

Abstract
Decades of work have elucidated cytokine signalling and transcriptional pathways that control T cell differentiation and have led the way to targeted biologic therapies that are effective in a range of autoimmune, allergic and inflammatory diseases. Recent evidence indicates that obesity and metabolic disease can also influence the immune system1–7, although the mechanisms and effects on immunotherapy outcomes remain largely unknown. Here, using two models of atopic dermatitis, we show that lean and obese mice mount markedly different immune responses. Obesity converted the classical type 2 T helper (TH2)-predominant disease associated with atopic dermatitis to a more severe disease with prominent TH17 inflammation. We also observed divergent responses to biologic therapies targeting TH2 cytokines, which robustly protected lean mice but exacerbated disease in obese mice. Single-cell RNA sequencing coupled with genome-wide binding analyses revealed decreased activity of nuclear receptor peroxisome proliferator-activated receptor-γ (PPARγ) in TH2 cells from obese mice relative to lean mice. Conditional ablation of PPARγ in T cells revealed that PPARγ is required to focus the in vivo TH response towards a TH2-predominant state and prevent aberrant non-TH2 inflammation. Treatment of obese mice with a small-molecule PPARγ agonist limited development of TH17 pathology and unlocked therapeutic responsiveness to targeted anti-TH2 biologic therapies. These studies reveal the effects of obesity on immunological disease and suggest a precision medicine approach to target the immune dysregulation caused by obesity.
Emerging clinical data of multiple immunological diseases including atopy and asthma indicate that obese patients have more severe disease and exhibit resistance to therapies that are effective in lean patients8–13, although the mechanisms underlying these observations remain unclear. To gain mechanistic insights into the immunopathological effects of obesity, we employed a well-characterized mouse model of atopic dermatitis14,15 (AD). We challenged obese mice fed high-fat diet (HFD) and lean controls with the vitamin D3 analogue MC903 to induce AD on the ear (Fig. 1a). The obese mice displayed a markedly increased inflammatory response, evidenced by an approximately twofold to fourfold increase in ear thickness relative to lean mice that received the same MC903 treatment (Fig. 1b). The MC903-treated ears of the obese mice had more severe erythema and scale, hallmarks of dermal inflammation (Fig. 1c). Histological evaluation demonstrated greater expansions of the epidermal and dermal layers in obese mice, along with a marked increase in leukocytic infiltration (Fig. 1d, dashed line). Of note, studies modulating duration of the HFD (Extended Data Fig. 1a–d) and using monogenic models of obesity on normal or HFD (Extended data Fig. 1e–j) suggested that the increased ear thickness and inflammation seen in obesity was at least partially dependent on HFD. Further, we observed a persistent inflammatory effect of obesity, even after weight loss (Extended Data Fig. 1k–m). Additionally, obesity increased disease severity, including after weight loss, in a second model of AD (Extended Data Fig. 2a–g), involving sensitization to ovalbumin (OVA) followed by serial tape-stripping and exposure to a mixture of OVA and papain (TOP). Finally, this increased inflammatory response was not limited to atopic diseases of the skin, as challenging lean and obese mice in an experimental model of allergic airway disease (ovalbumin sensitization and challenge) yielded increased cellular infiltration across multiple immune subsets in the bronchial–alveolar lavage fluid of obese mice and increased CD4+ and CD8+ T cells in the draining lymph node of obese mice (Extended Data Fig. 2h–j). Together, these results show that obesity exacerbates multiple mouse models of atopic disease.
Fig. 1 |. Obesity converts a classically TH2-driven inflammatory disease to a more severe TH17-driven disease that is worsened upon anti-TH2 antibody treatment.
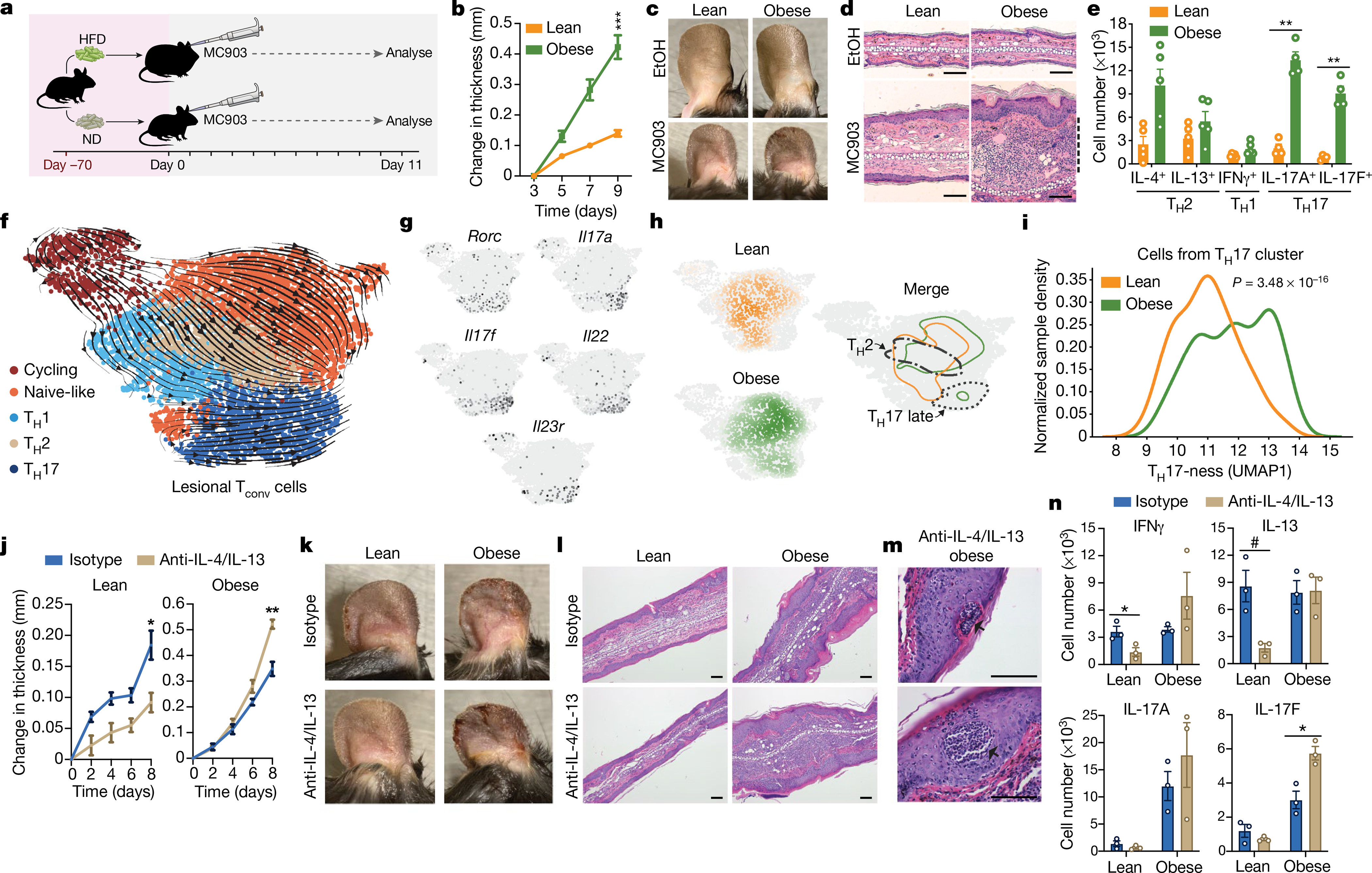
a, MC903-induced AD model with either high-fat diet (HFD) or normal diet (ND). b, Change in ear thickness during development of AD. c, Representative images of ears of mice treated with ethanol (EtOH) or MC903, on day 11. d, Representative haematoxylin and eosin (H&E) ear histology on day 11. Dashed line, leukocytic expansion of dermis. Original magnification, ×200. e, Total cytokine-competent CD4+ T cell numbers from whole ear, on day 11. f, RNA velocity visualization of transcriptional trajectories in UMAP space with Leiden clustering of scRNA-seq of Tconv cells from AD-challenged ears on day 10. g, Gene-expression heat maps of Rorc, Il17a, Il17f, Il22 and Il23r, with the highest-expressing cells in black. h, Distribution of Tconv cells in UMAP space (from f) from lean and obese mice after AD challenge. Contour lines are set at identical thresholds. Borders of TH2 and TH17 late clusters are outlined. TH17 late contour outlines cells in the top 50th percentile of Il17a, Il17f, Il22 and Il23r expression. i, TH17 cluster continuous probability density curves, scaled by sample. j–l, Change in ear thickness (j), with representative images from day 10 (k) and representative H&E ear histology from day 11 (l; original magnification, ×100) of lean or obese mice with induced AD treated with anti-IL-4/IL-13 or isotype control. m, Representative epidermal pustules (arrows) in obese mice with induced AD and treated with anti-IL-4/IL-13. Original magnification, ×400. n, Number of lesional CD4+ T cells with indicated cytokine competence (detected by intracellular cytokine staining and flow cytometry) from lean and obese mice treated with anti-IL-4/IL-13 or isotype control. Scale bars, 100 μm. n = 5 (b–e) except n = 4 for IL-17A and IL-17F CD4+ T cell measurements (e); n = 1 (pooled from 5 mice per group) for scRNA-seq in f–i; n = 5 (j–m) except n = 4 for lean mice with isotype control (j); n = 3 (pooled from 1–2 mice per sample) (n). Data are mean ± s.e.m. Peak values were tested with Welch’s t-test (b, j). P values adjusted for multiple comparisons using Holm-Šídák method (e); Mann–Whitney U test (i). #P < 0.06, *P < 0.05, **P < 0.01, ***P < 0.001.
The marked difference in atopic disease severity between lean and obese mice prompted us to profile the infiltrating T cell populations in the AD lesions using flow cytometry. Numbers of conventional CD4+ T (Tconv) cells were significantly increased in the obese mice, with trending increases in the regulatory T (Treg) and CD8+ T cell populations, suggestive of an overall increased inflammatory response (Extended Data Fig. 2k). Intracellular staining for cytokine competence revealed trending approximately 3.9- and 1.7-fold increases in CD4+ T cells from obese mice positive for the TH2 cytokines IL-4 and IL-13, respectively, in this cohort; however, we also observed an unexpected and prominent 6.5- and 11.5-fold increase in CD4+ T cells positive for the TH17 cytokines IL-17A and IL-17F, respectively (Fig. 1e, Extended Data Fig. 3a, b). We observed similar increases in TH17 cytokines in obese mice relative to lean mice challenged with TOP (Extended Data Fig. 4a, b) and in a model of allergic airway disease (Extended Data Fig. 4c).
More prominent TH17 inflammation may also occur in obese humans with allergic disease. Using a previously collected dataset of targeted serum proteomes of patients with AD, we identified two markers of cutaneous TH17 inflammation16,17 that correlated positively with body mass index (BMI) (Extended Data Fig. 5a–c). Separately, an analysis of lean, overweight, obese and morbidly obese individuals with asthma from a large, longitudinal, multi-centre study (SARP)18 demonstrated a clear decrease in sputum eosinophil percentage, a defining marker of the TH2-high endotype19, as a function of BMI (Extended Data Fig. 5d, e).
Although previous studies have demonstrated increased disease severity in obesity in TH17-dependent disease models such as multiple sclerosis and colitis20,21, our findings were surprising as experimental AD is classically a TH2-driven model of cutaneous autoimmunity, and suggested that obesity may shift the TH-responsive landscape away from TH2-directed inflammation towards an aberrant TH17 or unfocused inflammatory response. To further characterize this shift, we systematically profiled the lesional Tconv cells using single-cell RNA-sequencing (scRNA-seq) (Fig. 1f). Analysis revealed four Tconv cell populations (annotated here as naive-like, TH1, TH2 and TH17) and a cluster of cycling cells along with associated RNA velocity22,23 transcriptional trajectories visualized with uniform manifold approximation and projection (UMAP)24,25 (Fig. 1f; Methods). The gene-expression heatmaps of lineage-defining transcription factors (Gata3, Tbx21 and Rorc), lineage-defining effector cytokines and cytokine receptors (Il4, Il13, Ifng, Il17a, Il17f, Il22 and Il23r) and other prominent markers of activation, quiescence and memory (Ccr7, S1pr1, Sell, Klf2 and Bcl2) validated these population assignments (Fig. 1f, g, Extended Data Fig. 6a–c). Cells from lean and obese mice showed marked differences in distribution across the cell populations (Fig. 1h). In particular, we observed clear differences in the degree of TH17 differentiation between lean and obese mice (Fig. 1h, i). A proportion of the Tconv cells from the lean mice were assigned to an ‘early’ position (expressing the TH17 lineage-defining transcription factor Rorc, but not the effector cytokines Il17a, Il17f or Il22, or the cytokine receptor Il23r). By contrast, a majority of the TH17 cells from the obese mice were assigned to a ‘late’ position (expressing both Rorc, the effector cytokines and the cytokine receptor Il23r) (Fig. 1f–i). Of note, the emergence of these more-mature-appearing TH17 cells in the inflamed skin of obese animals probably depended on egress of lymphocytes from secondary lymphoid organs, as blocking lymphocyte egress from secondary lymphoid organs with FTY720 (an agonist of the sphingosine 1-phosphate receptor) decreased disease severity in obese mice and caused a marked increase in CD4+ T cells expressing IL-17A and IL-17F in the draining lymph node (Extended Data Fig. 7a–c). Together, these findings demonstrate that obesity alters the TH-response in AD and asthma to include substantial TH17 inflammation in addition to the canonical TH2 inflammation found in lean mice.
Biologic treatments blocking the signalling of the TH2 effector cytokines IL-4 and IL-13 have proved remarkably successful in managing TH2-driven severe allergic diseases such as asthma, sinusitis and AD26–29. Owing to the aberrant TH17 inflammation in AD in obese mice, we questioned to what extent neutralizing IL-4 and IL-13 would limit AD in these animals. We treated lean and obese mice with anti-IL-4 and anti-IL-13 neutralizing antibodies (anti-IL-4/IL-13) while eliciting experimental AD. As expected, anti-IL-4/IL-13 treatment strongly protected lean mice from AD (Fig. 1j–l). Notably, anti-IL-4/IL-13 treatment was not just ineffective in obese mice, it also worsened disease in both MC903-induced AD (Fig. 1j–l) and the TOP AD model (Extended Data Fig. 8a–d). Additionally, anti-IL-4/IL-13 treatment of obese mice with MC903-induced AD resulted in the emergence of small pustules, consistent with severe AD (Fig. 1m, arrows). Flow cytometric analyses of cytokine-competence demonstrated decreased IL-13-positive and IFNγ-positive CD4+ T cells after anti-IL-4/IL-13 treatment in lean but not obese mice (Fig. 1n). Whereas the small numbers of IL-7A-positive and IL-17F-positive CD4+ T cells tended to be even further decreased in lean mice after anti-IL-4/IL-13 treatment, the numbers of IL-17F-positive cells were increased in obese mice (with the IL-17A-positive and IFNγ-positive cells also trending higher in this cohort), suggesting that anti-IL-4/IL-13 TH2 blockade exacerbates non-TH2 inflammation in the obese animals and worsens disease.
The nature of a TH host response to a given immune challenge can shape the course of disease. Our observation of increased TH17 inflammation in the more severe manifestation of AD in obese mice led us to hypothesize that this immunological misfiring in obesity was an important causal step in disease pathophysiology and the negative response to anti-IL-4/IL-13 treatment. We aimed to identify a factor in vivo whose function was to protect the dominant TH2 response to experimental AD in lean mice. Such a factor likely would be expressed in TH2 cells and have dysregulated or decreased activity in obesity, and its T cell-specific ablation in lean mice might enable the emergence of non-TH2 inflammation in AD and lead to worsened disease upon anti-IL-4/IL-13 treatment. We focused on the nuclear hormone receptor (NHR) superfamily, a class of transcription factors that can be sensitive to systemic changes in physiologic and metabolic state. Among TH1, TH2 and TH17 cells, PPARγ30–33 (and its heterodimeric partner RXRα) stood out among NHRs as highly and differentially expressed in TH2 cells relative to TH1 and TH17 cells (Extended Data Fig. 9a–e). PPARγ is a transcription factor that was recently demonstrated to be important for regulating transcriptional networks in TH2 cells34. However, its physiological, in vivo relevance and functional role are not well understood35–41, with different studies demonstrating seemingly contradictory results regarding the effect of PPARγ activation or repression on TH2-associated diseases. Of note, one study demonstrated a role for PPARγ agonists in blunting the TH17 response and protecting mice against experimental autoimmune encephalomyelitis, a mouse model of multiple sclerosis, even though expression of Pparg in TH17 cells was not demonstrated42 (Extended Data Fig. 9a–e).
We assessed how obesity affects gene expression downstream of PPARγ in T cells. We performed chromatin immunoprecipitation with sequencing (ChIP–seq) of PPARγ in TH2 cells to identify its genome-wide binding sites (Extended Data Fig. 9f, g, Supplementary Information Table 1). We then interrogated our scRNA-seq dataset to determine whether the expression of the genes composing this PPARγ cistrome was altered in the lesional T cell subsets of lean and obese mice. In lean mice, genes in the PPARγ cistrome were most highly expressed in TH2 cells, as expected (Fig. 2a, b). However, expression of these genes in obese mice was decreased in TH2 cells and the related naive-like cells (and to a lesser degree in TH1 cells) compared with the corresponding subsets from lean mice (Fig. 2b), suggesting decreased PPARγ activity in T cells in obesity. By contrast, there was no change in PPARγ cistrome gene expression in TH17 and cycling cells between lean and obese mice. Together, these findings suggest that PPARγ activity is impaired in T cells in obesity. We hypothesized that PPARγ may act to preserve TH2 inflammatory predominance and that in vivo PPARγ deficiency could unfocus TH2 responses in favour of other TH cell responses.
Fig. 2 |. Mice with T cell-specific PPARγ deficiency largely phenocopy obese PPARγ-sufficient mice upon experimental AD challenge.

a, Heat map of PPARγ activity (ChIP–seq cistrome expression) using UMAP visualization of scRNA-seq data of Tconv cells from Fig. 1f. b, Violin plots representing T cell-PPARγ cistrome expression across distinct Leiden clusters from lean and obese mice. c, Change in ear thickness during MC903-induced AD development of PPARγ-TKO or control mice. d, Representative images of ears of mice as in c on day 10. e, Representative H&E ear histology of mice as in c on day 10. Dashed line highlights leukocytic expansion of dermis. Original magnification, ×200. f, Total cytokine-competent CD4+ T cell numbers assessed by flow cytometry from whole ear of mice as described in c on day 11. g, Distribution of Tconv cells in UMAP space (Fig. 1f) from lean control and PPARγ-TKO mice after AD challenge. Contour lines are set at identical thresholds. Borders of TH2 and TH17 late clusters are outlined. TH17 late contour defined as in Fig 1h. h, TH17 cluster continuous probability density curves of control and PPARγ TKO, scaled by sample. i–k, Change in ear thickness (i), representative images from day 10 (j), and representative H&E ear histology from day 11 (k; original magnification, ×100), of control or PPARγ-TKO mice with induced AD and treated with anti-IL-4/IL-13 or isotype control. l, Representative epidermal pustules (arrows) of PPARγ-TKO mice with induced AD and treated with anti-IL-4/IL-13. Original magnification, ×400. m, Lesional CD4+ T cell numbers with indicated cytokine competence. n, Proposed PPARγ function as focusing factor for TH2 responses in vivo. Control, Cd4cre; PPARγ TKO, Cd4crePpargfl/fl. Scale bars, 100 μm. n = 5 (c–e); n = 7 (f) except n = 4 for IL-17A/F-competent T cells; n = 1 (pooled from 5 mice per group) for scRNA-seq (g, h), run with Fig. 1 samples (batch 1 in Methods, ‘scRNA-seq’); n = 6 (i–l) except n = 5 for PPARγ TKO + isotype control; n = 3 (each pooled from 2 mice) (m). Data are mean ± s.e.m. Only peak values were tested with Welch’s t-test in (c, i). P values adjusted for multiple comparisons using Holm-Šídák method (f). Mann–Whitney U test (h). #P < 0.07, *P < 0.05, ***P < 0.001.
We tested this hypothesis by generating T cell-specific PPARγ-deficient mice (Cd4crePpargfl/fl (PPARγ TKO)). T cell-specific loss of PPARγ did not elicit overt systemic inflammation associated with spontaneous T cell dysregulation; PPARγ-TKO mice had normally sized spleens and lymph nodes (Extended Data Fig. 10a, b) and equivalent splenic and lymph node Treg (CD25+FOXP3+) and activated (CD62LloCD44hi) CD4+ T cell populations compared with controls (Cd4crePparg+/+) (Extended Data Fig. 10c, d). Additionally, PPARγ-TKO mice exhibited normal thymic T cell development, as measured by CD4 and CD8 expression of thymic T cells (Extended Data Fig. 10e). When challenged with experimental MC903 AD, the PPARγ-TKO lean mice developed more severe disease, similar but less extreme to that observed in wild-type obese mice (approximately 80% increase in change in ear thickness compared with control mice) (Fig. 2c, d). Histological examination in PPARγ-TKO mice also recapitulated many features of AD in obese Pparg-sufficient mice including the most distinctive histologic feature—leukocytic expansion of the dermis (Fig. 2e, dashed line). Flow cytometric analyses of cytokine competence revealed that the PPARγ-TKO mice had an increase in IL-17A-and IL-17F-positive CD4+ T cells while additionally demonstrating an increase in IL-13-positive CD4+ T cells and a trending increase in IL-4-and IFNγ-positive CD4+ T cells in this cohort (Fig. 2f). Supplementing the flow cytometry data with single-cell RNA-seq of lesional Tconv cells provided a more holistic picture, revealing that PPARγ-TKO mice exhibited a generally increased inflammatory response across all TH subtypes with a clear loss of TH2 selectivity (Fig. 2g). The Tconv cells from control mice largely overlapped with the TH2 cluster (Fig. 2g, merge), whereas cells from the PPARγ-TKO mice were more distributed among the naive-like, TH1 and TH17 clusters, and comparatively diminished in the TH2 cluster. Similar to what we observed in obese relative to lean Pparg-sufficient mice, the TH17 cells from the PPARγ-TKO mice were also further along their differentiation trajectory compared with the relatively few TH17 cells from control mice (Fig. 2g, h).
Given that T cell-specific ablation of PPARγ was associated with the emergence of non-TH2 inflammation and more severe disease in AD, we next investigated whether treatment with anti-IL-4/IL-13 would worsen AD in PPARγ-TKO mice. Indeed, anti-IL-4/IL-13 treatment exacerbated AD (Fig. 2i–k), with the emergence of small pustules (Fig. 2l, arrows) along with trending increases in IL-17A, IL-17F and IFNγ-positive lesional CD4+ T cells in treated PPARγ-TKO mice in this cohort (Fig. 2m). Of note, Treg cell-specific ablation of PPARγ43 (Foxp3crePpargfl/fl) did not lead to a significant difference in AD development and severity relative to controls (Foxp3crePparg+/+, Extended Data Fig. 10f–j). Together, these results strongly support the role of PPARγ as a focusing factor crucial to the in vivo TH2 response, serving to maintain TH2 responses against competing TH programs during an inflammatory challenge – a role not previously identified by in vitro investigations of PPARγ in TH2 cells (Fig. 2n).
Thiazolidinediones (TZDs) are an FDA-approved class of potent PPARγ agonists that are used as insulin-sensitizing medications to manage type 2 diabetes. Having observed that T cell-specific ablation of Pparg causes the aberrant emergence of non-TH2 inflammation and conversion of anti-IL-4/IL-13 treatment from therapy to anti-therapy, we hypothesized that pharmacological enforcement of PPARγ action via TZD treatment in obese mice may promote TH2 selective responses and suppress TH17 inflammation, reduce disease severity and perhaps even restore the therapeutic efficacy of anti-IL-4/IL-13 treatment. We treated obese control or PPARγ-TKO mice with rosiglitazone (the archetypal TZD) or DMSO and challenged the obese mice with AD. Rosiglitazone treatment significantly reduced AD severity in obese control mice, as measured by an approximately 40% reduction in the ear thickness increase (Fig. 3a) and a marked reduction in leukocytic infiltration (Fig. 3b, dashed line). These effects were dependent on T cell-specific PPARγ expression. In the PPARγ-TKO mice, rosiglitazone modestly increased disease severity (Fig. 3a, b), possibly related to T cell-extrinsic mechanisms of PPARγ action. Of note, although rosiglitazone treatment did not protect PPARγ-TKO mice from AD, its insulin-sensitizing effects were intact (Extended Data Fig. 11a–d), which argues for distinct therapeutic mechanisms of action for rosiglitazone in restoring organismal insulin sensitivity and protection from AD in obesity.
Fig. 3 |. Treatment with PPARγ agonist reduces TH17-inflammation and restores efficacy of anti-TH2 antibody treatment in obese mice challenged with AD.

a, Change in ear thickness during AD development of obese PPARγ-TKO or control mice treated with rosiglitazone (Rosi) or DMSO. b, Representative H&E ear histology on day 10 of cells treated as in a. c, RNA velocity visualization of transcriptional trajectories in UMAP space with Leiden clustering of scRNA-seq data of Tconv cells from AD-challenged ears on day 10. d, Distribution of Tconv cells in UMAP space from c isolated from obese control or PPARγ-TKO mice treated with DMSO or rosiglitazone after AD challenge. Contour lines set at identical thresholds. Borders of TH2 and TH17 clusters are outlined. e, TH17 cluster continuous probability density curves of obese control and PPARγ-TKO mice fed with DMSO or rosiglitazone-mixed HFD, scaled by sample. f, g, Violin plots representing T cell-PPARγ activity (ChIP–seq cistrome expression) in TH2 (f) and TH17 (g) cells from obese control or PPARγ-TKO mice treated with rosiglitazone or DMSO, using scRNA-seq data from c. h, i, Change in ear thickness (h) and representative H&E ear histology on day 10 (i; original magnification, ×100) of obese mice with induced AD fed DMSO- or rosiglitazone-mixed HFD, treated with anti-IL-4/IL-13 or isotype control. j, Representative epidermal pustules (arrows) of obese Bl6 mice with induced AD fed with DMSO-mixed HFD and treated with anti-IL-4/IL-13. Original magnification, ×400). k, Lesional CD4+ T cell number with indicated cytokine competence. l, Model of PPARγ agonists as immunopathological modifiers in obese mice to enable targeted therapy against a classically TH2-driven disease. Control, Cd4cre (except for scRNA-seq, when wild-type Bl6 mice co-housed with the PPARγ-TKO mice were used). Scale bars, 100 μm. Rosiglitazone-mixed HFD was introduced four weeks before initiation of experimental AD (h–k). n = 5 (a, b, h–j); n = 1 (pooled from 5 mice per group) for scRNA-seq (d, e); n = 3 (each pooled from 2 mice) (k). Data are mean ± s.e.m. Peak values tested with Welch’s t-test (a, h). Mann–Whitney U test (e). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Next, we investigated the effect of rosiglitazone treatment on the TH response profile in AD. We performed scRNA-seq to analyse the distribution of lesional Tconv cell states from obese control or PPARγ-TKO mice, with or without rosiglitazone treatment (Fig. 3c, Extended Data Fig. 12a–c). We observed that Tconv cells from obese control mice were distributed across the naive-like, TH1, TH2 and TH17 populations. Rosiglitazone treatment of obese control mice seemed to collapse their CD4+ T cell compartment to the TH2 cluster, a finding that was partially observed in the obese PPARγ-TKO mice (Fig. 3d). Further, rosiglitazone treatment strongly reduced the TH17 population in the obese control mice (Fig. 3d), although the rare TH17 cells present were somewhat more differentiated along the TH17 pathway relative to the TH17 cells from mice treated with DMSO (Fig. 3e). Additionally, PPARγ activity as measured by collective expression of the PPARγ T cell cistrome was increased by rosiglitazone in the TH2 and, to a lesser extent, TH17 cells (Fig. 3f, g, Supplementary Information Table 1), suggesting that rosiglitazone may activate PPARγ in TH2 and TH17 cells in vivo in concert with the consequent suppression of TH17 differentiation. Notably, the rosiglitazone-induced increase in PPARγ cistromic activity required PPARγ expression in T cells, and rosiglitazone treatment did not affect the degree of differentiation along the TH17 pathway among the TH17 cells in PPARγ-TKO mice (Fig. 3e–g). Finally, we hypothesized that if rosiglitazone treatment decreases immunological misfiring in obesity, enforcing a TH2-predominant immune response in AD, then it may be able to restore the efficacy of anti-IL-4/IL-13 treatment. Using regimens of rosiglitazone treatments in obese mice that did not on their own reduce AD severity, rosiglitazone indeed improved the outcomes of anti-IL-4/IL-13 for treatment of AD in obese mice (Fig. 3h–k, Extended Data Fig. 13a–c).
Obesity is a systemic pathophysiological state that is increasing in prevalence and incidence worldwide44–46, yet how obesity changes the architecture of immune responses is not well understood. Here, by studying the pathology of allergic inflammation in lean and obese mice with matched genetic backgrounds, we demonstrate that obese mice develop more severe disease associated with immunological ‘misfiring’47, converting an effective, targeted immunological therapy to an anti-therapy (Fig. 3l). Our demonstration that a PPARγ agonist can help to enforce an ‘on-target’ immunological response and restore efficacy for a targeted immunological therapy that would otherwise be harmful in this setting suggests a new strategy for precision immunotherapies to overcome altered pathology resulting from high-fat diet and obesity.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-022-04536-0.
Methods
Mice
Mouse studies were conducted at two separate academic research institutions: The Salk Institute for Biological Studies, USA and University of California, San Francisco (UCSF), USA. Mice were housed in specific pathogen-free facilities. When required, mice were purchased from The Jackson Laboratory. By default, mice within The Salk Institute for Biological Studies were fed with autoclaved normal chow (MI laboratory rodent diet 5001, Harlan Teklad) whereas mice fed at UCSF were given irradiated PicoLab Rodent Diet 20 5053. For experiments requiring the use of special diets, at the Salk Institute for Biological Studies and UCSF, diets were irradiated and purchased from Research Diets. The HFD used was 60 kcal% fat (D12492), the control normal diet was 10 kcal% fat (D12450J). Rosiglitazone was provided at 15 mg kg−1 of food. Unless specified, mice treated with rosiglitazone began treatment at the introduction of high-fat diet feeding. Mice designated as obese in this study were fed high-fat diet for at least 9 weeks, starting at an age of 6–7 weeks, and weighed at least 38 g (as an average within a cage). During the high-fat withdrawal studies, the high-fat diet (Research Diets, D12492) was replaced with control normal diet (D12450J), and the always obese and always lean control groups were fed with the corresponding high-fat diet or control normal diet from Research Diets. To definitively identify Tconv cells for FACS analyses, we used the Foxp3Thy1.1 (ref. 48) reporter mice. PPARγ-TKO mice were generated by crossing Cd4cre (ref. 49) transgenic mice and Pparg fl/fl (ref. 50) mice. PPARγ Treg cKO mice were generated by crossing Foxp3cre (Foxp3-Ires-YFP-cre)51 and Pparg fl/fl mice. All mice used for studies were male. All procedures involving animals were performed in accordance with protocols approved by the respective Institutional Animal Care and Use Committee (IACUC) of the Salk Institute for Biological Studies and UCSF.
MC903-induced experimental atopic dermatitis
Mice were anaesthetized with isoflurane and MC903 solution (0.1 mM in ethanol, 10 μl per ear, R&D Systems) was applied to mouse ears daily for 9–13 days. Ear thickness was assessed with a micrometer (Mitutoyo, 227–211). After collection, ears were dissected from the mice and prepared for histological analyses or flow cytometry.
TOP-induced experimental atopic dermatitis
Mice were injected intraperitoneally with an OVA-Imject Alum mixture (OVA (Sigma-Aldrich); Imject Alum Adjuvant (Thermo Fisher); mixture made per manufacturer instructions) on day −7. Seven days later, on day 0, mice were subjected to a treatment of tape-stripping (Shurtape, HP-500, 3 tape strips on both sides of the ear) and application of OVA–papain in PBS (250 μg OVA and 125 μg papain (Sigma-Aldrich, P3375) per 25 μl per mouse), which was repeated on days 2, 4 and 6. Ear thickness was assessed with a micrometer (Mitutoyo, 227–211). Upon collection on day 8, ears were dissected from the mice and prepared for histological analyses or flow cytometry.
Reanalyses of atopic dermatitis patient cohort
Peripheral blood serum from patients with moderate-to-severe AD (n = 59) were previously analysed by Olink Proseek multiplex assay using inflammation I, cardiovascular disease/CVD II, and CVD III multiplex panels. The proteins that were identified previously as differentially expressed genes from non-lesional keratinocytes treated with IL-13 or IL-17A were then correlated with patient BMI. Correlations between BMI and protein expression levels were assessed using Spearman correlation coefficients, and data are presented in scatter plots.
OVA-induced acute asthma
Mice were sensitized on days 0, 7 and 14 by intraperitoneal injection of 50 μg of OVA (Sigma-Aldrich) emulsified in 1 mg of aluminium potassium sulfate. One week after the last sensitization, mice were intranasally challenged on 3 consecutive days with 100 μg OVA in 40 μl saline. Twenty-four hours after the last OVA challenge, mice were sacrificed for downstream analyses including cell counting of bronchoalveolar lavage fluid and flow cytometric analyses of the lung-draining lymph nodes.
Reanalyses of asthma patient cohort
Clinical data were originally collected as part of the NHLBI Severe Asthma Research Program52 and reanalysed for correlation with patient BMI. One-way ordinary ANOVA followed by a test of linear trend was used to determine statistical significance.
FTY720 treatments
Mice were injected intraperitoneally, daily with FTY720 (1 mg kg−1) or water starting one day before initiation of the MC903 challenge through conclusion of challenge.
Antibody treatments
The following antibodies were used: anti-mouse IL-4 (BioXCell, 11B11), anti-mouse IL-13 (InvivoGen, 8H8), and isotype control (IgG1, BioXCell, MOPC-21). The antibodies were injected intraperitoneally at days 0, 3, 6, and 8 of the MC903-induced AD challenge at a dose of 0.25 mg per mouse for anti-IL-4, 0.1 mg per mouse for anti-IL-13, and 0.35 mg per mouse for isotype control (IgG1). For the TOP–AD model, the antibodies were injected intraperitoneally at days 0, 2, 4 and 6 with the same dosage as for the MC903-AD model. Injection volumes never exceeded 150 μl.
Single-cell suspension of ear-infiltrating immune cells
Dissected ears were minced into fine pieces (1–2 mm3), and digested in a stromal vascular isolation buffer (HBSS with calcium and magnesium, 20 mg ml−1 BSA, 20 mg ml−1 penicillin, 20 mg ml−1 streptomycin) containing 2 mg ml−1 Collagenase D (Roche) at 37 °C with intermittent shaking for 2 h. The suspension was then passed through a 100-μm mesh to remove undigested clumps and debris. The flow-through was centrifuged at 400 RCF for 10 min. The pellet containing the stromal vascular fraction was washed once in 10 ml RPMI, and the resultant isolated cells were prepared for FACS analysis directly or first stimulated with PMA and ionomycin in the presence of brefeldin A (GolgiPlug; BD) for 5 h at 37 °C for subsequent intracellular cytokine staining and then prepared for FACS analysis.
Histological analyses
Sections (5 μm) of fixed tissues were stained with haematoxylin and eosin according to standard procedures. Histopathological analyses were conducted on blinded samples for severity and extent of inflammation and morphological changes by a pathologist.
Flow cytometry
The following antibodies were used. Biolegend: CD3 (145-2C11), CD4 (RM4-5), CD8 (53–6.7), CD25 (7D4), CD44 (IM7), CD45.2 (104), CD62L (MEL-14), CD90.1 (Thy1.1, OX-7) and IFN-γ (XMG1.2), IL-17F (9D3.1C8), TCRγ/δ (GL3); eBioscience: FOXP3 (FJK-16s), IL-4 (11B11), IL-13 (eBio13A), IL-17A (eBio17B7). We used the eBioscience Fixable Viability Dye eFluor780 or DAPI to distinguish live from dying or dead cells. For intracellular staining, cells were treated with fixation and permeabilization reagents from eBioscience and labeled with appropriate antibodies before being analysed. Data were analysed using a LSRII, LSRFortessa, or FACSAria instrument (Becton Dickinson) and FlowJo software (FlowJo LLC). Cells were sorted on a BD FACSAria cell sorter.
scRNA-seq
scRNA-seq was performed in two batches on separate samples with the Chromium Single Cell 3′ Reagent Kit, v3 chemistry (10x Genomics, PN-1000092) following the manufacturer’s protocol. The same procedure was followed for both Chromium runs unless otherwise specified. In brief, cells were sorted by FACS (sorting on live, CD45.2+TCRγδ−CD4+ cells on a BD FACS Aria cell sorter), resuspended at 500–900 cells per μl in PBS + 1% FBS, and loaded into the Chromium Controller (10x Genomics, PN-1000202) for an anticipated targeted recovery of 3,000–6,000 cells depending on sample recovery from FACS. We performed 11 cycles of PCR for cDNA amplification after GEM recovery, and 25% of each cDNA sample was carried into transcriptome library preparation. We performed 13 cycles of PCR to introduce Chromium i7 multiplex indices (10x Genomics, PN-120262). cDNA was diluted 1:5 in buffer EB and quantified by Bioanalyzer DNA High Sensitivity (Agilent, 5067–4626) and Qubit dsDNA High Sensitivity (Thermo Fisher, Q32854) reagents. Samples were pooled equally and sequenced on a HiSeq 4000 (Illumina) using read parameters 28 × 8 × 98.
Raw fastq files were mapped to the mouse transcriptome (‘refdata-cellranger-mm10-3.0.0’) using Cell Ranger (10x Genomics, version 3.0.2) and ‘filtered_feature_bc_matrix.h5’ files were loaded into Scanpy (version 1.6.0)53 for processing. For batch 1 (lean, obese, Cd4cre and PPARγ TKO), 2,500–4,500 cell barcodes were recovered per sample, totalling 12,597 cells before filtering. For batch 2 (control-DMSO, control-Rosi, PPARγ TKO-DMSO and PPARγ TKO-Rosi), 3,100–5,500 cell barcodes were recovered per sample, totalling 15,867 cells before filtering. Cells with fewer than 200 detected genes, more than 6,000 detected genes, and/or mitochondrial read fraction higher than 25% were removed from the complete dataset. After filtering, 12,034 and 15,658 cells from batches 1 and 2, respectively, were carried through for subsequent analyses. Counts were log normalized and scaled before regressing out total UMI counts and mitochondrial fraction per cell. Principal component analysis (PCA) was performed on highly variable genes, and the first 20 PCA components were used to construct a UMAP24,25. Cell clustering was performed using the Leiden algorithm54 (version 0.7.0, resolution 0.9). Two (batch 1) or three (batch 2) small clusters identified as contaminating antigen presenting cells were excluded from downstream analyses. Additionally, Foxp3-expressing cells were excluded from analyses in this study. We performed Gaussian kernel density estimation (Scanpy ‘embedding_density’) to visualize the distribution of samples in UMAP space. Continuous probability density curves were generated using Seaborn (version 0.10.0) kdeplot function with ‘common_norm=False’ to account for differences in sample number. Mann–Whitney U statistics were calculated using Scipy.stats (version 1.4.1) mannwhitneyu function. The PPARγ activity score was calculated with the score_genes function (Scanpy) using filtered genes associated with the top 50% of peaks annotated by HOMER (see ‘ChIP–seq library generation’) compared against a random, size-matched control gene set.
RNA velocity
Cellranger bam files were processed with Velocyto (version 0.17.17) using the default Cell Ranger mm10 gtf reference file (version 3.0.0) with repeat masking (UCSC). Output files were loaded into scVelo (version 0.2.2) (https://scvelo.readthedocs.io) and merged with the gene-expression anndata object from Scanpy. Low abundance genes (less than 30 total counts) were filtered from the merged dataset. Cell moments were calculated considering 30 nearest neighbors and 30 principal components and used to estimate RNA velocities using default settings. The ‘TH17-ness’ metric was inferred from (1) the directionality of RNA Velocity vectors along the UMAP1 coordinate (x-axis) and (2) a composite TH17 score calculated from Il17a, Il17f, Il22 and Il23r expression using the score_genes function (Scanpy).
In vitro CD4+ T cell differentiation
CD4+ T cells were isolated from the spleen and lymph nodes using the EasySep Mouse CD4 Positive Selection Kit II (Stemcell Technologies). Naive (CD25−CD44hiCD62Llo) CD4+ T cells were sorted by flow cytometry from the bead purified CD4+ T cells. The naive CD4+ T cells were resuspended in Click’s medium (Irvine Scientific) at 1 million cells per ml, and then plated on day 0 in 24 well plates coated with goat anti-hamster IgG antibody (200 ng ml−1; MP Biomedicals) with the addition of soluble anti-CD3 (1 μg ml−1; 145-2C11) and anti-CD28 (1 μg ml−1; 37.51) from Bio X Cell. Polarizing conditions for different T helper subsets are as following: TH1: human IL-2 (100 U ml−1; PeproTech), mouse IL-12 (20 ng ml−1; PeproTech) and anti-IL-4 (5 μg ml−1; Bio X Cell); TH2: human IL-2 (100 U ml−1; PeproTech), mouse IL-4 (20 ng ml−1; Biolegend), anti-IFN-γ and anti-IL-12 (5 μg ml−1; Bio X Cell); TH17: mouse IL-6 (20 ng ml−1; Biolegend), human TGF-β (2 ng ml−1; PeproTech), anti-IFN-γ and anti-IL-12 (5 μg ml−1; Bio X Cell).
RNA-seq library generation and sequencing analysis
Total RNA was extracted from TH1, TH2 and TH17 cells approximately 96 h after initiation of in vitro differentiation. RNA-sequencing libraries were prepared from 100 ng total RNA (TrueSeq v2, Illumina) and single-end sequencing was performed on the Illumina HiSeq 2500, using bar-coded multiplexing and a 100 bp read length, yielding a median of 34.1 million reads per sample. Read alignment and junction finding was accomplished using STAR55 and differential gene expression with Cuffdiff 2 (ref. 56), utilizing UCSC mm10 as the reference sequence. Transcript expression was calculated as gene-level relative abundance in fragments per kilobase of transcript per million mapped fragments and employed correction for transcript abundance bias57.
Primers for quantitative PCR.
Pparg: forward, CACAATGCCATCAGGTTTGGG; reverse: GAAATGCTTTGCCAGGGCTC. Gata3: forward, CTTCCCACCCAGCAGCCTGC, reverse: CGGTACCATCTCGCCGCCAC. Tbx21: forward, GTCGCGCTCAGCAACCACCT, reverse: CGGCCACGGTGAAGGACAGG. Rorc: forward, CCGGACATCTCGGGAGCTGC, reverse: CGGCGGAAGAAGCCCTTGCA. Hprt: forward, GTCATGCCGACCCGCAGTCC, reverse: GGCCACAATGTGATGGCCTCCC.
Antibodies for western blot
Rabbit anti-PPARγ mAb (81B8, Cell Signaling), mouse anti-tubulin (DM1A, Sigma).
ChIP–seq library generation
Naive CD4+ T cells were activated and polarized in TH2 conditions. On days 1 and 2, T cells were transduced with a retroviral vector expressing TY1-tagged Pparg. On day 4, transduced T cells were collected for ChIP as described previously58 using a TY1 antibody (Sigma, SAB4800032). ChIP–seq libraries were constructed and sequenced (100 bp single end reads) as described previously13. Short DNA reads were demultiplexed using Illumina CASAVA v1.8.2. Reads were aligned against the mouse mm10 reference genome using the Bowtie2 aligner with standard parameters that allow up to 2 mismatches per read. Peak calling, motif analyses, and other data analysis were performed using HOMER, a software suite for ChIP–seq analysis as described previously58. Visualization of ChIP–seq results was achieved by uploading custom tracks onto the UCSC genome browser.
Glucose tolerance test
Fasting was induced for 6 h. Glucose (1.5 g kg−1, intraperitoneally) was injected and serum was collected for glucose and insulin measurements. Blood glucose was monitored using a Nova Max Plus glucometer. Serum insulin levels (Ultra Sensitive Insulin, Crystal Chem) were measured by ELISAs.
Statistical analyses
Statistical analyses were performed with Prism 9 (GraphPad). P values were calculated using Welch’s t-test unless otherwise noted. Mice cohort size was designed to be sufficient to enable statistical significance to be accurately determined. When applicable, mice were randomly assigned to treatment or control groups. Experimental data was not excluded from the statistical analyses, except for exclusions due to technical errors or loss of confidence in appropriate controls. The investigators were not blinded in the studies unless otherwise stated. Appropriate statistical analyses were applied, assuming a normal sample distribution, unless otherwise specified. No estimate of variance was made between each group. All in vivo experiments were conducted with at least two independent cohorts unless otherwise specified. All in vitro TH differentiation experiments were conducted at least three times. Unless otherwise noted, data from one instance of the repeated experiments are represented in this manuscript. Bulk and single-cell RNA-seq experiments and ChIP–seq experiments were conducted once using multiple biological samples per group (as indicated in figure legends).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The bulk RNA-sequencing and ChIP–seq data can be accessed in the NCBI Sequence Read Archive under the accession PRJNA553761. Additionally, the ChIP–seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under the accession code GSE189216. The single cell-RNA sequencing data used in this study are deposited in the GEO under the accession code GSE189476. Other relevant data are available from the corresponding authors upon reasonable request. Source data are provided with this paper.
Code availability
Code used in this manuscript can be provided by reasonable request to the corresponding authors.
Extended Data
Extended Data Fig. 1 |. Diet-induced and monogenic models of obesity provoke increased disease severity upon MC903-AD challenge.
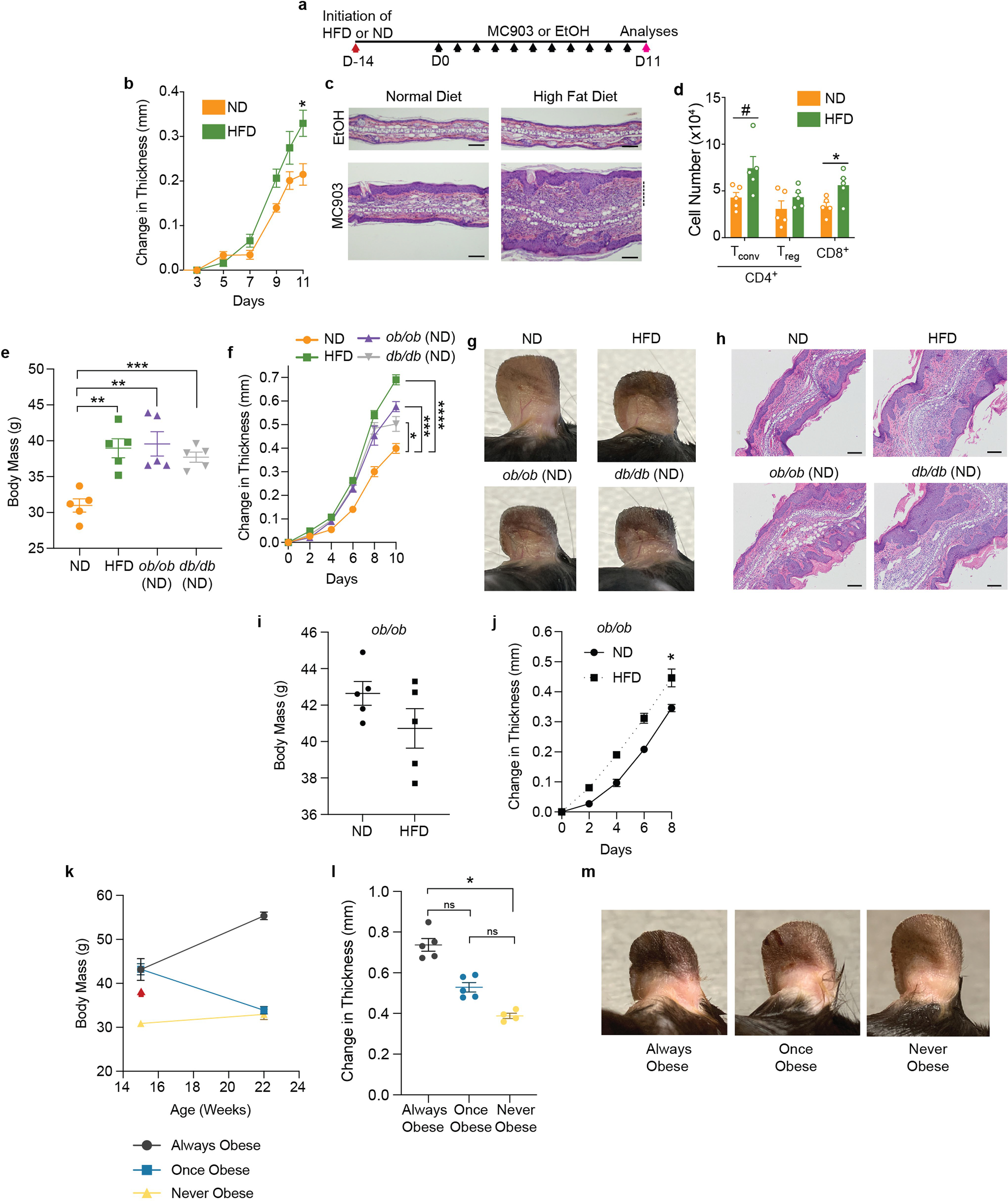
a, Scheme of MC903-induced atopic dermatitis disease model where animals are fed either high fat diet (HFD) or normal diet (ND) for two weeks before initiating MC903 treatments. Black arrows indicate MC903 or EtOH administration to ear. b, Change in ear thickness during AD development as in (a). c, Representative H&E ear histology, Day 11. Scale bars, 100 μm. Dashed line, leukocytic expansion of the dermis. d, Total Tconv, Treg, and CD8+ T cell number from whole ear, Day 11. e–h, Body mass (e), change in ear thickness during AD challenge (f), representative images at Day 10 of challenge (g), and representative H&E ear histology, Day 10 (h) of ND-fed mice, HFD-fed obese mice, or ND-fed ob/ob or db/db mice. i, j, Body mass at Day 0 (i) and change in ear thickness upon AD challenge (j) of 9 week old ob/ob mice fed ND or HFD for three weeks. k–m, Timeline of body mass (k), change in ear thickness (l), and representative images at Day 10 of challenge (m) of always obese (AO), once obese/now lean (OO), and never obese (NO) mice. Red arrow (k) indicates replacement of HFD with control lean diet for OO mice. n = 5 for all groups in (b–m) except 4 never obese (NO) mice were used in (l) and (m). Data are mean ± s.e.m. Peak values were tested with Welch’s t-test in (b, j) and ordinary one-way ANOVA with all groups tested against ND in (f). Ordinary one-way ANOVA with all groups tested against ND in (e) and with all groups tested against all groups in (l). P values were adjusted for multiple comparisons in (d) using Holm-Šídák method and Šídák’s multiple comparisons test in (e, f, l). ns – not significant, #P < 0.07, *P < 0.05, **P < 0.01, ***P < 0.001.
Extended Data Fig. 2 |. Increased inflammation in obesity in multiple models of allergic inflammatory disease.
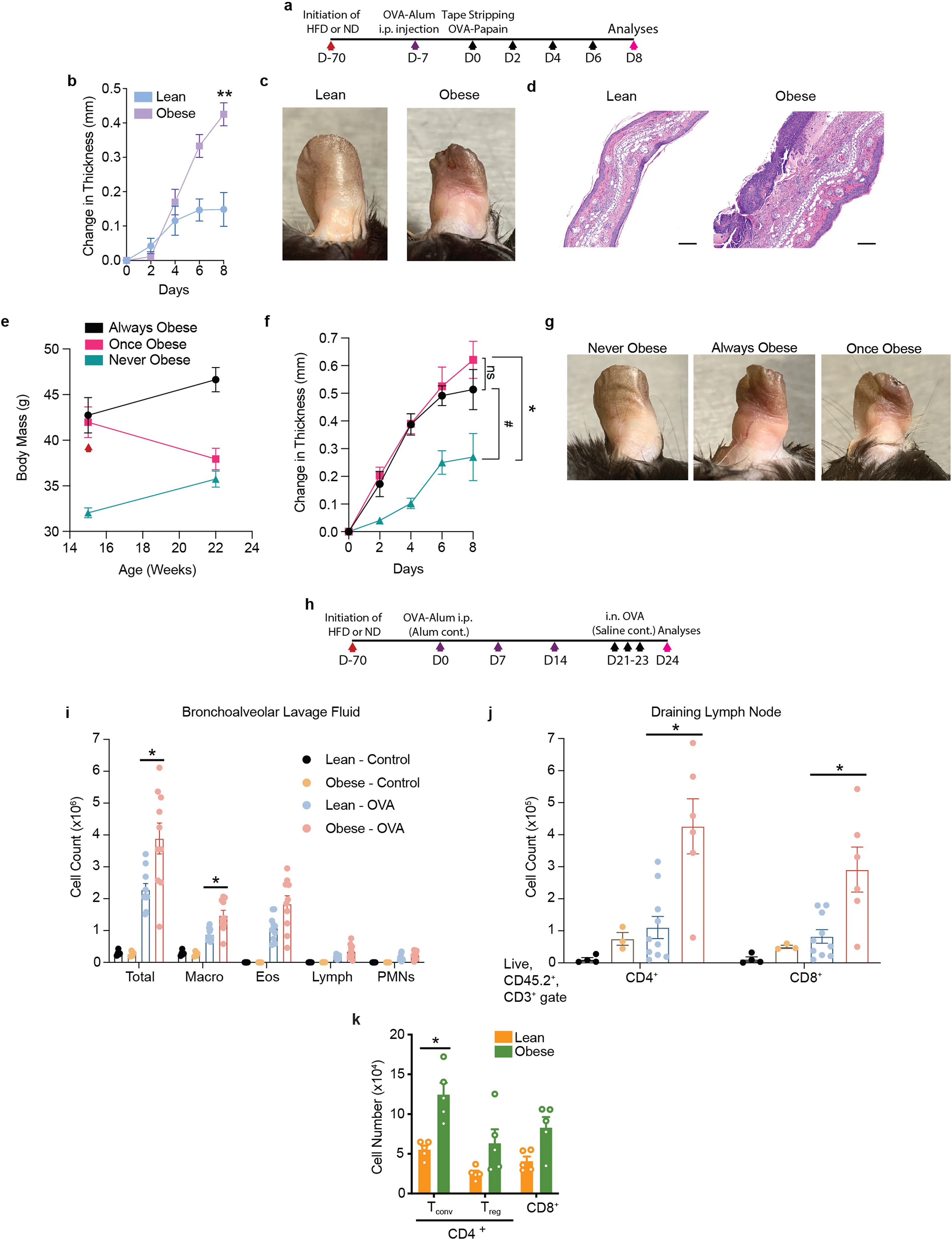
a, Scheme of TOP (Tape stripping followed by Ova-Papain exposure)-induced atopic dermatitis disease model with either high fat diet (HFD) or normal diet (ND). Black arrows indicate tape stripping followed by OVA-papain administration. b, Change in ear thickness during AD development. c, Representative pictures of ears at Day 8 of challenge. d, Representative H&E ear histology, Day 8. Scale bars, 100 μm. e–g, Timeline of body mass (e), change in ear thickness (f), and representative pictures of ears (g) at Day 8 of challenge of always obese (AO), once obese/now lean (OO), and never obese (NO) mice. Red arrow (e) indicates replacement of HFD with control lean diet for OO mice. h, Scheme of OVA-Alum allergic asthma model. Purple arrows indicate OVA-Alum i.p. injection. i, Total immune cell numbers from bronchoalveolar lavage fluid upon sacrifice on Day 24. j, Total CD4+ and CD8+ T cell numbers from lung draining lymph node, Day 24. k, Total Tconv, Treg, and CD8+ T cell numbers from whole ear of lean and obese mice challenged with MC903-AD, Day 11. Asthma model was conducted once. Macro, macrophages; Eos, eosinophils; Lymph, lymphocytes; PMNs, polymorphonuclear leukocytes; n = 5 for (b–g); n = 10 for Lean-OVA and Obese-OVA, n = 5 for Lean-Control, and n = 4 for Obese-Control in (i); n = 10 for Lean-OVA, n = 6 for Obese-OVA, n = 4 for Lean-Control, and n = 3 for Obese-Control in (j). n = 5 for all groups in (k). Data are mean ± s.e.m. Peak values were tested with Welch’s t-test in (b) and ordinary one-way ANOVA with preselected followup tests as indicated in (f). P values were adjusted for multiple comparisons using Šídák’s multiple comparisons test in (f) and the Holm-Šídák method in (i–k) where only the Lean-OVA and Obese-OVA groups were compared in (i, j). #P < 0.12, *P < 0.05, **P < 0.01.
Extended Data Fig. 3 |. Selected flow cytometry gating strategies for evaluating cytokine-competence.
a, b, Different T cell subsets were identified through the use of distinct antibody cocktails. Here, using obese mice, we show how the skin-resident/infiltrating hematopoietic cells can be analyzed by flow cytometry to identify CD4+ T cells (a) and selected cytokine competence of those CD4+ T cells (b).
Extended Data Fig. 4 |. Multiple models of allergic inflammatory disease demonstrate increased TH17-driven inflammation in obesity.

a, b, Total IL4+, IL13+, IFNγ+, IL17A+, and IL17F+ cell numbers of CD4+ T cells from whole ear (a) or ear skin draining lymph node (b) at Day 8 of mice challenged with TOP-induced AD. c, Total IL4+, IL13+, IFNγ+, IL17A+, and IL17F+ cell numbers of CD4+ T cells from bronchoalveolar lavage fluid upon sacrifice on Day 24 of mice challenged with experimental allergic airway disease (ovalbumin sensitization and challenge). n = 5 for all groups in (a, b); n = 10 for Lean and n = 6 for Obese in (c). Mann-Whitney tests were conducted in (a, b) and P values were adjusted for multiple comparisons using Holm-Šídák method in (a–c). *P < 0.05, **P < 0.01.
Extended Data Fig. 5 |. Evidence of increased TH17 inflammation or decreased TH2-associated pathology in obese patients with allergic disease.

a, Scheme demonstrating workflow and integration of human AD patient serum proteomics with cytokine-induced gene expression studies of human keratinocytes (KCs) correlated with patient BMI. b, c, Scatterplots depicting the serum protein levels from AD patients of genes specifically induced by IL17A (b) or IL13 (c) in human KCs versus patient BMI. n = 59 moderate-to-severe AD patients for scatter plots in (b, c). r and p values for scatter plots in (b, c) were obtained using Spearman rank correlation. d, e, Sputum eosinophil percentage from human severe asthma patients across a range of BMIs, on their first clinical baseline visit as part of SARP, represented irrespective of age of onset (d) or broken down by age of onset (e). n = 272, 211, and 95 for patients with BMI <30, 30–40, and >40, respectively in (d). n = 113, 83, and 52 (pediatric-onset patients) and n = 70, 66, and 23 (adult-onset patients) with BMI <30, 30–40, and >40, respectively in (e). P value from test of linear trend (post-test after statistically significant ordinary one-way ANOVA) in (d, e).
Extended Data Fig. 6 |. Gene heatmaps from scRNA-Seq data used in Fig. 1 and Fig. 2.

Heatmaps of transcription factors (a), cytokines (b), and markers of activation, quiescence, and memory (c) overlain on UMAP plot from Fig. 1f to assign names to clusters. Grayscale indicates gene expression, with the highest expressing cells in black.
Extended Data Fig. 7 |. Blocking lymphocyte egress from secondary lymphoid organs during AD challenge reduces disease severity and TH17 inflammation in the lesions of obese mice.

a, Change in ear thickness during development of MC903-AD of lean or obese mice treated with vehicle (water) or FTY720. b, Representative pictures of ears at Day 10 of challenge. c, Total IL4+, IL13+, IFNγ+, IL17A+, and IL17F+ CD4+ T cell numbers assessed by flow cytometry from draining lymph node of lean (left) and obese (right) mice treated with vehicle or FTY720. n = 5 for all groups in (a–c). Data are mean ± s.e.m. Peak values were tested with Welch’s t-test in (a). P values were adjusted for multiple comparisons using Holm-Šídák method in (c). *P < 0.05, **P < 0.01, ****P < 0.0001.
Extended Data Fig. 8 |. Targeted anti-IL-4/IL-13 blockade is ineffective in obese mice challenged with TOP.
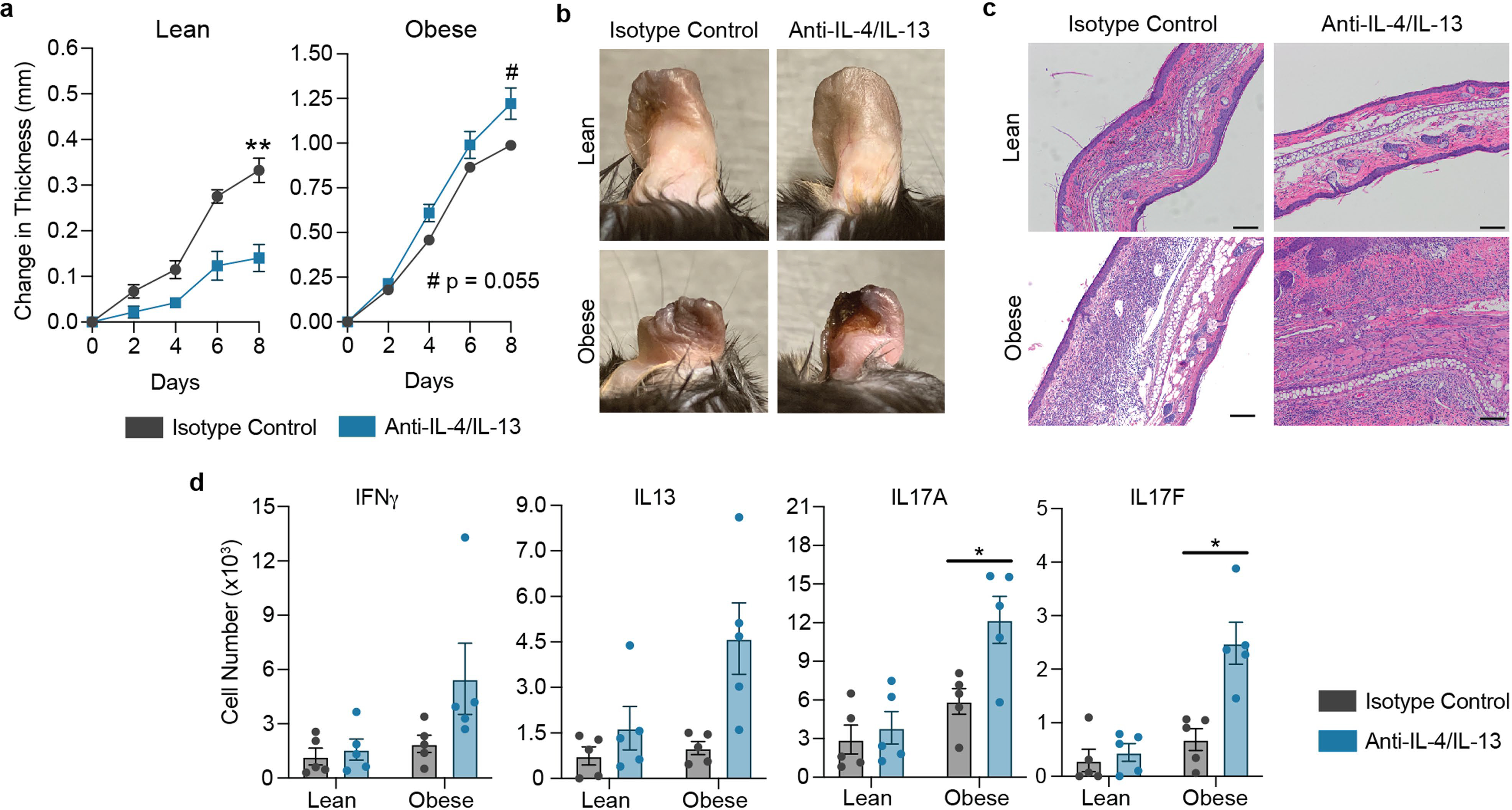
a, Change in ear thickness during development of TOP-AD of lean or obese mice treated with anti-IL-4/IL-13 or IgG1 isotype control. b, Representative pictures of ears at Day 8 of challenge. c, Representative images of H&E-stained histology of ears at Day 8. Scale bars, 100 μm. d, Lesional number of CD4+ T cells with the indicated cytokine competence as measured by intracellular cytokine staining via flow cytometry from lean and obese mice treated with anti-IL-4/IL-13 or isotype control. n = 5 for all groups in (a–d). Data are mean ± s.e.m. Peak values were tested with Welch’s t-test in (a). P values were adjusted for multiple comparisons using Holm-Šídák method in (d). #P < 0.06, *P < 0.05, **P < 0.01.
Extended Data Fig. 9 |. PPARγ is differentially expressed in in vitro differentiated TH2 cells.
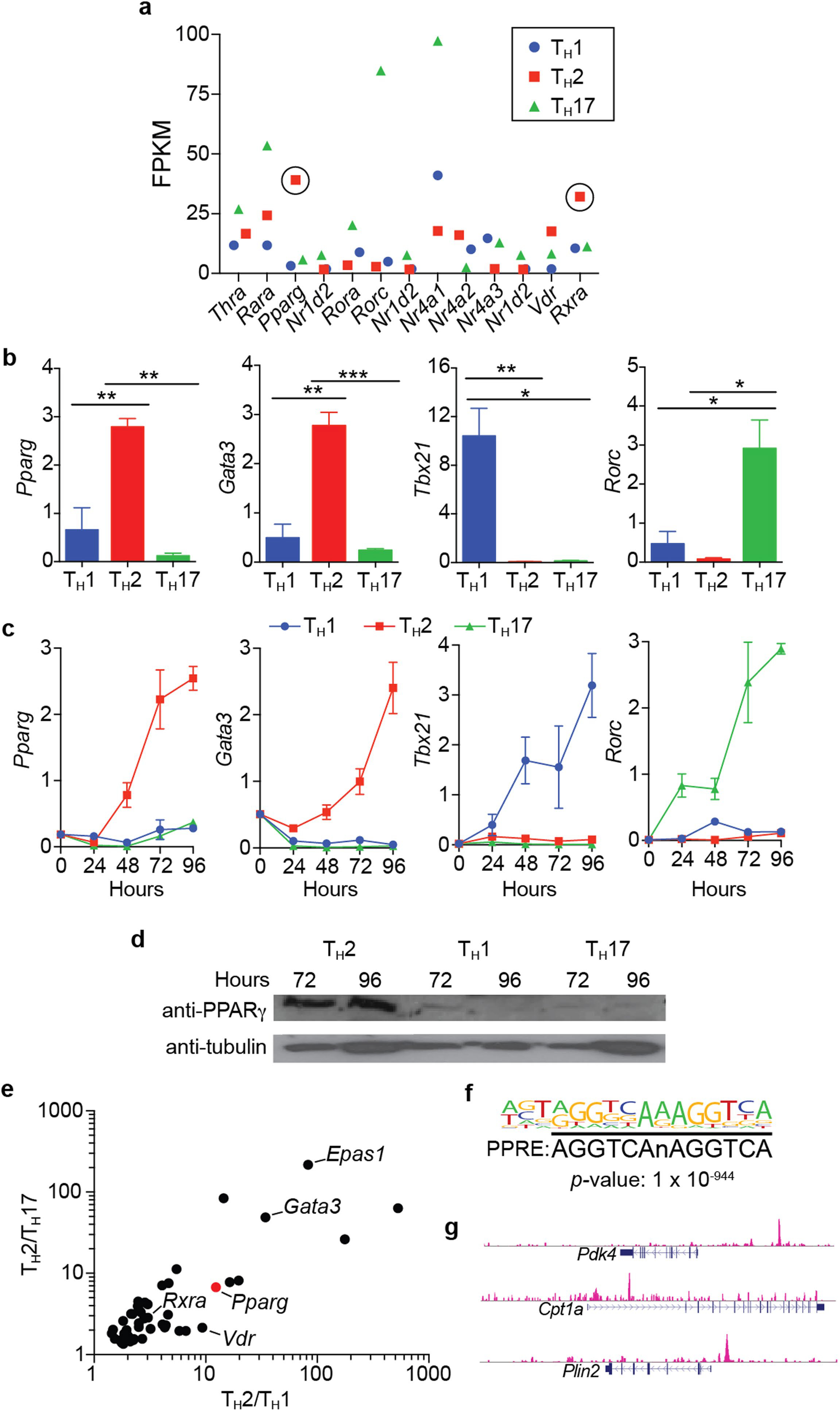
a, Fragments per kilobase of transcripts per million mapped reads (FPKM) values of nuclear hormone receptor (NHR) superfamily genes differentially expressed in in vitro differentiated TH1, TH2, and TH17 cells. NHR genes that are differentially expressed in TH2 cells are encircled (cells pooled from 4 mice before inducing differentiation in triplicate, same data set used in (e)). b, c, Relative expression (using Hprt expression as housekeeping gene) of indicated genes in in vitro differentiated TH1, TH2, and TH17 cells (cells pooled from 4 mice before inducing differentiation in triplicate). b, Gene expression determined at Hour 120 post induction of differentiation. c, Time course of gene expression from Hours 0–96 post induction of differentiation. d, Western blot of PPARγ and tubulin at Hours 72 and 96 in in vitro differentiated TH1, TH2, and TH17 cells (cells pooled from 3 mice before inducing differentiation). e, FPKM values of genes that are differentially expressed in TH2 cells and involved in transcriptional regulation. Position of Pparg is marked with a red dot. f, Top scoring DNA motif of PPARγ ChIP-Seq peaks in in vitro differentiated TH2 cells via de novo analysis (cells pooled from 4 mice). g, Visualization of PPARγ ChIP-Seq experiment utilizing UCSC genome browser across following genomic loci: Pdk4, Cpt1a, Plin2. Data are mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, Student’s t-test.
Extended Data Fig. 10 |. PPARγ TKO mice display neither overt systemic inflammation nor altered T cell development, and PPARγ Treg cKO (Foxp3Cre Ppargfl/fl) mice do not display an overt difference in AD severity relative to controls.

a–e, Comparison between PPARγ TKO and Control (Cd4Cre Pparg+/+) mice. a, Picture of control and PPARγ TKO spleens and lymph nodes. b, Spleen mass. c, d, Treg (c) and activated Tconv (d) cell frequency in spleen and LN. e, Developing T cell subsets in thymus. f–j, Comparison between PPARγ Treg cKO and Control (Foxp3Cre Pparg+/+) mice. f, Change in ear thickness during development of atopic dermatitis. g, Representative images of H&E-stained histology of ears at Day 10. Scale bars, 100μm. Dashed line, leukocytic expansion of the dermis. h, Absolute lesional leukocyte number assessed by flow cytometry at Day 13. i, Lesional activated Tconv cells as a percentage of Tconv cells assessed by flow cytometry at Day 13. j, Lesional Treg cells as a percentage of CD4+ T cells assessed by flow cytometry at Day 13. Control, Foxp3Cre Pparg+/+. LN, lymph node. For (a–e), n = 3 mice per group. For (f–i), n = 4 mice per group. Data are mean ± s.e.m.
Extended Data Fig. 11 |. PPARγ in T cells is dispensable for the insulin-sensitizing action of TZDs.
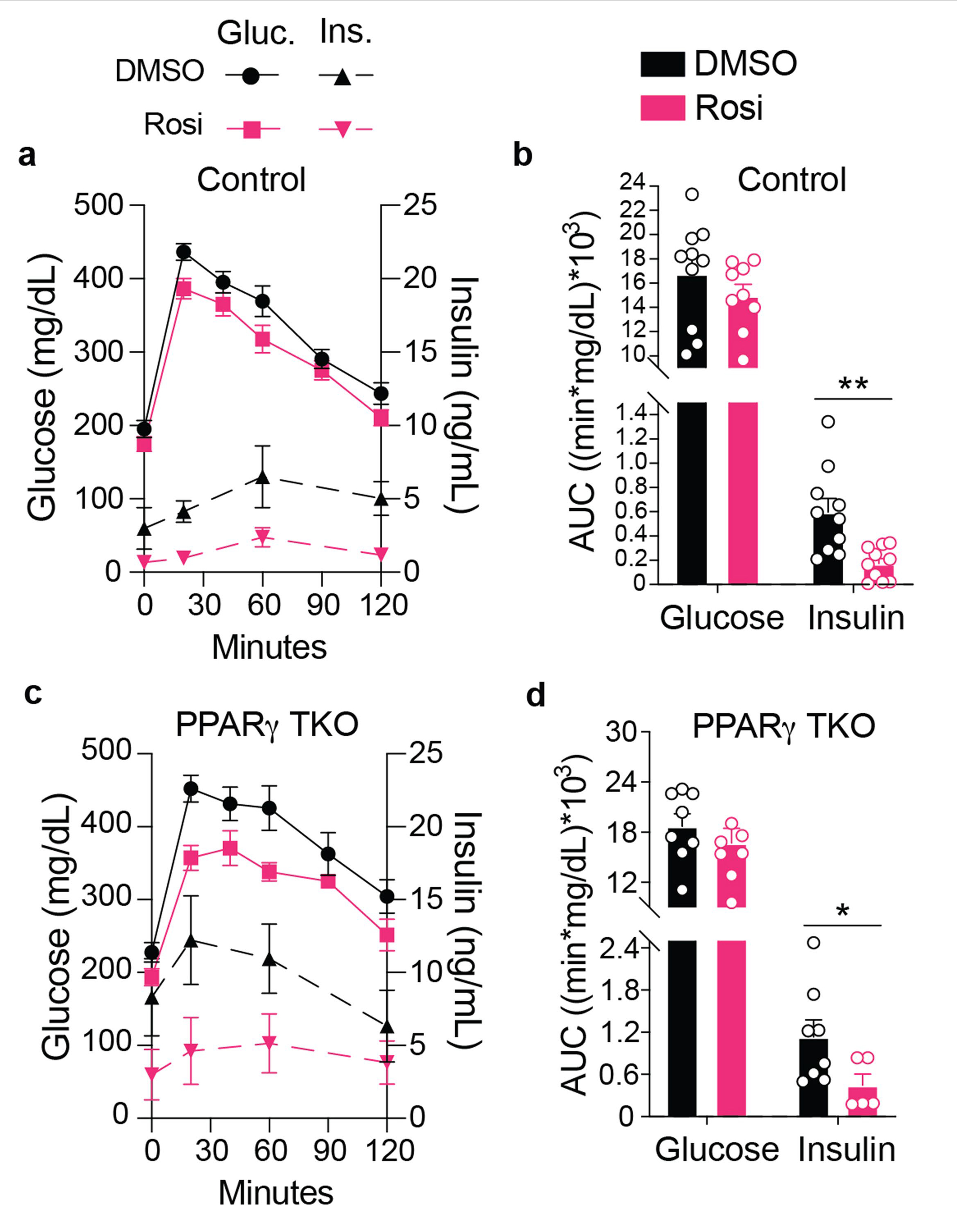
a–d, Glucose tolerance tests (GTTs) of Control (a, b) or PPARγ TKO (c, d) mice that have been treated with or without Rosi. Glucose and insulin traces (a, c) and corresponding area under the curves (AUCs) (b, d) are presented. Control, Cd4Cre; PPARγ TKO, Cd4Cre Ppargfl/fl; For (a–d) n = 10 for all Control mice with exception in (a, b) where n = 9 for the glucose measurements for mice treated with Rosi; n = 8 for PPARγ TKO mice with exception in (c, d) where n = 7 for the PPARγ TKO Rosi-treated mice that were sampled for the serum glucose measurements and n = 5 for the PPARγ TKO Rosi-treated mice that were sampled for the serum insulin measurements. Gluc., Glucose; Ins., Insulin; Rosi, Rosiglitazone. This experiment was conducted once with a weight- and age-matched cohort. Data are mean ± s.e.m. *P < 0.05, **P < 0.01.
Extended Data Fig. 12 |. Gene heatmaps from scRNA-Seq data used in Fig. 3.

Heatmaps of transcription factors (a), cytokines/cytokine receptors (b), and markers of activation, quiescence, and memory (c) overlain on UMAP plot from Fig. 3c to assign names to clusters. Grayscale indicates gene expression, with the highest expressing cells in black.
Extended Data Fig. 13 |. Pictures of the ears of obese DMSO- or Rosi-HFD fed mice challenged with AD while treated with anti-IL-4/IL-13 or IgG1 isotype control (a), and initiating Rosi treatment at AD-challenge onset prevents worsening of disease upon treatment with anti-IL-4/IL-13 in obese mice (b, c).
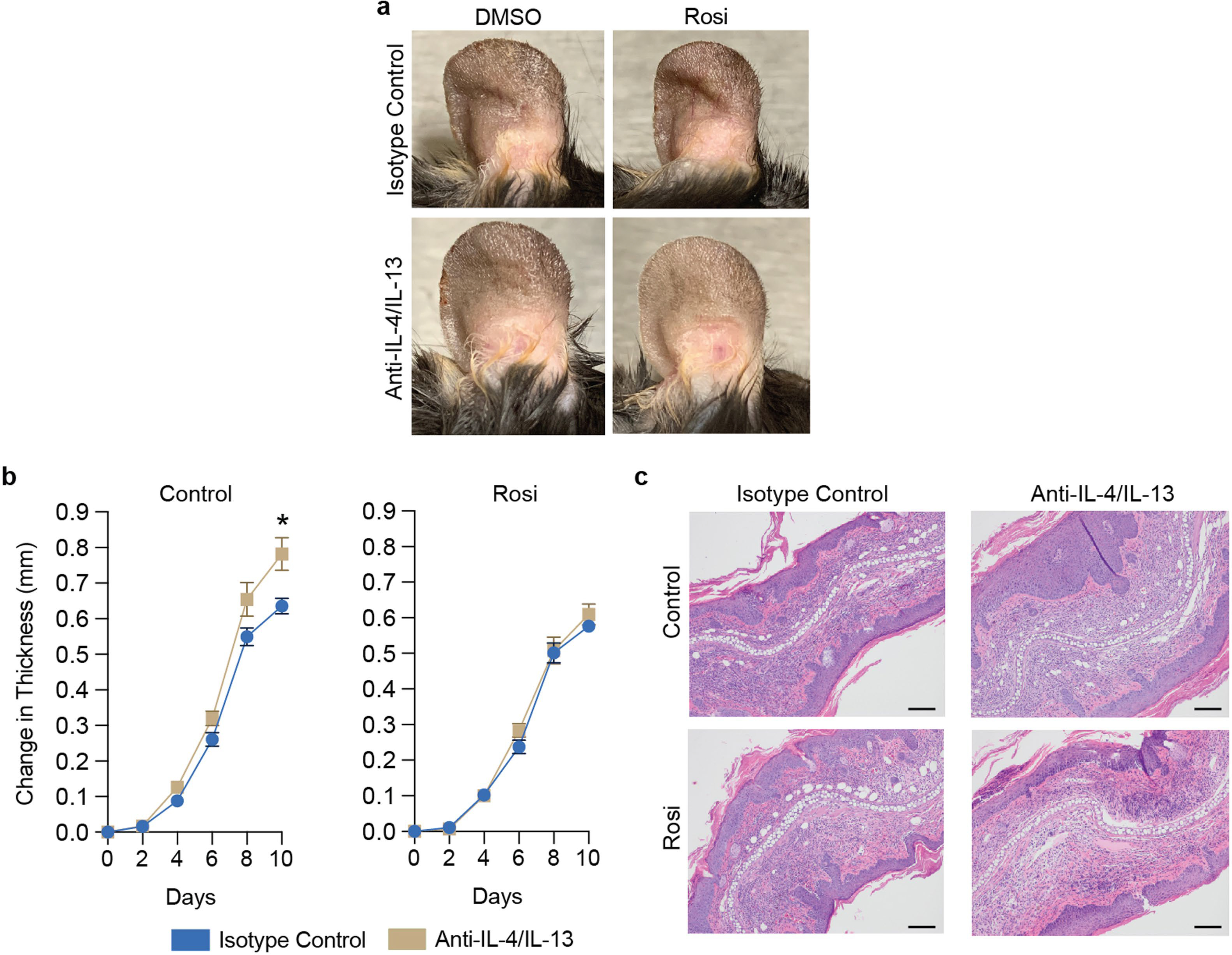
a, Representative pictures of ears at Day 10. b, Change in ear thickness during development of MC903-AD of obese mice treated with Rosi or DMSO with anti-IL-4/IL-13 or IgG1 isotype control. c, Representative images of H&E-stained histology of ears at Day 10. Scale bars, 100 μm. n = 5 for all groups in (a–c). Rosi-mixed HFD introduced four weeks prior to initiation of experimental AD in (a) and upon AD-challenge onset in (b, c). Data are mean ± s.e.m. Peak values were tested with Welch’s t-test in (b). *P < 0.05.
Supplementary Material
Acknowledgements
We thank A. Levine for advice and critical review of the manuscript; E. Marsh, B. Shy, S. Dodgson and B. Schaar for review of the manuscript; S. Pyle for scientific graphic illustration; Y. Dai, J. Alvarez, A. Cheng and Y. Zhang for technical assistance; and L. Ong, C. Brondos, J. Woo, J. Okano and J. Sawin for administrative assistance. We thank the Severe Asthma Research Program (SARP) for access to their patient data. S.P.B. was supported by U.S. National Institutes of Health (NIH) grants F30 DK096828, T32 GM007198, R38 HL143581 and K38 HL154202. J.L.S. and B.S.N. were supported by U.S. NIH grant P30GM127211. J.M.S. was supported by KAIST (N11210257) and the National Research Foundation of Korea (2017K1A1A2013124 and 2021R1A2C200757311). I.-K.L was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2017R1A2B3006406). R.L.G. is supported by NIH grants U01AI52038, R01AI53185, R01AR076082, R01DK121760 and R37AI052453. L.E.C.A. was supported by an American Heart Association grant 16BGIA27790079 and a VA BLR&D Career Development Award 1IK2BX001313. R.M.E. holds the March of Dimes Chair in Molecular and Developmental Biology at the Salk Institute, a SWCRF Investigator Award, and is supported by the NOMIS Foundation–Science of Health, and by U.S. NIH grants HL147835, HL105278, and CA014195. This research was made possible by funding from HHMI, the Foundation Leducq, the Don and Lorraine Freeberg Foundation, the Larry L. Hillblom foundation, and David C. Copley Foundation. Y.Z. was supported by the NOMIS Foundation, the Crohn’s and Colitis Foundation, the Leona M. and Harry B. Helmsley Charitable Trust and the National Institute of Health (R01-AI107027, R01-AI1511123, R21-AI154919, S10-OD023689 and NCI CCSG P30-014195). A.M. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund, is an investigator at the Chan Zuckerberg Biohub, and is a recipient of the Cancer Research Institute (CRI) Lloyd J. Old STAR grant. The Marson laboratory has received funds from the Innovative Genomics Institute (IGI), the Simons Foundation, and the Parker Institute for Cancer Immunotherapy (PICI). The Marson laboratory has received gifts from the Byers family, B. Bakar, K. Jordan, and E. Radutzky. This work was also supported by National Cancer Institute funded Salk Institute Cancer Center core facilities (CA014195) and the James B. Pendleton Charitable Trust as well as the UCSF Parnassus Flow Cytometry Core funded in part by the Diabetes Research Center grant from the National Institutes of Health (P30 DK063720). Research reported in this publication was supported by the National Institute of Environmental Health Sciences of the National Institutes of Health under Award Number P42ES010337 and by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Numbers R01DK057978 and R01DK120480 (all to R.M.E.). The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing interests
E.G.Y. has received institutional research funds from and is a consultant for Abbvie, Almirall, Amgen, Asana Biosciences, AstraZeneca, Boerhinger-Ingelhiem, Cara Therapeutics, Celgene, Elli Lilly, Galderma, Glenmark/Ichnos Sciences, Janssen, Kyowa Kirin, Leo Pharma, Pfizer, Regeneron, and UCB. E.G.Y. has received institutional research funds from AnaptysBio, Innovaderm, KAO, Kiniksa, Novan, Novartis, and Ralexar. E.G.Y. is a consultant for Arena, Aslan Pharmaceuticals, Bristol-Myers Squibb, Connect Pharma, EMD Serono, Evidera, Incyte, Pandion Therapeutics, RAPT Therapeutics, Sanofi, SATO Pharmaceutical, Siolta Therapeutics, Target Pharma Solutions, and Ventyx Biosciences. R.L.G. is a consultant and has equity interest in MatriSys Biosciences and Sente Inc. C.J.Y. is a Scientific Advisory Board member for and holds equity in Related Sciences and ImmunAI, a consultant for and holds equity in Maze Therapeutics, and a consultant for TReX Bio. C.J.Y. has received research support from Chan Zuckerberg Initiative, Chan Zuckerberg Biohub, and Genentech. A.M. is a compensated cofounder, member of the boards of directors and a member of the scientific advisory boards of Spotlight Therapeutics and Arsenal Biosciences. A.M. is a cofounder, member of the boards of directors and a member of the scientific advisory board of Survey Genomics. A.M. is a compensated member of the scientific advisory board of NewLimit. A.M. owns stock in Arsenal Biosciences, Spotlight Therapeutics, NewLimit, Survey Genomics, PACT Pharma, and Merck. A.M. has received fees from PACT Pharma, Juno Therapeutics, Trizell, Vertex, Merck, Amgen, Genentech, AlphaSights, 23andMe, ALDA, Rupert Case Management and Bernstein. A.M. is an investor in and informal advisor to Offline Ventures and a client of EPIQ. The Marson lab has received research support from Juno Therapeutics, Epinomics, Sanofi, GlaxoSmithKline, Gilead and Anthem.
Footnotes
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-022-04536-0.
Peer review information Nature thanks Percy Knolle, Ruslan Medzhitov and Sarah Teichmann for their contribution to the peer review of this work.
Reprints and permissions information is available at http://www.nature.com/reprints.
References
- 1.Buck MD, Sowell RT, Kaech SM & Pearce EL Metabolic instruction of immunity. Cell 169, 570–586 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Man K, Kutyavin VI & Chawla A Tissue immunometabolism: development, physiology, and pathobiology. Cell Metab. 25, 11–26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geltink RIK, Kyle RL & Pearce EL Unraveling the complex interplay between T cell metabolism and function. Annu. Rev. Immunol 36, 461–488 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Michelet X et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol 19, 1330–1640 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Nicholas DA et al. Fatty acid metabolites combine with reduced β-oxidation to activate Th17 inflammation in human type 2 diabetes. Cell Metab. 30, 447–461.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ringel AE et al. Obesity shapes metabolism in the tumor microenvironment to suppress anti-tumor immunity. Cell 183, 1848–1866.e26 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang C et al. STAT3 activation-induced fatty acid oxidation in CD8+ T effector cells is critical for obesity-promoted breast tumor growth. Cell Metab. 31, 148–161.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hersoug LG & Linneberg A The link between the epidemics of obesity and allergic diseases: does obesity induce decreased immune tolerance? Allergy 62, 1205–1213 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Wenzel SE Asthma phenotypes: the evolution from clinical to molecular approaches. Nat. Med 18, 716–725 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Zhang A & Silverberg JI Association of atopic dermatitis with being overweight and obese: a systematic review and metaanalysis. J. Am. Acad. Dermatol 72, 606–616.e4 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Fahy JV Type 2 inflammation in asthma—present in most, absent in many. Nat. Rev. Immunol 15, 57–65 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bieber T Atopic Dermatitis. New Engl. J. Med 358, 1483–1494 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Eckert L et al. The burden of atopic dermatitis in US adults: health care resource utilization data from the 2013 National Health and Wellness Survey. J. Am. Acad. Dermatol 78, 54–61.e1 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Li M et al. Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc. Natl Acad. Sci. USA 103, 11736–11741 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leyva-Castillo JM et al. Skin thymic stromal lymphopoietin initiates Th2 responses through an orchestrated immune cascade. Nat. Commun 4, 2847 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Swindell WR et al. Heterogeneity of inflammatory and cytokine networks in chronic plaque psoriasis. PLoS ONE 7, e34594 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiricozzi A et al. Integrative responses to IL-17 and TNF-α in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J. Invest. Dermatol 131, 677–687 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Hastie AT et al. Baseline sputum eosinophil 1 neutrophil subgroups’ clinical characteristics and longitudinal trajectories for NHLBI Severe Asthma Research Program (SARP 3) cohort. J. Allergy Clin. Immunol 146, 222–226 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuruvilla ME, Lee FE-H & Lee GB Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin. Rev. Allerg. Immuol 56, 219–233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winer S et al. Obesity predisposes to Th17 bias. Eur. J. Immunol 39, 2629–2635 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Endo Y et al. Obesity drives Th17 cell differentiation by inducing the lipid metabolic kinase ACC1. Cell Rep. 12, 1042–1055 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Manno GL et al. RNA velocity of single cells. Nature 560, 494–498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergen V, Lange M, Peidli S, Wolf FA & Theis FJ Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol 38, 1408–1414 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Becht E et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 37, 38–44 (2018). [DOI] [PubMed] [Google Scholar]
- 25.McInnes L, Healy J & Melville J UMAP: uniform manifold approximation and projection for dimension reduction. Preprint at 10.48550/arXiv.1802.03426 (2018). [DOI]
- 26.Gooderham MJ et al. Dupilumab: A review of its use in the treatment of atopic dermatitis. J. Am. Acad. Dermatol 78, S28–S36 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Hearn AP, Kent BD & Jackson DJ Biologic treatment options for severe asthma. Curr. Opin. Immunol 66, 151–160 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Tubau C & Puig L Therapeutic targeting of the IL-13 pathway in skin inflammation. Expert Rev. Clin. Immunol 17, 15–25 (2021). [DOI] [PubMed] [Google Scholar]
- 29.Laidlaw TM et al. Dupilumab improves upper- and lower-airway disease control in chronic rhinosinusitis with nasal polyps and asthma. Ann. Allergy Asthma Immunol. 126, 584–592.e1 (2021). [DOI] [PubMed] [Google Scholar]
- 30.Rosen ED & Spiegelman BM PPARγ: a nuclear regulator of metabolism, differentiation, and cell growth. J. Biol. Chem 276, 37731–37734 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Tontonoz P & Spiegelman BM Fat and beyond: the diverse biology of PPARγ. Annu. Rev. Biochem 77, 289–312 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Ahmadian M et al. PPARγ signaling and metabolism: the good, the bad and the future. Nat. Med 19, 557–566 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lazar MA Maturing of the nuclear receptor family. J. Clin. Invest 127, 1123–1125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henriksson J et al. Genome-wide CRISPR screens in T helper cells reveal pervasive crosstalk between activation and differentiation. Cell 176, 882–896.e18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woerly G et al. Peroxisome proliferator-activated receptors α and γ down-regulate allergic inflammation and eosinophil activation. J. Exp. Med 198, 411–421 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Honda K, Marquillies P, Capron M & Dombrowicz D Peroxisome proliferator–activated receptor γ is expressed in airways and inhibits features of airway remodeling in a mouse asthma model. J. Allergy Clin. Immunol 113, 882–888 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Hammad H et al. Activation of peroxisome proliferator-activated receptor-γ in dendritic cells inhibits the development of eosinophilic airway inflammation in a mouse model of asthma. Am. J. Pathol 164, 263–271 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee KS et al. Modulation of airway remodeling and airway inflammation by peroxisome proliferator-activated receptor γ in a murine model of toluene diisocyanate-induced asthma. J. Immunol 177, 5248–5257 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Chen T et al. PPAR-γ promotes type 2 immune responses in allergy and nematode infection. Sci. Immunol 2, eaal5196 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Nobs SP et al. PPARγ in dendritic cells and T cells drives pathogenic type-2 effector responses in lung inflammation. J. Exp. Med 214, 3015–3035 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stark JM, Tibbitt CA & Coquet JM The metabolic requirements of Th2 cell differentiation. Front. Immunol 10, 2318 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klotz L et al. The nuclear receptor PPARγ selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J. Exp. Med 206, 2079–2089 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bapat SP et al. Depletion of fat-resident Treg cells prevents age-associated insulin resistance. Nature 528, 137–141 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.NCD-RisC. Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19·2 million participants. Lancet 387, 1377–1396 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.The GBD 2015 Obesity Collaborators. Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med 377, 13–27 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abarca-Gómez L et al. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet 390, 2627–2642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lucas C et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 584, 463–469 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liston A et al. Differentiation of regulatory Foxp3+ T cells in the thymic cortex. Proc. Natl Acad. Sci. USA 105, 11903–11908 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee PP et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 15, 763–774 (2001). [DOI] [PubMed] [Google Scholar]
- 50.He W Adipose-specific peroxisome proliferator-activated receptor knockout causes insulin resistance in fat and liver but not in muscle. Proc. Natl Acad. Sci. USA 100, 15712–15717 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rubtsov YP et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Hastie AT et al. Mixed sputum granulocyte longitudinal impact on lung function in the severe asthma research program. Am. J. Resp. Crit. Care 203, 882–892 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolf FA, Angerer P & Theis FJ SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Traag VA, Waltman L & Eck NJV From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep 9, 5233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trapnell C et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol 31, 46–53 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roberts A, Pimentel H, Trapnell C & Pachter L Identification of novel transcripts in annotated genomes using RNA-seq. Bioinformatics 27, 2325–2329 (2011). [DOI] [PubMed] [Google Scholar]
- 58.Cho H et al. Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature 485, 123–127 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The bulk RNA-sequencing and ChIP–seq data can be accessed in the NCBI Sequence Read Archive under the accession PRJNA553761. Additionally, the ChIP–seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under the accession code GSE189216. The single cell-RNA sequencing data used in this study are deposited in the GEO under the accession code GSE189476. Other relevant data are available from the corresponding authors upon reasonable request. Source data are provided with this paper.