

Abstract
Type 2 diabetes mellitus (T2DM) is associated with impaired skeletal muscle function and degeneration of the skeletal muscles. However, the mechanisms underlying the degeneration are not well described in human skeletal muscle. Here we show that skeletal muscle of T2DM patients exhibit degenerative remodeling of the extracellular matrix that was associated with a selective increase of a subpopulation of fibro-adipogenic progenitors (FAPs) marked by expression of THY1 (CD90) - the FAPCD90+. We identified Platelet-derived growth factor (PDGF) as a key FAP regulator, as it promotes proliferation and collagen production at the expense of adipogenesis. FAPsCD90+ showed a PDGF-mimetic phenotype, with high proliferative activity, clonogenicity and production of extracellular matrix. FAPCD90+ proliferation was reduced by in vitro treatment with metformin. Furthermore, metformin treatment reduced FAP content in T2DM patients. These data identify a PDGF-driven conversion of a sub-population of FAPs as a key event in the fibrosis development in T2DM muscle.
Keywords: Skeletal muscle, Mesenchymal stem cells, Fibro adipogenic progenitors, Fibroblast, Type 2 diabetes, extracellular matrix, adipocytes
INTRODUCTION
Skeletal muscle is a major organ comprising ~40% of human body mass, accountable for ~30% of basal metabolic rate (Zurlo et al., 1990) and solely responsible for generating mechanical forces to enable breathing and locomotion. In addition, skeletal muscle is a major storage organ for glucose (glycogen) and the primary target for peripheral insulin stimulated glucose uptake (Baron et al., 1988). With the importance of skeletal muscle for overall body health, it is of no surprise that degeneration of skeletal muscle and loss of muscle mass is associated with a multitude of different diseases and ultimately shortened life span (Srikanthan and Karlamangla, 2014). Degeneration of skeletal muscle has often been investigated in models of severe genetic diseases or trauma-induced paralysis. Under such circumstances, the parenchymal muscle cells are progressively replaced by non-contractile tissue including extracellular matrix (ECM) proteins and adipocytes (fibro-fatty degeneration), resulting in loss of contractile and metabolic function.
Age-related diseases, such as type 2 diabetes mellitus (T2DM), are also associated with fibro-fatty degeneration of human skeletal muscle. In contrast to the aggressive degenerative progression of the muscle environment in Duchenne’s muscle dystrophy or following trauma, muscle fibro-fatty degeneration in T2DM develops gradually and is accompanied by muscle atrophy (Goodpaster et al., 2000; Moore et al., 2016; Rasmussen et al., 2018; Richardson et al., 2005).
Ectopic deposition of excessive ECM and adipocytes in different organs including liver, kidney, fat and skeletal muscle is a common pathogenic feature of many diseases, which can ultimately result in multi-organ failure and death (Zhao et al., 2019). In T2DM an excessive deposition of ectopic ECM and adipocytes has been observed in viscera, pancreas and skeletal muscle (Addison et al., 2014; Conte et al., 2019; Goodpaster et al., 2000; Moore et al., 2016). Rodent models of obesity and insulin resistance have revealed fibro-fatty degeneration in skeletal muscle to result in impaired metabolic and contractile muscle function (Buras et al., 2019; Inoue et al., 2013; Kang et al., 2011). Importantly, similar features have been reported in insulin resistant subjects or patients affected by diabetic polyneuropathy (Goodpaster et al., 2000; Moore et al., 2016). Understanding the mechanism driving such pathological muscle remodeling is fundamental to provide interventions toward preventing muscle fibro-fatty degeneration in T2DM patients. Moreover, it will be important to understand to which degree these changes are causally related to the disease or a consequence of the condition.
In order to understand and potentially reduce or prevent the fibro-fatty degeneration of skeletal muscle, it is essential to determine the cellular origin of fibrosis and adipocytes. In mice, skeletal muscle resident cells with fibro- and adipogenic potential (i.e. FAPs) were identified a decade ago by two independent reports as stem cell antigen 1+, CD34+, Platelet Derived Growth Factor Receptor α (PDGFRα)+ cells (Joe et al., 2010; Uezumi et al., 2010). Notably, several reports have shown that FAPs are cellular targets of therapeutic interventions toward reducing or preventing fibro-fatty degeneration in mouse models of regeneration, disease or aging (Heredia et al., 2013; Lemos et al., 2015; Mueller et al., 2016). In particular, the tyrosine kinase receptor and highly specific FAP marker, PDGFRα, has been shown to play a key role in FAP activation and induction of tissue fibrosis in several organs including adipose tissue and skeletal muscle (Iwayama et al., 2015; Mueller et al., 2016).
In models of insulin resistance, aberrant FAP activity underlie fibro-fatty degeneration of the diaphragm muscle during a high fat diet, which ultimately impaired mechanical muscle function (Buras et al., 2019). Unfortunately, translation of these findings into human is challenged by the lack of in vivo human FAP markers and the general scarcity of human skeletal muscle tissue biopsies. However, studies utilizing cultured cells from muscle homogenates suggest that similar cells are present in human muscle (Uezumi et al., 2014), indicating that these cells may represent a novel target for preventing fibro-fatty tissue degeneration in T2DM. Here we used FACS-mediated isolation and transcriptomic profiling at population and single-cell level of FAPs from human T2DM patient-derived biopsies to identify a subpopulation of FAPs, defined by their expression of THY1 (CD90), as putative cellular source of the fibro-fatty infiltration of muscles from T2DM patients and potential target of therapeutic interventions.
RESULTS
T2DM is associated with degenerative remodeling of the extracellular matrix
To investigate if T2DM is associated with fibro-fatty degeneration of skeletal muscle, we collected muscle biopsies (Study 1) from age and overweight-matched subjects (OBS), T2DM patients (T2D) and insulin treated T2DM patients (itT2D) with severe insulin resistance (treated with 196±26 IU insulin/day, (Kampmann et al., 2011a)). This allowed us to investigate potential alterations in skeletal muscle as a consequence of T2DM and the severity of insulin resistance. To perform an unbiased evaluation of major transcriptional alterations, RNA was isolated from crude muscle tissue, subjected to RNA-sequencing (Moller et al., 2017) and followed by differential gene expression (DE) and pathway enrichment analysis (Fig 1A). In addition to a dysregulated muscle morphology and inflammation, our analysis revealed that DE genes were substantially enriched in biological processes and pathways related to ECM turnover and remodeling in itT2D patients (Fig 1B,C, Fig S1A,B). This was particularly evident as the severity of insulin resistance increased (OBS<T2D<itT2D), which was supported by qPCR of common genes associated with fibrosis (Fig 1D–G). These findings support earlier studies reporting that obesity and T2DM are associated with fibro-fatty infiltration of skeletal muscle (Goodpaster et al., 2000; Moore et al., 2016; Rasmussen et al., 2018). Downregulated genes in itT2D patients were enriched in pathways related to insulin signaling and glucose response, consistent with the insulin resistant state (Fig S1C). We extended our analysis to tissue sections of skeletal muscle biopsies obtained from T2DM patients and patients without T2DM undergoing coronary artery bypass surgery (Study 2). These also revealed an increased deposition of Collagen1 in T2DM patients, as compared to patients without T2DM (Fig 1H, I). Collectively, our findings support the hypothesis that T2DM is associated with degeneration of skeletal muscle niche.
Fig 1. Type 2 diabetes is associated with degenerative remodeling.
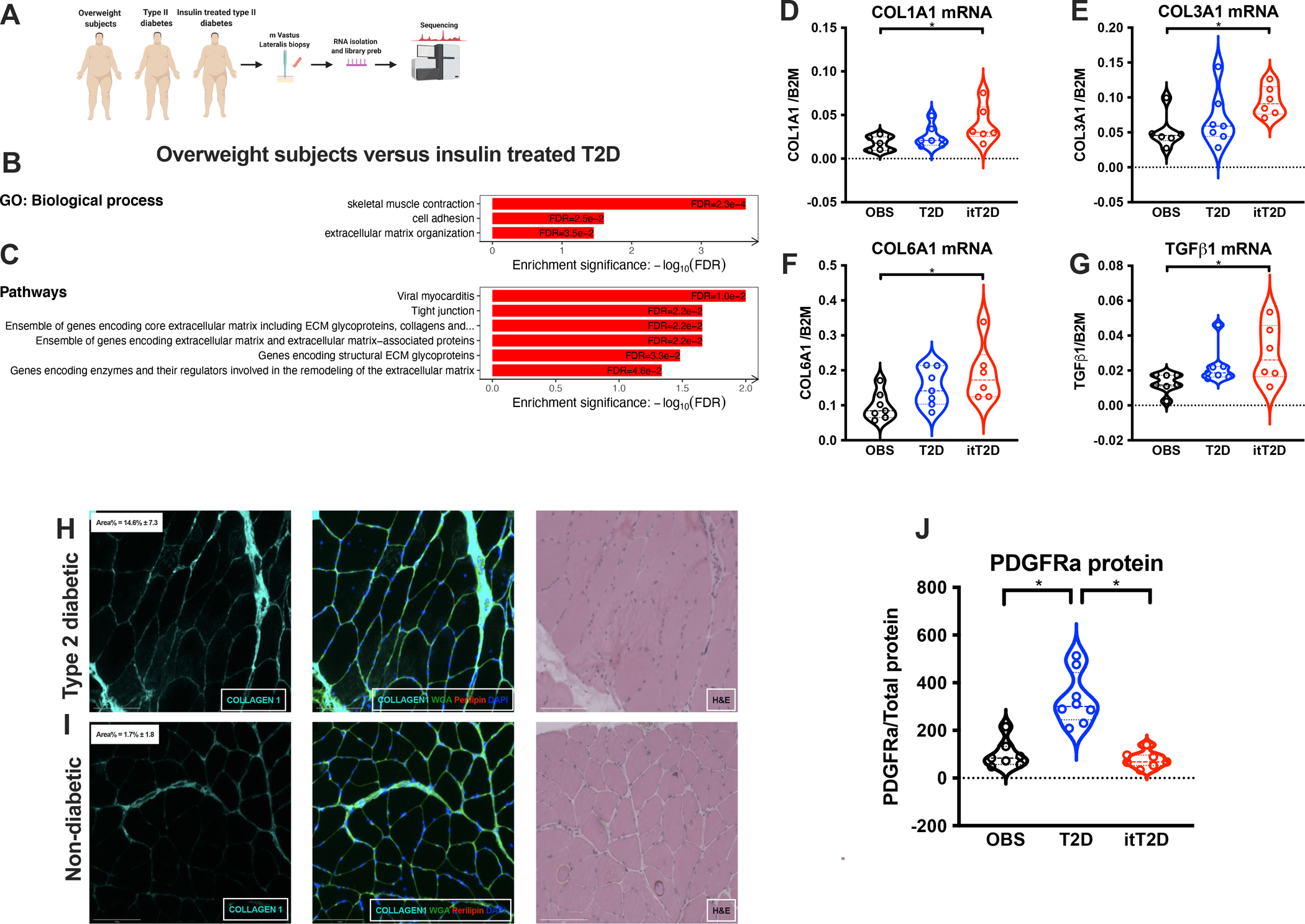
A, Work-flow of study 1 (biopsies from m. Vastus Lateralis) of overweight subjects (n=7), T2D (n=7) and insulin treated T2D (n=6); B, Biological processes altered in itT2D; C, Enriched pathways in itT2D; D, COL1A1 E, COL3A1; F, COL6A1; G, TGFB1 mRNA expression in OBS, T2D and itT2D; H+I, IHC staining of Collagen1, WGA and Perilipin1 from patients with (H) and without T2DM (I); J, PDGFRα expression (study 1). *p<0.05.
Human skeletal muscle FAPs can be prospectively isolated as CD34+CD56−CD45−CD31−
In mice, skeletal muscle fibrosis and fat accumulation is thought to primarily originate from FAPs, which have been shown to specifically express PDGFRα (Joe et al., 2010; Uezumi et al., 2010). To examine if FAPs could be the cellular source of fibrosis and adipocytes in T2DM, we quantified protein content of PDGFRα at whole tissue level. We found a specific increase in the T2D patients (Fig 1K, S1 D), which declined to OBS levels in the itT2D group (Study 1).
To further understand the cellular mechanism underlying fibro-fatty infiltration of skeletal muscles in T2DM patients, we set out to isolate FAPs from human skeletal muscle by FACS (non-T2DM, Study 2). We tested mesenchymal stem cell and FAP markers previously used to isolate mouse FAPs, including PDGFRα, ALCAM (CD166), CD34 and THY1 (CD90) (Joe et al., 2010; Madaro et al., 2018). While no signal was obtained from PDGFRα or CD166 (not shown), a population of CD34+CD56−CD45−CD31− cells (Fig 2A i, S1E i–iv) could be isolated, which could be further subdivided into a CD34+CD90− and a CD34+CD90+ population (Fig 2A ii, S1E v). For comparison, we also identified MuSCs based on expression of CD56 as well as CD82 (Fig 2A iii, S1E vi). Endothelial cells and hematopoietic cells were initially selected on CD31/CD45 expression and could then be discriminated based on CD34 expression by endothelial cells (Fig S1 iv; Fig S2E vii)(Crisan et al., 2008). For sorting, we included PI, fluorescence minus one controls were used to set the gates (Fig S1F i–iv) and purity was generally >95% (Fig S1 G–I).
Fig 2. Identification and characterization of human fibro-adipogenic progenitors.
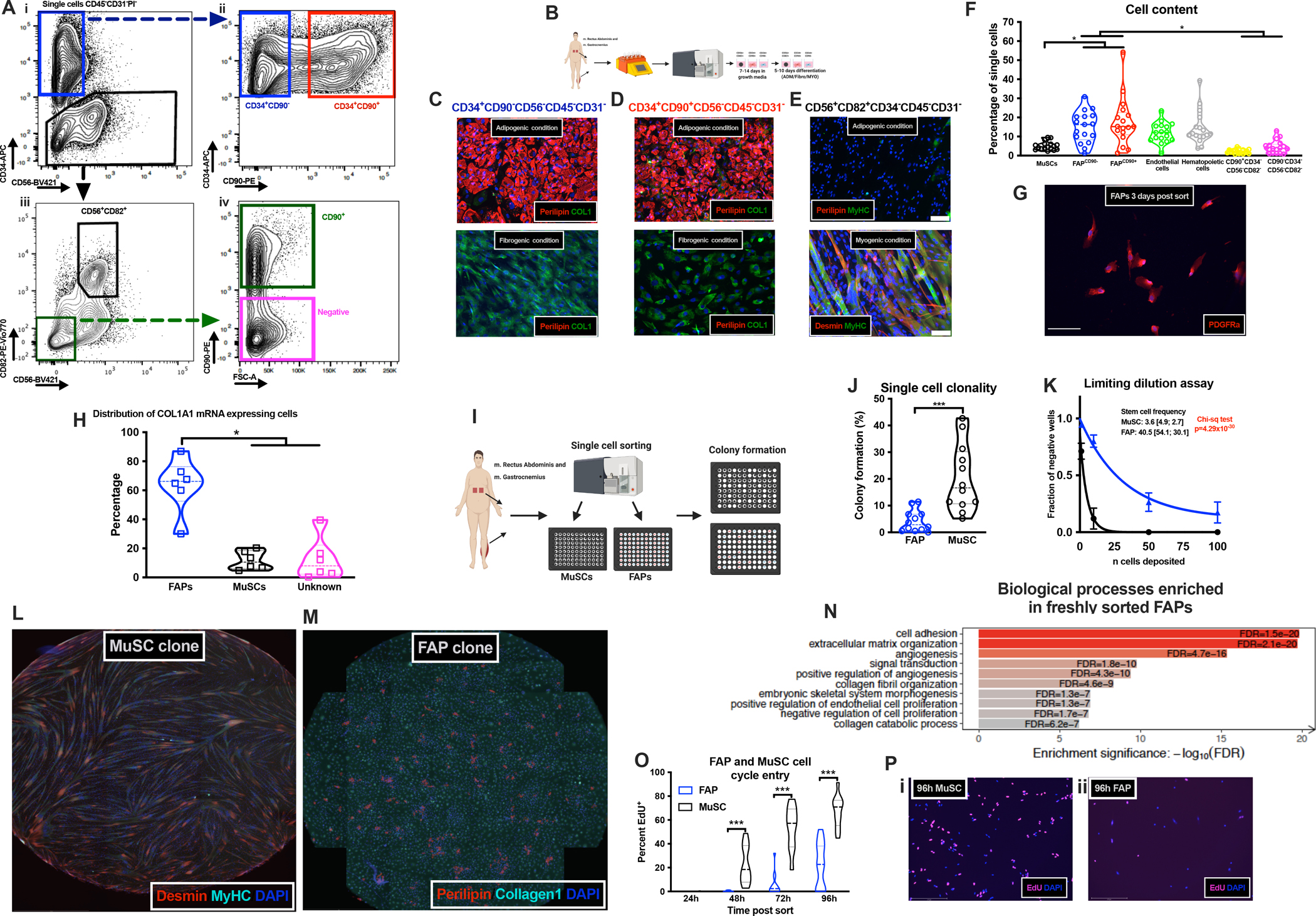
A-i, Sorting strategy (CD45−CD31−) identifying CD34+CD56− or CD34−; A-ii, CD34+CD90− and CD34+CD90+ cells; A-iii, CD56+CD82+ cells; A-iv, CD90+ cells; B, Work-flow; C-E, adipogenic, fibrogenic and myogenic differentiation of CD34+CD90−CD56− (C), CD34+CD90+CD56− (D) and CD56+CD82+CD34− (E) populations by IHC; F, % mononuclear cells in human skeletal muscle (n=17, study 2, non-T2DM); G, PDGFRα in FAPs (scalebar 50 μm); H, COL1A1 expressing CD90+CD56− (FAPs), CD56+ (MuSCs) and CD90−CD56− (Unidentified) cells (n=6, study 2, non-T2DM) after 3–9 days in vitro; I, Clonal experiment; J, Colony formation (%) of single sorted FAPs and MuSCs (n=12, study 2, non-T2DM); K, Limiting dilution assay on FAPs and MuSCs (n=3, study 2, non-T2DM), Solid line is non-linear fit; L, MyHC+/Desmin+ myotubes from single sorted MuSCs; M, Perilipin1+/Collagen1+ cells from single sorted FAPs; N, Biological processes in FAPs (n=3, study 2, non-T2DM); O, % of EdU+ MuSCs and FAPs (n=10–16, study 2, non-T2DM); P, EdU+ MuSCs (i) and FAPs (ii) 96h post sort. Cells obtained from of mm. Rectus Abdominus/Gastrocnemius. *p<0.05, **p<0.01 and ***p<0.001.
The FACS-isolated populations were tested for their adipogenic, fibrogenic and myogenic capacity in culture. Only the two CD34+ cell populations (CD90− and CD90+) gave rise to both adipocytes and collagen/α-smooth muscle actin expressing cells when cultured for 5–10 days under appropriate conditions (Fig 2C–E; Fig S2 A i–ii, B, C i–ii). In contrast, the MuSCs only rarely gave rise to adipocytes (Fig 2E), whereas these robustly differentiated into myosin heavy chain (MyHC) and Desmin expressing myotubes (Fig 2E). Moreover, we found no adipogenic differentiation and very little collagen expression in endothelial or hematopoietic cells (Fig S2D i–ii). The two minor populations (Fig 2A iv) grew poorly and we were therefore unable to induce adipogenesis in these. For FAPs, we also noted osteogenic capacity (Fig S2E,F indicated by calcium deposit formation) after 10 days in osteogenic induction medium, as previously shown in rodent FAPs (Wosczyna et al., 2012). This confirms the mesenchymal nature of FAPs, although heterotopic ossification is only rarely observed in humans (Shore and Kaplan, 2008). We therefore chose to adhere to the FAP nomenclature.
Using flow-data, we quantified cell populations from which it appeared that FAPs (FAPCD90− and FAPCD90+ combined) constitute a major proportion of the mononuclear (~40%) cells in our patient derived human skeletal muscle (Fig 2F). Staining of FACS isolated FAPs three days post isolation confirmed the expression of PDGFRα (Fig 2G) as well as TCF7L2 and TE7 (Fig S2G i–ii, H). To understand if FAPs are the major collagen producing cells in human skeletal muscle, we digested human skeletal muscle biopsies (Study 2, non-T2DM) and plated the entire content, allowing all adherent cells to attach and activate. After one week we detached the cells and probed for expression of COL1A1 mRNA in activated FAPs (CD90+CD56−, CD34 is not expressed in ex vivo activated FAPs), activated MuSCs (CD56+) and unidentified cells (CD90−CD56−) (Porichis et al., 2014). When selecting cells expressing COL1A1 (Fig S2 I i–ii, J, K), it was apparent that FAPs was the major collagen producing population with only a minority of MuSCs and unidentified cells expressing COL1A1 (Fig 2 H). Similar results were obtained when identifying the in vitro activated FAPs with a PDGFRα antibody (Fig S2L). Furthermore, when selecting the entire population of FAPs, MuSCs or unidentified cells, it was clear that FAPs were the primary COL1A1 producing cell type (Fig S2M). Collectively, our results suggest that human skeletal muscle FAPs are likely the most represented cell population in human skeletal muscle and a putative source of fibro-fatty degeneration.
Human FAPs are clonally and transcriptionally a distinct cell population
To understand if human FAPs have clonal (progenitor) potential and if these give rise to both adipocytes and fibroblasts, we performed a clonal assay, as previously described in mice (Joe et al., 2010). FAPs and MuSCs were FACS isolated and immediately re-sorted in 96-wells with one cell per well (Fig 2I). Notably, the MuSCs had approximately four times greater clonal potential (~17% in MuSCs vs ~4% in FAPs) than the FAP population (Fig 2J). To substantiate these findings, we performed a limiting dilution assay, which predicted a markedly greater stem cell frequency (~10 times) of MuSCs compared to FAPs (Fig 2K). In contrast, Joe et al. (2010) reported similar levels of clonality between FAPs and MuSCs, which, besides species-related differences, may be explained by the age of our donors compared to the age of the mice. Aging has recently been shown to alter the phenotype of FAPs in mice, including the ability to enter the cell cycle (Lukjanenko et al., 2019). When differentiating the single sorted MuSC more than 92% of these clones gave rise to Desmin+/MyHC+ myoblasts/myotubes (Fig 2L) similar to mouse MuSCs (Joe et al., 2010). For FAP colonies more than 72% contained Perilipin1+ adipocytes and Collagen1+ cells (Fig 2M), while the remaining were non-adipogenic Collagen1+ fibroblasts.
Next, in order to understand the nature of FAPs in vivo, we flow-sorted FAPs, MuSCs and the negative population and immediately processed these for transcriptomic analysis (RNA-seq). Principal component analysis, based on the expression of all detected genes, revealed that FAPs cluster separated from the other two sorted populations (Fig S2N) and are enriched in common FAP markers (Fig S2O–S). Gene enrichment analysis of FAPs (CD90− and CD90+ combined) revealed that FAPs in vivo display a gene expression pattern associated to ECM production, turnover and signaling, while also indicating potential contribution to angiogenic events (Fig 2N,S3A). In contrast, the MuSCs were enriched for straited muscle development and cell cycle control (Fig S3A,B). The negative population was devoid of MuSC markers, however, there was a specific expression of other myogenic genes. Enrichment analysis confirmed this and revealed that these were myonuclei (Fig S3C–F) and also highly diverse from the identified FAPs.
MuSCs are known to be quiescent in vivo under homeostatic conditions, however, it has not been described if this is also the case for FAPs. Given their continuous role in ECM maintenance one might speculate that FAPs possess higher mitotic turnover, as compared to MuSCs. We also reasoned that FAPs implicated in fibro-fatty degeneration of T2DM muscles could have higher proliferative activity. To address this issue, we flow-sorted MuSCs and FAPs from human skeletal muscle biopsies and cultured them with EdU (de Morree et al., 2019). At 24h post sort very limited EdU incorporation was detected in MuSCs as well as FAPs (0.0–0.8%), suggesting that the cells were quiescent at the time of extraction (Fig 2O,P i–ii). Moreover, while MuSC started to enter the S-phase between 24h and 48h, FAPs remained inactive until 48–72h and then slowly progressed (Fig 2O). This suggest that human FAPs are mitotically inactive in vivo (as MuSCs) and upon activation they display a slow kinetic of progression into S-phase, compared to MuSCs.
Single-cell RNA-sequencing confirms the identity of FAPs
We further investigated transcriptional identity of human FAPs by performing single-cell transcriptome analysis (scRNA-seq) on whole human muscle (Fig 3A). After quality control (Fig S3G–L), we performed an unsupervised clustering and initially identified 15 cell populations. Utilizing markers from mouse (Tabula Muris et al., 2018) as well as human skeletal muscle (Mackey et al., 2017; Uezumi et al., 2014) we annotated nine major cell populations, with some containing one or several subpopulations (Fig 3B). As expected, we found a population highly enriched in PDGFRA and CD34 as well as partly expressing THY1/CD90 (Fig 3C–E) and robust expression of COL1A1 (Fig 3F). This population had high expression of collagens, laminins and other matrix related proteins consistent with our FACS identified FAP population (Fig S3L–N, S4). By using FAP specific genes as input for gene set enrichment analysis (Fig S3AD), this revealed agreement with pathways observed in population-based RNA-seq data (Fig 2N), including PDGF signaling. Interestingly, the PDGF pathway has been implicated in the regulation of FAP quiescence (Mueller et al., 2016) rendering it likely that the PDGF pathway is also important in human FAPs.
Fig 3. Single cell RNA sequencing confirms unique FAP population.
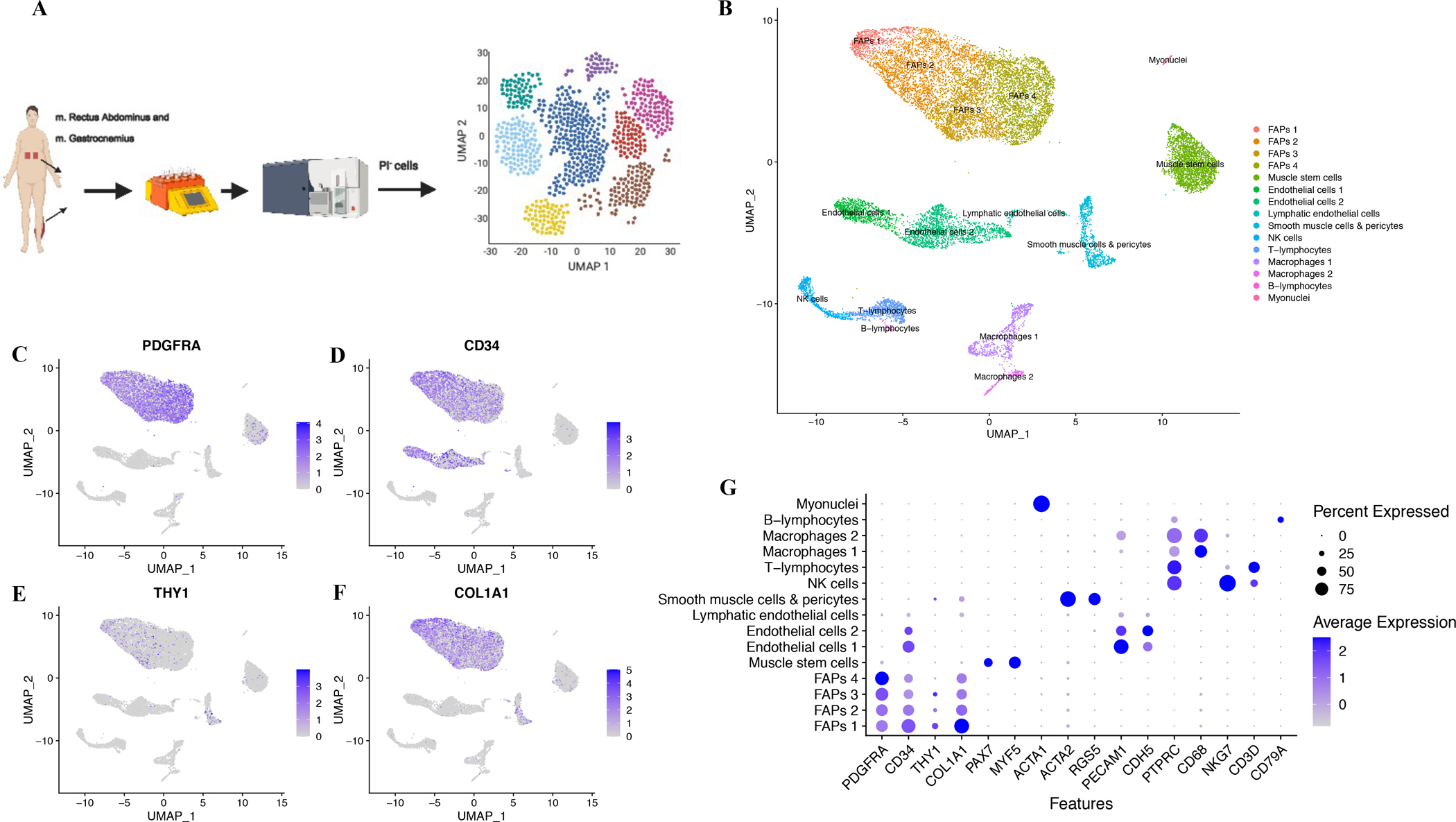
A, Single-cell RNA-seq experiment; B, UMAP clusters (n=4, study 2, 3 non-T2DM and 1 T2DM, mm. Rectus Abdominus/Gastrocnemius) revealed 15 populations; PDGFRA (C), CD34 (D), THY1 (CD90, E), COL1A1 (F) expression; G, canonical cell markers in 15 population, coloring indicates mean expression and dot-size % of cells in which the gene is expressed.
Our initial clustering resulted in four FAP subpopulations, which could indicate that FAPs contain subsets with potential to carry out distinct functions or at least sub-fractions at different stages. We noted one other population high in collagens and ECM proteins, as well as alpha smooth muscle actin (ACTA2), but negative for CD34 and PDGFRA (Fig 3G, Fig S4, S3AG). A similar population was recently described by Giordani et al. (2019) in which they were designated as smooth muscle-mesenchymal cells. In agreement, the population is distinct from FAPs and given the much greater presence of FAPs (Fig 2F, 3B), we find it likely that FAPs constitute a more potent contributor to skeletal muscle fibrosis and adipocyte formation.
PDGF controls FAP fibro- and adipogenic fate
Prompted by the finding that PDGFRA mRNA is specifically enriched in sorted FAPs, we next asked what the role of PDGF signaling was in human FAPs. From our in-situ RNA hybridization data, we noticed that PDGFRα expression was associated with COL1A1 expression in FAPs (Fig 4A,B). To test if PDGF stimulation increased collagen expression in freshly isolated FAPs, we isolated FAPs, exposed them to PDGF-AA (Fig 4C) and found that Collagen1 expression per cell was increased in response to PDGF-AA (Fig 4D,E).
Fig 4. PDGF controls FAP differentiation.

A, COL1A1 expression in PDGFRα+CD90+ and PDGFRα−CD90+ FAPs (n=4, study 2, non-T2DM); B, PDGFRα expression in COL1A1+ and COL1A1low/−; C, Workflow; D-E, Collagen1 protein in control or PDGF-AA stimulated FAPs (n=5, study 2, non-T2DM, scalebar 50 μm); F, Workflow; G-H, Perilipin1 expression in adipogenic control or PDGF-AA treated FAPs (n=6, study 2, non-T2DM, scalebar 50 μm); I, COL1A1 expression in control or PDGF-AA treated adipogenic FAPs (n=4, study 2, non-T2DM); J, Work-flow; K-L, Perilipin1 expression in control or Imatinib treated adipogenic FAPs (n=4, study 2, non-T2DM); M+N, FAP lactate production and glucose consumption in response to 3 days PDGF-AA treatment (n=5, study 2, non-T2DM); O, Work-flow; P, GlycoPER (pmol/min) in adipogenic or PDGF-AA treated FAPs (n=7, technical replicates, study 2, non-T2DM); Q+R, Basal and maximal glycoPER (pmol/min); S, GlycoPER control or PDGF-AA treated adipogenic FAPs; T, Maximal glycoPER in control or PDGF-AA treated adipogenic FAPs; T+V, Pathways enriched in FAP adipogenic or fibrogenic induced FAPs, respectively (study 2, non-T2DM); Cells from m. Rectus Abdominus. *p<0.05.
In addition to collagen production, we confirmed the mitogenic effect of PDGF-AA in our human FAPs (Fig S5A). In mice, constitutively active PDGF-signaling is associated with widespread fibrosis, also in adipose tissue (Marcelin et al., 2017; Olson and Soriano, 2009). Thus, we next asked whether PDGF-AA could affect adipogenic differentiation of human FAPs. To address this, we cultured FAPs in adipogenic medium, while exposing them to PDGF-AA (Fig 4F). Interestingly, PDGF-AA markedly reduced the ability to form Perilipin-1+ adipocytes from human FAPs (Fig 4G,H) while maintaining/inducing a more fibrogenic nature as evidenced by increased collagen expression (Fig 4G,I). Conversely, when inhibiting the PDGF pathway, using a tyrosine-kinase inhibitor (Imatinib), we could further increase FAP adipogenesis (Fig 4J,K,L). Collectively, our data reveal that PDGF signaling is a key determinant of human FAP cell fate, by promoting the expansion of FAPs with fibrogenic potential at the expense of the adipogenic fate. Considering these findings, it is interesting to speculate as to what the cellular source of PDGF-AA or PDGF-CC is in vivo. From our population-based RNA-seq data we found that the mature muscle fibers are particular high in PDGFA (Fig S5B) and less in PDGFC (Fig S5C). Moreover, examining our scRNA-seq data set indicate that also smooth muscle cells/pericytes may be a source of this mitogen (Fig S5D) in human skeletal muscle.
Increased reliance on glycolysis and lactate production is associated with FAP fibrogenesis
In addition to its role in fibrosis, PDGF-signaling has been associated with increased cellular reliance on glycolysis and lactate production for energy and substrate generation (Xiao et al., 2017). Thus, we investigated the potential relationship between metabolism and PDGF-signaling in cell fate determination of human FAPs (Study 2, non-T2DM). Exposure to PDGF increased lactate production (Fig 4M) and glucose consumption (Fig 4N), in human FAPs, as compared to untreated cells. This prompted us to do a more detailed metabolic profiling using real-time bioenergetics analysis. Before bioenergetic analysis FAPs were induced for 24h towards adipogenesis or fibrogenesis (PDGF-AA stimulation, Fig 4O). We found no difference in basal or maximal oxygen consumption (Fig S5E). In contrast, we observed a robust difference in basal and maximal glycolysis (Fig 4P,Q,R), suggesting a functional link between activation of glycolysis (lactate fermentation) and the fibrogenic phenotype of human FAPs. When adding PDGF to FAPs induced for adipogenesis for 24h this also increased maximal glycolysis (Fig 4S,T) suggesting that this metabolic switch may also be involved in the concurrent reduction of adipogenesis in FAP stimulated with PDGF.
Since no previous studies have isolated human skeletal muscle FAPs without prior culture, limited knowledge exists on the transcriptome of human FAPs undergoing differentiation. To provide greater detail of this process we performed RNA-seq on FAPs cultured for six days in adipogenic or fibrogenic (PDGF stimulation) differentiation media. Indeed, altered transcripts related to the PPARα, a major lipid metabolism regulator (Derosa et al., 2018; Pawlak et al., 2015), were enriched in the adipogenic FAPs (Fig 4U; S5F), along with common genes enriched during adipogenic differentiation (Fig S5G,H,I,J). By contrast fibrogenic FAPs exhibited strong enrichment for gene implicated in ECM production, turnover and signaling (Fig 4V, S5F). Interestingly, in fibrogenic FAPs, we noted a strong enrichment in Periostin (POSTN, Fig S5F), an extracellular matrix protein acting as a ligand and previously associated with fibrosis and wound healing (Walker et al., 2016). More recently, Periostin has been shown to increase in activated FAPs during regeneration in mouse skeletal muscle along with lysyl oxidase enzymes (LOX) (Scott et al., 2019). Notably, LOXL4 was the most differentially expressed gene in our fibrogenic FAPs (Fig S5F). This finding strongly supports the ability of PDGF signaling to stimulate a fibroblast or myofibroblast conversion of FAPs.
We next speculated if different subsets of FAPs exist in vivo with varying ability to transform into adipocyte or fibroblasts (Malecova et al., 2018; Merrick et al., 2019) based on PDGF signaling. To address this question, we took advantage of our scRNA-seq data. We re-clustered the FAP population (Fig 3B, FAPs 1–4), which resulted in seven subpopulations (Fig 5A, S5K), from which we generated a heatmap of the top-twenty differentially expressed genes in each cluster (Fig S5L). We immediately noted the high density of THY1 (CD90) positive cells mainly present in the cluster 2 (Fig S5L–N). This was of interest, since we had already noted that CD90 marks a subset of FAPs (Fig 2A ii). We therefore decided to investigate if the expression of CD90 defined a specific human FAP population with a distinct phenotype.
Fig 5. FAPs expressing THY1 (CD90) represent a distinct subpopulation.

A, UMAP of FAPs with interpolated lineage prediction (n=4, study 2); B, Cell size of MuSCs, FAPCD90− and FAPCD90+ (n=16, study 2, non-T2DM); C, EdU+ of FAPCD90− and FAPCD90+ post FACS (n=9–13, study 2, non-T2DM); D, Limiting dilution assay of FAPCD90− and FAPCD90+ (n=3), solid line is non-linear fit; E+F, % adipogenic or fibrogenic clones from single sorted FAPCD90−/+; G, PCA-plot based on DE genes in FAPCD90− and FAPCD90+ (n=3, study 2, non-T2DM); H+I, Biological processes in FAPCD90− and FAPCD90+, respectively (n=3, study 2, non-T2DM); J+K, Collagen-1 protein expression post sort (n=5, study 2, non-T2DM); L, Work-flow; M, OCR (pmol/min) of MuSCs, FAPCD90− and FAPCD90+ (n=7, study 2, non-T2DM); N, ECAR, (mpH/min) of MuSCs, FAPCD90− and FAPCD90+; O, EACR/OCR ratio of MuSCs, FAPCD90− and FAPCD90+; P, Skeletal muscle IHC of CD34 (Red), CD90 (Cyan) and WGA (green). Blue arrow is CD34+ CD90+, red arrows is CD34+CD90−. Cells from mm. Rectus Abdominus/Gastrocnemius. *p<0.05.
THY1 marks a specific subpopulation of skeletal muscle FAPs in vivo
To determine potential clinical relevancy, we first compared DE genes in T2DM (itT2D group) with DE genes upregulated in FAPCD90+ and FAPCD90− (Fig S6A). This revealed that genes upregulated in FAPCD90− tended to be downregulated in itT2D, whereas genes upregulated in itT2D tended to be upregulated in FAPCD90+ (Fig S6A). This indicates that FAPCD90+ may have a central role in the development of the fibro-fatty phenotype observed in itT2D muscle.
To understand the nature of FAPCD90+, we extracted and re-analyzed all FAPs from the initial scRNA-seq analysis and performed slingshot pseudo-time inference analysis to predict lineage patterns within the FAP population (Fig 5A, S5O). This indicated an origin in cluster two, where a high proportion of THY1 positive cells were observed. The lineage then progressed through cluster one, three and four. From here, two lineage trajectories arose progressing into either cluster six (CD90− cells) or cluster five and zero (mix of CD90− and CD90+ cells) (Fig 5A, S5O). Thus, FAPs expressing CD90 could represent progenitor or stem cell subpopulation within the larger pool of FAPs. To investigate this further we inspected our flow-data and noted that FAPCD90+ (in patients without T2DM) were larger in cell size than FAPCD90− (Fig 5A, Study 2). We then asked if the larger cell size could be related to the higher mitotic activity of FAPCD90+. Following isolation both FAPCD90+ and FAPCD90− were completely EdU negative for the first 24h (Fig 5B), suggesting that both were quiescent in vivo. However, at 48h, 72h and in particular 96h, the FAPCD90+ displayed an increased number of EdU+ cells compared to FAPCD90−, suggesting that FAPCD90+enter the cell cycle more rapidly than FAPCD90− (Fig 5B). As such, although inactive in vivo, FAPCD90+ have an increased propensity to enter the cell cycle, which is associated with a larger cell size. This finding suggests that FAPCD90+ could be a subset of progenitor cells within the FAP population that are poised for entry into cell cycle and further fibrogenic activation, as proposed for cardiac FAPs (Soliman et al., 2020). To examine this further, we decided to perform the limiting dilution and single-cell clonal assay. In agreement with our EdU data, FAPCD90+ had a greater level of clonality/progenitor formation than FAPCD90− (Fig 5C, S6B) and the single-hit Poisson model predicted a robust difference in stem cell frequency between the two FAP populations. In contrast, the clones formed by FAPCD90− had a greater proportion of clones becoming adipogenic, while in FAPCD90+ the content of adipogenic and non-adipogenic clones was similar (Fig 5D,E i–ii, S6C,D). Collectively, our data support a model in which FAPCD90+ have a progenitor phenotype, whereas FAPCD90− are prone towards adipocyte differentiation or have other stromal support functions.
FAPCD90+ are poised for extracellular matrix production associated with an increased glycolytic flux
Given the phenotypic difference between the two FAP populations, we decided to directly compare the population-based transcriptome of sorted FAPCD90− and FAPCD90+ cells. PCA confirmed the distinct transcriptomes of the two populations (Fig 5G), supporting that CD90 expression defines a unique population in agreement with the scRNA-seq data. Gene set enrichment analysis revealed a marked enrichment in processes related to angiogenesis and regulation of endothelial cell proliferation/migration in FAPCD90− (Fig 5H, S5E). In contrast, FAPCD90+ genes were enriched in processes related to ECM development, collagen formation and response to TGFβ (Fig 5I, S5E). We confirmed this using our generated scRNA-seq data by dividing the FAPs into CD90+ and CD90− (Fig S6F,G). To validate our transcriptomic findings, we flow-sorted FAPCD90− and FAPCD90+ and stained for Collagen1 24h post sort (Fig 5J,K). As expected, FAPCD90+ showed a higher Collagen1 protein expression compared to FAPCD90− (Fig 5J,K).
We next sought to understand if the phenotype of FAPCD90+ was associated with increased activity of PDGF-signaling in FAPCD90+. Indeed, from population-based RNA-seq we found increased expression of genes related to PDGF-signaling in FAPCD90+ (Fig S6G). This was corroborated by increased phosphorylation of PDGFRα in freshly sorted FAPCD90+ compared to FAPCD90− (Fig S6H). Finally, freshly sorted FAPCD90+ were resistant to further PDGF-AA stimulation, whereas FAPCD90− did increase EdU incorporation in response to PDGF-AA (Fig S6 I), indicating that further PDGF-signaling in FAPCD90+ does not increase proliferation at this stage. Thus, PDGF signaling is increased in FAPCD90+ compared to FAPCD90− in vivo. Given the PDGF-phenotype of FAPCD90+ and the metabolic changes associated with PDGF-AA treatment, we sought to further investigate FAPCD90+ metabolism ex vivo. To ensure that FAPs were as close as possible to in vivo, we performed the bioenergetic analysis 10–12h post isolation, a time at which none of FAP populations would be engaged in cell cycle (Fig 5L). At baseline, FAP populations were similar in oxygen consumption (Fig 5M; Fig S6G). In contrast, glycolysis was greater in FAPCD90+ compared to FAPCD90− (Fig 5N) supporting the hypothesis that ECM producing FAPCD90+ phenotype is associated with an increased reliance on glycolysis. This was supported by greater expression of LDHA in FAPCD90+ compared to FAPCD90− (Fig S6H). The difference in glycolysis was further exacerbated when calculating a glycolysis/oxygen consumption ratio (Fig 5O). In addition, maximal oxygen consumption was also higher in FAPCD90+ compared to FAPCD90− (Fig 5 M; S6H), suggesting a higher capacity for energy generation in FAPCD90+.
Staining for CD90 and CD34 in skeletal muscle sections confirmed the presence of both CD90+CD34+ and CD90−CD34+ cells (Fig 5P) in vivo in the muscle tissue, excluding that these are circulating cells, although we could unfortunately not combine CD90 and CD34 with CD31 to exclude endothelial cells from the stain. In summary, at least two distinct population of FAPs exist in human skeletal muscle with FAPCD90+ being more primed for producing progeny and delivering ECM than FAPCD90− (Fig S6I).
FAPCD90+ accumulate in skeletal muscle of T2DM patients
Based on the phenotype of FAPCD90+ we next hypothesized that FAPCD90+ would accumulate in muscles of T2DM patients and contribute to the muscle degeneration. We collected biopsies from patients (with or without T2DM) undergoing either abdominal aneurysm or coronary bypass surgery (Supplementary Table 1). When comparing total FAP content, we noted that patients with T2DM tended to have a higher content of FAPs (Fig 6A,B). However, the difference between patients with and without T2DM became even more pronounced, when we divided FAPs into FAPCD90+ and FAPCD90− and noted a selective increase in FAPCD90+ (Fig 6C–E). In fact, the only population in which we identified significant changes was FAPs and in particular FAPCD90+ (Fig S6 J–M). These findings support a specific upregulation of FAPCD90+ in T2DM. PDGF has previously been shown to be increased in T2DM (Abderrahmani et al., 2018; Marcelin et al., 2017) and our data suggest that this is a modulator of human FAPs, and specifically FAPCD90+.
Fig 6. FAPCD90+ accumulate in T2DM and respond to metformin treatment.
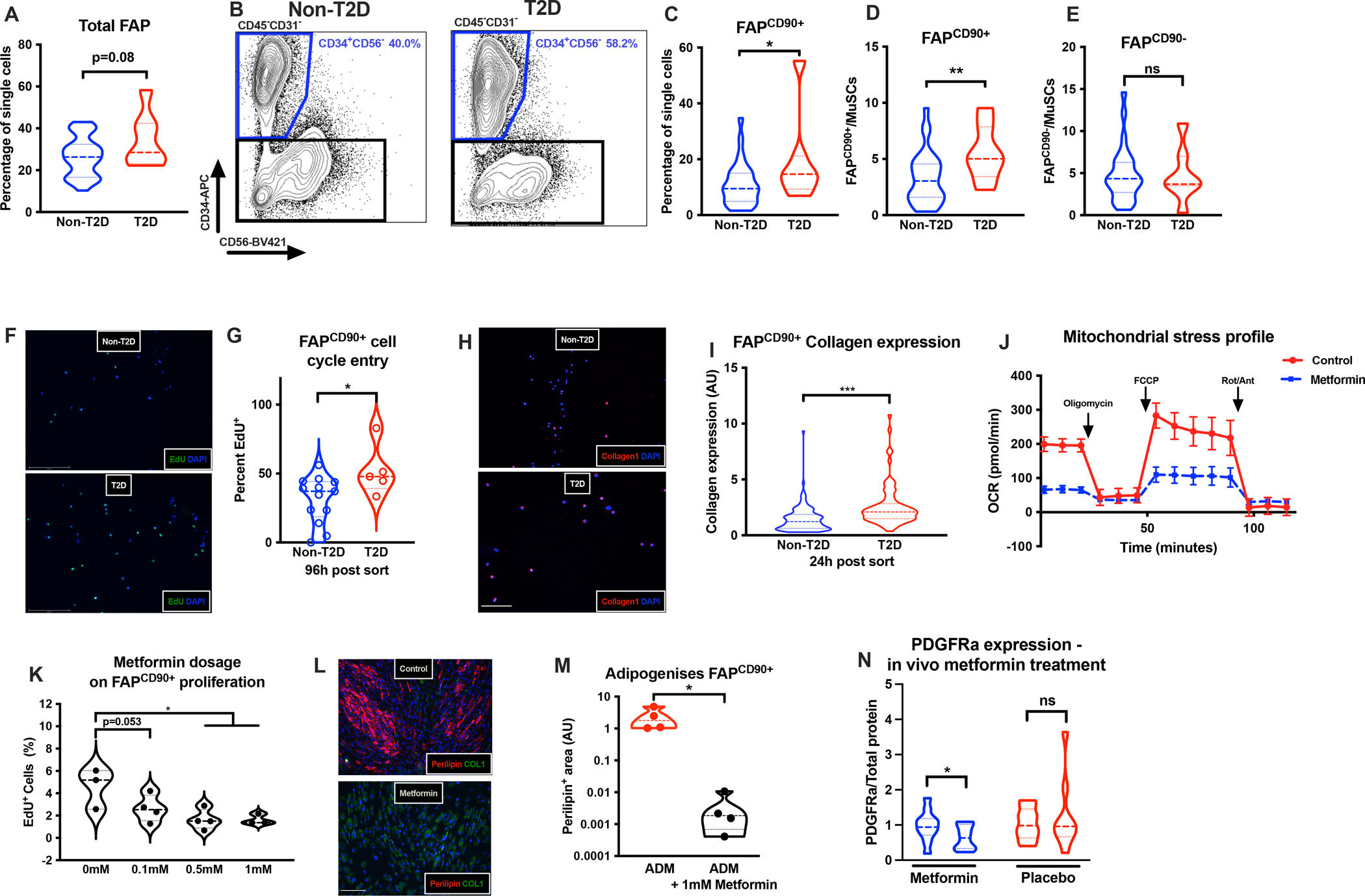
A+B, Total FAP content in non-T2DM (n=27–33, study 2) or T2DM (n=7–8) patients; C, % FAPCD90+ in non-T2DM and T2DM patients; D+E, FAPCD90+ and FAPCD90− (per MuSC) content, respectively, in non-T2DM and T2DM; F+G, EdU+ FAPs in non-T2DM and T2DM (n=4–12, study 2); H+I, Collagen1 expression in FAPCD90+ in non-T2DM or T2DM (scalebar 50μm, n=2–3, study 2); J, OCR (pmol/min) on control or metformin treated FAPCD90+ (24h, n=5–6, technical replicates, study 2); K, EdU+ FAPCD90+ following control or 0.1mM, 0.5mM or 1 mM metformin treatment; L-M, Adipogenesis in control or metformin treated FAPCD90+ (scalebar 50μm); N, PDGFRα expression pre/post metformin or placebo treatment (n=11–12, Study 3). Cells from mm. Rectus Abdominus/Gastrocnemius. *p<0.05, ** p<0.01, ***p<0.001.
Two other candidates for controlling FAP fate, given their central role in T2DM, is glucose and insulin. Interestingly, we found that insulin does increase FAP proliferation in general (Fig S6N) as well as first entry into the cell cycle post FACS isolation (Fig S6O). In contrast, we found no effect of high glucose on FAP cell-cycle entry (Fig S6O). Importantly, we found that flow-sorted FAPCD90+ from patients with T2DM showed higher propensity to enter cell cycle (Fig 6F,G) and displayed an increased Collagen1 expression 24h post sort (Fig 6H,I), as compared to FAPs from patients without T2DM. This suggests that T2DM muscles provide a permissive environment for expansion of FAPCD90+, leading to their accumulation and subsequent fibro-fatty degeneration.
FAPCD90+ can be targeted with metformin to decrease proliferation and inhibit adipogenesis
As a final question we asked if we could pharmacologically target FAPCD90+ to prevent excessive FAP accumulation in muscles of T2DM patients. Several receptor tyrosine kinase inhibitors have been shown to effectively reduce FAP proliferation in mice including nilotinib and imatinib (Fiore et al., 2016; Lemos et al., 2015). The latter is interesting since imatinib is transported across the cell membrane using cation transporters (SLC22A1/SLC22A3) that are also essential for transport of the T2DM drug; metformin (Sundelin et al., 2017). Metformin is, among other mechanisms, thought to increase cellular AMPK levels by inhibiting complex 1 in the respiratory chain and thereby limiting ATP production from oxidative phosphorylation (Zhou et al., 2001). To examine if FAPCD90+ metabolism was responsive to metformin treatment, we incubated FAPCD90+ with metformin (1mM) for 24 hours prior to measuring oxygen consumption and glycolytic rate (no metformin in the assay media). As predicted metformin markedly reduced oxygen consumption in FAPCD90+ (Fig 6J). Conversely, metformin increased glycolysis in FAPCD90+ (Fig S6P). Considering these findings, we predicted that metformin would also efficiently lower FAPCD90+ proliferation. Indeed, when exposing FAPCD90+ to a titration of metformin dosages cell proliferation decreased (Fig 6K). We therefore speculate that metformin may also reduce FAPCD90+ expansion in vivo. Moreover, metformin completely blocked the ability of FAPCD90+ to form adipocytes (Fig 6L, M), while increasing Collagen1 expression (Fig S6Q). However, although metformin may enhance reliance on glycolysis and Collagen1 production, the major effect on FAP proliferation is likely to reduce the overall development of fibrosis. To understand if metformin indeed decrease FAP proliferation in vivo, we obtained biopsy material from T2DM patients and matched controls randomized to three months of metformin or placebo treatment. Importantly, before treatment FAP content was indeed increased among these newly diagnosed T2DM patients compared to healthy controls (Fig S6S), confirming our results in Fig 1J. Secondly, three months metformin treatment reduced FAP content in T2DM patients (Fig 6N). Thus, our data support that metformin may modify the FAP response in T2DM patients and reduce activity of FAPCD90+ to ultimately reduce tissue fibrosis.
DISCUSSION
In the present study, we show that human skeletal muscle contains several populations of transcriptionally and functionally distinct FAPs and that a specific subpopulation of the FAPs is a key mediator of the muscle fibro-fatty degeneration observed among patients with T2DM. The identification of the cellular drivers of this fibro-fatty degeneration provides the opportunity to target these cells to ultimately preserve muscle health and function.
As a proof-of-concept we included two groups of T2DM patients, to display how the muscle remodeling is altered in the context of less (T2D) and more severe (itT2D) states of insulin resistance and T2DM. We initially found the ECM remodeling to be particular evident in patients treated with high doses of insulin and with a long disease duration (itT2D group). This could indicate that itT2D patients may represent a sub-group of patients with severe insulin resistance, which are more prone to diabetic complications. Severe insulin resistance is a defining feature of a sub-group of T2DM patients identified earlier in an unbiased approach (Ahlqvist et al., 2018). This group of patients carry an increased risk for complications such as kidney fibrosis and ultimately kidney failure (Ahlqvist et al., 2018). Interestingly, kidney failure in T2DM patients is also associated with elevated whole-body collagen production (Collagen 6 formation (Rasmussen et al., 2018)), which is also highly expressed in skeletal muscle FAPs. The itT2D patients have many similarities to the insulin resistant patient cluster (Ahlqvist et al., 2018) and may carry a similar risk profile for diabetic complications such as fibro-fatty degeneration of skeletal muscle as well as kidney failure.
Despite greater fibro-fatty remodeling in itT2D patients, protein expression of PDGFRα in muscle homogenates, indicative of FAP content, was only increased in the T2D group with a shorter disease duration. In agreement, we found increased FAP content (PDGFRα expression) in another cohort of early stages of T2DM patients (S6X) (Gormsen et al., 2018). The lower FAP content in itT2D patients likely reflects differentiation of FAPs into myofibroblasts and adipocytes, a process in which FAPs reduce expression of PDGFRα (Contreras et al., 2019). This is supported by extensive fibrogenic remodeling observed in itT2D patients. As such, muscle remodeling as a function of disease duration is consistent with progression from initial increased FAP content in T2D, to FAP differentiation in itT2D. Earlier reports have shown muscle fibro-fatty remodeling to be present in obese pre-diabetic subjects at whole tissue level (Goodpaster et al., 2000; Kang et al., 2011; Miljkovic et al., 2009). Thus, it is possible that expansion of the FAP pool is initiated at very early stages of the disease and that the degenerative profile in itT2D patients is related to longer disease duration with differentiation of the expanded FAP pool (Gormsen et al., 2018; Kampmann et al., 2011a). None-the-less, still we view the fibro-fatty remodeling as a complication to T2DM, rather than an underlying etiology to the initial development of insulin resistance and later T2DM (Warram et al., 1990).
In addition to identifying CD34+ FAPs we show that that mitogen; PDGF-AA increases FAP proliferation and collagen production, while also effectively reducing FAP adipogenesis. Importantly, the in vivo identified FAPCD90+ sub-population carried a phenotype with similar traits; i.e. enrichment for ECM production, high ability to proliferate and increased reliance on aerobic glycolysis. Moreover, clones derived from FAPCD90+ were less adipogenic, consistent with the metabolic phenotype (Xiao et al., 2017). This effect is likely not restricted to PDGF-AA but true for other stimulators of ECM protein formation such as TGFβ1 (Zhao et al., 2019) thereby linking ECM protein synthesis to enhanced reliance on glycolysis. Increasing glycolytic flux and lactate production not only delivers ATP, but is also key for essential aspects of collagen formation such as glycine synthesis and collagen hydroxylation (de Paz-Lugo et al., 2018; Zhao et al., 2019). In contrast, cell proliferation does not seem to be dependent on glycolysis per se (Ran et al., 2013).
The novel finding that human FAPs consist of at least two transcriptionally, metabolically and functionally distinct subsets is particular intriguing since we found these subsets to be altered in the context of T2DM. To our knowledge, this is the first time that functionally distinct FAP subpopulations have been described in human skeletal muscle and furthermore shown to be altered in the context of disease. Subpopulations of muscle FAPs have been described in mice in which VCAM1+ FAPs increased in acute and chronic muscle injury (Malecova et al., 2018). While the functional role of these subpopulation in mice are yet to be clearly defined, the VCAM1+ FAPs were associated with a pro-proliferative and pro-fibrotic phenotype (Malecova et al., 2018). We did not find enrichment for VCAM1 protein expression in neither of the FAP populations described here (data not shown), suggesting that the phenotypes observed in our samples are different from those seen in muscle injury per se.
In conclusion, human skeletal muscle FAPs are the primary cell population responsible for fibro-fatty degeneration. In vivo, human skeletal muscle contains a subpopulation of FAPs marked by expression of CD90. This population is highly primed for entering the cell cycle, producing clonal progeny, ECM production and increased utilization of glycolysis, likely driven by PDGF. FAPCD90+ accumulate in muscles from T2DMs and constitute a likely driver of the fibrotic remodeling of the muscle niche. Finally, the proliferative FAPCD90+ can be targeted with metformin in vitro and three months of in vivo metformin treatment in T2DM patients lowers the skeletal muscle FAP content.
LIMITATIONS OF STUDY
In this study we used several independent T2DM cohorts to cover different aspects of this complicated condition, but the selection and sizes of these cohorts pose limitations to the extent to which aspects of T2DM are covered by this study. Therefore, to further explore FAPs in disease settings larger cohorts of patients are needed in order to solidify the role of the FAPs in T2DM and the consequences for muscle function. Importantly, the present study has provided the framework to investigate such hypothesis in well-controlled clinical settings. Moreover, while we aimed to keep the FAP phenotype as close to in vivo as possible, we cannot exclude the possibility for in vitro derived effects on the isolated FAPs.
STAR METHODS
RESOURCES AVAILABLE
Lead contact
Further information, not already presented in the figures, and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Niels Jessen (niels.jessen@biomed.au.dk).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The whole muscle, population-based and single-cell RNA-seq data reported in this study are available at the European Genome-phenome Archive (EGA, https://ega-archive.org). The Accession numbers are listed in the key resource table. Microscopy data, original western blot images etc. are available from the Lead Contact on request.
No original code was produced in this paper. Code for bioinformatics analyses on whole muscle, population and single-cell RNA-seq is available through download of the specified (key resource table/STAR Methods), publicly available packages.
Any additional information required to reanalyse the data reported in this paper is available from the Lead Contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Collagen 1 (mouse) | Sigma | Cat# C2456, RRID:AB_476836 |
| Anti-Perilipin-1 (rabbit) | Cell Signaling Technologies | Cat# 9349, RRID:AB_10829911 |
| Anti-CD34 (rabbit) | Abcam | Cat# ab81289, RRID:AB_1640331 |
| Anti-PDGFRa (goat) | R&D Systems | Cat# AF-307-NA, RRID:AB_354459 |
| Anti-PDGFRa (recombinant, rabbit) | Abcam | Cat# ab134123 RRID: N/A |
| TE-7 (mouse) | Merck Millipore | Cat# CBL271, RRID:AB_93449 |
| Anti-Pax7 (mouse) | Developmental Studies Hybridoma Bank | Cat# pax7, RRID:AB_528428 |
| Anti-Desmin (rabbit) | Cell Signalling Technologies | Cat# 5332, RRID:AB_1903947 |
| Anti-Myosin Heavy Chain (mouse) | Developmental Studies Hybridoma Bank | Cat# MF 20, RRID:AB_2147781 |
| Anti-alpha-smooth muscle actin (mouse) | Sigma | Cat# A5228, RRID:AB_262054 |
| Anti-pPDGFRa (Y754) (rabbit) | Abcam | Cat# AB5460, RRID:AB_ 304906 |
| Anti-Collagen 1 (rabbit) | Meridian Life Science Inc | Cat# T40777R, RRID:AB_151099 |
| Anti-Collagen 3 (rabbit) | Genway Biotec Inc | Cat# GWB-7D650E, RRID:AB_10517236 |
| Anti-human-CD45-FITC | Miltenyi Biotec | Cat# 130-114-567, RRID:AB_2726699 |
| Anti-human-CD31-FITC | Miltenyi Biotec | Cat# 130-110-668, RRID:AB_2657279 |
| Anti-human-CD90-PE | Thermo Fisher Scientific | Cat# 12-0909-42, RRID:AB_10670624 |
| Anti-human-CD56-BV421 | BD Bioscience | Cat# 562751, RRID:AB_2732054 |
| Anti-human-CD82-PE-vio770 | Miltenyi Biotec | Cat# 130-101-302, RRID:AB_2659303 |
| Anti-mouse-CD34-APC | BD Bioscience | Cat# 555824, RRID:AB_398614 |
| Goat-anti-mouse IgG (H+L) Alexafluor 647 | Thermo Fisher Scientific | Cat# A-21235, RRID:AB_2535804 |
| Goat-anti-rabbit IgG (H+L) Alexafluor 568 | Thermo Fisher Scientific | Cat# A-11011, RRID:AB_143157 |
| Goat-anti-mouse IgG (H+L) Alexafluor 488 | Thermo Fisher Scientific | Cat# A-11001, RRID:AB_2534069 |
| Donkey-anti-goat IgG (H+L) Alexafluor 568 | Thermo Fisher Scientific | Cat# A-11057, RRID:AB_2534104 |
| Goat-anti-rabbit IgG (H+L) HRP | Thermo Fisher Scientific | Cat# 31460, RRID:AB_228341 |
| Goat-anti-mouse IgG (H+L) HRP | Thermo Fisher Scientific | Cat# G-21040, RRID:AB_2536527 |
| Patient characteristics presented in supplementary table 1 (study 2) | This paper | N/A |
| Patient characteristics from study 1 | Kampmann, U. et al. 2011a | N/A |
| Patient characteristics from study 3 | Gormsen, L.C. et al. 2018 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Propidium Iodide | BD Bioscience | Cat# 556463 |
| Hams F10 media incl. glutamine and bicarbonate | Sigma | Cat# N6908 |
| Horse serum | Thermo Fisher Scientific | Cat#26050088 |
| 100X Penstrep | Thermo Fisher Scientific | Cat#15140122 |
| Collagenase II | Worthington | Cat#46D16552 |
| Dispase II | Roche Diagnostics | Cat#04 942 078 001 |
| StemMACS cryopreservation buffer | Miltenyi Biotec | Cat#130-109-558 |
| Human FcR blocking solution | Miltenyi Biotec | Cat#130-059-901 |
| Compensation beads | Thermo Fisher Scientific | Cat#01-2222-41 |
| Collagen | Sigma | Cat#C8919 |
| Laminin | Thermo Fisher Scientific | Cat#23017-015 |
| Fetal Bovine serum | Thermo Fisher Scientific | Cat#16000044 |
| Recombinant human bFGF | Sigma | Cat# F0291 |
| DMEM (4.5 g/L glucose) incl glutamine and bicarbonate | Thermo Fisher Scientific | Cat#11965092 |
| Adipogenic differentiation media | Miltenyi Biotec | Cat#130-091-677 |
| Extra-cellular matrix coating gel | Sigma | Cat# E1270 |
| Poly-D-Lysine | Merck Millipore | Cat#A-003-E |
| Bio-AMF 2 growth media | Biological Industries | Cat#01-194-1A |
| Recombinant human Insulin | Sigma | Cat#91077C |
| Dexamethasone | Sigma | Cat# D4902 |
| 3-isobutyl-1-methylxanthine (IBMX) | Sigma | Cat# I7018 |
| Rosaglitazone | Sigma | Cat# R2408 |
| Recombinant human TGFb | Sigma | Cat# T7039 |
| Recombinant human PDGF-AA | Miltenyi Biotec | Cat#130-108-983 |
| Metformin | Sigma | Cat#D150959 |
| Imatinib | Sigma | Cat#SML1027 |
| Osteogenic differentiation media | Miltenyi Biotec | Cat#130-091-678 |
| Histofix | Histolabs | Cat#01000 |
| Normal Goat serum | Thermo Fisher Scientific | Cat31873 |
| Wheat-germ-agglutinin | Thermo Fisher Scientific | Cat# W1126 |
| Alzirian red staining solution | Sigma | Cat# A5533 |
| Phenol red and bicarbonate free DMEM Seahorse Media | Agilent | Cat#103575-100 |
| Glucose | Sigma | Cat#G8769 |
| Sodium-pyruvate | Thermo Fisher Scientific | Cat#11360-039 |
| L-glutamine | Sigma | Cat# G7513 |
| Hoechst 33342 | Thermo Fisher Scientific | Cat# H3570 |
| 2-Deoxy-glucose | Sigma | Cat# D8375 |
| Oligomycin | Sigma | Cat#75351 |
| Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) | Sigma | Cat#C2920 |
| Antimycin | Sigma | Cat#A8674 |
| Rotenone | Sigma | Cat#R8875 |
| Critical commercial assays | ||
| Click-iT™ EdU cell proliferation kit for imaging | Thermo Fisher Scientific | Cat#C10337 |
| Click-iT™ EdU cell proliferation kit for imaging | Thermo Fisher Scientific | Cat#C10340 |
| PrimeFlow™ RNA assay kit | Thermo Fisher Scientific | Cat#88-18005-210 |
| QiaSymphony RNA Mini kit | Qiagen | Cat#931636 |
| Seahorse Mito-stress test kit | Agilent | Cat#103015-100 |
| Takara SMARTer Stranded Total RNA-Seq Kit v2 pico kit | Takara Bio | Cat#634411 |
| TruSeq. Stranded mRNA Sample preparation kit | Illumina Inc | Cat#20020594 |
| Chromium Single Cell 3’ Reagent Kits (v3 Chemistry) | 10X Genomics | Cat#1000075 |
| Deposited data | ||
| Population-based RNA-sequencing data | This paper www.ega-archive.org | EGAS00001005599 |
| Single cell RNA-sequencing data | This paper www.ega-archive.org | EGAS00001005599 |
| Whole muscle RNA-sequencing data | This paper www.ega-archive.org | EGAS00001005599 |
| Oligonucleotides | ||
| B2M: Sense AATGTCGGATGGATGAAACC, Antisense TCTCTCTTTCTGGCCTGGAG | HotStar Taq, Qiagen | N/A |
| TGFB1: Sense GGACACCAACTATTGCTTCAGCTC, Antisense AAGTTGGCATGGTAGCCCTTGG | HotStar Taq, Qiagen | N/A |
| COL1A1: Sense TGCGATGACGTGATCTGTGACG, Antisense TTTCTTGGTCGGTGGGTGACTCTG | HotStar Taq, Qiagen | N/A |
| COL3A1: Sense ATTGCTGGGATCACTGGAGCAC, Antisense CCTGGTTTCCCACTTTCACCCTTG | HotStar Taq, Qiagen | N/A |
| COL6A1: Sense ATCAGCCAGACCATCGACACCATC, Antisense TTCGAAGGAGCAGCACACTTGC | HotStar Taq, Qiagen | N/A |
| COL1A1 (PrimeFlow type 4 probe) | Thermo Fisher Scientific | NM_000088.3 |
| RPL13A (PrimeFlow type 1 probe) | Thermo Fisher Scientific | NM_001270491.1 |
| Software and algorithms | ||
| ImageJ (for fluoresent image analysis) | NIH | https://imagej.nih.gov/ij/ |
| FlowJo (10.6.1) for FACS and flow cytometry analysis | BD Bioscience | https://www.flowjo.com/ |
| Prism 8 (version 9.0) for data presentation and statistics | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Limiting dilution Analysis | Walter and Eliza Hall Institute of Medical Research | http://bioinf.wehi.edu.au/software/limdil/ |
| Trimgalore (v 0.4.1) for adapter and quality trimming of RNA sequencing reads | Babraham Bioinformatics, Babraham Institute | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Hisat2 (v 2.1.0) for alignment of RNA sequencing reads to the reference genome | Kim D et al., 2015 | http://daehwankimlab.github.io/hisat2/ |
| Samtools (v 1.9) for sorting the alignment files | Li H et al., 2009 | http://www.htslib.org/ |
| htseq-count (v 0.6.1) for feature number estimation (gene counts) | Anders S et al., 2014 | https://htseq.readthedocs.io/en/master/count.html |
| R (v 4.0.3) for bioinformatics and statistical analysis | The R project for statistical computing | https://www.r-project.org/ |
| R package DESeq2 (v 1.26.0) for differential expression analysis of RNAseq data | Love MI., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| R package Seurat (v 4.0.1) for scRNAseq analysis | Hao et al., 2020 | https://satijalab.org/seurat/index.html |
| R package Doubletfinder (v 2.0.3) for doublet removal in the scRNAseq dataset | McGinnis CS et al., 2019 | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| R package XGR (v 1.1.7) for gene set and pathway enrichment analysis of differentially expressed genes | Fang et al., 2016 | http://galahad.well.ox.ac.uk:3030/ |
| R package Slingshot (v. 1.8.0) for pseudotime inference analysis | Street et al., 2018 | https://bustools.github.io/BUS_notebooks_R/slingshot.html |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Ethical approval
All participants and patients were given oral and written information, and gave written consent to participate in accordance with the declaration of Helsinki. The studies were approved by the Local Ethical Committee of Central Denmark Region.
Patients and protocol
Study one included seven age-matched (six males and one female; age (mean±SE): 59±2 years; BMI (mean±SE): 28.0±1.5 kg/m2) healthy overweight subjects (OBS), seven (three males and four females; age (mean±SE): 64±3 years; BMI (mean±SE): 31.8±2.4 kg/m2) type 2 diabetic patients (T2D) and six (four males and two females; age (mean±SE): 59±3 years; BMI (mean±SE): 35.7±2.1 kg/m2) insulin treated type 2 diabetic patients (itT2D)). Patients had their oral antidiabetic treatments (metformin) withdrawn two day before the study and their usual insulin treatment were replaced with a continuous infusion of short acting insulin (Actrapid, Novo Nordisk, Denmark) and glucose one day before the study. The rates of insulin and glucose infusions were adjusted to reach a plasma glucose level of 8 mM. The samples were collected in 2008–2010, as part of a clinical trial (ID: NCT00654056), and the data analyses related to this paper were performed post-hoc. More detailed patient information is described previously (Kampmann et al., 2011a; Kampmann et al., 2011b).
Study two included patients admitted to aortic aneurism surgery (m. rectus abdominis) or coronary artery by-pass surgery (CABG, m. gastrocnemius). Inclusion criteria were age >50 years and <80 years and BMI >20. Patients with Type 1 diabetes, Polymyalgia, Lipodystrophy, active chemo or radiation therapy, Thiazalidinedione treatment, current haemodialysis treatment or glucocorticoid treatment with six months prior to inclusion were excluded from participation. Furthermore, patients were classified into patients without T2DM (31 males and 2 females; age (mean [SD]): 71 [7] years; BMI (mean [SD]): 27.8 [3.5] kg/m2) and T2DM (8 males and 0 female; age (mean [SD]): 67 [5] years; BMI (mean [SD]): 29.1 [2.2] kg/m2) patients. The samples were collected in 2017–2020 and the data analyses related to this paper were primary endpoints in this trial. The study was not registered as a clinical trial. Additional patient information is provided in Supplementary table 1.
Study three included 12 healthy controls (6 males and 6 females; age (mean[SD]): 62 [6] years; BMI (mean [SD]): 27.3 [4.1] kg/m2; normal glucose tolerance), 12 Type 2 diabetic patients receiving placebo (9 males and 3 females; age (mean[SD]): 60 [5] years; BMI (mean [SD]): 31.2 [4.3] kg/m2; patients had type 2 diabetes as defined by American Diabetes Association criteria) and 12 Type 2 diabetic patients receiving metformin (6 males and 6 females; age (mean[SD]): 64 [5] years; BMI (mean [SD]): 30.3 [5.7] kg/m2; patients had type 2 diabetes as defined by American Diabetes Association criteria)(Gormsen et al., 2018). If patients with T2DM were receiving metformin treatment at the time of screening, treatment was discontinued for four weeks before initiation of the study. The study was a randomized, placebo-controlled, parallel-group study, in which patients with T2DM were randomized to receive either placebo or 1000 mg metformin twice daily for three months. Skeletal muscle biopsies were obtained from m Vastus Lateralis. The samples were collected in 2013–2016, as part of a clinical trial (ID: NCT01729156), and the data analyses related to this paper were performed post-hoc. More detailed patient information is available in earlier publications (Gormsen et al., 2018).
Muscle Biopsies
For study one the same skilled physician obtained all skeletal muscle biopsies from m. vastus lateralis under local anesthesia with a Bergström needle after an overnight fast. The biopsies were collected early in the morning in a resting and non-stimulated situation. Muscle biopsies were immediately frozen in liquid-nitrogen and thereafter stored at −80°C until further analyses.
For study two all skeletal muscle biopsies were obtained from m. rectus abdominis or m. gastrocnemius under full anesthesia by a skilled surgeon. The biopsies for tissue dissociation were immediately submerged into 4°C wash-buffer [Hams F10 incl. glutamine and bicarbonate (Cat no N6908 Sigma, Sigma-Aldrich, Denmark) 10% Horse serum (cat no 26050088, Gibco, Thermo Fisher Scientific, MA, USA), 1% Penstrep (cat no 15140122, Gibco)]. For histology the samples were dissected free of visible fat and connective tissue. A well-aligned portion of the biopsy was immediately mounted in Tissue-Tek (Qiagen, Valencia, CA, USA), frozen in isopentane pre-cooled with liquid nitrogen and stored at −80°C until further analysis.
METHOD DETAILS
Tissue dissociation
Muscle was transported from the operation suite to the laboratory within 15 min in ice-cold wash-buffer. Upon arrival the muscle biopsy was initially dissected free of visible tendon/connective tissue and fat. The biopsy was then divided into pieces of up to 0.5–0.8 grams and briefly mechanically minced with sterile scissors. The muscle slurry was then transferred to C-tubes (cat no . 130–093-237, Miltenyi Biotec, Lund, Sweden) containing 8ml wash-buffer including 700 U/ml Collagenase II (lot 46D16552, Worthington, Lakewood, NJ, USA) and 3.27 U/ml Dispase II (cat no . 04 942 078 001, Roche Diagnostics, Basel, Switzerland). Mechanical and enzymatic muscle digestion was then performed at 37°C on the gentleMACS with heaters (cat no . 130–096-427, Miltenyi Biotec) for 60 min using a skeletal muscle digestion program (37C_mr_SMDK1). When digestion was complete 8 ml wash buffer was added to the single cell solution and this was filtered through a 70μm cell strainer and washed twice to collect any remaining cells. The suspension was centrifuged at 500g for 5 min and the supernatant removed. The cell pellet was resuspended in freezing buffer (StemMACS, cat no . 130–109-558, Miltenyi Biotec) and stored 1–3 weeks at −80.
Flow cytometry and FACS
Approximately 1.5 hour before FACS the frozen cell suspension was thawed until a small amount of ice was left and resuspended in 10ml wash-buffer. The solution was centrifuged at 500g for 5 min and the supernatant removed to clear the freezing buffer. The cells were then resuspended in wash buffer and incubated in MACS human FcR blocking solution (20μl/sample, cat no . 130–059-901, Miltenyi Biotec) and primary antibodies against CD45-FITC (12μl/sample, Cat no . 130–114-567, Clone 5B1, Miltenyi Biotec), CD31-FITC (4μl/sample, Cat no . 130–110-668, Clone REA730, Miltenyi Biotec), CD90-PE (3.2μl/sample, Clone 5E10, eBioscience, Thermo Fisher Scientific), CD56-BV421 (1:100, Cat no . 562752, BD Bioscience, San Jose, CA, USA), CD82-PE-vio770 (Cat no . 130–101-302 10μl/sample, Miltenyi Biotec) and CD34-APC (20μl/sample, clone 581, BD Bioscience) in darkness at 4°C for 30 min. Propidium iodide (PI, 10μl/sample, cat 556463, BD Bioscience) immediately before sorting to exclude non-viable cells. The suspension was washed in 10 ml wash-buffer and centrifuged at 500g. Finally, the sample resuspended in wash-buffer and filtered through a 30μm filter to remove any remaining debris/aggregates. Non-stained cells and single-color controls were prepared in combination with the primary (full color) samples. To ensure bright single-color controls for compensation, compensation beads (cat no . 01–2222-41, eBioscience, Thermo Fisher Scientific) was utilized. Cell sorting was performed using a FACS-AriaIII cell sorter (BD Bioscience) with 405nm, 488nm, 561nm and 633nm lasers. A 100μm nozzle at 20 psi was utilized to lower pressure/stress on the cells as well as prevent clogging. Gating strategies were optimized through multiple earlier experiments, which included various full color samples (Fig S2 A i–vi), unstained sample, single color samples and fluorescence minus one (FMO) controls for CD34, CD90, CD82 and CD56 (Fig S1 F i–iv). Cells were sorted into 4 °C cooled collection tubes containing wash-buffer. Data was collected in FACSdiva software and later analyzed in FlowJo (FlowJo 10.6.1, BD)
The following populations were FACS isolated; CD56+CD82+CD34− Lin−(CD45 and CD31) PI− (MuSCs), CD34+CD90−CD56−Lin−PI− (FAPsCD90−), CD34+CD90+CD56−Lin−PI− (FAPsCD90+), Lin+CD34+PI− (Endothelial cells), Lin+CD34−PI− (Hematopoietic cells), CD90+CD34−CD56−Lin−PI− (smooth muscle cells/pericytes) and CD90−CD34−CD56−Lin−PI− (unidentified). The purity FAP and MuSC populations were checked following the sort by re-running the samples which yielded >96% pure populations (Fig S1 G–I) and later by immunocytochemistry (ICC) when cells were plated (Fig 2 G, S2 G–H).
Single cell sorting and limiting dilution assay
For both single cell and limiting dilution assay the cell populations (MuSCs and FAPs) were first sorted in bulk and immediately resorted depositing 1, 10, 50 or 100 cells in individual wells of a 96-well half-area plate. The number of wells was 96 for single cell sorting, 36 for 10 cells, 36 for 50 cells and 24 for 100 cells. Plates were coated 1:1 with collagen (Cat no C8919, Sigma) and laminin (Cat no 23017–015, Gibco, Thermo Fisher Scientific). Media was wash-buffer + 20% FBS (Cat. no . 16000044, Thermo Fisher Scientific) + 5ng freshly added bFGF (human rbFGF, Cat no . F0291, Sigma). After two to three weeks the wells were scored for colonies (> 8 cells). Wells with colonies were allowed to grown to confluency before initiating differentiation for myogenic (DMEM, (Cat. no . 11965092, Thermo Fisher Scientific, 4.5 g/L glucose), 5% FBS, 1% Penstrep) or adipogenic (Adipogenic differentiation media; ADM, Cat. no . 130–091–677, Miltenyi Biotec) linage. Wells were scored for being myogenic (Myosin heavy chain/Desmin expressing cells and myotubes), Adipogenic (containing >3 Perilipin-1 positive adipocytes) or fibrogenic (containing Collagen-1 positive cells and < Perilipin-1 positive adipocytes). Limiting dilution analysis calculations were based on the single hit Poisson model (see quantification and statistical analysis).
Cell culture
Most cell culture experiments were performed without passages to limit cell-culture artefacts. If passages were performed this is indicated in the specific figure. All cell culture experiments were performed at 37°C and 5% CO2. All tissue-culture plates or chamberslides were treated with extra-cellular matrix (ECM gel, Cat no . E1270, Sigma) and chamberslides were in additions pre-treated with Poly-D-Lysine (Cat no A-003-E, Millipore, Sigma) to increase adherence of the ECM. Cells from FACS were initially plated at a density of approximately 1×104/cm2. All cells were plated in wash-buffer and after 24 h this was switched to a growth media (GM, Bio-AMF 2, Cat. no . 01–194-1A, Biological Industries, CT, USA). Media was changed every 2–3 days. If cells were passaged, they were maintained <60% confluency. When reaching >95% confluency, the media was switched to a differentiation media, depending on the cell type. For myogenic differentiation we used DMEM (4.5g/L glucose), 5% FBS and 1% penstrep for 5–8 days. For adipogenic differentiation we used either a complete adipogenic differentiation media (ADM, Miltenyi Biotec) or DMEM (4.5g/L glucose) + 20% FBS + 1μg/ml Insulin (Cat no . 91077C, Sigma) + 0.25 μM Dexamethasone (Cat no . D4902, Sigma) + 0.5 mM 3-isobutyl-1-methylxanthine (IBMX, Cat no I7018, Sigma) + 5 μM Rosaglitazone (Cat no . R2408, Sigma) and 1% Penstrep for 10 days. For fibrogenic stimulation we utilized DMEM (4.5g/L glucose), 10% FBS, 1% Penstrep and 1 ng/ml TGFβ (Cat no . T7039, Merck, Sigma) or 20 ng/ml PDGF-AA (human PDGF-AA, Cat no . 130–108-983, Miltenyi Biotec) for 5–6 days. Inhibition of PDGF-signaling was performed by addition of 1μM imatinib (Cat no . SML1027, Sigma) during adipogenic differentiation. To examine the effect of metformin on FAP proliferation and differentiation, FAP were exposed to metformin (Cat no . D150959, Sigma) in either growth conditions or during adipogenic differentiation. Osteogenic differentiation was induced utilizing a mesenchymal osteogenic differentiation media (Cat no . 130–091-678, Miltenyi Biotec).
Immunohisto- and cytochemistry and imaging
For whole skeletal muscle tissue cryosections (8 μm) were cut at −18°C and placed on a slide and stored at −80°C. Before staining sections were allowed to reach room temperature. Sections were fixed in Histofix (Histolab, Gothenborg, Sweden) followed by 1 hour in blocking buffer (10% goat serum, 0.2% Triton X in PBS). Sections were stained for Collagen 1 (mouse, 1:1000; Cat. no . C2456, Sigma), Perilipin-1 (rabbit, 1:200, Cat. no . 9349, Cell Signaling Technologies), CD34 (rabbit, 1:100, Cat no ab81289, Abcam, Cambridge, UK), CD90 (mouse, Cat no 14–0909-80, eBioscience, Thermo Fisher Scientific) and incubated at 4°C overnight in blocking buffer. This was followed by incubation with the secondary antibody Alexa-fluor 647 goat-anti-mouse and Alexa-fluor 568 goat-anti-rabbit (1:500; Cat no A-21235 and Cat no A-11011, Invitrogen, Thermo Fisher) combined with Wheat-germ-agglutinin (WGA; Cat no . W1126, Thermo Fisher Scientific) conjungated to Alexa-fluor 488 for 1.5 hours at room temparature. Finally, sections were washed 3×5 min, with one wash containing DAPI (1:50000, D3571, Invitrogen, Thermo Fisher Scientific), mounted with mouting media and stored in darkness at 4°C.
For cells in plates or chamberslides, these were fixed for 10 min in 4% paraformaldehyde, washed twice in PBS and stored in PBS until staining. Cells were then incubated for 30 min in blocking buffer (10% goat serum, 0.2% Triton X in PBS). Afterwards cells were incubated with primary antibody against PDGFRa (goat, 1:200, AF-307-NA, R&D Systems, MN, USA), TE-7 (mouse, 1:200, Cat no CBBL271, Merck, Sigma), Pax7 (mouse, 1:50, Pax7, Developmental Studies Hybridoma Bank, IA, USA), Desmin (rabbit, 1:200, Clone D93F5, Cat no . 5332, Cell Signalling Technologies), Myosin Heavy Chain (MyHC, mouse, 1:5, MF20, DHSB), Collagen 1 (1:500, Cat no Cat. no . C2456, Sigma), alpha-smooth muscle actin (1:150, Cat no . A5228, Sigma) or pPDGFRa (Y754, rabbit, 1:20, ab5460, abcam) at 4°C overnight in blocking buffer. This was followed by incubation with the secondary antibody Alexa-fluor 647 goat-anti-mouse, Alexa-fluor 488 goat-anti-mouse, Alexa-fluor 568 donkey-anti goat and Alexa-fluor 568 goat-anti-rabbit (1:500; Cat no A-21235, cat no A11001, cat no A-11057 and cat no A-11011, Invitrogen, Thermo Fisher Scientific) for 1.5 hours at room temperature. Finally, cells were washed 3×5 min, with one wash containing DAPI, and either maintained in PBS or mounted in mounted media. Minus primary controls were included for all stains during optimization to ensure specificity.
Alzirian red (2g/100ml dH2O, pH 4.2, Cat no . A5533, Sigma) staining was performed for two min and subsequently washed extensive with dH2O to stain for calcium depots (osteogenic differentiation) for 5 min and thoroughly was afterwards before imaging. Cells were stored in dH2O.
Images were acquired using a Leica DM2000 fluorescent microscope and a Leica Hi-resolution Color DFC camera (Leica, Stockholm, Sweden) or EVOS M7000 automated imaging system (Thermo Fisher Scientific). ImageJ was utilized for threshold and quantification of percentage positive area/foci.
Primeflow (RNA-flow cytometry)
For primeflow, muscle tissue was digested as previously described and all cells were plated in GM in a 25 cm2 tissue culture flask precoated with gelatine (0.2%). All cells were allowed to adhere for 3 days before the media was changed, to allow for more slowly or poorly adhering cells to attach. After 6–8 days the cells reached 80–90% confluency and were trypsinized and the primeflow protocol was initiated. Initially the cells were stained with antibodies against CD90-PE (3.2ul/sample, Clone 5E10, eBioscience), CD56-BV421 (1:100, Cat No.562752, BD Bioscience or PDGFRa-BV421 (1:100, Cat no 562799, BD Bioscience) for 40 min in wash-buffer. The cell suspension was then washed, centrifuged at 500g for 5 min, the supernatant was removed and cells were resuspended in residual volume. Here after we followed the instructions provided by the manufacturer (PrimeFlow™ RNA assay kit, Cat no 88–18005-210, eBioscience/Thermo Fisher). In brief the samples were incubated with the first fixation buffer (PrimeFlow fixation buffer 1) for 30min in dark at 4°C. Samples were centrifuged for 5 min at 500g, supernatant removed and resuspended in residual volume. Primeflow RNA permeabilization buffer including RNAase inhibitors were added, samples gently mixed and then centrifuged for 5 min at 500g, supernatant removed and samples resuspended in residual volume and then repeated once more. Samples were the incubated with a second fixation buffer (PrimeFlow fixation buffer 2) for 1h at room temp at in dark. The samples were then washed twice (PrimeFlow RNA wash buffer). Prewarmed target probe (1:20, COL1A1, NM_000088.3, Type 4 probe; RPL13A, NM_001270491.1, Type 1 probe) was then added to the samples and then placed in a hybridization oven at 40°C for 2h. Samples were again washed twice (PrimeFlow RNA wash buffer) with the final wash including RNAase inhibitors. Samples were left in final wash-buffer overnight in dark at 4°C.
On day 2 samples were initially incubated with a pre-amplification mix (1:1 PrimeFlow RNA Pre-AMP mix) for 1.5h in hybridization oven at 40°C. Samples were washed three times in wash-buffer (PrimeFlow RNA wash buffer) and then incubated for 1.5h in the hybridization oven at 40°C (1:1, PrimeFlow RNA AMP mix). Samples were washed twice (PrimeFlow RNA wash buffer) and finally incubated with label probes (1:100, PrimeFlow RNA label probes) for 1h in hybridization oven at 40°C. Samples were the washed twice (PrimeFlow RNA wash buffer) and then analyzed using a BD LSR Fortessa (equipped with 405, 488, 561 and 640 nm lacers and 18 detectors). We only included one fluorophore per laser to minimize then need for compensation. Compensations were performed using compensation beads specific to the PrimeFlow kit. We performed negative controls by omitting the target probes but preforming the rest of the protocol as specified (Fig S2K). Positive controls for “housekeeping” genes were included (Fig S2 K). Samples were initially gated on forward- and side scatter to exclude debris and dead cells, as well as doublets using forward scatter height versus area.
5-ethynyl-2’-deoxyuridine (EdU) incorporation
To detect proliferation/cell cycle entry we utilized the EdU/click-it detection platform to visualize cells that contained newly synthesized DNA (Cat no . C10337 or C10340, Invitrogen, Thermo Fisher Scientific). For experiments performed immediately post FACS, cells were plated in 96-well half-area tissue culture mircroplates (Corning, Sigma) coated with ECM (Sigma). Cells were plated at approximately 1×104/cm2 in wash-buffer + 10μM EdU and the fixed for 10 min in 4% PFA at various timepoints post FACS. After two washes in PBS cells were stored in PBS in dark at 4°C.
For experiments in which we tested the effect of PDGF-AA (Miltenyi Biotec) or Insulin (Sigma) on FAP proliferation we serum starved (0.5% Horse serum) FAPs that were in the growth phase for 18–20h to synchronize the cells. After this we changed the media with media (F10 or DMEM and 0.5% horse serum) added control or PDGF-AA/Insulin to the cells + 10μM EdU. After 24h we fixed the cells for 10 min in PFA, washed them twice with PBS and stored them in PBS in dark at 4°C. To study the effect of glucose (1 g/L versus 4.5g/L) and Insulin (PBS versus 1ng/ml Insulin) on FAP activation following FACS we cultured the FAPs for 96h in DMEM + 0.5% Horse serum + 10uM EdU. To investigate the effect of PDGF-AA on FAPCD90− and FAPCD90+ cell cycle entry, PDGF-AA (20ng/ml) was added to the media in low serum conditions (0.5% Horse serum) post sort. Cells were fixed at 96h post sort.
To detect EdU incorporation we followed the manufacturer instructions and counter-stained the cells with DAPI. Images were acquired using Images were acquired using an EVOS M7000 automated imaging system (Thermo Fisher Scientific) and performed automatically to ensure similar treatment of all wells.
Real time qPCR on whole skeletal muscle
Skeletal muscle (20 mg) was homogenized in TriZol reagent (Gibco BRL, Life Technologies, Roskilde, Denmark). RNA was quantified by measuring absorbance at 260 nm and 280 nm and the integrity of the RNA was checked by visual inspection of the two ribosomal RNAs on an ethidium bromide stained agarose gel. Reverse transcription was performed using random hexamer primers as described by the manufacturer (GeneAmp RNA PCR Kit from Perkin Elmer Cetus, Norwalk, CT, USA). PCR-mastermix containing the specific primers and Taq DNA polymerase (HotStar Taq, Qiagen Inc. USA) was added. The primers were designed using the primer analysis software Oligo version 6.64. See Key resource table for sequences.
Real time quantitation of target gene to B2M mRNA was performed with a SYBR-Green real-time PCR assay using an ICycler from BioRad. The threshold cycle (Ct) was calculated, and the relative gene-expression was calculated essentially as described in the User Bulletin #2, 1997 from Perkin Elmer (Perkin Elmer Cetus, Norwalk, CT, USA).
Western blotting
Frozen crude muscle tissue were homogenized in ice-cold lysis buffer (50 mM HEPES, 137 mM NaCl, 10 mM Na4P2O7, 10 mM NaF, 1 mM MgCl2, 2 mM EDTA, 1% NP-40, 10% glycerol (vol/vol), 1 mM CaCl2, 2 mM Na3VO4, 100 mM AEBSF [4-(2-aminoethyl) benzenesulfonyl fluoride], hydrochloride, pH 7.4) using a Precellys homogenizer (Bertin Technologies, France). Insoluble materials were removed by centrifugation at 14,000g for 20 minutes at 4 °C. Protein concentration of the supernatant was determined using a Bradford assay (BioRad, CA, USA). Samples were adjusted to equal concentrations with milli-Q water and denatured by mixing with 4x Laemmli’s buffer and heating at 95 °C for 5 minutes.
For cells, each well was washed twice in ice-cold PBS and the cells were scraped off in ice-cold PBS with added protease inhibitors (1:100 Halt, cat no . 78429, Thermo Fisher Scientific; 5 mM NAM; 2 mM NaOV). The suspension was transferred to a tube and centrifuged for 10 min at 2000g at 4 °C. The supernatant was removed and the pellet placed in a −80°C freezer for 4 hours to lyse cells after which ice-cold lysis buffer was added.
Equal amounts of protein were separated by SDS-PAGE using the BioRad Criterion system, and proteins were electroblotted onto PVDF membranes (BioRad). Control for equal loading was performed using the Stain-Free technology that allows visualization of total protein amount loaded to each lane and has been shown to be superior to beta-actin and GAPDH in human skeletal muscle (Gurtler et al., 2013; Vigelso et al., 2015). Membranes were blocked for 2 hours in a 2% bovine serum albumin solution (Sigma-Aldrich, MO, USA) and incubated overnight with primary antibodies PDGFRα (1:1000, AF-307-NA, R&D Systems, USA) or PDGFRα (1:1000, Cat no ab134123, Abcam), Collagen 1 (Cat no . T40777R, Meridian Life Science Inc, TN, USA)) and Collagen 3 (Cat no GWB-7D650E, Genway Biotec Inc, CA, USA). After incubation in primary antibodies the membranes were incubated 1 hour with HRP-conjugated secondary antibodies (goat anti-rabbit, IgG, Cat no . 31460, goat anti-mouse, IgG, Cat nb. G21040, mouse anti-goat, IgG, Cat no . 31400, Invitrogen, Thermo Fisher Scientific). Proteins were visualized by chemiluminescence (Pierce Supersignal West Dura, Thermo Scientific, IL, USA) and quantified with ChemiDocTM MP imaging system (BioRad). Protein Plus Precision All Blue standards were used as marker of molecular weight (BioRad).
Glucose and lactate analysis
Pre-conditioned media from FAP cell culture stimulated 3 days with PDGF-AA (20 ng/ml, human PDGF-AA, Cat no . 130–108-983, Miltenyi Biotec) or control was collected and stored at −20°C. For analysis samples were quickly defrosted and glucose and lactate concentrations were determined in triplicates on an YSI Select (YSI Life Sciences, Yellow Springs, OH, USA).
Bioenergetic analysis
Realtime analysis of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), as a proxy of cellular glycolysis, was performed using the Seahorse technology.
For freshly FACS isolated cells, we plated 1×105 cells in ECM (Sigma) coated Seahorse XF96 cell culture microplate (part no 101085–004, Agilent) in wash-buffer (to minimize change in cell bioenergetics related to high concentration of growth-factors etc). The number of cells was optimized before running the experiment. MuSCs and FAPs were allowed to adhere for 12h before running the bioenergetic analysis. One hour before the analysis the cells were washed twice in phenol red and bicarbonate free DMEM Seahorse Media (SM) (part no 103575–100, Agilent) containing 25 mM glucose (Cat no . G8769, Sigma), 1 mM sodium-pyruvate (Cat no . 11360–039, Gibco) and 2 mM L-glutamine (Cat no . G7513, Sigma), (pH 7.4) and allowed to equilibrate in a non-Co2 incubator for an hour. During this time brightfield images of all wells were captured to ensure a sufficient monolayer of cells. For freshly sorted cells, we performed the mitochondrial stress test using the Seahorse Mito-stress test kit (part no 103015–100, Agilent) with a final concentration of 4mM Oligomycin, 5mM FCCP and 2.5 mM Antimycin A/Rotenone diluted into the SM media. Bioenergetic analysis was performed on the Seahorse XFe96 Analyzer (Agilent). Following the bioenergetic analysis Hoechst 33342 (Cat no . H3570, Invitrogen) was added to all wells and images obtained of all wells to count the number of cells in each well. The number of cells per well was directly incorporated into the Wave software (Seahorse Wave Controller 2.4 software, Agilent) to normalize OCR and ECAR values. When possible, we included up to three replicates for each cell type from each donor, however, for some donors/cell types it was only possible to include one or two replicates.
To assess bioenergetics during stimulation towards fibro- or adipogeneses in cultured FAPs we plated them in a XF24 cell culture plate at a density of 6×104 per well resulting in a confluent monolayer in GM (several cell titrations were initially performed). Twenty-four hours before the bioenergetic profiling the media was switched to adipogenic (ADM, Miltenyi Biotec), ADM+20ng/ml PDGF-AA or fibrogenic (DMEM+5%FBS+20ng/ml PDGF-AA) differentiation media. One hour before the analysis the cells were washed twice in SM phenol red free and bicarbonate free DMEM (part no 103335–100, Agilent), 25 mM glucose, 1 mM sodium-bicarbonate and 2 mM L-glutamine, (pH 7.4) and allowed to equilibrate in a non-Co2 incubator. For the glycolytic rate assay HEPES (part no 103337–100, Agilent) was added to a final concentration of 5 mM. Before bioenergetic analysis the wells were examined to ensure a confluent monolayer of cells in all wells, otherwise these were excluded from the final analysis. To asses mitochondrial function we utilized the strategy described above, by sequentially injecting Oligomycin (4mM), FCCP (5mM) and Antimycin/Rotenone (2.5 mM) diluted into the SM media. To assess the glycolytic rate and profile this we sequentially injected Antimycin/Rotenone (2.5 mM) and 2-deoxy glucose (2-DG, 100mM). For this analysis all chemicals were purchased from sigma. Initial data analysis was performed in Wave software (Agilent).
RNA sequencing on tissue and FACS isolated cell populations
For whole tissue in study one the protocol has been described previously (Moller et al., 2017). In brief, total RNA was purified from frozen biopsies using the QiaSymphony robot in combination with the QiaSymphony RNA Mini kit (Qiagen, CA, USA) according to the Manufacturers protocol including DNase treatment. We were not able to isolate muscle RNA from one of the itT2D patients and one of the OBS subjects, leaving six patients/subjects in each of the groups for RNA-sequencing. RNA concentration was determined using a spectrophotometer with absorbance at 260 nM (NanoDrop ND-1000) and RNA integrity was assessed using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Whole transcriptome, strand-specific RNA-Seq libraries facilitating multiplexed paired-end sequencing were prepared from 500 ng total-RNA using the Ribo-Zero Magnetic Gold technology (Epicentre, an Illumina company) for depletion of rRNA followed by library preparation using the ScriptSeq v2 technology (Epicentre). The RNA-Seq libraries were combined into 2 nM pooled stocks, denatured and diluted to 10 pM with pre-chilled hybridization buffer and loaded into TruSeq
PE v3 flowcells on an Illumina cBot followed by indexed paired-end sequencing (101 + 7 + 101 bp) on an Illumina HiSeq 2000 using TruSeq SBS Kit v3 chemistry (Illumina). Paired de-multiplexed fastq files were generated using CASAVA software (Illumina) and processed using tools from CLC Bio (QIAGEN).
For sorted cells, these were FACS isolated into tubes containing wash-buffer as previously described. After sorting the cells were transferred to RNAase free Eppendorf tubes and centrifuged at 800g after which the supernatant was removed and the cell pellet frozen in liquid nitrogen and stored at −80°C. Following RNA extraction RNA was quality controlled using the Agilent Bioanalyzer 2100. Sequencing libraries were prepared using Takara SMARTer Stranded Total RNA-Seq Kit v2 pico kit. Libraries were quality controlled using the Agilent Bioanalyzer 2100 and quantified using qPCR. Equimolar amounts of libraries were pooled and sequenced on an Illumina HiSeq 4000 lane as Paired end 100bp.
For FAPs stimulated towards adipogenic (ADM + 1% penstrep) and fibrogenic (DMEM+20ng/ml PDGF-AA, 10% FBS, 1% penstrep) differentiation for six days. The cells were then washed in PBS, trypsinized, transferred to RNAase free Eppendorf tubes, centrifuged at 500g and supernatant was removed. The cell pellet was frozen in liquid N2 and stored at −80°C. The library preparation was done using TruSeq® Stranded mRNA Sample preparation kit (Illumina inc). After RNA extraction 100 ng of total RNA was mRNA enriched using the oligodT bead system. The isolated mRNA was subsequently fragmented using enzymatic fragmentation. Then first strand synthesis and second strand synthesis were performed and the double stranded cDNA was purified (AMPure XP, Beckman Coulter). The cDNA was end repaired, 3’ adenylated and Illumina sequencing adaptors ligated onto the fragments ends, and the library was purified (AMPure XP). The mRNA stranded libraries were pre-amplified with PCR (15 cycles) and purified (AMPure XP). The libraries size distribution was validated and quality inspected on a Bioanalyzer 2100 or BioAnalyzer 4200 tapeStation (Agilent Technologies). High quality libraries are pooled based in equimolar concentrations based on the Bioanalyzer Smear Analysis tool (Agilent Technologies). The library pool(s) were quantified using qPCR and optimal concentration of the library pool used to generate the clusters on the surface of a flowcell before sequencing on a NextSeq500 instrument (2×51 cycles, single read) according to the manufacturer instructions (Illumina Inc.).
Single-cell RNA seq (10x Genomics)
Digestion of four whole human skeletal muscle was performed as described previously and cell suspension was frozen. The cryopreserved samples were thawed in a water bath at 37°C before dilution in 10 ml wash-buffer followed by centrifugation at 500g for 5 min. The samples were resuspended in wash-buffer, stained with antibodies (see FACS section for additional information). Following antibody incubation, the cells were washed in wah-buffer, centrifuged at 500g and resuspended in wash-buffer. The cells were then filtered through a 30 μm cell strainer before sorting. PI was added immediately before sorting. Cells were sorted into DMEM, 10% Horse serum and 1% Penstrep. The PI-negative cells were loaded on the Chromium system (10x Genomics) using manufactures protocol (Chromium Single Cell 3’ Reagent Kits (v3 Chemistry)) for a targeted recovery of 5000 cells from each donor. The four libraries were sequenced on an Illumina Hiseq4000 and mapped to the human genome (build GRCh38) using Cell Ranger software (10x Genomics, version 3.1.0). Following normalization and quality control, we captured >18000 cells that we included in the analysis with an average of 129000 reads and 1397 identified genes per cell from four biological replicates (Fig S3 G–K).
Bioinformatics
Bulk RNAseq analysis
For all paired-end reads, trim galore was used for adapter removal and to trim low-quality ends (Babraham Bioinformatics). A Phred33 cut-off score of 20 was applied before adaptor trimming, and all sequences below 20 bp were subsequently removed. The trimmed reads were aligned to the human genome (hg19) by HISAT2 using default settings (Kim et al., 2015), and then sorted by gene name using samtools (Li et al., 2009). The overlap between read alignments and genomic annotations were quantified by htseq-count (Anders et al., 2015). The counts were then normalised and tested for differential expression (DE) using the R Bioconductor package DESeq2 (Love et al., 2014). When the different cell types had been isolated from the same individual, a model design was implemented to correct for inter-person variability. Differentially expressed genes were considered significant when the FDR adjusted p-value were < 0.05 and had a log2fold change > ± 1. For principal component analysis, all gene counts were regularized log2 transformed and was then used as input for principal component analysis by the plotPCA function of DESeq2.
Shared differentially expressed genes (FDR adjusted p-value<0.05, DESeq2) between the comparisons FAPCD90+ vs FAPCD90− and itT2D vs OBS were presented in a Venn plot. Furthermore, the log2fold change of the overlapping genes were presented as a bar plot.
Gene ontology and pathway enrichment analysis
The differentially expressed genes were used as input for ontology and pathway enrichment analysis by the R Bioconductor package XGR(Fang et al., 2016). Functional pathway analysis on selected pathway gene sets (MsigdbC2CPall) and Gene Ontology (Biological Process) analysis was assessed with a minimum overlap of 3 target genes. The significance was determined using a cut-off of FDR adjusted p-value < 0.05 and visualised according to the adjusted p-value.
Shared genes between the gene ontology term “Extracellular Matrix” (GO:0031012) and genes up-regulated in FAPCD90+ compared to FAPCD90− were merged with shared genes between the gene ontology term “Angiogenesis” (GO: 0001525) and genes up-regulated in FAPCD90− compared to FAPCD90+. The merged data was presented as a volcano plot created in the R Bioconductor package EnhancedVolcano (Blighe K (2019). EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. R package version 1.2.0)
Single cell sequencing analysis
The scRNAseq count data was analysed in R using the Seurat package and workflow (Stuart et al., 2019). Shortly, The four 10X datasets from the four subjects were individually read into the Seurat environment in R, and individually turned into Seurat objects. Then, all four objects were merged into a single Seurat object. The cells with unique feature counts above 200 and below 2500, and in addition, had a mitochondrial gene content < 10% were selected for further analysis. The gene counts for each cell were normalized to the cells’ total gene count, scaled by 10000, and log-transformed. Next, variable features were identified by the FindVariableFeatures function using the “vst” selection method and returning 2000 variable genes per dataset. The gene counts were then scaled and dimensional reduction by principal component analysis taking the variable features as input was performed. The dimensionality of the dataset was identified using the JackStraw procedure and the ElbowPlot function and Seurats graph-based clustering approach, FindNeighbors and FindClusters, was subsequently used to cluster the cells based on the previously determined dimensionality of the dataset. The resolution was set to 0.5 in the FindClusters function. To predict and remove doublets, the pre-processed Seurat object, as prepared above, were used as input for the R package DoubletFinder (McGinnis et al., 2019). The optimal pK (PC neighbourhood size) and pN (number of generated artificial doublets) values were estimated using a principal component number of 20 and assuming a 7.5% doublet formation rate. The doubletFinder function was then evoked and the predicted doublets were removed from the Seurat object (Fig S3 K). The clusters were then visualized in a Uniform Manifold Approximation and Projection (UMAP) plot. Differential expression analysis by the function FindAllMarkers was used to identify specific cluster biomarkers. As an inclusion criterion, a gene had to be detected in minimum 25% of the cells in either of the compared groups and have at least ±0.25 log2fold difference between the compared groups of cells. The top-10 differentially expressed genes from each cluster were then presented in a heatmap using the DoHeatmap function.
To investigate FAPs in further detail, all cells in the FAPs clusters were subsequently extracted and re-analysed. To block out inter-person differences, we integrated FAPs from our four different subjects as described previously (Stuart et al., 2019). Shortly, normalization and identification of variable features were done for each dataset independently. Next, variable features were identified and subsequently used to identify anchors for integrating the four datasets. The integrated dataset was then analysed as described above. Pseudotime trajectory inference analysis was carried out on the resulting FAP clusters, using the Slingshot method (Street et al., 2018). The slingshot function was evoked with default parameters using the reduced-dimensional PCA data as input. No cluster of origin was assigned.
Furthermore, FAPCD90+ and FAPCD90− cells were extracted and the FindAllMarkers function was used to identify differentially expressed genes. Subsequently, the DE genes were used as input for gene set enrichment analysis as described above.
QUANTIFICATION AND STATISTICAL ANALYSIS
The images presented in the manuscript are representative of the data and the image/staining quality. All n’s are true biological replicates (separate biological donors) unless otherwise stated. In line with nature of our data and the number of biological replicates, we have assumed parametric distribution when performing parametric-based analyses (i.e. no statistical test of normality was performed). Statistical tests, including paired/unpaired Student’s t-test (two-sided), non-paired Mann-Whitney non-parametric test (one/two-sided), one-way ANOVA and non-linear fit analyses were performed using Prism 8 (Version 8.4.1, GraphPad Software). Analysis of limiting dilution data were performed using a web application made available by the Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia (http://bioinf.wehi.edu.au/software/limdil/46)(Joe et al., 2010). This software tests departures from the single-hit Poisson model using a generalized linear model. Statistical parameters can be found in the specific figures and figure legends. All data, including supplementary figures, are presented as Violin plots with median and quartiles with superimposed individual values or mean ± SEM.
Supplementary Material
ACKNOWLEDGEMENTS
Flow cytometry was performed at the FACS-Core Facility, Aarhus University. Illustrations were created in Biorender.com. Funding from A.P. Møller Foundation, Riisfort Foundation, Toyota Foundation, The Independent Research Fund Denmark (DFF–5053–00195to JF) and the Steno Diabetes Center Aarhus, which is partially funded by an unrestricted donation from the Novo Nordisk Foundation, Novo Nordisk Foundation (NNF17OC0027242 to NJ and NNF16OC0021496 to THP), the Lundbeck Foundation (R190–2014-3904 to THP) and the Roche per la Ricerca 2019 to LM.
Footnotes
DECLARATION OF INTEREST
The authors declare no competing interests.
REFERENCES
- Abderrahmani A, Yengo L, Caiazzo R, Canouil M, Cauchi S, Raverdy V, Plaisance V, Pawlowski V, Lobbens S, Maillet J, et al. (2018). Increased Hepatic PDGF-AA Signaling Mediates Liver Insulin Resistance in Obesity-Associated Type 2 Diabetes. Diabetes 67, 1310–1321. 10.2337/db17-1539. [DOI] [PubMed] [Google Scholar]
- Addison O, Marcus RL, Lastayo PC, and Ryan AS (2014). Intermuscular Fat: A Review of the Consequences and Causes. International journal of endocrinology 2014, 309570. 10.1155/2014/309570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlqvist E, Storm P, Käräjämäki A, Martinell M, Dorkhan M, Carlsson A, Vikman P, Prasad RB, Aly DM, Almgren P, et al. (2018). Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. The Lancet Diabetes & Endocrinology 6, 361–369. 10.1016/s2213-8587(18)30051-2. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron AD, Brechtel G, Wallace P, and Edelman SV (1988). Rates and tissue sites of non-insulin- and insulin-mediated glucose uptake in humans. Am J Physiol 255, E769–774. 10.1152/ajpendo.1988.255.6.E769. [DOI] [PubMed] [Google Scholar]
- Buras ED, Converso-Baran K, Davis CS, Akama T, Hikage F, Michele DE, Brooks SV, and Chun TH (2019). Fibro-Adipogenic Remodeling of the Diaphragm in Obesity-Associated Respiratory Dysfunction. Diabetes 68, 45–56. 10.2337/db18-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte M, Martucci M, Sandri M, Franceschi C, and Salvioli S (2019). The Dual Role of the Pervasive “Fattish” Tissue Remodeling With Age. Frontiers in Endocrinology 10. 10.3389/fendo.2019.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras O, Cruz-Soca M, Theret M, Soliman H, Tung LW, Groppa E, Rossi FM, and Brandan E (2019). Cross-talk between TGF-beta and PDGFRalpha signaling pathways regulates the fate of stromal fibro-adipogenic progenitors. J Cell Sci 132. 10.1242/jcs.232157. [DOI] [PubMed] [Google Scholar]
- Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, Zhang L, et al. (2008). A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 3, 301–313. 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- de Morree A, Klein JDD, Gan Q, Farup J, Urtasun A, Kanugovi A, Bilen B, van Velthoven CTJ, Quarta M, and Rando TA (2019). Alternative polyadenylation of Pax3 controls muscle stem cell fate and muscle function. Science 366, 734–738. 10.1126/science.aax1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Paz-Lugo P, Lupianez JA, and Melendez-Hevia E (2018). High glycine concentration increases collagen synthesis by articular chondrocytes in vitro: acute glycine deficiency could be an important cause of osteoarthritis. Amino Acids 50, 1357–1365. 10.1007/s00726-018-2611-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derosa G, Sahebkar A, and Maffioli P (2018). The role of various peroxisome proliferator-activated receptors and their ligands in clinical practice. J Cell Physiol 233, 153–161. 10.1002/jcp.25804. [DOI] [PubMed] [Google Scholar]
- Fang H, Knezevic B, Burnham KL, and Knight JC (2016). XGR software for enhanced interpretation of genomic summary data, illustrated by application to immunological traits. Genome Med 8, 129. 10.1186/s13073-016-0384-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiore D, Judson RN, Low M, Lee S, Zhang E, Hopkins C, Xu P, Lenzi A, Rossi FM, and Lemos DR (2016). Pharmacological blockage of fibro/adipogenic progenitor expansion and suppression of regenerative fibrogenesis is associated with impaired skeletal muscle regeneration. Stem cell research 17, 161–169. 10.1016/j.scr.2016.06.007. [DOI] [PubMed] [Google Scholar]
- Giordani L, He GJ, Negroni E, Sakai H, Law JYC, Siu MM, Wan R, Corneau A, Tajbakhsh S, Cheung TH, and Le Grand F (2019). High-Dimensional Single-Cell Cartography Reveals Novel Skeletal Muscle-Resident Cell Populations. Mol Cell. 10.1016/j.molcel.2019.02.026. [DOI] [PubMed] [Google Scholar]
- Goodpaster BH, Thaete FL, and Kelley DE (2000). Thigh adipose tissue distribution is associated with insulin resistance in obesity and in type 2 diabetes mellitus. Am J Clin Nutr 71, 885–892. 10.1093/ajcn/71.4.885. [DOI] [PubMed] [Google Scholar]
- Gormsen LC, Sondergaard E, Christensen NL, Jakobsen S, Nielsen EHT, Munk OL, Tolbod LP, Jessen N, and Nielsen S (2018). Metformin does not affect postabsorptive hepatic free fatty acid uptake, oxidation or resecretion in humans: A 3-month placebo-controlled clinical trial in patients with type 2 diabetes and healthy controls. Diabetes Obes Metab 20, 1435–1444. 10.1111/dom.13244. [DOI] [PubMed] [Google Scholar]
- Gurtler A, Kunz N, Gomolka M, Hornhardt S, Friedl AA, McDonald K, Kohn JE, and Posch A (2013). Stain-Free technology as a normalization tool in Western blot analysis. Anal Biochem 433, 105–111. 10.1016/j.ab.2012.10.010. [DOI] [PubMed] [Google Scholar]
- Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, Rando TA, and Chawla A (2013). Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 153, 376–388. 10.1016/j.cell.2013.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Jiang Y, Barnes RH 2nd, Tokunaga M, Martinez-Santibanez G, Geletka L, Lumeng CN, Buchner DA, and Chun TH (2013). Thrombospondin 1 mediates high-fat diet-induced muscle fibrosis and insulin resistance in male mice. Endocrinology 154, 4548–4559. 10.1210/en.2013-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwayama T, Steele C, Yao L, Dozmorov MG, Karamichos D, Wren JD, and Olson LE (2015). PDGFRalpha signaling drives adipose tissue fibrosis by targeting progenitor cell plasticity. Genes Dev 29, 1106–1119. 10.1101/gad.260554.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, and Rossi FM (2010). Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol 12, 153–163. 10.1038/ncb2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampmann U, Christensen B, Nielsen TS, Pedersen SB, Orskov L, Lund S, Moller N, and Jessen N (2011a). GLUT4 and UBC9 protein expression is reduced in muscle from type 2 diabetic patients with severe insulin resistance. PLoS One 6, e27854. 10.1371/journal.pone.0027854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampmann U, Hoeyem P, Mengel A, Schmitz O, Rungby J, Orskov L, and Moller N (2011b). Insulin dose-response studies in severely insulin-resistant type 2 diabetes--evidence for effectiveness of very high insulin doses. Diabetes Obes Metab 13, 511–516. 10.1111/j.1463-1326.2011.01373.x. [DOI] [PubMed] [Google Scholar]
- Kang L, Ayala JE, Lee-Young RS, Zhang Z, James FD, Neufer PD, Pozzi A, Zutter MM, and Wasserman DH (2011). Diet-induced muscle insulin resistance is associated with extracellular matrix remodeling and interaction with integrin alpha2beta1 in mice. Diabetes 60, 416–426. 10.2337/db10-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360. 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos DR, Babaeijandaghi F, Low M, Chang CK, Lee ST, Fiore D, Zhang RH, Natarajan A, Nedospasov SA, and Rossi FM (2015). Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat Med 21, 786–794. 10.1038/nm.3869. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing S (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukjanenko L, Karaz S, Stuelsatz P, Gurriaran-Rodriguez U, Michaud J, Dammone G, Sizzano F, Mashinchian O, Ancel S, Migliavacca E, et al. (2019). Aging Disrupts Muscle Stem Cell Function by Impairing Matricellular WISP1 Secretion from Fibro-Adipogenic Progenitors. Cell Stem Cell. 10.1016/j.stem.2018.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey AL, Magnan M, Chazaud B, and Kjaer M (2017). Human skeletal muscle fibroblasts stimulate in vitro myogenesis and in vivo muscle regeneration. J Physiol. 10.1113/JP273997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madaro L, Passafaro M, Sala D, Etxaniz U, Lugarini F, Proietti D, Alfonsi MV, Nicoletti C, Gatto S, De Bardi M, et al. (2018). Denervation-activated STAT3-IL-6 signalling in fibro-adipogenic progenitors promotes myofibres atrophy and fibrosis. Nat Cell Biol 20, 917–927. 10.1038/s41556-018-0151-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malecova B, Gatto S, Etxaniz U, Passafaro M, Cortez A, Nicoletti C, Giordani L, Torcinaro A, De Bardi M, Bicciato S, et al. (2018). Dynamics of cellular states of fibro-adipogenic progenitors during myogenesis and muscular dystrophy. Nature communications 9. 10.1038/s41467-018-06068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelin G, Ferreira A, Liu Y, Atlan M, Aron-Wisnewsky J, Pelloux V, Botbol Y, Ambrosini M, Fradet M, Rouault C, et al. (2017). A PDGFRα-Mediated Switch toward CD9high Adipocyte Progenitors Controls Obesity-Induced Adipose Tissue Fibrosis. Cell Metabolism. 10.1016/j.cmet.2017.01.010. [DOI] [PubMed] [Google Scholar]
- McGinnis CS, Murrow LM, and Gartner ZJ (2019). DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst 8, 329–337 e324. 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick D, Sakers A, Irgebay Z, Okada C, Calvert C, Morley MP, Percec I, and Seale P (2019). Identification of a mesenchymal progenitor cell hierarchy in adipose tissue. Science 364. 10.1126/science.aav2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miljkovic I, Cauley JA, Petit MA, Ensrud KE, Strotmeyer E, Sheu Y, Gordon CL, Goodpaster BH, Bunker CH, Patrick AL, et al. (2009). Greater adipose tissue infiltration in skeletal muscle among older men of African ancestry. J Clin Endocrinol Metab 94, 2735–2742. 10.1210/jc.2008-2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller AB, Kampmann U, Hedegaard J, Thorsen K, Nordentoft I, Vendelbo MH, Moller N, and Jessen N (2017). Altered gene expression and repressed markers of autophagy in skeletal muscle of insulin resistant patients with type 2 diabetes. Scientific reports 7, 43775. 10.1038/srep43775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CW, Allen MD, Kimpinski K, Doherty TJ, and Rice CL (2016). Reduced skeletal muscle quantity and quality in patients with diabetic polyneuropathy assessed by magnetic resonance imaging. Muscle Nerve 53, 726–732. 10.1002/mus.24779. [DOI] [PubMed] [Google Scholar]
- Mueller AA, van Velthoven CT, Fukumoto KD, Cheung TH, and Rando TA (2016). Intronic polyadenylation of PDGFRalpha in resident stem cells attenuates muscle fibrosis. Nature 540, 276–279. 10.1038/nature20160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson LE, and Soriano P (2009). Increased PDGFRα Activation Disrupts Connective Tissue Development and Drives Systemic Fibrosis. Developmental Cell 16, 303–313. 10.1016/j.devcel.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak M, Lefebvre P, and Staels B (2015). Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol 62, 720–733. 10.1016/j.jhep.2014.10.039. [DOI] [PubMed] [Google Scholar]
- Porichis F, Hart MG, Griesbeck M, Everett HL, Hassan M, Baxter AE, Lindqvist M, Miller SM, Soghoian DZ, Kavanagh DG, et al. (2014). High-throughput detection of miRNAs and gene-specific mRNA at the single-cell level by flow cytometry. Nature communications 5, 5641. 10.1038/ncomms6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran C, Liu H, Hitoshi Y, and Israel MA (2013). Proliferation-independent control of tumor glycolysis by PDGFR-mediated AKT activation. Cancer Res 73, 1831–1843. 10.1158/0008-5472.CAN-12-2460. [DOI] [PubMed] [Google Scholar]
- Rasmussen DGK, Hansen TW, von Scholten BJ, Nielsen SH, Reinhard H, Parving HH, Tepel M, Karsdal MA, Jacobsen PK, Genovese F, and Rossing P (2018). Higher Collagen VI Formation Is Associated With All-Cause Mortality in Patients With Type 2 Diabetes and Microalbuminuria. Diabetes Care 41, 1493–1500. 10.2337/dc17-2392. [DOI] [PubMed] [Google Scholar]
- Richardson DK, Kashyap S, Bajaj M, Cusi K, Mandarino SJ, Finlayson J, DeFronzo RA, Jenkinson CP, and Mandarino LJ (2005). Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J Biol Chem 280, 10290–10297. 10.1074/jbc.M408985200. [DOI] [PubMed] [Google Scholar]
- Scott RW, Arostegui M, Schweitzer R, Rossi FMV, and Underhill TM (2019). Hic1 Defines Quiescent Mesenchymal Progenitor Subpopulations with Distinct Functions and Fates in Skeletal Muscle Regeneration. Cell Stem Cell 25, 797–813 e799. 10.1016/j.stem.2019.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore EM, and Kaplan FS (2008). Insights from a rare genetic disorder of extra-skeletal bone formation, fibrodysplasia ossificans progressiva (FOP). Bone 43, 427–433. 10.1016/j.bone.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman H, Paylor B, Scott RW, Lemos DR, Chang C, Arostegui M, Low M, Lee C, Fiore D, Braghetta P, et al. (2020). Pathogenic Potential of Hic1-Expressing Cardiac Stromal Progenitors. Cell Stem Cell 26, 205–220 e208. 10.1016/j.stem.2019.12.008. [DOI] [PubMed] [Google Scholar]
- Srikanthan P, and Karlamangla AS (2014). Muscle mass index as a predictor of longevity in older adults. Am J Med 127, 547–553. 10.1016/j.amjmed.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, and Dudoit S (2018). Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477. 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundelin E, Gormsen LC, Jensen JB, Vendelbo MH, Jakobsen S, Munk OL, Christensen M, Brosen K, Frokiaer J, and Jessen N (2017). Genetic Polymorphisms in Organic Cation Transporter 1 Attenuates Hepatic Metformin Exposure in Humans. Clin Pharmacol Ther 102, 841–848. 10.1002/cpt.701. [DOI] [PubMed] [Google Scholar]
- Tabula Muris C, Overall c., Logistical c., Organ c., processing, Library p., sequencing, Computational data a., Cell type a., Writing g., et al. (2018). Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372. 10.1038/s41586-018-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uezumi A, Fukada S, Yamamoto N, Ikemoto-Uezumi M, Nakatani M, Morita M, Yamaguchi A, Yamada H, Nishino I, Hamada Y, and Tsuchida K (2014). Identification and characterization of PDGFRalpha+ mesenchymal progenitors in human skeletal muscle. Cell death & disease 5, e1186. 10.1038/cddis.2014.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uezumi A, Fukada S, Yamamoto N, Takeda S, and Tsuchida K (2010). Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol 12, 143–152. 10.1038/ncb2014. [DOI] [PubMed] [Google Scholar]
- Vigelso A, Dybboe R, Hansen CN, Dela F, Helge JW, and Guadalupe Grau A (2015). GAPDH and beta-actin protein decreases with aging, making Stain-Free technology a superior loading control in Western blotting of human skeletal muscle. J Appl Physiol (1985) 118, 386–394. 10.1152/japplphysiol.00840.2014. [DOI] [PubMed] [Google Scholar]
- Walker JT, McLeod K, Kim S, Conway SJ, and Hamilton DW (2016). Periostin as a multifunctional modulator of the wound healing response. Cell Tissue Res 365, 453–465. 10.1007/s00441-016-2426-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warram JH, Martin BC, Krolewski AS, Soeldner JS, and Kahn CR (1990). Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offspring of diabetic parents. Ann Intern Med 113, 909–915. 10.7326/0003-4819-113-12-909. [DOI] [PubMed] [Google Scholar]
- Wosczyna MN, Biswas AA, Cogswell CA, and Goldhamer DJ (2012). Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J Bone Miner Res 27, 1004–1017. 10.1002/jbmr.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Peng H, Hong C, Chen Z, Deng X, Wang A, Yang F, Yang L, Chen C, and Qin X (2017). PDGF Promotes the Warburg Effect in Pulmonary Arterial Smooth Muscle Cells via Activation of the PI3K/AKT/mTOR/HIF-1alpha Signaling Pathway. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology 42, 1603–1613. 10.1159/000479401. [DOI] [PubMed] [Google Scholar]
- Zhao X, Kwan JYY, Yip K, Liu PP, and Liu FF (2019). Targeting metabolic dysregulation for fibrosis therapy. Nature reviews. Drug discovery. 10.1038/s41573-019-0040-5. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. (2001). Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108, 1167–1174. 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurlo F, Larson K, Bogardus C, and Ravussin E (1990). Skeletal muscle metabolism is a major determinant of resting energy expenditure. J Clin Invest 86, 1423–1427. 10.1172/JCI114857. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The whole muscle, population-based and single-cell RNA-seq data reported in this study are available at the European Genome-phenome Archive (EGA, https://ega-archive.org). The Accession numbers are listed in the key resource table. Microscopy data, original western blot images etc. are available from the Lead Contact on request.
No original code was produced in this paper. Code for bioinformatics analyses on whole muscle, population and single-cell RNA-seq is available through download of the specified (key resource table/STAR Methods), publicly available packages.
Any additional information required to reanalyse the data reported in this paper is available from the Lead Contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Collagen 1 (mouse) | Sigma | Cat# C2456, RRID:AB_476836 |
| Anti-Perilipin-1 (rabbit) | Cell Signaling Technologies | Cat# 9349, RRID:AB_10829911 |
| Anti-CD34 (rabbit) | Abcam | Cat# ab81289, RRID:AB_1640331 |
| Anti-PDGFRa (goat) | R&D Systems | Cat# AF-307-NA, RRID:AB_354459 |
| Anti-PDGFRa (recombinant, rabbit) | Abcam | Cat# ab134123 RRID: N/A |
| TE-7 (mouse) | Merck Millipore | Cat# CBL271, RRID:AB_93449 |
| Anti-Pax7 (mouse) | Developmental Studies Hybridoma Bank | Cat# pax7, RRID:AB_528428 |
| Anti-Desmin (rabbit) | Cell Signalling Technologies | Cat# 5332, RRID:AB_1903947 |
| Anti-Myosin Heavy Chain (mouse) | Developmental Studies Hybridoma Bank | Cat# MF 20, RRID:AB_2147781 |
| Anti-alpha-smooth muscle actin (mouse) | Sigma | Cat# A5228, RRID:AB_262054 |
| Anti-pPDGFRa (Y754) (rabbit) | Abcam | Cat# AB5460, RRID:AB_ 304906 |
| Anti-Collagen 1 (rabbit) | Meridian Life Science Inc | Cat# T40777R, RRID:AB_151099 |
| Anti-Collagen 3 (rabbit) | Genway Biotec Inc | Cat# GWB-7D650E, RRID:AB_10517236 |
| Anti-human-CD45-FITC | Miltenyi Biotec | Cat# 130-114-567, RRID:AB_2726699 |
| Anti-human-CD31-FITC | Miltenyi Biotec | Cat# 130-110-668, RRID:AB_2657279 |
| Anti-human-CD90-PE | Thermo Fisher Scientific | Cat# 12-0909-42, RRID:AB_10670624 |
| Anti-human-CD56-BV421 | BD Bioscience | Cat# 562751, RRID:AB_2732054 |
| Anti-human-CD82-PE-vio770 | Miltenyi Biotec | Cat# 130-101-302, RRID:AB_2659303 |
| Anti-mouse-CD34-APC | BD Bioscience | Cat# 555824, RRID:AB_398614 |
| Goat-anti-mouse IgG (H+L) Alexafluor 647 | Thermo Fisher Scientific | Cat# A-21235, RRID:AB_2535804 |
| Goat-anti-rabbit IgG (H+L) Alexafluor 568 | Thermo Fisher Scientific | Cat# A-11011, RRID:AB_143157 |
| Goat-anti-mouse IgG (H+L) Alexafluor 488 | Thermo Fisher Scientific | Cat# A-11001, RRID:AB_2534069 |
| Donkey-anti-goat IgG (H+L) Alexafluor 568 | Thermo Fisher Scientific | Cat# A-11057, RRID:AB_2534104 |
| Goat-anti-rabbit IgG (H+L) HRP | Thermo Fisher Scientific | Cat# 31460, RRID:AB_228341 |
| Goat-anti-mouse IgG (H+L) HRP | Thermo Fisher Scientific | Cat# G-21040, RRID:AB_2536527 |
| Patient characteristics presented in supplementary table 1 (study 2) | This paper | N/A |
| Patient characteristics from study 1 | Kampmann, U. et al. 2011a | N/A |
| Patient characteristics from study 3 | Gormsen, L.C. et al. 2018 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Propidium Iodide | BD Bioscience | Cat# 556463 |
| Hams F10 media incl. glutamine and bicarbonate | Sigma | Cat# N6908 |
| Horse serum | Thermo Fisher Scientific | Cat#26050088 |
| 100X Penstrep | Thermo Fisher Scientific | Cat#15140122 |
| Collagenase II | Worthington | Cat#46D16552 |
| Dispase II | Roche Diagnostics | Cat#04 942 078 001 |
| StemMACS cryopreservation buffer | Miltenyi Biotec | Cat#130-109-558 |
| Human FcR blocking solution | Miltenyi Biotec | Cat#130-059-901 |
| Compensation beads | Thermo Fisher Scientific | Cat#01-2222-41 |
| Collagen | Sigma | Cat#C8919 |
| Laminin | Thermo Fisher Scientific | Cat#23017-015 |
| Fetal Bovine serum | Thermo Fisher Scientific | Cat#16000044 |
| Recombinant human bFGF | Sigma | Cat# F0291 |
| DMEM (4.5 g/L glucose) incl glutamine and bicarbonate | Thermo Fisher Scientific | Cat#11965092 |
| Adipogenic differentiation media | Miltenyi Biotec | Cat#130-091-677 |
| Extra-cellular matrix coating gel | Sigma | Cat# E1270 |
| Poly-D-Lysine | Merck Millipore | Cat#A-003-E |
| Bio-AMF 2 growth media | Biological Industries | Cat#01-194-1A |
| Recombinant human Insulin | Sigma | Cat#91077C |
| Dexamethasone | Sigma | Cat# D4902 |
| 3-isobutyl-1-methylxanthine (IBMX) | Sigma | Cat# I7018 |
| Rosaglitazone | Sigma | Cat# R2408 |
| Recombinant human TGFb | Sigma | Cat# T7039 |
| Recombinant human PDGF-AA | Miltenyi Biotec | Cat#130-108-983 |
| Metformin | Sigma | Cat#D150959 |
| Imatinib | Sigma | Cat#SML1027 |
| Osteogenic differentiation media | Miltenyi Biotec | Cat#130-091-678 |
| Histofix | Histolabs | Cat#01000 |
| Normal Goat serum | Thermo Fisher Scientific | Cat31873 |
| Wheat-germ-agglutinin | Thermo Fisher Scientific | Cat# W1126 |
| Alzirian red staining solution | Sigma | Cat# A5533 |
| Phenol red and bicarbonate free DMEM Seahorse Media | Agilent | Cat#103575-100 |
| Glucose | Sigma | Cat#G8769 |
| Sodium-pyruvate | Thermo Fisher Scientific | Cat#11360-039 |
| L-glutamine | Sigma | Cat# G7513 |
| Hoechst 33342 | Thermo Fisher Scientific | Cat# H3570 |
| 2-Deoxy-glucose | Sigma | Cat# D8375 |
| Oligomycin | Sigma | Cat#75351 |
| Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) | Sigma | Cat#C2920 |
| Antimycin | Sigma | Cat#A8674 |
| Rotenone | Sigma | Cat#R8875 |
| Critical commercial assays | ||
| Click-iT™ EdU cell proliferation kit for imaging | Thermo Fisher Scientific | Cat#C10337 |
| Click-iT™ EdU cell proliferation kit for imaging | Thermo Fisher Scientific | Cat#C10340 |
| PrimeFlow™ RNA assay kit | Thermo Fisher Scientific | Cat#88-18005-210 |
| QiaSymphony RNA Mini kit | Qiagen | Cat#931636 |
| Seahorse Mito-stress test kit | Agilent | Cat#103015-100 |
| Takara SMARTer Stranded Total RNA-Seq Kit v2 pico kit | Takara Bio | Cat#634411 |
| TruSeq. Stranded mRNA Sample preparation kit | Illumina Inc | Cat#20020594 |
| Chromium Single Cell 3’ Reagent Kits (v3 Chemistry) | 10X Genomics | Cat#1000075 |
| Deposited data | ||
| Population-based RNA-sequencing data | This paper www.ega-archive.org | EGAS00001005599 |
| Single cell RNA-sequencing data | This paper www.ega-archive.org | EGAS00001005599 |
| Whole muscle RNA-sequencing data | This paper www.ega-archive.org | EGAS00001005599 |
| Oligonucleotides | ||
| B2M: Sense AATGTCGGATGGATGAAACC, Antisense TCTCTCTTTCTGGCCTGGAG | HotStar Taq, Qiagen | N/A |
| TGFB1: Sense GGACACCAACTATTGCTTCAGCTC, Antisense AAGTTGGCATGGTAGCCCTTGG | HotStar Taq, Qiagen | N/A |
| COL1A1: Sense TGCGATGACGTGATCTGTGACG, Antisense TTTCTTGGTCGGTGGGTGACTCTG | HotStar Taq, Qiagen | N/A |
| COL3A1: Sense ATTGCTGGGATCACTGGAGCAC, Antisense CCTGGTTTCCCACTTTCACCCTTG | HotStar Taq, Qiagen | N/A |
| COL6A1: Sense ATCAGCCAGACCATCGACACCATC, Antisense TTCGAAGGAGCAGCACACTTGC | HotStar Taq, Qiagen | N/A |
| COL1A1 (PrimeFlow type 4 probe) | Thermo Fisher Scientific | NM_000088.3 |
| RPL13A (PrimeFlow type 1 probe) | Thermo Fisher Scientific | NM_001270491.1 |
| Software and algorithms | ||
| ImageJ (for fluoresent image analysis) | NIH | https://imagej.nih.gov/ij/ |
| FlowJo (10.6.1) for FACS and flow cytometry analysis | BD Bioscience | https://www.flowjo.com/ |
| Prism 8 (version 9.0) for data presentation and statistics | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Limiting dilution Analysis | Walter and Eliza Hall Institute of Medical Research | http://bioinf.wehi.edu.au/software/limdil/ |
| Trimgalore (v 0.4.1) for adapter and quality trimming of RNA sequencing reads | Babraham Bioinformatics, Babraham Institute | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Hisat2 (v 2.1.0) for alignment of RNA sequencing reads to the reference genome | Kim D et al., 2015 | http://daehwankimlab.github.io/hisat2/ |
| Samtools (v 1.9) for sorting the alignment files | Li H et al., 2009 | http://www.htslib.org/ |
| htseq-count (v 0.6.1) for feature number estimation (gene counts) | Anders S et al., 2014 | https://htseq.readthedocs.io/en/master/count.html |
| R (v 4.0.3) for bioinformatics and statistical analysis | The R project for statistical computing | https://www.r-project.org/ |
| R package DESeq2 (v 1.26.0) for differential expression analysis of RNAseq data | Love MI., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| R package Seurat (v 4.0.1) for scRNAseq analysis | Hao et al., 2020 | https://satijalab.org/seurat/index.html |
| R package Doubletfinder (v 2.0.3) for doublet removal in the scRNAseq dataset | McGinnis CS et al., 2019 | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| R package XGR (v 1.1.7) for gene set and pathway enrichment analysis of differentially expressed genes | Fang et al., 2016 | http://galahad.well.ox.ac.uk:3030/ |
| R package Slingshot (v. 1.8.0) for pseudotime inference analysis | Street et al., 2018 | https://bustools.github.io/BUS_notebooks_R/slingshot.html |