz, 2H), 3.88–3.80 (m, 2H), 3.80–3.57 (m, 4H), 3.21 (p, J = 6.3 Hz, 1H), 2.92–2.80 (m, 1H), 2.51 (dd, J = 12.0, 10.8 Hz, 1H), 2.26 (td, J = 12.0, 2.7 Hz, 1H), 1.99 (dt, J = 12.7, 6.1 Hz, 2H), 1.89–1.79 (m, 2H), 1.79–1.65 (m, 5H), 1.63 (d, J = 2.2 Hz, 2H), 1.54–1.39 (m, 1H), 1.05 (t, J = 7.4 Hz, 6H).
(R)-(1-((4-(Cyclopropylthio)phenyl)sulfonyl)piperidin-3-yl)(4,4-difluoropiperidin-1-yl)methanone (42d).
The title compound was synthesized using a similar procedure as described for 34 by employing 41 (40 mg, 0.089 mmol) and cyclopropanethiol (6.6 mg, 0.089 mmol) to give a white solid (26 mg, 65%). 1H NMR (300 MHz, CDCl3) δ 7.63 (d, J = 7.0 Hz, 2H), 7.47 (d, J = 8.3 Hz, 2H), 3.89–3.76 (m, 2H), 3.77 – 3.56 (m, 4H), 2.88 (t, J = 11.6 Hz, 1H), 2.47 (t, J = 11.1 Hz, 1H), 2.28–2.17 (m, 2H), 2.10–1.88 (m, 4H), 1.89–1.67 (m, 3H), 1.52–1.34 (m, 1H), 1.18 (d, J = 6.9 Hz, 2H), 0.80–0.70 (m, 2H).
(R)-(1-((4-(Cyclobutylthio)phenyl)sulfonyl)piperidin-3-yl)(4,4-difluoropiperidin-1-yl)methanone (42e).
The title compound was synthesized using a similar procedure as described for 34 by employing 41 (40 mg, 0.089 mmol) and cyclobutanethiol (7.8 mg, 0.089 mmol) to give a white solid (36 mg, 88%). 1H NMR (300 MHz, CDCl3) δ 7.59 (d, J = 8.5 Hz, 2H), 7.24 (d, J = 8.5 Hz, 2H), 4.05–3.94 (m, 1H), 3.86–3.75 (m, 2H), 3.74–3.58 (m, 4H), 2.93–2.78 (m, 1H), 2.64–2.51 (m, 1H), 2.45 (t, J = 11.3 Hz, 1H), 2.26–1.92 (m, 10H), 1.87–1.64 (m, 3H), 1.48–1.37 (m, 1H).
(R)-(4,4-Difluoropiperidin-1-yl)(1-((4-(oxetan-3-ylthio)phenyl)sulfonyl)piperidin-3-yl)methanone (42f).
The title compound was synthesized using a similar procedure as described for 34 by employing 41 (40 mg, 0.089 mmol) and oxetane-3-thiol (8.0 mg, 0.089 mmol) to give a white solid (16 mg, 39%). 1H NMR (300 MHz, CDCl3) δ 7.64 (d, J = 8.4 Hz, 2H), 7.20 (d, J = 8.4 Hz, 2H), 5.15 (t, J = 6.6 Hz, 2H), 4.69 (t, J = 6.3 Hz, 2H), 4.65–4.58 (m, 1H), 3.90–3.58 (m, 6H), 2.94–2.83 (m, 1H), 2.47 (t, J = 11.3 Hz, 1H), 2.22 (td, J = 11.8, 2.7 Hz, 1H), 2.12–1.89 (m, 4H), 1.88–1.66 (m, 3H), 1.50–1.35 (m, 1H).
(R)-(4,4-Difluoropiperidin-1-yl)(1-((4-(isopropylsulfonyl)phenyl)sulfonyl)piperidin-3-yl)methanone (23).
To a solution of 42a (317 mg, 0.71 mmol) in DCM (15 mL) was added mCPBA (492 mg, 2.84 mmol) and stirred at room temperature overnight. Saturated NaHCO3 was added, and the mixture was extracted with EtOAc. The organic layer was washed with brine, dried with Na2SO4, filtered, and concentrated. The residue was purified with flash chromatography (20% EtOAc in hexane) to give 23 as a white solid (221 mg, 65%). 1H NMR (300 MHz, CDCl3) δ 8.08 (d, J = 8.1 Hz, 2H), 7.96 (d, J = 8.1 Hz, 2H), 3.95–3.84 (m, 2H), 3.78–3.58 (m, 4H), 3.26 (p, J = 6.8 Hz, 1H), 2.89 (t, J = 11.6 Hz, 1H), 2.59 (t, J = 11.4 Hz, 1H), 2.31 (td, J = 12.0, 2.7 Hz, 1H), 2.14–1.83 (m, 6H), 1.80–1.69 (m, 1H), 1.55–1.44 (m, 1H), 1.35 (dd, J = 6.9, 2.2 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 170.97, 141.74, 141.37, 129.99, 128.09, 121.15 (t, J = 243.2 Hz), 55.70, 48.49, 46.26, 42.23, 38.79, 38.64, 34.86 (t, J = 22.7 Hz), 33.74 (t, J = 25.2 Hz), 27.30, 24.46, 15.63, 15.56. LC-MS (ESI) m/z 479.0 [M + H]+. Purity: 99.1%.
(R)-(4,4-Difluoropiperidin-1-yl)(1-((4-(isobutylsulfonyl)phenyl)sulfonyl)piperidin-3-yl)methanone (24).
The title compound was synthesized using a similar procedure as described for 23 by employing 42b (21 mg, 0.046 mmol) and mCPBA (31 mg, 0.18 mmol) to give a white solid (8 mg, 35%). 1H NMR (300 MHz, CDCl3) δ 8.10 (d, J = 8.2 Hz, 2H), 7.96 (d, J = 8.4 Hz, 2H), 3.89 (d, J = 11.8 Hz, 2H), 3.79–3.59 (m, 4H), 3.05 (d, J = 6.5 Hz, 2H), 2.89 (t, J = 11.4 Hz, 1H), 2.58 (t, J = 11.4 Hz, 1H), 2.40–2.24 (m, 2H), 2.16–1.82 (m, 6H), 1.81–1.66 (m, 1H), 1.56–1.41 (m, 1H), 1.12 (d, J = 6.7 Hz, 6H). LC-MS (ESI) m/z 491.3 [M - H]−. Purity: 96.4%.
(R)-(4,4-Difluoropiperidin-1-yl)(1-((4-(pentan-3-ylsulfonyl)phenyl)sulfonyl)piperidin-3-yl)methanone (25).
The title compound was synthesized using a similar procedure as described for 23 by employing 42c (22 mg, 0.046 mmol) and mCPBA (31 mg, 0.18 mmol) to give a white solid (12 mg, 52%). 1H NMR (300 MHz, CDCl3) δ 8.07 (d, J = 8.5 Hz, 2H), 7.95 (d, J = 8.4 Hz, 2H), 3.90 (d, J = 11.7 Hz, 2H), 3.79–3.58 (m, 4H), 2.96–2.81 (m, 2H), 2.60 (t, J = 11.4 Hz, 1H), 2.37–2.26 (m, 1H), 2.13–1.81 (m, 8H), 1.83–1.67 (m, 3H), 1.55–1.41 (m, 1H), 1.05 (t, J = 7.5 Hz, 6H). LC-MS (ESI) m/z 507.3 [M + H]+. Purity: >98%.
(R)-(1-((4-(Cyclopropylsulfonyl)phenyl)sulfonyl)piperidin-3-yl)(4,4-difluoropiperidin-1-yl)methanone (26).
The title compound was synthesized using a similar procedure as described for 23 by employing 42d (26 mg, 0.058 mmol) and mCPBA (42 mg, 0.24 mmol) to give a white solid (18 mg, 64%). 1H NMR (300 MHz, CDCl3) δ 8.09 (d, J = 8.5 Hz, 2H), 7.95 (d, J = 8.3 Hz, 2H), 3.99–3.83 (m, 2H), 3.80–3.57 (m, 4H), 2.90 (t, J = 11.5 Hz, 1H), 2.66–2.46 (m, 2H), 2.39–2.24 (m, 1H), 2.14–1.82 (m, 6H), 1.73 (t, J = 13.0 Hz, 1H), 1.56–1.37 (m, 3H), 1.20–1.08 (m, 2H). LC-MS (ESI) m/z 476.7 [M + H]+. Purity: 98.4%.
(R)-(1-((4-(Cyclobutylsulfonyl)phenyl)sulfonyl)piperidin-3-yl)(4,4-difluoropiperidin-1-yl)methanone (27).
The title compound was synthesized using a similar procedure as described for 23 by employing 42e (18 mg, 0.039 mmol) and mCPBA (27 mg, 0.16 mmol) to give a white solid (13 mg, 68%). 1H NMR (300 MHz, CDCl3) δ 8.06 (d, J = 8.3 Hz, 2H), 7.94 (d, J = 8.4 Hz, 2H), 3.96–3.80 (m, 3H), 3.77–3.55 (m, 4H), 2.89 (t, J = 11.6 Hz, 1H), 2.74–2.51 (m, 3H), 2.35–2.19 (m, 3H), 2.10–1.86 (m, 8H), 1.80–1.69 (m, 1H), 1.52–1.40 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 170.99, 142.46, 141.59, 129.16, 128.25, 121.17 (t, J = 242.3 Hz), 56.81, 48.46, 46.25, 42.22, 38.75, 38.63, 34.84 (t, J = 24.1 Hz), 33.73 (t, J = 23.1 Hz), 27.29, 24.44, 22.77, 22.74, 16.90. LC-MS (ESI) m/z 491.1 [M + H]+. Purity: >98%.
(R)-(4,4-Difluoropiperidin-1-yl)(1-((4-(oxetan-3-ylsulfonyl)phenyl)sulfonyl)piperidin-3-yl)methanone (28).
The title compound was synthesized using a similar procedure as described for 23 by employing 42f (16 mg, 0.035 mmol) and mCPBA (24 mg, 0.14 mmol) to give a white solid (10 mg, 59%). 1H NMR (300 MHz, CDCl3) δ 8.09 (d, J = 8.2 Hz, 2H), 7.98 (d, J = 8.4 Hz, 2H), 5.07 – 4.97 (m, 2H), 4.87 (td, J = 7.7, 3.5 Hz, 2H), 4.52 (tt, J = 7.9, 6.1 Hz, 1H), 3.95–3.82 (m, 2H), 3.79–3.58 (m, 4H), 2.89 (t, J = 11.5 Hz, 1H), 2.59 (t, J = 11.3 Hz, 1H), 2.32 (td, J = 11.9, 2.7 Hz, 1H), 2.14–1.81 (m, 6H), 1.81–1.62 (m, 1H), 1.53–1.38 (m, 1H). LC-MS (ESI) m/z 493.1 [M + H]+. Purity: 98.4%.
Synthetic procedures for 29–31 (Scheme 4).
Scheme 4. Synthetic Route for 29-31.
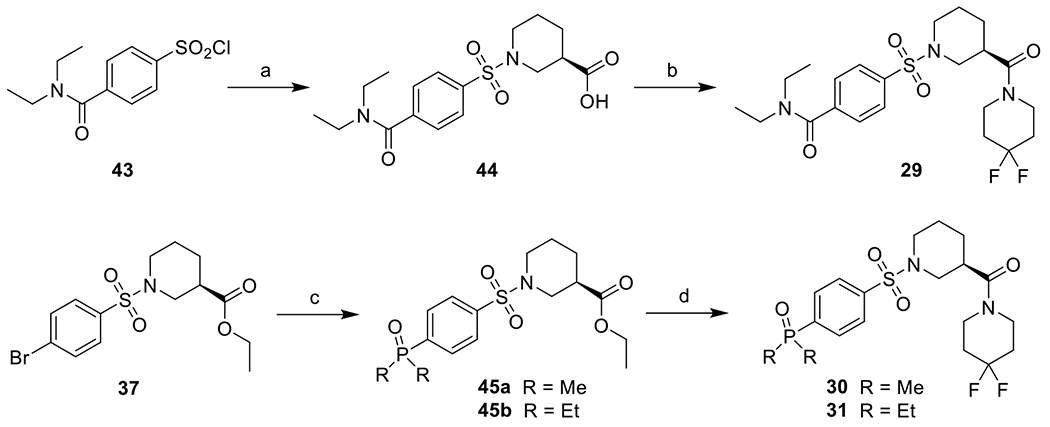
Reagents and conditions: a) (R)-piperidine-3-carboxylic acid, Na2CO3, THF, H2O, rt, 98%; b) 4,4-difluoropiperidine, HATU, DIEA, DMF, rt, 32%; c) dimethylphosphine oxide or diethylphosphine oxide, Pd(OAc)2, XantPhos, K3PO4, DMF, 120 °C, 88-95%; d) i. LiOH, THF, H2O, rt; ii. 4,4-difluoropiperidine, HATU, DIEA, DMF, rt, 83-95%.
(R)-1-((4-(Diethylcarbamoyl)phenyl)sulfonyl)piperidine-3-carboxylic acid (44).
To a solution of 43 (100 mg, 0.36 mmol) and Na2CO3 (114 mg, 1.08 mmol) in H2O (1.5 mL) was added a solution of (R)-piperidine-3-carboxylic acid (47 mg, 0.36 mmol) in THF (1.5 mL) dropwise at 0 °C and stirred at room temperature for 3h. THF was removed by evaporation and the pH was adjusted to 3 by 1N HCl solution. It was extracted with EtOAc, and the organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated to give 44 as a white solid (130 mg, 98%). 1H NMR (300 MHz, CDCl3) δ 7.83 (d, J = 8.0 Hz, 2H), 7.55 (d, J = 7.9 Hz, 2H), 3.85 (d, J = 11.1 Hz, 1H), 3.67–3.51 (m, 3H), 3.24 (q, J = 6.7 Hz, 2H), 2.73–2.49 (m, 2H), 2.49–2.36 (m, 1H), 2.01 (d, J = 13.5 Hz, 1H), 1.88–1.76 (m, 1H), 1.68 (q, J = 13.9, 12.4 Hz, 1H), 1.47–1.35 (m, 1H), 1.29 (t, J = 7.1 Hz, 3H), 1.14 (t, J = 6.9 Hz, 3H).
(R)-4-((3-(4,4-difluoropiperidine-1-carbonyl)piperidin-1-yl)sulfonyl)-N,N-diethylbenzamide (29).
To a solution of 44 (25 mg, 0.068 mmol) and HATU (39 mg, 0.102 mmol) in DMF (1 mL) was added 4,4-difluoropiperidine (8.2 mg, 0.068 mmol) and DIEA (26 mg, 0.204 mmol). The mixture was stirred at room temperature overnight. The mixture was then diluted with EtOAc, washed with brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified with flash chromatography (10% MeOH in DCM) to give 29 as a white solid (18 mg, 32%). 1H NMR (300 MHz, CDCl3) δ 7.81 (d, J = 8.1 Hz, 2H), 7.54 (d, J = 8.2 Hz, 2H), 3.93–3.81 (m, 2H), 3.78–3.52 (m, 6H), 3.30–3.16 (m, 2H), 2.86 (tt, J = 11.2, 3.4 Hz, 1H), 2.56 (t, J = 11.4 Hz, 1H), 2.29 (td, J = 11.9, 2.8 Hz, 1H), 2.13–1.79 (m, 6H), 1.78–1.64 (m, 1H), 1.55–1.37 (m, 1H), 1.28 (t, J = 7.1 Hz, 3H), 1.14 (t, J = 7.1 Hz, 3H). LC-MS (ESI) m/z 494.2 [M + Na]+. Purity: 99.4%.
Ethyl (R)-1-((4-(dimethylphosphoryl)phenyl)sulfonyl)piperidine-3-carboxylate (45a).
To a suspension of 37 (60 mg, 0.16 mmol), dimethylphosphine oxide (14 mg, 0.18 mmol) and K3PO4 (0.19 mmol) in DMF (4 mL) was added Pd(OAc)2 (1.8 mg, 0.008 mmol) and XantPhos (5.6 mg, 0.0096 mmol) under argon. The mixture was heated at 120 °C under argon for 5h. The mixture was then partitioned between EtOAc and water, and the organic layer was separated, washed with brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified with silica chromatography (DCM:MeOH = 10:1) to give 45a as a white solid (51 mg, 88%). 1H NMR (300 MHz, CDCl3) δ 7.86–7.66 (m, 4H), 3.97 (q, J = 7.1 Hz, 2H), 3.64 (d, J = 7.4 Hz, 1H), 3.43 (dt, J = 11.8, 4.1 Hz, 1H), 2.53–2.36 (m, 2H), 2.27 (td, J = 11.2, 3.0 Hz, 1H), 1.64 (d, J = 13.1 Hz, 7H), 1.58 – 1.37 (m, 1H), 1.35–1.16 (m, 1H), 1.09 (t, J = 7.1 Hz, 3H). LC-MS (ESI) m/z 396.0 [M + Na]+.
Ethyl (R)-1-((4-(diethylphosphoryl)phenyl)sulfonyl)piperidine-3-carboxylate (45b).
The title compound was synthesized using a similar procedure as described for 45a by employing 37 (80 mg, 0.21 mmol) and diethylphosphine oxide (25 mg, 0.23 mmol) to give a white solid (80 mg, 95%). 1H NMR (300 MHz, CDCl3) δ 7.88 (d, J = 6.2 Hz, 4H), 4.13 (q, J = 7.1 Hz, 2H), 3.85 (d, J = 8.5 Hz, 1H), 3.70–3.58 (m, 1H), 2.67–2.53 (m, 2H), 2.41 (td, J = 11.4, 3.1 Hz, 1H), 2.12–1.85 (m, 5H), 1.89–1.75 (m, 1H), 1.75–1.56 (m, 1H), 1.50–1.34 (m, 1H), 1.25 (t, J = 7.1 Hz, 3H), 1.21–1.05 (m, 6H). LC-MS (ESI) m/z 424.1 [M + Na]+.
(R)-(4,4-Difluoropiperidin-1-yl)(1-((4-(dimethylphosphoryl)phenyl)sulfonyl)piperidin-3-yl)methanone (30).
The title compound was synthesized using a similar procedure as described for 39 by employing 45a (19 mg, 0.052 mmol) and 4,4-difluoropiperidine (7.6 mg, 0.063 mmol) to give a white solid (19 mg, 83%). 1H NMR (300 MHz, CDCl3) δ 7.99–7.86 (m, 4H), 3.87 (t, J = 11.3 Hz, 2H), 3.80–3.59 (m, 4H), 2.90 (t, J = 11.7 Hz, 1H), 2.53 (t, J = 11.3 Hz, 1H), 2.29 (td, J = 11.8, 2.7 Hz, 1H), 2.14–1.90 (m, 4H), 1.89–1.72 (m, 9H), 1.51–1.40 (m, 1H). LC-MS (ESI) m/z 470.9 [M + Na]+. Purity: 97.3%.
(R)-(1-((4-(diethylphosphoryl)phenyl)sulfonyl)piperidin-3-yl)(4,4-difluoropiperidin-1-yl)methanone (31).
The title compound was synthesized using a similar procedure as described for 39 by employing 45b (16 mg, 0.040 mmol) and 4,4-difluoropiperidine (5.8 mg, 0.048 mmol) to give a white solid (18 mg, 95%). 1H NMR (300 MHz, CDCl3) δ 7.89 (d, J = 6.2 Hz, 4H), 3.96–3.83 (m, 2H), 3.78–3.60 (m, 4H), 3.51–3.42 (m, 1H), 2.96–2.83 (m, 1H), 2.56 (t, J = 11.3 Hz, 1H), 2.30 (t, J = f11.8 Hz, 1H), 2.18–1.87 (m, 9H), 1.79–1.70 (m, 1H), 1.55–1.41 (m, 1H), 1.26–1.06 (m, 6H). LC-MS (ESI) m/z 499.0 [M + Na]+. Purity: 99.1%.
Biological assays
Growth Inhibition Assay.
Cell growth inhibition was assessed by MTT assay. MIA PaCa-2 cells were seeded in 96-well plates at 200 cells/well (glucose-containing medium) or 8000 cells/well (galactose-containing medium) while BxPC-3 cells were seeded at density of 300 cells/well (glucose-containing medium). After overnight incubation at 37 °C and 5% CO2, cells were treated with indicated compounds. The plates were incubated with compound or DMSO control for 7 days at 37 °C and 5% CO2. MTT solution (20 μL, 3 mg/mL) was added to the wells, and the cells were incubated for 4 h at 37 °C. Supernatant was removed, and DMSO (100 μL) was added to each well. The plates were shaken for 15 min at room temperature, and absorbance of the formazan crystals was measured at 570 nm. Cell growth inhibition was assessed by the cell viability rate as [1 − (At −Ab)/(Ac − Ab)] × 100 (At, Ac, and Ab are the absorbance values from cells which were treated with compound, cells which were not treated with compound, and blank, respectively).
Quantification of total cellular ATP in glucose- and galactose-containing medium.
Glucose-containing medium represents an RPMI 1640 medium (Thermo Fisher, 11875101) containing 10% FBS (Thermo Fisher, 10082147). Galactose-containing medium was prepared by adding 10 mM galactose (#G0750, Sigma-Aldrich) to glucose-free RPMI 1640 medium (Thermo Fisher, 11879020) containing 10% FBS. Cells were seeded into 96-well plates (Corning, 3603) at the density of 10000 cells/well. The total cellular ATP content was determined 24 hours after compound treatment using the CellTiter-Glo® Luminescence Cell Viability Assay according to the manufacturer’s protocol (Promega Corporation, G7570). Luminescence signals were quantified using a Synergy H1 Hybrid Multi-Mode Reader (BioTek, Winooski, VT).
Determination of NAD+/NADH ratio.
MIA PaCa-2 cells were seeded at a density of 5 × 103 cells/well in flat bottom 96-well plates. The next day, cells were treated with indicated concentration of compounds diluted in RPMI 1640 with 10% FBS for 48 hours. The medium was aspirated, and cells were washed with PBS. After addition of 100 µL PBS, cells were lysed by adding 100 µL of bicarbonate base buffer with 1% DTAB. Samples were split into separate wells for acid and base treatments as described in the NAD/NADH-Glo™ Assay protocol (G9071). Briefly, to measure NAD+, 25 µL of 0.4 N HCl was added to 50 µL of cell lysate and heated at 60 °C for 15 minutes. To measure NADH, 50 µl of cell lysate was heated at 60 °C for 15 minutes directly. After heating, both samples were incubated at room temperature for 10 minutes. Trizma® base (25 µL) was added to each well of 0.4 N HCl-treated samples (NAD+) and HCl/Trizma® solution (50 µL) was added to each well of untreated samples (NADH). The intensity of NAD+ or NADH were measured using the NAD/NADH-Glo™ Assay. Both NAD+ and NADH samples (10 µL) were incubated with 10 µL of NAD/NADH-Glo™ Detection Reagent in white 384-well luminometer plates. After a 30-minute incubation, luminescence was measured on a Synergy H1 Hybrid Multi-Mode Reader (BioTek, Winooski, VT).
Animal studies.
Female C57BL/6 mice, 6-10 week old, were used for Pan02 allograft mice model. 5×106 Pan02 cells were implanted subcutaneously into right flank regions of mice. Once the tumors grew to 100 mm3, animals were randomized and treated with the indicated compounds. The compounds were prepared in the following formulations: 7.5 mg/kg of 20, 10 mg/kg of 23, and 2.5 mg/kg of 27 were dissolved in 10% DMSO, 50% PG, and 40% saline, while 15 mg/kg of 21 was dissolved in 10% DMSO, 60% PG, 30% saline. Compounds were dosed daily for 28 consecutive days. Tumor growth was monitored twice a week by digital caliper and tumor volumes were calculated by the V = (length x width2)/2 equation. The body weights of mice were monitored before the dosing and twice a week. Mice were sacrificed after 28 days of compounds administration. The University of Michigan Institutional Animal Care and Use Committee approved all animal experiments.
Statistics.
The 50% inhibitory concentration values (IC50) were determined by analyzing the log of the concentration response curves by nonlinear regression analysis using GraphPad Prism (version 5). All experiments were performed at least three separate times unless otherwise noted.
Microsomal Stability Test.
10 mM stock solutions of test compounds, and verapamil (positive control) were prepared in DMSO. Solutions were diluted to 1 mM with acetonitrile and further diluted to 10 μM with 0.1 M phosphate buffer (3.3 mM MgCl2). NADPH (1 mg) was dissolved in 60 μL of 0.1 M phosphate buffer (3.3 mM MgCl2). 330 μL of 0.1 M phosphate buffer (3.3 mM MgCl2) and 40 μL of 10 μM test compounds were added to 10 μL of microsomes (20 mg/mL). Data points were collected at 0, 5, 10, 15, 30, 45, and 60 min. Solutions were centrifuged at 3500 g for 10 min to pellet precipitated proteins. The supernatant was used for LC−MS/ MS analysis. Natural log peak area ratios (compound peak area/ internal standard peak area) were plotted against time, and the gradient of the line was determined to calculate the half-life (t1/2) of the test compound in microsomes.
Supplementary Material
ACKNOWLEDGEMENT
This work was supported by the NIH grant R01 CA188252.
ABBREVATION USED
- ATP
Adenosine triphosphate
- CYP
Cytochromes P450
- ENO1
enolase 1
- FADH2
flavin adenine dinucleotide hydroquinone form
- HLM
human liver microsome
- MLM
mouse liver microsome
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NAD+
nicotinamide adenine dinucleotide
- ND1, NADH
ubiquinone oxidoreductase chain 1
- OXPHOS
oxidative phosphorylation
- ROS
reactive oxygen species
- tPSA
topological polar surface area
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
NMR and LC/MS copies of the final products.
Molecular formula strings and the associated biological data (CSV)
REFERENCE
- (1).Ashton TM; McKenna WG; Kunz-Schughart LA; Higgins GS Oxidative phosphorylation as an emerging target in cancer therapy. Clin. Cancer Res 2018, 24, 2482–2490. [DOI] [PubMed] [Google Scholar]
- (2).Lissanu Deribe Y; Sun Y; Terranova C; Khan F; Martinez-Ledesma J; Gay J; Gao G; Mullinax RA; Khor T; Feng N; Lin YH; Wu CC; Reyes C; Peng Q; Robinson F; Inoue A; Kochat V; Liu CG; Asara JM; Moran C; Muller F; Wang J; Fang B; Papadimitrakopoulou V; Wistuba II; Rai K; Marszalek J; Futreal PA Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat. Med 2018, 24, 1047–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Boreel DF; Span PN; Heskamp S; Adema GJ; Bussink J Targeting oxidative phosphorylation to increase the efficacy of radio- and immune-combination therapy. Clin. Cancer Res 2021, 27, 2970–2978. [DOI] [PubMed] [Google Scholar]
- (4).Molina JR; Sun Y; Protopopova M; Gera S; Bandi M; Bristow C; McAfoos T; Morlacchi P; Ackroyd J; Agip AA; Al-Atrash G; Asara J; Bardenhagen J; Carrillo CC; Carroll C; Chang E; Ciurea S; Cross JB; Czako B; Deem A; Daver N; de Groot JF; Dong JW; Feng N; Gao G; Gay J; Do MG; Greer J; Giuliani V; Han J; Han L; Henry VK; Hirst J; Huang S; Jiang Y; Kang Z; Khor T; Konoplev S; Lin YH; Liu G; Lodi A; Lofton T; Ma H; Mahendra M; Matre P; Mullinax R; Peoples M; Petrocchi A; Rodriguez-Canale J; Serreli R; Shi T; Smith M; Tabe Y; Theroff J; Tiziani S; Xu Q; Zhang Q; Muller F; DePinho RA; Toniatti C; Draetta GF; Heffernan TP; Konopleva M; Jones P; Di Francesco ME; Marszalek JR An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med 2018, 24, 1036–1046. [DOI] [PubMed] [Google Scholar]
- (5).Sun Y; Bandi M; Lofton T; Smith M; Bristow CA; Carugo A; Rogers N; Leonard P; Chang Q; Mullinax R; Han J; Shi X; Seth S; Meyers BA; Miller M; Miao L; Ma X; Feng N; Giuliani V; Geck Do M; Czako B; Palmer WS; Mseeh F; Asara JM; Jiang Y; Morlacchi P; Zhao S; Peoples M; Tieu TN; Warmoes MO; Lorenzi PL; Muller FL; DePinho RA; Draetta GF; Toniatti C; Jones P; Heffernan TP; Marszalek JR Functional genomics reveals synthetic lethality between phosphogluconate dehydrogenase and oxidative phosphorylation. Cell Rep. 2019, 26, 469–482 e5. [DOI] [PubMed] [Google Scholar]
- (6).Zhang L; Yao Y; Zhang S; Liu Y; Guo H; Ahmed M; Bell T; Zhang H; Han G; Lorence E; Badillo M; Zhou S; Sun Y; Di Francesco ME; Feng N; Haun R; Lan R; Mackintosh SG; Mao X; Song X; Zhang J; Pham LV; Lorenzi PL; Marszalek J; Heffernan T; Draetta G; Jones P; Futreal A; Nomie K; Wang L; Wang M Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci. Transl. Med 2019, 11. [DOI] [PubMed] [Google Scholar]
- (7).Farge T; Saland E; de Toni F; Aroua N; Hosseini M; Perry R; Bosc C; Sugita M; Stuani L; Fraisse M; Scotland S; Larrue C; Boutzen H; Feliu V; Nicolau-Travers ML; Cassant-Sourdy S; Broin N; David M; Serhan N; Sarry A; Tavitian S; Kaoma T; Vallar L; Iacovoni J; Linares LK; Montersino C; Castellano R; Griessinger E; Collette Y; Duchamp O; Barreira Y; Hirsch P; Palama T; Gales L; Delhommeau F; Garmy-Susini BH; Portais JC; Vergez F; Selak M; Danet-Desnoyers G; Carroll M; Recher C; Sarry JE Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017, 7, 716–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bajpai R; Sharma A; Achreja A; Edgar CL; Wei C; Siddiqa AA; Gupta VA; Matulis SM; McBrayer SK; Mittal A; Rupji M; Barwick BG; Lonial S; Nooka AK; Boise LH; Nagrath D; Shanmugam M Electron transport chain activity is a predictor and target for venetoclax sensitivity in multiple myeloma. Nat. Commun 2020, 11, 1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Haq R; Shoag J; Andreu-Perez P; Yokoyama S; Edelman H; Rowe GC; Frederick DT; Hurley AD; Nellore A; Kung AL; Wargo JA; Song JS; Fisher DE; Arany Z; Widlund HR Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell 2013, 23, 302–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Vitiello GA; Medina BD; Zeng S; Bowler TG; Zhang JQ; Loo JK; Param NJ; Liu M; Moral AJ; Zhao JN; Rossi F; Antonescu CR; Balachandran VP; Cross JR; DeMatteo RP Mitochondrial inhibition augments the efficacy of imatinib by resetting the metabolic phenotype of gastrointestinal stromal tumor. Clin. Cancer Res 2018, 24, 972–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Xu Y; Xue D; Bankhead A 3rd; Neamati N Why all the fuss about oxidative phosphorylation (OXPHOS)? J. Med. Chem 2020, 63, 14276–14307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ye XQ; Li Q; Wang GH; Sun FF; Huang GJ; Bian XW; Yu SC; Qian GS Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int. J. Cancer 2011, 129, 820–831. [DOI] [PubMed] [Google Scholar]
- (13).Janiszewska M; Suva ML; Riggi N; Houtkooper RH; Auwerx J; Clement-Schatlo V; Radovanovic I; Rheinbay E; Provero P; Stamenkovic I Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Sancho P; Burgos-Ramos E; Tavera A; Bou Kheir T; Jagust P; Schoenhals M; Barneda D; Sellers K; Campos-Olivas R; Grana O; Viera CR; Yuneva M; Sainz B Jr.; Heeschen C MYC/PGC-1alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015, 22, 590–605. [DOI] [PubMed] [Google Scholar]
- (15).Valle S; Alcala S; Martin-Hijano L; Cabezas-Sainz P; Navarro D; Munoz ER; Yuste L; Tiwary K; Walter K; Ruiz-Canas L; Alonso-Nocelo M; Rubiolo JA; Gonzalez-Arnay E; Heeschen C; Garcia-Bermejo L; Hermann PC; Sanchez L; Sancho P; Fernandez-Moreno MA; Sainz B Jr. Exploiting oxidative phosphorylation to promote the stem and immunoevasive properties of pancreatic cancer stem cells. Nat. Commun 2020, 11, 5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Viale A; Pettazzoni P; Lyssiotis CA; Ying H; Sanchez N; Marchesini M; Carugo A; Green T; Seth S; Giuliani V; Kost-Alimova M; Muller F; Colla S; Nezi L; Genovese G; Deem AK; Kapoor A; Yao W; Brunetto E; Kang Y; Yuan M; Asara JM; Wang YA; Heffernan TP; Kimmelman AC; Wang H; Fleming JB; Cantley LC; DePinho RA; Draetta GF Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tataranni T; Agriesti F; Pacelli C; Ruggieri V; Laurenzana I; Mazzoccoli C; Sala GD; Panebianco C; Pazienza V; Capitanio N; Piccoli C Dichloroacetate affects mitochondrial function and stemness-associated properties in pancreatic cancer cell lines. Cells 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wang Q; Li M; Gan Y; Jiang S; Qiao J; Zhang W; Fan Y; Shen Y; Song Y; Meng Z; Yao M; Gu J; Zhang Z; Tu H Mitochondrial protein UQCRC1 is oncogenic and a potential therapeutic target for pancreatic cancer. Theranostics 2020, 10, 2141–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Broadhurst PJ; Hart AR Metformin as an adjunctive therapy for pancreatic cancer: a review of the literature on its potential therapeutic use. Dig. Dis. Sci 2018, 63, 2840–2852. [DOI] [PubMed] [Google Scholar]
- (20).Chaban Y; Boekema EJ; Dudkina NV Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim. Biophys. Acta 2014, 1837, 418–426. [DOI] [PubMed] [Google Scholar]
- (21).Sharma LK; Lu J; Bai Y Mitochondrial respiratory complex I: structure, function and implication in human diseases. Curr. Med. Chem 2009, 16, 1266–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Naguib A; Mathew G; Reczek CR; Watrud K; Ambrico A; Herzka T; Salas IC; Lee MF; El-Amine N; Zheng W; Di Francesco ME; Marszalek JR; Pappin DJ; Chandel NS; Trotman LC Mitochondrial Complex I inhibitors expose a vulnerability for selective killing of Pten-Null cells. Cell Rep. 2018, 23, 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Tocilescu MA; Fendel U; Zwicker K; Kerscher S; Brandt U Exploring the ubiquinone binding cavity of respiratory complex I. J. Biol. Chem 2007, 282, 29514–29520. [DOI] [PubMed] [Google Scholar]
- (24).Fendel U; Tocilescu MA; Kerscher S; Brandt U Exploring the inhibitor binding pocket of respiratory complex I. Biochim. Biophys. Acta 2008, 1777, 660–665. [DOI] [PubMed] [Google Scholar]
- (25).Tsuji A; Akao T; Masuya T; Murai M; Miyoshi H IACS-010759, a potent inhibitor of glycolysis-deficient hypoxic tumor cells, inhibits mitochondrial respiratory complex I through a unique mechanism. J. Biol. Chem 2020, 295, 7481–7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bridges HR; Jones AJ; Pollak MN; Hirst J Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J 2014, 462, 475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Pollak MN Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov. 2012, 2, 778–790. [DOI] [PubMed] [Google Scholar]
- (28).Pollak M Potential applications for biguanides in oncology. J. Clin. Invest 2013, 123, 3693–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ellinghaus P; Heisler I; Unterschemmann K; Haerter M; Beck H; Greschat S; Ehrmann A; Summer H; Flamme I; Oehme F; Thierauch K; Michels M; Hess-Stumpp H; Ziegelbauer K BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med. 2013, 2, 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Schockel L; Glasauer A; Basit F; Bitschar K; Truong H; Erdmann G; Algire C; Hagebarth A; Willems PH; Kopitz C; Koopman WJ; Heroult M Targeting mitochondrial complex I using BAY 87-2243 reduces melanoma tumor growth. Cancer Metab. 2015, 3, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Helbig L; Koi L; Bruchner K; Gurtner K; Hess-Stumpp H; Unterschemmann K; Baumann M; Zips D; Yaromina A BAY 87-2243, a novel inhibitor of hypoxia-induced gene activation, improves local tumor control after fractionated irradiation in a schedule-dependent manner in head and neck human xenografts. Radiat. Oncol 2014, 9, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Vashisht Gopal YN; Gammon S; Prasad R; Knighton B; Pisaneschi F; Roszik J; Feng N; Johnson S; Pramanik S; Sudderth J; Sui D; Hudgens C; Fischer GM; Deng W; Reuben A; Peng W; Wang J; McQuade JL; Tetzlaff MT; Di Francesco ME; Marszalek J; Piwnica-Worms D; DeBerardinis RJ; Davies MA A novel mitochondrial inhibitor blocks MAPK pathway and overcomes MAPK inhibitor resistance in melanoma. Clin. Cancer Res 2019, 25, 6429–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33). ClinicalTrials.gov identifier: NCT01297530, NCT02882321 and NCT03291938.
- (34).Yap TA; Ahnert JR; Piha-Paul SA; Fu S; Janku F; Karp DD; Naing A; Dumbrava EEI; Pant S; Subbiah V; Tsimberidou AM; Hong DS; Rose KM; Xu Q; Vellano CP; Mahendra M; Jones P; Francesco MED; Marszalek JR; Meric-Bernstam F Phase I trial of IACS-010759 (IACS), a potent, selective inhibitor of complex I of the mitochondrial electron transport chain, in patients (pts) with advanced solid tumors. J. Clin. Oncol 2019, 37, 3014–3014. [Google Scholar]
- (35).Xue D; Xu Y; Kyani A; Roy J; Dai L; Sun D; Neamati N Discovery and lead optimization of benzene-1,4-disulfonamides as oxidative phosphorylation inhibitors. J. Med. Chem 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Nassar AE; Kamel AM; Clarimont C Improving the decision-making process in the structural modification of drug candidates: enhancing metabolic stability. Drug Discov. Today 2004, 9, 1020–1028. [DOI] [PubMed] [Google Scholar]
- (37).Pu Y; Christesen A; Ku Y A simple and highly effective oxidative chlorination protocol for the preparation of arenesulfonyl chlorides. Tetrahedron Lett. 2021, 51, 418–421. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.