

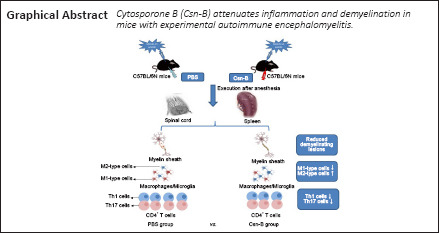
Key Words: cytosporone B (Csn-B), experimental autoimmune encephalomyelitis, macrophages, microglia, multiple sclerosis, NR4A1 agonist, NR4A1, Th1, Th17, treatment
Abstract
Nuclear receptor subfamily 4 group A1 (NR4A1) is an orphan nuclear receptor, which is expressed in the majority of cells. NR4A1 expression in peripheral blood mononuclear cells is low during the preclinical stage of multiple sclerosis. Knockout of the Nr4a1 gene in mice can aggravate the symptoms of experimental autoimmune encephalomyelitis (EAE), which is an animal model of multiple sclerosis. In this study, we intragastrically administered the NR4A1 agonist cytosporone B (Csn-B) to mice after inducing EAE. After treatment with Csn-B, the clinical symptoms in the EAE mice were substantially attenuated compared with that in PBS-treated control mice. The percentages of CD4+ T cells and F4/80+ cells in the central nervous system were decreased. In addition, interferon-γ and interleukin-17 production by proinflammatory Th1/Th17 cells in the central nervous system and interferon-γ levels in splenocytes were decreased after Csn-B treatment. These findings suggest that the NR4A1 agonist Csn-B can alleviate nerve injury after EAE induction, and, therefore, may be useful as a potential treatment for multiple sclerosis.
Introduction
Multiple sclerosis (MS) is a chronic inflammatory, demyelinating disease with a general tendency to relapse in the central nervous system (CNS) (Stadelmann et al., 2011). MS has a high global prevalence and is a frequent cause of non-traumatic disability in young individuals, resulting in a significant medical, psychosocial, and economic burden (Weinshenker, 1994; Rommer and Stüve, 2013). While the etiology has not yet been fully elucidated, MS is generally considered to be caused by abnormal immune regulation induced by a combination of genetic susceptibility and environmental factors (Tselis, 2011). Several empirical studies based on MS and experimental autoimmune encephalomyelitis (EAE), an animal model of MS (Constantinescu et al., 2011), have suggested that activated CD4+ T cells and macrophages infiltrate the CNS (Zamvil and Steinman, 1990; Park et al., 2005; Prat and Antel, 2005; Okun et al., 2010; Rawji and Yong, 2013) and produce a substantial amount of pro-inflammatory factors, which result in demyelination and eventual axonal degeneration (Noseworthy et al., 2000). Hence, inflammation is of critical importance in the development of MS. However, the roles that these specific pathological processes play in the occurrence and progression of MS remain unclear. At present, there is no cure available for patients with MS, and multiple relapses and cumulative disability lead to a poor prognosis.
The orphan nuclear receptors NR4A1 (NUR77), NR4A2 (NRR1), and NR4A3 (NOR1) constitute the NR4A subfamily of transcription factors (Germain et al., 2006). Orphan nuclear receptors are immediate-early response gene products (Maxwell and Muscat, 2006), which can regulate glucose metabolism (Fu et al., 2007; Miao et al., 2019), lipid homeostasis (Pols et al., 2008; Miao et al., 2020), cancer development (He et al., 2020), inflammation (Hu et al., 2017; Xiong et al., 2020), and vascular remodeling (Nomiyama et al., 2009; Li et al., 2018; Kang et al., 2019). Additionally, NR4A family members regulate gene expression that is related to cell differentiation, proliferation, and programmed death (Winoto and Littman, 2002; Liu et al., 2019; He et al., 2020). The latter process is characterized by receptor migration to the mitochondria and subsequent release of cytochrome c into the cytoplasm (Li et al., 2000). Achiron et al. (2010) conducted a study to compare gene expression profiles of peripheral blood mononuclear cells from nine patients who developed MS (MS2b) and 11 matched patients without MS. The expression of NR4A1 was significantly down-regulated in patients with MS2b (Achiron et al., 2010), and the expression of NR4A1 and NR4A2 genes in 31 patients with clinically isolated syndrome was significantly lower than that in 13 healthy subjects matched for gender and age. The expression of the NR4A1 protein in peripheral blood mononuclear cells decreased significantly in patients with MS2b and clinically isolated syndrome (Achiron et al., 2010). Gene-array analysis (Achiron et al., 2010) indicated that NR4A1 was down-regulated during the MS pre-disease state. Our previous report showed that the absence of the Nr4a1 gene in mice increased EAE severity, which was characterized by substantially enhanced inflammation and demyelination (Wang et al., 2018). Judging from the current evidence, it can be inferred that Nr4a1 gene expression may be related to the regulatory mechanisms of EAE and MS.
Cytosporone B (Csn-B) was isolated from the endophytic fungus Dothiorella sp. HTF3. Csn-B is a natural agonist for NR4A1 (Brady et al., 2000) and demonstrates strong binding affinity that upregulates NR4A1 transactivational activity (Zhan et al., 2008). One study found that Csn-B induced apoptosis by targeting the mitochondria (Zhan et al., 2008). Csn-B administration decreased the formation of atherosclerotic plaques in mice that consumed fat-rich diets (Hu et al., 2014) and mitigated inflammation in a colitis mouse model by reducing pro-inflammatory factors (Wu et al., 2016). By increasing NR4A1-mediated apoptosis, Csn-B suppressed xenograft tumor growth in nude mice (Zhan et al., 2008). Csn-B also inhibited excessive inflammation and improved osteoarthritis by activating NR4A1 (Xiong et al., 2020). Together, these findings suggest that targeting NR4A1 may have a potential curative effect on inflammatory diseases. This study evaluated the functions of the NR4A1 agonist Csn-B and the mechanisms by which Csn-B potentially regulated the development of EAE.
Materials and Methods
Animals
Female mice are more sensitive than male mice, as manifested by a higher incidence of EAE and greater severity of clinical signs (Voskuhl et al., 1996). In previous studies (Zhang et al., 2015; Wang et al., 2020), female mice were mainly used. Six to eight-week-old, female C57BL/6N mice (n = 49, weighing 18–20 g) were acquired from Zhejiang Vital River Laboratory Animal Technology Co., Ltd., China (license No. SCXK (Zhe) 2019-0001). The mice were maintained in a specific pathogen-free environment at the Zhengzhou University Experimental Animal Center (Zhengzhou, China). Forty-four mice were used for the EAE modeling, and the other five mice were used as a source of naïve CD4+ T cells. The experimental procedures and protocols were approved by the Animal Ethics Committee of Zhengzhou University (approval No. 2019-KY-142) on June 12, 2019.
EAE induction
EAE was induced in the C57BL/6N mice as described previously (Stromnes and Goverman, 2006). Briefly, the myelin oligodendrocyte glycoprotein peptide 35–55 (MOG35-55; 250 µg; R&D Systems, Minneapolis, MN, USA) was dissolved in 100 µL phosphate buffer saline (PBS). The peptide was mixed thoroughly with 100 µL Freund′s complete adjuvant (MilliporeSigma, Burlington, MA, USA) containing 5 mg/mL of Mycobacterium tuberculosis H37Ra (Difco Laboratories, Inc.; Detroit, MI, USA) to prepare a water-in-oil emulsion of complete antigen. Each mouse was subcutaneously injected at the neck and back with 100 µL complete antigen/site. Pertussis toxin (200 ng; List Biological Lab; Epsom, UK) was dissolved in 200 µL PBS and injected intraperitoneally at 0 and 48 hours post-immunization as a co-adjuvant to induce EAE. Subsequently, EAE mice were randomly assigned to the Csn-B (n = 27) and PBS (n = 17) groups.
Drug administration
The NR4A1 agonist Csn-B (MilliporeSigma) was dissolved in 200 µL PBS. The mice with EAE were intragastrically administered 2.5, 10, or 20 mg/kg/d Csn-B based on a previous study (Egarnes et al., 2017). Five mice were in each group. Csn-B was administered once per day beginning on day 10 post-immunization as a preventative agent in the dose optimization study or on day 14 post-immunization as a treatment in the follow-up experiments. The Csn-B was administered until day 21. Control mice were intragastrically administered 200 µL PBS at the same time points as the Csn-B administration (Figure 1).
Figure 1.
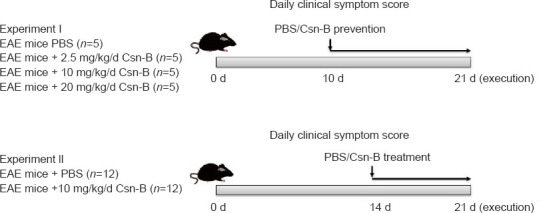
Experimental flow chart.
Csn-B: Cytosporone B; EAE: experimental autoimmune encephalomyelitis; PBS: phosphate buffer saline.
EAE clinical score
Beginning at the time of EAE induction in the mice, the symptoms were assessed each day based on the Clinical Grading of Mice with Classic Encephalomyelitis (Stromnes and Goverman, 2006) by an investigator who was blinded to the groups. Additional Table 1 summarizes the criteria for the assessment of clinical symptoms in mice with EAE. A higher score indicates greater severity of symptoms and a worse prognosis for the mice.
Additional Table 1.
Clinical grading of mice with experimental autoimmune encephalomyelitis
| Grade | Clinical |
|---|---|
| 0 | Normal |
| 0.5 | The tip of the tail droops |
| 1 | Tail paralysis |
| 2 | Staggering gait; hind limb paresis |
| 2.5 | One hind limb paralyzed |
| 3 | Paralysis of hind limb |
| 3.5 | Hind limbs paralyzed; weakness of forelimb |
| 4 | Paralysis of all limbs |
| 5 | Dying |
Induction of Th1 and Th17 cell differentiation in vitro
Th1 and Th17 cell differentiation was induced in vitro utilizing protocols that were previously reported (Zhang et al., 2015). In summary, mice were sacrificed after intraperitioneal injection of 2% pentobarbital (40 mg/kg; Notlas Bio Co., Ltd.; Beijing, China). Naïve CD4+ T cells were isolated from the spleens of C57BL/6N mice (n = 5) by negative selection with mouse CD4 microbeads (Miltenyi Bio Co., Somerville, MA, USA). Purified cells were cultured with 1 µg/mL soluble rat anti-CD3 (Abcam, Hong Kong, China, Cat# ab24948, RRID: AB_470375) and 1 µg/mL hamster anti-CD28 (Abcam, Cat# ab25234, RRID: AB_470416) antibodies at 37°C for 3 days. Five ng/mL mouse interleukin-12 (IL-12, R&D Systems) was added to stimulate Th1 differentiation, and 2 ng/mL transforming growth factor-β1 (Beyotime Bio Co., Ltd., Nantong, Jiangsu, China), 20 ng/mL interleukin-6 (IL-6, R&D Systems), 10 ng/mL interleukin-1β (IL-1β, R&D Systems), 10 µg/mL rabbit anti-interleukin-4 (anti-IL-4, Abcam, Cat# ab62351, RRID: AB_943835), and 10 µg/mL rat anti-interferon-γ (anti-IFN-γ, Miltenyi Biotec, Cat#130-102-388, RRID: AB_2659990) were added to stimulate Th17 differentiation (Zhang et al., 2015). The culture medium also contained Csn-B at different concentrations (5 and 10 µg/mL) for the intervention groups, while the control group was treated with PBS. Flow cytometry was used to evaluate cells that had been incubated for 3 days.
Histopathology
On day 21 post-immunization, mice were anesthetized by intraperitioneal injection of 2% pentobarbital, and 30 mL cold PBS was perfused into the heart to clear the blood. The spinal cord tissue was obtained immediately and fixed using 4% paraformaldehyde for 24 hours, then placed in ethanol and xylene. The tissue was then embedded in paraffin and cut into 5-mm-thick sections. The spinal cord sections were stained with hematoxylin and eosin (HE) to evaluate the degree of inflammation and luxol fast blue (LFB) to evaluate the degree of demyelination. For HE staining, the paraffin sections obtained in the previous step were first deparaffinized in xylene, and anhydrous ethanol was used to remove the xylene. The sections were hydrated by immersing in solutions with decreasing concentrations of alcohol. The sections were incubated in hematoxylin and eosin staining solutions and then washed in running water. Finally, the sections were dehydrated with ethanol and xylene and sealed. For LFB staining, the paraffin sections were first deparaffinized as described, stained with LFB staining solution overnight at room temperature, and washed with ethanol and distilled water. The sections were incubated in saturated lithium carbonate solution and washed again. Finally, the sections were dehydrated with ethanol and xylene and sealed. Tissue sections were observed by microscopy. The inflammation and demyelination of spinal cords after HE and LFB staining, respectively, were evaluated under blinded conditions using a scale from 0 to 3 as previously described (Yang et al., 2010). Additional Table 2 summarizes the criteria for the assessment of inflammation and demyelination. A higher score indicated that the tissue displayed greater inflammation and demyelination than a lower score.
Additional Table 2.
Criteria for the assessment of inflammation and demyelination.
| Grade | Inflammation | Demyelination |
|---|---|---|
| 0 | None inflammatory cells | None demyelination site |
| 1 | Some inflammatory cells | One or two demyelination sites |
| 2 | Inflammatory cells infiltrate perivascular tissue | Three or more demyelination sites |
| 3 | Severe perivascular cuffing and extend into the surrounding tissues | Large demyelination sites |
Flow cytometry
As described above, the mice were perfused, and the spinal cords and the spleen were obtained on day 21 post-immunization. The mononuclear cells infiltrating the spinal cord and spleen were isolated using a 70 µm nylon cell strainer and enzymatic digestion with Liberase™ (Roche; Basel, Switzerland) for 20–30 minutes at 37°C. Then mononuclear cells from spinal cord were isolated using a Percoll gradient (70/37%). These collected cells were washed, and all surviving cells were counted using a light microscope (CKX31; Huyu Trading Co., Ltd.; Shanghai, China). Mononuclear cells isolated from the spinal cord and spleen tissues were suspended at a concentration of 2 × 106/mL in complete RPMI 1640 medium containing 10% fetal bovine serum (FBS) and then washed in PBS containing 3% FBS and 0.02% NaN3 for the purpose of surface marker staining. Fc receptors were blocked using Fc receptor Blocker (MK Bio Co., Ltd., Shanghai, China). The cells were incubated on ice with the appropriate fluorochrome-conjugated antibodies for 30 minutes. In brief, after washing these cells in fluorescence-activated cell sorter (FACS) buffer, anti-CD4 (Abcam, Cat# ab53420, RRID: AB_2075566), anti-F4/80 (MyBioSource, Cat# MBS607610, RRID: AB_10888718), anti-CD40 (LSBio (LifeSpan) Cat# LS-C43310-250, RRID: AB_1051259), anti-CD86 (US Biological, Cat# C2438-01H, RRID: AB_2074980), anti-CD206 (Thermo Fisher Scientific, Cat# MA5-28581, RRID: AB_2745540) or anti-I-region associated antigen (anti-Ia, Abcam, Cat#ab51262, RRID: AB_869913) antibodies were added for surface marker staining. Prior to intracellular cytokine staining, phorbol 12-myristate 13-acetate (50 ng/mL), ionomycin (500 ng/mL), and Golgi-Stop (1 mg/106 cells) were added to the cells, which were then incubated in RPMI 1640 medium containing 10% FBS at 37°C for 4 hours. The cells were fixed and permeabilized using Fix/Perm reagents (BD Biosciences) and then incubated with fluorescent-labeled antibodies against intracellular cytokines. Intracellular staining was performed in accordance with the manufacturer’s instructions. M1 macrophages were labeled with anti-IL-12 (MBL International, Cat# JM-5162-100, RRID: AB_591999). M2 macrophages were labeled with anti-IL-4 (Abcam, Cat# ab62351, RRID: AB_943835). Th1 cells were labeled with anti-IFN-γ (Miltenyi Biotec, Cat# 130-102-388, RRID: AB_2659990) and Th17 cells were labeled with ani-IL-17 (US Biological, Cat# 18439-10B, RRID: AB_2124882). After the last wash, the cells were collected on a FACSAria flow cytometer (BD Biosciences) and analyzed using FlowJo™ software v.10.0 (Tree Star, Ashland, OR, USA).
Cytokine levels in splenocytes detected by enzyme-linked immunosorbent assays
The mice were perfused on day 21 post-immunization. Single-cell suspensions were prepared from the spleen tissues using sterile 70 µm nylon cell strainers. Erythrocytes from the spleens were dissolved by adding NH4Cl-Tris buffer at room temperature for 5 minutes. Then, the single-cell suspensions were washed, re-suspended, and all surviving cells were counted. Splenocytes were incubated in RPMI 1640 (Gibco) with 10% FBS in triplicate in 24-well plates at a concentration of 2 × 106/mL. Cells were re-stimulated for 48 and 72 hours with 25 µg/mL MOG35-55. Cell-free supernatants were collected, and the cytokines interleukin-17 (IL-17) and IFN-γ, were measured using enzyme-linked immunosorbent assay kits (R&D Systems) in accordance with the manufacturer’s instructions.
Statistical analysis
The sample size was selected based on previous literature and a prior study by our team (Wang et al., 2018). No animals were missing in any experimental group, and the evaluator assessed disease symptoms in the mice using a blinded method. GraphPad Prism v.9.0 software (GraphPad Software, Inc.; La Jolla, CA, USA, www.graphpad.com) was used for statistical analyses. Data are expressed as the mean ± standard error of mean (SEM). Student’s t-tests were used to compare the means of two groups. Comparisons of more than two groups were conducted using one-way analysis of variance with Bonferroni post hoc tests. Statistical significance was defined as a P-value less than 0.05.
Results
NR4A1 agonist Csn-B affects the differentiation of Th1 and Th17 cells in vitro
To elucidate the role of Csn-B in the differentiation of CD4+ T cells in vitro, naïve T cells were collected from wild-type mice and induced to differentiate into Th1 or Th17 cells. Under the conditions for Th1 differentiation, the percentage of CD4+IFN-γ+ cells significantly decreased after Csn-B treatment compared with that in the PBS control group. Under conditions for Th17 differentiation, the percentage of CD4+IL-17+ cells was also significantly decreased compared with that in the control group (Figure 2). These results suggest that Csn-B has a significant impact upon Th1 and Th17 cell differentiation in vitro.
Figure 2.
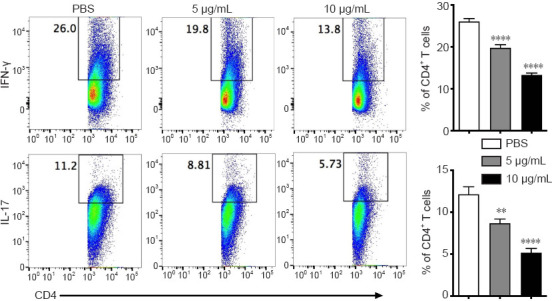
Th1 and Th17 cell differentiation is decreased in vitro after treatment of naïve CD4+ T cells with cytosporone B (Csn-B).
Naïve CD4+ T cells were separated by microbeads and differentiation was induced to generate T helper (Th)1 or Th17 cells. The cells were treated with 5 or 10 µg/mL Csn-B, and PBS was used in the control group. Intracellular levels of interferon (IFN)-γ (for Th1) and interleukin (IL)-17 (for Th17) in differentiated CD4+ T cells were determined using flow cytometry based on the gated populations (left), and the percentages of IFN-γ+ and IL-17+ cells within the CD4+ T cell population were analyzed (right). Data are expressed as means ± SEM. **P < 0.01, ****P < 0.001 (one-way analysis of variance followed by Bonferroni post hoc test). Csn-B: cytosporone B; IFN-γ: interferon-γ; IL-17: interleukin-17; PBS: phosphate buffer saline.
Determination of the optimal doses of Csn-B in EAE suppression
To determine the optimal Csn-B dose for treatment of EAE, the EAE mice were intragastrically administered 2.5, 10, or 20 mg/kg Csn-B per day as a preventive intervention. The control group was treated with PBS. Symptom onset was observed beginning on days 12–13 after immunization. The clinical function scores showed no significant difference between the mice treated with 2.5 mg/kg/d Csn-B and the control groups; however, the clinical function scores of the 10 and 20 mg/kg/d Csn-B groups were substantially lower than those in the PBS-treated mice (Figure 3). No difference was observed between the 10 and 20 mg/kg/d Csn-B groups (Figure 3). The results indicated that oral treatment with Csn-B reduced the severity of EAE symptoms in vivo, and the Csn-B dose of 10 mg/kg per day was chosen as the therapeutic dose for all subsequent experiments.
Figure 3.
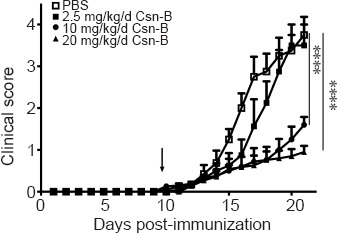
The NR4A1 agonist cytosporone B (Csn-B) can suppress the development of experimental autoimmune encephalomyelitis symptoms in vivo.
After induction of experimental autoimmune encephalomyelitis with myelin oligodendrocyte glycoprotein peptide 35–55 and pertussis toxin, 2.5, 10, or 20 mg/kg Csn-B or PBS was administered to the mice once per day from day 10 to 21 post-immunization (the arrow represents the time of administration). The clinical function score showed no significant difference between the mice treated with 2.5 mg/kg/d Csn-B and the PBS control group, whereas the clinical function scores for the 10 and 20 mg/kg/d Csn-B groups were significantly lower than the scores for mice treated with PBS. Data are presented as means ± SEM (n = 5/group). ****P < 0.001 (one-way analysis of variance followed by Bonferroni post hoc test). Csn-B: cytosporone B; MOG35–55: myelin oligodendrocyte glycoprotein-35–55; PBS: phosphate buffer saline.
Csn-B attenuates EAE clinical symptoms by reducing demyelination and inflammatory cell infiltration in the spinal cord
To determine the impact of Csn-B on EAE, mice were administered the MOG35-55 peptide to induce EAE. EAE clinical symptoms were observed beginning on days 12–13 after induction. Based on the above study, mice were intragastrically administered 10 mg/kg Csn-B per day as a treatment beginning on day 14 post-immunization. Compared with the scores in the control group, the clinical function scores of the Csn-B group were significantly decreased (Figure 4A). The Csn-B treatment significantly alleviated demyelination and inflammation in L3 spinal cord tissue compared with that in the control group on day 21 (Figure 4B). The pathological scores for inflammation and demyelination were significantly lower in the Csn-B group of mice than in the control group (Figure 4B). Flow cytometry demonstrated that the percentages and absolute numbers of CD4+ T cells (P < 0.001) and F4/80+ macrophages/microglia (P < 0.01) isolated from the CNS were significantly decreased in the Csn-B-treated mice compared with those in the PBS control group (Figure 4C). These findings illustrated that the NR4A1 agonist Csn-B attenuated the clinical symptoms of EAE by reducing demyelination lesions and inflammatory cell infiltration in the CNS.
Figure 4.
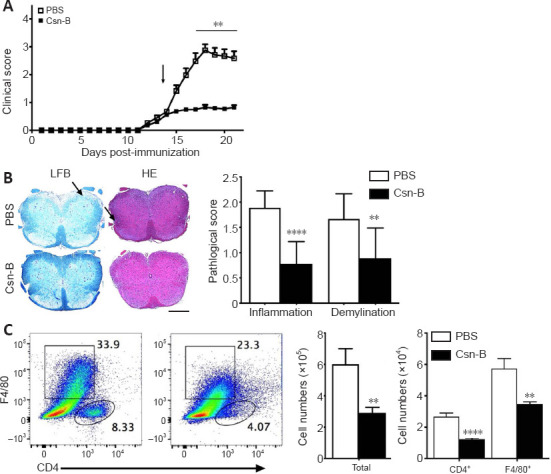
Cytosporone B (Csn-B) attenuates clinical symptoms and reduces demyelination and inflammatory cell infiltration in the central nervous system.
After induction of experimental autoimmune encephalomyelitis (EAE) with myelin oligodendrocyte glycoprotein peptide 35–55 and pertussis toxin, 10 mg/kg Csn-B or PBS was administered to the mice once per day from day 14 to 21 post-immunization. L3 spinal cords were harvested on day 21. (A) Clinical symptom scores. The arrow represents the time of Csn-B or PBS administration. (B) Luxol fast blue (LFB) staining shows demyelination, and hematoxylin-eosin (HE) staining shows inflammation (left panel). PBS-treated EAE mice demonstrated more severe demyelination and greater inflammatory cell infiltration (arrows) compared with that observed in the Csn-B-treated EAE mice. The pathological scores for inflammation and demyelination are presented in the bar graphs (right panel). Scale bars: 200 µm. (C) Flow cytometry was used to detect CD4+ T and F4/80+ cells in the central nervous system of PBS-treated (left plot) and Csn-B-treated (right plot) based on the gated populations. The black boxes represent the distribution and percentage of F4/80+ cells, and the oval boxes represent the distribution and percentage of CD4+ cells. Absolute numbers of CD4+ T and F4/80+ cells are presented in the bar graphs (right panel). Data are presented as means ± SEM (n = 4 per group). **P < 0.01, ****P < 0.001 (Student’s t-test). Csn-B: Cytosporone B; HE: hematoxylin-eosin; LFB: Luxol fast blue; PBS: phosphate buffer saline.
Macrophages/microglia display an enhanced anti-inflammatory cell phenotype in the CNS and spleen after Csn-B treatment
To investigate the immunological mechanisms associated with the reduction in EAE severity in Csn-B-treated mice, we evaluated the polarization of macrophages/microglia that were isolated from the CNS and spleens by flow cytometry. As shown in Figure 5, compared with that in mice given PBS, the Csn-B group showed significantly decreased expression of M1 markers. The expression of Ia, CD86, CD40, and IL-12 in the CNS and Ia and CD86 in splenocytes was decreased in mice treated with Csn-B. In contrast, the expression of the M2 markers CD206 and IL-4 were elevated in the CNS-infiltrating cells and splenocytes of Csn-B treated mice compared with that in the PBS group (Figure 5). These results indicated that macrophages/microglia displayed an enhanced M2 (anti-inflammatory) phenotype after treatment with Csn-B.
Figure 5.
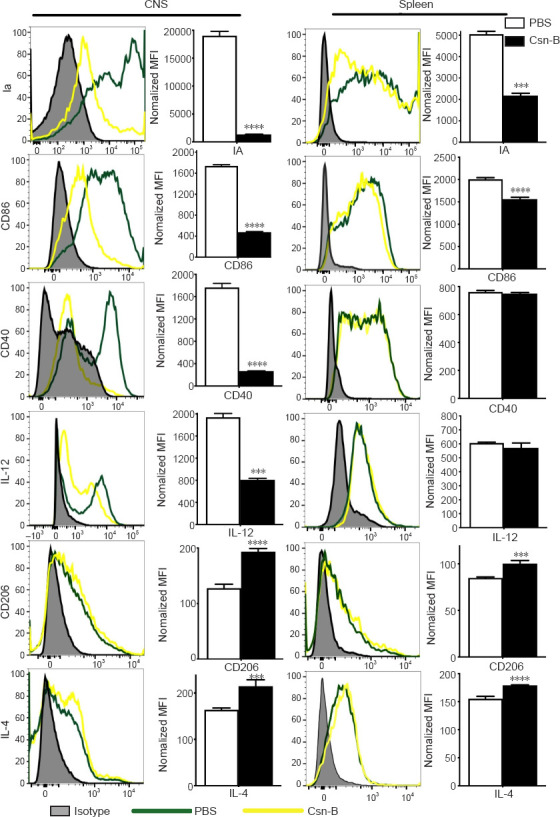
Macrophages/microglia display an enhanced anti-inflammatory cell phenotype after cytosporone B (Csn-B) treatment.
After induction of experimental autoimmune encephalomyelitis, 10 mg/kg Csn-B or PBS was administered once per day from day 14 to 21 post-immunization. The mice were sacrificed on day 21. Infiltrating cells in the central nervous system (CNS) and splenocytes were isolated and stained with the pan-macrophage marker F4/80; M1 markers Ia, CD86, CD40, and IL-12; and the M2 markers CD206 and IL-4. The cells were analyzed using flow cytometry. Expression of these markers in the CNS and spleens was presented as the mean fluorescence intensity (MFI) (left), and the quantitative results are shown in the bar graphs (right). Data are expressed as means ± SEM. ***P < 0.005, ****P < 0.001 (Student’s t-test). Csn-B: Cytosporone B; IA: I region associated antigen; IL: interleukin; MOG35–55: myelin oligodendrocyte glycoprotein-35–55; PBS: phosphate buffer saline.
Csn-B inhibits Th1 and Th17 cell responses in the CNS and spleens of EAE mice
To explore the effect of Csn-B on differentiation of CD4+ T cells in vivo, T cell phenotypes in Csn-B- and PBS-treated mice were examined. Compared with that in the PBS group, the percentages of IFN-γ+ cells and IL-17+ cells in the CNS of Csn-B treated mice were significantly reduced. The percentage of IFN-γ+ cells was also reduced in the spleen; however, the numbers of IL-17+ cells in the spleen were not influenced by Csn-B treatment (Figure 6A). Enzyme-linked immunosorbent assay kits were used to detect the secretion of cytokines from splenocytes cultured in vitro for 48 and 72 hours with MOG35-55. Splenocytes in the Csn-B group secreted lower levels of IFN-γ and IL-17 than cells in the PBS control group in response to MOG35-55 (Figure 6B). These results were similar to the results using flow cytometry observed above, and, thus, demonstrated that Csn-B effectively reduced secretion of the pro-inflammatory cytokines IFN-γ and IL-17 by Th1/Th17 cells in mice with EAE.
Figure 6.
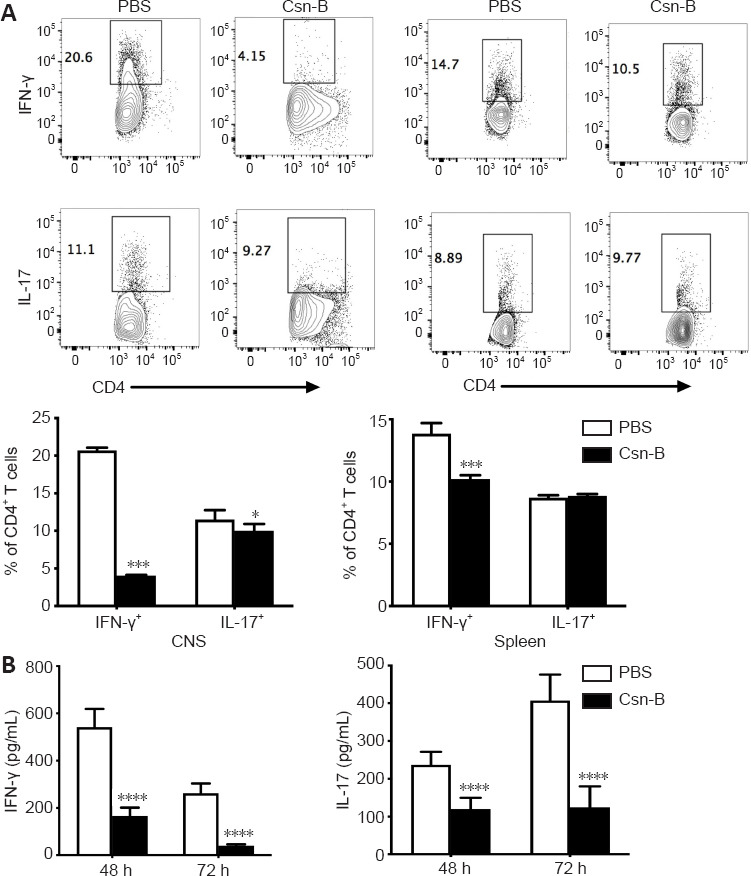
Cytosporone B (Csn-B) decreases T helper (Th)1 and Th17 cell responses in vivo.
After induction of experimental autoimmune encephalomyelitis, 10 mg/kg Csn-B or PBS was administered once per day from day 14 to 21 post-immunization. The mice were sacrificed on day 21. Infiltrating cells in the central nervous system (CNS) and splenocytes were isolated. (A) Upper: CD4+ T cells were gated within the CNS cell or splenocyte populations and analyzed by flow cytometry. The black boxes represent the distribution and percentages of IFN-γ+ and IL-17+ cells. Lower: Percentages of IFN-γ+ and IL-17+ cells. (B) Splenocytes were cultured in medium containing the myelin oligodendrocyte glycoprotein peptide 35–55 (25 µg/mL) for 48 and 72 hours. Supernatants were collected and cytokine levels were analyzed by enzyme-linked immunosorbent assays. Data are presented as means ± SEM (n = 4 per group). *P < 0.05, ***P < 0.005, ****P < 0.001 (Student’s t-test). Csn-B: Cytosporone B; IL-17: interleukin-17; INF-γ: interferon-γ; MOG35–55: myelin oligodendrocyte glycoprotein-35-55; PBS: phosphate buffer saline.
Discussion
The present study revealed that Csn-B can attenuate the clinical severity of EAE with a potential therapeutic effect. First, we provided evidence that Csn-B suppressed Th1/Th17 differentiation of CD4+ T cells in vitro. Second, we demonstrated that Csn-B ameliorated the severity of EAE at the pathological level using an appropriate dose compared with that observed in the control group. The reduction in EAE severity was manifested mainly by decreases in inflammatory cell infiltration and formation of demyelination lesions in the CNS. Furthermore, the percentages and absolute numbers of CD4+ T cells and F4/80+ cells in the CNS were reduced in the Csn-B-treated mice compared with that in the control group. Third, we also discovered that Csn-B decreased pro-inflammatory Th1/Th17 cell responses and related cytokine production, such as IFN-γ and IL-17. We further validated that macrophage/microglial differentiation was directed toward an anti-inflammatory M2 phenotype after Csn-B treatment. These results suggest that Csn-B may be a potential novel therapy for EAE and may represent a safe and effective candidate drug for MS patients in the future.
In the physiological state, NR4A1 plays a crucial role in regulating inflammatory responses in vivo; however, reduced expression has been reported in chronic diseases, such as cancer and neurodegenerative disease (Wu and Chen, 2018; Yan et al., 2020), and upregulating the activity of NR4A1 can reduce the severity of these diseases (Rouillard et al., 2018; Yang et al., 2020). Over-expression of the Nr4a1 gene suppressed the secretion of pro-inflammatory cytokines mediated by oxidized low-density lipoprotein; however, Nr4a1 knockdown resulted in elevated cytokine secretion in animal models of atherosclerosis (Shao et al., 2010). Inflammatory gene expression was reduced when NR4A1 was upregulated in atherosclerotic lesions, including the expression of IL-1β, IL-6, and IL-8 (Bonta et al., 2006). Thus, correcting NR4A1 dysfunction may be beneficial for improving inflammatory disease prognoses. In accordance with these findings, the current study demonstrated a reduction in inflammation and demyelination in mice with EAE after treatment with the NR4A1 agonist Csn-B.
MS is characterized by multi-focal inflammatory lesions causing demyelination and axonal degeneration (Constantinescu et al., 2011). Therefore, inflammation plays a crucial role in the pathological mechanism of MS. The clinical signs of inflammation in nerve tissue are controlled in part by the infiltration of pro-inflammatory CD4+ T cells and activation of macrophages/microglia. Previous studies have shown an imbalance between Th1/Th17 and regulatory T cells in EAE (Becher and Segal, 2011; Zhan et al., 2020). Th1/Th17 cells are characterized as pathogenic T cells (Langrish et al., 2005; Park et al., 2005; Weiner, 2008), whereas regulatory T cells can modulate the immune system and reduce inflammation in EAE (Danikowski et al., 2017). Th17 cells infiltrating the CNS were observed not only in EAE mice but also in patients with MS (Balasa et al., 2020). In addition, clinical scores and inflammation were significantly reduced when IL-17 receptors were lacking or IL-17-specific inhibitors were used in animal models of MS and rheumatoid arthritis (Pöllinger, 2012). Overexpression of NR4A1 can inhibit T cell differentiation, whereas NR4A1 silencing reduces T cell tolerance and increases inflammatory effects in mice; thus, this protein may be a potential target for tumor immunotherapy (Liu et al., 2019).
Macrophages/microglia are important effector cells in EAE lesions (Jack et al., 2005; Lassmann, 2010; Rawji and Yong, 2013), which release IL-1β and TNF-α resulting in demyelination and axonal injury (Ledeen and Chakraborty, 1998; Bogie et al., 2014). However, the mechanism of this process is unclear. Studies have shown that the mechanism may be related to triggering uncontrolled synaptic activation and excitotoxicity in the EAE animal model (Musella et al., 2016; Rizzo et al., 2018). Microglia, as resident macrophages of the CNS, play a role in immune surveillance and facilitating repair mechanisms after brain injury or inflammation-associated tissue damage during CNS diseases when at rest (Benarroch, 2013; Yamasaki et al., 2014). Under pathological conditions, microglia are activated and undergo morphological and molecular changes (Wu et al., 2014; Wolf et al., 2017) and participate in the EAE autoimmune reaction via antigen presentation, secretion of various cytotoxic substances, and phagocytosis (Lassmann and van Horssen, 2011; Dendrou et al., 2015). Previous studies have demonstrated that microglia/macrophages display two activation phenotypes, M1 (pro-inflammation) and M2 (anti-inflammation) (Gordon, 2003), which participate in the pathogenesis of EAE. It has been shown that inhibiting M1 and promoting M2 cells can significantly reduce EAE symptoms (Qin et al., 2012; Liu et al., 2015). M1-type cells secrete a large number of pro-inflammatory cytokines, which lead to the central recruitment of other pro-inflammatory cells, while M2-type cells can secrete anti-inflammatory cytokines and promote the differentiation of oligodendrocytes to accelerate the re-myelination process (Kigerl et al., 2009; Mikita et al., 2011; Chu et al., 2018).
Different mechanisms may be involved in Csn-B-mediated reduction in the clinical severity of EAE in mice, but these mechanisms synergistically reduce inflammation. Our previous study showed that NR4A1 inhibited Th1/Th17 cell differentiation (Wang et al., 2018), and the current study demonstrated that upregulating the activity of NR4A1 by Csn-B treatment suppressed pro-inflammatory CD4+ T cell responses as well as cytokine production. It seems reasonable that, in our study, mice treated with Csn-B showed a similar effect. One study has shown that Csn-B treatments affected the regulation of macrophage/microglial activation and polarization in EAE (Rothe et al., 2017) and increased M2-type cells. Our results are in line with the findings of this study and demonstrated that the number of anti-inflammatory cells increased, while pro-inflammatory cells decreased after Csn-B treatment. A previous study illustrated that NR4A1 regulated the expression of pro-inflammatory genes by controlling the nuclear factor κB pathway, thereby reducing the secretion of inflammatory cytokines in an osteoarthritis model (Xiong et al., 2020). Another study indicated that regulation of the Janus kinase-signal transducer and activator of transcription signaling pathway can prevent the development of EAE by reducing levels of pathogenic Th17 cells and its key effector molecule IL-17A (Ma et al., 2016). However, there is no consensus on these mechanisms.
In conclusion, our study provides several novel findings that indicate Csn-B can effectively alleviate inflammation and demyelination in an animal model of MS during the acute phase. Csn-B may be a novel therapy and may provide a theoretical and experimental basis for the selection of effective drugs that achieve clinical benefits in patients with MS and other autoimmune diseases. This study was a preliminary exploration of the mechanisms by which Csn-B prevented EAE in mice. The limitation is that the study only focuses on tissue, however, the unique signaling pathway(s) downstream of NR4A1 that prevents inflammation at the molecular level and the effect of Csn-B on long-term prognoses for MS and other diseases both require further investigation.
Additional files:
Additional Table 1: Clinical grading of mice with experimental autoimmune encephalomyelitis.
Additional Table 2: Criteria for the assessment of inflammation and demyelination.
Footnotes
Conflicts of interest: The authors have no conflicts of interest to disclose.
Availability of data and materials: All data generated or analyzed during this study are included in this published article and its supplementary information files.
C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Zunino S, Yu J, Song LP; T-Editor: Jia Y
Funding: This study was supported by the National Natural Science Foundation of China, Nos. U1804178 (to LMW) and 31870334 (to LZ).
References
- 1.Achiron A, Grotto I, Balicer R, Magalashvili D, Feldman A, Gurevich M. Microarray analysis identifies altered regulation of nuclear receptor family members in the pre-disease state of multiple sclerosis. Neurobiol Dis. 2010;38:201–209. doi: 10.1016/j.nbd.2009.12.029. [DOI] [PubMed] [Google Scholar]
- 2.Balasa R, Barcutean L, Balasa A, Motataianu A, Roman-Filip C, Manu D. The action of TH17 cells on blood brain barrier in multiple sclerosis and experimental autoimmune encephalomyelitis. Hum Immunol. 2020;81:237–243. doi: 10.1016/j.humimm.2020.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Becher B, Segal BM. T(H)17 cytokines in autoimmune neuro-inflammation. Curr Opin Immunol. 2011;23:707–712. doi: 10.1016/j.coi.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benarroch EE. Microglia:Multiple roles in surveillance, circuit shaping, and response to injury. Neurology. 2013;81:1079–1088. doi: 10.1212/WNL.0b013e3182a4a577. [DOI] [PubMed] [Google Scholar]
- 5.Bogie JF, Stinissen P, Hendriks JJ. Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol. 2014;128:191–213. doi: 10.1007/s00401-014-1310-2. [DOI] [PubMed] [Google Scholar]
- 6.Bonta PI, van Tiel CM, Vos M, Pols TW, van Thienen JV, Ferreira V, Arkenbout EK, Seppen J, Spek CA, van der Poll T, Pannekoek H, de Vries CJ. Nuclear receptors Nur77, Nurr1, and NOR-1 expressed in atherosclerotic lesion macrophages reduce lipid loading and inflammatory responses. Arterioscler Thromb Vasc Biol. 2006;26:2288–2294. doi: 10.1161/01.ATV.0000238346.84458.5d. [DOI] [PubMed] [Google Scholar]
- 7.Brady SF, Wagenaar MM, Singh MP, Janso JE, Clardy J. The cytosporones, new octaketide antibiotics isolated from an endophytic fungus. Org Lett. 2000;2:4043–4046. doi: 10.1021/ol006680s. [DOI] [PubMed] [Google Scholar]
- 8.Chu F, Shi M, Zheng C, Shen D, Zhu J, Zheng X, Cui L. The roles of macrophages and microglia in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neuroimmunol. 2018;318:1–7. doi: 10.1016/j.jneuroim.2018.02.015. [DOI] [PubMed] [Google Scholar]
- 9.Constantinescu CS, Farooqi N, O'Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS) Br J Pharmacol. 2011;164:1079–1106. doi: 10.1111/j.1476-5381.2011.01302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Danikowski KM, Jayaraman S, Prabhakar BS. Regulatory T cells in multiple sclerosis and myasthenia gravis. J Neuroinflammation. 2017;14:117. doi: 10.1186/s12974-017-0892-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15:545–558. doi: 10.1038/nri3871. [DOI] [PubMed] [Google Scholar]
- 12.Egarnes B, Blanchet MR, Gosselin J. Treatment with the NR4A1 agonist cytosporone B controls influenza virus infection and improves pulmonary function in infected mice. PLoS One. 2017;12:e0186639. doi: 10.1371/journal.pone.0186639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu Y, Luo L, Luo N, Zhu X, Garvey WT. NR4A orphan nuclear receptors modulate insulin action and the glucose transport system:potential role in insulin resistance. J Biol Chem. 2007;282:31525–31533. doi: 10.1074/jbc.M701132200. [DOI] [PubMed] [Google Scholar]
- 14.Germain P, Staels B, Dacquet C, Spedding M, Laudet V. Overview of nomenclature of nuclear receptors. Pharmacol Rev. 2006;58:685–704. doi: 10.1124/pr.58.4.2. [DOI] [PubMed] [Google Scholar]
- 15.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 16.He L, Yuan L, Yu W, Sun Y, Jiang D, Wang X, Feng X, Wang Z, Xu J, Yang R, Zhang W, Feng H, Chen HZ, Zeng YA, Hui L, Wu Q, Zhang Y, Zhang L. A regulation loop between yap and nr4a1 balances cell proliferation and apoptosis. Cell Rep. 2020;33:108284. doi: 10.1016/j.celrep.2020.108284. [DOI] [PubMed] [Google Scholar]
- 17.Hu M, Luo Q, Alitongbieke G, Chong S, Xu C, Xie L, Chen X, Zhang D, Zhou Y, Wang Z, Ye X, Cai L, Zhang F, Chen H, Jiang F, Fang H, Yang S, Liu J, Diaz-Meco MT, Su Y, et al. Celastrol-induced Nur77 interaction with TRAF2 alleviates inflammation by promoting mitochondrial ubiquitination and autophagy. Mol Cell. 2017;66:141–153.e6. doi: 10.1016/j.molcel.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu YW, Zhang P, Yang JY, Huang JL, Ma X, Li SF, Zhao JY, Hu YR, Wang YC, Gao JJ, Sha YH, Zheng L, Wang Q. Nur77 decreases atherosclerosis progression in apoE(-/-) mice fed a high-fat/high-cholesterol diet. PLoS One. 2014;9:e87313. doi: 10.1371/journal.pone.0087313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jack C, Ruffini F, Bar-Or A, Antel JP. Microglia and multiple sclerosis. J Neurosci Res. 2005;81:363–373. doi: 10.1002/jnr.20482. [DOI] [PubMed] [Google Scholar]
- 20.Kang JI, Choi Y, Cui CH, Lee D, Kim SC, Kim HM. Pro-angiogenic ginsenosides F1 and Rh1 inhibit vascular leakage by modulating NR4A1. Sci Rep. 2019;9:4502. doi: 10.1038/s41598-019-41115-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lassmann H. Axonal and neuronal pathology in multiple sclerosis:what have we learnt from animal models. Exp Neurol. 2010;225:2–8. doi: 10.1016/j.expneurol.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 24.Lassmann H, van Horssen J. The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett. 2011;585:3715–3723. doi: 10.1016/j.febslet.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 25.Ledeen RW, Chakraborty G. Cytokines, signal transduction, and inflammatory demyelination:review and hypothesis. Neurochem Res. 1998;23:277–289. doi: 10.1023/a:1022493013904. [DOI] [PubMed] [Google Scholar]
- 26.Li H, Kolluri SK, Gu J, Dawson MI, Cao X, Hobbs PD, Lin B, Chen G, Lu J, Lin F, Xie Z, Fontana JA, Reed JC, Zhang X. Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3. Science. 2000;289:1159–1164. doi: 10.1126/science.289.5482.1159. [DOI] [PubMed] [Google Scholar]
- 27.Li P, Bai Y, Zhao X, Tian T, Tang L, Ru J, An Y, Wang J. NR4A1 contributes to high-fat associated endothelial dysfunction by promoting CaMKII-Parkin-mitophagy pathways. Cell Stress Chaperones. 2018;23:749–761. doi: 10.1007/s12192-018-0886-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu CY, Guo SD, Yu JZ, Li YH, Zhang H, Feng L, Chai Z, Yuan HJ, Yang WF, Feng QJ, Xiao BG, Ma CG. Fasudil mediates cell therapy of EAE by immunomodulating encephalomyelitic T cells and macrophages. Eur J Immunol. 2015;45:142–152. doi: 10.1002/eji.201344429. [DOI] [PubMed] [Google Scholar]
- 29.Liu X, Wang Y, Lu H, Li J, Yan X, Xiao M, Hao J, Alekseev A, Khong H, Chen T, Huang R, Wu J, Zhao Q, Wu Q, Xu S, Wang X, Jin W, Yu S, Wang Y, Wei L, et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature. 2019;567:525–529. doi: 10.1038/s41586-019-0979-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma K, Chen X, Chen JC, Wang Y, Zhang XM, Huang F, Zheng JJ, Chen X, Yu W, Cheng KL, Feng YQ, Gu HY. Rifampicin attenuates experimental autoimmune encephalomyelitis by inhibiting pathogenic Th17 cells responses. J Neurochem. 2016;139:1151–1162. doi: 10.1111/jnc.13871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maxwell MA, Muscat GE. The NR4A subgroup:immediate early response genes with pleiotropic physiological roles. Nuclear receptor signaling. 2006;4:e002. doi: 10.1621/nrs.04002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miao L, Yang Y, Liu Y, Lai L, Wang L, Zhan Y, Yin R, Yu M, Li C, Yang X, Ge C. Glycerol kinase interacts with nuclear receptor NR4A1 and regulates glucose metabolism in the liver. FASEB J. 2019;33:6736–6747. doi: 10.1096/fj.201800945RR. [DOI] [PubMed] [Google Scholar]
- 33.Miao L, Su F, Yang Y, Liu Y, Wang L, Zhan Y, Yin R, Yu M, Li C, Yang X, Ge C. Glycerol kinase enhances hepatic lipid metabolism by repressing nuclear receptor subfamily 4 group A1 in the nucleus. Biochem Cell Biol. 2020;98:370–377. doi: 10.1139/bcb-2019-0317. [DOI] [PubMed] [Google Scholar]
- 34.Mikita J, Dubourdieu-Cassagno N, Deloire MS, Vekris A, Biran M, Raffard G, Brochet B, Canron MH, Franconi JM, Boiziau C, Petry KG. Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of multiple sclerosis. Amelioration of clinical status by M2 activated monocyte administration. Mult Scler. 2011;17:2–15. doi: 10.1177/1352458510379243. [DOI] [PubMed] [Google Scholar]
- 35.Musella A, Mandolesi G, Mori F, Gentile A, Centonze D. Linking synaptopathy and gray matter damage in multiple sclerosis. Mult Scler. 2016;22:146–149. doi: 10.1177/1352458515581875. [DOI] [PubMed] [Google Scholar]
- 36.Nomiyama T, Zhao Y, Gizard F, Findeisen HM, Heywood EB, Jones KL, Conneely OM, Bruemmer D. Deficiency of the NR4A neuron-derived orphan receptor-1 attenuates neointima formation after vascular injury. Circulation. 2009;119:577–586. doi: 10.1161/CIRCULATIONAHA.108.822056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343:938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 38.Okun E, Mattson MP, Arumugam TV. Involvement of Fc receptors in disorders of the central nervous system. Neuromolecular Med. 2010;12:164–178. doi: 10.1007/s12017-009-8099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pöllinger B. IL-17 producing T cells in mouse models of multiple sclerosis and rheumatoid arthritis. J Mol Med (Berl) 2012;90:613–624. doi: 10.1007/s00109-011-0841-4. [DOI] [PubMed] [Google Scholar]
- 41.Pols TW, Ottenhoff R, Vos M, Levels JH, Quax PH, Meijers JC, Pannekoek H, Groen AK, de Vries CJ. Nur77 modulates hepatic lipid metabolism through suppression of SREBP1c activity. Biochem Biophys Res Commun. 2008;366:910–916. doi: 10.1016/j.bbrc.2007.12.039. [DOI] [PubMed] [Google Scholar]
- 42.Prat A, Antel J. Pathogenesis of multiple sclerosis. Curr Opin Neurol. 2005;18:225–230. doi: 10.1097/01.wco.0000169737.99040.31. [DOI] [PubMed] [Google Scholar]
- 43.Qin H, Yeh WI, De Sarno P, Holdbrooks AT, Liu Y, Muldowney MT, Reynolds SL, Yanagisawa LL, Fox TH, 3rd, Park K, Harrington LE, Raman C, Benveniste EN. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc Natl Acad Sci U S A. 2012;109:5004–5009. doi: 10.1073/pnas.1117218109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rawji KS, Yong VW. The benefits and detriments of macrophages/microglia in models of multiple sclerosis. Clin Dev Immunol. 2013;2013:948976. doi: 10.1155/2013/948976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rizzo FR, Musella A, De Vito F, Fresegna D, Bullitta S, Vanni V, Guadalupi L, Stampanoni Bassi M, Buttari F, Mandolesi G, Centonze D, Gentile A. Tumor necrosis factor and interleukin-1βmodulate synaptic plasticity during neuroinflammation. Neural Plast. 2018;2018:8430123. doi: 10.1155/2018/8430123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rommer PS, Stüve O. Management of secondary progressive multiple sclerosis:prophylactic treatment-past, present, and future aspects. Curr Treat Options Neurol. 2013;15:241–258. doi: 10.1007/s11940-013-0233-x. [DOI] [PubMed] [Google Scholar]
- 47.Rothe T, Ipseiz N, Faas M, Lang S, Perez-Branguli F, Metzger D, Ichinose H, Winner B, Schett G, Krönke G. The nuclear receptor Nr4a1 acts as a microglia rheostat and serves as a therapeutic target in autoimmune-driven central nervous system inflammation. J Immunol. 2017;198:3878–3885. doi: 10.4049/jimmunol.1600638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rouillard C, Baillargeon J, Paquet B, St-Hilaire M, Maheux J, Lévesque C, Darlix N, Majeur S, Lévesque D. Genetic disruption of the nuclear receptor Nur77 (Nr4a1) in rat reduces dopamine cell loss and l-Dopa-induced dyskinesia in experimental Parkinson's disease. Exp Neurol. 2018;304:143–153. doi: 10.1016/j.expneurol.2018.03.008. [DOI] [PubMed] [Google Scholar]
- 49.Shao Q, Shen LH, Hu LH, Pu J, Qi MY, Li WQ, Tian FJ, Jing Q, He B. Nuclear receptor Nur77 suppresses inflammatory response dependent on COX-2 in macrophages induced by oxLDL. J Mol Cell Cardiol. 2010;49:304–311. doi: 10.1016/j.yjmcc.2010.03.023. [DOI] [PubMed] [Google Scholar]
- 50.Stadelmann C, Wegner C, Brück W. Inflammation, demyelination, and degeneration - recent insights from MS pathology. Biochim Biophys Acta. 2011;1812:275–282. doi: 10.1016/j.bbadis.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 51.Stromnes IM, Goverman JM. Active induction of experimental allergic encephalomyelitis. Nat Protoc. 2006;1:1810–1819. doi: 10.1038/nprot.2006.285. [DOI] [PubMed] [Google Scholar]
- 52.Tselis A. Evidence for viral etiology of multiple sclerosis. Semin Neurol. 2011;31:307–316. doi: 10.1055/s-0031-1287656. [DOI] [PubMed] [Google Scholar]
- 53.Voskuhl RR, Pitchekian-Halabi H, MacKenzie-Graham A, McFarland HF, Raine CS. Gender differences in autoimmune demyelination in the mouse:implications for multiple sclerosis. Ann Neurol. 1996;39:724–733. doi: 10.1002/ana.410390608. [DOI] [PubMed] [Google Scholar]
- 54.Wang D, Zhu B, Liu X, Han Q, Ge W, Zhang W, Lu Y, Wu Q, Shi L. Daphnetin ameliorates experimental autoimmune encephalomyelitis through regulating heme oxygenase-1. Neurochem Res. 2020;45:872–881. doi: 10.1007/s11064-020-02960-0. [DOI] [PubMed] [Google Scholar]
- 55.Wang LM, Zhang Y, Li X, Zhang ML, Zhu L, Zhang GX, Xu YM. Nr4a1 plays a crucial modulatory role in Th1/Th17 cell responses and CNS autoimmunity. Brain Behav Immun. 2018;68:44–55. doi: 10.1016/j.bbi.2017.09.015. [DOI] [PubMed] [Google Scholar]
- 56.Weiner HL. A shift from adaptive to innate immunity:a potential mechanism of disease progression in multiple sclerosis. J Neurol 255 Suppl. 2008;1:3–11. doi: 10.1007/s00415-008-1002-8. [DOI] [PubMed] [Google Scholar]
- 57.Weinshenker BG. Natural history of multiple sclerosis. Ann Neurol. 1994;36(Suppl):S6–11. doi: 10.1002/ana.410360704. [DOI] [PubMed] [Google Scholar]
- 58.Winoto A, Littman DR. Nuclear hormone receptors in T lymphocytes. Cell. 2002;109(Suppl):S57–66. doi: 10.1016/s0092-8674(02)00710-9. [DOI] [PubMed] [Google Scholar]
- 59.Wolf SA, Boddeke HW, Kettenmann H. Microglia in physiology and disease. Annu Rev Physiol. 2017;79:619–643. doi: 10.1146/annurev-physiol-022516-034406. [DOI] [PubMed] [Google Scholar]
- 60.Wu H, Li XM, Wang JR, Gan WJ, Jiang FQ, Liu Y, Zhang XD, He XS, Zhao YY, Lu XX, Guo YB, Zhang XK, Li JM. NUR77 exerts a protective effect against inflammatory bowel disease by negatively regulating the TRAF6/TLR-IL-1R signalling axis. J Pathol. 2016;238:457–469. doi: 10.1002/path.4670. [DOI] [PubMed] [Google Scholar]
- 61.Wu L, Chen L. Characteristics of Nur77 and its ligands as potential anticancer compounds (Review) Mol Med Rep. 2018;18:4793–4801. doi: 10.3892/mmr.2018.9515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu W, Shao J, Lu H, Xu J, Zhu A, Fang W, Hui G. Guard of delinquency?A role of microglia in inflammatory neurodegenerative diseases of the CNS. Cell Biochem Biophys. 2014;70:1–8. doi: 10.1007/s12013-014-9872-0. [DOI] [PubMed] [Google Scholar]
- 63.Xiong Y, Ran J, Xu L, Tong Z, Adel Abdo MS, Ma C, Xu K, He Y, Wu Z, Chen Z, Hu P, Jiang L, Bao J, Chen W, Wu L. Reactivation of NR4A1 restrains chondrocyte inflammation and ameliorates osteoarthritis in rats. Front Cell Dev Biol. 2020;8:158. doi: 10.3389/fcell.2020.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, Wu PM, Doykan CE, Lin J, Cotleur AC, Kidd G, Zorlu MM, Sun N, Hu W, Liu L, Lee JC, Taylor SE, Uehlein L, Dixon D, Gu J, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med. 2014;211:1533–1549. doi: 10.1084/jem.20132477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yan J, Huang J, Wu J, Fan H, Liu A, Qiao L, Shen M, Lai X. Nur77 attenuates inflammatory responses and oxidative stress by inhibiting phosphorylated IκB-αin Parkinson's disease cell model. Aging (Albany NY) 2020;12:8107–8119. doi: 10.18632/aging.103128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang J, Yan Y, Ciric B, Yu S, Guan Y, Xu H, Rostami A, Zhang GX. Evaluation of bone marrow- and brain-derived neural stem cells in therapy of central nervous system autoimmunity. Am J Pathol. 2010;177:1989–2001. doi: 10.2353/ajpath.2010.091203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang PB, Hou PP, Liu FY, Hong WB, Chen HZ, Sun XY, Li P, Zhang Y, Ju CY, Luo LJ, Wu SF, Zhou JX, Wang ZJ, He JP, Li L, Zhao TJ, Deng X, Lin T, Wu Q. Blocking PPARγinteraction facilitates Nur77 interdiction of fatty acid uptake and suppresses breast cancer progression. Proc Natl Acad Sci U S A. 2020;117:27412–27422. doi: 10.1073/pnas.2002997117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zamvil SS, Steinman L. The T lymphocyte in experimental allergic encephalomyelitis. Annu Rev Immunol. 1990;8:579–621. doi: 10.1146/annurev.iy.08.040190.003051. [DOI] [PubMed] [Google Scholar]
- 69.Zhan T, Wang X, Ouyang Z, Yao Y, Xu J, Liu S, Liu K, Deng Q, Wang Y, Zhao Y. Rotating magnetic field ameliorates experimental autoimmune encephalomyelitis by promoting T cell peripheral accumulation and regulating the balance of Treg and Th1/Th17. Aging (Albany NY) 2020;12:6225–6239. doi: 10.18632/aging.103018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhan Y, Du X, Chen H, Liu J, Zhao B, Huang D, Li G, Xu Q, Zhang M, Weimer BC, Chen D, Cheng Z, Zhang L, Li Q, Li S, Zheng Z, Song S, Huang Y, Ye Z, Su W, et al. Cytosporone B is an agonist for nuclear orphan receptor Nur77. Nat Chem Biol. 2008;4:548–556. doi: 10.1038/nchembio.106. [DOI] [PubMed] [Google Scholar]
- 71.Zhang Y, Li X, Ciric B, Ma CG, Gran B, Rostami A, Zhang GX. Therapeutic effect of baicalin on experimental autoimmune encephalomyelitis is mediated by SOCS3 regulatory pathway. Sci Rep. 2015;5:17407. doi: 10.1038/srep17407. [DOI] [PMC free article] [PubMed] [Google Scholar]