

Abstract
In the technique presented here, dubbed ‘qMRS’, we quantify the change in 1H MRS signal following administration of 2H-labeled glucose. As in recent human DMRS studies, we administer [6,6′−2H2]-glucose orally to healthy subjects. Since 2H is not detectable by 1H MRS, the transfer of the 2H label from glucose to a downstream metabolite leads to a reduction in the corresponding 1H MRS resonance of the metabolite, even if the total concentration of both isoforms remains constant. Moreover, introduction of the deuterium label alters the splitting pattern of the proton resonances, making indirect detection of the deuterated forms– as well as the direct detection of the decrease in unlabeled form– possible even without a 2H coil. Because qMRS requires only standard 1H MRS acquisition methods, it can be performed using commonly implemented single voxel spectroscopy (SVS) and chemical shift imaging (CSI) sequences. In this work, we implement qMRS in semi-LASER based CSI, generating dynamic maps arising from the fitted spectra, and demonstrating the feasibility of using qMRS and qCSI to monitor dynamic metabolism in the human brain using a 7T scanner with no auxiliary hardware.
Keywords: Deuterium, Glucose, Metabolism, MRI, Spectroscopy, Chemical shift imaging, Glutamate
1. Introduction
For the past century, the study of cellular metabolism has revolutionized our understanding of biological energy production, phenotypic variation, and disease etiology. With the advent of non-invasive medical imaging technologies, continued efforts have focused on expanding the capabilities of such technology to provide information beyond structural and mechanistic aspects and into the realm of molecular biochemical and physiological insights (Fuss and Cheng, 2016).
Despite the obvious appeal of such goals, the only metabolic imaging technique used routinely in the clinic is positron emission tomography (PET), which provides information reflecting tissue glucose uptake after infusion of the radioactive glucose analog 2–18F-fluoro-2-deoxy-D-glucose (18FDG) (Fuss and Cheng, 2016; Kelloff et al., 2005). PET is most commonly applied in clinical oncology, where elevated glycolytic metabolism in cancer cells enables visualization of both primary and metastatic lesions in patients (Heiden et al., 2009). However, while PET provides insight into tissue glucose uptake, it does not provide any further information about downstream glucose metabolism. An alternate metabolic imaging method both capable of monitoring downstream metabolism and not reliant on ionizing radiation would be preferable in many instances.
While conventional magnetic resonance imaging (MRI) is nonionizing and provides exceptional anatomical information, it offers only limited insight with regard to metabolism. Chemical exchange saturation transfer (CEST) MRI is an emerging MRI method capable of detecting endogenous metabolite levels in both normal and diseased tissues (Van Zijl and Yadav, 2011). However, CEST is also limited in its ability to measure the dynamics of metabolite turnover (Van Zijl and Yadav, 2011; Cai et al., 2012; Kogan et al., 2013).
Proton Magnetic resonance spectroscopy (1H MRS), a technique built on the same fundamental physics as magnetic resonance imaging, allows for chemically specific detection of small molecule metabolites (Morris, 1986; Gujar et al., 2005; Öz et al., 2020). While standard clinical MRI measures the signal generated from protons (1H) on water and fat to generate bulk structural images of the body, 1H MRS generally suppresses these signals in order to measure much weaker signals generated from protons on less abundant molecules, including key metabolites such as N-acetyl-aspartate (tNAA), choline (tCho), creatine (tCr), glutamate (Glu), glutamine (Gln), γ-aminobutyric acid (GABA), and lactate (Lac). Still, a major limitation of current 1H MRS based approaches is that experimental set-ups suitable for human measurements provide only static metabolite concentration, and as a result are unable to assess changes in tissue metabolic rates that are not reflected in a change in steady state concentration.
To date, the primary strategy to generate dynamic information using MRS in human beings has been to introduce exogenous (non-radioactive) isotope-labeled substrates administered to the patient or subject. 13C MRS using costly 13C labeled substrates like glucose and acetate has been used extensively to measure metabolic flux both in isolated cells and in vivo (Shulman and Rothman, 2001; Beckmann et al., 1991; de Graaf et al., 2003). Despite this, the clinical application of 13C MRS has been limited owing to the requirement for additional scanner hardware. Moreover, while hyperpolarization techniques can be employed to achieve improved sensitivity of 13C (Merritt et al., 2007; Brindle, 2015), these approaches require further specialized equipment and technical expertise. Recently, there has been growing interest in the use of deuterium (2H) as an alternative to 13C for metabolite labeling studies (Lu et al., 2017; De Feyter et al., 2018; Kreis et al., 2020). In deuterium MRS (DMRS), the protons on glucose or acetate are replaced by deuterium, and as these substrates are metabolized the deuterium label is transferred to the downstream metabolites which can then be detected by DMRS.
Although 13C MRS and DMRS offer unique solutions to measure metabolite turnover, the capability to detect any nucleus other than 1H is generally not available on clinical MRI systems (van Zijl and Brindle, 2020). Detection of these nuclei requires specialized coils for transmission and reception that must be designed with additional expertise or otherwise are purchased at an additional cost. Hardware availability aside, there are also inherent physical advantages of proton spectroscopy over DMRS: 1H has a gyromagnetic ratio (γ) almost seven times higher than 2H. Because both the spin energy gap itself and the Larmor frequency are functions of γ, both the sensitivity and the spectral resolution of the 1H spectrum are higher than for 2H. Thus, while DMRS has the advantage of very high specificity for the introduced substrate and lack of nuisance signals, it is difficult to resolve the plurality of metabolites which are generated further downstream upon absorption of glucose or acetate.
Given these limitations, we sought to develop a 1H MRS method that increases the sensitivity and versatility of MRS for measuring metabolic dynamics without the need for specialized hardware or radioactive tracers. To this end, we recently introduced quantitative exchanged-label turnover (QELT) MRS or qMRS, a method that detects deuterium labeling of metabolites by measuring the reduction in 1H MRS signal after administration of deuterium labeled substrates (Rich et al., 2020 a). Building on our preclinical qMRS studies, here we demonstrate the potential of the analogous spectroscopic imaging technique, qCSI, for monitoring the dynamics of neural metabolism in healthy human subjects. Since deuterium labeled glucose is non-toxic (De Feyter et al., 2018; Macallan et al., 2009) and can be easily administered orally, this approach is safe and straightforward for use in human subjects. Given the universal availability of 1H MRS on clinical scanners and its ability to detect several biologically relevant metabolites, we envision an expansive translational potential for this technique.
2. Materials and methods
2.1. Phantom experiments
A solution of glutamate (L-glutamic acid, Sigma-Aldrich) at 10 mM concentration were prepared in 20% D2O with the addition of TMS (D2O, 0.1% TMS, Sigma-Aldrich) and 80% PBS at a pH of 7.4. The D2O was used for a lock signal and the TMS was used as an internal standard for chemical shift referencing. A solution of glutamate, at 8 mM concentration, and deuterated glutamate (2,2,4-L-glutamic acid, Cambridge Isotopes), at 2 mM concentration, were prepared in the same conditions. These solutions were used for proton spectroscopy. The same solutions, with the same concentrations, were prepared in 100% PBS at a pH of 7.4 for deuterium spectroscopy so as to avoid a large water signal from D2O. The 1H NMR spectra were collected at 37 °C on a 400 MHz spectrometer (Varian).
The 1H NMR spectra for both solutions of glutamate were acquired with 32000 data points, a spectral width of 6000 Hz, 64 averages, and a recycle delay of 2.0 s. Water suppression was implemented, with a saturation delay of 6.0 s. The deuterium NMR spectra for both solutions of glutamate were acquired with 1024 points, a spectral width of 1000 Hz, 1024 averages, and a recycle delay of 300 ms. The acquisition time for the proton spectra was ~10 min, with a similar acquisition time for the deuterium spectra of ~13 min. For both the For spectra, automatic phase correction and an automatic Whitaker Smoother baseline correction was applied. For proton spectra, a line broadening of 0.2 Hz was used, and for the deuterium spectra, a line broadening of 5 Hz was used.
2.2. Human subjects information
This protocol was approved by the Institutional Review Board at the University of Pennsylvania, with informed consent obtained prior to the initial scan. Eight subjects participated in this study: four male and four female, ranging in age from 23 to 52 years, with a mean age of 32 years. Full chemical shift imaging timecourses were collected on four subjects, two of each sex. Remaining subjects participated in SVS, single-timepoint CSI acquisitions, or non-deuterated glucose experiments.
Volunteers were scanned during two sessions on separate days: one ‘baseline’ measurement, and one measurement after oral deuterated glucose ingestion. For both sessions, the volunteers fasted overnight before undergoing studies in the morning. Baseline scanning sessions lasted approximately 45 min. In the second session, subjects were scanned for approximately two hours, beginning directly after oral ingestion 0.8 g/kg of body weight of [6,6-2H2]-glucose dissolved in water. This oral preparation was provided by the pharmacy service of the Hospital of the University of Pennsylvania, based on the self-reported body weight of each participant.
To ensure normoglycemia, blood glucose testing was performed on all subjects before the baseline session, and both before and after the glucose-ingestion session. To evaluate the choice of glucose dosage, in four subjects a full time-course of blood glucose measurements was also performed upon ingestion of an equivalent amount of non-deuterated glucose (Glucon-D, Dabur, Inc.). Testing was performed using standard home blood glucose monitoring equipment (Accu-check, Roche Diabetes). Results shown in Supplementary Fig. 4 show some intersubject variability in blood glucose levels, but with all subjects returning to baseline glucose levels within 2 h.
2.3. MRI acquisition methods
MR experiments were performed on a 7T scanner (MAGNETOM Terra, Siemens Healthcare, Erlangen, Germany) equipped with a 1Tx/32Rx head coil (Nova Medical, Wilmington, MA, USA). Axial T1-weighted FLASH images were obtained to enable localization of the cortex. Following localization, spectroscopy data were acquired using custom sequences for CSI with sLASER localization (MRSI) (Scheenen et al., 2008; Brown et al., 1982) and SVS with PRESS localization (Bottomley, 1987). Localized shimming was performed to obtain water line widths of 0.08 ppm or less. Water suppression was achieved using variable pulse power and optimization relaxation (VAPOR) pulse cluster (Tkáč and Gruetter, 2005) pre-encoded to the PRESS sequence.
Specifically, SVS in two voxels and two MRSI measurements of the encompassing slab were performed in the baseline session. In the second (post-glucose) session, SVS measurements in two voxels were performed directly upon positioning the subject in the scanner (t = 20–30 min post- ingestion), with six subsequent MRSI acquisitions (t = 50, 60, 70, 80, 90, 100 min) and one additional pair of SVS acquisitions (t = 120 min) at the end of the experiment. Voxel sizes for MRSI and SVS were 10 mm × 10 mm × 10 mm (before interpolation) and 40 mm × 10 mm × 10 mm, respectively. The acquisition time for each MRSI volume was 9 min 55 s. Intrasubject registration between session was accomplished using an in-house co-registration program, ImScribe (available at https://www.med.upenn.edu/cmroi/imscribe.html), as described in previous work (Nanga et al., 2018).
Sequence parameters were as follows:
SVS: TR/TE = 3000/23 ms, spectral width = 4 kHz, averages = 64, scan time = 5 min. In each instance, an additional spectrum with 8 averages was acquired without water suppression to obtain a water reference signal for quantification and eddy current correction.
CSI: TR/TE = 2050/40 ms, spectral width = 4 kHz with 2048 points, averages = 4, FOV = 160×160 mm × 10 mm slice thickness on a 16×16 grid, interpolated to 32×32. The sequence incorporated ellip tical weighting with Hamming-windowed spatial filtering. This resulted in a true voxel size ~2.7x larger than the acquired volume, but removed Gibbs ringing associated with the lower resolution scanning.
2.4. Quantification and data analysis
Prior knowledge fitting was done to determine metabolite concentrations measured by in vivo 1H MRS using LCModel software (v.6.3) (Provencher, 1993). Custom basis sets were simulated using specific timings and pulse shapes of the refocusing pulses in the custom sequences (Soher et al., 2011; Simpson et al., 2017). For the PRESS sequence, a spatial distribution of 20×20 locations covering the voxel were simulated and summed to take into account the effects of chemical shift artifacts caused by relatively low bandwidth refocusing pulses.
In order to account for the difference in spectral patterns that occur when glutamate becomes deuterated, two additional metabolites representing single proton replacement and double proton replacement were also included. For the former, it has been noted (De Feyter et al., 2018) that replacement can occur on either the H4 or the H4’ glutamate proton with equal probability. Here we have chosen to include the H4 proton replacement simulation as the spectral patterns of both are very similar for typical in vivo linewidths (shown in Fig. S1).
For the double proton replacement case, both the H4 and H4’ glutamate protons were replaced with deuterium. Exact coupling constants involving deuterium nuclei were not known, though 2H-1H couplings are typically 1,2 Hz. In Fig. S1, we show that the spectral differences at in vivo linewidths are minimally dependent on 2H-1H coupling over this range and chose 1.5 Hz for the basis set. All fits were performed over the spectral range from 0.5 to 4.2 ppm. While our previous paper presenting experimental data from rats showed changes in GABA and Gln, preliminary analysis in humans did not observe those same effects. Therefore, no deuterated versions of these metabolites were included in the basis set.
First attempts at fitting found that the default soft constraints imposed by LCModel were not satisfactory for this basis set and data. In particular, there was a tendency for the fit to overestimate deuterated glutamate and underestimate the macromolecule peak around 2.0–2.2 ppm (called ‘MM20’ in the software) in baseline scans. Empirically, it was determined that adding several soft constraints on the ratio of MM20 to total creatine in the range of 0.1–1.0 mitigated the problem and produced more consistent macromolecule maps.
Metabolite ratios to tNAA were reported, as tNAA does not show signal changes after deuterated glucose ingestion (Cember et al., 2020). For CSI, region of interests (ROIs) representing primarily gray matter (GM) and primarily white matter (WM) were manually drawn on the Glu to tNAA ratio map, and individual fits over the ROIs were averaged.
3. Results
The very first experiments in which we observed the qMRS effect upon subject ingestion of deuterated glucose were single voxel spectroscopy experiments at 3T and 7T (Bagga et al., 2020). Comparing only one pre-ingestion and one post-ingestion acquisition, we observed visually apparent decrease of the main glucose resonance at 2.3 ppm in mostly gray matter voxels. An example pair of such spectra is shown in Fig. 1 C. We sought to extend this measurement to a form which proffers some degree of spatial and temporal resolution, and continued experiments using a semi-LASER based CSI sequence at 7T, collecting sequential post-ingestion timepoints to create metabolic maps which can be viewed with respect to any of the fitted resonances in the basis set. Fig. 1 shows spectra from selected voxels intended to be mostly gray (A) or most white (B) matter and illustrates the presence of the same decrease observed in the SVS data. Fig. 1 D illustrates the simulated proton resonances of deuterated glutamate species which were included in the basis set used to fit the qCSI data.
Fig. 1.
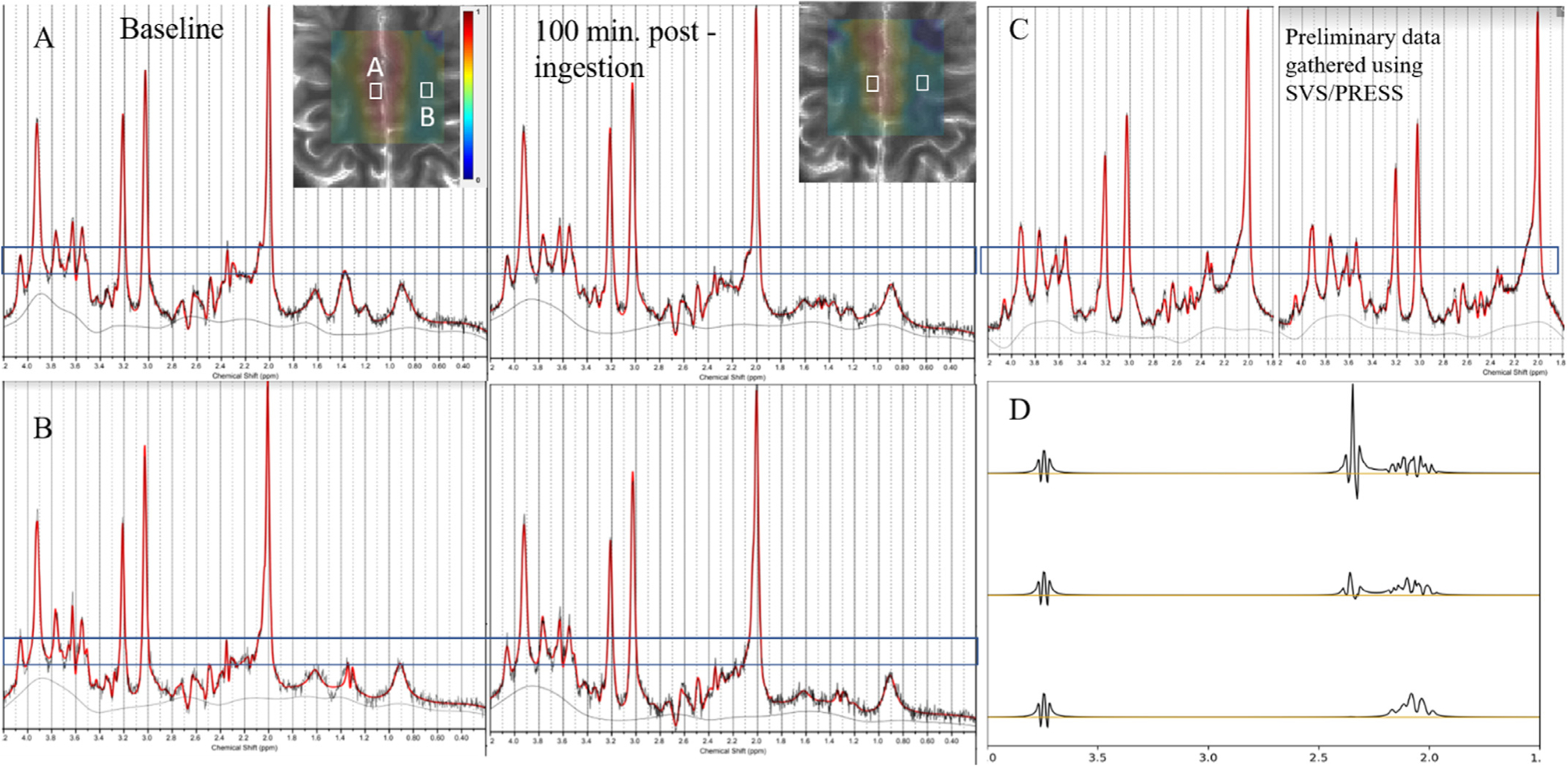
Spectroscopic underpinnings of qCSI. (A,B) Example spectra from two CSI voxels (A,B, as shown in white boxes overlaid on image) showing decrease in main Glu resonance. (C) Corresponding decrease observed in experiments with a larger SVS voxel. (D) Simulations of glutamate elements in the basis set for fitting of semi-LASER data, including unlabeled Glu (top), singly-labeled [2H, 1H]-glutamate-4 (middle), and doubly-labeled [2H2]-glutamate-4 (bottom).
The resulting timecourses from qCSI can be visualized as in Fig. 2, in which the colormap corresponds to the ratio [Glu]/[tNAA] as fit in each voxel. This subject exhibits a strong decrease in the unlabeled Glu signal in the central gray matter region as well as rather dramatic decreases in certain regions of white matter. This reflects that neural glutamate present at baseline has been replaced by newly synthesized, labeled glutamate upon metabolism of the ingested glucose. Timecourses of the [Glu]/[tNAA] map for the remaining subjects are available in Fig. S2.
Fig. 2.
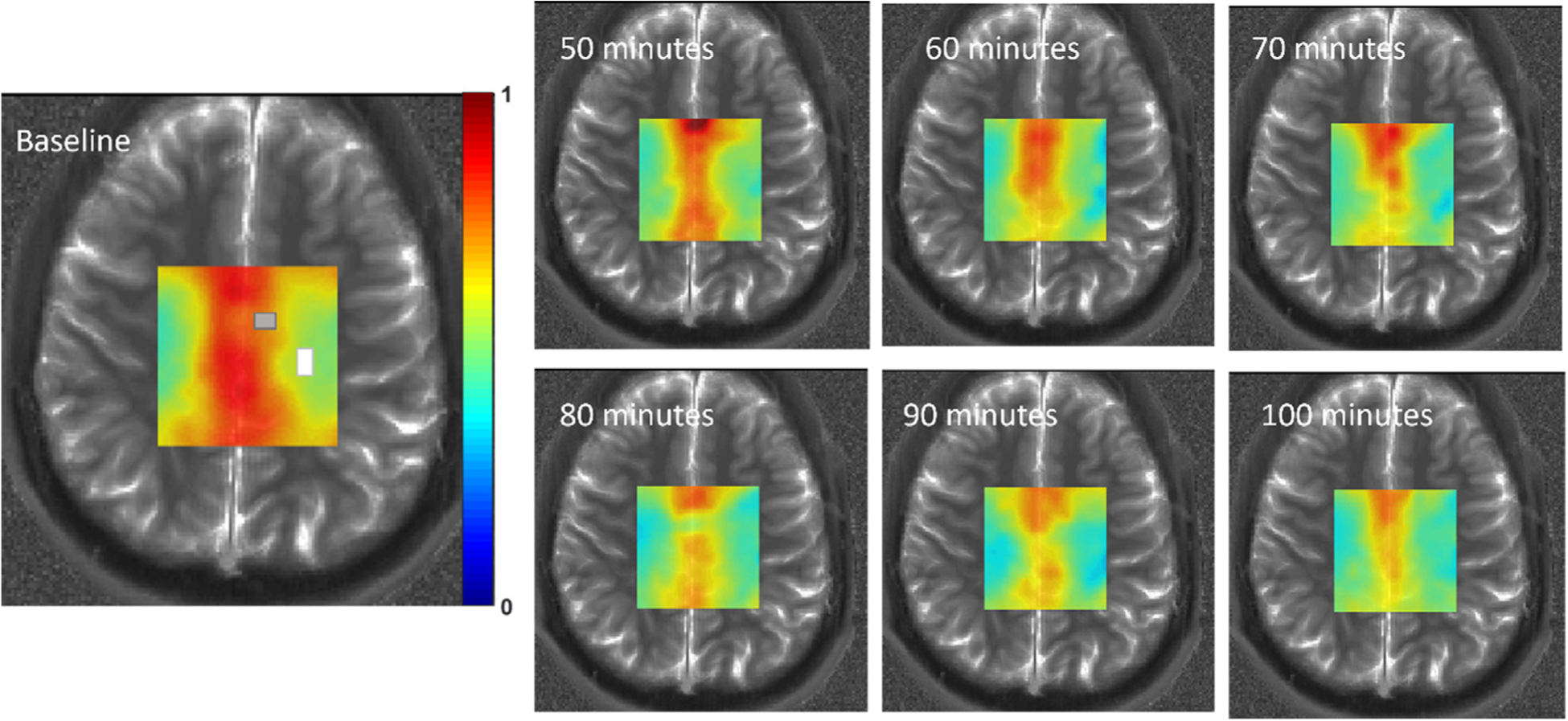
Example CSI metabolic maps of [Glu]/[tNAA] from a single subject. Approximate gray and white matter ROIs as analyzed further are indicated on the baseline image with corresponding colors. ROI sizes and positions varied slightly between subjects (see SI). Colorbar is 0:1, representing the ratio [Glu]:[tNAA].
Fig. 3 shows data from the gray and white matter ROIs from all subjects, derived from the CSI maps corresponding to [Glu], [2H, 1H]- glutamate-4 and [2H2]-glutamate-4. As expected, the concentration of glutamate is higher in gray matter, and its replacement by the deuterated version upon glucose ingestion is greater. However, our present data does not detect that the appearance of the labeled versions of glutamate in the gray matter was sufficient to account for the large (~18%) decrease in the unlabeled version. This discrepancy is not present to the same degree in the white matter data, as the change in the total of all three glutamate species clearly corresponds to a spurious measurement of approximately that concentration of [2H, 1H]-glutamate-4 at baseline (See Table 1).
Fig. 3. Line plots of glutamate concentrations normalized to [tNAA] over time, average of all subjects.
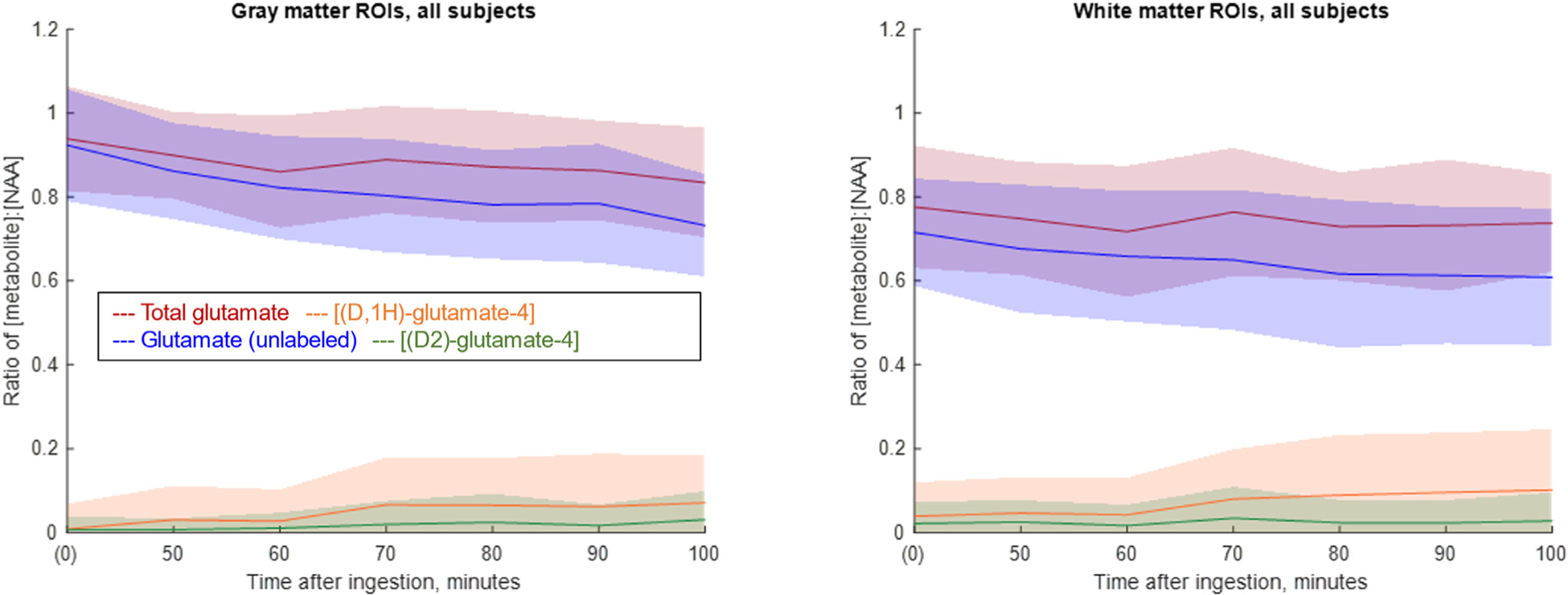
Left: Gray matter ROIs. Right: White matter ROIs. The solid line indicates the mean value, taken over all pixels in the ROIs from four subjects. The shaded area correspondences to one standard deviation. Blue: Glutamate (unlabeled). Orange: (2H, 1H)-glutamate-4 (singly-labeled glutamate). Green: (2H2)-glutamate-4 (doubly-labeled glutamate) Red: Total glutamate (sum of other three contributions). Quantification is by LCModel analysis of CSI data, as described in Methods.
Table 1.
[Met]:[tNAA] ratios from white matter ROIs of all subjects.
| Baseline | t = 50 min | t = 60 min | t = 70 min | t = 80 min | t = 90 min | t = 100 min | ||
|---|---|---|---|---|---|---|---|---|
| Glu | Mean | 0.7155 | 0.6765 | 0.6584 | 0.6498 | 0.6165 | 0.6131 | 0.6088 |
| St. Dev.. | 0.1281 | 0.1522 | 0.1554 | 0.1672 | 0.176 | 0.1633 | 0.1638 | |
| [2H, 1H]-glutamate-4 | 0.039 | 0.0465 | 0.0424 | 0.0799 | 0.0892 | 0.0958 | 0.1013 | |
| 0.0806 | 0.0853 | 0.0879 | 0.1184 | 0.1429 | 0.1421 | 0.1446 | ||
| [2H2]-glutamate-4 | 0.0216 | 0.0253 | 0.0166 | 0.0344 | 0.0237 | 0.0231 | 0.0276 | |
| 0.0519 | 0.0514 | 0.0494 | 0.0744 | 0.0543 | 0.0542 | 0.0677 | ||
| Total glutamate | 0.7761 | 0.7483 | 0.7174 | 0.7641 | 0.7294 | 0.732 | 0.7377 | |
| 0.1457 | 0.1347 | 0.1562 | 0.1524 | 0.1288 | 0.1565 | 0.1169 |
Tables 1 and 2 tabulate the results presented in Fig. 3, listing as a ratio of concentration relative to [tNAA] the quantity of each of the glutamate versions by timepoint, as well as their standard deviation. Fig. S3 exhibits these plots for the individual subjects separately. Fig. S5 provides barplots of tNAA and Glu data for the control experiment involving ingestion of undeuterated D-glucose, showing that, in the absence of a label, these quantities both remain stable.
Table 2.
[Met]:[tNAA] ratios from gray matter ROIs of all subjects.
| Baseline | t = 50 min | t = 60 min | t = 70 min | t = 80 min | t = 90 min | t = 100 min | ||
|---|---|---|---|---|---|---|---|---|
| Glu | Mean | 0.9232 | 0.8613 | 0.8217 | 0.8032 | 0.7817 | 0.7842 | 0.7322 |
| St. Dev | 0.134 | 0.1146 | 0.1224 | 0.1349 | 0.1297 | 0.1414 | 0.122 | |
| [2H, 1H]-glutamate-4 | 0.0083 | 0.0306 | 0.0273 | 0.0663 | 0.0651 | 0.0618 | 0.0709 | |
| 0.0598 | 0.0805 | 0.0749 | 0.1124 | 0.1121 | 0.1255 | 0.1134 | ||
| [2H2]-glutamate-4 | 0.007 | 0.0071 | 0.0107 | 0.0194 | 0.0246 | 0.0167 | 0.0309 | |
| 0.031 | 0.026 | 0.0364 | 0.0553 | 0.0677 | 0.0496 | 0.0677 | ||
| Total glutamate | 0.9385 | 0.899 | 0.8597 | 0.8889 | 0.8714 | 0.8627 | 0.834 | |
| 0.1249 | 0.1035 | 0.1337 | 0.1278 | 0.1337 | 0.1189 | 0.1306 |
4. Discussion
Building on our previously proposed method for indirectly measuring 2H metabolism using 1H MRS in rats, the present work extends the method into human beings and uses CSI for greater spatial coverage. While we are primarily interested in showing comparisons of gray and white matter, CSI has the advantages of larger coverage with smaller voxel sizes compared to SVS and could potentially provide more specifically localized information. However, due to nuisance signals from skull lipids, we restricted our excited volume to a limited portion covering less than half the brain. Also, we restricted our acquisition to a single slice to keep adequate temporal resolution and to ensure more homogenous B0 and B1 fields over the ROI. This is particularly important at ultra high field (UHF), where whole-brain spectroscopic imaging is more challenging.
Some of the uncertainty in the analysis presented here arises from the challenge to accurately draw gray and white matter ROIs on a map of this resolution while minimizing partial voluming, hence the relatively small ROIs chosen for quantitative analysis. In future improvements to the qCSI methodology, fast spectroscopic imaging techniques optimized for UHF could be used to acquire higher resolution metabolic images that allow for larger coverage and better separation of gray and white matter (Moser et al., 2019; Coello et al., 2018). However, because of the coarse-grained visible differences in the map corresponding to these tissue types, we nonetheless decided to perform regional analysis in this manner.
As expected, average qCSI-quantified glutamate values are consistently higher in gray matter than in white matter. Our results indicate that the turnover from glucose to glutamate is also higher in gray matter than in white matter, although the standard deviation is higher in our white matter measurements, making it difficult to quantify this relationship with our present results. Roughly, the labeling detected in gray matter at 100 min is ~18%, while in white matter it is ~15%. The origin of the strong decrease in [Glu] relative to the appearance of the labeled versions in the gray matter data remains unclear. However, given the observed sensitivity to fitting parameters (see Introduction), it is likely that suboptimal LC Model constraints continue to plague the quantification of the gray matter data.
The trend of continuous unlabeled Glu decrease visible in our gray matter data suggest that during this 100 min acquisition window, brain metabolism of orally ingested glucose has not yet reached steady state. This result would seem to agree with the timecourses measured in previous studies: for example, Lu et al. (2017), who studied Glx labeling in rats using DMRS, found that the labeled Glx signal plateaued at around 100 min post-infusion. As we were using oral administration rather than intravenous (IV) infusion, the additional time required for absorption of glucose through the digestive system (see Fig. S4) would lead one to predict that the dynamics of downstream labeling would shift correspondingly in time. Thus, it seems to be consistent with existing data that we would still be observing the appearance of deuterium-labeled glutamate dynamically on this timescale. Of course, future procedural improvements allowing for capturing earlier post-ingestion timepoints (t = < 50 min) would be valuable for making quantitative kinetic measurements.
In the present work, we were not able to detect changes in GABA or glutamine in human subjects. This could be due to the reliance on oral ingestion of deuterated glucose, as opposed to the continuous infusion used in animal experiments, or to reduced spectral sensitivity to GABA and glutamine as compared to glutamate. The use of a GABA-edited MEGA-PRESS sequence (Mullins et al., 2014) or other specialized acquisition may improve quantification of these less salient components. Future work would also consider incorporating deuterated versions of these metabolites into the basis set.
Another spectroscopic approach for detecting metabolic dynamics using the 1 H signal would be to introduce glucose labeled not with deuterium, but instead with 13C (van Eijsden et al., 2010; de Graaf et al., 2011; Dehghani et al., 2020). Once metabolized, 13C would end up on the fourth carbon of glutamate. This carbon has two coupled protons whose J-coupling would then split by ~80 Hz on either side of the original resonance resulting in a spectrum that exhibits only 85–90% of the original signal at 2.35 ppm (arising from protons that are coupled 12C4) and about 10–15% of the signal (those coupled to 13C4) would be trans ferred to split peaks around the center frequency. Since protons attached to 13C4 labeled glutamate would be split and moved away from the glutamate proton resonance on unlabeled 12C4, the main glutamate resonance intensity would be reduced in proportion to deuterium labeling, not unlike in qMRS. However, it is important to note that leveraging splitting due to J-coupling would also interfere with other resonances, making spectral quantification more difficult. 13C-labeled glucose is also 4,5x more expensive than that labeled with 2H. Accordingly, from the perspective of cost and robustness, qMRS based on 2H metabolites would therefore appear to have a greater potential for clinical translation.
Investigators in the area of oncology may be particularly interested in monitoring lactate changes after ingestion of [6,6′−2H2]-glucose as a measure of the Warburg effect (Pavlova and Thompson, 2016). Based on successful preclinical work using the qMRS lactate signal to monitor glycolytic metabolism in glioblastoma (Rich et al., 2020b), we are currently pursuing human studies correlating anaerobic metabolism quantified by qMRS with pathological tumor grade and prognosis.
While the feasibility studies presented in this work are performed at 7T, there is potential to extend the scope of these measurements to the much more available 3T MRI scanners. At 3T, field inhomogeneity is less of an issue, thus facilitating full brain coverage. However, in addition to a reduction in SNR, spectral resolution is compromized, making unambiguous detection of Glu more difficult, especially for an extended basis set that includes deuterated versions of metabolites. We have initiated these studies to a limited degree, and preliminary results suggest that SVS-based qMRS to detect an minimum the decrease of the main glucose resonance is feasible at 3T (Bagga et al., 2020). In the realm of qCSI, sequence development is still underway to find an optimal compromise between spatial and temporal resolution at this field strength.
In summary, we demonstrated the feasibility of qCSI upon oral ingestion of deuterated glucose in human brain studies with modest temporal and spatial resolution. We were able to detect both the decrease in the unlabeled glutamate and the increase in the labeled derivatives by taking advantage of their specific splitting patterns in the proton spectrum. Given the centrality of glucose-to-glutamate turnover to neural metabolism and the novel technical simplicity of the experiment, qCSI may find many uses for studying glucose utilization in the healthy brain as well as in pathological conditions.
Supplementary Material
Acknowledgments
The authors would like to thank D. Reddy and B. Benyard for their assistance in coordination and handling of human subjects.
Funding
This work was carried out at a NIH National Institute of Biomedical Imaging and Bioengineering (NIBIB)-supported Resource Centers, under award numbers P41 EB015893 (RR), P41 EB029460 (RR). Additional funding was provided by National Institute of Neurological Disorders and Stroke Award Number R01NS087516 (RR) and National Institute of Aging Award Number R01AG063869 (RR) and NIH training grant no. T32EB020087-02.
Footnotes
Declaration of Competing Interest
Neil Wilson was an employee of Siemens Medical Solutions during some duration of this manuscript preparation.
Credit authorship contribution statement
Abigail T.J. Cember: Investigation, Formal analysis, Visualization, Writing – original draft. Neil E. Wilson: Methodology, Software, Formal analysis, Writing – original draft. Laurie J. Rich: Conceptualization, Investigation, Writing – original draft. Puneet Bagga: Conceptualization, Investigation. Ravi P. R. Nanga: Investigation, Writing review & editing. Sophia Swago: Investigation. Anshuman Swain: Investigation. Deepa Thakuri: Investigation. Mark Elliot: Methodology, Software. Mitchell D. Schnall: Supervision, Funding acquisition, Writing review & editing. John A. Detre: Supervision, Funding acquisition, Writing review & editing. Ravinder Reddy: Conceptualization, Supervision, Project administration, Funding acquisition, Writing review & editing.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi: 10.1016/j.neuroimage.2022.118977.
Data availability statement
Authors will make all original data available upon reasonable request. All software used in image processing and analysis is commercially available.
References
- Fuss TL, Cheng LL, 2016. Metabolic imaging in humans. Top. Magn. Reson. Imaging 25 (5), 223–235. doi: 10.1097/RMR.0000000000000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelloff GJ, Hoffman JM, Johnson B, et al. , 2005. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clin. Cancer Res 11 (8), 2785–2808. doi: 10.1158/1078-0432.CCR-04-2626. [DOI] [PubMed] [Google Scholar]
- Heiden MGV, Cantley LC, Thompson CB, 2009. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science 324 (5930), 1029–1033. doi: 10.1126/science.1160809, (80-). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Zijl PCM, Yadav NN, 2011. Chemical exchange saturation transfer (CEST): what is in a name and what isn’t? Magn. Reson. Med 65 (4), 927–948. doi: 10.1002/mrm.22761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai K, Haris M, Singh A, et al. , 2012. Magnetic resonance imaging of glutamate. Nat. Med 18 (2), 302–306. doi: 10.1038/nm.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogan F, Hariharan H, Reddy R, 2013. Chemical exchange saturation transfer (CEST) imaging: description of technique and potential clinical applications. Curr. Radiol. Rep 1 (2), 102–114. doi: 10.1007/s40134-013-0010-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris PG, 1986. Nuclear Magnetic Resonance Imaging in Medicine and Biology. Clarendon Press. [Google Scholar]
- Gujar SK, Maheshwari S, Bjorkman-Burtscher I, Sundgren PC, 2005. Magnetic resonance spectroscopy. J Neuro Ophthalmol. 25 (3), 217–226. doi: 10.1097/01.wno.0000177307.21081.81. [DOI] [PubMed] [Google Scholar]
- Öz G, Deelchand DK, Wijnen JP, et al. , 2020. Advanced single voxel 1H magnetic resonance spectroscopy techniques in humans: experts’ consensus recommendations. NMR Biomed. doi: 10.1002/nbm.4236, Published online January 10,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman RG, Rothman DL, 2001. 13C NMR of intermediary metabolism: implications for systemic physiology. Annu. Rev. Physiol 63 (1), 15–48. doi: 10.1146/annurev.physiol.63.1.15. [DOI] [PubMed] [Google Scholar]
- Beckmann N, Turkalj I, Seelig J, Keller U, 1991. 13C NMR for the assessment of human brain glucose metabolism in vivo. Biochemistry 30 (26), 6362–6366. doi: 10.1021/bi00240a002. [DOI] [PubMed] [Google Scholar]
- de Graaf RA, Mason GF, Patel AB, Behar KL, Rothman DL, 2003. In vivo 1H-[13 C]-NMR spectroscopy of cerebral metabolism. NMR Biomed. 16 (6–7), 339–357. doi: 10.1002/nbm.847. [DOI] [PubMed] [Google Scholar]
- Merritt ME, Harrison C, Storey C, Jeffrey FM, Sherry AD, Malloy CR, 2007. Hyperpolarized 13C allows a direct measure of flux through a single enzyme-catalyzed step by NMR. Proc. Natl. Acad. Sci. USA 104 (50), 19773–19777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindle KM, 2015. Imaging metabolism with hyperpolarized 13C-labelled cell substrates. J. Am. Chem. Soc 20, 6418–6427. [DOI] [PubMed] [Google Scholar]
- Lu M, Zhu XH, Zhang Y, Mateescu G, Chen W, 2017. Quantitative assessment of brain glucose metabolic rates using in vivo deuterium magnetic resonance spectroscopy. J. Cereb. Blood Flow Metab 37 (11), 3518–3530 10.1177/0271678X17706444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Feyter HM, Behar KL, Corbin ZA, et al. , 2018. Deuterium metabolic imaging (DMI) for MRI-based 3D mapping of metabolism in vivo. Sci. Adv 4 (8), 7314–7336. doi: 10.1126/sciadv.aat7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis F, Wright AJ, Hesse F, Fala M, Hu DE, Brindle KM, 2020. Measuring tumor glycolytic flux in vivo by using fast deuterium MRI. Radiology 294 (2), 289–296. doi: 10.1148/radiol.2019191242. [DOI] [PubMed] [Google Scholar]
- van Zijl PCM, Brindle KM, 2020. Spectroscopic measurements of metabolic fluxes. Nat. Biomed. Eng 4 (3), 254–256. doi: 10.1038/s41551-020-0535-8. [DOI] [PubMed] [Google Scholar]
- Rich LJ, Bagga P, Wilson NE, et al. , 2020a. 1H magnetic resonance spectroscopy of 2H-to-1H exchange quantifies the dynamics of cellular metabolism in vivo. Nat. Biomed. Eng 4 (3), 335–342. doi: 10.1038/s41551-019-0499-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macallan DC, Asquith B, Zhang Y, de Lara C, Ghattas H, Defoiche J, Beverley PCL, 2009. Measurement of proliferation and disappearance of rapid turnover cell populations in human studies using deuterium-labeled glucose. Nature Protocols 4 (9), 1313–1327. doi: 10.1038/nprot.2009.117. [DOI] [PubMed] [Google Scholar]
- Scheenen TWJ, Heerschap A, Dennis WJK, et al. , 2008. Towards 1 H-MRSI of the human brain at 7T with slice-selective adiabatic refocusing pulses. Magn. Reson. Mater. Phys 21, 95–101. doi: 10.1007/s10334-007-0094-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TR, Kincaid BM, Ugurbil K, 1982. NMR chemical shift imaging in three dimensions. Proc. Natl. Acad. Sci. U. S. A 79 (11 I), 3523–3526. doi: 10.1073/pnas.79.11.3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottomley PA, 1987. Spatial localization in NMR spectroscopy in vivo. Ann. N. Y. Acad. Sci 508 (1), 333–348. [DOI] [PubMed] [Google Scholar]
- Soher BJ, Semanchuk P, Todd D, Steinberg J, Young K, 2011. VeSPA: integrated applications for RF pulse design, spectral simulation and MRS data analysis. Proceedings of the International Society for Magnetic Resonance in Medicine 19 (19), 1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkáč I, Gruetter R, 2005. Methodology of 1H NMR spectroscopy of the human brain at very high magnetic fields. Appl. Magn. Reson 29 (1), 139–157. doi: 10.1007/BF03166960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanga RPR, DeBrosse C, Kumar D, et al. , 2018. Reproducibility of 2D GluCEST in healthy human volunteers at 7 T. Magn. Reson. Med 80 (5), 2033–2039. doi: 10.1002/mrm.27362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provencher SW, 1993. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn. Reson. Med 30 (6), 672–679. doi: 10.1002/mrm.1910300604. [DOI] [PubMed] [Google Scholar]
- Simpson R, Devenyi GA, Jezzard P, Hennessy TJ, Near J, 2017. Advanced processing and simulation of MRS data using the FID appliance (FID-A) —an open source, MATLAB-based toolkit. Magn. Reson. Med 77 (1), 23–33. doi: 10.1002/mrm.26091. [DOI] [PubMed] [Google Scholar]
- Rich LJ, Bagga P, Miszei G, et al. Combining 1MRS with deuterium labeled glucose: a new strategy to assess dynamics of neural metabolism in vivo. Proc Annu Meet Int Soc Magn Reson Med. Published online 2020. [Google Scholar]
- Moser P, Bogner W, Hingerl L, et al. , 2019. Non-cartesian GRAPPA and coil combination using interleaved calibration data – application to concentric-ring MRSI of the human brain at 7T. Magn. Reson. Med 82 (5), 1587–1603. doi: 10.1002/mrm.27822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coello E, Noeske R, Burns BL, et al. , 2018. High-resolution echo-planar spectroscopic imaging at ultra-high field. NMR Biomed. 31 (11), e3950. doi: 10.1002/nbm.3950. [DOI] [PubMed] [Google Scholar]
- Mullins PG, McGonigle DJ, O’Gorman RL, et al. , 2014. Current practice in the use of MEGA-PRESS spectroscopy for the detection of GABA. Neuroimage 86, 43–52. doi: 10.1016/j.neuroimage.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eijsden P, Behar KL, Mason GF, Braun KPJ, de Graaf RA, 2010. In vivo neurochemical profiling of rat brain by 1 H-[ 13C] NMR spectroscopy: cerebral energetics and glutamatergic/GABAergic neurotransmission. J. Neurochem 112 (1), 24–33. doi: 10.1111/j.1471-4159.2009.06428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf RA, Rothman DL, Behar KL, 2011. State of the art direct 13C and indirect 1H-[13C] NMR spectroscopy in vivo. A practical guide. NMR Biomed 24 (8), 958–972. doi: 10.1002/nbm.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehghani M, Do KQ, Magistretti P, Xin L, 2020. Lactate measurement by neurochemical profiling in the dorsolateral prefrontal cortex at 7T: accuracy, precision, and relaxation times. Magn. Reson. Med 83 (6), 1895–1908. doi: 10.1002/mrm.28066. [DOI] [PubMed] [Google Scholar]
- Pavlova NN, Thompson CB, 2016. The emerging hallmarks of cancer metabolism. Cell Metab. 23 (1), 27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagga P, Rich LJ, Cember ATJ, Nanga RPR, Thakuri D, Elliott M, Haris M, Detre JA, Reddy R, 2020. Assessing gray and white matter glutamatergic turnover in human brain non-invasivley using 1H MRS and deuterated glucose. Proceedings of the International Society for Magnetic Resonance in Medicine. [Google Scholar]
- Rich LJ, Bagga P, Mizsei G, Schnall MD, Detre JA, Haris M, Reddy R 2020b. Detecting glycolytic metabolism in glioblastoma using a new 1H MRS and [6,6–2H2]glucose infusion based approach.Proceedings of the International Society for Magnetic Resonance in Medicine
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Authors will make all original data available upon reasonable request. All software used in image processing and analysis is commercially available.