

Abstract
The preset neurodegenerations in Alzheimer disease (AD) are due to several mechanisms such as amyloidogenic proteolysis, neuroinflammation, mitochondrial dysfunction, neurofibrillary tangles, cholinergic dysfunction, among others. The aim of this work was to develop multitarget molecules for the treatment of AD. Therefore, a family of 64 molecules was designed based on ligand structure pharmacophores able to inhibit the activity of beta secretase (BACE1) and acetylcholinesterase (AChE) as well as to avoid amyloid beta (Aβ1–42) oligomerization. The backbone of designed molecules consisted of a trisubstituted aromatic ring, one of the substituents was a heterocyclic amine (piperidine, morpholine, pyrrolidine or N-methyl pyrrolidine) separated from the aromatic system by three carbon atoms. The set of compounds was screened in silico employing molecular docking calculations and chemoinformatic analyses. Based on Gibbs free energy of binding, binding mode and in silico predicted toxicity results, three of the best candidates were selected, synthesized, and evaluated in vitro; F3S4-m, F2S4-m, and F2S4-p. All three compounds prevented Aβ1–42 aggregation (F3S4-m in 30.5%, F2S4-p in 42.1%, and F2S4-m in 60.9%). Additionally, inhibitory activity against AChE (ki 0.40 μM and 0.19 μM) and BACE1 (IC50 15.97 μM and 8.38 μM) was also observed for compounds F2S4-m and F3S4-m, respectively. Despite the BACE IC50 results demonstrated that all compounds are very less potent respect to peptidomimetic inhibitor (PI-IV IC50 3.20 nM), we can still say that F3S4-m is capable to inhibit AChE and BACE1.
1. Introduction
Alzheimer’s disease (AD) is a cognitive disorder produced by the decrease of neurons and synaptic connectivity [1]. Over the years, and with the help of numerous investigations, the multifactorial nature of AD has been confirmed and many theories to understand its development have been raised [2]. While some theories focus on how gut-microbiota and oxidative stress play a key role in neurological conditions [3, 4], others study receptor proteins involved in the pathogenesis of AD [5], such as the amyloidogenic pathway, the hyperphosphorylation of Tau protein pathway, and some other theories attempt to get an understanding of the molecular mechanism of the cholinergic system which is involved in the failure of cognition [6–10].
The amyloid hypothesis involves the presence of extracellular plaques formed by amyloid beta peptide (Aβ) of lengths between 39 to 43 amino acid (aa) residues, with molecular weight 4–6 KDa. However, Aβ of 42 aa residues (Aβ1–42) represents the most amyloidogenic and toxic form [11]. The Aβ is the natural product of the metabolism of the amyloid precursor protein (APP) which is present in neurons and can be processed by the action of several proteases by two pathways. The most common route is the non-amyloidogenic pathway, where the APP is processed by the catalysis of α-secretase and γ-secretase. However, in the AD most of the APP is processed by the amyloidogenic pathway where APP is hydrolyzed by β-secretase-1 (BACE1) and γ-secretase. The hydrolysis of APP by BACE1 allows the Aβ peptide formation [12]. For this reason, several of the current therapies are focused on the inhibition of BACE1 to decrease Aβ production [13]. Once Aβ1–42 is released, it undergoes conformational changes from α-helix to β-sheet conformation, where the limiting event is the formation of an electrostatic interaction between the lateral chains of Asp23 or Ala42 and Lys28 acquiring a β-turn or S-shape which allow intermonomer interaction forming oligomers and fibrils of Aβ1–42 [14, 15]. The Aβ1–42 oligomers and fibrils deposited in the brain induce cytotoxicity at pre-frontal cortex, the hippocampus, the amygdala, and the Basal nucleus of Meynert [16]. Some of these brain areas are related to memory where an important cholinergic innervation is found acting principally the Acetylcholine (ACh) neurotransmitter hydrolyzed by acetylcholinesterase enzyme (AChE) [17]. Then, several AChE inhibitors have been approved by the Food and Drug Administration for AD treatment since AChE is responsible for degrading the ACh [18–20].
Considering that AChE and BACE1 are important targets for the treatment of AD, the catalytic sites of both enzymes had been fully elucidated [21, 22]. The main active site of AChE is the catalytic triad (His447, Glu334 and Ser203), and behind this, there is a secondary site or peripheral anion site (PAS) containing Tyr337 [23–25]. The catalytic site of BACE1 is formed by Asp32, Asp228 and Glu125 that contribute to the anionic charges observed on the interaction surface of the enzyme. Amino acids such as Tyr68 and Tyr71 are also found on BACE1, giving aromatic characteristics to the catalytic site to establish π-π interactions with aromatic rings [26, 27].
It has been shown that the majority of Aβ1–42 oligomerization inhibitors are those containing a tertiary amine and an aromatic ring [28]. Additionally, it has been suggested that the vast majority of BACE1 inhibitors establish tight hydrogen bonding network with aspartic residues (Asp32 and Asp228) located in the catalytic cleft of the enzyme and are also capable of forming other hydrogen bonds and π-π interactions with the aromatic residues also located at the catalytic site [26]. Having in mind the type of interactions required to inhibit BACE1 and Aβ1–42 oligomerization and considering that glutamic and aspartic residues present on the active sites of BACE1 and AChE which are negatively charged, a family of 64 new molecules were designed with a tertiary amine on its structure which is capable to protonate at physiological pH and stablish electrostatic interactions with the negative catalytic site of the enzymes. Other chemical moieties were also incorporated in the structure of ligands aiming to establish hydrogen bonding, hydrophobic and π-π interactions. This family of compounds contains a heterocyclic amine such as piperidine, morpholine, pyrrolidine or N-methyl pyrrolidine linked three atoms away from a trisubstituted aromatic ring with different functional groups such as secondary or primary amines, aliphatic moieties, ester, or alcohol groups. In addition, the molecular weight of designed molecules was less than 300 g/mol. All designed molecules were screened in silico by molecular docking studies towards the three selected molecular targets (AChE, BACE1 and Aβ1–42) based on the Gibbs free energy results (ΔG) and in the binding mode, as well as in the toxicity results, and the Blood Brain Barrier (BBB) permeability measured under chemoinformatic studies, three of the best compounds named F2S4-m, F2S4-p and F3S4-m were selected for synthesis and evaluated in vitro. The results showed that F3S4-m was the best compound to inhibit both BACE1 and AChE activities and was able to avoid the aggregation of Aβ1–42, despite not having the best performance to inhibit Aβ1–42 aggregation as the other two compounds.
2. Materials and methods
2.1. In silico studies
2.1.1. Target selections
The 3D structures of the proteins were retrieved from Protein Data Bank (PDB: http://www.rcsb.org/pdb/home/home.do) for docking studies; BACE1 (PDB ID: 2QP8), AChE (PDB ID: 4PQE) and Aβ1–42 in α-helix with PDB ID: IZ0Q, Aβ1–42 in β-Sheet with PDB ID: 2BEG and the Random Coil (RC) conformation reported by our research group [29]. These proteins were prepared by adding polar hydrogens and Kollman charges, meanwhile, water molecules and other ligands were removed to be then prepared for docking studies employing the AutoDock 4.4 software (AutoDock. http://autodock.scripps.edu/2019).
2.1.2 Ligands design
Ligands were designed with pharmacophores capable of generating either a positive electronic density or a positive charge able to interact with the anionic region of the catalytic target sites. This strategy was expected to induce not only inhibition of BACE1 and AChE but also to avoid Aβ1–42 oligomerization. The chemical moieties groups employed in ligands contain a tertiary amine such as, N-methyl pyrrolidine (F1), pyrrolidine (F3), piperidine (F2) and morpholine (F4) able to protonate at physiological pH and attached three atoms away from an aromatic ring backbone. In addition, the aromatic could allow the establishment of π-π interactions, also the aromatic ring was substituted with secondary or primary amines, aliphatic moieties, ester, or alcohol groups; these substituents were attached in a 1,3-(-m) or 1,4 (-p) position regarding the amine. These substitutions led to 64 compounds grouped in four families of molecules named as FxSy-m/p where Fx is the type of tertiary amine employed, Sy is the substituent attached to the aromatic ring and -m/p is the substituent in meta or para position relative to the tertiary amine (Fig 1). For the in-silico studies a positive control for each protein was employed; peptidomimetic inhibitor IV (IP-IV) [30], galantamine and curcumin, for BACE1, AChE and Aβ, respectively.
Fig 1. Chemical structures of ligands designed with heterocyclic amines; methyl pyrrolidine (F1), piperidine (F2), pyrrolidine (F3) and morpholine (F4) distanced 3 atoms away from the aromatic backbone.

Substitution of the aromatic system with benzoic acid (S1 and S8), hydroxybenzoic acid (S2), phenylmetanimine (S3), phenylmethanamine (S4), butylbenzene (S5), methyl benzoate (S6) and phenyl ethanol (S7) in a -m or -p position related to the heterocyclic amine.
2.1.3 Docking analyses
All ligands were drawn in 2D using ChemBioDraw Ultra 12.0 (ChemBioDraw Ultra. http://chembiodraw-ultra.software.informer.com/, 2019), and their geometry was pre-optimized using Hyperchem (HyperChem, HYPERCUBE, INC. http://www.hyper.com/, 2019) at the molecular mechanic level. AutoDock tools 1.5.6 (AutoDock. http://autodock.scripps.edu/, 2019) were used to prepare docking studies with a grid box of 60 Å3 on the catalytic site for each protein. The Glu22 and Asp23 are exposed in all the three different conformations of Aβ1–42; whereas for BACE1, Asp32 and Asp228 are key residues in the catalytic site [26]. In the case of AChE, a midpoint between the primary catalytic site comprised by His447, Glu334, Ser203 and a secondary cavity containing Tyr337 was considered for the docking studies. Lamarckian genetic algorithm was employed to perform the search with an initial population of 100 random individuals and 1x107 iterations were executed with AutoDock tools software, the remaining parameters were kept at their default values (AutoDock. http://autodock.scripps.edu/, 2019). The free energy values results were analyzed with AutoDockTools (AutoDock. http://autodock.scripps.edu/, 2019) software, meanwhile the biding poses were analyzed using PyMOL software (PyMOL, A Molecular Visualization System on an Open-Source Foundation, Maintained and Distributed by Schrodinger, 2019 (http://www.pymol.org). To validate the docking procedure, each enzyme was docked with their corresponding inhibitors: Galantamine for AChE, curcumin for Aβ1–42 and PI-IV for BACE1 [30].
2.1.4 Physicochemical properties evaluated in silico
The OSIRIS Property Explorer (Osiris property explorer server “http://www.organic-chemistry.org) and Molinspiration Cheminformatics 2019 (“http://www.molinspiration.com”) servers were employed for chemoinformatic studies to predict the physicochemical properties of ligands according to Lipinski´s rule. Those ligands with characteristics favoring the absorption and distribution after oral administration were taken into consideration [31].
2.1.5 Blood-brain-barrier permeability prediction
To predict nervous system permeability, the Blood Brain Barrier Permeability Predictor server (http://www.cbligand.org/BBB/index.php) was employed using an AdaBoost algorithm, in combination with the MACSS fingerprint. Additionally we employed a Light BBB (http://ssbio.cau.ac.kr/software/bbb) server based on previously known structures through a machine learning algorithm and SwissADME (http://www.swissadme.ch/) server based on the boiled egg experiment for brain permeability prediction [32–34]. For each server, the protonation state at pH = 7.4 of each structure was predicted and then the SMILEs code was used.
2.2 Chemical synthesis
All chemical starting materials purchased from Sigma-Aldrich were directly used without further purification. All solvents were distilled prior to use and acetone was dried following standard procedures.
Mass spectra were recorded by staff of Centro de Nanociencias y Micro y Nanotecnologías at the National Polytechnic Institute (https://www.ipn.mx/nanocentro/). High resolution mass spectra were recorded on a micOTOF-Q mass spectrometer. Thin layer chromatography (TLC) was performed using commercially available pre-coated plates (TLC Silica gel 60 F₂₅₄). Column chromatography was conducted using Merck Silica gel 60 (0.063–0.100 mm). Preparative thin layer chromatography (prep-TLC) was performed using commercially available pre-coated plates (TLC Aluminum oxide 60 F₂₅₄, neutral).
The synthesized compounds were characterized by 1H and 13C NMR spectroscopy. All spectra were recorded on a Varian 300 MHz spectrometer. Samples of compounds were dissolved in the indicated deuterated solvent; chemical shifts (δ) were reported in ppm downfield to tetramethyl silane. Coupling constants (J) are reported in Hertz and rounded to 0.1 Hz. Splitting patterns are abbreviated as follows: singlet (s), doublet (d), triplet (t), quintet (q), multiplet (m), broad (br) or a combination of these.
Based on the in-silico results which include docking and the best physicochemical properties according to the Lipinsky rules, compounds F3S4-m, F2S4-m and F2S4-p were the most promissory to be selected for synthesis following the methodology shown in Scheme 1. The methenamine compounds F3S4-m and F2S4-m were obtained employing the Williamson ether synthesis, which involves the reaction of an alkoxide with a primary alkyl halide via a nucleophilic substitution reaction. The synthesis of compound F2S4-p was performed following a different approach to obtain the amine group in para position relative to the alkoxy group. Initially, the Williamson ether synthesis was performed employing the 4-hydroxy-3-methoxybenzaldehyde (Vanillin), as the starting material. The next step required the formation of the appropriate oxime which ultimately was further reduced to the desired amine employing a catalytic hydrogenation procedure.
Scheme 1. General reaction for F3S4-m, F2S4-m, and F2S4-p formation.

Reagents (K2CO3, dry acetone) and conditions: (a) reflux 48 h, (b) reflux 12 h, (c) reflux 8 h; (d) NaHCO3, EtOH, reflux 4 h; (e) Pd/C 10%, 70 psi, absolute EtOH, 4 h.
2.2.1. Synthesis of (4-methoxy-3-(2-(pyrrolidin-1-yl) ethoxy) phenyl) methanamine (F3S4-m)
Finely grounded K2CO3 (2914 mg, 21 mmol) was added to a mixture of 3-hydroxy-4-methoxybenzylamine hydrochloride (2000 mg, 10.5 mmol) and 1-(2-chloroethyl) pyrrolidine hydrochloride (1790 mg, 10.5 mmol) dissolved in dry acetone (30 mL). The progress of reaction was monitored by TLC until completion. The mixture reacted under refluxing conditions for 48 h protected from moisture employing a calcium chloride trap. After cooling the reaction to room temperature, the K2CO3 was removed by vacuum-filtration and the solvent was removed under reduced pressure. The resulting viscous residue was then purified by alumina gel column chromatography using MetOH as eluent, obtaining a light-brown sticky residue which then was subjected to solvent removal in a Schlenk line under vacuum overnight, to afford F3S4-m (1324 mg) as a light-brown powder, solubility MetOH, EtOH, Acetone (75.6% yield, 96% purity). IR (cm-1): 2929, 2957, 1575, 1136, 1442, 1260. 1H NMR (300 MHz, CDCl3) δ 7.20 (s, 1H, H-2), 6.94 (dd, 3J = 8.4, 1.3, 1H, H-6), 6.81 (d, 3J = 8.3, 1H, H-5), 4.59 (a, 3H, NH3), 4.36 (t, 3J = 5.7, 2H, H-7), 3.92 (s, 2H, H-11), 3.80 (s, 3H, OCH3), 3.30 (t, 2H, H-8), 3.08 (br, 4H, H-9), 1.97 (br, 4H, H-10). 13C NMR (75.45 MHz, CDCl3) δ 148.5, 148.0, 120.2 112.9, 111.6, 67.2, 55.9, 54.7, 54.5, 45.4, 23.4. HR-MS m/z for C14H22N2O2, calculated 250.1681, found; 251.1622 [M+H]+. Decomposition was observed after 250°C. compound (1324 mg, yield 75%).
2.2.2. Synthesis of (4-methoxy-3-(2-(piperidin-1-yl) ethoxy) phenyl) methanamine (F2S4-m)
For the syntheses of F3S4-m the following quantities: K2CO3 (435 mg, 3.15 mmol), 3-hydroxy-4-methoxybenzylamine hydrochloride (300 mg, 1.60 mmol), 1-(2-chloroethyl) piperidine hydrochloride (316 mg, 1.70 mmol), dry acetone (40 mL). The mixture was reacted for 12 h under nitrogen atmosphere. The obtained yellow viscous residue (565 mg) was purified by preparative thin layer chromatography (TLC). Four aluminium oxide plates (20 X 20 cm) were employed to purify 200 mg of crude mixture. The plates were eluted with EtOAc/MetOH (8:2). The layer with an Rf around 0.2 (observed under UV-light) was scrapped out from the plates, the aluminium oxide was then stirred with methanol for at least 30 min and then vacuum-aid filtrated. The organic solvent was removed under reduced pressure to yield a light-yellow sticky slurry which then was subjected to solvent removal in a Schlenk line under vacuum overnight to finally obtaining compound F2S4-m (109 mg, 0.412 mmol, 54.5% yield, 95% purity). Solubility; MetOH, EtOH, Acetone, CHCl3, EtOAc, H2O. IR (cm-1): 2930.6, 2852–2832, 1513, 1441, 1259, 1136, 806.6. 1H NMR (300 MHz, CDCl3) δ 6.88 (s, 1H, H-2), 6.83 (br, 2H, H-5, H-6), 4.15 (t, 3J = 6.2, 1H, H7), 3.84 (s, 3H, OMe), 3.78 (s, 2H, PhCH2), 2.81 (t, 3J = 6.3, 2H, H-8), 2.50 (br, 4H, H-9), 1.60 (q, 3J = 5.5, 4H, H-10), 1.44 (q, 3J = 5.9, 2H, H-11). 13C NMR (75.45 MHz, CDCl3) δ 148.4 and 148.3 (C-3 y C-4), 136.2 (C-1), 119.4 (C-6), 112.3 and 111.7 (C-2 y C-5), 66.4 (C-7), 57.9 (OMe), 56.2 (PhCH2), 55.1 (C-9), 46.3 (C-8), 26.0 (C-10), 24.3 (C-11). MS (ESI): m/z calculated for C15H25ClN2O2: 300.1605 (hydrochloride), C15H24N2O2: 264.1838 (free amine); found: 265.1911 g/mol [M+H]+.
2.2.3. Synthesis of (3-methoxy-4-(2-(piperidin-1-yl) ethoxy) benzaldehyde (1)
Synthesized as described for F3S4-m using the following quantities: K2CO3 (276 mg, 20.0 mmol), 4-hydroxy-3-methoxy benzaldehyde (152 mg, 10.0 mmol) and 1-(2-chloroethyl) piperidine hydrochloride (276 mg, 15.0 mmol), dry acetone (200 mL). The mixture reacted for 8 h. The desired product was obtained as an oily, brown-coloured residue which then remained in a Schlenk line under vacuum overnight to remove solvent traces and finally obtaining compound 1 (65% yield, 92.5% purity). No further purification was required. Solubility; MetOH, EtOH, CHCl3, CH2Cl2. 1H NMR (300 MHz, CDCl3) δ 9.79 (s.1H, CHO), 7.37 (d.1H, 3J = 3 Hz, H-5), 7.35 (dd, 1H, 3J = 3, 9 Hz, H-6), 6.96 (d, 1H, 3J = 9 Hz, H-2), 4.20 (t, 2H, 3J = 6 Hz, H-7), 3.87 (s, 3H, OMe), 2.82 (t, 2H, 3J = 6 Hz, H-8), 2.5 (t, 4H, 3J = 6 Hz, H-9), 1.58 (q, 4H, 3J = 6 Hz, H-10), 1.41 (q, 2H, 3J = 3 Hz, H-11). MS (ESI): m/z calculated for C15H21N O3: 263.1521, found 264.1598 [M+H]+.
2.2.4. Synthesis of (E)-3-methoxy-4-(2-(piperidin-1-yl) ethoxy) benzaldehyde oxime (2)
Finely grounded NaHCO3 (143 mg, 17.0 mmol) was added to a mixture of 3-methoxy-4-(2-(piperidin-1-yl) ethoxy) benzaldehyde (278 mg, 10.0 mmol) and hydroxylamine hydrochloride (111 mg, 16.0 mmol) dissolved in EtOH (10 mL). The mixture reacted under refluxing conditions for 4 h. The progress of reaction was monitored by TLC, once completed, the mixture was allowed to cool down to room temperature followed by addition of distilled water (100 mL). The resulting white precipitate was filtered with the aid of vacuum-filtration. The desired product was then dried in a Schlenk line under vacuum overnight to obtain a white solid (60% yield, 97% purity). MetOH, EtOH, CHCl3, CH2Cl2. mp 119.6–120.8°C. 1H NMR (300 MHz, CDCl3) δ 10.77 (s.1H, NOH), 8.02 (s, 1H CH = NOH), 7.00 (dd.1H, 3J = 3, 9 Hz, H-6), 6.95 (d, 1H, 3J = 3 Hz, H-2), 6.87 (d, 1H, 3J = 9 Hz, H-5), 4.29 (t, 1H, 3J = 6 Hz, H-7), 3.78 (s, 3H, OMe), 2.85 (t, 2H, 3J = 6 Hz, H-8), 2.58 (t, 4H, 3J = 6 Hz, H-9), 1.63 (q, 4H, 3J = 6 Hz, H-10), 1.43 (c, 2H, 3J = 3 Hz, H-11). MS (ESI): m/z calculated for C15H22N2 O3: 278.1630, found 279.1709 [M+H]+.
2.2.5. Synthesis of (3-methoxy-4-(2-(piperidin-1-yl) ethoxy) phenyl) methanamine (F2S4-p)
To a solution of (E)-3-methoxy-4-(2-(piperidin-1-yl) ethoxy) benzaldehyde oxime (1.8 g, 6.5 mmol) dissolved in absolute EtOH (162.5 mL) was added 325 mg of Pd/C (10%). The mixture was allowed to react under catalytic hydrogenation (70 psi, 4h). The chemical reaction was monitored by TLC, once completed; the reducing agent was removed by filtration. The solvent was removed under reduced pressure to afford the desired product as a pale-yellow powder (94% yield, 95% purity). Ssolubility; MetOH, EtOH, water. mp 230.2–231.5°C. IR (cm-1): 3000–2800 2640. 1H NMR (300 MHz, CD3OD) δ 7.20 (d, 1H, 3J = 3, H-2), 7.05 (dd, 1H, 4J = 3, 3J = 6, H-5), 7.04 (dd, 1H, 4J = 3, 3J = 6, H-6), 4.97 (t, 2H, 3J = 6, Ar-CH2-NH2), 4.33 (s, 2H, 3J = 6, H-7), 4.08 (t, 2H, 3J = 6, Ar-CH2-NH2), 3.91 (s, 3H, OMe), 3.36 (t, 2H, 3J = 6, H-8), 3.18 (t, 4H, 3J = 6, H-9), 1.86 (q, 4H, 3J = 6, H-10), 1.64 (q, 2H, 3J = 3, H-11). 13C NMR (75 MHz, CD3OD) 151.52 (6 CHAr), 149.49, 129.03, 123.00, 116.16 and 114.18, 66.81 (N-CH2-CH2-O) and 57.56 (N-CH2-CH2-O), δ = 56.81 (OCH3), 55.43 (CH2-(CH2-CH2)2-N), 44.39 (Ar-CH2-NH2), 25.05 (CH2-(CH2-CH2)2-N), δ 23.49 (CH2-(CH2-CH2)2-N). MS (ESI): m/z calculated for C15H24N2 O2: 264.1838, found 265.1923 [M+Na].
All spectra are shown in the supporting information as S1-S12 Figs in S1 File.
2.3 In vitro studies
2.3.1 Inhibition of Aβ1–42 aggregation by Thioflavin T (ThT) assay
Evaluation of ligands as Aβ1–42 fibril formation inhibitors was performed as follows: a solution of Aβ1–42 (Calbiochem, Cat. No. PP69) at 0.25 μg/μL in milliQ water, was incubated with or without each one of the selected compounds (F2S4-p, F2S4-m, F3S4-m) at 100 μM in a quartz cell at 37°C, the mixture (300 μL) was constantly shaken over 48 h. Aliquots (150 μL) from this solution were taken at 48 h. 25 μL of ThT at 3.0 mM was added and diluted to a final volume of 600 μL with water. The increase in ThT fluorescence was measured at λ emission = 480 nm and λ excitation = 445 nm [28]. Fluorescence emission was recorded using an LS-55 Spectrofluorometer (PerkinElmer), equipped with a water jacketed cell holder for temperature control. All the experiments were performed using cells with a path-length of 0.5 cm, at 37°C.
2.3.2 Inhibition of BACE 1 activity
The enzymatic activity of BACE 1 was measured using a fluorescence resonance energy transfer (FRET) kit (Thermo Scientific, MA, US). All compounds were dissolved in 50 mM sodium acetate buffer pH 4.5 with 0.3% v/v of MeOH HPLC grade. Measurements were taken 1 h later of being shaken at room temperature. The synthesized inhibitors were compared against a β-Secretase IP-IV (Cayman Chemichal, MI, US) as positive control [30].
The fluorescence was measured in Tecan M1000 pro multimode microplate reader (Tecan Trading, ZRH, Switzerland). After blank subtraction, activity was determined from Eq 1.
| (1) |
Replacing the fluorescence intensity (FI) obtained in the completed reaction with each compound and the fluorescence intensity of the negative control fluorescence (NCF) corresponding to BACE1 and substrate without inhibitor.
2.3.3 Acetylcholinesterase inhibition assays
The Bonting-Featherstone method [35, 36] was used to quantify the remaining ACh in the assay. A standard curve of ACh at 0.2, 0.4, 0.8, 1.6, 3.2, 6.4 and 12.8 mM diluted in 0.1M potassium phosphate buffer pH 8 was done. Samples were incubated over 1 h at 37°C under constant shaking. After 25 μL of alkaline hydroxylamine (1:1) with NaOH 14% w/v was added. Each sample was diluted 1:4 using a solution of FeCl3 78 mM in 3.8 mM HCl, thereafter each sample was centrifuged at 5000 rpm/10 min, and finally the optical density of the supernatant was measured at λ = 500 nm, using the spectrophotometer multiskan sky microplate (Thermo Fisher Scientific, CA, US). Determination of AChE catalytic activity was conducted in the presence of compounds at 25, 50 and 100 μM of each inhibitor using Galantamine as positive control. For this purpose, 0.0006 U/μl of electric eel (Electrophorus electricus) AChE C3389 (Merck, Darmstadt, Germany) was preincubated in the presence of compounds for 1 h under constant shaking. Subsequently, the respective ACh solution was added and re-incubated for 1 h at 37°C, giving the same treatment previously reported for the standard curve. The enzyme activity was expressed as the difference in optical density between the inhibitor assays and the respective standard curve labeled as ΔAbs. ΔAbs = (Absorbance of ACh without AChE—Absorbance of blank)- (Absorbance of ACh with AChE—Absorbance of blank). Once obtained 1/ΔAbs, and 1/[ACh], a Lineweaver Burk plot (LWP) was drawn. The LWP was performed for each inhibitor concentration and the slope was plotted in function of their increase to determine inhibition constant (Ki) values.
2.3.4 Evaluation of cytotoxic effect of compounds on astrocytes, PC12, and NIH3T3 fibroblasts cultures
The cell lines of astrocytes, PC12 and NIH3T3 fibroblasts were grown in essential DMEM medium with fetal bovine serum, 10% and 1X antibiotic-antifungal (penicillin G, sodium salt and 1% streptomycin sulfate) using a 5% CO2 atmosphere at 37°C. For manipulation and visualization, a biosafety level 2 vertical laminar flow cabinet (NUAIRE A2 NU-543-400) and an inverted binocular microscope (MOTIC AE-20) were used, respectively. To detach the cells, a PBS-trypsin solution was used. The cells were seeded in a 96 plate well with 10x103 cells in each well. After, 24 h the medium was replaced, by different treatments: medium only, medium with 0.01% of DMSO and medium with F2S4-p, F2S4-m, or F3S4-m at 12.5, 25, 50, 100, and 200 μM. Two independent experiments were performed with n = 8 for each treatment. For the viability test, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazole (MTT) was employed as follows; the medium was replaced with 100 μL of a MTT solution (0.5 mg of MTT /1 ml of medium). The 96 plate well was incubated for 4 h using a 5% CO2 atmosphere at 37°C. After, the MTT was removed and 100 μL of DMSO was added, to solubilize the formazan salts. The plate was then read in the spectrophotometer multiskan sky microplate (Thermo Fisher Scientific, CA, US) at 550 nm. Cell viability of astrocytes cell cultures in the presence of Cadmium chloride (0.01, 0.1, 1, 10, 100 μM) as control for cytotoxicity effect is shown in the supporting information as S13 Fig in S1 File. It was found that astrocytes viability diminished up to 25% at 100 μM, at concentrations 1 μM ≤ on the other hand, cell viability remained above 80%.
2.3.5. Statistical analysis
The results are presented as the mean ± SE. All analyses were performed using the statistical program Sigma Stat for Windows Version 2.03 software (SPSS Inc) and the graphs were realized using GraphPad Prism Version 5.00 software.
3. Results
3.1. The compounds with methanamine substituent in the aromatic ring have better affinity on Aβ1–42, AChE and BACE1 by in silico studies
According to in silico studies, the compounds with the best theoretical energy values were those in which the methanamine substituent is bounded to the aromatic ring backbone (S4 group) in -m and -p position relative to the heterocyclic amine (Fig 2A and 2B). Compounds F2S4-m (Fig 2C), F3S4-m (Fig 2D), F2S4-p (Fig 2E) and F3S4-p (Fig 2F) showed a greater affinity to the α-helix conformation of Aβ1–42 with ΔG values as lowest as -7.35, -7.44, -7.74, -7.58 kcal /mol respectively, in comparison with Aβ1–42 in β-sheet and RC conformations, showing higher ΔG values (Fig 2A and 2B). In addition, compounds with S4 group showed not only better ΔG values than curcumin, but also were more selective for Aβ1–42 in α-helix conformation Fig 2A and 2B. All molecules established an electrostatic interaction with Asp23 and Glu22 in the monomeric form of Aβ1–42 (Fig 2C–2F).
Fig 2. Docking results for designed compounds towards three Aβ1–42 monomer secondary conformations (α-helix, β-sheet, and random coil).

Free energy values obtained for; A) 1,3 substituted (-meta) compounds, B) 1,4 substituted (-para) compounds. Methanamine (S4) bearing compounds in both substitution positions (red points in chart) containing piperidine (F2) and pyrrolidine (F3) were the best compounds. Binding analysis of containing methanamine compounds (S4) and Aβ1–42: C) F2S4-p, D) F3S4-m, E) F2S4-p, F) F3S4-p. All compounds were able to place one of amino groups in an interaction with Asp 23 and Glu 22 and interact with aromatic residues (Phe20) by π-π stackings.
Besides the electrostatic interaction between the heterocyclic charged amine of compounds with Asp23/Glu22 in the α-helix Aβ1–42 conformation, other types of non-bond interaction were observed. All the nonbond interactions with the residues are shown in the S1 Table in S1 File. For example, compounds containing carboxylic acid, as observed in the in the S1 and S8 series, exhibited H-bond interactions with Lys28 and electrostatic interaction with Lys26. Compounds with aliphatic chains as in the S5 series showed H-bond interactions, electrostatic interactions with Glu22 and Asp23 and more specifically with His14 for F4S5-m.
Compounds bearing the hydroxybenzoic acid, as in the S2 series, established H-bond interactions with Ser26 and several Van der Waals forces with its lateral chain. Those compounds of the S7 group, containing phenyl ethanol as substituent, formed H-bonds and electrostatic interactions with Phe20 or Asp23, respectively. In the case of F4S7-m/-p an electrostatic interaction between the NH+ group and the carboxyl group of His14 was observed to happen in an analogous way to what occurs in methanamine compounds (S3-m/-p family) in which H-bonds and electrostatic interactions with Asp23 and Phe20 are stablished. However, none of F4S7-m/-p showed better ΔG values than the methanamine substituent (S4) in which interaction between Asp23, Glu22 and the NH2 group are observed leaving the aromatic ring or the NH+ group available to establish π-π or π-cation stacking with Phe19 and Phe20. This feature could not be seen in F1S4-m/p compounds due to distancing between NH+ group with Phe19/20 or Asp23/Glu22 carboxylic acid groups measuring 10.5/12.5 and 7.4/10.2 Ǻ, respectively. Similar distancing features were observed in the F4S4-m/p for Asp23/Glu22 lateral chains with the aromatic eter contained in the morpholine group. Otherwise, the F2S4 and F3S4 -m or -p compounds presented similar interactions with distancing ranges varying from 1.5 to 3.5 Å for the NH2 methanamine group with the lateral chain of Asp23 and Glu22; and 4–10 Ǻ with lateral chain of Phe19 or Phe20 with NH+ or aromatic ring (Fig 2C–2F).
The results from the nonbond interaction between all compounds with BACE1 are shown in S2 Table in S1 File. The lowest free energy values obtained for F2S4-p, F3S4-m, F1S4-p, F3S4-p, F1S4-m, and F2S4-m were -10, -9.89, -9.81, -9.76, -9.73 and -9.5 Kcal/mol respectively (Fig 3A and 3B). These values are like those reported elsewhere for a series of PDB-based ligands against AChE, BACE1 and γ-secretase among other promising drug-targeted enzymes for the AD treatment [37–39]. The best results obtained from the docking interaction between ligands and Aβ1–42 were obtained for compounds F2S4-m / p and F3S4-m, therefore, we focused on describing their binding mode to BACE1.
Fig 3. Docking results of design and known compounds towards BACE1 and human AChE.

Free energy values obtained for: A) 1,3 substituted (-meta) and B) 1,4 substituted (-para) compounds toward BACE1(squares) and AChE (triangles). The methanamine (S4) bearing compounds (red squares and triangles) showed the best values towards the two proteins, beating galantamine but do not IP-IV. Binding analysis of containing methanamine compounds (S4) and PI-IV toward: C1-5) BACE1 (red) and D1-5) human AChE.
Regarding the binding mode analysis towards BACE1 F2S4-m (Fig 3C1), F3S4-m (Fig 3C2), F2S4-p (Fig 3C3) and F3S4-p (Fig 3C4) compounds were able to reach the binding pocket where the aspartic dyad is located at 2–3 Ǻ due to an electrostatic interaction with the amine groups of compounds. In the case of peptidomimetic inhibitor (Fig 3C5), only H-bonds interactions could be established by its hydroxyl group.
Likewise, compounds F3S4-m, F3S4-p and F2S4-m have in common the establishment of at least one of the following type of interactions; π-σ, π-cation or π-π between the aromatic or polar rings of the molecule in question with Tyr71 (Fig 3C1–3C3). These types of interactions agree with those reported elsewhere in which phenolic compounds interact with amino residues of AChE [40]. On the other hand, compound F2S4-p, which showed the lowest free energy values, establishes an intramolecular π-cation interaction causing the NH+ group approaching to Gly230 and establishing an H-bonding interaction. The polar groups of IP-IV were unable to establish electrostatic interactions with the aspartic dyad or other acidic residue; instead, hydrogen bond interactions between Gly74, Thr232, Phe108 Asp228 and the amino, carbonyl, sulfonamide and hydroxyl groups of IP-IV were observed, respectively (Fig 3C5).
Therefore, those molecules designed with an extra amino group were selected, corresponding to the substituents S3-m/p and S4-m/p of the four families of compounds to be explored by docking studies on human AChE. The best results obtained were those from compounds with methanamine (S4) substituent over those containing methanamine (S3) (Fig 3A and 3B). Four of the best compounds were F2S4-p, F3S4-m, F3S4-p, and F2S4-m with free energy values of -9.13, -8.89, -8.81 and -8.73 kcal/mol respectively, being these values better than the control compound galantamine with a free energy value of -7.66 kcal/mol.
Analyzing the binding mode of F2S4-m (Fig 3D1), F3S4-m (Fig 3D2), F2S4-p (Fig 3D3), F3S4-p (Fig 3D4) and galantamine (Fig 3D5) towards the AChE binding pocket, it is observed that none of them had the ability to reach Glu334 (S2 Table in S1 File). However, all the designed compounds were placed at 3–5 Ǻ to establish π-π or π-cation interactions with the aromatic ring of Tyr337. It is observed that despite not having the ability to interact with Glu334, several π-π, π-σ, π-cation or electrostatic interactions with different polar and aromatic residues were established.
F2S4-m and F3S4-m established π-π interactions with Trp86 and Tyr337 through their aromatic ring, π-σ with Trp86 and Tyr337 with the two ether groups and π-cation with Trp86 through their heterocyclic amines, as well as Asp74, His447 and Glu202 and numerous H-bonds with them. Compound F2S4-p established the same type of interactions as the other two compounds, with the addition of a π-π and π-cation interaction with His447 through its aromatic group and NH2, respectively.
3.2. The compounds with methanamine substituent complying with all Lipinsky rules and can cross the BBB predicted by in silico studies
The molecules with better results from molecular docking (ΔG and binding mode) and the reported inhibitors employed as controls were submitted to physicochemical properties evaluation using OSIRIS property explorer and molinspiration server. The results from all compounds according to the Lipinsky rules (MW < 500 g /mol, Logp<5, H-bonds donator < 5 and H-bond acceptor <10) are presented in Table 1. All molecules evaluated from the S4 series (F1S4, F2S4, F3S4 and F2S4) both in meta and para substitution position complied with all the Lipinsky rules, this outcome allowed us to hypothesize that these compounds would have a good behavior and bioavailability at a systemic level after oral administration. In computational prediction of biological effects none of the compounds shown properties as tumorigenic or mutagenic, F3S4-m shown medium risk as a teratogenic due to the presence of N-(-2-hydroxyethyl)-pyrrolidine. Furthermore, the in silico BBB permeability predictors, showed that all compounds from the S4 family can cross the BBB, showing that curcumin and the PI-IV inhibitor not. It is important to mention that these parameters were calculated considering the negative and/or positive ionization of the molecule at physiological pH.
Table 1. In silico properties, biological activity prediction and Blood Brain Barrier (BBB) permeability prediction Log P (bipartition coefficient) MW (molecular weight); nON, (number of hydrogen bond acceptors); nOHNH (number of hydrogen bond donors); Mt (mutagenicity); Tg, (teratogenicity), T (tumorigenic).
| Physicochemical properties | BBB permeability prediction | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | LogP | MW | nON | nOHNH | M | T | Tg | BBB predictor server | Swiiss ADME | Light BBB | |
| Score | Result | Result | Result | ||||||||
| F1S4-m | 1.19 | 250.34 | 4 | 2 | - | - | - | 0.081 | + | + | + |
| F2S4-m | 1.71 | 264.37 | 4 | 2 | - | - | - | 0.081 | + | + | + |
| F3S4-m | 1.2 | 250.34 | 4 | 2 | - | - | M | 0.088 | + | + | + |
| F4S4-m | 0.64 | 266.34 | 5 | 2 | - | - | - | 0.088 | + | + | + |
| F1S4-p | 1.19 | 250.34 | 4 | 2 | - | - | - | 0.092 | + | + | + |
| F2S4-p | 1.71 | 264.37 | 4 | 2 | - | - | - | 0.092 | + | + | + |
| F3S4-p | 1.2 | 250.34 | 4 | 2 | - | - | M | 0.079 | + | + | + |
| F4S4-p | 0.64 | 266.34 | 5 | 2 | - | - | - | 0.079 | + | + | + |
| Galantamine | 1.54 | 287.36 | 4 | 1 | - | - | - | 0.142 | + | + | + |
| Curcumin | 2.3 | 368.38 | 6 | 2 | - | - | - | 0.017 | - | - | - |
| PI-IV | 2.98 | 578.74 | 9 | 4 | - | - | - | 0.053 | + | - | - |
3.3. Compound F2S4-m inhibit the Aβ1–42 oligomerization better than F2S4-p and F3S4-m compounds
The inhibition of Aβ1–42 oligomerization results is shown in Fig 4. The ThT intensity when is bonded to fibrillated Aβ1–42 is shown in Fig 4A. The highest intensity is observed because the presence of Aβ1–42, a reduction in intensity indicates that fibrillated Aβ1–42 has been diminished by the presence of the evaluated compounds. The fluorescence intensity was determined as shown in Fig 4A, the mean of percentage of Aβ1–42 fibrillization was obtained and plotted in Fig 4B, where is possible to observe that F3S4-m inhibited in 30.5%, F2S4-p in 42.1%, and F2S4-m in 60.9% the Aβ1–42 fibrillization. The average of the inhibition percentages with their respective standard error is shown in Fig 4.
Fig 4. Percentage of inhibition of Aβ1–42 fibrillation.

A) ThT assay using compounds F2S4-m, F2S4-p, and F3S4-m at 100μM B) Plot of percentage of inhibition of Aβ1–42 fibrillation by the compounds.
3.4. F3S4-m has better BACE1 inhibition activity than F2S4-m and F2S4-p
The IC50 values for the BACE1 inhibition were obtained from linear regression plots using the logarithmic function being plotting the % inhibition against log [inhibitor (μM)] or log % inhibition against log [inhibitor (μM)] (Fig 5A). The double logarithm function is done to fit the data to a straight line since, as the inhibitor is more powerful, the data would not fit if we only plotted the log of the concentrations.
Fig 5.

Inhibition of the BACE1 enzyme by: (A) known inhibitor IV, (B) F3S4-m, (C) F2S4-m and (D) F2S4-p. The standard error was calculated from the fluorescence measurements of the duplicate of two independent assays with n = 3.
For F2S4-m, F3S4-m, and F2S4-p, the IC50 was obtained from linear regression plot of inhibition ratio against the log of inhibitor concentration, Fig 5B and 5C. Then, substituting in the plot equation, when the enzyme has the 50% of inhibition the concentration corresponding to the IC50 values was determined as: 3.20 nM for PI-IV, 8.38 μM and 15.97 μM for F3S4-m and F2S4-m, respectively. Compound F2S4-p showed a proportional relationship between its concentration and the fluorescence emission; or, inversely proportional to the inhibition of BACE1 enzyme (Fig 5D). Therefore, it was not possible to determine its IC50 value.
3.5. F3S4-m has better AChE inhibition activity than F2S4-m and F2S4-p
A Lineweaver Burk representation was made by plotting [1/ΔAbs] versus [1/ACh] for each case; Galantamine (Fig 6A), F3S4-m (Fig 6B), F2S4-p (Fig 6C) and F2S4-m (Fig 6D). As can be seen in Fig 6C compound F2S4-p showed no significant effect as an AChE inhibitor.
Fig 6.
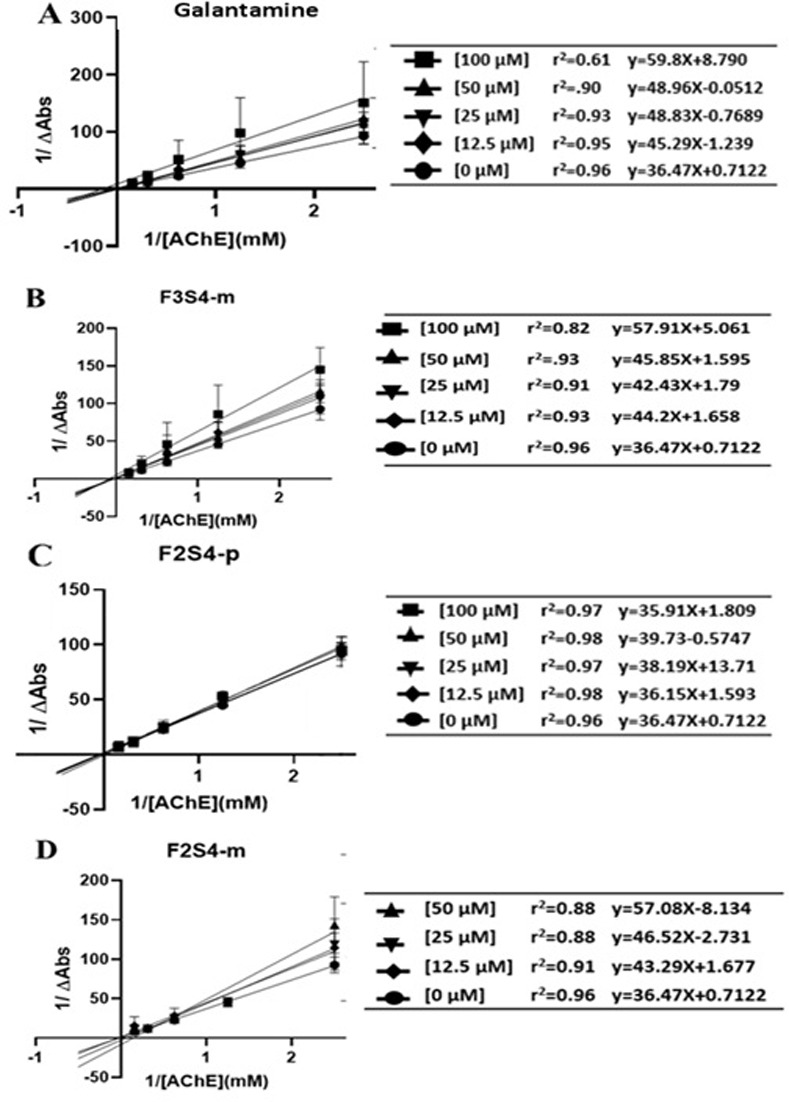
Lineweaver-Burk kinetic model of AChE inhibition by A) Galantamine, B) F3S4-m, C) F2S4-p and D) F2S4-m.
Subsequently, the graphic determination of the inhibition constant (Ki) was carried out by means of the expression of the slopes obtained from the representations of double reciprocals as a function of the concentration of each inhibitor. Ki (Galantamine) = 0.20 μM, Ki (F2S4-p) = NS, Ki (F2S4-m) = 0.40 μM, Ki (F3S4-m) = 0.19 μM (Fig 7) were obtained. The compound with similar Ki to galantamine was F3S4-m.
Fig 7. Determination of the inhibition constant (Ki) of compounds F2S4-m, F3S4-m, F2S4-p and galantamine by graphing the slope of lineaweaver-burk model against the respective inhibitor concentration.

3.6. F2S4-p was more cytotoxic than F2S4-m and F3S4-m evaluated in cell cultures
The cytotoxicity of compounds was evaluated in fibroblast cells, astrocyte and PC12 cell cultures. As can be seen in Fig 8, only compound F2S4-p showed cytotoxic in fibroblast cultures but not in astrocyte cultures, neither in PC12 cell cultures (Fig 8B and 8C). Compounds F3S4-m and F2S4-m were not cytotoxic in any of the evaluated cell lines up to 200 μM.
Fig 8.

Cytotoxic evaluation of compounds F3S4-m, F2S4-m, F2S4-p in different cell lines by MTT assay: A) Fibroblasts, B) Primary culture of astrocytes and C) PC12 cells.
4. Discussion
Even though, in recent years numerous advances have been made to understanding the pathological mechanisms involved in AD, to date there is no treatment capable of stopping the course of the disease [41] for that reason, it is important to continue the search for new molecules that can be able to recognize and inhibit more than one target of the pathological processes causing AD [42]. Given that the amyloidogenic hypothesis continues to be one of the main accepted hypotheses, it is important to focus on the design and use of new compounds towards molecules involved with this pathway [43]. Thus, the BACE1 and aggregation of Aβ1–42 in oligomers and fibrils have become important pharmacological targets since the production and aggregation of Aβ1–42 can cause neuronal death [44]. Accumulation of Aβ1–42 is also related to cholinergic deficit [28, 45]. Therefore, the AChE is also one of the main therapeutic targets in AD, because its inhibition increases ACh levels and thus retards cognitive deficit [46].
Therefore, it is of foremost importance to design compounds that may have inhibitory activity on AChE, BACE1 and decrease the aggregation of Aβ1–42, all these biomolecules involved in AD [44]. Consequently, in this study there were designed new compounds considering the presence of a heterocyclic tertiary amine capable of protonation at physiological pH and other chemical moieties capable to establish electrostatic interactions, hydrogen bonding, hydrophobic interactions as well as π-π interactions with the aminoacid residues located inside the catalytic site of the targets.
Results from in silico studies confirmed that F3S4-m, F2S4-m, and F2S4-p established at least one polar interaction with one of the aa residues from the active sites in the three targets employed. Regarding the Aβ1–42 aggregation, compounds were able to stablish nonbond interaction with Asp23 and Glu22 which prevents the interaction of this residues with Lys 28 leading to blockage of the subsequent Aβ1–42 aggregation. However, the presence of a methylene bound to the heterocyclic amine (Family F1) seems to keep the NH+ group away from the acid dyad and the Phe19 and Phe20 residues based on the analysis of the pyrrolidine added compounds (F1S4-m and F3S4-m), a considerable difference in their free energy was found due to was better F3S4-m than F1S4-m. There are reported studies about the binding capacity of nicotine and two of its main fragments: pyridine and methyl pyrrolidine, towards Aβ1–28 in its monomeric form with a remarkable inhibition of β-folded formation and precipitation based on interaction with His residues [47]. Therefore, the interaction of pyridine and methyl pyrrolidine could be different depending on the Aβ length as in Aβ1–28 the acid dyad is the terminal carboxyl in the peptide and the folding in the Aβ1–28 is different from Aβ1–42.
The presence of a -NH2 group at the aromatic ring allows the molecule to be placed between acidic residues in Aβ1–42 without distancing away the pyrrolidine from the acid dyad or from the aromatic rings of Phe19 and Phe20 in Aβ1–42, allowing the establishment of π-cation interactions; for this reason the presence of three atoms of distance between the aromatic ring and the amine heterocycle seems to be fundamental to maintain the amine within < 6 Å of interaction with the Phe ring.
There are chemical differences and similarities between S4 and S3 family compounds, both containing diamino groups located at the same distance; is the presence of an extra alkyl group decreasing its basicity and therefore the ability to favor the reaction with the acid dyad. The size of compounds does not seem to be a limitation to reach the active site of BACE1 or Aβ1–42, at least in its monomeric form. In the same way, the docking studies performed with BACE1, the meta-substituted amine group of molecules showed better qualities allowing to place the molecule in the middle of the catalytic dyad forming electrostatic interactions, leaving the heterocyclic amine available to establish extra polar interactions with Asp228 at 5–6 Å and cation-π with Tyr71 at 6Å distance or even the aromatic ring with Tyr71 through α-π interactions. In addition, the compounds with pyrrolidine and the amine group in the aromatic ring were the best compounds for AChE inhibition, having less ΔG values means favored affinity on the enzyme.
It was observed an outstanding performance of all compounds in the in silico pharmacokinetic studies, complied with the Lipinsky’s rule. Although the F3S4-m compound showed the lowest free energy by docking studies, possible teratogenic effects were observed due to the presence of N-(2-hydroxyethyl)-pyrrolidine. Nevertheless, this substituent can be found in drugs already approved for in vivo use such as Levetiracetam and brivaracetam diclofenamic acid [48]. In addition, among the all three synthesized compounds, F2S4-m demonstrated to reduce by 60.9% the amount of fibrillated Aβ1–42 by ThT assay.
F2S4-m and F3S4-m showed inhibitory activity against AChE and BACE1 due to these molecules have an amine group in their structures like donepezil, a current AChE inhibitor in the market [49]. However, the results obtained suggest that the amine position within the molecule is relevant to the inhibitory activity, due to F2S4-p compound did not show inhibitory activity on the tested enzymes.
Additionally, few studies suggest that pyrrolidine could be better group to inhibit AChE than piperidine, however, other studies have even focused on the design of 1,3-linked benzylpiperidine compounds, dimethylbenzimidazoline as cholinesterase inhibitors of both AChE and BChE [50, 51]. In addition, some multitarget therapeutic strategies for AD proposed dual compounds that would allow the inhibition of ChE and BACE1 or that can inhibit ChE and Glycogen synthase kinase-3β (GSK-3β) or ChE and monoamine oxidase (MAO) enzyme, and other combinations of compounds that may influence ChE and receptors. But it is not mentioned that they inhibit the Aβ aggregation, so the compounds designed here identified from the chemical characteristics of the active site of three important targets in AD could be a multitarget tool on more than one pharmacological target in the AD [10, 44]. In addition, F3S4-m, F2S4-m are not cytotoxic in the PC12, astrocytes and fibroblast, the compound F2S4-p shows cytotoxic effect against astrocytes cells. Furthermore, more experiments need to be done to explore the toxicity of F3S4-m as in silico analyses showed that it might has teratogenic effects.
Therefore, the compounds herein designed and obtained could be a good promise for the treatment of AD as the combination of the pharmacophore groups in a small molecule could be important to reach the brain. As these compounds have a small molecular weight, they can be incorporated in different nanotechnology-based drug delivery approaches (e.g., functionalized nanoparticles, nano micellar water formulations, lipid-based nanoparticles, nanogels, etc.) to help crossing the BBB and reach directly into the brain [52].
5. Conclusion
Newly developed compounds F2S4-m and F3S4-m are proposed as potential multitarget therapeutics for the AD as these molecules have shown to inhibit three important pharmacological targets in the AD, Aβ1–42 aggregation, and AChE and BACE1 enzyme activities in vitro. Despite compound F2S4-p did not show inhibition of AChE and BACE1 activity, but it showed Aβ1–42 aggregation, this molecule could be employed in combination with any of the compounds as an interesting approach employing different mechanisms of action as a more effective way to tackle a complex neurological disease.
Supporting information
Tables of free energy values (kcal/mol) and spectroscopic characterisation of synthesised compounds.
(DOCX)
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
This research was funded by grants from Consejo Nacional de Ciencia y Tecnología CB286653 for MCRH and 317214 for JCB. Secretaria de investigacion y Posgrado, Instituto Politécnico Nacional SIP20200211 for MCRH and SEP-CONACYT-ANUIES-ECOS Francia: 296636 for JCB. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Zarini-Gakiye E, Amini J, Sanadgol N, Vaezi G, Parivar K. Recent updates in the Alzheimer’s disease etiopathology and possible treatment approaches: A narrative review of current clinical trials. Curr Mol Pharmacol. 2020;13(4):273–294. doi: 10.2174/1874467213666200422090135 . [DOI] [PubMed] [Google Scholar]
- 2.Fan L, Mao C, Hu X, Zhang S, Yang Z, Hu Z, et al. New insights into the pathogenesis of alzheimer’s disease. Front Neurol. 2020. Jan 10; 10:1312. doi: 10.3389/fneur.2019.01312 ; pmcid: pmc6965067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Obrenovich M, Tabrez S, Siddiqui B, McCloskey B, Perry G. The Microbiota-Gut-Brain Axis-Heart Shunt Part II: Prosaic Foods and the Brain-Heart Connection in Alzheimer Disease. Microorganisms. 2020. Mar;8(4):493. doi: 10.3390/microorganisms8040493 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Islam BU, Jabir NR, Tabrez S. The role of mitochondrial defects and oxidative stress in Alzheimer’s disease. J Drug Target. 2019;27(9):932–942. doi: 10.1080/1061186X.2019.1584808 . [DOI] [PubMed] [Google Scholar]
- 5.Islam BU, Tabrez S. Management of Alzheimer’s disease—An insight of the enzymatic and other novel potential targets. Int. J. Biol. Macromol. 2017; 97: 700–709. doi: 10.1016/j.ijbiomac.2017.01.076 [DOI] [PubMed] [Google Scholar]
- 6.Rao CV, Asch AS, Carr DJJ, Yamada HY. "Amyloid-beta accumulation cycle" as a prevention and/or therapy target for Alzheimer’s disease. Aging Cell. 2020. Mar;19(3):e13109. Epub 2020 Jan 25. doi: 10.1111/acel.13109 ; pmcid: pmc7059149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castro MA, Hadziselimovic A, Sanders CR. The vexing complexity of the amyloidogenic pathway. Protein Sci. 2019. Jul;28(7):1177–1193. Epub 2019 Apr 11. doi: 10.1002/pro.3606 ; pmcid: pmc6566549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castro A, Martinez A. Targeting beta-amyloid pathogenesis through acetylcholinesterase inhibitors. Curr Pharm Des. 2006;12(33):4377–87. doi: 10.2174/138161206778792985 . [DOI] [PubMed] [Google Scholar]
- 9.Wysocka A, Palasz E, Steczkowska M, Niewiadomska G. Dangerous liaisons: tau interaction with muscarinic receptors. Curr Alzheimer Res. 2020;17(3):224–237. doi: 10.2174/1567205017666200424134311 ; pmcid: pmc7509759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cummings JL. Treatment of Alzheimer’s disease: current and future therapeutic approaches. Rev Neurol Dis. 2004. Spring;1(2):60–9. . [PubMed] [Google Scholar]
- 11.Fan Q, Liu Y, Wang X, Zhang Z, Fu Y, Liu L, et al. Ginnalin a inhibits aggregation, reverses fibrillogenesis, and alleviates cytotoxicity of amyloid β(1–42). ACS Chem Neurosci. 2020. Feb 19;11(4):638–647. Epub 2020 Feb 3. doi: 10.1021/acschemneuro.9b00673 . [DOI] [PubMed] [Google Scholar]
- 12.Hampel H, Vassar R, De Strooper B, Hardy J, Willem M, Singh N, et al. The β-Secretase BACE1 in Alzheimer’s disease. Biol Psychiatry. 2021. Apr 15;89(8):745–756. Epub 2020 Feb 13. doi: 10.1016/j.biopsych.2020.02.001 ; pmcid: pmc7533042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coimbra JRM, Baptista SJ, Dinis TCP, Silva MMC, Moreira PI, Santos AE, et al. Combining virtual screening protocol and in vitro evaluation towards the discovery of bace1 inhibitors. Biomolecules. 2020. Apr 1;10(4):535. doi: 10.3390/biom10040535 ; pmcid: pmc7226079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Penke B, Szűcs M, Bogár F. Oligomerization and conformational change turn monomeric β-amyloid and tau proteins toxic: their role in alzheimer’s pathogenesis. Molecules. 2020. Apr 3;25(7):1659. doi: 10.3390/molecules25071659 ; pmcid: pmc7180792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blinov N, Wishart DS, Kovalenko A. Solvent composition effects on the structural properties of the aβ42 monomer from the 3d-rism-kh molecular theory of solvation. J Phys Chem B. 2019. Mar 21;123(11):2491–2506. Epub 2019 Mar 12. doi: 10.1021/acs.jpcb.9b00480 . [DOI] [PubMed] [Google Scholar]
- 16.Li H, Guo Q, Inoue T, Polito VA, Tabuchi K, Hammer RE, et al. Vascular and parenchymal amyloid pathology in an Alzheimer disease knock-in mouse model: interplay with cerebral blood flow. Mol Neurodegener. 2014. Aug 9; 9:28. doi: 10.1186/1750-1326-9-28 ; pmcid: pmc4132280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orta-Salazar E, Cuellar-Lemus CA, Díaz-Cintra S, Feria-Velasco AI. Cholinergic markers in the cortex and hippocampus of some animal species and their correlation to Alzheimer’s disease. Neurologia. 2014. Oct;29(8):497–503. English, Spanish. Epub 2013 Feb 22. doi: 10.1016/j.nrl.2012.10.007 . [DOI] [PubMed] [Google Scholar]
- 18.Nemy M, Cedres N, Grothe MJ, Muehlboeck JS, Lindberg O, Nedelska Z, et al. Cholinergic white matter pathways make a stronger contribution to attention and memory in normal aging than cerebrovascular health and nucleus basalis of Meynert. Neuroimage. 2020. May 1;211:116607. Epub 2020 Feb 6. doi: 10.1016/j.neuroimage.2020.116607 . [DOI] [PubMed] [Google Scholar]
- 19.Saxena M, Dubey R. Target Enzyme in Alzheimer’s Disease: Acetylcholinesterase Inhibitors. Curr Top Med Chem. 2019;19(4):264–275. doi: 10.2174/1568026619666190128125912 . [DOI] [PubMed] [Google Scholar]
- 20.Islam BU, Khan MS, Jabir NR, Kamal MA, Tabrez S. Elucidating Treatment of Alzheimer’s Disease via Different Receptors. Curr Top Med Chem. 2017;17(12):1400–1407. doi: 10.2174/1568026617666170103163715 [DOI] [PubMed] [Google Scholar]
- 21.Anand P, Singh B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch Pharm Res. 2013. Apr;36(4):375–99. Epub 2013 Feb 24. doi: 10.1007/s12272-013-0036-3 . [DOI] [PubMed] [Google Scholar]
- 22.Li S, Zhao H, Li J, Hao J, Yu H. A series of molecular modeling techniques to reveal selective mechanisms of inhibitors to β-Site amyloid precursor protein cleaving enzyme 1 (BACE1) and β-site amyloid precursor protein cleaving enzyme 2 (BACE2). J Biomol Struct Dyn. 2021. May;39(8):2824–2837. Epub 2020 Apr 20. doi: 10.1080/07391102.2020.1754917 . [DOI] [PubMed] [Google Scholar]
- 23.Thapa S, Lv M, Xu H. Acetylcholinesterase: a primary target for drugs and insecticides. Mini Rev Med Chem. 2017;17(17):1665–1676. doi: 10.2174/1389557517666170120153930 . [DOI] [PubMed] [Google Scholar]
- 24.Iman K, Mirza MU, Mazhar N, Vanmeert M, Irshad I, Kamal MA. In silico structure-based identification of novel acetylcholinesterase inhibitors against alzheimer’s disease. CNS Neurol Disord Drug Targets. 2018. Apr 26;17(1):54–68. doi: 10.2174/1871527317666180115162422 . [DOI] [PubMed] [Google Scholar]
- 25.Cheung J, Gary EN, Shiomi K, Rosenberry TL. Structures of human acetylcholinesterase bound to dihydrotanshinone I and territrem B show peripheral site flexibility. ACS Med Chem Lett. 2013. Sep 23;4(11):1091–6. doi: 10.1021/ml400304w ; pmcid: pmc4027152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hernández-Rodríguez M, Correa-Basurto J, Gutiérrez A, Vitorica J, Rosales-Hernández MC. Asp32 and Asp228 determine the selective inhibition of BACE1 as shown by docking and molecular dynamics simulations. Eur J Med Chem. 2016. Nov 29; 124:1142–1154. Epub 2016 Aug 16. doi: 10.1016/j.ejmech.2016.08.028 . [DOI] [PubMed] [Google Scholar]
- 27.Haghighijoo Z, Hemmateenejad B, Edraki N, Miri R, Emami S. Multi-structure docking analysis of BACE1 crystal structures and non-peptidic ligands. J Mol Graph Model. 2017. Sep;76:128–135. Epub 2017 Jun 28. doi: 10.1016/j.jmgm.2017.06.023 . [DOI] [PubMed] [Google Scholar]
- 28.Hernández-Rodríguez M, Correa-Basurto J, Nicolás-Vázquez MI, Miranda-Ruvalcaba R, Benítez-Cardoza CG, Reséndiz-Albor AA, et al. Virtual and in vitro screens reveal a potential pharmacophore that avoids the fibrillization of aβ1–42. PLoS One. 2015. Jul 14;10(7): e0130263. doi: 10.1371/journal.pone.0130263 ; pmcid: pmc4501547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hernández-Rodríguez M, Correa-Basurto J, Benitez-Cardoza CG, Resendiz-Albor AA, Rosales-Hernández MC. In silico and in vitro studies to elucidate the role of Cu2+ and galanthamine as the limiting step in the amyloid beta (1–42) fibrillation process. Protein Sci. 2013. Oct;22(10):1320–35. Epub 2013 Aug 19. doi: 10.1002/pro.2319 ; pmcid: pmc3795491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stachel SJ, Coburn CA, Steele TG, Jones KG, Loutzenhiser EF, Gregro AR, et al. Structure-based design of potent and selective cell-permeable inhibitors of human beta-secretase (BACE-1). J Med Chem. 2004. Dec 16;47(26):6447–50. doi: 10.1021/jm049379g . [DOI] [PubMed] [Google Scholar]
- 31.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001. Mar 1;46(1–3):3–26. doi: 10.1016/s0169-409x(00)00129-0 . [DOI] [PubMed] [Google Scholar]
- 32.Shaker B, Yu MS, Song JS, Ahn S, Ryu JY, Oh KS, et al. LightBBB: computational prediction model of blood-brain-barrier penetration based on LightGBM. Bioinformatics. 2021;37: 1135–1139 doi: 10.1093/bioinformatics/btaa918 [DOI] [PubMed] [Google Scholar]
- 33.Daina A, Michielin O, Zoete V. SwissTargetPrediction: updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019;47: W357–W3664. doi: 10.1093/nar/gkz382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daina A, Zoete V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem. 2016;11: 1117–1121. doi: 10.1002/cmdc.201600182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonting SL, Featherstone RM. Ultramicro assay of the cholinesterases. Arch Biochem Biophys. 1956. Mar;61(1):89–98. doi: 10.1016/0003-9861(56)90319-8 . [DOI] [PubMed] [Google Scholar]
- 36.Méndez-Garrido A, Hernández-Rodríguez M, Zamorano-Ulloa R, Correa-Basurto J, Mendieta-Wejebe JE, Ramírez-Rosales D, et al. In vitro effect of H2O2, some transition metals and hydroxyl radical produced via fenton and fenton-like reactions, on the catalytic activity of AChE and the hydrolysis of ACh. Neurochem Res. 2014. Nov;39(11):2093–104. Epub 2014 Aug 6. doi: 10.1007/s11064-014-1400-5 . [DOI] [PubMed] [Google Scholar]
- 37.NR, Rehman MT, Alsolami K, Shakil S, Zughaibi TA, Alserihi RF, et al. Concatenation of molecular docking and molecular simulation of BACE-1, γ-secretase targeted ligands: in pursuit of Alzheimer’s treatment. Ann Med. 2021;53(1):2332–2344. doi: 10.1080/07853890.2021.2009124 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jabir NR, Rehman MT, Tabrez S, Alserihi RF, AlAjmi MF, Khan M, et al. Identification of Butyrylcholinesterase and Monoamine Oxidase B Targeted Ligands and their Putative Application in Alzheimer’s Treatment: A Computational Strategy, Curr Pharm Des 2021;27(20):2425–2434. 10.2174/1381612827666210226123240 [DOI] [PubMed] [Google Scholar]
- 39.Jabir NR, Shakil S, Tabrez S, Khan MS, Rehman RT & Ahmed AB. In silico screening of glycogen synthase kinase-3β targeted ligands against acetylcholinesterase and its probable relevance to Alzheimer’s disease. J. Biomol. Struct. Dyn. 2021. 39(14); 5083–5092. doi: 10.1080/07391102.2020.1784796 [DOI] [PubMed] [Google Scholar]
- 40.Jabir NR, Khan FR, Tabrez S. Cholinesterase targeting by polyphenols: A therapeutic approach for the treatment of Alzheimer’s disease. CNS Neurosci Ther. 2018;24(9):753–762. doi: 10.1111/cns.12971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.López-Locanto Ó. Tratamiento farmacológico de la enfermedad de Alzheimer y otras demencias. Arch Med Int. 2015;37(2):61–67. Available from: "http://www.scielo.edu.uy/scielo.php?script=sci_arttext&pid=S1688-423X2015000200003&lng=es" [Google Scholar]
- 42.Grill JD, Cummings JL. Current therapeutic targets for the treatment of Alzheimer’s disease. Expert Rev Neurother. 2010. May;10(5):711–28. doi: 10.1586/ern.10.29 ; pmcid: pmc4140224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Masters CL, Selkoe DJ. Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb Perspect Med. 2012. Jun;2(6): a006262. doi: 10.1101/cshperspect.a006262 ; pmcid: pmc3367542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.León R, Garcia AG, Marco-Contelles J. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease. Med Res Rev. 2013. Jan;33(1):139–89. Epub 2011 Jul 26. doi: 10.1002/med.20248 . [DOI] [PubMed] [Google Scholar]
- 45.Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet. 1976. Dec 25;2(8000):1403. doi: 10.1016/s0140-6736(76)91936-x . [DOI] [PubMed] [Google Scholar]
- 46.Mendiola-Precoma J, Berumen LC, Padilla K, Garcia-Alcocer G. Therapies for prevention and treatment of alzheimer’s disease. Biomed Res Int. 2016;2016:2589276. Epub 2016 Jul 28. doi: 10.1155/2016/2589276 ; pmcid: pmc4980501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salomon AR, Marcinowski KJ, Friedland RP, Zagorski MG. Nicotine inhibits amyloid formation by the beta-peptide. Biochemistry. 1996. Oct 22;35(42):13568–78. doi: 10.1021/bi9617264 . [DOI] [PubMed] [Google Scholar]
- 48.Stephen LJ, Brodie MJ. Brivaracetam: a novel antiepileptic drug for focal-onset seizures. Ther Adv Neurol Disord. 2017. Nov 23;11:1756285617742081. doi: 10.1177/1756285617742081 ; pmcid: pmc5784556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hampel H, Mesulam MM, Cuello AC, Farlow MR, Giacobini E, Grossberg GT, et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain. 2018. Jul 1;141(7):1917–1933. doi: 10.1093/brain/awy132 ; pmcid: pmc6022632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mo J, Chen T, Yang H, Guo Y, Li Q, Qiao Y, et al. Design, synthesis, in vitro and in vivo evaluation of benzylpiperidine-linked 1,3-dimethylbenzimidazolinones as cholinesterase inhibitors against Alzheimer’s disease. J Enzyme Inhib Med Chem. 2020. Dec;35(1):330–343. doi: 10.1080/14756366.2019.1699553 ; pmcid: pmc6968383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hiremathad A, Piemontese L. Heterocyclic compounds as key structures for the interaction with old and new targets in Alzheimer’s disease therapy. Neural Regen Res. 2017. Aug;12(8):1256–1261. doi: 10.4103/1673-5374.213541 ; pmcid: pmc5607816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khan NH, Mir M, Ngowi EE, Zafar U, Khakwani MMAK, Khattak S, et al. Nanomedicine: A Promising Way to Manage Alzheimer’s Disease. Front. Bioeng. Biotechnol. 2021. Apr; 9: 1–20. doi: 10.3389/fbioe.2021.630055 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables of free energy values (kcal/mol) and spectroscopic characterisation of synthesised compounds.
(DOCX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.