

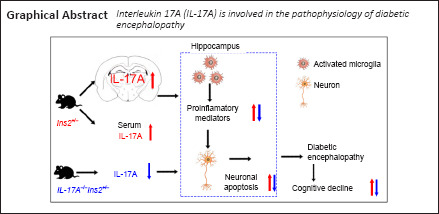
Key Words: Akita mice, apoptosis, cognitive impairment, diabetic encephalopathy, hippocampus, interleukin 17A, mice, microglia, neuroinflammation, neuron, targeted treatment
Abstract
Interleukin 17A (IL-17A) was previously shown to be a key pro-inflammatory factor in diabetes mellitus and associated complications. However, the role of IL-17A in diabetic encephalopathy remains poorly understood. In this study, we established a mouse model of diabetic encephalopathy that was deficient in IL-17A by crossing Il17a–/– mice with spontaneously diabetic Ins2Akita (Akita) mice. Blood glucose levels and body weights were monitored from 2–32 weeks of age. When mice were 32 weeks of age, behavioral tests were performed, including a novel object recognition test for assessing short-term memory and learning and a Morris water maze test for evaluating hippocampus-dependent spatial learning and memory. IL-17A levels in the serum, cerebrospinal fluid, and hippocampus were detected with enzyme-linked immunosorbent assays and real-time quantitative polymerase chain reaction. Moreover, proteins related to cognitive dysfunction (amyloid precursor protein, β-amyloid cleavage enzyme 1, p-tau, and tau), apoptosis (caspase-3 and -9), inflammation (inducible nitric oxide synthase and cyclooxygenase 2), and occludin were detected by western blot assays. Pro-inflammatory cytokines including tumor necrosis factor-α, interleukin-1β, and interferon-γ in serum and hippocampal tissues were measured by enzyme-linked immunosorbent assays. Microglial activation and hippocampal neuronal apoptosis were detected by immunofluorescent staining. Compared with that in wild-type mice, mice with diabetic encephalopathy had higher IL-17A levels in the serum, cerebrospinal fluid, and hippocampus; downregulation of occludin expression; lower cognitive ability; greater loss of hippocampal neurons; increased microglial activation; and higher expression of inflammatory factors in the serum and hippocampus. IL-17A knockout attenuated the abovementioned changes in mice with diabetic encephalopathy. These findings suggest that IL-17A participates in the pathological process of diabetic encephalopathy. Furthermore, IL-17A deficiency reduces diabetic encephalopathy-mediated neuroinflammation and cognitive defects. These results highlight a role for IL-17A as a mediator of diabetic encephalopathy and potential target for the treatment of cognitive impairment induced by diabetic encephalopathy.
Introduction
Diabetes mellitus (DM) is a complex metabolic disorder that results in diverse complications, including diabetic encephalopathy (DE). DE manifests as cognitive impairment that is associated with both structural and electrophysiological changes in the brain (Bhusal et al., 2019; Kim, 2019). The precise etiology of DE is not fully understood; however, it is driven at least in part by oxidative stress, mitochondrial dysfunction, inflammation, and autophagy (Giacco and Brownlee, 2010; Muriach et al., 2014; Bhusal et al., 2019).
Neuroinflammation has been identified as a particularly potent driver of DE progression (Esposito et al., 2002; Muriach et al., 2014). Neuroinflammation is generally driven by astrocytes, microglia, and other types of glial cells following their activation (Gogoleva et al., 2019). In DM rodent model systems, persistent DM initiated glial cell activation and a form of chronic inflammation wherein hippocampal cytokine levels were elevated (Hwang et al., 2014; Oliveira et al., 2016). The neuroinflammatory activity is associated with damage to hippocampal neurons (Kadłubowska et al., 2016; Wang et al., 2017) and cognitive impairment in diabetic mice (Oliveira et al., 2016). Given the close link between neuroinflammation and cognitive impairment, it is essential that microglial activation be carefully controlled to prevent DE onset or progression.
The interleukin (IL)-17 family of cytokines includes six closely related proteins (IL-17A–F) (Qiu et al., 2017). Of these, IL-17A has been comprehensively studied and analyzed as a pro-inflammatory mediator of autoimmune diseases, including multiple sclerosis and type 1 DM (T1DM) (Wang et al., 2013; Qiu et al., 2021). Patients diagnosed with DM exhibit increased IL-17A levels in their blood and pancreatic islets with similar findings in a rodent T1DM model (Baharlou et al., 2016; Shruthi et al., 2016; Sigurdardottir et al., 2019). IL-17A can also exacerbate DM-associated complications, such as oxidative stress, vascular leakage, and retinal inflammation (Qiu et al., 2016, 2017; Sigurdardottir et al., 2019). IL-17A-knockout (KO) mice exhibited reduction in insulitis and hyperglycemia following treatment with streptozotocin (Tong et al., 2015), and IL-17A blockade reduced the severity of diabetic retinopathy in rodent models (Qiu et al., 2017). These findings suggest that IL-17A is a key pro-inflammatory mediator of DM and associated complications. However, the specific relationship between IL-17A and DE remains to be clarified.
Ins2Akita (Akita; Ins2C96Y) mice harbor a spontaneous single-nucleotide substitution in the insulin 2 (Ins2) gene. This mutation results in improper proinsulin processing and, ultimately, in increased toxic injury to the pancreatic β cells and impaired secretion of insulin, which renders these animals more susceptible to developing T1DM (Al-Awar et al., 2016). Because the Ins2C96Y mutation can result in spontaneous DM development, Akita mice are an ideal model for exploring the incidence of DM-related complications (Mathews et al., 2002; Zhou et al., 2011). In this study, we aimed to explore the involvement of IL-17A in DE pathophysiology and whether IL-17A deficiency alleviated neuroinflammation and cognitive impairment in a mouse model of DE.
Materials and Methods
Animal models
Specific pathogen-free level Akita (Ins2, RRID: MGI: 3625075) and IL-17A-KO (Il17a–/–, RRID: IMSR_NM-KO-00131) mice on the C57BL/6 background (body weight, 16–22 g) were obtained originally from the Model Animal Research Center of Nanjing University (Nanjing, China) and Dr. Yang (Nanjing Medical University, Nanjing, China), respectively. IL-17A–/– Akita mice (Il17a–/–Ins2+/–) were generated by cross-breeding these two strains as detailed in a prior report (Chavali et al., 2014), and a breeding colony was established at the Experimental Animal Center of Nantong University (Nantong, China; license No. SYXK [Su] 2017-0046). Briefly, male wild-type (WT) C57BL/6 mice and female Ins2+/– mice were crossed to obtain male Ins2+/– mice, which were used in the DE model group at 32 weeks of age following 28 weeks of hyperglycemia. Age-matched WT littermates were used as controls for the DE mice. In addition, female Ins2+/– mice and male Il17a–/– mice were crossed to obtain heterozygous Il17a+/–Ins2+/– F1 mice. Female F1 mice were then crossed with male Il17a–/– mice to generate Il17a–/–Ins2+/– F2 animals (Figure 1A). Male Il17a–/–Ins2+/– mice with hyperglycemia for 28 weeks were used in the IL-17A-KO DE group for experimental analyses. Polymerase chain reaction (PCR) amplification was used to genotype all mice at 3 weeks of age, and PCR products were separated via 1.5% agarose gel electrophoresis. The mice were housed at 21 ± 1°C with a 12-hour light/dark cycle and ad libitum access to food and water. National Institutes of Health guidelines were used to conduct all animal studies, which received approval from the Institutional Animal Care and Use Committee of Nantong University (approval No. 20200323-329) on March 23, 2020. All experiments were designed and reported according to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines (Percie du Sert et al., 2020).
Figure 1.

Schematic experimental overview.
(A) Scheme for generating Ins2+/− and Il17a−/−Ins2+/− mice from WT, Ins2+/− and Il17a−/− mice. (B) Flow chart of the study design. CSF: Cerebrospinal fluid; DE: diabetic encephalopathy; ELISA: enzyme-linked immunosorbent assay; IL-17A: interleukin 17A; MWM: morris water maze; NOR: novel object recognition; PFA: paraformaldehyde; qPCR: real-time polymerase chain reaction; WT: wild type.
Experimental design
All mice were generated by cross-breeding as shown in Figure 1A. The mice were genotyped at 3 weeks of age and grouped according to genotype. The mice in the WT (n = 16), DE (n = 19), and IL-17A-KO DE (n = 18) groups were selected at random for use in different experiments. Blood glucose levels and body weights were monitored beginning at the age of 2 weeks and every 2 or 4 weeks thereafter until 32 weeks of age. Behavioral tests were performed at 32 weeks of age. These tests included a novel object recognition test for assessing short-term memory and learning and a Morris water maze test for evaluating hippocampus-dependent spatial learning and memory. Because two behavioral tests were performed per mouse, the less stressful test (novel object recognition) preceded the more stressful test (Morris water maze). Behavioral tests in different groups of mice were conducted at a relatively fixed time every day to avoid nuisance variables, such as inconsistent testing time and day-to-day variability. After behavioral testing, mice were deeply anesthetized, and the cerebrospinal fluid (CSF) and serum were collected for enzyme-linked immunosorbent assay (ELISA) analyses; then, the mice were sacrificed, and hippocampal tissues were isolated for real-time PCR (qPCR), western blotting, and ELISA. In addition, five mice per group were perfused with saline solution followed by 4% paraformaldehyde, after which immunofluorescent analyses were performed to detect hippocampal neuronal apoptosis and microglial activation (Figure 1B).
Morris water maze
The Morris water maze test was used as a means of assessing spatial learning and memory associated with hippocampal function as described previously (Powell et al., 2004; Tabuchi et al., 2007). Briefly, a white pool (diameter: 180 cm) was filled with water at 22 ± 1°C. A circular platform (diameter: 10 cm) was positioned 1 cm below the surface of the water. In navigation tests, mice underwent four trials each day on five consecutive days. Pools were separated into four quadrants, and mice were placed in a random quadrant within the pool for each trial, after which they were allowed to swim freely until they had located the hidden platform. If the platform was not located within 90 seconds, the experimenter guided the mouse to the platform. The mice were allowed to rest on the platform for 15 seconds and then returned to their home cage to await subsequent trials. At 24 hours after the final training session, a probe trial was conducted. For this trial, the hidden platform was removed and mice were allowed to swim in the pool for 60 seconds. During this trial, the escape latency (time to cross the platform position), swimming speed, number of platform crossings, and time spent in the target quadrant were recorded with an automated video tracking system (XR-XM101; Shanghai Xinruan Information Technology Co. Ltd., Shanghai, China).
Novel object recognition test
The novel object recognition test can be used to evaluate hippocampus-dependent object recognition memory based on the tendency for rodents to spend more time investigating a novel object than a familiar one (Wu et al., 2020). The test can also be used to assess short-term memory in rodents with a shortened retention time of 15 minutes (Stover et al., 2015; Reza-Zaldivar et al., 2019) and was performed in this study using a modified version of the protocols detailed in prior reports (Stover et al., 2015; Lueptow, 2017; Reza-Zaldivar et al., 2019). Briefly, mice were first allowed to habituate to a 50 cm × 50 cm × 50 cm open-field area for 5 minutes 1 day before the novel object recognition test. Training was then performed 24 hours later by placing two identical objects in the arena and allowing the mice to explore freely for a 5-minute period. The mice were then returned to their home cages, and the objects were cleaned using 70% ethanol. At 15 minutes following the end of this training session, the mice were returned to the arena for an additional 5 minutes during which one of these two objects had been exchanged for a novel object with a distinct size and shape. The time that the mice spent sniffing or touching this novel object was assessed with the XR-XM101 video tracking system. A discrimination index was established based upon the ratio of time spent exploring this novel object relative to the total time spent exploring both objects during the trial.
Blood glucose and body weight monitoring
Beginning at the age of 2 weeks and every 2 or 4 weeks thereafter, blood glucose levels were measured in samples collected from mouse tail veins using a blood glucose meter (Roche, Basel, Switzerland) until 32 weeks of age. Before detecting blood glucose, the body weight of each mouse was measured with a balance beginning at 4 weeks of age.
qPCR
Mice were anesthetized at 32 weeks of age by intraperitoneal injection of sodium pentobarbital (55 mg/kg; Sigma-Aldrich, St. Louis, MO, USA), and the hippocampal tissues were rapidly dissected. Total RNA was isolated using Trizol (Invitrogen, Carlsbad, CA, USA). A Transcriptor First Strand cDNA Synthesis Kit (Roche) was then used to prepare complementary DNA based on the manufacturer’s directions. SYBR Green I reagent, Universal SYBR Green Master Mix (Roche), and a Rotor-Gene 3000 Real-time Cycler (Corbett Research, Sydney, Australia) were used to conduct qPCR analyses. Each reaction contained 0.4 μM primers and 2 μL complementary DNA, and the thermocycler settings were as follows: 95°C for 10 minutes; 35 cycles of 94°C for 15 seconds and 60°C for 60 seconds. The Rotor-Gene 6.0 software (Corbett Research) was used to analyze the resultant data, and melting curves were used to verify the amplification of a single product. The 2–ΔΔCt method (Livak and Schmittgen, 2001) was used to assess relative gene expression, and β-actin was used for normalization. Primer sequences were as follows: Il17a, 5’-TGG ACT CTG AGC CGC A-3’ and 5’-GGC GGA CAA TAG AGG A-3’; β-actin, 5’-CTG TCC CTG TAT GCC TCT G-3’ and 5’-ATG TCA CGC ACG ATT TCC-3’.
Western blot assay
Proteins related to cognitive dysfunction, apoptosis, and inflammation were detected by western blot assays. Radio-immunoprecipitation assay buffer (Thermo Scientific, Waltham, MA, USA) was used to extract proteins from hippocampal tissue samples that were isolated as described above. A total of 20–40 μg protein per sample was separated using 10–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). The blots were blocked using 5% non-fat milk in Tris-buffered saline containing Tween 20 followed by overnight incubation at 4°C with one of the following antibodies: rabbit anti-amyloid precursor protein (APP; 1:1000; Millipore, Cat# AB5352, RRID: AB_91793), rabbit anti-occludin (1:2000; Abcam, Cambridge, UK, Cat# ab167161, RRID: AB_2756463), rabbit anti-β-amyloid cleavage enzyme 1 (BACE1; 1:1000; Abcam Cat# ab2077, RRID: AB_302817), rabbit anti-tau (phospho S396) (1:1000; Abcam Cat# ab109390, RRID:AB_10860822), rabbit anti-tau (1:1000; Abcam, Cat# ab76128, RRID: AB_1524475), rabbit anti-caspase-3 (1:1000; Abcam, Cat# ab184787, RRID: AB_2827742), rabbit anti-caspase-9 (1:1000; Abcam, Cat# ab202068, RRID: AB_2889070), rabbit anti-CD11b (1:1000; Abcam, Cat# ab52478, RRID: AB_868788), rabbit anti-inducible nitric oxide synthase (iNOS; 1:300; Abcam, Cat# ab15323, RRID: AB_301857), rabbit anti-cyclooxygenase 2 (COX-2; 1:300; Abcam, Cat# ab179800, RRID: AB_2894871), or mouse anti-β-actin (1:5000; Abcam, Cat# ab6276, RRID: AB_2223210). IRDye 800-conjugated goat anti-rabbit IgG (1:5000; Rockland, Gilbertsville, PA, USA, Cat# 611-145-003, RRID: AB_1057621) or IRDye 680-conjugated goat anti-mouse IgG (1:5000; Rockland, Cat# 610-144-007, RRID: AB_1660925) were then utilized to detect the protein bands, which were visualized using the Odyssey laser scanning system (LI-COR Inc., Lincoln, NE, USA) as previously described (Huang et al., 2020). The intensity of each protein band was quantified by densitometric analysis using ImageJ software (NIH, Bethesda, MD, USA) (Schneider et al., 2012) and expressed as a relative value normalized to β-actin.
CSF and serum collection
CSF samples were taken from the cisterna magna as previously described (Liu and Duff, 2008). Briefly, mice were deeply anesthetized as indicated above. The skin was sagittally incised inferior to the occiput. The subcutaneous tissues and muscles were carefully separated by blunt dissection with forceps. Then, the glistening surface of the dura covering the cisterna magna was exposed. The dura mater was punctured using borosilicate glass pipettes with a tip diameter of 20 μm, and CSF flowed into the glass pipettes. Whole blood was collected by cardiac puncture and placed at 37°C for 1 hour, and the supernatant (serum) was obtained by centrifugation at 1800 × g for 15 minutes at 4°C. The CSF and serum samples were stored at –80°C.
ELISA
When mice were 32 weeks of age, samples of serum, CSF, and hippocampal tissues were isolated and the levels of pro-inflammatory cytokines were measured, including IL-17A, tumor necrosis factor (TNF)-α, IL-1β, and interferon (IFN)-γ, using commercial ELISA kits (eBioscience, San Diego, CA, USA) based on the manufacturer’s directions.
Immunofluorescent result analyses
Under deep anesthesia as described above, mice were sequentially perfused with saline solution and 4% paraformaldehyde. The brains were collected, immersed for 24 hours in 4% paraformaldehyde at 4°C, and cryopreserved for 48 hours in 30% sucrose at 4°C. Next, 25 μm-thick coronal brain sections were prepared using a chilled microtome (CM 1900; Leica; Wetzlar, Germany). The sections were incubated for 30 minutes in 10% goat serum and 0.1% Triton X-100 followed by staining overnight with antibodies specific for rabbit anti-ionized calcium binding adapter molecule 1 (IBA1; 1:1000; FUJIFILM Wako Shibayagi, Osaka, Japan, Cat# 019-19741, RRID: AB_839504) or rabbit anti-neuronal nuclei (NeuN; 1:400; Abcam, Cat# ab177487, RRID: AB_2532109) at 4°C. Sections were then incubated for 4 hours at room temperature with an Alexa Fluor 594-conjugated goat anti-rabbit IgG (1:500; Abcam, Cat# ab150080, RRID: AB_2650602). Apoptotic cell death was assessed via terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining conducted with an in situ Cell Death Detection Kit (Roche) following the manufacturer’s instructions. Nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI). A laser scanning confocal microscope (TCS-SPE; Leica) was used to image the sections, and positively stained cells in three different sections of the murine brain were selected. The numbers of IBA1-positive microglial cells and areas were determined, and the averages from three sections per brain and five brains per group were calculated.
Statistical analysis
The sample sizes for groups of mice were calculated based on published papers (Horakova et al., 2019; Liu et al., 2019) describing similar experiments and statistical requirements for a small sample size. Five animals died during the experimental period. We replenished the groups to ensure the sample size was sufficient. The evaluators were blinded to the mouse groups while monitoring blood glucose and body weights and conducting behavioral tests. Data are expressed as the mean ± standard deviation (SD) for the corresponding groups and were analyzed using SPSS 12.0 (SPSS, Chicago, IL, USA). Data were compared via one-way analysis of variance with Student-Newman-Keuls test for pairwise comparisons. P < 0.05 was the significance threshold for this study.
Results
IL-17A is upregulated in mice with DE, and IL-17A KO reduced DE-mediated elevation of blood glucose levels and downregulation of occludin expression
Body weights and blood glucose levels were measured beginning at 2 weeks of age. Significant spontaneous increases in blood glucose levels in the Akita mice began at the age of 4 weeks and reached a peak level after 12 weeks. These glucose levels were maintained through 32 weeks of age (corresponding to a 28-week hyperglycemic period). Thus, 32-week-old Akita mice were used as the experimental model of DE. Notably, significantly lower levels of blood glucose were observed in the IL-17A-KO DE mice from 10–32 weeks of age relative to the levels in DE mice (Figure 2A). Before testing blood glucose levels, the body weight of each mouse was measured. The body weights of the DE mice were decreased from 12–32 weeks of age relative to that of the WT mice, while IL-17A-KO DE mice exhibited body weights similar to those observed in the DE mice (Figure 2B). These results demonstrated that IL-17A KO attenuated the elevation of blood glucose observed in DE mice, but IL-17A deficiency did not alleviate weight loss. We also measured the expression of the tight junction protein occludin in hippocampal tissues. Expression of occludin was downregulated in DE mice relative to that in WT controls, and Il17a gene KO partially reversed the DE-mediated downregulation of occludin (Figure 2C). This result suggested that IL-17A deficiency attenuated the blood-brain barrier (BBB) breakdown in the hippocampus of DE mice.
Figure 2.

IL-17A is upregulated in DE mice, and IL-17A knockout attenuated the DE-mediated elevation in blood glucose levels and decrease in occludin expression.
(A) IL-17A knockout was associated with a reduction in blood glucose levels in mice with DE as determined by measuring blood glucose levels in non-fasted mice every 2–4 weeks over the 32-week study period (n = 11). (B) Body weights of DE mice were decreased from 12–32 weeks of age relative to those in WT mice. IL-17A-KO DE mice did not exhibit a reversal in body weight loss compared with that in the DE mice. (C) Il17a gene knockout attenuated the downregulation of occludin observed in the DE model mice (n = 3). The original images are shown in Additional Figure 1A (1.7MB, tif) and B (1.7MB, tif) . (D) IL-17A levels in the serum and CSF were upregulated in 32-week-old DE mice relative to that in WT controls as measured via ELISA (n = 10, serum samples; n = 7, CSF samples). (E) IL-17A mRNA and protein levels were increased in hippocampal samples collected from DE mice relative to those in WT mice at 32 weeks of age as assessed via qPCR (n = 5) and ELISA (n = 10), respectively. Data are expressed as means ± SD. *P < 0.05, **P < 0.01, vs. WT mice; #P < 0.05, ##P < 0.01, vs. DE mice (one-way analysis of variance followed by Student-Newman-Keuls test). CSF: Cerebrospinal fluid; DE: diabetic encephalopathy; ELISA: enzyme-linked immunosorbent assay; IL-17A: interleukin 17A; qPCR: real-time quantitative polymerase chain reaction; WT: wild type.
To explore the role of IL-17A as a mediator of DE pathogenesis, we assessed IL-17A levels in serum and CSF samples from WT and DE mice. Marked increases in IL-17A were observed in both biofluids at 32 weeks of age in the DE mice compared with that in the WT controls (Figure 2D). Consistently, IL-17A mRNA and protein levels were increased in the hippocampus of DE mice relative to that in the WT mice (Figure 2E), suggesting that DE is associated with increased IL-17A production in the serum, hippocampus, and CSF of Akita mice.
IL-17A deficiency alleviates cognitive impairment in DE mice
To establish whether DE mice exhibited cognitive impairment and whether such impairment was affected by IL-17A KO, we utilized novel object recognition and Morris water maze tests to evaluate 32-week-old mice in our different experimental groups. The novel object recognition test was used to study short-term learning and memory, while the Morris water maze test was performed to assess spatial learning and memory.
To examine the effects of IL-17A deficiency on cognitive dysfunction in DE mice, the novel object recognition test was conducted. On the day before novel object recognition testing, the mice were habituated to the open-field environment, and the numbers of grid crossings were recorded. There were no significant differences in crossing numbers among the mouse groups (Figure 3A), indicating that DE mice with or without IL-17A did not exhibit motor impairment. At 24 hours post-habituation, training and testing sessions were conducted. These results revealed that discrimination index values in DE mice were significantly lower than those for WT mice, whereas IL-17A-KO DE mice exhibited a significant increase in the average discrimination index value relative to those for DE mice (Figure 3B). These data suggested that IL-17A deficiency was sufficient to partially reduce DE-mediated impairment of short-term learning and memory.
Figure 3.

IL-17A deficiency alleviates DE-induced cognitive impairment as detected in the novel object recognition test.
(A) The habituation of mice in an open-field environment was performed 1 day before the novel object recognition testing. Representative grid crossing paths are shown, and no significant differences in crossing numbers were observed among the groups. (B) Il17a gene knockout significantly improved discrimination index values relative to those for DE mice. Representative tracking paths for mice in the novel object recognition test are shown. The upper and lower rows show the training and testing tracks, respectively. The IL-17A-KO DE mice spent more time exploring novel objects than the DE mice. Mice were analyzed at 32 weeks of age. Data are expressed as means ± SD (n = 8). *P < 0.05, **P < 0.01 (one-way analysis of variance followed by Student-Newman-Keuls test). DE: Diabetic encephalopathy; IL-17A: interleukin 17A; KO: knockout; N: novel object; O: old object; WT: wild type.
Morris water maze testing was then used to evaluate spatial learning and memory. DE mice exhibited a significant increase in escape latency relative to WT mice during the navigational training test, while this effect was largely reversed in IL-17A-KO DE mice (Figure 4A). Similarly, in probe trials, DE mice exhibited reductions in the time spent in the target quadrant, numbers of platform crossings, and swimming speed, while the latency to the initial crossing of the escape platform was increased. IL-17A-KO DE mice demonstrated markedly improved time spent in the target quadrant, numbers of platform crossings, and swimming speed and decreased latency to the initial crossing of the escape platform relative to DE mice (Figure 4B). The results of these two behavioral tests suggested that a loss of IL-17A expression was sufficient to protect against DE-related cognitive impairment.
Figure 4.
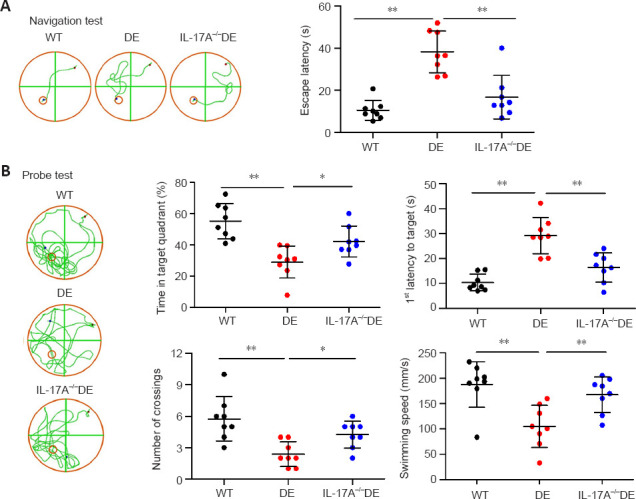
IL-17A deficiency alleviates DE-induced cognitive impairment as detected in the Morris water maze test.
(A) Loss of IL-17A expression was associated with an increase in escape latency in DE mice. Representative tracking paths are shown for mice on the fifth (final) day of navigation training. The escape platforms are represented by the small orange circles. The red and blue dots denote the beginning and ending points for the mice. (B) IL-17A knockout significantly improved DE-related cognitive impairment in mice during a probe test. Representative tracking paths are shown for probe tests without the presence of a hidden platform that were performed 24 hours after the final training session. Mice were analyzed at 32 weeks of age. Data are expressed as means ± SD (n = 8). *P < 0.05, **P < 0.01 (one-way analysis of variance followed by Student-Newman-Keuls test). DE: Diabetic encephalopathy; IL-17A: interleukin 17A; WT: wild type.
IL-17A KO reduces the upregulation of APP and BACE1 as well as tau phosphorylation in the hippocampus of DE mice
Growing evidence suggests that neuroinflammation is associated with the upregulation of BACE1 and APP, which are associated with cognitive impairment (Lee et al., 2008; Sun et al., 2015). Thus, we assessed the expression of these proteins in our mice. In the hippocampus of DE mice, the protein levels of APP and BACE1 were increased relative to those in the WT mice. Upregulation of these proteins in the DE mice was partially reversed when IL-17A was knocked out (Figure 5A). Tau hyperphosphorylation and accumulation are also closely associated with impaired memory and learning (Egervari et al., 2020), and we found that DE mice exhibited elevated levels of tau protein that was phosphorylated at Ser396 compared with that in the WT mice (Figure 5B). IL-17A-KO DE mice exhibited reduced levels of phosphorylated tau relative to DE mice. The total tau expression levels did not differ significantly among these three groups (Figure 5B). These data suggest that IL-17A KO can prevent, at least partially, the DE-related upregulation of key proteins associated with cognitive dysfunction.
Figure 5.

IL-17A knockout reduces the upregulation of APP, BACE1, and phosphorylated tau in mice with DE.
(A) IL-17A knockout attenuated DE-mediated APP and BACE1 upregulation. The original images are shown in Additional Figure 1C (1.7MB, tif) –F (1.7MB, tif) . (B) IL-17A knockout reduced phosphorylated tau (ser396) levels in DE mice. Hippocampal brain samples from mice in the indicated groups were homogenized at 32 weeks of age and analyzed via western blots. The original images of western blot bands are shown in Additional Figure 1G (1.7MB, tif) –I (1.7MB, tif) . Data are expressed as means ± SD (n = 4). **P < 0.01, vs. WT mice; ##P < 0.01, vs. DE mice (one-way analysis of variance followed by Student-Newman-Keuls test). APP: Amyloid precursor protein; BACE1: β-amyloid cleavage enzyme 1; DE: diabetic encephalopathy; IL-17A: interleukin 17A; p-tau: phosphorylated tau; WT: wild type.
IL-17A KO reduces apoptotic death of hippocampal neurons in DE mice
To evaluate hippocampal apoptosis, we conducted TUNEL and DAPI staining of hippocampal tissue sections of DE mice and demonstrated a marked increase in the number of apoptotic cells within the CA1 region of the hippocampus compared with that in the WT group (Figure 6A). NeuN staining also revealed that there was a reduction in the number of neurons in the hippocampal CA1 region in DE mice relative to that in WT controls (Figure 6B). These phenotypes were partially reversed in the IL-17A-KO DE mice. Most caspase proteins are promoters and effectors of apoptosis, playing an important role in this form of cell death (Van Opdenbosch and Lamkanfi, 2019). Therefore, we assessed caspase-3 and -9 levels in the hippocampus via western blots. Caspase-3 and -9 levels in the hippocampus were significantly elevated in DE mice but were reduced in the IL-17A-KO DE group (Figure 6C). These data suggest that IL-17A KO can protect against DE-related apoptotic death of hippocampal neurons in Akita mice.
Figure 6.

IL-17A knockout reduces apoptotic death of hippocampal neurons in mice with DE.
(A) TUNEL staining was conducted to evaluate hippocampal neuron apoptosis. Apoptotic cells and DAPI-stained nuclei are shown in green and blue, respectively (n = 5). (B) IL-17A knockout reversed the reduction in neuron numbers in the hippocampal CA1 region in DE mice. Representative images of NeuN+-labeled neurons (red, stained with Alexa Fluor 594) in the CA1 region of the hippocampus. DAPI (blue) was used to stain nuclei, and NeuN+DAPI+ neurons in each section were counted (n = 5). (C) IL-17A knockout reduced DE-related increases in caspase-3 and -9 levels in mice, which were measured via western blots (n = 4). The original images of western blot bands are shown in Additional Figure 1J (1.7MB, tif) –L (1.7MB, tif) . Data are expressed as means ± SD. *P < 0.05, **P < 0.01, vs. WT mice; #P < 0.05, ##P < 0.01, vs. DE mice (one-way analysis of variance followed by Student-Newman-Keuls test). DAPI: 4′,6-Diamidino-2-phenylindole; DE: diabetic encephalopathy; IL-17A: interleukin 17A; NeuN: neuronal nuclei; TUNEL: terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling; WT: wild type.
IL-17A KO reduces microglial activation in the hippocampus of DE mice
To explore the functional importance of IL-17A as a mediator of DE-related changes in glial characteristics, anti-IBA1 immunostaining of brain sections was performed to analyze microglia. This approach revealed significant increases in hippocampal IBA1-positive microglial cell numbers and somal areas in DE mice. The microglia exhibited increased IBA1 immunoreactivity, larger somal size, and retracted processes relative to those observed in WT mice. Notably, these DE-related morphological changes were largely attenuated in the IL-17A-KO DE mice (Figure 7A). Consistently, CD11b expression levels were enhanced in DE mice compared with that in WT mice, while this change was reversed in IL-17A-KO DE mice (Figure 7B). Moreover, levels of pro-inflammatory iNOS and COX-2 were elevated in DE mice; however, this upregulation was notably reduced in the IL-17A-KO DE group (Figure 7C). Levels of pro-inflammatory cytokines (TNF-α, IL-1β, IFN-γ) in serum and hippocampal samples were also assessed via ELISA. These cytokines were upregulated in DE mice compared with that in WT mice, whereas this upregulation was diminished in IL-17A-KO DE mice. Thus, IL-17A KO alleviated DE-induced neuroinflammation in mice (Figure 7D and E).
Figure 7.

IL-17A knockout reduces microglial activation in mice with DE.
(A) Significant increases in the numbers and cell body areas of hippocampal IBA1-positive microglia (arrows; red, stained with Alexa Fluor 594) were observed in DE mice relative to WT mice. Microglia exhibited significantly enhanced anti-IBA1 immunoreactivity, increased somal sizes, and retracted processes in the DE mice. These hippocampal changes were attenuated in the IL-17A-KO DE mice (n = 5). Scale bar: 50 μm. (B) IL-17A knockout ablated DE-related increases in CD11b immunoreactivity (n = 4). The original images are shown in Additional Figure 1M (1.7MB, tif) and N (1.7MB, tif) . (C) IL-17A knockout blunted the upregulation of inflammatory proteins iNOS and COX-2 in DE mice as assessed by western blots (n = 4). The original images of western blot bands are shown in Additional Figure 1O (1.7MB, tif) –Q (1.7MB, tif) . (D, E) IL-17A-KO DE animals exhibited reductions in serum and hippocampal pro-inflammatory cytokine levels (TNF-α, IL-1β, IFN-γ) relative to those in DE mice as assessed by ELISA (n = 5–7). Data are expressed as means ± SD. *P < 0.05, **P < 0.01, vs. WT mice; #P < 0.05, ##P < 0.01, vs. DE mice (one-way analysis of variance followed by Student-Newman-Keuls test). COX-2: Cyclooxygenase 2; DAPI: 4′,6-diamidino-2-phenylindole; DE: diabetic encephalopathy; IBA1: ionized calcium binding adapter molecule 1; IFN-γ: interferon γ; IL-17A: interleukin 17A; IL-1β: interleukin-1β; KO: knockout; iNOS: inducible nitric oxide synthase; TNF-α: tumor necrosis factor-α; WT: wild type.
Discussion
Cognitive dysfunction is a common and serious comorbidity that is observed in some patients suffering from type 1 and type 2 DM (He et al., 2018; Zhang et al., 2020). T1DM is an autoimmune disease in which pancreatic β-cells undergo immune-mediated destruction. There is growing evidence that DM-related cognitive impairment is associated with the entwined processes of neuroinflammation and neuronal cell damage (Zilliox et al., 2016; Pei and Sun, 2018), suggesting that targeting neuroinflammation may represent a viable approach to treating DE. IL-17A has been closely linked to chronic inflammation and the pathogenesis of DM, multiple sclerosis, and other autoimmune conditions (Wang et al., 2013; Ma et al., 2019). For this study, we modeled DE using Akita mice that experienced spontaneous hyperglycemia beginning at 4 weeks of age and evaluated cognitive impairment in these animals via the Morris water maze and novel object recognition tests at 32 weeks of age. IL-17A levels in these DE mice and the functional role of this inflammatory cytokine in DE were then assessed. Serum, CSF, and hippocampal levels of IL-17A were elevated in DE mice relative to those in WT controls, and the DE mice exhibited an increase in hippocampal TUNEL-positivity and caspase-3/9 expression. When IL-17A was knocked out in DE model animals, a reduction in neuronal apoptosis and concomitant improvements in cognitive function were observed. Hyperglycemia-driven microglial activation and increases in pro-inflammatory cytokine production observed in the context of DE were also significantly reduced in IL-17A-KO DE mice. Together, these data suggest a mechanism whereby hyperglycemia-driven IL-17A upregulation can induce neuroinflammation and neurodegeneration and, ultimately, contribute to the development of DE and consequent cognitive impairment.
The pathogenic role of IL-17A in DM is well documented. T1DM patients exhibit marked elevation of IL-17A expression in plasma, serum, and activated peripheral blood T cells (Marwaha et al., 2010; Hang et al., 2014; Baharlou et al., 2016). In our study, we observed increased IL-17A levels in the serum of Akita DE mice. IL-17A-KO DE mice exhibited significantly lower levels of blood glucose compared with that in DE mice, suggesting that IL-17A deficiency can alleviate diabetes development induced by an insulin gene mutation. A prior study reported that Il17a–/– mice exhibited attenuated hyperglycemia and islet inflammation following treatment with streptozotocin (Tong et al., 2015). However, the neutralization of IL-17 with monoclonal antibodies or IL-17 silencing did not prevent disease progression in non-obese diabetic mice (Emamaullee et al., 2009; Joseph et al., 2012). These discrepancies are likely attributable to differences among T1DM models. IL-17 has been shown to exhibit negative feedback in T helper (Th)17 cell populations. Blocking IL-17A in autoreactive Th17 cells may enhance other Th17-lineage cytokines to compensate, thereby reversing the benefits that might result from the loss of IL-17A (Smith et al., 2008; Chong et al., 2020). Whether this negative feedback loop can completely counteract the effects of IL-17A deficiency is uncertain. A recent report indicated that IL-17A-defective mice were highly resistant to the induction of experimental autoimmune encephalomyelitis (McGinley et al., 2020). Our present study also demonstrated that IL-17A KO had a protective effect against the development of DE. These results demonstrate that IL-17A-related negative feedback is not sufficient to fully counteract the effects of IL-17A genetic ablation in certain autoimmune diseases.
IL-17A was upregulated in the CSF and hippocampus in DE mice. IL-17A deficiency has been reported to alleviate BBB disruption, neuropathology, and motor impairment in a mouse model of Parkinson’s disease (Liu et al., 2019). Moreover, IL-17 can disrupt BBB tight junctions and promote human Th17 cell infiltration into the brain (Kebir et al., 2007). In our study, the interendothelial tight junction protein occludin was downregulated in DE mice, and Il17a gene KO attenuated occludin downregulation in these mice. Taken together, our results suggest that high levels of IL-17A in the central nervous system (CNS) are likely correlated with an impaired BBB, and IL-17A deficiency may alleviate BBB breakdown in the hippocampus of DE mice.
In the present study, DE mice exhibited impaired memory and learning as demonstrated by the novel object recognition and Morris water maze tests. IL-17A-KO DE mice exhibited less severe cognitive decline, which supports a role for excess IL-17A production as a driver of DE progression and cognitive impairment. In addition, APP and BACE1 have been identified as facilitators of cognitive dysfunction (Lee et al., 2008; Sun et al., 2015). APP serves as the source of extracellular amyloid-β plaques that are thought to be responsible for synaptic damage (Shahani et al., 2014), and BACE1 serves as the rate-limiting enzyme responsible for amyloid-β formation within neurons (Oehlrich et al., 2014). Tau hyperphosphorylation can also drive impairment of synaptic functionality, neurodegeneration, and cognitive dysfunction (Egervari et al., 2020). We found that the KO of IL-17A was sufficient to suppress DE-related increases in APP, BACE1, and phosphorylated tau levels in the hippocampus, thus further supporting a role for IL-17A as a functional mediator of reduced cognitive function and a potentially viable target for therapeutic intervention.
A majority of the available evidence to date has suggested that IL-17A production in the brain is primarily deleterious. Some studies having demonstrated that IL-17A can drive neurodegenerative activity in rodent models of stroke and multiple sclerosis (Wang et al., 2013; Dai et al., 2020). DM-related cognitive impairment is tied to neuronal cell death (Zilliox et al., 2016; Pei and Sun, 2018). In a rodent model of Parkinson’s disease, Liu et al. (2019) found that IL-17A deficiency was sufficient to alleviate dopaminergic neurodegeneration. Caspases are primarily involved in cell death and inflammatory responses. Caspase-9 and caspase-3 are initiator and effector caspases, respectively, and are involved in apoptosis (Van Opdenbosch and Lamkanfi, 2019). Abnormal increases of proapoptotic proteins lead to the excessive loss of neurons, which can result in neurodegeneration and cognitive impairment (Ghoneum and El Sayed, 2021). In our study, we observed a reduction in the number of neurons in the hippocampal CA1 region in DE mice together with increased hippocampal cellular apoptosis as evidenced by the upregulation of TUNEL/DAPI double-positive cells and caspase-3 and caspase-9 protein expression relative to WT controls. IL-17A KO ablated the hyperglycemia-related neuronal damage. These results suggest that increases in hippocampal IL-17A may directly drive hippocampal neurodegeneration, and loss of this inflammatory cytokine was protective against DE.
Microglial activation and associated neuroinflammatory processes are closely tied to multiple sclerosis, DM, and other diseases (Wang et al., 2013; Lo et al., 2016; Xu et al., 2021). One recent study demonstrated hippocampal microglial activation in diabetic mice (Bhusal et al., 2019). IL-17A can stimulate microglia to produce pro-inflammatory cytokines, chemokines, and other factors (Yu et al., 2016), whereupon they can potentially damage surrounding neurons and facilitate neurodegeneration (Bhusal et al., 2019; Rom et al., 2019). The inflammatory mediators COX-2 and iNOS are also closely associated with inflammation within the CNS (Zhao et al., 2019). Our data revealed that DE mice exhibited increased hippocampal neuroinflammation as evidenced by increased IBA1-positive cell body area; CD11b, iNOS, and COX-2 expression; and TNF-α, IL-1β, and IFN-γ production. This DE-related neuroinflammatory activity was notably attenuated in the IL-17A-KO DE mice, suggesting a role for IL-17A in this deleterious neurodegenerative process.
Peripheral blood samples from DE mice demonstrated increased levels of pro-inflammatory cytokines, which was consistent with systemic DM-related inflammatory activity. These cytokines were primarily derived from immune cells, including monocytes, macrophages, and Th1 cells. In patients and mice with T1DM, higher serum levels of TNF-α, IL-1β, and IFN-γ have been reported (Cnop et al., 2005; Shehata et al., 2011). We detected increased serum IL-17A levels in our Akita mice. Thus, we suggest that IL-17A can synergistically promote deleterious innate immune responses during T1DM development given that the KO of IL-17A was sufficient to decrease the production of pro-inflammatory cytokines and alleviate DE-related symptoms. Our results suggest that microglial- and adaptive immune cell-derived IL-17A and other factors can synergistically damage hippocampal neurons and further aggravate DE-related neurodegeneration.
The current study is subject to certain limitations. First, the specific mechanisms whereby IL-17A regulates neuroinflammation and DE require further clarification. Second, the utilization of the IL17A-KO Akita model as the sole experimental system may not be fully representative of the physiologic parameters quantified herein given the central role IL17A in the pathogenesis of diabetes and DE. These findings would be strengthened by adoptively transferring Th17 cells from WT mice into the KO mice to establish the degree to which this KO model influenced the study results. Third, IL-17 can profoundly stimulate the hematopoietic system with a particularly pronounced effect on granulopoiesis. In a recent study, Il17a–/– mice or mice treated with anti-IL-17A antibodies exhibited significantly reduced numbers of neutrophils and inflammatory monocytes in the spleen and lymph nodes after the induction of EAE, indicating that early recruitment of these innate inflammatory populations is IL-17A-dependent (McGinley et al., 2020). Moreover, Il17a–/– mice exhibited significant reductions in the numbers of IL-1β-secreting neutrophils and inflammatory monocytes in the CNS during EAE development. These findings suggested that a key function of IL-17A in the context of CNS autoimmunity was to recruit IL-1β-secreting myeloid cells that primed pathogenic Th17 cells (McGinley et al., 2020). Therefore, we hypothesize that the IL-17A-KO DE mice in our study likely exhibit modulations in peripheral myeloid cell populations. Additional experiments to observe the changes in these cell populations are warranted in the future.
In summary, the present study suggests that IL-17A deficiency alleviates cognitive impairment, cognition-related protein upregulation, neuronal apoptosis, and neuroinflammation in mice with DE. These findings demonstrate that adaptive immune inflammation may be involved in the pathophysiology of DE. Thus, blocking IL-17A expression or function is likely to represent a beneficial means of alleviating the symptoms of DE and highlights a promising new avenue for future therapeutic intervention efforts.
Additional files:
Additional file 1: Open peer review report 1 (134KB, pdf) .
Additional Figure 1 (1.7MB, tif) : Original images of western blot bands.
Original images of western blot bands.
(A, B) Original images of occludin (A) and β-actin (B) in Figure 2C. (C–F) Original images of APP (C), β-actin (upper, D), BACE1 (F) and β-actin (lower, F) in Figure 5A. (G–I) Original images of p-tau (G), tau (H), and β-actin (I) in Figure 5B. (J–L) Original images of caspase-3 (J), caspase-9 (K), and β-actin (L) in Figure 6C. (M, N) Original images of CD11b (M), and β-actin (N) in Figure 7B. (O–Q) Original images of iNOS (O), COX-2 (P), and β-actin (Q) in Figure 7C.
Footnotes
Conflicts of interest: All authors declare no conflicts of interest.
Availability of data and materials: All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open peer reviewer: Gabriela de Paula, Lund University, Sweden.
P-Reviewer: de Paula G; C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Zunino S, Yu J, Song LP; T-Editor: Jia Y
Funding: This work was supported by the Natural Science Foundation of Jiangsu Province of China, No. BK20180948 (to ZL), Nantong Applied Research Program of China, No. MS12019011 (to XXF), and Science and Technology Project of Nantong University of China, No. TDYXY2019007 (to XXF).
References
- 1.Al-Awar A, Kupai K, Veszelka M, Szűcs G, Attieh Z, Murlasits Z, Török S, Pósa A, Varga C. Experimental diabetes mellitus in different animal models. J Diabetes Res. 2016;2016:9051426. doi: 10.1155/2016/9051426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baharlou R, Ahmadi-Vasmehjani A, Davami MH, Faraji F, Atashzar MR, Karimipour F, Sadeghi A, Asadi MA, Khoubyari M. Elevated levels of T-helper 17-associated cytokines in diabetes type I patients:indicators for following the course of disease. Immunol Invest. 2016;45:641–651. doi: 10.1080/08820139.2016.1197243. [DOI] [PubMed] [Google Scholar]
- 3.Bhusal A, Rahman MH, Lee IK, Suk K. Role of hippocampal lipocalin-2 in experimental diabetic encephalopathy. Front Endocrinol (Lausanne) 2019;10:25. doi: 10.3389/fendo.2019.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chavali V, Nandi SS, Singh SR, Mishra PK. Generating double knockout mice to model genetic intervention for diabetic cardiomyopathy in humans. Methods Mol Biol. 2014;1194:385–400. doi: 10.1007/978-1-4939-1215-5_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chong WP, Mattapallil MJ, Raychaudhuri K, Bing SJ, Wu S, Zhong Y, Wang W, Chen Z, Silver PB, Jittayasothorn Y, Chan CC, Chen J, Horai R, Caspi RR. The cytokine IL-17A limits Th17 pathogenicity via a negative feedback loop driven by autocrine induction of IL-24. Immunity. 2020;53:384–397.e385. doi: 10.1016/j.immuni.2020.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes:many differences, few similarities. Diabetes 54 Suppl. 2005;2:S97–107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- 7.Dai Q, Han S, Liu T, Zheng J, Liu C, Li J, Li S. IL-17A neutralization improves the neurological outcome of mice with ischemic stroke and inhibits caspase-12-dependent apoptosis. Front Aging Neurosci. 2020;12:274. doi: 10.3389/fnagi.2020.00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Egervari G, Akpoyibo D, Rahman T, Fullard JF, Callens JE, Landry JA, Ly A, Zhou X, Warren N, Hauberg ME, Hoffman G, Ellis R, Ferland JN, Miller ML, Keller E, Zhang B, Roussos P, Hurd YL. Chromatin accessibility mapping of the striatum identifies tyrosine kinase FYN as a therapeutic target for heroin use disorder. Nat Commun. 2020;11:4634. doi: 10.1038/s41467-020-18114-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Emamaullee JA, Davis J, Merani S, Toso C, Elliott JF, Thiesen A, Shapiro AM. Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes. 2009;58:1302–1311. doi: 10.2337/db08-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Esposito K, Nappo F, Marfella R, Giugliano G, Giugliano F, Ciotola M, Quagliaro L, Ceriello A, Giugliano D. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans:role of oxidative stress. Circulation. 2002;106:2067–2072. doi: 10.1161/01.cir.0000034509.14906.ae. [DOI] [PubMed] [Google Scholar]
- 11.Ghoneum MH, El Sayed NS. Protective effect of Biobran/MGN-3 against sporadic Alzheimer's disease mouse model:possible role of oxidative stress and apoptotic pathways. Oxid Med Cell Longev. 2021;2021:8845064. doi: 10.1155/2021/8845064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gogoleva VS, Drutskaya MS, Atretkhany KS. The role of microglia in the homeostasis of the central nervous system and neuroinflammation. Mol Biol (Mosk) 2019;53:790–798. doi: 10.1134/S0026898419050057. [DOI] [PubMed] [Google Scholar]
- 14.Hang H, Yuan S, Yang Q, Yuan D, Liu Q. Multiplex bead array assay of plasma cytokines in type 2 diabetes mellitus with diabetic retinopathy. Mol Vis. 2014;20:1137–1145. [PMC free article] [PubMed] [Google Scholar]
- 15.He J, Ryder AG, Li S, Liu W, Zhu X. Glycemic extremes are related to cognitive dysfunction in children with type 1 diabetes:A meta-analysis. J Diabetes Investig. 2018;9:1342–1353. doi: 10.1111/jdi.12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horakova O, Kroupova P, Bardova K, Buresova J, Janovska P, Kopecky J, Rossmeisl M. Metformin acutely lowers blood glucose levels by inhibition of intestinal glucose transport. Sci Rep. 2019;9:6156. doi: 10.1038/s41598-019-42531-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang Y, Liu Z, Cao BB, Qiu YH, Peng YP. Treg cells attenuate neuroinflammation and protect neurons in a mouse model of Parkinson's disease. J Neuroimmune Pharmacol. 2020;15:224–237. doi: 10.1007/s11481-019-09888-5. [DOI] [PubMed] [Google Scholar]
- 18.Hwang IK, Choi JH, Nam SM, Park OK, Yoo DY, Kim W, Yi SS, Won MH, Seong JK, Yoon YS. Activation of microglia and induction of pro-inflammatory cytokines in the hippocampus of type 2 diabetic rats. Neurol Res. 2014;36:824–832. doi: 10.1179/1743132814Y.0000000330. [DOI] [PubMed] [Google Scholar]
- 19.Joseph J, Bittner S, Kaiser FM, Wiendl H, Kissler S. IL-17 silencing does not protect nonobese diabetic mice from autoimmune diabetes. J Immunol. 2012;188:216–221. doi: 10.4049/jimmunol.1101215. [DOI] [PubMed] [Google Scholar]
- 20.Kadłubowska J, Malaguarnera L, Wąż P, Zorena K. Neurodegeneration and neuroinflammation in diabetic retinopathy:potential approaches to delay neuronal loss. Curr Neuropharmacol. 2016;14:831–839. doi: 10.2174/1570159X14666160614095559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim HG. Cognitive dysfunctions in individuals with diabetes mellitus. Yeungnam Univ J Med. 2019;36:183–191. doi: 10.12701/yujm.2019.00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, Hong JT. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008;5:37. doi: 10.1186/1742-2094-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu L, Duff K. A technique for serial collection of cerebrospinal fluid from the cisterna magna in mouse. J Vis Exp. 2008:960. doi: 10.3791/960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Z, Qiu AW, Huang Y, Yang Y, Chen JN, Gu TT, Cao BB, Qiu YH, Peng YP. IL-17A exacerbates neuroinflammation and neurodegeneration by activating microglia in rodent models of Parkinson's disease. Brain Behav Immun. 2019;81:630–645. doi: 10.1016/j.bbi.2019.07.026. [DOI] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 27.Lo W, O'Donnell M, Tancredi D, Orgain M, Glaser N. Diabetic ketoacidosis in juvenile rats is associated with reactive gliosis and activation of microglia in the hippocampus. Pediatr Diabetes. 2016;17:127–139. doi: 10.1111/pedi.12251. [DOI] [PubMed] [Google Scholar]
- 28.Lueptow LM. Novel object recognition test for the investigation of learning and memory in mice. J Vis Exp. 2017:55718. doi: 10.3791/55718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma J, Li YJ, Chen X, Kwan T, Chadban SJ, Wu H. Interleukin 17A promotes diabetic kidney injury. Sci Rep. 2019;9:2264. doi: 10.1038/s41598-019-38811-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marwaha AK, Crome SQ, Panagiotopoulos C, Berg KB, Qin H, Ouyang Q, Xu L, Priatel JJ, Levings MK, Tan R. Cutting edge:Increased IL-17-secreting T cells in children with new-onset type 1 diabetes. J Immunol. 2010;185:3814–3818. doi: 10.4049/jimmunol.1001860. [DOI] [PubMed] [Google Scholar]
- 31.Mathews CE, Langley SH, Leiter EH. New mouse model to study islet transplantation in insulin-dependent diabetes mellitus. Transplantation. 2002;73:1333–1336. doi: 10.1097/00007890-200204270-00024. [DOI] [PubMed] [Google Scholar]
- 32.McGinley AM, Sutton CE, Edwards SC, Leane CM, DeCourcey J, Teijeiro A, Hamilton JA, Boon L, Djouder N, Mills KHG. Interleukin-17A serves a priming role in autoimmunity by recruiting IL-1β-producing myeloid cells that promote pathogenic T cells. Immunity. 2020;52:342–356.e6. doi: 10.1016/j.immuni.2020.01.002. [DOI] [PubMed] [Google Scholar]
- 33.Muriach M, Flores-Bellver M, Romero FJ, Barcia JM. Diabetes and the brain:oxidative stress, inflammation, and autophagy. Oxid Med Cell Longev. 2014;2014:102158. doi: 10.1155/2014/102158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oehlrich D, Prokopcova H, Gijsen HJ. The evolution of amidine-based brain penetrant BACE1 inhibitors. Bioorg Med Chem Lett. 2014;24:2033–2045. doi: 10.1016/j.bmcl.2014.03.025. [DOI] [PubMed] [Google Scholar]
- 35.Oliveira WH, Nunes AK, França ME, Santos LA, Lós DB, Rocha SW, Barbosa KP, Rodrigues GB, Peixoto CA. Effects of metformin on inflammation and short-term memory in streptozotocin-induced diabetic mice. Brain Res. 2016;1644:149–160. doi: 10.1016/j.brainres.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 36.Pei B, Sun J. Pinocembrin alleviates cognition deficits by inhibiting inflammation in diabetic mice. J Neuroimmunol. 2018;314:42–49. doi: 10.1016/j.jneuroim.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 37.Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, Emerson M, Garner P, Holgate ST, Howells DW, Karp NA, Lazic SE, Lidster K, MacCallum CJ, Macleod M, Pearl EJ, et al. The ARRIVE guidelines 2.0:Updated guidelines for reporting animal research. PLoS Biol. 2020;18:e3000410. doi: 10.1371/journal.pbio.3000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Powell CM, Schoch S, Monteggia L, Barrot M, Matos MF, Feldmann N, Südhof TC, Nestler EJ. The presynaptic active zone protein RIM1alpha is critical for normal learning and memory. Neuron. 2004;42:143–153. doi: 10.1016/s0896-6273(04)00146-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qiu AW, Bian Z, Mao PA, Liu QH. IL-17A exacerbates diabetic retinopathy by impairing Müller cell function via Act1 signaling. Exp Mol Med. 2016;48:e280. doi: 10.1038/emm.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qiu AW, Liu QH, Wang JL. Blocking IL-17A alleviates diabetic retinopathy in rodents. Cell Physiol Biochem. 2017;41:960–972. doi: 10.1159/000460514. [DOI] [PubMed] [Google Scholar]
- 41.Qiu AW, Cao X, Zhang WW, Liu QH. IL-17A is involved in diabetic inflammatory pathogenesis by its receptor IL-17RA. Exp Biol Med (Maywood) 2021;246:57–65. doi: 10.1177/1535370220956943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reza-Zaldivar EE, Hernández-Sapiéns MA, Gutiérrez-Mercado YK, Sandoval-Ávila S, Gomez-Pinedo U, Márquez-Aguirre AL, Vázquez-Méndez E, Padilla-Camberos E, Canales-Aguirre AA. Mesenchymal stem cell-derived exosomes promote neurogenesis and cognitive function recovery in a mouse model of Alzheimer's disease. Neural Regen Res. 2019;14:1626–1634. doi: 10.4103/1673-5374.255978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rom S, Zuluaga-Ramirez V, Gajghate S, Seliga A, Winfield M, Heldt NA, Kolpakov MA, Bashkirova YV, Sabri AK, Persidsky Y. Hyperglycemia-driven neuroinflammation compromises BBB leading to memory loss in both diabetes mellitus (DM) type 1 and type 2 mouse models. Mol Neurobiol. 2019;56:1883–1896. doi: 10.1007/s12035-018-1195-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ:25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shahani N, Pryor W, Swarnkar S, Kholodilov N, Thinakaran G, Burke RE, Subramaniam S. Rheb GTPase regulates β-secretase levels and amyloid βgeneration. J Biol Chem. 2014;289:5799–5808. doi: 10.1074/jbc.M113.532713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shehata AM, Quintanilla-Fend L, Bettio S, Singh CB, Ammon HP. Prevention of multiple low-dose streptozotocin (MLD-STZ) diabetes in mice by an extract from gum resin of Boswellia serrata (BE) Phytomedicine. 2011;18:1037–1044. doi: 10.1016/j.phymed.2011.06.035. [DOI] [PubMed] [Google Scholar]
- 47.Shruthi S, Mohan V, Amutha A, Aravindhan V. Increased serum levels of novel T cell cytokines IL-33, IL-9 and IL-17 in subjects with type-1 diabetes. Cytokine. 2016;86:6–9. doi: 10.1016/j.cyto.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 48.Sigurdardottir S, Zapadka TE, Lindstrom SI, Liu H, Taylor BE, Lee CA, Kern TS, Taylor PR. Diabetes-mediated IL-17A enhances retinal inflammation, oxidative stress, and vascular permeability. Cell Immunol. 2019;341:103921. doi: 10.1016/j.cellimm.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith E, Stark MA, Zarbock A, Burcin TL, Bruce AC, Vaswani D, Foley P, Ley K. IL-17A inhibits the expansion of IL-17A-producing T cells in mice through “short-loop”inhibition via IL-17 receptor. J Immunol. 2008;181:1357–1364. doi: 10.4049/jimmunol.181.2.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stover KR, Campbell MA, Van Winssen CM, Brown RE. Early detection of cognitive deficits in the 3xTg-AD mouse model of Alzheimer's disease. Behav Brain Res. 2015;289:29–38. doi: 10.1016/j.bbr.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 51.Sun J, Zhang S, Zhang X, Zhang X, Dong H, Qian Y. IL-17A is implicated in lipopolysaccharide-induced neuroinflammation and cognitive impairment in aged rats via microglial activation. J Neuroinflammation. 2015;12:165. doi: 10.1186/s12974-015-0394-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, Südhof TC. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318:71–76. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tong Z, Liu W, Yan H, Dong C. Interleukin-17A deficiency ameliorates streptozotocin-induced diabetes. Immunology. 2015;146:339–346. doi: 10.1111/imm.12512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity. 2019;50:1352–1364. doi: 10.1016/j.immuni.2019.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang D, Zhang J, Jiang W, Cao Z, Zhao F, Cai T, Aschner M, Luo W. The role of NLRP3-CASP1 in inflammasome-mediated neuroinflammation and autophagy dysfunction in manganese-induced, hippocampal-dependent impairment of learning and memory ability. Autophagy. 2017;13:914–927. doi: 10.1080/15548627.2017.1293766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang X, Ma C, Wu J, Zhu J. Roles of T helper 17 cells and interleukin-17 in neuroautoimmune diseases with emphasis on multiple sclerosis and Guillain-Barrésyndrome as well as their animal models. J Neurosci Res. 2013;91:871–881. doi: 10.1002/jnr.23233. [DOI] [PubMed] [Google Scholar]
- 57.Wu C, Yang L, Li Y, Dong Y, Yang B, Tucker LD, Zong X, Zhang Q. Effects of exercise training on anxious-depressive-like behavior in Alzheimer rat. Med Sci Sports Exerc. 2020;52:1456–1469. doi: 10.1249/MSS.0000000000002294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu Y, Jin MZ, Yang ZY, Jin WL. Microglia in neurodegenerative diseases. Neural Regen Res. 2021;16:270–280. doi: 10.4103/1673-5374.290881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu A, Duan H, Zhang T, Pan Y, Kou Z, Zhang X, Lu Y, Wang S, Yang Z. IL-17A promotes microglial activation and neuroinflammation in mouse models of intracerebral haemorrhage. Mol Immunol. 2016;73:151–157. doi: 10.1016/j.molimm.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 60.Zhang D, Qi F, Gao J, Yan X, Wang Y, Tang M, Zhe X, Cheng M, Wang M, Xie Q, Su Y, Zhang X. Altered cerebellar-cerebral circuits in patients with type 2 diabetes mellitus. Front Neurosci. 2020;14:571210. doi: 10.3389/fnins.2020.571210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao J, Bi W, Xiao S, Lan X, Cheng X, Zhang J, Lu D, Wei W, Wang Y, Li H, Fu Y, Zhu L. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci Rep. 2019;9:5790. doi: 10.1038/s41598-019-42286-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou C, Pridgen B, King N, Xu J, Breslow JL. Hyperglycemic Ins2AkitaLdlr–/– mice show severely elevated lipid levels and increased atherosclerosis:a model of type 1 diabetic macrovascular disease. J Lipid Res. 2011;52:1483–1493. doi: 10.1194/jlr.M014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zilliox LA, Chadrasekaran K, Kwan JY, Russell JW. Diabetes and cognitive impairment. Curr Diab Rep. 2016;16:87. doi: 10.1007/s11892-016-0775-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Original images of western blot bands.
(A, B) Original images of occludin (A) and β-actin (B) in Figure 2C. (C–F) Original images of APP (C), β-actin (upper, D), BACE1 (F) and β-actin (lower, F) in Figure 5A. (G–I) Original images of p-tau (G), tau (H), and β-actin (I) in Figure 5B. (J–L) Original images of caspase-3 (J), caspase-9 (K), and β-actin (L) in Figure 6C. (M, N) Original images of CD11b (M), and β-actin (N) in Figure 7B. (O–Q) Original images of iNOS (O), COX-2 (P), and β-actin (Q) in Figure 7C.