

Abstract
Acetylation of histone lysine residues by histone acetyltransferase (HAT) p300 and its paralog CBP play important roles in gene regulation in health and diseases. The HAT domain of p300/CBP has been found to be a potential drug target for cancer. Compound screening followed by structure-activity relationship studies yielded a novel series of 1,4-pyrazine-containing inhibitors of p300/CBP HAT with their IC50s as low as 1.4 μM. Enzyme kinetics and other studies support the most potent compound 29 is a competitive inhibitor of p300 HAT against the substrate histone. It exhibited a high selectivity for p300 and CBP, with negligible activity on other classes of HATs in human. Compound 29 inhibited cellular acetylation of several histone lysine residues and showed strong activity against proliferation of a panel of solid and blood cancer cells. These results indicate it is a novel pharmacological lead for drug development targeting these cancers as well as a useful chemical probe for biological studies of p300/CBP.
Keywords: Histone acetyltransferase, p300/CBP, small-molecule inhibitor, cancer therapy
INTRODUCTION
Post-translational modifications of histones play critical roles in gene regulation in health and diseases, among which acetylation of a histone lysine sidechain is one of the most important. Histone proteins, including histone H2A, H2B, H3 and H4, are enriched with basic amino acid residues lysine and arginine, which are protonated at the physiological pH with strong electrostatic and hydrogen-bond interactions with the negatively charged DNAs. Acetylation of the amino group in a histone lysine sidechain partially neutralizes the positive charges and renders a more open DNA conformation. This facilitates the binding of transcription factors as well as other proteins to DNA for gene transcription and other actions, such as DNA replication and repair 1, 2. Moreover, an acetylated histone lysine can be recognized by certain epigenetic “reader” proteins, such as bromodomain-containing proteins, to form a transcription complex for gene expression 3, 4.
Human p300 (E1A binding protein p300) and its paralog CBP [CREB (cAMP-response element binding protein) binding protein] represent a distinct class of histone acetyltransferase (HAT) 5–7, playing important roles in gene expression and regulation. P300/CBP with ~2,400 amino acids consist of multiple structured domains connected with intrinsically disordered regions (IDR). These structured domains of p300 and CBP including their HAT domains are highly conserved. The IDRs of p300/CBP can interact with a number of transcription factors (e.g., CREB, p53 and HIF-1) and other transcription proteins (e.g., steroid receptor coactivators) to form large transcription complexes 8–10. In addition, p300/CBP not only acetylates histones, such as histone H3 lysine 9 or 27 (H3K9 or H3K27), they can also acetylate certain transcription factors (e.g., p53 and Myc) and regulate their functions 7, 11, 12.
HAT activity of p300/CBP has been found to be essential for many nuclear receptor-mediated gene transcription pathways, such as those of estrogen receptor (ER) in females and androgen receptor (AR) in males. P300 HAT is required for ER- or AR-mediated gene expression 8, 10, 13 and therefore, it is of importance in female and male development as well as in breast and prostate cancer. HAT of p300/CBP is a potential drug target for cancer therapy. Inhibitors of p300/CBP HAT have been found to inhibit ER- and AR-mediated gene expression as well as proliferation of breast and prostate cancer cells 14, 15. Evidence has also shown that p300/CBP HAT is critical to other cancers with p300 overexpression or harboring a p300/CBP fusion oncogene 7, 16, 17.
Several distinct chemo-types of p300/CBP HAT inhibitors (Figure 1) have been reported 6, 7. However, many of these compounds have poor cell-permeability (e.g., Lys-CoA and its analogs 18, 19) or contain a “PAINS” (pan-assay interfering compound) 20 or related structure (e.g., C646 21, L002 22 and Cpd-2c 23), which limit their applications in cellular and in vivo studies. Compound A-485 is a potent, drug-like inhibitor of p300/CBP HAT, which is competitive against the enzyme cofactor acetyl coenzyme A (Ac-CoA) 15. It showed strong activity against proliferation of several cancers including prostate cancer. Our recently disclosed compound SYC-1171 is also a drug-like inhibitor of p300/CBP HAT 14, which is competitive against the substrate histone with a different mechanism of action from A-485. SYC-1171 inhibited ER-mediated gene transcription pathways and suppressed proliferation of several cancer cell lines. Here, we report discovery, biochemical and biological activities of 1,4-pyrazine-containing inhibitors of p300/CBP HAT.
Figure 1.
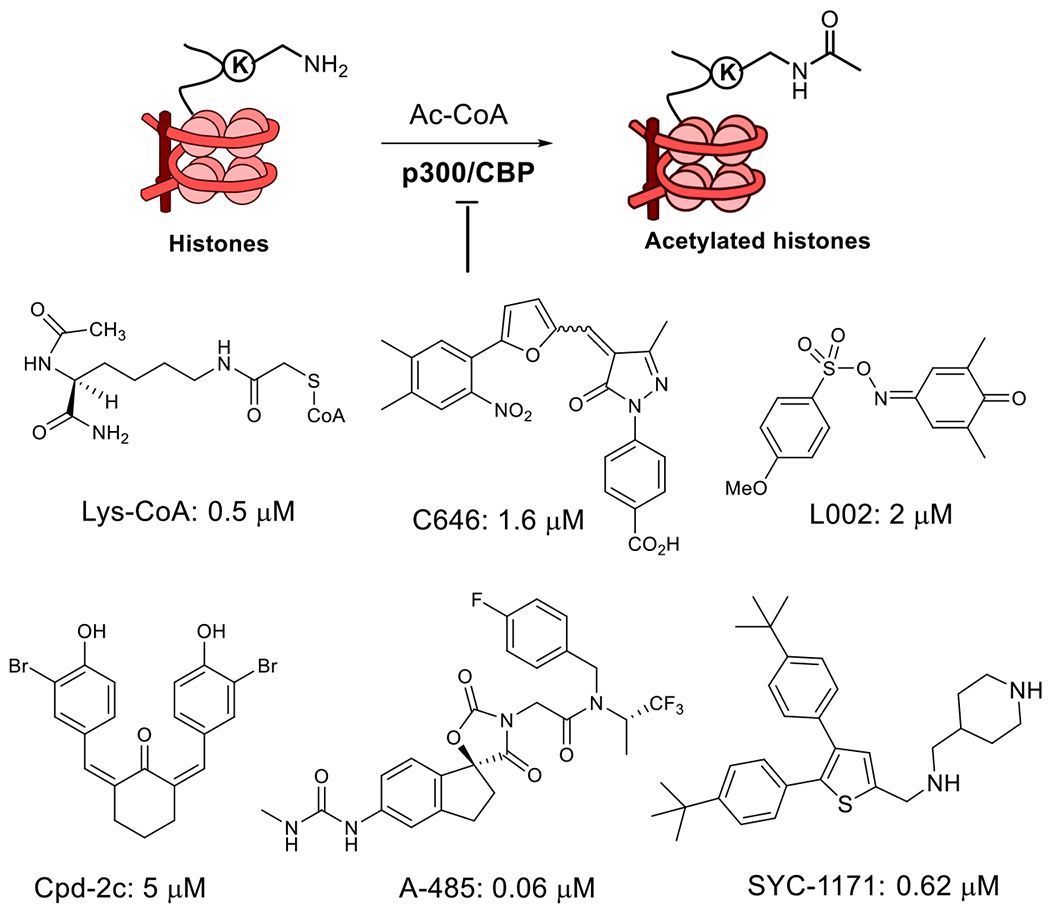
P300/CBP HAT catalyzed reaction and representative inhibitors.
RESULTS and DISCUSSION
Inhibitor discovery.
We have developed a biochemical assay to determine the activity and inhibition of recombinant HAT domain of human p300 14, using the substrate histone H3 (1-21) peptide and the 3H-labeled cofactor Ac-CoA. The p300 HAT catalyzed reaction transfers the 3H-acetyl group to the peptide substrate, which is then harvested, washed and subjected to scintillation counting. An inhibitor of p300 HAT can dose-dependently reduce the scintillation counts. We screened our proprietary compound library containing ~1,500 compounds, which were synthesized for structure-activity relationship (SAR) studies of lysine specific demethylase 1 (LSD1, an H3K4 demethylase) 24, Flavivirus protease 25, 26 and several other proteins with a peptide substrate 27. Given the substrate similarities between p300 HAT (e.g., H3K9 or H3K27) and these proteins, there could be a higher likelihood of finding an inhibitor. Several 1,4-pyrazine or related pyridine compounds 1-3 (Figure 2), originally synthesized targeting LSD1, were found to inhibit p300 HAT with IC50 values of 5.7-62.6 μM.
Figure 2.
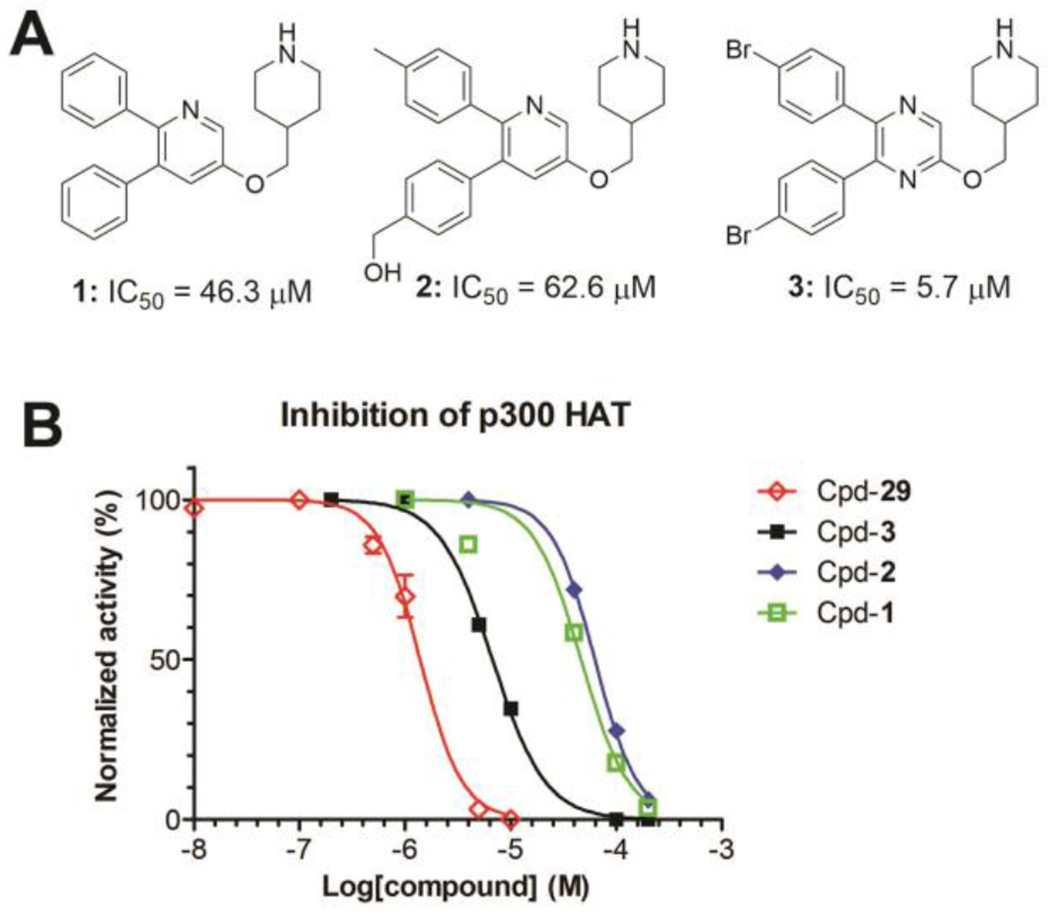
(A) Structures and (B) dose-responsive curves of representative inhibitors of p300 HAT.
While 5,6-diphenyl-3-(piperidin-4-ylmethoxy)pyridine (1) and compound 2 with additional hydroxymethyl and methyl groups at the 5- and 6-phenyl rings are weak inhibitors of p300 HAT (IC50 = 46.3 ± 6.1 and 62.6 ± 1.0 μM, respectively), 1,4-pyrazine compound 3 with para-Br substituents at the 5- and 6-phenyl rings exhibited a good inhibitory activity with an IC50 value of 5.7 ± 0.2 μM. In addition, compounds 1-3 had no or weak activity against LSD1 (IC50 = 52.4 μM for 3 and >100 μM for 1 and 2) 24, showing good selectivity.
Chemistry.
Based on the structure of compound 3, we performed medicinal chemistry studies to find compounds with improved potency. Schemes 1 and 2 show the general synthetic methods for compounds 4-32. A commercially available benzil compound 34 was reacted with a 2-aminoacetamide (35) in the presence of sodium hydroxide to give 2-hydroxy-5,6-diphenylpyrazine (36) (Scheme 1), which was subjected to a Mitsunobu reaction with a BOC (tert-butyloxycarbonyl) protected amino-alcohol to produce (upon deprotection) target compounds 3-12 and 14. Converting the 2-OH group of compound 36 to a -Cl by treatment with POCl3 yielded compound 37, which was reacted with a sodium salt of BOC protected 4-(hydroxymethyl)piperidine to (upon deprotection) give pyrazine containing compounds 16-18, 22 and 23.
Scheme 1.
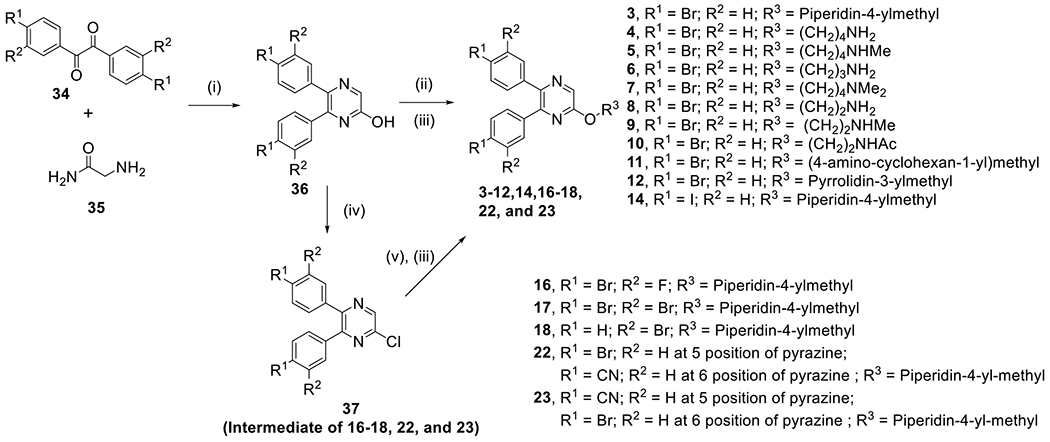
Synthesis of compounds 3-12, 14, 16-18, 22 and 23.a
aReagents and conditions: (i) NaOH, MeOH, reflux; (ii) an alcohol, PPh3, diisopropyl azodicarboxylate, THF, room temperature; (iii) 4 M HCl (in p-dioxane), CH2Cl2, 0 °C; (iv) POCl3, 80 °C; (v) N-Boc-piperidin-4-ylmethanol, NaH, DMF, 0 °C to room temperature.
Scheme 2.
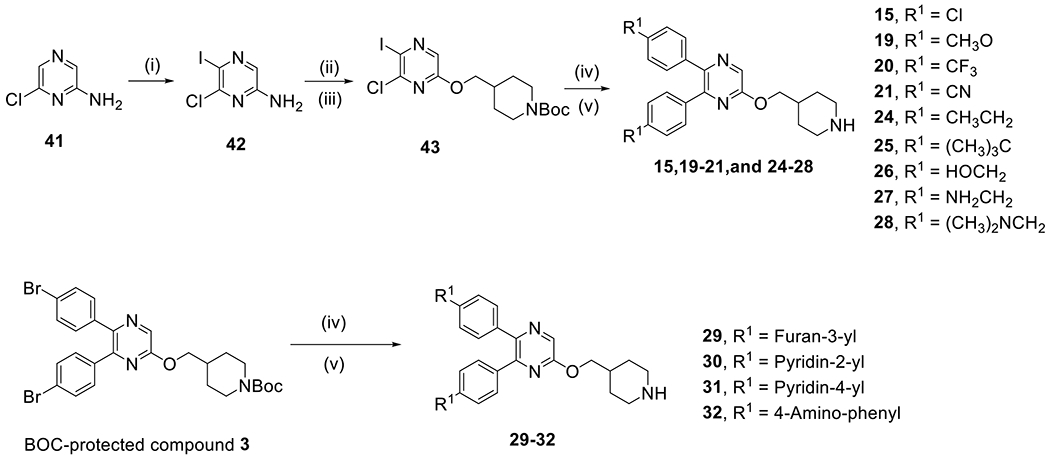
Synthesis of compounds 15, 19-21 and 24-32.a
aReagents and conditions: (i) N-Iodosuccinimide, DMSO, room temperature, 80%; (ii) NaNO2, H2SO4(Conc.); (iii) N-Boc-piperidin-4-ylmethanol, PPh3, diisopropyl azodicarboxylate, THF; (iv) Aryl-boronic acid or Aryl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane, Pd(PPh3)4, Na2CO3, p-dioxane-H2O, 110 °C; (v) 4 M HCl (in p-dioxane), CH2Cl2, 0 °C.
Scheme 2 shows the synthesis of compounds 15, 19-21 and 24-32. 6-chloro-2-aminopyrazine (41) was iodized with N-iodosuccinimide to give 6-chloro-5-iodo-2-aminopyrazine (42) and its amino group converted to a hydroxy by treatment with NaNO2 in H2SO4. A Mitsunobu reaction between the hydroxy group and a Boc-protected piperidine-containing alcohol gave compound 43. Palladium-catalyzed Suzuki reactions followed by deprotection of the BOC produced the target compounds 15, 19-21 and 24-28. Similar reactions starting from the BOC-protected compound 3 yielded compounds 29-32.
Structure-activity relationships.
Compounds 4-13 with a variety of the R2 substituent were synthesized in an effort to find a more favorable group at this position. Due to the relatively high costs as well as long procedure of the p300 HAT assay, inhibitory activities of these compounds were initially screened at 10 μM, together with compound 3 as a positive control. Equally or more active compounds were chosen for IC50 determination. The structures and inhibitory activity against p300 HAT of compounds 4-13 are shown in Table 1. Compound 4 with a linear, 4-carbon alkyl spacer between the -O- and amino group exhibited a 21.2% inhibition at 10 μM, significantly weaker than compound 3 (72.5% inhibition at 10 μM or IC50 of 5.7 μM) with a cyclic 4-carbon spacer. Compound 5 with a terminal N-methyl substituent showed a similarly weak activity (19.1% inhibition). Compound 6 with a linear, 3-carbon alkyl spacer is slightly more inhibitory (35.4% inhibition at 10 μM), while 7 with two terminal N-methyl substituents almost loses the inhibitory activity (4.8% inhibition). Analogous compounds 8 and 9 with a 2-carbon spacer are also weak inhibitors. In addition, compound 10 bearing a terminal N-acetyl group is inactive. Compound 11 with a cyclohexyl-containing 5-carbon spacer between the -O- and amino group showed a comparable inhibitory activity (59.1% inhibition at 10 μM) to compound 3, while compound 12 having a 5-membered pyrrolidine moiety is weaker (42.7% inhibition). Compound 13 with a -COOH R2 substituent is also a weak inhibitor (30% inhibition at 10 μM). These results indicate that the piperidin-4-ylmethoxy group is the most favored R2 substituent among these compounds.
Table 1.
Structures and inhibitory activities of compounds 3-13.
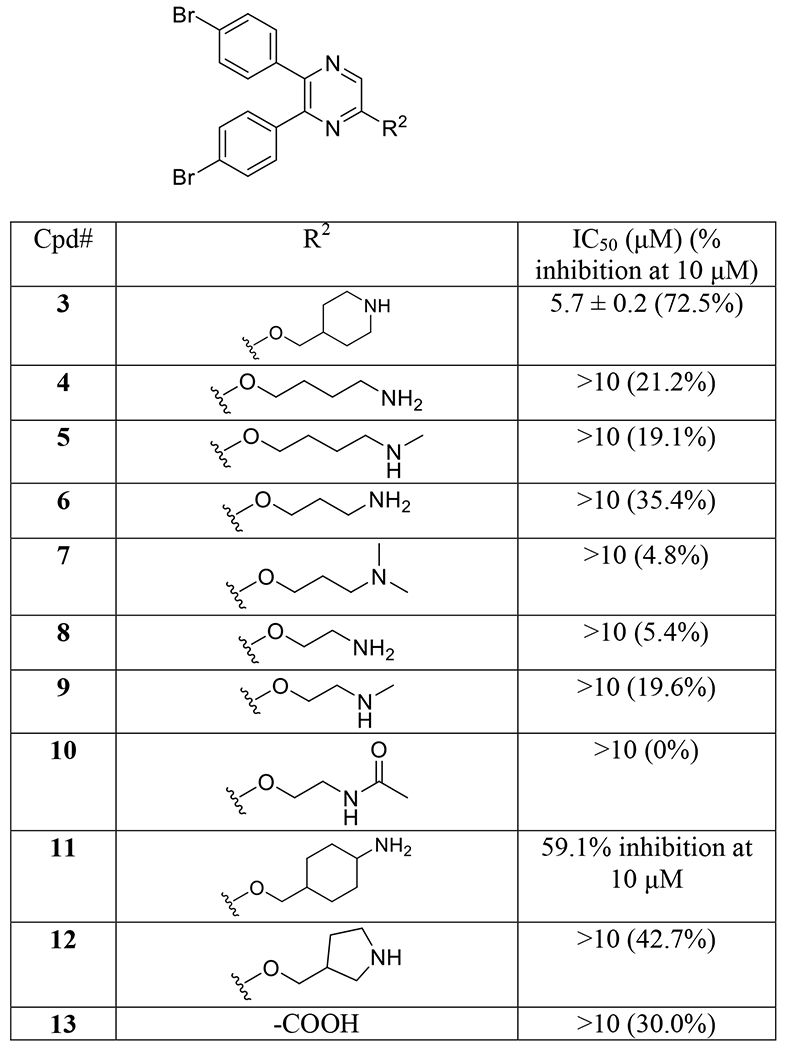
|
Compounds 14-32 (Table 2) were synthesized to optimize the R5 and R6 substituents in compound 3. Replacing the para-Br groups in 3 with -I in compound 14 slightly reduced the inhibitory activity against p300 HAT with an IC50 of 7.8 μM, while compound 15 with -Cl groups showed considerably decreased activity (IC50 > 10 μM). As compared to compound 3, compounds 16 and 17 bearing additional meta-F and -Br groups, respectively, in their R5 and R6 are also weaker inhibitors (IC50 ≥ 10 μM). Moving the para-Br groups in 3 to the meta-position in compound 18 (IC50 = 9.9 μM) is disfavored. Changing the para-Br groups to electron-releasing -OMe, or electron-withdrawing -CF3 and -CN groups with comparable sizes in compounds 19-21 resulted in more activity losses (38%−0% inhibition at 10 μM). Replacing only one -Br group with a -CN for compounds 22 and 23 also failed to improve the inhibitory activity. In addition, compound 24 (55% inhibition at 10 μM) or 25 (IC50 = 17.6 μM) with an ethyl or a tert-butyl group, respectively, at this position is less inhibitory than compound 3. While a polar hydroxymethyl group in compound 26 (48.2% inhibition at 10 μM) is also less active, compound 27 with an aminomethyl group at this position exhibited ~2-fold activity enhancement with an IC50 value of 2.8 μM. Masking the primary amino group with two methyl groups in compound 28 (46.3% inhibition at 10 μM) significantly reduces inhibitory activity against p300 HAT.
Table 2.
Structures and inhibitory activities of compounds 14-32.
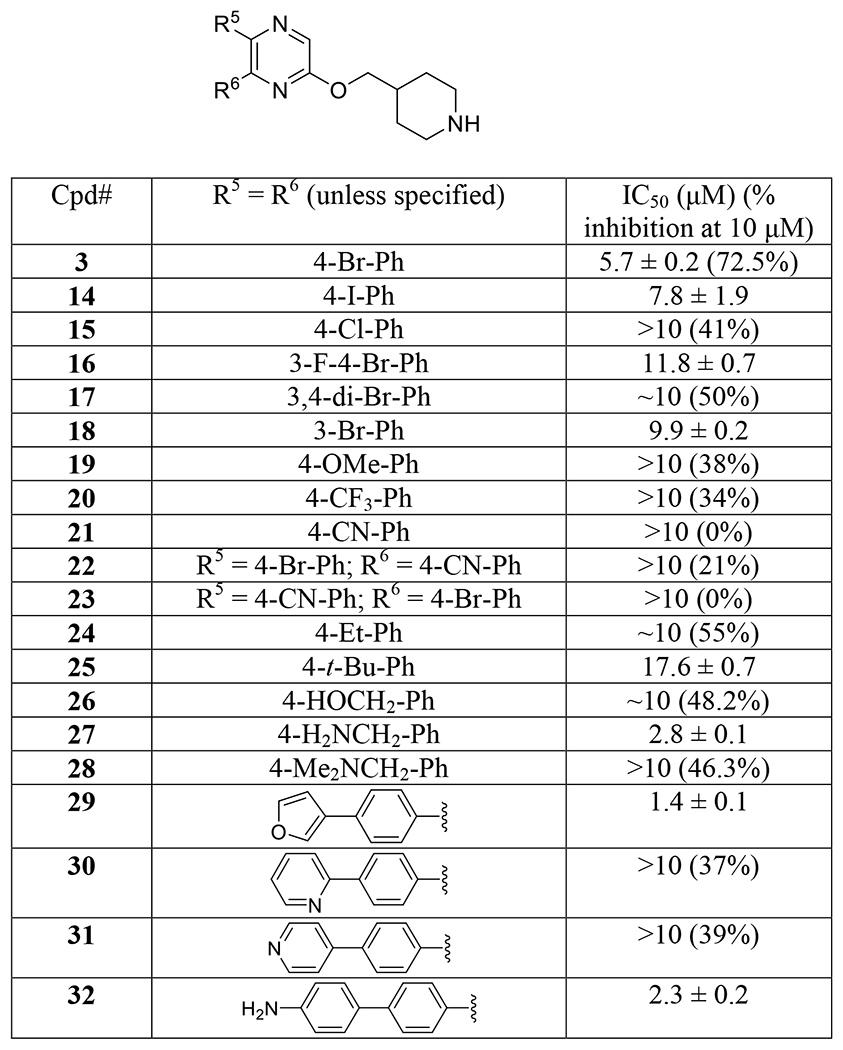
|
Compounds 29-32 (Table 2) contain bicyclic R5 and R6 substituents. Compound 29 having furan-3-ylphenyl groups is a strong inhibitor with an IC50 of 1.4 μM, exhibiting >4× more activity than the initial lead compound 3. Compounds 30 and 31 having pyridinylphenyl substituents are weak (37-39% inhibition at 10 μM). Compound 32 with para-aminobiphenyl groups represents another strong inhibitor of p300 HAT with an IC50 value of 2.3 μM.
Mode of inhibition against p300 HAT.
Next, steady-state enzyme kinetics studies were performed to investigate a possible mode of action for compound 29, the most potent inhibitor of p300 HAT in this series of compounds. Inhibitory activities of compound 29 were determined using increasing concentrations of the substrate histone peptide and the cofactor Ac-CoA. As shown in Figure 3A, the IC50 values of compound 29 against p300 HAT did not significantly increase with the increasing concentrations of Ac-CoA from 1 - 50 μM (0.14 - 7.1× Km 28) following the Cheng-Prusoff equation (IC50 = Ki + Ki×[Substrate]/Km), showing it is likely non-competitive against the enzyme cofactor Ac-CoA. In addition, the IC50 values of compound 29 linearly increase (R2 = 0.93) when the higher concentrations of the histone peptide (2 - 100 μM) were used in the assay (Figure 3B). These results suggest compound 29 is competitive against the substrate histone.
Figure 3.
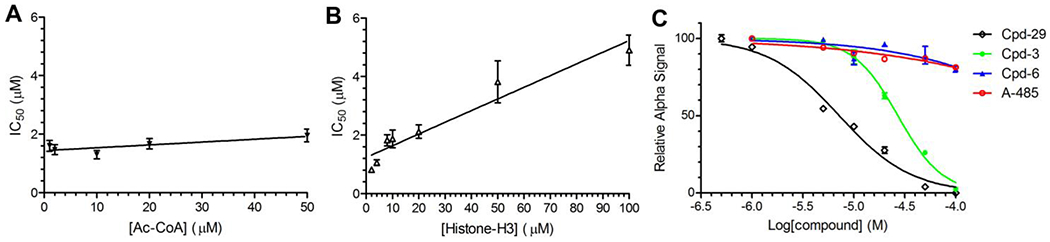
Mode of inhibition studies for inhibitors of p300 HAT. (A, B) Plots of IC50 values of compound 29 versus increasing concentrations of (A) Ac-CoA (1 - 50 μM, or 0.14 - 7.1× Km) and (B) histone H3 (1-100 μM) suggest the inhibitor is non-competitive against Ac-CoA and competitive against histone; (C) ALPHA assay results show inhibitors 29 and 3 can dose-dependently disrupt the binding between p300 HAT and histone H4, while such binding was not significantly inhibited by inactive compound 6 as well as A-485 (an Ac-CoA competitive inhibitor).
An ALPHA (amplified luminescent proximity homogeneous assay) assay15 was used to further probe the mode of inhibition for compound 29. The assay is based on the fact that upon excitation by a laser beam at 680 nm, donor beads generate singlet oxygen radicals, which can only travel for a very short distance (<200 nm) in the solution and cannot reach free acceptor beads. The association of histone H4(1-21) peptide with p300 HAT brings their coated donor and acceptor beads to a close proximity, which allows the radicals to activate the p300 HAT coated acceptor beads and produce luminescence at ~570 nm. Histone H4(1-21) was used 14, 15 because development of a similar assay with the histone H3 peptide had not been successful, presumably due to a weaker binding between p300 HAT and histone H3. As shown in Figure 3C, compound 29 inhibited the binding between the protein and histone H4 and dose-dependently reduced the ALPHA-signal with an IC50 of 7.2 μM. The weaker inhibitor compound 3 also inhibited the binding with an IC50 of 27.0 μM. However, compound 6 with a very weak activity did not significantly disrupt the binding at 100 μM. Moreover, although with a potent activity, Ac-CoA-competitive inhibitor A-485 did not affect the p300 HAT-histone H4 interaction, as it occupies the Ac-CoA binding pocket of the protein 15.
Taken together, the above enzyme kinetics and ALPHA results strongly support compound 29 is a competitive inhibitor of p300 HAT against the substrate histone, similar to our previously disclosed inhibitor SYC-1171 14. Both compounds exhibit a different mode of inhibition from compound A-485.
Selectivity among human histone acetyltransferases.
Three classes of histone or protein lysine acetyltransferases with distinct conserved motifs and structures have been found in humans, which include the p300/CBP, Gcn5-related N-acetyltransferase (GNAT) and MYST (MOZ, Ybf2, Sas2 and Tip60) family of HATs. To examine the enzyme selectivity, compound 29 was tested for its activities against a selected HAT from these three classes of HATs. CBP is a homolog of p300, with its HAT domain having 87% identity with that of p300. P300/CBP associating factor (PCAF) is a representative member of the GNAT family and MYST3 belongs to the MYST family of HATs. The GNAT and MYST HAT proteins have distinct sequences and 3-dimensional structures from those of p300/CBP HATs. Using similar HAT assays, compound 29 was found to exhibit comparable inhibitory activity against human CBP HAT with an IC50 of 2.2 μM (Table 3). However, compound 29 did not significantly inhibit the activity of PCAF and Myst3 even at 100 μM, showing a high selectivity for p300/CBP HAT. In addition, as with the initial lead compound 3, compound 29 did not significantly inhibit activity of LSD1 at 100 μM.
Table 3.
Inhibitory activities of compound 29 against selected HATs.
| IC50 (μM) | |
|---|---|
| P300-HAT | 1.4 ± 0.1 |
| CBP-HAT | 2.2 ± 0.1 |
| PCAF | >100 |
| Myst3 | >100 |
Inhibition of cellular histone acetylation.
Activity of selected compounds to inhibit acetylation of histone lysine residues in human cells was evaluated. Potent inhibitors 29, 27 and inactive compound 6 were included in the experiments. Kasumi-1 leukemia cells were incubated with these compounds at various concentrations for 12h. Histone proteins were extracted and subjected to electrophoresis separation, followed by Western blot to measure the levels of acetylation at histone H3 lysine 9, 18 and 27 residues (H3K9, H3K18 and H3K27), using their specific antibodies. As shown in Figure 4, the most potent compound 29 almost depleted acetylation of H3K9 and K27 at 5 and 10 μM, while it significantly inhibited that of H3K18 in a dose-dependent manner. Compound 27 can also dose-dependently inhibit acetylation of H3K9 and K27, but exhibited less activities as compared to compound 29. It seems that H3K18 acetylation was not significantly affected by compound 27 at 5 and 10 μM. In addition, H3K27 acetylation appears to be the most sensitive target for these inhibitors. Inactive compound 6 did not significantly reduce acetylation of these histone lysine residues, largely excluding possible off-target effects for these compounds.
Figure 4.
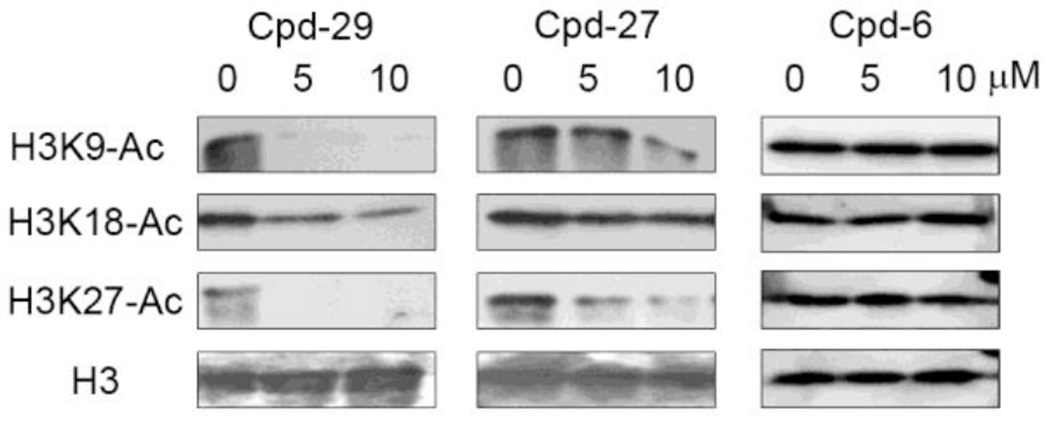
Inhibition of cellular H3K9, K18 and K27 acetylation by compounds 29, 27 and 6.
Antitumor activity.
Activity of the most potent compound 29 was tested against proliferation of a panel of tumor cell lines. Inactive compound 6 as well as known inhibitor A-485 was included in the assays. As shown in Table 4, compound 29 was found to exhibit strong activity with EC50 values of 5.3 and 6.2 μM against proliferation of MCF-7 (breast) and LNCaP (prostate) cancer cells, which are dependent on sex hormone estrogen (for MCF-7) or androgen (for LNCaP). These results are consistent with previous studies showing p300 HAT is critical to regulate ER- or AR-mediated signaling pathways and its inhibition suppresses growth of ER- or AR-positive breast or prostate cancer 14, 15. However, sex hormone-independent breast cancer MDA-MB231 and prostate cancer PC-3 cells were still sensitive to compound 29 with comparable EC50s of 8.5 and 4.4 μM, respectively. Moreover, compound 29 showed more potent antiproliferative activities against pancreatic cancer PANC-1 and MDA-PANC-28 cells with EC50s of 1.2 and 4.3 μM, which is in agreement with p300 HAT’s importance in pancreatic cancer 29. In addition to solid tumors, compound 29 had strong activity against growth of acute myeloid leukemia Molm-13 and MV4;11 and multiple myeloma RPMI-8226 cells (EC50: 3.6-8.7 μM). Inactive compound 6 was found to have no or negligible activities (EC50 >20 μM) against proliferation of these tumor cells, largely excluding possible off-target effects. With more potent biochemical activity (IC50 ~60 nM 15), A-485 exhibited more pronounced activities against proliferation of blood cancer (Molm-13, MV4;11 and RPMI-8226) and prostate cancer (LNCaP and PC-3) cells with EC50 values of 0.16-2.1 μM 15, while A-485 did not significantly inhibit proliferation of breast cancer (MCF-7 and MDA-MB231) and pancreatic cancer (PANC-1 and −28) cells (Table 4) 14, 15. These activity variations between compound 29 and A-485 might be due to different cell types or modes of inhibition (i.e., competitive against histone vs. Ac-CoA).
Table 4.
Antiproliferative activity EC50 (μM) of compounds 29 and 6.
Taken together, these results suggest p300/CBP HAT could be important to a wide range of solid and blood cancers. Pharmacological inhibition of p300/CBP HAT (e.g., by compound 29) represents a potentially useful approach to cancer therapy.
CONCLUSION
Histone acetyltransferase p300 and its paralog CBP acetylate histone lysine sidechains and play critical roles in regulating gene expression in normal physiology and in certain diseases, such as cancer. The HAT domain of p300/CBP has been recently validated to be a drug target for cancer 14, 15. Our compound screening identified 1,4-pyrazine and related pyridine compounds 1-3 to be a novel chemotype of inhibitors of p300 HAT. Structure-activity relationship studies yielded a more potent compound 29 with an IC50 of 1.4 μM. Enzyme kinetics and ALPHA assays strongly support compound 29 is a competitive inhibitor against the substrate histone. In addition, compound 29 exhibited a high selectivity for the HAT domain of p300 and CBP with negligible activity on two other classes of human HATs. Compound 29 inhibited acetylation of several lysine residues of histone in cells, with H3K27 and H3K9 being the most sensitive. It also exhibited strong activities against proliferation of a panel of solid and blood cancer cells with EC50s of 1.2-8.7 μM. These results demonstrate that compound 29 is a novel pharmacological lead for drug development targeting these cancers as well as a useful chemical probe for biological studies of p300/CBP.
Experimental Section
All chemicals for synthesis were purchased from Alfa Aesar (Ward Hill, MA) or Aldrich (Milwaukee, WI). Unless otherwise stated, all solvents and reagents used as received. All reactions were performed using a Teflon-coated magnetic stir bar at the indicated temperature and were conducted under an inert atmosphere when stated. The identity of the synthesized compounds was characterized by 1H and 13C NMR on a Varian (Palo Alto, CA) 400-MR spectrometer and mass spectrometer (Shimadzu LCMS-2020). Chemical shifts were reported in parts per million (ppm, δ) downfield from tetramethylsilane. Proton coupling patterns are described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad (br). The identity of the potent inhibitors was confirmed with high resolution mass spectra (HRMS) using an Agilent 6550 iFunnel quadrupole-time-of-flight (Q-TOF) mass spectrometer with electrospray ionization (ESI). The purities of the final compounds were determined to be >95% with a Shimadzu Prominence HPLC using a Zorbax C18 (or C8) column (4.6 × 250 mm) monitored by UV at 254 nm.
Chemical synthesis.
Synthesis of compounds 1, 2, 3, 13, 22, 23 and 26 was reported in our previous publications24, 26.
General synthetic methods for compounds 16-18:
NaOH solution (12.5 M, 3.2 mL, 40 mmol) was added over 30 min to a refluxing mixture of a commercially available compound 34 (20 mmol) and glycine amide hydrogen chloride (35, 2.21 g, 20 mmol) in methanol (50 mL). After refluxing for another 30 min, the reaction mixture was cooled down and treated with HCl (12 N, 2.5 mL), followed by KHCO3 (2 g), to give a yellow solid, which was collected by filtration, washed with water, and recrystallized in tert-BuOH to yield compound 36 as yellow needles (4.69 g, 58%). 1H NMR (400 MHz, CDCl3) δ 8.12 (s, 1H), 7.54 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 8.4 Hz, 2H), 7.26 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H).
The reaction mixture of compound 36 (1.22 g, 2.76 mmol) and phosphoryl trichloride (2 mL) was heated to 100°C for 2 h. It was added dropwise into water (100 mL) at 0°C and the pH adjusted to 8 with NaOH (5 M). The product was extracted with ethyl acetate (50 mL), dried over sodium sulfate and concentrated to give compound 37 (507 mg) which was used for the next step without further purification. A solution of compound 37 (507 mg, 1.1 mmol), potassium hydroxide (123 mg, 2.2 mmol) and tert-butyl 4-(hydroxymethyl)piperidine-1-carboxylate (237 mg, 1.1 mmol) in THF (4 mL) was stirred at 40°C for overnight. The solution was neutralized with HCl (1 M) and extracted with CH2Cl2 (15 mL × 3). The organic phases were concentrated and purified with column chromatography (silica gel, hexane/ethyl acetate 5:1-2:1) to afford BOC-protected a target compound as a pale yellow powder (575 mg, 97%), which in 1,4-dioxane (9 mL) was treated with HCl (4 M in dioxane, 9 mL) at 0°C and monitored with TLC. After reaction completion, the solvents were evaporated to dryness and the white solid washed successively with dioxane and CH2Cl2 to give the target compound in almost quantitative yield.
2,3-bis(4-bromo-3-fluorophenyl)-5-(piperidin-4-ylmethoxy)pyrazine hydrochloride (16).
1H NMR (400 MHz, DMSO-d6) δ 8.43 (s, 1H), 7.69 (dt, J = 12.4, 7.6 Hz, 2H), 7.51 (dd, J = 9.6, 1.6 Hz, 1H), 7.39 (dd, J = 9.6, 1.7 Hz, 1H), 7.12 (d, J = 8.4 Hz, 1H), 7.08 – 6.99 (m, 1H), 4.31 (d, J = 6.4 Hz, 2H), 3.24 (dd, J = 13.6, 8.4 Hz, 2H), 2.90 (t, J = 12.0 Hz, 2H), 2.14 (s, 1H), 1.92 (d, J = 13.2 Hz, 2H), 1.66 – 1.45 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 159.3, 158.0, 156.8, 145.8, 142.0, 139.7, 139.6, 139.4, 139.3, 133.5, 133.4, 127.3, 127.1, 117.9, 117.7, 117.6, 117.4, 109.1, 108.9, 108.1, 107.9, 69.8, 42.9, 32.9, 25.2. MS (ESI) [M+H]+ 538.0
2,3-bis(3,4-dibromophenyl)-5-(piperidin-4-ylmethoxy)pyrazine hydrochloride (17).
1H NMR (400 MHz, DMSO-d6) δ 8.43 (s, 1H), 7.87 (s, 1H), 7.80 (s, 1H), 7.74 (dd, J = 16.8, 8.0 Hz, 2H), 7.26 (d, J = 8.0 Hz, 1H), 7.16 (d, J = 8.0 Hz, 1H), 4.31 (d, J = 5.6 Hz, 2H), 3.15 – 3.08 (m, 2H), 2.96 – 2.86 (m, 2H), 2.14 (s, 1H), 1.93 (d, J = 12.8 Hz, 2H), 1.51 (d, J = 11.6 Hz, 2H). MS (ESI) [M+H]+ 657.8
2,3-bis(3-bromophenyl)-5-(piperidin-4-ylmethoxy)pyrazine hydrochloride (18).
1H NMR (400 MHz, DMSO-d6) δ 8.41 (s, 1H), 8.03 (s, 1H), 7.93 (d, J = 7.6 Hz, 1H), 7.82 (d, J = 7.6 Hz, 1H), 7.65 – 7.39 (m, 3H), 7.36 – 7.19 (m, 2H), 4.29 (s, 2H), 3.25 (d, J = 4.0 Hz, 2H), 2.80 (s, 2H), 2.14 (s, 1H), 1.92 (s, 2H), 1.58 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 166.0, 157.9, 146.5, 142.7, 140.3, 140.0, 135.5, 134.0, 133.2, 132.1, 132.0, 131.7, 131.6, 131.5, 130.9, 130.5, 130.4, 130.2, 128.7, 128.5, 128.3, 126.6, 121.7, 121.5, 121.4, 69.7, 42.9, 32.92, 25.2. MS (ESI) [M+H]+ 502.0
General synthetic methods for compounds 4-12, and 14:
A reaction mixture of 2-hydroxypyrazine 36 (1.12 g, 2.76 mmol), N-Boc-4-piperidinemethanol (594 mg, 2.76 mmol), triphenylphosphine (1.16 g, 4.42 mmol) and diisopropyl azodicarboxylate (894 mg, 4.42 mmol) in anhydrous THF (27 mL) was stirred at room temperature overnight. It was concentrated and purified with column chromatography (silica gel, hexane/ethyl acetate 5:1-2:1) to give BOC-protected target compound in ~90% yield. Deprotection was performed as described above to give the target compound as a white or pale yellow powder in ~100% yield.
4-((5,6-bis(4-bromophenyl)pyrazin-2-yl)oxy)butan-1-amine hydrochloride (4).
1H NMR (400 MHz, DMSO-d6) δ 8.37 (s, 1H), 8.05 (s, 3H), 7.55 – 7.34 (m, 8H), 4.41 (s, 2H), 2.84 (s, 2H), 1.83 – 1.76 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 157.7, 146.6, 142.8, 137.3, 137.0, 132.9, 131.6, 131.4, 131.3, 131.2, 122.4, 121.5, 65.6, 38.4, 25.3, 23.7. MS (ESI) [M+H]+ 476.0
4-((5,6-bis(4-bromophenyl)pyrazin-2-yl)oxy)-N-methylbutan-1-amine hydrochloride (5).
1H NMR (400 MHz, DMSO-d6) δ 8.98 (s, 2H), 8.38 (s, 1H), 7.56 (dd, J = 15.2, 8.4 Hz, 4H), 7.36 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 4.41 (d, J = 5.6 Hz, 2H), 2.92 (s, 2H), 2.50 (s, 3H), 1.96 – 1.71 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 157.8, 146.7, 142.8, 137.4, 137.1, 132.98, 131.7, 131.5, 131.4, 131.3, 122.4, 121.6, 65.7, 32.2, 29.3, 25.4, 22.2. MS (ESI) [M+H]+ 490.0
3-((5,6-bis(4-bromophenyl)pyrazin-2-yl)oxy)propan-1-amine hydrochloride (6).
1H NMR (400 MHz, DMSO-d6) δ 8.40 (s, 1H), 8.08 (s, 3H), 7.56 (dd, J = 14.4, 8.4 Hz, 4H), 7.37 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 4.48 (t, J = 6.0 Hz, 2H), 2.97 (t, J = 6.4 Hz, 2H), 2.17 – 2.06 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 157.6, 146.6, 142.9, 137.3, 137.0, 133.1, 131.7, 131.5, 131.4, 131.3, 122.5, 121.6, 63.6, 36.2, 26.5. MS (ESI) [M+H]+ 462.0.
3-((5,6-bis(4-bromophenyl)pyrazin-2-yl)oxy)-N,N-dimethylpropan-1-amine hydrochloride (7).
1H NMR (400 MHz, DMSO-d6) δ 10.75 (s, 2H), 8.38 (s, 1H), 7.61 – 7.49 (m, 4H), 7.38 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 4.48 (t, J = 6.0 Hz, 2H), 3.25 – 3.12 (m, 2H), 2.76 (s, 6H), 2.30 – 2.14 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 157.6, 146.6, 143.0, 137.3, 137.0, 133.0, 131.7, 131.5, 131.4, 131.3, 122.5, 121.6, 63.7, 53.8, 42.0, 23.5. MS (ESI) [M+H]+ 490.0.
2-((5,6-bis(4-bromophenyl)pyrazin-2-yl)oxy)ethan-1-amine hydrochloride (8).
1H NMR (400 MHz, DMSO-d6) δ 8.04 (s, 1H), 7.52 (d, J = 8.0 Hz, 2H), 7.45 (d, J = 8.0 Hz, 2H), 7.38 (s, 1H), 7.29 (d, J = 8.0 Hz, 2H), 7.19 (d, J = 8.0 Hz, 2H), 4.76 (s, 1H), 3.59 (s, 2H), 3.43 (d, J = 5.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 153.1, 147.1, 138.5, 138.4, 136.9, 131.5, 131.4, 131.2, 131.1, 130.9, 121.7, 120.3, 59.6, 43.0. MS (ESI) [M+H]+ 448.0.
2-((5,6-bis(4-bromophenyl)pyrazin-2-yl)oxy)-N-methylethan-1-amine hydrochloride (9).
1H NMR (400 MHz, DMSO-d6) δ 9.23 (s, 2H), 8.42 (s, 1H), 7.57 (dd, J = 13.2, 8.4 Hz, 4H), 7.38 (d, J = 8.4 Hz, 2H), 7.29 (d, J = 8.4 Hz, 2H), 4.69 (d, J = 4.4 Hz, 2H), 3.38 (d, J = 4.0 Hz, 2H), 2.61 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 157.0, 146.5, 143.4, 137.3, 136.8, 133.3, 131.7, 131.5, 131.4, 131.3, 122.6, 121.7, 61.8, 46.9, 32.6. MS (ESI) [M+H]+ 462.0.
N-(2-((5,6-bis(4-bromophenyl)pyrazin-2-yl)oxy)ethyl)acetamide hydrochloride (10).
1H NMR (400 MHz, DMSO-d6) δ 8.03 (s, 1H), 7.53 (d, J = 8.4 Hz, 3H), 7.46 (d, J = 8.4 Hz, 2H), 7.30 (d, J = 8.4 Hz, 2H), 7.21 (d, J = 8.4 Hz, 2H), 4.19 (t, J = 5.6 Hz, 2H), 3.60 (q, J = 5.6 Hz, 2H), 1.99 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.4, 152.8, 147.1, 138.3, 138.3, 137.4, 131.5, 131.3, 131.2, 131.1, 131.0, 121.8, 120.4, 62.5, 39.2, 20.7. MS (ESI) [M+H]+ 490.0.
4-(((5,6-bis(4-bromophenyl)pyrazin-2-yl)oxy)methyl)cyclohexan-1-amine hydrochloride (11).
1H NMR (400 MHz, DMSO-d6) δ 8.39 (s, 1H), 8.14 (s, 3H), 7.55 (dd, J = 16.8, 8.4 Hz, 4H), 7.36 (d, J = 8.4 Hz, 2H), 7.27 (d, J = 8.4 Hz, 2H), 4.31 (d, J = 7.2 Hz, 2H), 3.22 (s, 1H), 2.02 (s, 1H), 1.66 – 1.62 (m, 8H). 13C NMR (100 MHz, DMSO-d6) δ 158.0, 146.6, 142.8, 137.4, 137.1, 133.1, 131.7, 131.5, 131.4, 131.2, 122.4, 121.5, 68.7, 47.4, 33.2, 26.2, 23.2. MS (ESI) [M+H]+ 516.0.
2,3-bis(4-bromophenyl)-5-(pyrrolidin-3-ylmethoxy)pyrazine hydrochloride (12).
1H NMR (400 MHz, DMSO-d6) δ 9.47 (s, 2H), 8.41 (s, 1H), 7.55 (dd, J = 14.4, 8.4 Hz, 4H), 7.36 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 4.50 – 4.34 (m, 2H), 3.37 – 3.32 (m, 1H), 3.30 – 3.21 (m, 1H), 3.20 – 3.11 (m, 1H), 3.05 (dd, J = 11.1, 7.2 Hz, 1H), 2.82 (dt, J = 13.6, 6.8 Hz, 1H), 2.12 (td, J = 12.8, 7.2 Hz, 1H), 1.79 (td, J = 15.2, 7.6 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 157.6, 146.6, 143.1, 137.3, 137.0, 133.1, 131.7, 131.5, 131.4, 131.3, 122.5, 121.6, 67.0, 46.8, 44.2, 36.7, 26.8. MS (ESI) [M+H]+ 488.0.
2,3-bis(4-Iodophenyl)-5-(piperidin-4-ylmethoxy)pyrazine hydrochloride (14) was prepared from 4,4′-Iodobenzil, following the same procedure as compound 3, as a hydrochloric acid salt (pale yellow powder). 1H NMR (400 MHz, DMSO-d6) δ 9.07 (s, 1H), 8.74 (s, 1H), 8.34 (s, 1H), 7.69 (s, 3H), 7.44 – 6.91 (m, 4H), 4.26 (s, 2H), 3.18 – 3.04 (m, 2H), 2.87 (s, 2H), 2.12 (s, 1H), 1.89 (s, 2H), 1.52 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 156.8, 154.7, 152.5, 145.2, 137.9, 131.1, 129.7, 127.1, 126.8, 124.0, 116.7, 107.8, 68.6, 54.2, 30.3, 24.3. MS (ESI) [M+H]+ 598.0.
General synthetic methods for compounds 15, 19-21 and 24-32:
A reaction mixture of 2-amino-6-chloropyrazine (41, 8 g, 62 mmol) and N-iodosuccinimide (15.3 g, 68 mmol) in DMSO (50 mL) was stirred at room temperature for 72 h before quenching with sodium thiosulfate aqueous solution (50 mL). The product was extracted with ethyl acetate (3 × 100 mL) and the combined organic layers were washed with water and brine and dried over Na2SO4. Upon removal of the solvents, the residue oil was purified with column chromatography (silica gel, hexanes:ethyl acetate from 5:1 to 2:1) to afford compound 42 (12.7 g, 80%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.72 (s, 1H), 4.73 (br, 2H).
To a suspension of 42 (3.78 g, 14.9 mmol) in sulfuric acid (18 mL) at 0 °C was added sodium nitrite (1.09 g, 15.8 mmol) in 3 portions. The resulting reaction mixture was stirred at 0 °C for 1 h and then poured into a beaker with excessive ice. The resulting precipitate was collected by filtration, washed with water and dried under vacuum to afford 6-chloro-5-iodopyrazin-2-ol (3.6 g) as a pale yellow solid, which is used directly for the next step. 1H NMR (400 MHz, DMSO-d6) δ 7.98 (s, 1H). To a solution of the crude product, N-Boc-4-piperidinemethanol (3.1 g, 14.5 mmol) and triphenylphosphine (5.9 g, 22.5 mmol) in THF (40 mL) was added diisopropyl azodicarboxylate (4.55 g, 22.5 mmol) at 0 °C. The reaction mixture was warmed to room temperature and stirred for 12 h. Upon removal of the solvents, the residue oil was purified with column chromatography (silica gel, hexanes: ethyl acetate from 10:1 to 5:1) to afford compound 43 (5.2 g, 77% for 2 steps) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 7.96 (s, 1H), 4.16 (d, J = 6.4 Hz, 4H), 2.73 (t, J = 12.0 Hz, 2H), 1.96 (s, 1H), 1.77 (d, J = 12.8 Hz, 2H), 1.46 (s, 9H), 1.33 – 1.19 (m, 2H).
A mixture of compound 43 (1.94 mmol), an arylboronic acid or aryl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2.51 mmol), tetrakis(triphenylphosphine)palladium (110 mg, 0.095 mmol), and sodium carbonate (610 mg, 5.75 mmol) in p-dioxane/H2O (15/3 mL) was heated to 110 °C for 24 h in a sealed pressure tube. The reaction was cooled and quenched with brine (20 mL). The product was extracted with ethyl acetate (3 × 20 mL) and the combined organic layers were washed with water and brine and dried over Na2SO4. Upon removal of the solvents, the residue oil was purified with column chromatography (silica gel, hexanes: ethyl acetate from 5:1 to 1:2) to give, after deprotection of the BOC, target products 15, 19-21 and 24-28 as an off-white or pale yellow solid in 61-84% yield.
BOC-protected compound 3, which was synthesized and reported in our previous publications24, 26, was coupled with an arylboronic acid (2.51 mmol) in the presence of tetrakis(triphenylphosphine)palladium (110 mg, 0.095 mmol) and sodium carbonate (610 mg, 5.75 mmol) in p-dioxane/H2O (15/3 mL) under 110 °C for 24 h in a sealed tube. The reaction was cooled and quenched with brine (20 mL). The product was extracted with ethyl acetate (3 × 20 mL) and the combined organic layers were washed with water and brine and dried over Na2SO4. Upon removal of the solvents, the residue oil was purified with column chromatography (silica gel, hexanes: ethyl acetate from 5:1 to 1:2) to give, after deprotection of the BOC, target products 29-32 as an off-white or pale yellow solid in 68-79% yield.
2,3-bis(4-chlorophenyl)-5-(piperidin-4-ylmethoxy)pyrazine hydrochloride (15).
1H NMR (400 MHz, DMSO-d6) δ 8.92 (s, 3H), 8.37 (s, 1H), 7.46 – 7.37 (m, 6H), 7.33 (d, J = 8.4 Hz, 2H), 4.28 (d, J = 6.0 Hz, 2H), 3.28 (d, J = 11.6 Hz, 2H), 2.89 (t, J = 11.6 Hz, 2H), 2.14 (s, 1H), 1.92 (d, J = 12.8 Hz, 2H), 1.62 – 1.47 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 157.8, 146.6, 143.0, 137.0, 136.6, 133.7, 132.9, 131.4, 131.2, 128.4, 128.3, 69.6, 42.6, 32.9, 25.1. MS (ESI) [M+H]+ 414.1.
2,3-bis(4-methoxyphenyl)-5-(piperidin-4-ylmethoxy)pyrazine hydrochloride (19).
1H NMR (400 MHz, DMSO-d6) δ 9.23 (s, 1H), 8.91 (s, 1H), 8.25 (s, 1H), 7.35 (d, J = 8.4 Hz, 2H), 7.26 (d, J = 8.4 Hz, 2H), 6.88 (t, J = 7.6 Hz, 4H), 4.27 (d, J = 5.6 Hz, 2H), 3.75 (d, J = 3.6 Hz, 6H), 3.28 (d, J = 11.2 Hz, 2H), 2.89 (s, 2H), 2.13 (s, 1H), 1.92 (d, J = 12.8 Hz, 2H), 1.55 (d, J = 11.6 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 159.5, 158.8, 157.2, 146.9, 143.6, 131.4, 130.9, 130.8, 130.5, 130.4, 113.6, 113.6, 69.3, 55.2, 55.1, 42.6, 32.9, 25.2, 25.1. MS (ESI) [M+H]+ 406.2.
5-(piperidin-4-ylmethoxy)-2,3-bis(4-(trifluoromethyl)phenyl)pyrazine hydrochloride (20).
1H NMR (400 MHz, DMSO-d6) δ 8.78 (s, 1H), 8.56 (s, 1H), 8.48 (s, 1H), 7.74 (dd, J = 15.2, 8.0 Hz, 4H), 7.64 (d, J = 8.0 Hz, 2H), 7.56 (d, J = 8.0 Hz, 2H), 4.32 (d, J = 6.0 Hz, 2H), 3.30 – 3.25 (m, 2H), 2.91 (t, J = 11.6 Hz, 2H), 2.16 (s, 1H), 1.94 (d, J = 13.2 Hz, 2H), 1.52 (dd, J = 23.6, 11.6 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 158.1, 146.8, 142.9, 142.0, 141.8, 133.6, 130.5, 130.3, 125.3, 125.2, 69.8, 42.7, 32.8, 25.2. MS (ESI) [M+H]+ 482.2.
4,4’-(5-(piperidin-4-ylmethoxy)pyrazine-2,3-diyl)dibenzonitrile hydrochloride (21).
1H NMR NMR (400 MHz, DMSO-d6) δ 9.00 (bs, 1H), 8.67 (bs, 1H), 8.48 (s, 1H), 7.85 (d, J = 8.0 Hz, 2H), 7.82 (d, J = 8.0 Hz, 2H), 7.59 (d, J = 8.0 Hz, 2H), 7.51 (d, J = 8.0 Hz, 2H), 4.32 (d, J = 6.4 Hz, 2H), 3.34 – 3.25 (m, 2H), 2.90 (q, J = 11.6 Hz, 2H), 2.20 – 2.09 (m, 1H), 1.93 (d, J = 12.0 Hz, 2H), 1.50 (q, J = 11.6 Hz, 2H). MS (ESI) [M+H]+ 396.2.
2,3-bis(4-ethylphenyl)-5-(piperidin-4-ylmethoxy)pyrazine hydrochloride (24).
1H NMR (400 MHz, DMSO-d6) δ 9.21 (s, 1H), 8.88 (s, 1H), 8.22 (s, 1H), 7.40 – 6.72 (m, 7H), 4.19 (s, 2H), 3.15 – 2.95 (m, 2H), 2.80 (s, 2H), 2.50 (s, 4H), 2.06 (s, 1H), 1.84 (s, 2H), 1.49 (s, 2H), 1.08 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 156.5, 146.5, 143.3, 143.1, 142.4, 135.0, 134.6, 131.1, 128.6, 128.4, 126.7, 68.6, 42.0, 32.1, 27.1, 24.4, 14.6. MS (ESI) [M+H]+ 402.2.
2,3-bis(4-(tert-butyl)phenyl)-5-(piperidin-4-ylmethoxy)pyrazine hydrochloride (25).
1H NMR (400 MHz, DMSO-d6) δ 9.00 – 8.27 (br, 4H), 8.30 (s, 1H), 7.38 – 7.26 (m, 8H), 4.27 (d, J = 6.0 Hz, 2H), 3.33 – 3.24 (m, 2H), 2.90 (t, J = 11.6 Hz, 2H), 2.13 (br, 1H), 1.93 (d, J = 13.2 Hz, 2H), 1.58 – 1.48 (m, 2H), and 1.26 (s, 18H). 13C NMR (100 MHz, DMSO-d6) 157.4, 151.2, 150.3, 147.4, 144.0, 135.7, 135.4, 131.9, 129.2, 129.0, 125.0, 124.9, 69.4, 42.7, 34.4, 34.3, 33.0, 31.1, 31.0, 25.2; HRMS (ESI+) calcd for C30H40N3O [M+H]+ 458.3166, found 458.3179.
((5-(Piperidin-4-ylmethoxy)pyrazine-2,3-diyl)bis(4,1-phenylene))dimethanamine Hydrochloride (27).
1H NMR (400 MHz, D2O) δ 8.02 (s, 1H), 7.20 (d, J = 7.6 Hz, 2H), 7.11 (s, 6H), 4.10 (s, 2H), 3.89 (s, 4H), 3.22 (d, J = 10.0 Hz, 2H), 2.79 (t, J = 10.4 Hz, 2H), 1.99 (s, 1H), 1.84 (d, J = 11.6 Hz, 2H), 1.41 – 1.32 (m, 2H); MS (ESI) [M+H]+ 404.2.
1,1’-((5-(Piperidin-4-ylmethoxy)pyrazine-2,3-diyl)bis(4,1-phenylene))bis(N,N-dimethylmethanamine) Hydrochloride (28).
1H NMR (400 MHz, D2O) δ 8.20 (s, 1H), 7.38 (d, J = 7.2 Hz, 2H), 7.30 (s, 6H), 4.26 (d, J = 4.0 Hz, 2H), 4.18 (s, 4H), 3.37 (d, J = 11.6 Hz, 2H), 2.94 (t, J = 12.4 Hz, 2H), 2.71 (s, 12H), 2.15 (s, 1H), 2.00 (d, J = 12.8 Hz, 2H), 1.51 (dd, J = 26.4, 12.8 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 157.9, 147.4, 143.7, 139.2, 138.8, 132.9, 131.0, 130.8, 130.8, 130.2, 129.9, 129.8, 69.6, 58.9, 58.8, 42.6, 41.5, 41.4, 32.9, 25.1; MS (ESI) [M+H]+ 406.3.
2,3-bis(4-(Furan-3-yl)phenyl)-5-(piperidin-4-ylmethoxy)pyrazine hydrochloride (29) was prepared following the same procedure as a hydrochloric acid salt. 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 8.57 (s, 1H), 8.33 (s, 1H), 8.19 (d, J = 6.0 Hz, 2H), 7.75 – 7.67 (m, 2H), 7.61 – 7.49 (m, 4H), 7.44 (d, J = 8.0 Hz, 2H), 7.36 (d, J = 8.0 Hz, 2H), 6.95 (s, 2H), 4.30 (d, J = 6.0 Hz, 2H), 3.30 (d, J = 11.6 Hz, 2H), 2.91 (t, J = 12.4 Hz, 2H), 2.14 (s, 1H), 1.94 (d, J = 13.6 Hz, 2H), 1.51 (dd, J = 24.4, 12.0 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) 158.0, 147.7, 144.9, 144.8, 144.2, 140.3, 140.1, 137.3, 136.9, 132.6, 131.8, 130.5, 130.2, 125.7, 109.0, 69.9, 43.0, 33.4, 25.6. HRMS (ESI+) calcd for C30H28N3O3 [M + H]+, 478.2131; found, 478.2125.
5-(Piperidin-4-ylmethoxy)-2,3-bis(4-(pyridin-2-yl)phenyl)pyrazine hydrochloride (30).
1H NMR (400 MHz, DMSO-d6) δ 9.07 (s, 1H), 8.93 (s, 1H), 8.79 (s, 1H), 8.74 (d, J = 4.8 Hz, 2H), 8.51 – 8.35 (m, 2H), 8.33 – 8.16 (m, 4H), 8.00 (d, J = 8.4 Hz, 4H), 7.79 (t, J = 6.0 Hz, 2H), 7.65 (s, 1H), 7.02 (d, J = 8.4 Hz, 4H), 4.27 (d, J = 5.6 Hz, 2H), 3.29 (d, J = 11.6 Hz, 2H), 2.89 (d, J = 10.8 Hz, 2H), 2.13 (s, 1H), 1.92 (d, J = 12.4 Hz, 2H), 1.53 (d, J = 12.4 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.1, 152.0, 144.8, 142.7, 129.9, 128.0, 127.3, 126.3, 123.7, 123.6, 116.3, 69.7, 42.6, 32.8, 25.1. MS (ESI) [M+H]+ 500.2.
5-(Piperidin-4-ylmethoxy)-2,3-bis(4-(pyridin-4-yl)phenyl)pyrazine hydrochloride (31).
1H NMR (400 MHz, DMSO-d6) δ 9.19 (s, 1H), 8.92 (s, 4H), 8.48 (s, 1H), 8.33 (s, 3H), 8.04 (s, 3H), 7.66 (d, J = 29.6 Hz, 4H), 4.34 (s, 2H), 3.29 (s, 2H), 2.92 (s, 2H), 2.18 (s, 1H), 1.93 (s, 2H), 1.59 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 157.9, 153.0, 147.0, 143.1, 141.1, 140.6, 134.8, 133.9, 133.3, 130.7, 130.5, 127.8, 123.3, 69.7, 42.5, 32.9, 25.1. MS (ESI) [M+H]+ 500.2.
4′,4″-(5-(piperidin-4-ylmethoxy)pyrazine-2,3-diyl)bis(([1,1’-biphenyl]-4-amine)) hydrochloride (32).
1H NMR (400 MHz, DMSO-d6) δ 9.20 (s, 1H), 8.87 (s, 1H), 8.38 (s, 1H), 7.95 – 7.19 (m, 15H), 4.31 (s, 2H), 3.29 (s, 2H), 2.90 (s, 2H), 2.15 (s, 1H), 1.92 (s, 2H), 1.56 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 157.6, 147.1, 143.6, 139.1, 138.3, 137.5, 137.4, 137.1, 133.8, 133.3, 132.4, 130.2, 129.9, 127.7, 127.6, 126.2, 126.2, 122.7, 122.5, 69.5, 42.5, 32.8, 25.1. MS (ESI) [M+H]+ 528.3.
Inhibition of p300-HAT and other HATs.
The HAT domain (1195-1673) of human p300, HAT of CBP, PCAF and Myst3 HATs were obtained and their activity and inhibition assays performed using our previous methods 14
In brief, a compound with increasing concentrations was incubated with an HAT (10 nM) in 20 μL of 50 mM phosphate buffer (pH = 7.0) containing 0.01% Brij-35 for 10 min at 25 °C. Histone H3 peptide (ARTKQTARKSTGGKAPRKQLA) (20 μM) and Acetyl-CoA (1 μM 3H-Ac-CoA and 19 μM Ac-CoA) were added to initiate the reaction. After 30 min at 25 °C, the reaction was stopped by adding 6 N formic acid (5 μL). 20 μL of reaction mixture was then transferred to a small piece of P81 filter paper (Whatman) that binds histone H3 peptide. The filter paper was washed 3× with 50 mM NaHCO3, dried, and transferred into a scintillation vial containing scintillation cocktail (2 mL). Radioactivity on the filter paper was measured with a Beckman LS-6500 scintillation counter. IC50 values were obtained by using a standard sigmoidal dose response curve fitting in Prism (version 5.0, GraphPad Software, Inc., La Jolla, CA). IC50 values were the mean values from at least three experiments.
Alpha assay.
We followed our published method 14 to investigate whether a compound can disrupt the binding of p300 HAT and histone H4 peptide. In brief, the assay was performed in a 384-well plate using His6-tagged P300 HAT (125 nM) coated nickel chelate acceptor beads (5 μL, Perkin Elmer), biotinylated H4 peptide [SGRGKGGKGLGKGGAKRHRKVLRGG-K(Biotin)-NH2] (30 nM) coated streptavidin donor beads (5 μL, Perkin Elmer), and increasing concentrations of a compound in a PBS buffer with 0.5 % BSA (final volume of 25 μL). Upon incubation for 1h, Alpha signal was determined on a Tecan Spark microplate reader (excitation at 680 nm and reading at 570 nm). The IC50 values were determined using the sigmoidal dose-response fitting in the program of Prism 5.0 (GraphPad).
Western blot.
106Kasumi-1 cells/well were incubated with a variety of concentrations of a compound for 12 hours. Histones were extracted using EpiQuik total histone extraction kit (Epigentek), according to the manufacturer’s protocol. Equal amounts of the protein extract (2 μg) were separated using SDS-PAGE and transferred to PVDF membranes. The blots were probed with primary antibodies against H3K9Ac, H3K18Ac, H3K27Ac and H3 (Cell Signaling), followed by anti-rabbit IgG (Thermo Scientific) secondary antibodies.
Cell growth inhibition.
The anti-proliferation assays for cancer cells were performed using our previous method 31–33. In brief, for blood cancer cells, 106 cells per well were added into 96-well plates and cultured with increasing concentrations of a compound in RPMI-1640 medium supplemented with 20% fetal bovine serum and penicillin (100 U/mL) and streptomycin (100 μg/mL) at 37 °C in a 5% CO2 atmosphere with 100% humidity. For solid tumor cells, 105 cells per well were added into 96-well plates and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum and penicillin (100 U/mL) and streptomycin (100 μg/mL) overnight. Upon addition of increasing concentrations of a compound, cells were incubated for 5 days. Cell viability was assessed by using an XTT assay kit (Roche). Compound EC50 values were calculated from dose response curves using Prism 5.0.
Supplementary Material
Highlights:
Compound screening identified 1,4-pyrazine and related pyridine compounds to be novel inhibitors of human histone acetyltransferase p300.
Structure-activity relationship studies yielded a more potent compound 29.
Enzyme kinetics and other studies support compound 29 is competitive against the substrate histone.
Compound 29 exhibited a high selectivity for histone acetyltransferase p300 and its paralog CBP.
Compound 29 inhibited cellular histone acetylation and proliferation of a panel of solid and blood cancer cells.
Acknowledgement
This work was supported by a grant (RP180177) from Cancer Prevention and Research Institute of Texas, a grant (W81XWH-18-1-0368) from USAMRAA of the US Department of Defense, and a grant (R01CA266057) from the US National Institute of Health/National Cancer Institute to Y.S.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Jones PA; Baylin SB The epigenomics of cancer. Cell 2007, 128, 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kouzarides T Chromatin modifications and their function. Cell 2007, 128, 693–705. [DOI] [PubMed] [Google Scholar]
- 3.Belkina AC; Denis GV BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer 2012, 12, 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang Z; Yik JH; Chen R; He N; Jang MK; Ozato K; Zhou Q Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell 2005, 19, 535–545. [DOI] [PubMed] [Google Scholar]
- 5.Schiltz RL; Mizzen CA; Vassilev A; Cook RG; Allis CD; Nakatani Y Overlapping but distinct patterns of histone acetylation by the human coactivators p300 and PCAF within nucleosomal substrates. J Biol Chem 1999, 274, 1189–1192. [DOI] [PubMed] [Google Scholar]
- 6.Dancy BM; Cole PA Protein lysine acetylation by p300/CBP. Chem Rev 2015, 115, 2419–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang F; Marshall CB; Ikura M Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cell Mol Life Sci 2013, 70, 3989–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson AB; O’Malley BW Steroid receptor coactivators 1, 2, and 3: critical regulators of nuclear receptor activity and steroid receptor modulator (SRM)-based cancer therapy. Mol Cell Endocrinol 2012, 348, 430–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dilworth FJ; Chambon P Nuclear receptors coordinate the activities of chromatin remodeling complexes and coactivators to facilitate initiation of transcription. Oncogene 2001, 20, 3047–3054. [DOI] [PubMed] [Google Scholar]
- 10.Kraus WL; Manning ET; Kadonaga JT Biochemical analysis of distinct activation functions in p300 that enhance transcription initiation with chromatin templates. Mol Cell Biol 1999, 19, 8123–8135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu W; Shi XL; Roeder RG Synergistic activation of transcription by CBP and p53. Nature 1997, 387, 819–823. [DOI] [PubMed] [Google Scholar]
- 12.Vervoorts J; Luscher-Firzlaff JM; Rottmann S; Lilischkis R; Walsemann G; Dohmann K; Austen M; Luscher B Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep 2003, 4, 484–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma H; Hong H; Huang SM; Irvine RA; Webb P; Kushner PJ; Coetzee GA; Stallcup MR Multiple signal input and output domains of the 160-kilodalton nuclear receptor coactivator proteins. Mol Cell Biol 1999, 19, 6164–6173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu F; Hua Y; Kaochar S; Nie S; Lin YL; Yao Y; Wu J; Wu X; Fu X; Schiff R; Davis CM; Robertson M; Ehli EA; Coarfa C; Mitsiades N; Song Y Discovery, Structure-Activity Relationship, and Biological Activity of Histone-Competitive Inhibitors of Histone Acetyltransferases P300/CBP. J Med Chem 2020, 63, 4716–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lasko LM; Jakob CG; Edalji RP; Qiu W; Montgomery D; Digiammarino EL; Hansen TM; Risi RM; Frey R; Manaves V; Shaw B; Algire M; Hessler P; Lam LT; Uziel T; Faivre E; Ferguson D; Buchanan FG; Martin RL; Torrent M; Chiang GG; Karukurichi K; Langston JW; Weinert BT; Choudhary C; de Vries P; Van Drie JH; McElligott D; Kesicki E; Marmorstein R; Sun C; Cole PA; Rosenberg SH; Michaelides MR; Lai A; Bromberg KD Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lavau C; Du C; Thirman M; Zeleznik-Le N Chromatin-related properties of CBP fused to MLL generate a myelodysplastic-like syndrome that evolves into myeloid leukemia. EMBO J 2000, 19, 4655–4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gervais C; Murati A; Helias C; Struski S; Eischen A; Lippert E; Tigaud I; Penther D; Bastard C; Mugneret F; Poppe B; Speleman F; Talmant P; VanDen Akker J; Baranger L; Barin C; Luquet I; Nadal N; Nguyen-Khac F; Maarek O; Herens C; Sainty D; Flandrin G; Birnbaum D; Mozziconacci MJ; Lessard M Acute myeloid leukaemia with 8p11 (MYST3) rearrangement: an integrated cytologic, cytogenetic and molecular study by the groupe francophone de cytogenetique hematologique. Leukemia 2008, 22, 1567–1575. [DOI] [PubMed] [Google Scholar]
- 18.Lau OD; Kundu TK; Soccio RE; Ait-Si-Ali S; Khalil EM; Vassilev A; Wolffe AP; Nakatani Y; Roeder RG; Cole PA HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol Cell 2000, 5, 589–595. [DOI] [PubMed] [Google Scholar]
- 19.Kwie FH; Briet M; Soupaya D; Hoffmann P; Maturano M; Rodriguez F; Blonski C; Lherbet C; Baudoin-Dehoux C New potent bisubstrate inhibitors of histone acetyltransferase p300: design, synthesis and biological evaluation. Chem Biol Drug Des 2011, 77, 86–92. [DOI] [PubMed] [Google Scholar]
- 20.Baell J; Walters MA Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [DOI] [PubMed] [Google Scholar]
- 21.Bowers EM; Yan G; Mukherjee C; Orry A; Wang L; Holbert MA; Crump NT; Hazzalin CA; Liszczak G; Yuan H; Larocca C; Saldanha SA; Abagyan R; Sun Y; Meyers DJ; Marmorstein R; Mahadevan LC; Alani RM; Cole PA Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol 2010, 17, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang H; Pinello CE; Luo J; Li D; Wang Y; Zhao LY; Jahn SC; Saldanha SA; Chase P; Planck J; Geary KR; Ma H; Law BK; Roush WR; Hodder P; Liao D Small-molecule inhibitors of acetyltransferase p300 identified by high-throughput screening are potent anticancer agents. Mol Cancer Ther 2013, 12, 610–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Costi R; Di Santo R; Artico M; Miele G; Valentini P; Novellino E; Cereseto A Cinnamoyl compounds as simple molecules that inhibit p300 histone acetyltransferase. J Med Chem 2007, 50, 1973–1977. [DOI] [PubMed] [Google Scholar]
- 24.Wu F; Zhou C; Yao Y; Wei L; Feng Z; Deng L; Song Y 3-(Piperidin-4-ylmethoxy)pyridine Containing Compounds Are Potent Inhibitors of Lysine Specific Demethylase 1. J Med Chem 2016, 59, 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao Y; Huo T; Lin YL; Nie S; Wu F; Hua Y; Wu J; Kneubehl AR; Vogt MB; Rico-Hesse R; Song Y Discovery, X-ray Crystallography and Antiviral Activity of Allosteric Inhibitors of Flavivirus NS2B-NS3 Protease. J Am Chem Soc 2019, 141, 6832–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nie S; Yao Y; Wu F; Wu X; Zhao J; Hua Y; Wu J; Huo T; Lin YL; Kneubehl AR; Vogt MB; Ferreon J; Rico-Hesse R; Song Y Synthesis, Structure-Activity Relationships, and Antiviral Activity of Allosteric Inhibitors of Flavivirus NS2B-NS3 Protease. J Med Chem 2021, 64, 2777–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu F; Nie S; Yao Y; Huo T; Li X; Wu X; Zhao J; Lin YL; Zhang Y; Mo Q; Song Y Small-molecule inhibitor of AF9/ENL-DOT1L/AF4/AFF4 interactions suppresses malignant gene expression and tumor growth. Theranostics 2021, 11, 8172–8184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X; Wang L; Zhao K; Thompson PR; Hwang Y; Marmorstein R; Cole PA The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature 2008, 451, 846–850. [DOI] [PubMed] [Google Scholar]
- 29.Ono H; Basson MD; Ito H P300 inhibition enhances gemcitabine-induced apoptosis of pancreatic cancer. Oncotarget 2016, 7, 51301–51310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Y; Zhang R; Li Z; Mei L; Wan S; Ding H; Chen Z; Xing J; Feng H; Han J; Jiang H; Zheng M; Luo C; Zhou B Discovery of Highly Potent, Selective, and Orally Efficacious p300/CBP Histone Acetyltransferases Inhibitors. J Med Chem 2020, 63, 1337–1360. [DOI] [PubMed] [Google Scholar]
- 31.Wu F; Jiang H; Zheng B; Kogiso M; Yao Y; Zhou C; Li XN; Song Y Inhibition of Cancer-Associated Mutant Isocitrate Dehydrogenases by 2-Thiohydantoin Compounds. J Med Chem 2015, 58, 6899–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng Z; Yao Y; Zhou C; Chen F; Wu F; Wei L; Liu W; Dong S; Redell M; Mo Q; Song Y Pharmacological inhibition of LSD1 for the treatment of MLL-rearranged leukemia. J Hematol Oncol 2016, 9, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang L; Deng L; Chen F; Yao Y; Wu B; Wei L; Mo Q; Song Y Inhibition of histone H3K79 methylation selectively inhibits proliferation, self-renewal and metastatic potential of breast cancer. Oncotarget 2014, 5, 10665–10677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.