

Abstract
The ability to detect 2′-O-methylation sites (Nm) in high-throughput fashion is important, as increasing evidence points to a more diverse landscape for this RNA modification as well as the possibility of yet unidentified functions. Here we describe an optimized version of RibOxi-seq, which is built upon the original published method, that not only accurately profiles ribosomal RNA (rRNA) Nm sites with minimal RNA input but is also robust enough to identify mRNA intronic and exonic sites.
Keywords: 2′-O-methylation, Nm, Ribose methylation, rRNA modifications, mRNA modifications, RNA modifications
1. Introduction
RNA Nm modifications were discovered more than two decades ago, and since then most sites have been found on noncoding RNAs such as transfer RNAs (tRNAs), small nucleolar RNAs (snRNAs), and ribosomal RNAs (rRNAs) (reviewed in [1]). Although the exact functions of Nms have not been pinpointed, evidence suggests Nms can stabilize RNA alternative secondary structures by favoring C3’-endo sugar pucker conformation of the methylated ribose, as well as disrupting RNA–RNA interactions [2, 3]. In addition, both methylated ribonucleosides and fibrillarin (FBL), the methyltransferase involved in snoRNA guided 2′-O-methylation pathways, have been implicated in diseases [4, 5]. Furthermore, recent studies discovered that novel Nms can be found on mRNA transcripts and such sites within a gene alone can exert translational repression through disruption of mRNA–tRNA decoding [3, 6]. Thus, deciphering Nm landscape may provide important new insights on potentially generalizable regulatory roles.
Currently there are three underlying strategies for high throughput profiling of Nm sites. The first is 2OMe-seq, which couples primer extension with next-generation sequencing (NGS), to find global RT stop sites. The basis of this technique is that under some conditions reverse transcriptase pauses at sites of 2′-O-methylation [7]. The second is RiboMeth-seq, which takes advantage of the resistance of Nm bases to either enzymatic or chemical cleavage. NGS libraries generated from RNAs extensively fragmented either enzymatically or chemically and sequenced at very high depth followed by alignment and summarization/visualization of aligned 3′-end counts allow the identification of base positions where read-3′-mapping is significantly lower than the rest of the positions. These positions often correspond to sites of 2′-O-methylation [8, 9]; The third approach is RibOxi/Nm-seq, which were developed independently but published almost simultaneously, and takes advantage of Nm’s property of resistance to cleavage. Additional steps of oxidation, β-elimination, and dephosphorylation post-fragmentation allow the enrichment of Nm at 3′-end of fragmented RNAs. Owing to oxidation of nonmodified ends, only fragments with Nm ends in the final mixture can be ligated to linkers and subsequently converted to NGS libraries ([10, 11], Fig. 1).
Fig. 1.
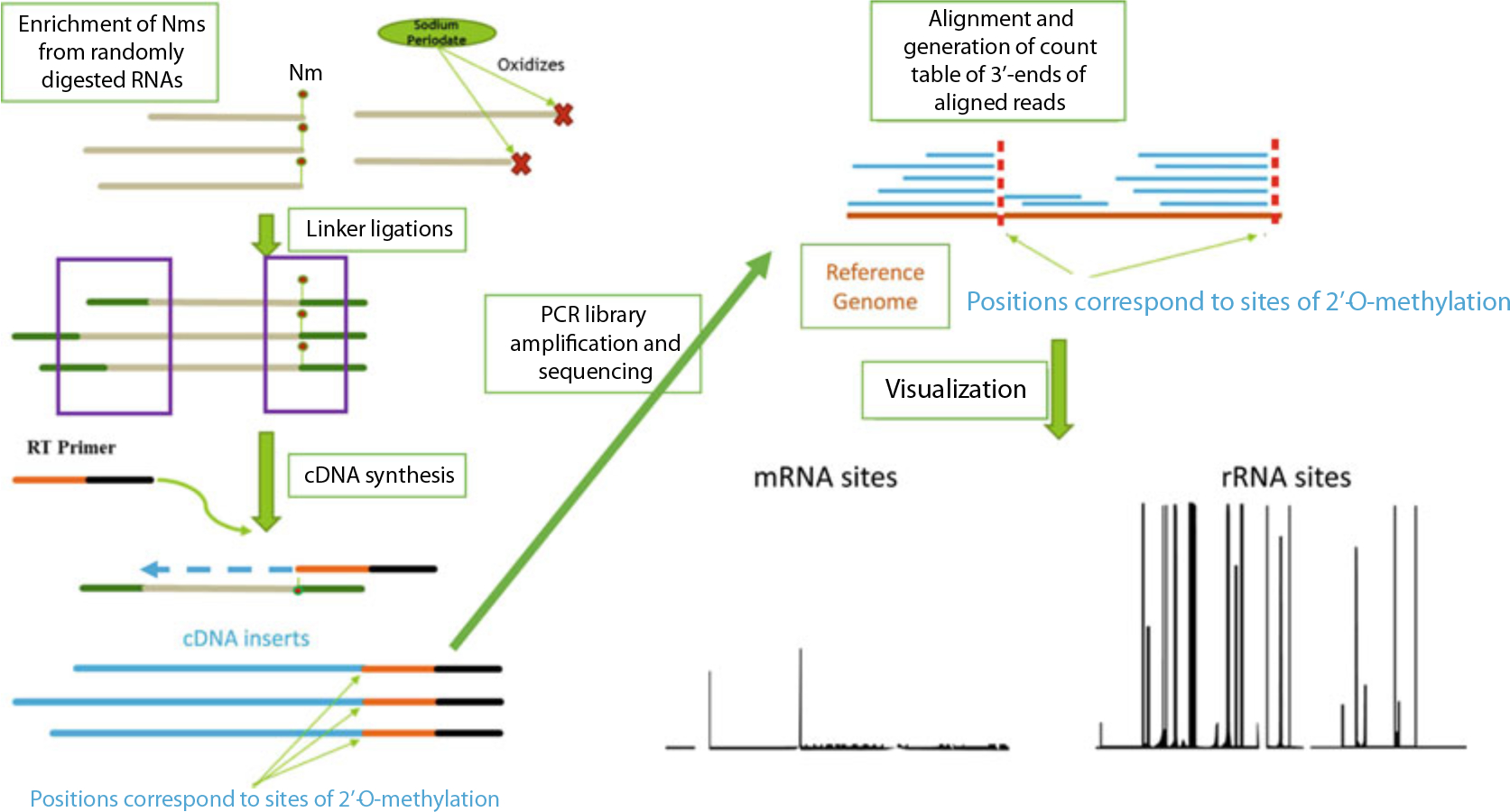
Concept of RibOxi-seq chemistry and computational pipeline. RNA input is randomly digested, followed by enrichment of Nm ends through oxidation of unmethylated 3′-ends by sodium periodate. Libraries containing only nonoxidized fragments are made through the ligation method. Libraries are sequenced on Illumina platforms of choice. Reads are then aligned to a reference genome to allow 3′-end pile-up counting. Positions with significant 3′-end counts corresponds to sites protected by Nm
RibOxi-seq, as initially developed, was ideal for profiling rRNA Nm sites using modest input RNA material and sequencing depth while maintaining good accuracy (reviewed in [12]). Since then, the protocol has gone through further extensive optimization and testing. In our experience, the current protocol requires significantly less input RNA, as well as much lower sequencing depth for profiling rRNA sites. More importantly, the method now allows the detection of mRNA sites. The principle and outline of the protocol remain very similar to the original method, where major procedures include RNA preparation and fragmentation, RNA oxidation, elimination and dephosphorylation, 3′-DNA linker ligation, 5′-RNA linker ligation, cDNA synthesis, library amplification, library QC, and data processing/analysis. One of the improvements in this optimized protocol is that the 5′-RNA linker now contains a random sequence for PCR deduplication instead of the RT primer in the original design. In addition, the 3′-linker design provides the ability to bioinformatically remove RT mispriming events, which occurs in RT reactions with extremely low input ([13], Fig. 2).
Fig. 2.

Illustration of RT mispriming events and mitigation strategy. Under the condition where there is a reasonable amount of RNA material, the RT primer (shown as the floating sequence) mostly anneals to a designated target sequence during an RT reaction as shown in top panel. However, when RNA material is scarce, RT primer can prime with as few as 4 bases as shown in middle panel, drastically increasing nonspecific RT products. The strategy to mitigate the mispriming is to include a constant barcode during 3′-linker ligation as shown in the bottom panel. This does not prevent mispriming; however, after sequencing, the mispriming events can be filtered out by checking whether the reads have the barcode preceding the linker sequence
2. Materials
2.1. RNA Fragmentation
Benzonase nuclease: ≥99% purity.
10× Benzonase buffer: 500 mM Tris–HCl pH 7.5, 200 mM NaCl, 20 mM MgCl2, 1 mg/mL BSA.
Linear polyacrylamide (LPA): 10 μg/μL.
Acid-phenol–chloroform: with IAA, 125:24:1, pH 4.5.
2.2. RNA Oxidation, β-Elimination, and Dephosphorylation
Oxidation-elimination buffer: 2 M lysine hydrochloride pH 8.5.
Oxidation-only buffer: 4.375 mM sodium borate, 50 mM boric acid, pH 8.6.
Alkaline phosphatase.
Alkaline phosphatase buffer: 10× from phosphatase manufacturer.
T4 PNK: 1 Unit/μL.
10× T4 PNK buffer: 700 mM Tris–HCl, 100 mM MgCl2, 50 mM DTT, pH 6.0.
10× T4 PNK buffer: 700 mM Tris–HCl, 100 mM MgCl2, 50 mM DTT, pH 7.6.
8 Buffer exchange columns.
RNA purification columns.
Sodium metaperiodate: ≥99%.
2.3. 3′-DNA Linker Ligation
5’ Preadenylated 3′ blocked DNA oligo: 5′–/5rApp/ATCAC GCTGTAGGCACCATCAATGACAG/3SpC3/–3′, 10 μM.
NEB T4 RNA ligase 2 truncated KQ.
10× NEB T4 RNA ligase buffer: without ATP.
RNase Inhibitor.
PEG 8000.
2.4. PAGE Gel Purification
TBE-Urea gel: 10%.
RNA PAGE gel loading dye: 2×.
SYBR Gold nucleic acid dye.
PAGE gel recovery kit.
2.5. 5′-RNA Linker Ligation
5′ blocked RNA oligo: 5′-/Biosg/ ACACUCUUUCCCUA CACGACGCUCUUCCGAUCUNNNN-3′, 50 μM (see Note 1).
NEB T4 RNA ligase 1.
10× NEB T4 RNA ligase buffer.
ATP: 10 mM.
DMSO: 100%.
RNase Inhibitor.
2.6. cDNA Synthesis
RT Primer: 5′- GTGACTGGAGTTCA GACGTGTGCTCTTCCGATCTGTCATTGATGGTGCC TACAG-3′, 10 μM.
NEB ProtoScript II RT kit.
dNTP: 10 mM.
RNase Inhibitor.
NaOH: 1 N solution.
Tris–HCl: 200 mM pH 7.5.
AMPure XP or equivalent SPRI beads.
2.7. Library Amplification
Illumina compatible I5 primer: 5′- AATGATACGGCGAC CACCGAGATCTACAC -(I5 index)- ACACTCTTTCCCTA CACGACGCTCTTCCGATCT-3′, 2.5 μM (see Note 2).
Illumina compatible I7 primer: 5′-CAAGCAGAAGACGGCA TACGAGAT -(I7 index)- GTGACTGGAGTTCA GACGTGTGCTCTTCCGATCT-3′, 2.5 μM (see Note 2).
High fidelity DNA polymerase: 2×.
Gel purification components: PAGE/agarose, DNA recovery reagents.
2.8. Library Quantification and Visualization
Broad range UV-spectrophotometer.
Qubit or equivalent.
Library size distribution QC equipment and reagents.
3. Methods
It is critical throughout the protocol to practice caution and avoid introducing RNase contamination. It is suggested that all incubation procedures that use a thermal cycler set lid temperature about 10 °C above block temperature. Since profiling rRNA sites is more efficient, it requires drastically less RNA input, thus the fragmentation reaction volume is significantly less. Thus, enzyme is further diluted to allow using larger volume while maintaining final units of enzyme per μL volume.
3.1. RNA Preparation and Fragmentation
To profile only rRNA sites, skip step 2. For the mRNA protocol (which also profiles rRNA sites at a lower depth), skip step 4.
Extract total RNA and prepare 1 μg total RNA in 9 μL water (or 600–800 μg mRNA in 500 μL H2O for mRNA protocol).
[Skip if profiling rRNA only] Perform poly(A) enrichment and obtain ~3–7 μg of poly(A) enriched RNA in 44 μL (see Note 3).
Prepare 0.25 U/μL benzonase working solution by mixing 100 μL 10× benzonase buffer, 899 μL RNase-free H2O, and 1 μL of 250 U/μL benzonase stock in a 1.5 mL Eppendorf tube.
[Skip if profiling mRNA] Further dilute benzonase working solution by mixing 100 μL of the 0.25 U/μL benzonase working solution, 40 μL 10× benzonase buffer, and 360 μL H2O. Final benzonase concentration is now 0.05 U/μL.
Transfer 8 μL (44 μL for mRNA protocol) of the 1 μg RNA input to a 0.2 mL PCR tube and incubate at 95 °C for 3 min in a thermal cycler. Immediately place the tube on ice for 10 s.
Add 1 μL (5 μL for mRNA protocol) of 10× benzonase buffer and 1 μL of 0.05 U/μL (use 0.25 U/μL for mRNA protocol) benzonase working solution to the sample (final RNA concentration ~100 ng/μL), vortex to mix and immediately incubate on ice for 80 min (see Note 4).
In a clean Eppendorf tube, add 90 μL H2O (50 μL for mRNA protocol) and 100 μL acid-phenol:chloroform. Transfer fragmentation reaction mixture from the previous step to the Eppendorf tube and vortex for 15–20 s at high intensity. Place at room temperature for 2 min.
Centrifuge at 18,000–20,000 × g for 5 min. Carefully transfer 90–95 μL aqueous phase to a new Eppendorf tube. Add 1 μL LPA to help precipitation. Ethanol precipitate RNA with 2.5 volumes of 100% EtOH, wash with 80% EtOH once, and resuspend into 33 μL H2O.
Use 1 μL of the fragmented RNA for QC. In our experience, there should be a sharp peak between 25–200 nt skewing to the right (Fig. 3c).
Fig. 3.
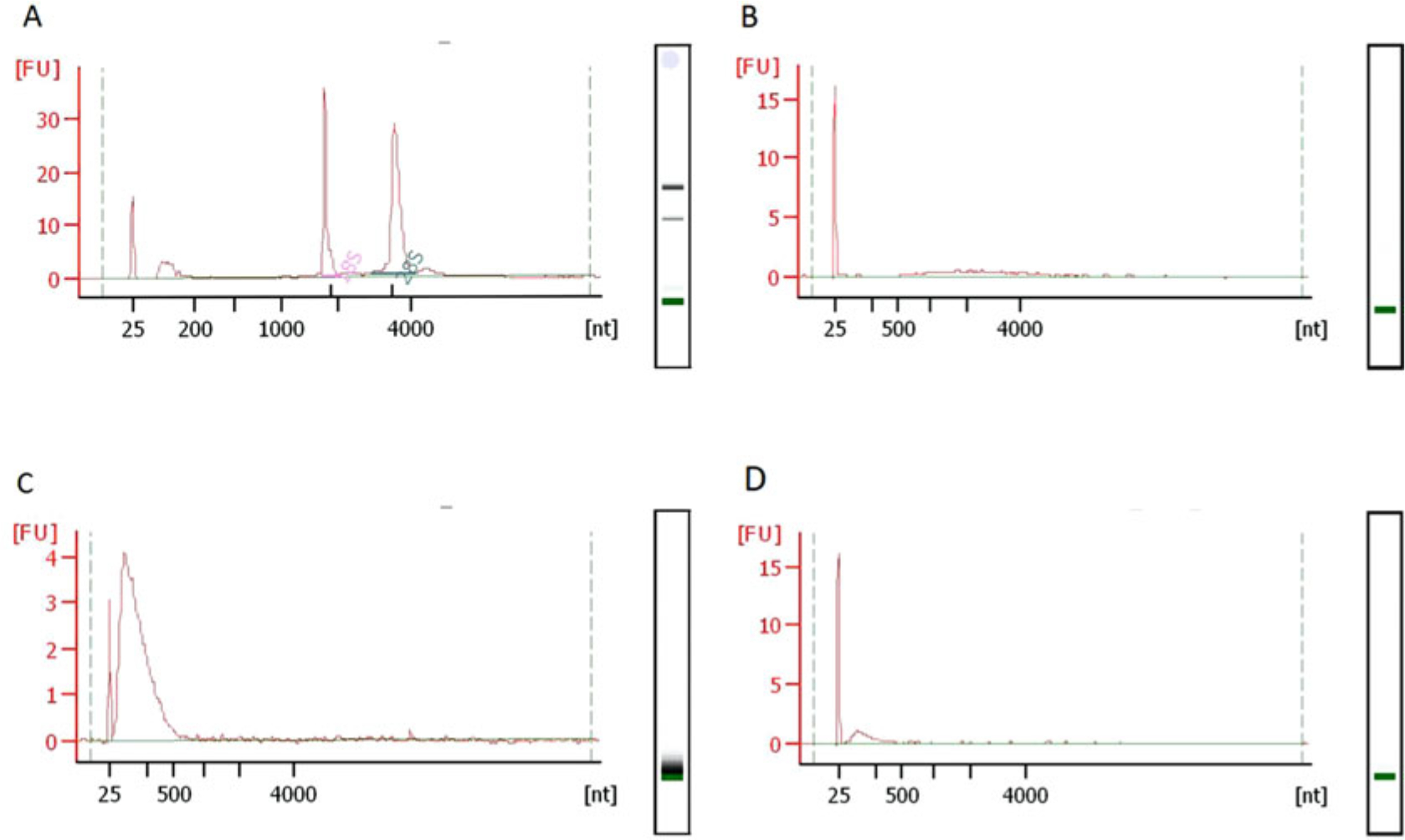
Typical RNA fragment size-distributions during RibOxi-seq. All QC in this figure was done using Bioanalyzer Nano 6000 chips. (a) Typical size distributions of total RNA extracted from HEK293T cells with RNA integrity number of 10. (b) After poly(A) enrichment twice, there should not be any major rRNA peaks left. (c) The RNA size distribution after benzonase fragmentation. (d) Size distribution after 3′-linker ligation. A size shift is not observable, most likely due to the fact that only a very small portion of the RNA was protected from oxidation by Nm and ligated with linkers
3.2. RNA Oxidation, β-Elimination, and Dephosphorylation
Prepare 200 mM NaIO4 solution by dissolving 42.78 mg of NaIO4 powder in 1 mL nuclease free H2O. Protect the solution from light and keep on ice and use only the same day.
Add 4 μL oxidation-elimination buffer and 4 μL NaIO4 to the 32 μL fragmented RNA. Mix well by vortexing, briefly spin-down, and incubate at 37 °C with shaking for 45 min. Purify and resuspend RNA in 42 μL H2O (see Note 5). If performing additional oxidation, β-elimination, and dephosphorylation cycles (a minimum of 3 for profiling mRNA sites), go to step 3, otherwise, continue from step 4.
Add 5 μL 10× alkaline phosphatase buffer, 1 μL RNase inhibitor, and 10 units of the phosphatase enzyme solution. Incubate according to manufacturer’s instructions followed by inactivation. Purify and resuspend in 32 μL H2O. Repeat from step 1 to achieve desired number of oxidation, β-elimination, and dephosphorylation cycles (see Note 6).
Add 5 μL 10× T4 PNK buffer (pH 6.0), 20 units T4 PNK and 1 μL RNase inhibitor to purified RNA from step 2. Mix well by vortexing, briefly spin-down, and incubate at 37 °C for 3 h. Add 5 μL 10× T4 PNK buffer (pH 7.6), 20 more units T4 PNK, 10 μL 10 mM ATP, 33 μL H2O to the reaction. Mix well by vortexing, briefly spin-down, and incubate at 37 °C for an additional 1 h. Inactivate the enzyme following the manufacturer’s instructions.
Ethanol precipitate the reaction as detailed in previous steps into a new Eppendorf tube with help of 1 μL LPA. Thoroughly resuspend the pellet in 35 μL oxidation-only buffer. Add 5 μL NaIO4 solution. Mix well by vortexing, briefly spin-down and incubate at 37 °C with shaking for 45 min.
Purify oxidized RNA and resuspend in 10 μL H2O (see Note 7).
3.3. 3′-DNA Linker Ligation
Transfer the oxidized RNA into a 0.2 mL PCR tube. Add 1 μL 3′ DNA linker, 1 μL RNase inhibitor, 8.5 μL PEG 8000, 2.5 μL 10× T4 RNA ligase buffer, and 2 μL (400 units) T4 RNA ligase 2 truncated KQ. Mix well, spin down, and incubate in a thermal cycler at 16 °C overnight for 18 h (see Note 8).
Cleanup and concentrate RNA to 15 μL. Perform gel purification to separate ligation products from free 3′ DNA linkers. Recover RNA with 11 μL H2O (see Note 9, Fig. 4).
Fig. 4.
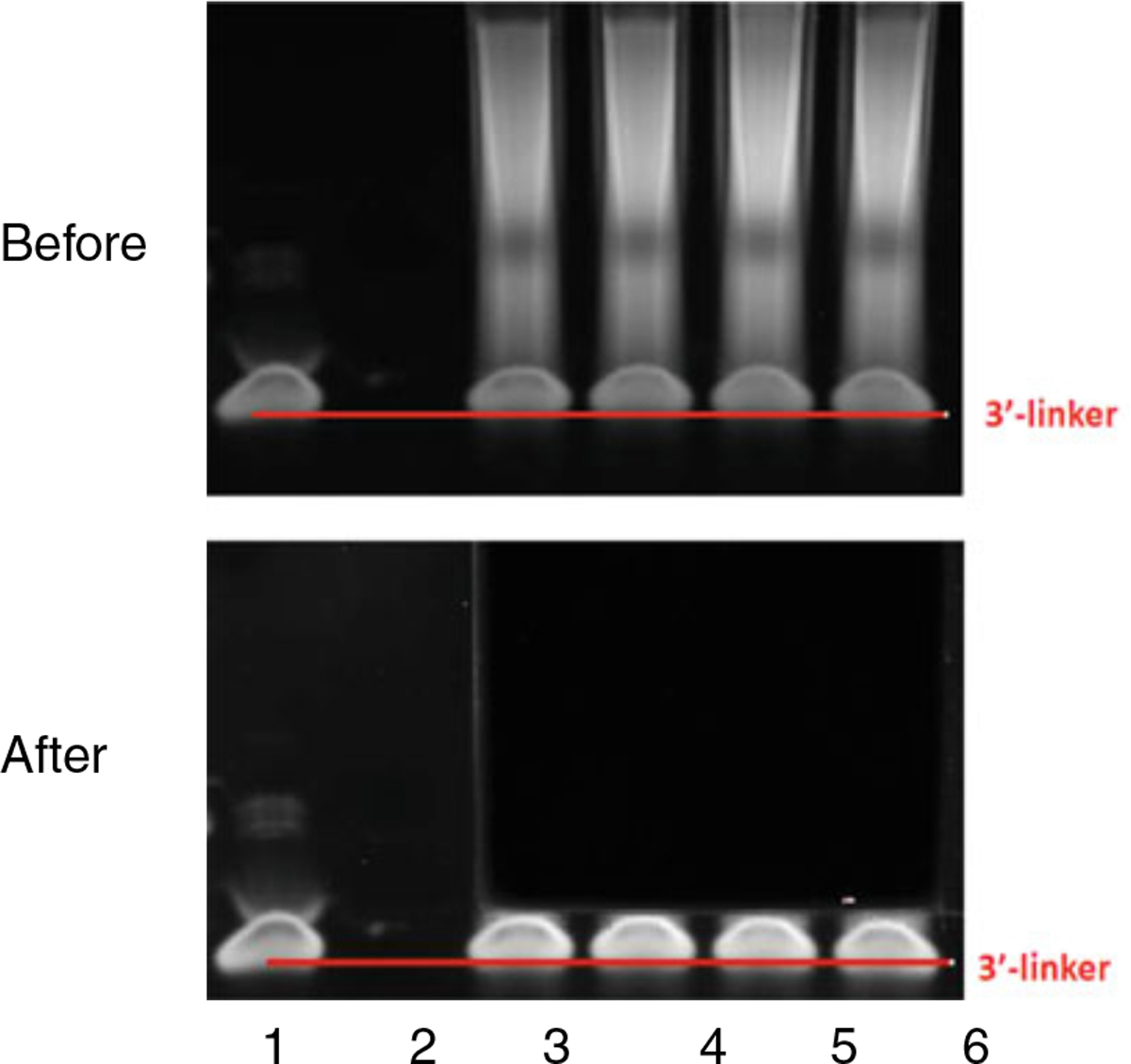
A typical PAGE gel pattern before and after cutting. Lane 1 shows RibOxi-seq 3′-linker. Lanes 3–6 contain reactions postligation. The goal is to recover RNAs and exclude free linkers. The top panel illustrates a typical electrophoresis. The bottom panel illustrates where we normally cut
3.4. 5′-RNA Linker Ligation
Thaw the 50 μM RNA linker, denature 1.3 μL of the linker in a 0.2 mL PCR tube in a thermal cycler at 72 °C for 2 min and return to ice.
Immediately after starting the denaturation of the RNA linker, prepare the ligation reaction by mixing 11 μL product from the previous step with 2 μL 100% DMSO, 2 μL T4 RNA ligase buffer, 2 μL 10 mM ATP, and 0.5 μL RNase inhibitor.
After placing denatured RNA linker on ice for 1 min, add 1 μL of the linker and 1.5 μL T4 RNA ligase I to the mixture in step 2. Mix well and spin down before incubating the reaction at 25 °C for 1 h. Inactivate the RNA ligase enzyme following the manufacturer’s instructions.
3.5. cDNA Synthesis
Denature the RT primer by mixing 20 μL ligated product, 2 μL 10 mM dNTP, 2 μL H2O, and 1 μL 10 μM RT primer in a 0.2 mL PCR tube and incubate under 65 °C in a thermal cycler for 5 min. During the 5 min, prepare reaction mix by mixing 8 μL Protoscript II buffer, 1 μL RNase inhibitor, 4 μL 10× DTT, and 2 μL Protoscript II enzyme mix (see Note 10).
Once the 5-min denaturation is finished, combine both mixtures, and mix well. In a thermal cycler, incubate the combined RT reaction at 50 °C for 1 h (see Note 10).
Hydrolyze RNA by adding 4.4 μL 1 N NaOH and incubating at 95 °C for 20 min in a thermal cycler. Immediately place on ice. Add 4.4 μL 1 M Tris–HCl pH 7.5 and mix well. Use AmpureXP beads or equivalent to clean up the reaction and elute with 20 μL H2O.
3.6. Library Amplification and Library QC
Add 2.5 μL of Illumina compatible forward and reverse primers with indexes of choice to the cDNA. Combine with 25 μL 2× high fidelity DNA polymerase.
Use an appropriate thermal cycler program to allow 3 cycles of initial amplification with low annealing temperature to account for partial complementarity between cDNA ends and PCR primers. This should be then followed by an additional 15 cycles (18 cycles for mRNA protocol) at appropriately higher annealing temperature depending on the polymerase of choice (see Note 11).
Clean up the library using methods of choice to avoid PCR reaction buffer interfering with the subsequent gel electrophoresis. Gel-purify the library, using a commercial or custom DNA ladder for referencing sub 1 kb sizes (see Note 12).
Confirm size distribution using method of choice and quantify concentration with UV spectrophotometer followed by quantification with Qubit or equivalent (see Note 12).
Library is sequenced on an Illumina platform. 3–5 million reads (10–25 million reads for mRNA protocol) per sample for sequencing depth and 1 × 150 cycle configuration are recommended (see Note 13).
3.7. Data Processing and Analysis
A computational pipeline available at: https://github.com/yz201906/RibOxi-seq. It is under development to add more functions, but it can currently be used to process the sequencing reads, align them to a reference genome and generate count tables and visualization in one command line. Here we will go through the individual steps. The read output from the sequencer contains randomer used for deduplication at 5′-ends, a linker sequence at 3′-ends where the first six bases are used for mispriming removal, thus requiring additional processing before alignment and downstream steps (Fig. 5).
The non-barcoded portion of the linker needs to be trimmed first using Cutadapt or a similar package (Fig. 6a, blue portion). Since this part of the read is at the very 3′-end, which can have low quality and base-call errors especially with low complexity libraries, we allow for 20% mismatch rate. Any read without the linker match and subsequent trimming is discarded, Fig. 6b represents the sequence structure after this round of trimming.
A custom python script move_umi.py, which is part of the riboxi-pipeline, is used to move the randomer sequence to the read-name/identifier line for each read. This allows PCR deduplication later on after alignment (Fig. 6c).
Final trimming occurs on the barcode region of the 3′-linker, by allowing 0 base mismatch and discarding untrimmed reads, thus ensuring thorough removal of RT mispriming events (Fig. 6d).
The processed reads are next aligned to a reference genome. The .bam output is the preferred format, which can then be directly indexed and deduplicated using umi_tools (Fig. 7).
The deduplicated alignment file can then be used with the bedtools genomecov function to generate a UCSC genome browser compatible file for visualization. In addition, after converting the deduplicated .bam file into .bed file, a 3′-end count table can be generated using riboxi_bed_parsing.py script from the riboxi_pipeline. The resulting table can be converted to .RData file that is loaded into the riboxi_shinyapp for further analysis and visualization.
Fig. 5.
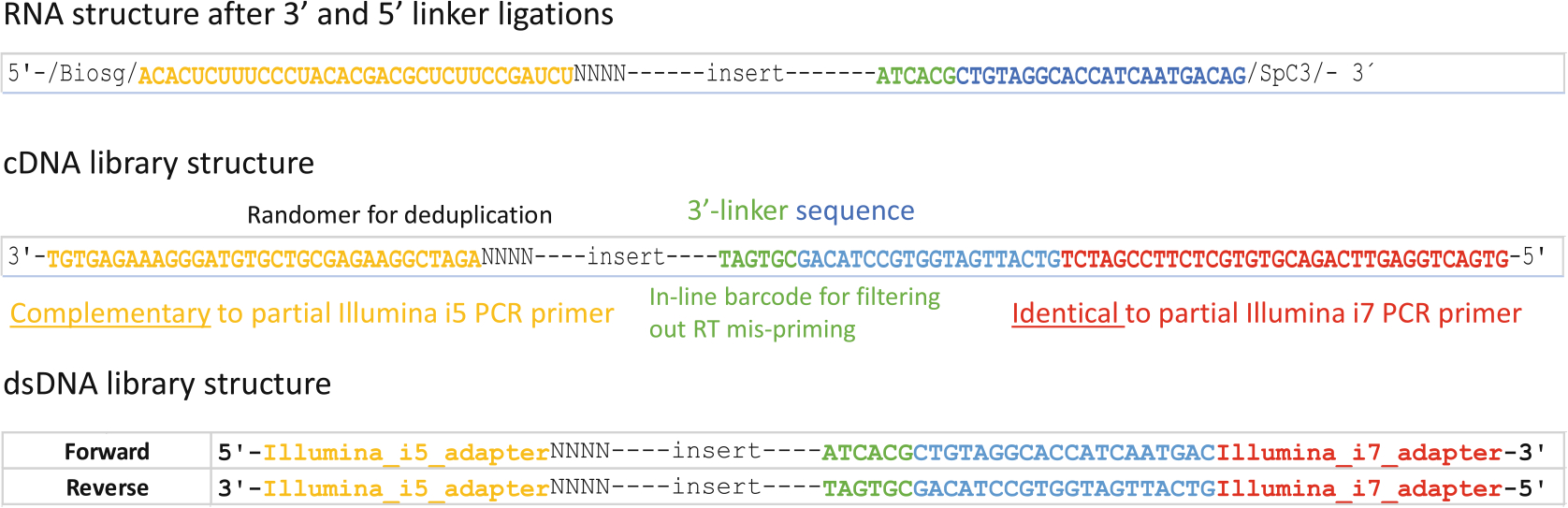
Pseudo-sequence demonstration of RNA, cDNA, and dsDNA library structures. The top panel shows RNA structure with both 3′ and 5′ linkers ligated. The blue portion functions as the RT primer annealing site. The middle panel shows cDNA structure, where yellow indicates sequence complementarity to a portion of the Illumina forward PCR primer and red marks the sequence identical to a portion of the Illumina reverse PCR primer. The bottom panel illustrates the structure of the final dsDNA library
Fig. 6.
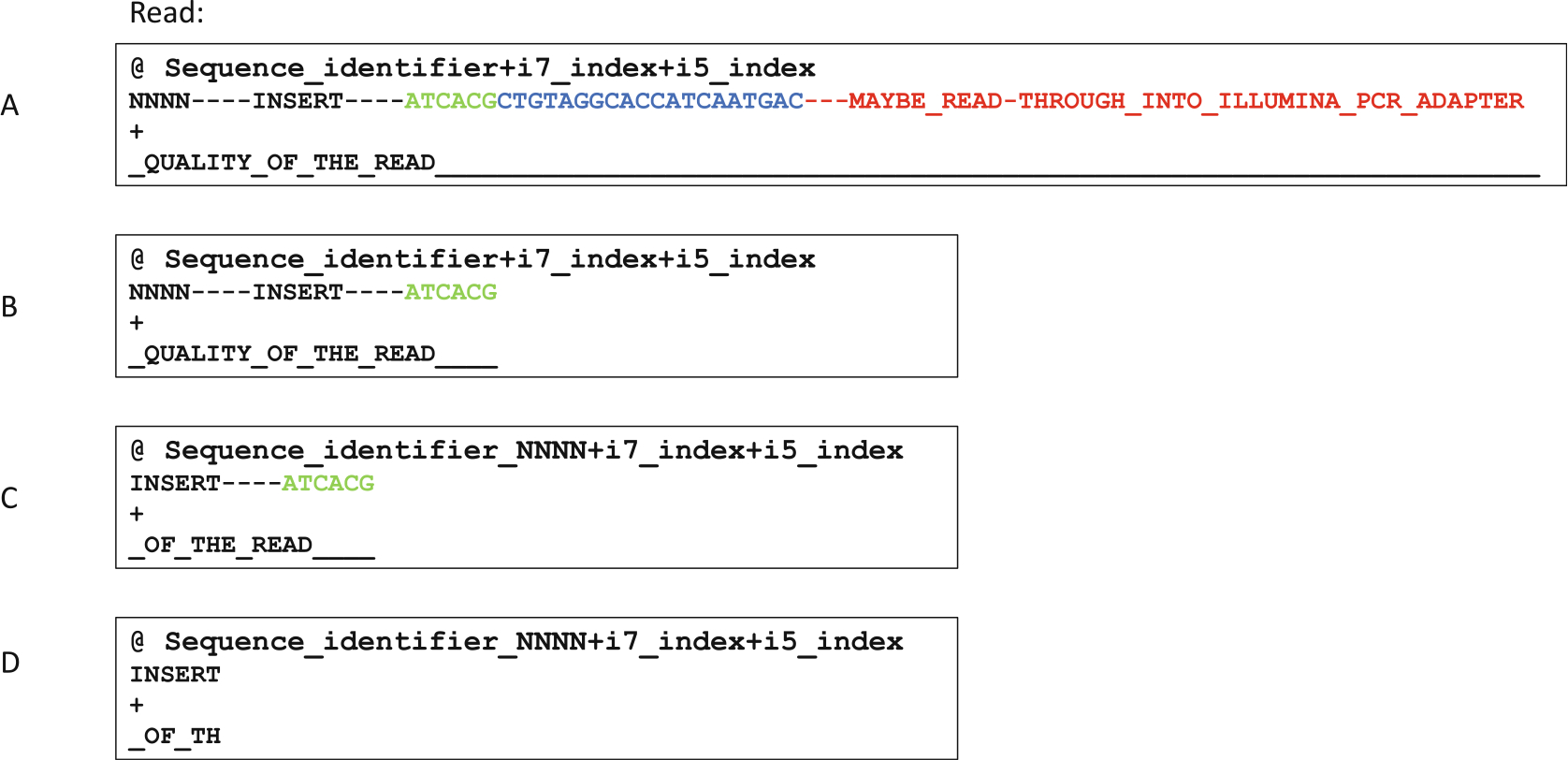
Pseudo-sequence demonstration of how sequencing reads are proccessed. The reads were generated with 1 × 150 cycle sequencing. (a) Illustration of a raw read before any processing. This is in the .fastq format where each read is represented by 4 lines. The first line contains read name and indexes. The second line contains actual sequences. The third line is for optional identifiers, but usually is left empty. The fourth line contains quality strings corresponding to each base in line 2. (b) Resulting read after removing the 3′-linker sequence and everything downstream of it. (c) Resulting read after moving the random sequence from line 2 to line 1. (d) The resulting read after final trimming to remove the barcode used for mispriming event filtering. Reads that do not contain this barcode sequence are discarded as mispriming events
Fig. 7.
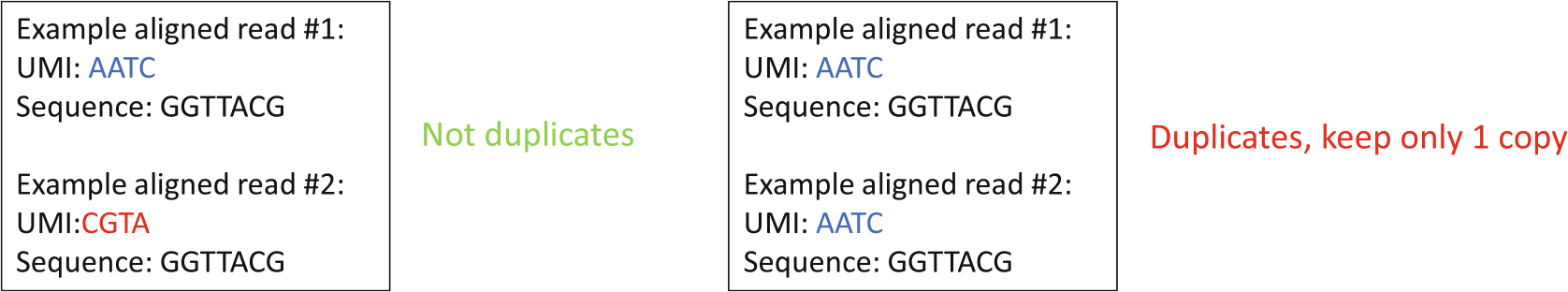
Demonstration of how PCR duplicates are removed. This process uses the random sequence that was moved to the read identifier line for deduplication as unique molecular identifiers (UMI). Each time an aligned read that is identical to another read that was previously seen is identified, UMIs of these two reads are compared to one another. If the UMIs are different, then both alignments are kept as shown on the left. On the other hand, if the UMIs are identical, then only one of the alignments is kept
4. Notes
Having a random UMI allows for distinguishing PCR duplicates vs. identical RNA fragments that physically exist. For example, a quadruple-mer can accommodate 9999 copies of identical fragments, which is not too common in random digestions. Thus, theoretically if 2 sequencing reads contain identical sequences but with different UMIs, then they should all be counted, while only 1 should be retained if the UMIs are the same. In our experience, a quadruple-mer is sufficient for deduplication for the mRNA protocol and low input and low depth rRNA protocol. We recommend 6–8 N bases for higher RNA input (>5 μg) and sequencing depth range (15–100 million clusters).
The i7 and i5 indexes portion can be any sequences of choice (normally a 6/8-base sequence). Be aware of best pooling practices, which can help with index sequence design and ensure index read quality [14].
For cost effectiveness, we used NEB Next polyA mRNA Magnetic Isolation Module with a modified and scaled up protocol. In our routine, we prepare 500 μL beads following original protocol, and use it for 600 μg HEK 293T total RNA that is in 500 μL H2O. For the mRNA protocol, if is critical to have as little rRNA content as possible. Thus, we perform a second poly(A) selection immediately after elution reusing the magnetic beads for each sample (Wash beads once with wash buffer, then twice with RNA dilution buffer). You should see a significant difference in rRNA content for each sample between 1-round and 2-round poly(A) selection (Fig. 3b).
The digestion time requires some optimization especially for different cell lines/tissue samples. Ideally, a time gradient for digestion time should be performed. Under shorter durations, shift in peak sizes should be observed. Eventually, the size of the peak no longer shifts, and reduction in the height of the peak is observed. This is the incubation time that should be chosen.
Initially, we tried either ethanol precipitation or Zymo RNA-Clean concentrator-5/25 after each oxidation–elimination reaction; however, we always see white precipitate that becomes cloudy when vortexed, presumably from high salt concentration. Although we have not verified whether this impacts the subsequent dephosphorylation reaction, it is safer to perform buffer exchange at this step instead. We use Ambion Nucaway columns, which did not result in the same white precipitate.
The goal of using non-T4 PNK phosphatase is to speed up dephosphorylation steps if multiple oxidation-elimination and dephosphorylation cycles are to be performed. Most alkaline phosphatase can be used at this step. We have tested with Antarctic phosphatase from NEB for multiple rounds of oxidation, elimination and phosphorylation cycles followed by mass spectrometry. We were able to confirm that the procedure is robust against at least 6 nmole RNA material (Supplemental Fig. 1).
Alternatively, elute in 11 μL H2O, and use 1 μL for concentration/distribution QC. It is normal to see significant loss of materials when compared to post-digestion concentration (Fig. 3d).
It is easiest to add PEG 8000 and oxidized RNA to the 0.2 mL tube first, then make a master mix for the rest of the reagents and add appropriate amount to each sample when multiple samples are involved.
Here we use a 10% TBE-Urea gel in a Bio-Rad mini-protean system with 1× TBE buffer. The wells of the gel are flushed, and gel prerun at 180 mV for 45 min. Prior to electrophoresis, 2× loading dye is added to sample and subsequently incubated at 72 °C for 2 min and returned to ice for 1 min. Electrophoresis is performed at 180 mV for 40 min. Gels are stained with SYBR Gold. RNA recovery was performed with ZR small RNA PAGE recovery kit.
To reduce mispriming events, it is preferable to use a reverse transcriptase that can sustain activity at higher temperatures. We have only extensively tested the NEB ProtoScript II enzyme, where 48–50 °C temperature range is achievable. Other enzymes that could function at or above such temperature should also be suitable. We minimize RNA and primer spent at low temperature by adding the reaction mix to the RNA–primer mixture immediately after the 5-min denaturation is finished. While thermal cycler is set to 48 °C, we quickly vortex and spin down the combined reaction before placing the tubes into the thermal cycler.
For this step, we have extensively tested the NEB Q5 DNA polymerase 2× master mix. We have eventually landed on the annealing temperature for initial cycles at 52 °C, while setting 62 °C for the remaining cycles.
After library amplification, a cleanup should be performed either with ethanol precipitation, SPRI beads, buffer exchange or column purification. A gel-purification step is also necessary here due to unavoidable empty ligation products that have sizes around 200 bp (Fig. 8a). Either PAGE-gel purification or agarose gel (4%) purification is suitable, but we have opted for agarose since DNA recovery efficiency is much higher. The trade-off is that the desired library peak and empty ligation product peak usually form broader peaks that require more than 1 round of gel-purification (Fig. 8b).
Even if the experimental goal is to detect mRNA sites, the procedure is still very sensitive to rRNA sites which can act as internal technical control. In our experience, libraries prepared with 2× poly(A) enriched samples were still sufficient for rRNA sites profiling even at 1–2 M/sample depth (Supplemental Data 1, 2, Supplemental Figs. 2, and 3).
Fig. 8.
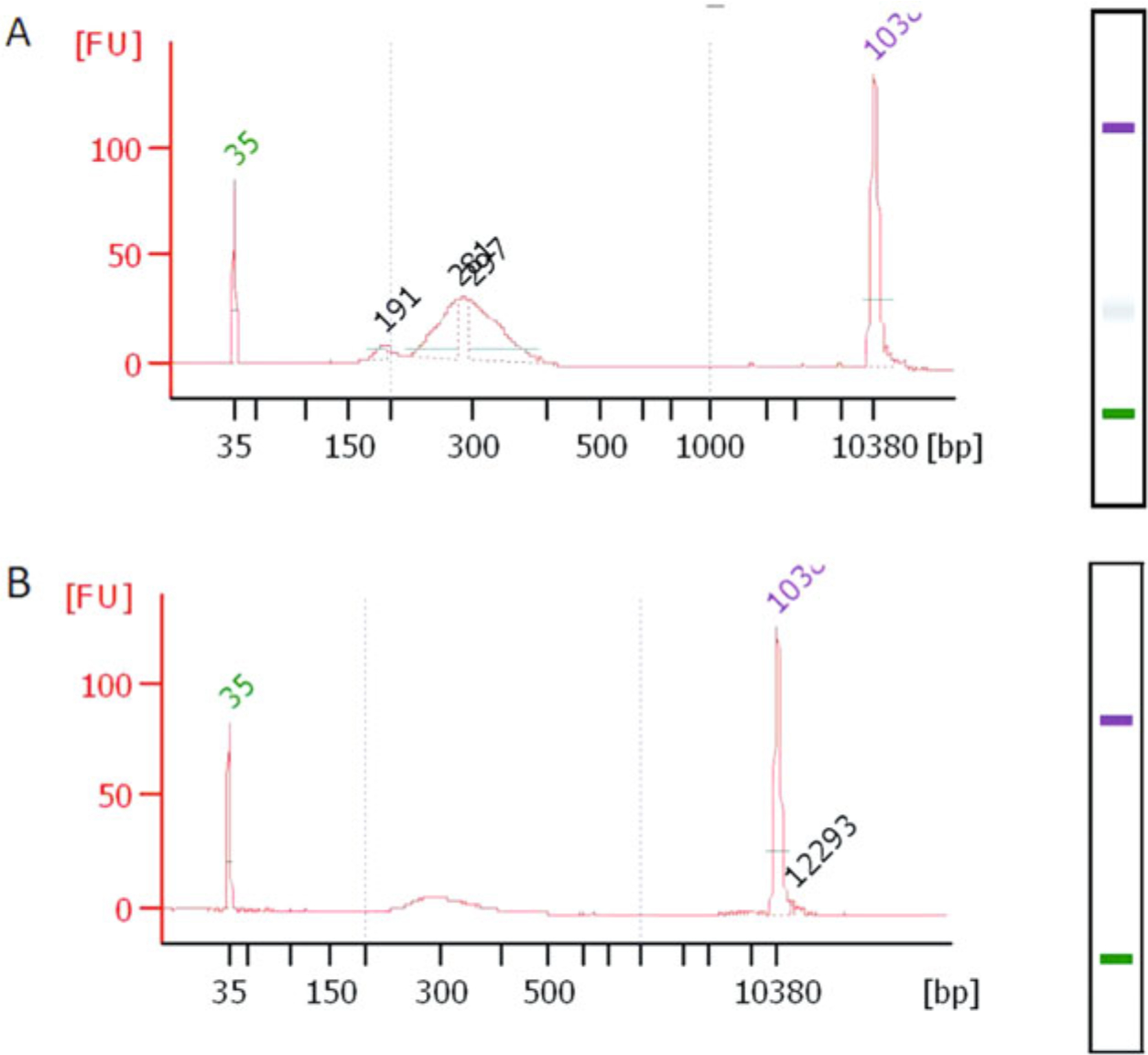
Final library distribution QC. The QCs shown here were done using Bioanalyzer DNA 1000 high sensitivity chips. (a) RibOxi-seq library after 1 round of agarose gel purification (cutting above 220–250 bp). There is still an empty ligation product peak that is at 200 bp. (b) In our experience, a second round of the purification is required to completely remove the 200 bp empty ligations
Supplementary Material
Acknowledgments
The extension work of the original RibOxi-seq is supported NIH grant GM135383 and Duke University Department of Medicine Strong Start to C.L.H. GGC was supported by grant HD099975 from NICHD.
We thank Dr. Disa Elisabet Tehler and Dr. Anders Lund for helpful comments on the initial publication and current development of the method.
Footnotes
Supplementary Information The online version of this chapter (https://doi.org/10.1007/978-1-0716-1851-6_22) contains supplementary material, which is available to authorized users.
References
- 1.Ayadi L, Galvanin A, Pichot F, Marchand V, Motorin Y (2019) RNA ribose methylation (2′-O-methylation): occurrence, biosynthesis and biological functions. Biochim Biophys Acta Gene Regul Mech 1862(3):253–269 [DOI] [PubMed] [Google Scholar]
- 2.Abou Assi H, Rangadurai AK, Shi H, Liu B, Clay MC, Erharter K, Kreutz C, Holley CL, Al-Hashimi HM (2020) 2′-O-methylation can increase the abundance and lifetime of alternative RNA conformational states. Nucleic Acids Res 48(21):12365–12379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi J, Indrisiunaite G, DeMirci H, Ieong KW, Wang J, Petrov A, Prabhakar A, Rechavi G, Dominissini D, He C, Ehrenberg M, Puglisi JD (2018) 2′-O-methylation in mRNA disrupts tRNA decoding during translation elongation. Nat Struct Mol Biol 25(3):208–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dimitrova DG, Teysset L, Carré C (2019) RNA 2′-O-methylation (Nm) modification in human diseases. Genes (Basel) 10(2):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rajan KS, Zhu Y, Adler K, Doniger T, Cohen-Chalamish S, Srivastava A, Shalev-Benami M, Matzov D, Unger R, Tschudi C, Günzl A, Carmichael GG, Michaeli S (2020) The large repertoire of 2′-O-methylation guided by C/D snoRNAs on Trypanosoma brucei rRNA. RNA Biol 17(7):1018–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elliott BA, Ho HT, Ranganathan SV, Vangaveti S, Ilkayeva O, Abou Assi H, Choi AK, Agris PF, Holley CL (2019) Modification of messenger RNA by 2′-O-methylation regulates gene expression in vivo. Nat Commun 10 (1):3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Incarnato D, Anselmi F, Morandi E, Neri F, Maldotti M, Rapelli S, Parlato C, Basile G, Oliviero S (2017) High-throughput single-base resolution mapping of RNA 2′-O-methylated residues. Nucleic Acids Res 45 (3):1433–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krogh N, Jansson MD, Häfner SJ, Tehler D, Birkedal U, Christensen-Dalsgaard M, Lund AH, Nielsen H (2016) Profiling of 2′-O-me in human rRNA reveals a subset of fractionally modified positions and provides evidence for ribosome heterogeneity. Nucleic Acids Res 44 (16):7884–7895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marchand V, Blanloeil-Oillo F, Helm M, Motorin Y (2016) Illuminabased RiboMeth-Seq approach for mapping of 2′-O-Me residues in RNA. Nucleic Acids Research 44(16): e135–e135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu Y, Pirnie SP, Carmichael GG (2017) High-throughput and site-specific identification of 2′-O-methylation sites using ribose oxidation sequencing (RibOxi-seq). RNA 23 (8):1303–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dai Q, Moshitch-Moshkovitz S, Han D, Kol N, Amariglio N, Rechavi G, Dominissini D, He C (2018) Corrigendum: nm-seq maps 2′-O-methylation sites in human mRNA with base precision. Nat Methods 15 (3):226–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krogh N, Nielsen H (2019) Sequencing-based methods for detection and quantitation of ribose methylations in RNA. Methods 156:5–15 [DOI] [PubMed] [Google Scholar]
- 13.Gillen AE, Yamamoto TM, Kline E, Hesselberth JR, Kabos P (2016) Improvements to the HITS-CLIP protocol eliminate widespread mispriming artifacts. BMC Genomics 17:338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Illumina (2020) https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/experiment-design/index-adapters-pooling-guide-1000000041074-10.pdf
- 15.Lestrade L, Weber MJ (2006) snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res 34:D158–D162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshihama M, Nakao A, Kenmochi N (2013) snOPY: a small nucleolar RNA orthological gene database. BMC Res Notes 6:426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.