

Abstract
Stereoselective synthesis of d-glycero- and l-glycero-β-d-mannoheptosides has been achieved by cesium carbonate-mediated β-selective anomeric O-alkylation of the corresponding d-mannoheptoses. In addition, this method has been utilized in the total synthesis of a tetrasaccharide repeat unit of Bacillus thermoaerophilus surface-layer glycoprotein.
Graphical Abstract
Introduction
Harmful bacteria often produce unique and structurally complex capsular polysaccharides (CPS) which are essential virulence factors for pathogen invasion and promising targets for the development of effective vaccines for preventing bacterial infections.1 Among a variety of rare microbial glycosides, β-d-manno-heptopyranosides are expressed in the capsular polysaccharides (CPS) or surface glycoproteins of invasive bacteria strains. For instance, 6-deoxy-β-d-manno-heptopyranosides2 exist in Campylobacter jejuni,3 Plesimonas shigelloides,4 as well as Burkholderia pseudomallei and Burkholderia mallei.5 In addition, β-d-glycero-d-manno-heptopyranosides are found in the glycan of surface-layer glycoprotein from Plesimonas shigelloides4 and Bacillus thermoaerophilus.6 In general, there are two synthetic obstacles for β-d-manno-heptopyranosides: 1) efficient synthesis of seven-carbon d-manno-heptopyranose monosaccharide building blocks; and 2) stereoselective construction of β-d-manno-heptopyranosidic linkages.
Syntheses of higher-carbon monosaccharides, such as 6-deoxy-d-manno-heptose, d-glycero-d-manno-heptose, and l-glycero-d-manno-heptose, pose a challenge in carbohydrate chemistry.7,8 Among various approaches, one-carbon homologation of readily available partially protected d-mannose derivatives bearing C6-aldehyde functionality has been the most popular strategy for the synthesis of d-mannoheptose.9 Efforts involving two-carbon homologation of pentodialdofuranose or aldopentose derivatives via Wittig olefination and osmium-catalyzed dihydroxylation were also reported.10 In addition, syntheses of l-glycero-d-manno-heptose and d-glycero-d-manno-heptose were achieved by indium-mediated acyloxyallylation of unprotected l-lyxose and d-ribose, respectively, followed by ozonolysis.11 Furthermore, l-glycero-d-manno-heptose was also prepared by de novo synthesis based on Mukaiyama-type aldol reaction.12 Recently, Yang and co-workers reported an efficient approach for the synthesis of d-glycero-d-manno-heptose and l-glycero-d-manno-heptose via Mukaiyama-type aldol reactions using d-ribose and l-lyxose as starting materials, respectively.13
As a class of 1,2-cis-glycosidic linkages,14 β-mannosides are difficult to construct due to the steric effect of the axial C2-substituents and the absence of anomeric effect.15 Compared to β-mannosides, synthesis of β-manno-heptopyranosides containing an additional carbon atom at C6 is considered even more challenging. Nevertheless, synthesis of 6-deoxy-β-d-manno-heptosides was accomplished via Crich β-mannosylation followed by radical reduction16 or the use of well-known intramolecular aglycon delivery strategy.17 In addition, Crich β-mannosylation involving the use of 4,6-O-benzylidene protecting strategy has been applied to stereoselective synthesis of d-glycero-β-d-manno-heptopyranosides and l-glycero-β-d-manno-heptopyranosides9a–b,16 Recently, our group developed an approach for stereoselective synthesis of β-mannosides involving Cs2CO3-mediated anomeric O-alkylation18,19 of partially protected mannoses with suitable electrophiles. As part of our program to explore anomeric O-alkylation for microbial glycan synthesis, we became interested in the stereoselective construction of β-d-manno-heptopyranosides. Herein, we report our findings in this direction as well as a total synthesis of tetrasaccharide repeat unit of Bacillus thermoaerophilus surface-layer glycoprotein.
Results and Discussion
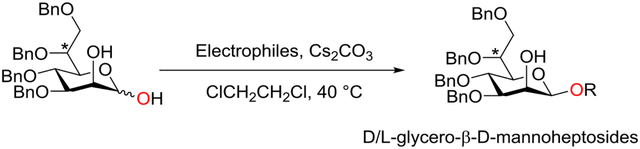
Starting from known readily available partially protected α-d-mannosyl thioglycoside 1,9b d-glycero-d-manno-heptose 7a was prepared in eight steps following Crich procedures (Scheme 1).9b In practice, regioselective protection of the C2-hydroxyl group of diol 1 as its 2-naphthylmethyl (Nap) ether afforded 2 in 93% yield. Swern-oxidation of the primary alcohol of 2 to the corresponding aldehyde followed by Wittig olefination with methyltriphenylphosphonium bromide in the presence of n-butyllithium as the base afforded alkene 3. OsO4-catalyzed dihydroxylation of the alkene of 3 afforded two diastereomers 4a and 4b (4a/4b = 2/1). The stereochemistry of diols 4a and 4b was determined based on comparison of the double doublet representing H5 with literature values.9b The l, d-series (4b) has a smaller 3J5,6 of 2.0 Hz when compared to the 3J5,6 coupling of 5.7 Hz observed in the d, d-series (4a). The major diastereomer 4a was then treated with trifluoroacetic acid to remove the bis-ketal and the resulting tetra-ol was globally protected as their benzyl ethers to furnish intermediate 5. Next, DDQ-mediated deprotection of 2-naphthylmethyl (Nap) ether gave the C2-free alcohol 6 which was then oxidized to the desired d-glycero-d-manno-heptose 7a using bromine20 as the oxidant. Following the same sequence of reactions, l-glycero-d-manno-heptose 7b was obtained from 4b.21
Scheme 1.
Synthesis of d- and l-glycero-d-manno-heptose 7a-b.
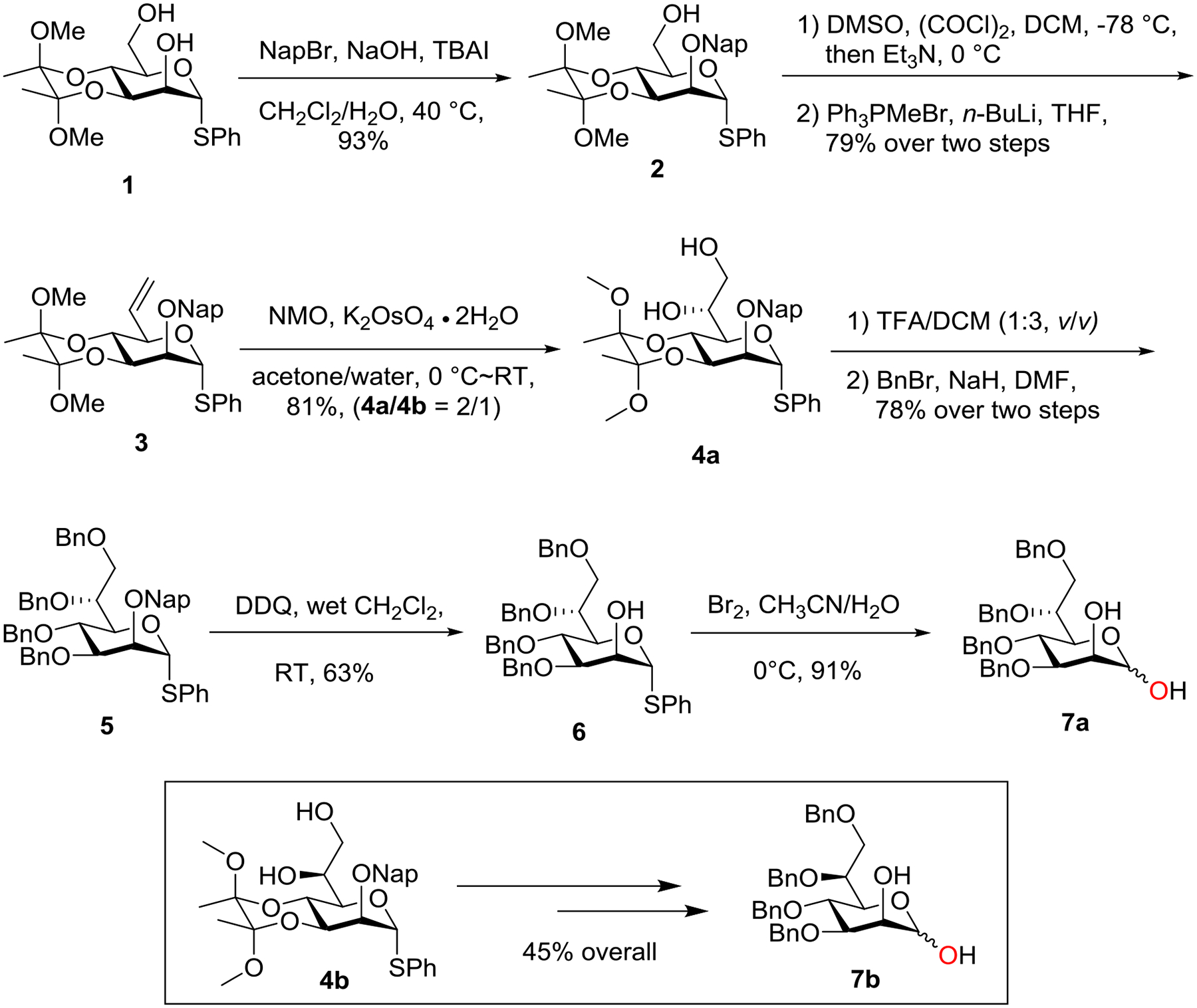
With d-glycero-d-manno-heptose 7a and l-glycero-d-manno-heptose 7b in hands, we investigated their reactivity towards cesium carbonate-mediated anomeric O-alkylation with various primary and secondary triflate electrophiles (Table 1). It was found that anomeric O-alkylation of d-glycero-d-manno-heptose 7a and l-glycero-d-manno-heptose 7b with known C4-triflate 818a (2.5 eq.) in the presence of Cs2CO3 (3.0 eq.) afforded corresponding d-glycero-β-d-manno-heptoside 13 and l-glycero-β-d-manno-heptoside 14 in 53% and 50% yield, respectively (β only). The resulting similar yields and anomeric selectivity between 13 and 14 indicated that the C6-stereochemsitry of the D-manno-heptose has little effect on this type of the β-selective anomeric alkylation. The moderate yields of 13 and 14 also suggest that d/l-glycero-d-manno-heptose 7a-b are not as reactive as their corresponding d-mannose in this type of reaction, as anomeric O-alkylation of 3,4,6-tri-O-benzyl-d-mannose with C4-triflate 8 under the same condition afforded corresponding β-d-mannoside in 75% yield (β only).18a Upon further optimization, d-glycero-β-d-manno-heptoside 13 was obtained in 65% yield when 3.0 eq. of C4-triflate 8 and 3.5 eq. of Cs2CO3 were used. Under the same condition, d-glycero-β-d-manno-heptoside 15 and 16 were obtained in 54% and 37% yields from anomeric O-alkylation of d-glycero-d-manno-heptose 7a involving C4-triflate 918a and C3-triflate 10,21 respectively. In all these cases, significant elimination of sugar-derived triflates were observed. Not surprisingly, when more reactive sugar-derived primary triflates 1118a and 1219 were employed as electrophiles, anomeric O-alkylation of d-glycero-d-manno-heptose 7a was efficient and d-glycero-β-d-manno-heptoside 17 and 18 were obtained in 78% and 72% yields, respectively (β only). It is worth noting that reduced amounts of primary triflates (2.0 eq.) or Cs2CO3 (2.5 eq.) sufficed this type of anomeric O-alkylation. This approach provides an alternative to the available method for stereoselective synthesis of both d-glycero- and l-glycero-β-d-manno-heptopyranosides.9a–b,16 In the Crich strategy, synthesis of 4,6-O-benzylidene protected l-glycero-d-manno-heptopyranosyl thioglycoside donor is difficult due to the development of 1,3-diaxial interaction in the acetal ring.16 In addition, a small amount of l-glycero-α-d-manno-heptopyranosides was obtained when less reactive acceptor was used.16
Table 1.
Synthesis of d- and l-glycero-β-d-manno-heptopyranosides via Cs2CO3-meidated anomeric O-alkylation.a
|
Isolated yield based on triflate (3.0 eq.) and Cs2CO3 (3.5 eq.) unless otherwise noted.
2.5 eq. of triflate and 3.0 eq. of Cs2CO3 were used.
2.0 eq. of triflate and 2.5 eq. of Cs2CO3 were used.
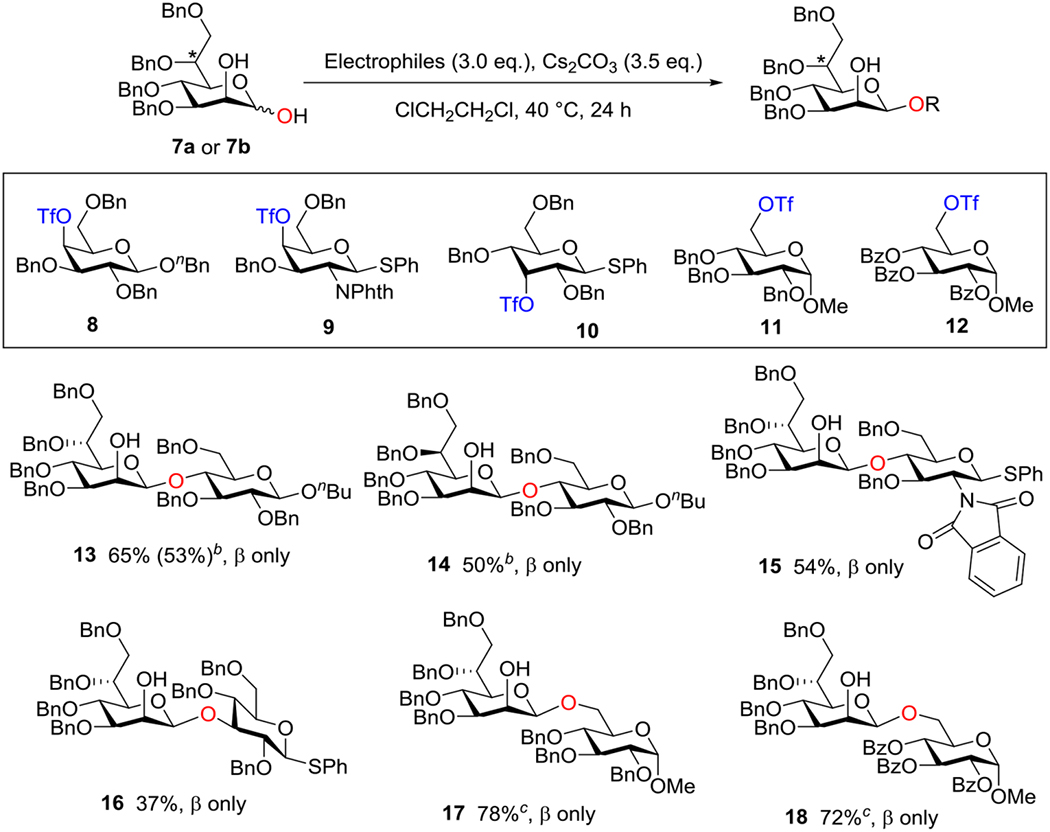
Next, we aimed at the utilization of this anomeric O-alkylation for the synthesis of a tetrasaccharide repeat unit of Bacillus thermoaerophilus surface-layer glycoprotein. In 1995, Kosma and co-workers described the characterization of the surface-layer glycoprotein from Bacillus thermoaerophilus and proposed the very unusual disaccharide motif →3)-β-d-glycero-d-mannoheptopyranosyl-(1→4)-α-l-rhamnopyranosyl-(1→ as the repeating unit of the glycan chain (cf. 19, Figure 1). Previously, Crich and Li reported the synthesis of tetra- and hexasaccharide repeat units of the glycan 19.9b
Figure 1.
Repeating unit of the glycan chain of the surface-layer glycoprotein from Bacillus thermoaerophilus.
Last year, we reported the synthesis of protected β-d-mannopyranosyl-(1→4)-α-l-rhamnopyranoside via cesium carbonate-mediated anomeric O-alkylation followed stereochemical inversion involving four steps:18f 1) anomeric O-alkylation of 3,4,6-tri-O-benzyl-d-mannose with an l-fucose-derived C4-triflate to afford a β-d-mannopyranosyl-(1→4)-α-l-quinovopyranoside; 2) liberation of the equatorial C2-alcohol of the resulting L-quinovose moiety; 3) Swern-oxidation of this C2-alcohol to the β-d-mannopyranosyl-(1→4)-α-d-rhamnopyranoside corresponding ketone; and 4) stereoselective reduction of this ketone to the axial C2-alcohol to achieve β-d-mannopyranosyl-(1→4)-α-l-rhamnopyranoside. However, when d-glycero-d-manno-heptose 7a was subjected to anomeric O-alkylation with the same l-fucose-derived triflate,18f corresponding disaccharide was obtained in low yield. Therefore, we decided to prepare an alternative electrophile, l-sugar-derived 2,3-unsaturated C4-nosylate 2021 (Scheme 2) and evaluate its reactivity. In practice, d-glycero-d-manno-heptose 7a was first treated with C4-nosylate 20 (3.0 eq.) in DCE in the presence of Cs2CO3 (3.5 eq.) at 40 °C for 24 h, and the crude mixture was quickly purified by a short silica gel chromatography to remove unreacted C4-nosylate 20. The crude product was then subjected to osmium tetroxide-catalyzed dihydroxylation and subsequent global benzoylation. Gratifyingly, desired disaccharide 21 was obtained in 46% (over three steps, β only). The 3J1,2, 3J2,3, 3J3,4 and 3J4,5 of the L-rhamnoside moiety of 21 was measured to be 1.8, 3.5, 9.7 and 9.7 Hz, respectively, which confirms the cis-2,3-dibenzoates (via per-benzoylation of the cis-2,3-diol obtained by the dihydroxylation) and structure of the L-rhamnoside.
Scheme 2.
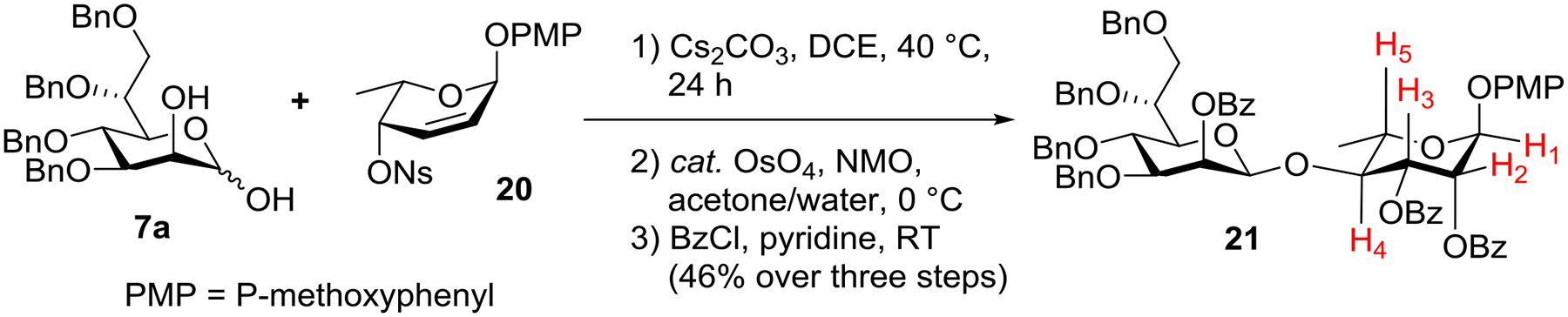
Anomeric O-alkylation of d-glycero-d-manno-heptose 7a with C4-nosylate 20.
Encouraged by this result, we initiated the synthesis of a tetrasaccharide repeat unit of Bacillus thermoaerophilus surface-layer glycoprotein. In order to access disaccharide acceptor 28, benzylation of the diol moiety in intermediate 4a followed by hydrolysis of the bis-ketal afforded diol 22 in 69% yield over two steps (Scheme 3). Next, oxidative deprotection of the Nap ether of 22 in dichloromethane gave intermediate 23 bearing a 2,3-O-naphthalylidene acetal which serves as a temporary protecting group. Thus, the C4-alcohol of 23 was benzylated followed by hydrolysis of 2,3-O-naphthalylidene to furnish diol 24 (68% yield over three steps). Dibutyltin oxide-mediated regioselective PMB-protection of diol 24 provided 25 which was converted into the lactol 26 using bromine as the oxidant.20 Similarly, cesium carbonate-mediated anomeric O-alkylation of d-glycero-d-manno-heptose 26 with C4-nosylate 20, followed by osmium tetroxide-catalyzed dihydroxylation and subsequent global benzylation, afforded disaccharide 27 (49% over three steps). The PMB ether of 27 was subsequent removed under acidic conditions to produce the disaccharide acceptor 28.
Scheme 3.
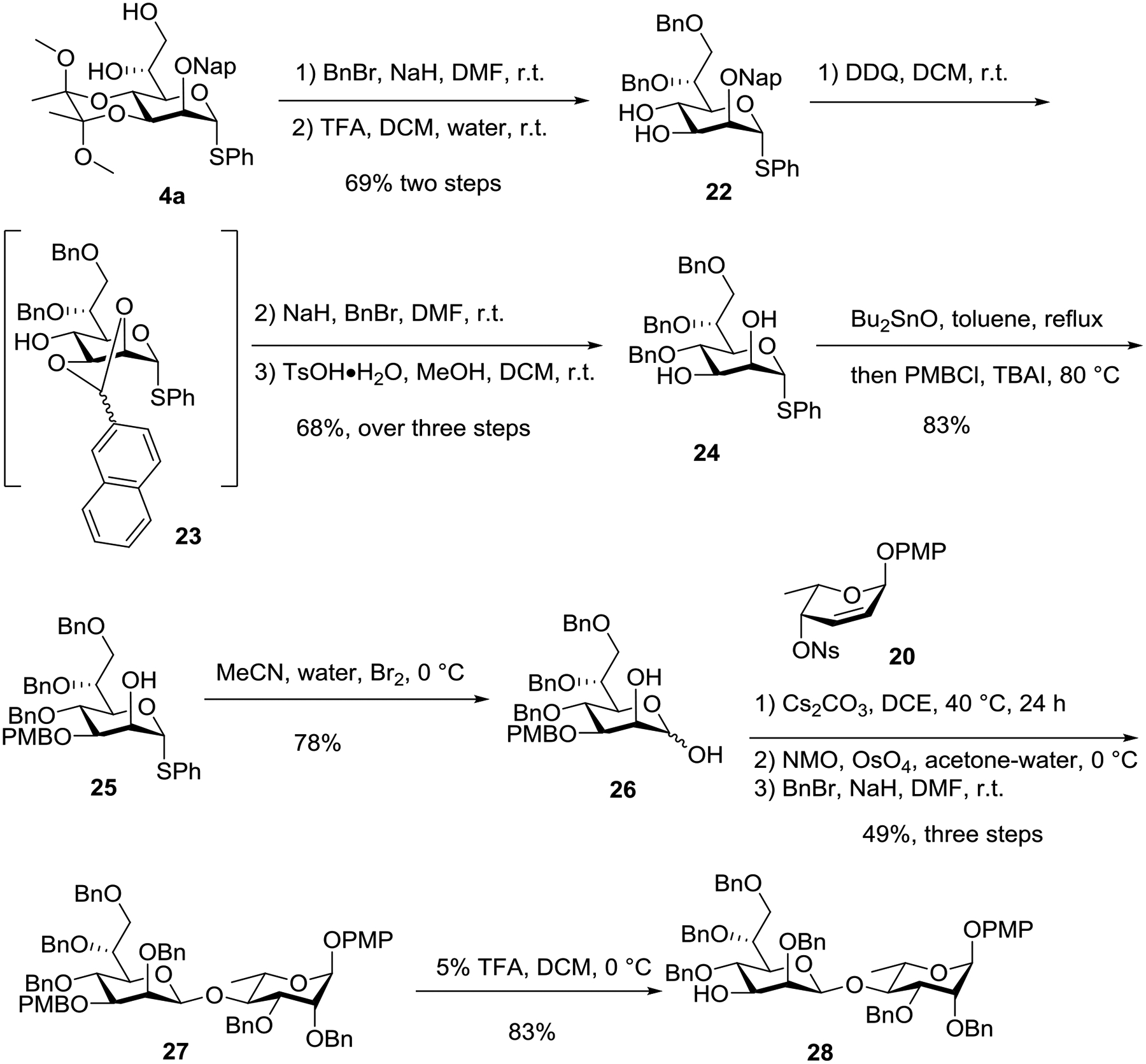
Synthesis of disaccharide acceptor 28.
In the endgame, disaccharide 21 was converted into its corresponding glycosyl trichloroacetimidate donor by two steps: 1) cerium (IV) ammonium nitrate (CAN)-mediated oxidative deprotection of the anomeric PMP ether gave lactol 29 (76%); and 2) treatment of this lactol with trichloroacetonitrile in the presence of DBU afforded glycosyl trichloroacetimidate donor 30 (89%, Scheme 4). Next, 2+2 glycosylation of donor 30 and acceptor 28 went smoothly in the presence of catalytic amount of TMSOTf to produce tetrasaccharide 31 in 90% yield. Finally, removal of the benzoates in 31 followed global debenzylation by Pd/C-catalyzed hydrogenolysis afforded tetrasaccharide repeat unit 32 of Bacillus thermoaerophilus surface-layer glycoprotein.
Scheme 4.
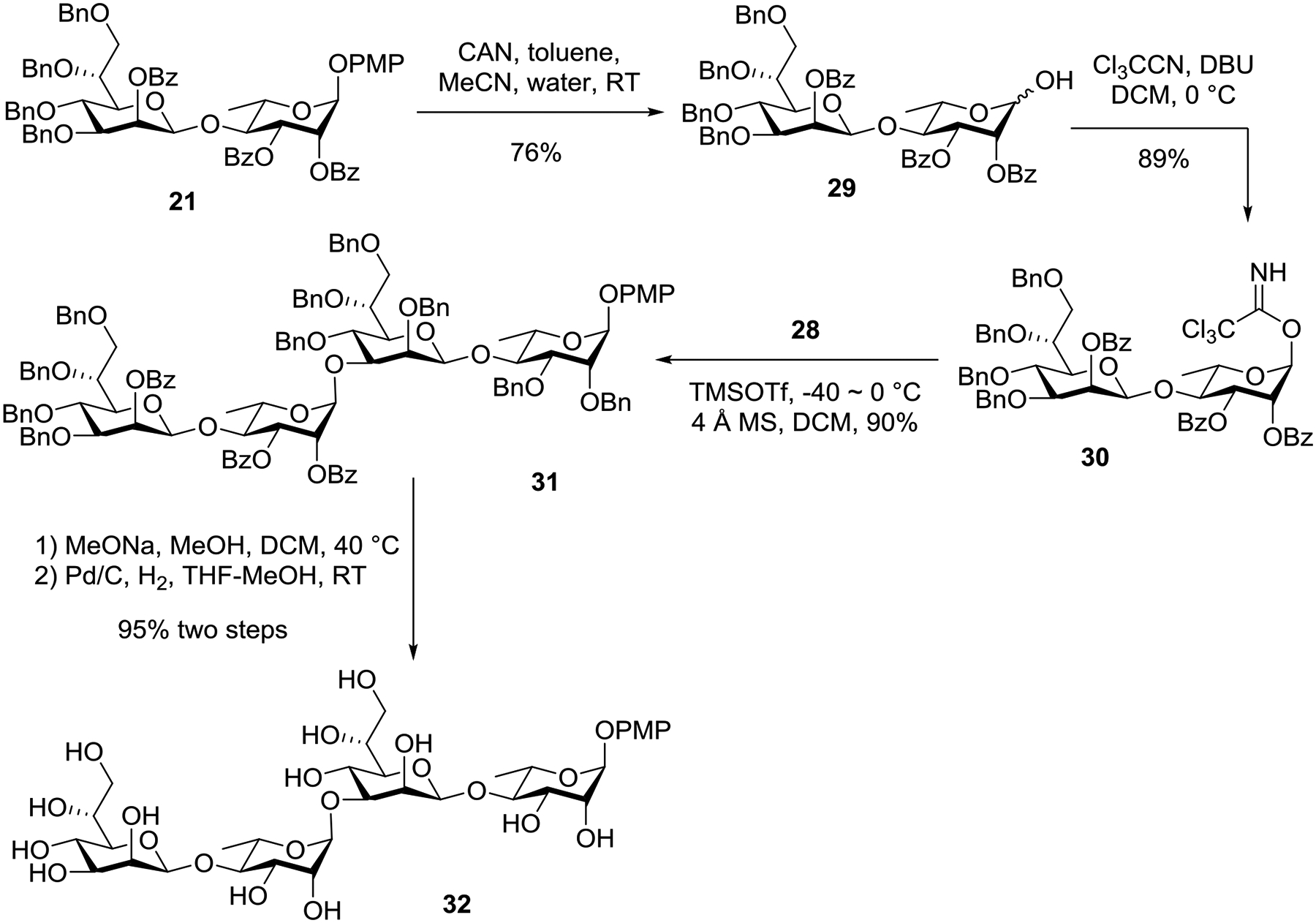
Synthesis of a tetrasaccharide repeat unit of Bacillus thermoaerophilus surface-layer glycoprotein.
Conclusion
We have described an approach for stereoselective synthesis of d/l-glycero-β-d-mannoheptosides via cesium carbonate-mediated anomeric O-alkylation. In addition, this method has been utilized in the total synthesis of a tetrasaccharide repeat unit of Bacillus thermoaerophilus surface-layer glycoprotein. A key step in the synthesis involves an anomeric O-alkylation of a d-glycero-d-manno-heptose with an L-sugar-derived 2,3-unsaturated C4-nosylate followed by a diastereoselective osmium tetroxide-catalyzed dihydroxylation to establish the desired d-glycero-β-d-manno-heptopyranosyl-(1→4)-α-l-rhamnopyranoside repeating unit.
Experimental Section
Materials and Methods.
All reagents and chemicals were purchased from Acros Organics, Sigma Aldrich, Fisher Scientific, Alfa Aesar, and Strem Chemicals and used without further purification. THF, dichloromethane, toluene, and diethyl ether were purified by passing through two packed columns of neutral alumina (Innovative Technology). Anhydrous DMF and benzene were purchased from Acros Organics and Sigma-Aldrich and used without further drying. All reactions were carried out in oven-dried glassware under an argon atmosphere unless otherwise noted. All reactions that require elevated temperature were carried out under traditional oil bath heating. Analytical thin layer chromatography was performed using 0.25 mm silica gel 60-F plates. Preparative thin layer chromatography was performed using 1 mm silica gel prep TLC plates. Flash column chromatography was performed using 200–400 mesh silica gel (Scientific Absorbents, Inc.). Yields refer to chromatographically and spectroscopically pure materials, unless otherwise stated.
Proton and carbon nuclear magnetic resonance spectra (1H NMR and 13C{1H} NMR) were recorded on either Bruker 600 (1H NMR-600 MHz; 13C{1H} NMR 150 MHz) at ambient temperature with CDCl3 as the solvent unless otherwise stated. Chemical shifts are reported in parts per million relative to residual protic solvent internal standard CDCl3: 1H NMR at δ 7.26, 13C{1H} NMR at δ 77.16. Data for 1H NMR are reported as follows: chemical shift, integration, multiplicity (app = apparent, par obsc = partially obscure, ovrlp = overlapping, s = singlet, d = doublet, dd = doublet of doublet, t = triplet, q = quartet, m = multiplet) and coupling constants in Hertz. All 13C{1H} NMR spectra were recorded with complete proton decoupling. Structural assignments were made with additional information from gCOSY, gHSQC, and gHMBC experiments. High resolution mass spectra (HRMS) were acquired on a Waters Acuity Premiere XE TOF LC-MS by electrospray ionization. Optical rotations were measured with Autopol-IV digital polarimeter; concentrations are expressed as g/100 mL.
Phenyl 2-O-(2-naphthalenyl)methyl-3,4-O-[(2S,3S)-2,3-dimethoxy-2,3-butanedily]-1-thio-α-d-mannopyranoside (2).
To a solution of known phenyl 3,4-O-[(2S,3S)-2,3-dimethoxy-2,3-butanedily]-1-thio-α-d-mannopyranoside (1)9b (10.0 g, 25.9 mmol) in CH2Cl2 (20 mL) was added (2-naphthalenyl)methyl bromide (6.3 g, 28.5 mmol), tetra-n-butylammonium iodide (1.91 g, 5.18 mmol) and a solution of NaOH (3.1 g) in water (20 mL). The mixture was stirred vigorously at 40 °C for 12 h. After the CH2Cl2 layer was separated, resulting aqueous solution was extracted with CH2Cl2 twice. The combined organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (Hexanes/EtOAc = 4/1) to give compound 2 (12.69 g, 24.09 mmol, 93%) as colorless syrup. = +144.9 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.89 – 7.80 (m, 4H, HAr), 7.57 (dd, J = 8.4, 1.7 Hz, 1H, HAr), 7.50 – 7.45 (m, 2H, HAr), 7.36 – 7.33 (m, 2H, HAr), 7.25 – 7.22 (m, 3H, HAr), 5.48 (d, J = 0.7 Hz, 1H, H-1), 5.05 (d, J = 12.1 Hz, 1H, -OCHHAr), 4.85 (d, J = 12.1 Hz, 1H, -OCHHAr), 4.32 (t, J = 10.0 Hz, 1H, H-4), 4.24 (ddd, J = 10.0, 4.4, 3.2 Hz, 1H, H-5), 4.09 (dd, J = 10.0, 2.9 Hz, 1H, H-3), 4.03 (dd, J = 2.9, 0.7 Hz, 1H, H-2), 3.87 – 3.76 (m, 2H, H-6), 3.33 (s, 3H, -OCH3), 3.32 (s, 3H, -OCH3), 1.84 (t, J = 6.5 Hz, 1H, C6-OH), 1.41 (s, 3H, -CH3), 1.35 (s, 3H, -CH3). 13C{1H} NMR (150 MHz, CDCl3) δ 135.9, 134.0, 133.40, 133.1, 132.0, 129.2, 128.2, 128.0, 127.8, 127.7, 126.9, 126.2, 126.1, 126.0, 100.2, 99.8, 87.7, 77.6, 73.4, 72.1, 69.5, 64.0, 61.7, 48.2, 48.1, 18.0, 17.9. HRMS (ESI) m/z: [M+Na]+ Calcd for C29H34NaO7S 549.1923; found 549.1927.
Phenyl 2-O-(2-naphthalenyl)methyl-6,7-dideoxy-3,4-O-[(2S,3S)-2,3-dimethoxy-2,3-butanedily]-1-thio-α-d-glycero-D-manno-hepto-6-enopyranoside (3).
To a solution of oxalyl chloride (1.8 mL, 21.3 mmol) in anhydrous CH2Cl2 (20 mL) cooled at −78 °C was added dry DMSO (3.0 mL, 42.2 mmol) dropwise. After the resulting mixture was stirred at −78 °C for 30 min, a solution of compound 2 (5.80 g, 11.0 mmol) in anhydrous CH2Cl2 (9 mL + 1 mL rinse) was added. The mixture was stirred at −78 °C for 1 h before being slowly warmed to −40 °C over a period of 2 h. Et3N (12.0 mL, 86.1 mmol) was added and the resulting mixture was slowly warmed to 0 °C. The mixture was stirred at 0 °C for 1 h and a lot of solids were seen in the flask. The mixture was diluted with CH2Cl2 (50 mL) and sequentially washed with water, saturated NaHCO3 solution and brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated, and dried under vacuum overnight to afford the aldehyde intermediate as yellow syrup which was directly used in the next step. To a suspension of methyltriphenylphosphonium bromide (8.84 g, 27.4 mmol) in anhydrous THF (30 mL) cooled at 0 °C was added a solution of n-BuLi in hexanes (2.5 M, 8.8 mL). The mixture was stirred at 0 °C for 30 min when all of the solids were dissolved completely. The resulting solution was cooled to −40 °C, and a solution of the aforementioned aldehyde intermediate in anhydrous THF (9 mL + 1 mL rinse) was added dropwise. The resulting mixture was stirred at −40 °C for 2 h and slowly warmed to room temperature overnight. The reaction mixture was quenched with water, and THF was removed under reduced pressure. The residue was diluted with CH2Cl2 and washed with water and brine. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (Hexanes/EtOAc = 20/1) to give compound 3 (4.54 g, 8.69 mmol, 79%) as light yellow syrup. = +191.0 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.95 – 7.78 (m, 4H, HAr), 7.60 (dd, J = 8.4, 1.7 Hz, 1H, HAr), 7.55 – 7.42 (m, 2H, HAr), 7.40 – 7.32 (m, 2H, HAr), 7.29 – 7.12 (m, 3H, HAr), 6.01 (ddd, J = 17.3, 10.7, 5.6 Hz, 1H, H-6), 5.55 (d, J = 0.8 Hz, 1H, H-1), 5.47 (dt, J = 17.3, 1.6 Hz, 1H, H-7a), 5.28 (dt, J = 10.7, 1.6 Hz, 1H, H-7b), 5.05 (d, J = 12.4 Hz, 1H, -OCHHAr), 4.90 (d, J = 12.4 Hz, 1H, -OCHHAr), 4.66 (m, 1H, H-5), 4.17 – 4.07 (m, 2H, H-3, H-4), 4.06 (m, 1H, H-2), 3.35 (s, 3H, -OCH3), 3.30 (s, 3H, -OCH3), 1.44 (s, 3H, -CH3), 1.38 (s, 3H, -CH3). 13C{1H} NMR (150 MHz, CDCl3) δ 136.0, 134.6, 133.8, 133.4, 133.1, 131.5, 129.0, 128.0, 127.9, 127.8, 127.4, 126.8, 126.2, 126.1, 125.9, 118.0, 100.1, 99.9, 87.5, 77.7, 73.1, 72.2, 69.6, 67.7, 48.1, 48.0, 18.0. HRMS (ESI) m/z: [M+Na]+ Calcd for C30H34NaO6S 545.1974; found 545.1963.
Phenyl 2-O-(2-naphthalenyl)methyl-3,4-O-[(2S,3S)-2,3-dimethoxy-2,3-butanedily]-1-thio-α-d-glycero-d-manno-heptopyranoside (4a) and phenyl 2-O-(2-naphthalenyl)methyl-3,4-O-[(2S,3S)-2,3-dimethoxy-2,3-butanedily]-1-thio-α-l-glycero-d-manno-heptopyranoside (4b).
To a solution of compound 3 (2.56 g, 4.90 mmol) in acetone/water (10 mL/1 mL) cooled at 0 °C was added potassium osmate(VI) dehydrate (9.0 mg, 0.0245 mmol) and an aqueous solution of NMO (50 wt%, 1.5 mL). The mixture was slowly warmed to room temperature and stirred overnight. The reaction mixture was quenched by NaHSO3. Acetone was removed under reduced pressure and the residue was diluted by CH2Cl2 and washed by water and brine. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (CH2Cl2/EtOAc = 10/1 → 4/1 with 1% methanol) to give compound 4a (1.48 g, 2.65 mmol, 54%) and 4b (736 mg, 1.32 mmol, 27%) as colorless syrup. Compound 4a: = +45.6 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.92 – 7.82 (m, 4H, HAr), 7.59 (dd, J = 8.4, 1.6 Hz, 1H, HAr), 7.53 – 7.48 (m, 2H, HAr), 7.43 – 7.36 (m, 2H, HAr), 7.31 – 7.23 (m, 3H, HAr), 5.47 (s, 1H, H-1), 5.07 (d, J = 12.2 Hz, 1H, -OCHHAr), 4.88 (d, J = 12.2 Hz, 1H, -OCHHAr), 4.44 (t, J = 10.0 Hz, 1H, H-4), 4.30 (dd, J = 10.0, 5.7 Hz, 1H, H-5), 4.12 (dd, J = 10.0, 2.8 Hz, 1H, H-3), 4.04 (d, J = 2.8 Hz, 1H, H-2), 3.99 (m, 1H, H-6), 3.73 (dd, J = 11.7, 5.0 Hz, 1H, H-7a), 3.68 (dd, J = 11.7, 3.8 Hz, 1H, H-7b), 3.38 (s, 3H, -OCH3), 3.35 (s, 3H, -OCH3), 3.19 (bs, 1H, -OH), 2.20 (bs, 1H, -OH), 1.44 (s, 3H, -CH3), 1.38 (s, 3H, -CH3). 13C{1H} NMR (150 MHz, CDCl3) δ 135.80, 133.55, 133.37, 133.14, 132.16, 129.24, 128.19, 127.99, 127.95, 127.82, 126.84, 126.20, 126.12, 126.01, 100.21, 100.16, 87.68, 77.40, 73.41, 73.37, 71.04, 69.28, 67.02, 63.14, 48.52, 48.25, 18.05, 17.93. HRMS (ESI) m/z: [M+Na]+ Calcd for C30H36NaO8S 579.2029; found 579.2025. Compound 4b: = +39.1 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.94 – 7.79 (m, 4H, HAr), 7.59 (dd, J = 8.4, 1.7 Hz, 1H, HAr), 7.51 – 7.46 (m, 2H, HAr), 7.36 – 7.21 (m, 5H, HAr), 5.53 (d, J = 1.2 Hz, 1H, H-1), 5.09 (d, J = 12.1 Hz, 1H, -OCHHAr), 4.86 (d, J = 12.1 Hz, 1H, -OCHHAr), 4.47 (t, J = 10.2 Hz, 1H, H-4), 4.16 (dd, J = 10.0, 2.0 Hz, 1H, H-5), 4.11 (dd, J = 10.4, 2.8 Hz, 1H, H-3), 4.03 (dd, J = 2.8, 1.2 Hz, 1H, H-2), 3.96 (m, 1H, H-6), 3.52 (d, J = 5.2 Hz, 2H, H-7), 3.36 (s, 3H, -OCH3), 3.33 (s, 3H, -OCH3), 2.32 (s, 1H, -OH), 1.80 (s, 1H, -OH), 1.41 (s, 3H, -CH3), 1.35 (s, 3H, -CH3). 13C{1H} NMR (150 MHz, CDCl3) δ 135.9, 133.4, 133.2, 132.1, 129.4, 128.2, 128.1, 128.0, 127.8, 126.9, 126.2, 126.0, 100.2, 99.9, 87.5, 77.2, 73.5, 72.9, 69.5, 68.6, 65.1, 63.3, 48.3, 48.2, 18.0, 17.9. HRMS (ESI) m/z: [M+Na]+ Calcd for C30H36NaO8S 579.2029; found 579.2025.
Phenyl 2-O-(2-naphthalenyl)methyl-3,4,6,7-tetra-O-benzyl-1-thio-α-d-glycero-d-manno-heptopyranoside (5).
To a solution of compound 4a (1.39 g, 2.50 mmol) in CH2Cl2 (30 mL) was added TFA (10 mL) and water (0.3 mL). The mixture was stirred at room temperature for 4 h. The mixture was neutralized with saturated NaHCO3 solution and extracted with CH2Cl2. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (CH2Cl2/MeOH = 30/1 → 10/1) to give the tetra-ol intermediate which was dissolved in anhydrous DMF (5 mL). To this DMF solution cooled at 0 °C was added NaH (60% on mineral oil, 800 mg), and the resulting mixture was stirred at 0 °C for 30 min and BnBr (1.8 mL) was added dropwise. The reaction mixture was slowly warmed to room temperature and stirred for 5 h. After being quenched with methanol (1 mL) at 0 °C, the reaction mixture was diluted with CH2Cl2 (100 mL), washed by water and brine, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (Hexanes/EtOAc = 10/1) to give compound 5 (1.57 g, 1.96 mmol, 78% over two steps from 4a) as colorless syrup. = +12.7 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.86 – 7.81 (m, 1H, HAr), 7.79 – 7.71 (m, 3H, HAr), 7.52 – 7.46 (m, 3H, HAr), 7.43 – 7.39 (m, 2H, HAr), 7.37 – 7.16 (m, 23H, HAr), 5.57 (d, J = 1.9 Hz, 1H, H-1), 4.90 (d, J = 10.8 Hz, 1H, -OCHHAr), 4.88 – 4.78 (m, 3H, -OCHHAr), 4.72 – 4.61 (m, 4H, -OCHHAr), 4.51 (d, J = 12.0 Hz, 1H, -OCHHAr), 4.47 (d, J = 12.0 Hz, 1H, -OCHHAr), 4.43 (dd, J = 9.4, 1.4 Hz, 1H, H-5), 4.19 (t, J = 9.4 Hz, 1H, H-4), 4.09 – 4.02 (m, 2H, H-6, H-2), 3.90 (dd, J = 9.4, 3.0 Hz, 1H, H-3), 3.82 (dd, J = 10.5, 4.4 Hz, 1H, H-7a), 3.76 (dd, J = 10.5, 6.9 Hz, 1H, H-7b). 13C{1H} NMR (150 MHz, CDCl3) δ 138.9, 138.6, 138.6, 138.3, 135.6, 134.4, 133.3, 133.1, 132.1, 129.1, 128.6, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 128.0, 127.9, 127.8, 127.7, 127.7, 127.6, 127.5, 127.4, 126.7, 126.3, 126.1, 126.0, 86.0, 80.6, 78.7, 76.5, 75.0, 74.9, 73.3, 73.2, 72.4, 72.3, 72.2, 71.2. HRMS (ESI) m/z: [M+Na]+ Calcd for C52H50NaO6S 825.3226; found 825.3227.
Phenyl 3,4,6,7-tetra-O-benzyl-1-thio-α-d-glycero-d-manno-heptopyranoside (6).
To compound 5 (1.20 g, 1.50 mmol) in a mixture of CH2Cl2 (10 mL) and water (1 mL) was added 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (681 mg, 3.00 mmol). The mixture was stirred at room temperature for 5 h and then quenched by 40% Na2S2O3 solution. After the organic layer was separated, aqueous solution was extracted by CH2Cl2 (100 mL). The combined organic layer was washed with saturated NaHCO3 solution and brine, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (Hexanes/EtOAc = 4/1) to give compound 6 (626 mg, 0.945 mmol, 63%) as colorless syrup. = +5.4 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.52 – 7.45 (m, 2H, HAr), 7.38 – 7.17 (m, 23H, HAr), 5.53 (d, J = 1.9 Hz, 1H, H-1), 4.82 (d, J = 10.8 Hz, 1H, -OCHHAr), 4.76 – 4.59 (m, 5H, -OCHHAr), 4.52 – 4.42 (m, 3H, -OCHHAr, H-5), 4.24 (td, J = 3.0, 1.9 Hz, 1H, H-2), 4.04 – 3.95 (m, 2H, H-6, H-4), 3.89 (dd, J = 8.8, 3.0 Hz, 1H, H-3), 3.76 (dd, J = 10.3, 4.8 Hz, 1H, H-7a), 3.70 (dd, J = 10.3, 6.8 Hz, 1H, H-7b), 2.57 (d, J = 3.0 Hz, 1H, C2-OH). 13C{1H} NMR (150 MHz, CDCl3) δ 138.8, 138.5, 138.4, 137.6, 133.9, 132.1, 129.1, 128.8, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 127.6, 127.6, 127.5, 87.5, 80.8, 78.3, 74.9, 74.6, 73.3, 72.7, 72.5, 72.3, 70.9, 69.8. HRMS (ESI) m/z: [M+Na]+ Calcd for C41H42NaO6S 685.2600, found 685.2593.
3,4,6,7-Tetra-O-benzyl-1-thio-α/β-d-glycero-d-manno-heptopyranose (7a).
To a solution of compound 6 (566 mg, 0.854 mmol) in MeCN/water (8 mL/0.8 mL) cooled at 0 °C was slowly added a solution of Br2 in CH2Cl2 (5 M, 512 μL). The mixture was stirred at 0 °C for 3 h before being quenched with 40% aqueous Na2S2O3 solution. After MeCN was removed under reduced pressure, the mixture was extracted with CH2Cl2 (100 mL). The organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (Hexanes/EtOAc = 1/1 with 1% methanol) to give compound 7a (443 mg, 0.777 mmol, 91%) as colorless syrup. α/β = 1/0.4. = +0.7 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.38 – 7.12 (m, 40H, α HAr × 20, β HAr × 20), 5.24 (d, J = 1.8 Hz, 1H, α H-1), 4.84 – 4.78 (m, 2H, α -OCHHAr, β -OCHHAr), 4.76 – 4.63 (m, 9H, α -OCHHAr × 4, β -OCHHAr × 4, β H-1), 4.61 (d, J = 10.8 Hz, 1H, β -OCHHAr), 4.57 (d, J = 10.9 Hz, 1H, α -OCHHAr), 4.51 – 4.42 (m, 4H, α -OCHHAr × 2, β -OCHHAr × 2), 4.14 (dd, J = 10.1, 1.4 Hz, 1H, α H-6), 4.05 – 3.99 (m, 3H, α H-2, α H-5, β H-6), 3.97 (dd, J = 3.3, 1.4 Hz, 1H, β H-2), 3.93 (dd, J = 9.0, 3.2 Hz, 1H, α H-3), 3.88 (dd, J = 9.7, 8.9 Hz, 1H, β H-4), 3.83 (dd, J = 10.1, 9.0 Hz, 1H, α H-4), 3.79 – 3.66 (m, 4H, α H-7a, β H-7a, β H-7b, α H-7b), 3.59 (dd, J = 8.9, 3.2 Hz, 1H, β H-3), 3.54 (dd, J = 9.7, 1.5 Hz, 1H, β H-5). 13C{1H} NMR (150 MHz, CDCl3) δ 138.7, 138.6, 138.5, 138.4, 138.3, 137.9, 137.7, 128.8, 128.7, 128.5, 128.5, 128.4, 128.2, 128.1, 128.1, 128.0, 128.0, 127.9, 127.8, 127.7, 127.7, 127.6, 127.6, 94.1 (β C-1), 93.9 (α C-1), 82.1 (β), 80.3, 78.2 (β), 77.5, 75.7 (β), 74.9, 74.8 (β), 74.4, 73.9 (β), 73.4, 73.3 (β), 72.8 (β), 72.5, 72.1, 72.0 (β), 71.9, 70.5 (β), 70.4, 68.6 (β), 68.5. HRMS (ESI) m/z: [M+Na]+ Calcd for C35H38NaO7 593.2515, found 593.2512.
3,4,6,7-Tetra-O-benzyl-α/β-L-glycero-d-manno-heptopyranose (7b).
3,4,6,7-Tetra-O-benzyl-α/β-L-glycero-D-manno-heptopyranose 7b (α/β = 1/0.4) was prepared from intermediate 4b (45% yield over 4 steps) following the same procedure described previously for the synthesis of 7a. = +9.2 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.49 – 6.97 (m, 40H, α HAr × 20, β HAr × 20), 5.22 (s, 1H, α H-1), 4.91 – 4.75 (m, 4H, α -OCHHAr × 2, β -OCHHAr × 2), 4.70 – 4.43 (m, 11H, α -OCHHAr × 5, β -OCHHAr × 5, β H-1), 4.36 – 4.29 (m, 2H, α -OCHHAr, β -OCHHAr), 4.23 (d, J = 11.4 Hz, 1H, β C1-OH), 4.15 – 4.06 (m, 2H, α H-6, β H-6), 4.04 – 3.92 (m, 5H, α H-2, α H-4, α H-3, α H-7, β H-4), 3.88 (m, 1H, β H-2), 3.84 (dd, J = 9.8, 6.2 Hz, 1H, β H-7a), 3.79 – 3.75 (m, 2H, α H-7a, β H-7b), 3.72 (dd, J = 9.8, 6.2 Hz, 1H, α H-7b), 3.59 (dd, J = 9.2, 3.3 Hz, 1H, β H-3), 3.41 (dd, J = 9.8, 1.7 Hz, 1H, β H-5), 2.94 (d, J = 4.9 Hz, 1H, β C2-OH), 2.70 (d, J = 3.5 Hz, 1H, α C1-OH), 2.48 (bs, 1H, α C2-OH). 13C{1H} NMR (150 MHz, CDCl3) δ 138.8, 138.7, 138.5, 138.3, 138.3, 137.9, 137.7, 128.7, 128.6, 128.5, 128.5, 128.5, 128.4, 128.3, 128.1, 128.1, 128.1, 128.0, 127.9, 127.9, 127.8, 127.8, 127.7, 127.7, 127.7, 127.6, 127.6, 94.9 (β C-1), 94.1 (α C-1), 82.3 (β), 80.3, 74.9 (β), 74.8 (β), 74.8, 74.7, 73.8, 73.5 (β), 73.4, 73.2 (β), 73.0 (β), 72.9, 71.8, 71.4 (β), 70.9, 70.4 (β), 70.1, 68.8 (β), 68.3. HRMS (ESI) m/z: [M+Na]+ Calcd for C35H38NaO7 593.2515, found 593.2512.
Phenyl 2,4,6-tri-O-benzyl-3-O-trifluoromethanesulfonyl-1-thio-β-d-allopyranoside (10).
To a solution of phenyl 2,4,6-tri-O-benzyl-1-thio-β-D-allopyranoside22 (270 mg, 0.5 mmol) in 2.5 mL methylene chloride and pyridine (407 μL, 5 mmol) cooled at 0 °C was added trifluoromethanesulfonic anhydride (168 μL, 1 mmol) dropwise. The resulting mixture was stirred at 0 °C for 2 h and then quenched with ice water. Organic layer was separated, and aqueous layer was extracted with CH2Cl2. The combined organic extracts were washed sequentially with saturated CuSO4 solution, water and brine, dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography with a short pad of silica gel (pure CH2Cl2 as eluent) to afford 239 mg (0.35 mmol, 70%) of phenyl 2,4,6-tri-O-benzyl-3-O-trifluoromethanesulfonyl-1-thio-β-D-allopyranoside (10). 1H NMR (600 MHz,CDCl3) δ 7.61 – 7.55 (m, 2H, HAr), 7.43 – 7.30 (m, 15H, HAr), 7.27 – 7.23 (m, 3H, HAr), 5.39 (d, J = 2.5 Hz, 1H, H-3), 4.96 (d, J = 9.7 Hz, 1H, H-1), 4.73 (d, J = 11.4 Hz, 1H, -OCH2Ar), 4.70 (d, J = 11.4 Hz, 1H, -OCH2Ar), 4.61 (d, J = 8.9 Hz, 1H, -OCH2Ar), 4.59 (d, J = 9.4 Hz, 1H, -OCH2Ar), 4.51 (d, J = 11.9 Hz, 1H, -OCH2Ar), 4.44 (d, J = 11.2 Hz, 1H, -OCH2Ar), 3.85 (ddd, J = 9.8, 3.7, 1.8 Hz, 1H, H-5), 3.77 (dd, J = 10.9, 1.8 Hz, 1H, H-6a), 3.73 (d, J = 3.7 Hz, 1H, H-6b), 3.71 – 3.67 (m, 1H, H-4), 3.42 (dd, J = 9.7, 2.6 Hz, 1H, H-2). 13C{1H} NMR (150 MHz, CDCl3) δ 138.1, 136.6, 136.5, 132.8, 132.6, 129.0, 128.7, 128.6, 128.6, 128.5, 128.4, 128.38, 128.3, 127.9, 127.8, 127.8, 119.6, 117.5, 84.3, 83.7, 75.0, 73.6, 73.6, 72.9, 72.4, 71.7, 68.4.
General Procedure for Cs2CO3-mediated β-mannosylation.
To a solution of lactol 7a (57 mg, 0.1 mmol) and sugar-derived triflate (8-12) (0.3 mmol for secondary triflates, 0.2 mmol for primary triflates) in anhydrous 1,2-dichloroethane (1.0 mL) was added Cs2CO3 (114 mg, 0.35 mmol for secondary triflates; 81 mg, 0.25 mmol for primary triflates). The mixture was stirred at 40 °C for 24 h, filtered, concentrated, and purified by preparative TLC to give corresponding disaccharide.
Butyl 2,3,6-tri-O-benzyl-4-O-(3,4,6,7-tetra-O-benzyl-β-d-glycero-d-manno-heptopyranosyl)-β-D-glucopyranoside (13).
Compound 13 was prepared from lactol 7a and triflate 8 as colourless syrup (68.9 mg, 65%) according to the general procedure. Eluent system for preparative TLC: Hexanes/EtOAc = 2/1 with 1% methanol. The yield of 13 dropped to 53% when 0.25 mmol of triflate 8 and 0.30 mmol of Cs2CO3 were used. JC1′,H1′ = 159.3 Hz. = −1.8 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.32 – 7.12 (m, 35H, HAr), 4.94 – 4.87 (m, 3H, -OCHHAr), 4.81 (d, J = 10.8 Hz, 1H, -OCHHAr), 4.71 – 4.47 (m, 8H, -OCHHAr, H-1′), 4.40 – 4.31 (m, 4H, -OCHHAr, H-1), 3.98 (t, J = 9.4 Hz, 1H, H-4), 3.96 – 3.91 (m, 3H, H-6′, -OCHHCH2CH2CH3, H-2′), 3.88 (t, J = 9.5 Hz, 1H, H-4′), 3.77 (dd, J = 11.1, 2.4 Hz, 1H, H-6a), 3.69 (dd, J = 11.1, 4.5 Hz, 1H, H-6b), 3.66 (t, J = 9.1 Hz, 1H, H-3), 3.61 (dd, J = 10.5, 7.0 Hz, 1H, H-7′a), 3.56 (dd, J = 10.4, 4.3 Hz, 1H, H-7′b), 3.52 (dt, J = 9.6, 6.8 Hz, 1H, -OCHHCH2CH2CH3), 3.49 – 3.43 (m, 2H, H-5, H-2), 3.35 (dd, J = 9.8, 1.3 Hz, 1H, H-5′), 3.32 (dd, J = 9.1, 3.0 Hz, 1H, H-3′), 2.83 (d, J = 2.8 Hz, 1H, C2′-OH), 1.70 – 1.59 (m, 2H, -OCH2CH2CH2CH3), 1.49 – 1.37 (m, 2H, -OCH2CH2CH2CH3), 0.94 (t, J = 7.4 Hz, 3H, -OCH2CH2CH2CH3). 13C{1H} NMR (150 MHz, CDCl3) δ 139.0, 138.7, 138.6, 138.5, 138.4, 138.2, 138.0, 128.6, 128.5, 128.5, 128.4, 128.4, 128.4, 128.3, 128.1, 128.0, 127.9, 127.8, 127.8, 127.7, 127.7, 127.7, 127.6, 127.6, 127.4, 103.8, 99.4, 82.7, 82.3, 81.9, 78.8, 76.5, 75.2, 75.2, 74.9, 74.9, 74.6, 73.8, 73.6, 73.2, 72.5, 71.17, 71.16, 69.9, 69.4, 67.7, 31.49, 19.5, 14.0. HRMS (ESI) m/z: [M+Na]+ Calcd for C66H74NaO12 1081.5078, found 1081.5088.
Butyl 2,3,6-tri-O-benzyl-4-O-(3,4,6,7-tetra-O-benzyl-β-l-glycero-d-manno-heptopyranosyl)-β-d-glucopyranoside (14).
To a solution of lactol 7b (57 mg, 0.1 mmol) and triflate 8 (141 mg, 0.25 mmol) in anhydrous 1,2-dichloroethane (1.0 mL) was added Cs2CO3 (98 mg, 0.30 mmol). The mixture was stirred at 40 °C for 24 h, filtered and purified by preparative TLC to give disaccharide 14 (53.0 mg, 50%) as colourless syrup. Eluent system for preparative TLC: Hexanes/EtOAc = 2/1 with 1% methanol. JC1′,H1′ = 157.9 Hz. = +1.9 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.36 – 7.12 (m, 35H, HAr), 5.03 (d, J = 11.5 Hz, 1H, -OCHHAr), 4.95 – 4.86 (m, 2H, -OCHHAr), 4.79 – 4.68 (m, 3H, -OCHHAr), 4.65 – 4.57 (m, 2H, -OCHHAr), 4.55 – 4.44 (m, 4H, -OCHHAr, H-1′), 4.43 – 4.35 (m, 4H, -OCHHAr, H-1), 4.10 (t, J = 9.4 Hz, 1H, H-4′), 4.03 (td, J = 6.0, 2.1 Hz, 1H, H-6′), 3.99 – 3.94 (m, 2H, H-2′, -OCHHCH2CH2CH3), 3.90 (dd, J = 9.8, 9.0 Hz, 1H, H-3), 3.79 (dd, J = 9.9, 6.4 Hz, 1H, H-7′a), 3.73 – 3.67 (m, 3H, H-6, H-7′b), 3.63 (t, J = 9.0 Hz, 1H, H-4), 3.56 (dt, J = 9.5, 6.8 Hz, 1H, -OCHHCH2CH2CH3), 3.45 – 3.39 (m, 2H, H-5, H-2), 3.38 (dd, J = 9.2, 3.0 Hz, 1H, H-3′), 3.32 (dd, J = 9.7, 2.1 Hz, 1H, H-5′), 2.44 (bs, 1H, C2′-OH), 1.75 – 1.58 (m, 2H, -OCH2CH2CH2CH3), 1.54 – 1.38 (m, 2H, -OCH2CH2CH2CH3), 0.96 (t, J = 7.4 Hz, 3H, -OCH2CH2CH2CH3). 13C{1H} NMR (150 MHz, CDCl3) δ 139.1, 138.8, 138.8, 138.6, 138.4, 138.0, 138., 128.6, 128.6, 128.5, 128.4, 128.4, 128.3, 128.3, 128.2, 128.0, 127.9, 127.9, 127.9, 127.7, 127.7, 127.7, 127.6, 127.5, 127.4, 103.8, 100.0, 82.8, 82.4, 82.2, 76.7, 75.5, 75.4, 75.4, 75.1, 74.8, 74.4, 73.6, 73.6, 73.5, 72.6, 71.2, 70.5, 69.9, 68.9, 68.0, 32.0, 19.5, 14.0. HRMS (ESI) m/z: [M+H]+ Calcd for C66H75O12 1059.5259, found 1059.5286.
Phenyl 3,6-di-O-benzyl-4-O-(3,4,6,7-tetra-O-benzyl-β-d-glycero-d-manno-heptopyranosyl)-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside (15).
Compound 15 was prepared from lactol 7a and triflate 9 as colourless syrup (61.3 mg, 54%) according to the general procedure. Eluent system for preparative TLC: Hexanes/EtOAc = 3/1 with 1% methanol. JC1′,H1′ = 158.0 Hz. = +4.9 (c 1.0, CHCl3). 1H NMR (600 MHz, CHCl3) δ 7.88 – 7.53 (m, 4H, HAr), 7.41 – 7.13 (m, 30H, HAr), 6.96 – 6.88 (m, 2H, HAr), 6.84 – 6.70 (m, 3H, HAr), 5.49 (d, J = 10.5 Hz, 1H, H-1), 4.86 – 4.79 (m, 2H, -OCHHAr), 4.68 – 4.44 (m, 9H, H-1′, -OCHHAr), 4.40 – 4.32 (m, 3H, H-3, -OCHHAr), 4.24 (t, J = 10.4 Hz, 1H, H-2), 4.08 (t, J = 9.3 Hz, 1H, H-4), 4.03 (d, J = 3.1 Hz, 1H, H-2′), 3.99 (td, J = 5.8, 1.2 Hz, 1H, H-6′), 3.91 (t, J = 9.4 Hz, 1H, H-4′), 3.83 (dd, J = 11.3, 3.5 Hz, 1H, H-6a), 3.79 (dd, J = 11.3, 2.1 Hz, 1H, H-6b), 3.70 – 3.67 (m, 1H, H-5), 3.62 (d, J = 5.8 Hz, 2H, H-7′), 3.43 (dd, J = 9.9, 1.2 Hz, 1H, H-5′), 3.40 (dd, J = 9.1, 3.1 Hz, 1H, H-3′), 2.50 (bs, 1H, C2′-OH). 13C{1H} NMR (150 MHz, CDCl3) δ 168.0, 167.4, 138.9, 138.5, 138.4, 138.3, 138.1, 137.9, 133.9, 133.8, 133.0, 131.9, 131.8, 131.7, 128.9, 128.6, 128.4, 128.4, 128.3, 128.1, 128.0, 128.0, 127.9, 127.7, 127.7, 127.6, 127.5, 127.3, 127.2, 123.5, 123.4, 100.7, 83.4, 82.2, 79.2, 79.0, 78.7, 77.9, 76.4, 75.0, 74.9, 74.0, 73.6, 73.2, 72.5, 71.4, 70.9, 68.9, 67.8, 54.94. HRMS (ESI) m/z: [M+Na]+ Calcd for C69H67NNaO12S 1156.4282, found 1156.4250.
Phenyl 2,4,6-tri-O-benzyl-3-O-(3,4,6,7-tetra-O-benzyl-β-d-glycero-d-manno-heptopyranosyl)-1-thio-β-d-glucopyranoside (16).
Compound 16 was prepared from lactol 7a and triflate 10 as colourless syrup (39.2 mg, 37%) according to the general procedure. Eluent system for preparative TLC: Hexanes/EtOAc = 3/1 with 1% methanol. JC1′,H1′ = 162.0 Hz. = +1.1 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.62 – 7.55 (m, 2H, HAr), 7.39 – 7.06 (m, 38H, HAr), 5.13 – 5.04 (m, 2H, -OCHHAr), 4.81 – 4.75 (m, 2H, -OCHHAr, H-1′), 4.65 (d, J = 9.7 Hz, 1H, H-1), 4.62 – 4.57 (m, 2H, -OCHHAr), 4.56 – 4.47 (m, 6H, -OCHHAr), 4.44 (d, J = 11.6 Hz, 1H, -OCHHAr), 4.22 (d, J = 12.2 Hz, 1H, -OCHHAr), 4.19 (d, J = 12.2 Hz, 1H, -OCHHAr), 4.02 (t, J = 8.9 Hz, 1H, H-4), 3.94 – 3.90 (m, 2H, H-2′, H-6′), 3.84 (t, J = 9.5 Hz, 1H, H-4′), 3.77 (dd, J = 10.8, 1.9 Hz, 1H, H-6a), 3.66 (dd, J = 10.7, 4.9 Hz, 1H, H-6b), 3.61 (t, J = 9.4 Hz, 1H, H-3), 3.57 – 3.50 (m, 4H, H-2, H-7′, H-5), 3.37 (d, J = 9.9 Hz, 1H, H-5′), 3.27 (dd, J = 9.1, 3.0 Hz, 1H, H-3′), 2.28 (bs, 1H, C2′-OH). 13C{1H} NMR (150 MHz, CDCl3) δ 139.0, 138.9, 138.6, 138.5, 138.3, 138.0, 137.9, 133.9, 131.8, 129.1, 128.8, 128.7, 128.5, 128.4, 128.4, 128.4, 128.3, 128.2, 128.2, 128.0, 127.9, 127.9, 127.8, 127.7, 127.6, 127.6, 127.6, 127.5, 127.5, 127.4, 127.3, 100.8, 87.4, 83.9, 82.6, 81.4, 79.1, 78.6, 76.3, 76.1, 75.3, 75.1, 74.6, 74.3, 73.4, 73.0, 72.3, 71.5, 70.8, 69.4, 68.2. HRMS (ESI) m/z: [M+H]+ Calcd for C68H71O11S 1095.4717, found 1095.4689.
Methyl 2,3,4-tri-O-benzyl-6-O-(3,4,6,7-tetra-O-benzyl-β-d-glycero-d-manno-heptopyranosyl)-α-d-glucopyranoside (17).
Compound 17 was prepared from lactol 7a and triflate 11 as colourless syrup (79.3 mg, 78%). Eluent system for preparative TLC: Hexanes/EtOAc = 2/1 with 1% methanol. JC1′,H1′ = 158.1 Hz. = +4.9 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.44 – 7.13 (m, 35H, HAr), 5.02 (d, J = 10.9 Hz, 1H, -OCHHAr), 4.91 – 4.77 (m, 4H, -OCHHAr), 4.76 – 4.52 (m, 8H, -OCHHAr, H-1), 4.47 (d, J = 12.1 Hz, 1H, -OCHHAr), 4.43 (d, J = 12.1 Hz, 1H, -OCHHAr), 4.10 (dd, J = 10.9, 2.0 Hz, 1H, H-6a), 4.06 (d, J = 1.0 Hz, 1H, H-1′), 4.04 – 3.97 (m, 2H, H-6′, H-3), 3.96 – 3.88 (m, 2H, H-4′, H-2′), 3.78 (ddd, J = 10.1, 5.2, 2.0 Hz, 1H, H-5), 3.74 – 3.67 (m, 2H, H-7′), 3.58 – 3.51 (m, 2H, H-2, H-6b), 3.49 – 3.41 (m, 3H, H-4, H-3′, H-5′), 3.36 (s, 3H, -OCH3), 2.30 (d, J = 2.5 Hz, 1H, C2′-OH). 13C{1H} NMR (150 MHz, CDCl3) δ 138.9, 138.8, 138.5, 138.4, 138.2, 137.9, 128.6, 128.6, 128.6, 128.5, 128.4, 128.3, 128.3, 128.1, 128.1, 128.1, 128.0, 127.9, 127.8, 127.8, 127.7, 127.7, 127.6, 127.5, 100.2, 98.0, 82.3, 81.8, 79.9, 78.5, 77.5, 75.9, 75.9, 75.0, 74.9, 74.2, 73.5, 73.3, 72.5, 71.5, 70.8, 69.9, 68.3, 68.2, 55.3. HRMS (ESI) m/z: [M+Na]+ Calcd for C63H68NaO12 1039.4608, found 1039.4617.
Methyl 2,3,4-tri-O-benzoyl-6-O-(3,4,6,7-tetra-O-benzyl-β-d-glycero-d-manno-heptopyranosyl) -α-d-glucopyranoside (18).
Compound 18 was prepared from donor 7a and triflate 12 as colourless syrup (76.3 mg, 72%) according to the general procedure. Eluent system for preparative TLC: Hexanes/EtOAc = 2/1 with 1% methanol. JC1′,H1′ = 158.2 Hz. = +6.1 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 8.11 – 7.78 (m, 6H, HAr), 7.58 – 7.15 (m, 29H, HAr), 6.17 (t, J = 9.9 Hz, 1H, H-3), 5.61 (t, J = 9.9 Hz, 1H, H-4), 5.27 (dd, J = 10.2, 3.7 Hz, 1H, H-2), 5.22 (d, J = 3.7 Hz, 1H, H-1), 4.88 (d, J = 10.8 Hz, 1H, -OCHHAr), 4.78 (d, J = 11.7 Hz, 1H, -OCHHAr), 4.72 – 4.57 (m, 4H, -OCHHAr), 4.45 (d, J = 12.1 Hz, 1H, -OCHHAr), 4.42 (d, J = 12.1 Hz, 1H, -OCHHAr), 4.37 (d, J = 1.1 Hz, 1H, H-1′), 4.25 (ddd, J = 10.3, 5.5, 2.1 Hz, 1H, H-5), 4.20 (dd, J = 3.1, 1.1 Hz, 1H, H-2′), 4.12 (dd, J = 11.5, 2.1 Hz, 1H, H-6a), 4.02 – 3.96 (m, 2H, H-4′, H-6′), 3.71 – 3.63 (m, 3H, H-6b, H-7′), 3.57 (dd, J = 8.8, 3.1 Hz, 1H, H-3′), 3.50 (dd, J = 9.4, 1.7 Hz, 1H, H-5′), 3.45 (s, 3H, -OCH3), 2.45 (bs, 1H, C2′-OH). 13C{1H} NMR (150 MHz, CDCl3) δ 165.9, 165.8, 165.5, 138.9, 138.5, 138.4, 137.9, 133.6, 133.5, 133.2, 130.1, 130.0, 129.8, 129.4, 129.2, 129.0, 128.6, 128.6, 128.6, 128.4, 128.4, 128.3, 128.1, 128.1, 128.0, 127.8, 127.7, 127.6, 127.4, 100.8, 97.0, 81.7, 78.5, 75.9, 74.8, 74.0, 73.3, 72.5, 72.2, 71.5, 70.7, 70.6, 69.3, 68.9, 68.1, 67.7, 55.7. HRMS (ESI) m/z: [M+Na]+ Calcd for C63H62NaO15 1081.3986, found 1081.3975.
4-Methoxyphenyl 2,3,6-trideoxy-4-O-(4-nitrophenyl)sulfonyl-α-l-threo-hex-2-enopyranoside (20).
To a solution of 4-methoxyphenyl 2,3,6-trideoxy-α-L-threo-hex-2-enopyranoside23 (236 mg, 1.00 mmol) in anhydrous CH2Cl2 (5 mL) was added 4-dimethylaminopyridine (12.2 mg, 0.10 mmol), followed by the addition of pyridine (121 μL, 1.50 mmol). The mixture was cooled to −15 °C and (4-nitrophenyl)sulfonyl chloride (288 mg, 1.30 mmol) was added. After the reaction mixture was stirred at −15 °C for 20 h, it was diluted by CH2Cl2 (20 mL), washed by CuSO4 solution and brine, dried over anhydrous Na2SO4, filtered, and concentrated (Note: CH2Cl2, CuSO4 solution and brine were all cooled in fridge to about 4 °C before use. The workup should be completed quickly). The residue was purified by flash column chromatography (CH2Cl2) to give compound 20 (282 mg, 0.67 mmol, 67%) as dark green syrup (Note: this nosylate 20 is found to be unstable. Severe decomposition was observed during concentration of fractions. The concentration should be conducted at less than 30 °C and completed quickly). 1H NMR (600 MHz, CDCl3) δ 8.41 (d, J = 8.9 Hz, 2H, HAr), 8.14 (d, J = 9.0 Hz, 2H, HAr), 6.99 (d, J = 9.1 Hz, 2H, HAr), 6.82 (d, J = 9.1 Hz, 2H, HAr), 6.20 (dd, J = 10.0, 3.2 Hz, 1H, H-2), 6.07 (dd, J = 10.0, 5.5 Hz, 1H, H-3), 5.56 (d, J = 3.2 Hz, 1H, H-1), 4.80 (dd, J = 5.5, 2.2 Hz, 1H, H-4), 4.41 (qd, J = 6.6, 2.2 Hz, 1H, H-5), 3.77 (s, 3H, -OCH3), 1.23 (d, J = 6.6 Hz, 3H, H-6). 13C{1H} NMR (150 MHz, CDCl3) δ 155.2, 151.2, 150.9, 142.9, 132.0, 129.2, 125.0, 124.6, 118.2, 114.7, 93.6, 73.8, 65.3, 55.8, 16.5.
4-Methoxyphenyl 2,3-di-O-benzoyl-4-O-(2-O-benzoyl-3,4,6,7-tetra-O-benzyl-β-d-glycero-d-manno-heptopyranosyl)-α-l-rhamnopyranoside (21).
To a solution of lactol 7a (57 mg, 0.1 mmol) and nosylate 20 (126 mg, 0.30 mmol) in anhydrous 1,2-dichloroethane (1.0 mL) was added Cs2CO3 (114 mg, 0.35 mmol). The mixture was stirred at 40 °C for 24 h and filtered. The filtrate was concentrated and loaded on silica gel flash column chromatography. After unreacted nosylate 20 was quickly removed by DCM, the crude product was collected and concentrated. The residue was dissolved in acetone/water (2 mL/0.2 mL) and cooled to 0 °C, osmium tetroxide (2.5 mg, 0.0098 mmol) was added followed by addition of an aqueous solution of NMO (50 wt%, 60 μL). The mixture was stirred at 0 °C overnight and quenched with NaHSO3. After acetone was removed under reduced pressure, the residue was diluted with CH2Cl2 and washed with water and brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated. The residue was dissolved in anhydrous pyridine (1.0 mL), and benzoyl chloride (35 μL, 0.3 mmol) was added. The mixture was stirred at room temperature for 1 h before being quenched with methanol (0.1 mL). The mixture was diluted by CH2Cl2, washed sequentially with 1 M HCl solution, saturated NaHCO3 solution and brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (Hexanes/EtOAc = 4/1) to give compound 21 (52.2 mg, 0.046 mmol, 46% over three steps) as light yellow syrup. JC1′,H1′ = 155.8 Hz. = −23.6 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 8.08 – 7.94 (m, 6H, HAr), 7.58 – 7.48 (m, 3H, HAr), 7.44 – 7.24 (m, 19H, HAr), 7.20 – 7.13 (m, 3H, HAr), 7.13 – 7.08 (m, 4H, HAr), 7.03 (d, J = 9.0 Hz, 2H, HAr), 6.80 (d, J = 8.9 Hz, 2H, HAr), 5.83 (dd, J = 9.7, 3.5 Hz, 1H, H-3), 5.79 (dd, J = 3.5, 1.8 Hz, 1H, H-2), 5.56 (d, J = 2.9 Hz, 1H, H-2′), 5.49 (d, J = 1.8 Hz, 1H, H-1), 5.00 (d, J = 12.2 Hz, 1H, -OCHHAr), 4.83 – 4.73 (m, 3H, -OCHHAr, H-1′), 4.58 – 4.45 (m, 4H, -OCHHAr), 4.19 – 4.08 (m, 3H, H-4, H-6′, -OCHHAr), 4.05 – 3.96 (m, 2H, H-4′, H-5), 3.76 (s, 5H, -OCH3, H-7), 3.60 (d, J = 9.8 Hz, 1H, H-5′), 3.43 (dd, J = 9.3, 2.9 Hz, 1H, H-3′), 1.35 (d, J = 6.2 Hz, 3H, H-6). 13C{1H} NMR (150 MHz, CDCl3) δ 165.6, 165.6, 165.1, 155.2, 150.2, 139.1, 138.3, 138.2, 137.5, 133.6, 133.5, 133.1, 130.1, 130.0, 130.0, 129.8, 129.6, 129.6, 128.8, 128.7, 128.5, 128.5, 128.4, 128.4, 128.3, 128.1, 127.8, 127.8, 127.8, 127.5, 127.3, 117.9, 114.7, 99.4, 96.7, 81.1, 79.7, 76.8, 76.4, 75.1, 73.9, 73.5, 73.2, 72.6, 71.3, 71.2, 71.0, 68.2, 67.9, 55.8, 18.1. HRMS (ESI) m/z: [M+Na]+ Calcd for C69H66NaO15 1157.4299, found 1157.4291.
Synthesis of tetrasaccharide 32.
Phenyl 2-O-(2-naphthalenyl)methyl-6,7-di-O-benzyl-1-thio-α-d-glycero-d-manno-hepto-pyranoside (22).
To a solution of compound 4a (2.23 g, 4.0 mmol) in anhydrous DMF (8 mL) cooled at 0 °C was added NaH (60% on mineral oil, 640 mg). The resulting mixture was stirred at 0 °C for 30 min before BnBr (1.4 mL) was added dropwise. The reaction mixture was slowly warmed up and stirred at room temperature for 4 h. The mixture was cooled to 0 °C and methanol (1 mL) was added to quench this reaction. The resulting mixture was diluted with CH2Cl2 (100 mL), washed by water and brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was dissolved in CH2Cl2 (10 mL), and TFA (5 mL) and water (0.2 mL) were added. After the mixture was stirred at room temperature for 4 h, it was neutralized by saturated NaHCO3 solution and extracted with CH2Cl2. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated. Purification by silica gel flash column chromatography (Hexanes/EtOAc = 5/1 → 3/2) afforded compound 22 (1.72 g, 2.76 mmol, 69% over two steps from 4a) as yellow syrup. = +31.1 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.89 – 7.75 (m, 4H, HAr), 7.52 – 7.45 (m, 3H, HAr), 7.37 – 7.27 (m, 12H, HAr), 7.24 – 7.16 (m, 3H, HAr), 5.61 (d, J = 1.4 Hz, 1H, H-1), 4.89 (d, J = 11.8 Hz, 1H, -OCHHAr), 4.83 – 4.73 (m, 2H, -OCHHAr), 4.66 (d, J = 11.3 Hz, 1H, -OCHHAr), 4.50 (d, J = 12.0 Hz, 1H, -OCHHAr), 4.45 (d, J = 12.0 Hz, 1H, -OCHHAr), 4.17 (dd, J = 9.4, 5.6 Hz, 1H,), 4.07 (dd, J = 3.5, 1.4 Hz, 1H, H-2), 4.01 (t, J = 9.3 Hz, 1H, H-4), 3.93 (td, J = 5.3, 3.3 Hz, 1H, H-6), 3.89 (dd, J = 9.3, 3.5 Hz, 1H, H-3), 3.74 – 3.65 (m, 2H, H-7a, C4-OH), 3.60 (dd, J = 10.6, 5.2 Hz, 1H, H-7b), 2.51 (s, 1H, C3-OH). 13C{1H} NMR (150 MHz, CDCl3) δ 138.1, 137.9, 135.1, 134.1, 133.3, 133.2, 131.6, 129.2, 128.6, 128.6, 128.5, 128.2, 128.1, 128.0, 127.9, 127.8, 127.8, 127.5, 126.9, 126.4, 126.3, 125.9, 85.5, 80.5, 78.9, 73.6, 73.0, 72.9, 72.1, 71.5, 70.6, 70.4. HRMS (ESI) m/z: [M+Na]+ Calcd for C38H38NaO6S 645.2281, found 645.2316.
Phenyl 4,6,7-tri-O-benzyl-1-thio-α-d-glycero-d-manno-heptopyranoside (24).
To a solution of compound 22 (1.56 g, 2.50 mmol) in CH2Cl2/water (10 mL/1 mL) was added 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (851 mg, 3.75 mmol). The mixture was stirred at room temperature for 4 h and quenched with 40% Na2S2O3 solution. After the CH2Cl2 layer was separated, resulting aqueous solution was extracted with CH2Cl2. Combined organic layer was washed by saturated NaHCO3 solution and brine, dried over anhydrous Na2SO4, filtered and concentrated to afford crude compound 23. Next, a solution of 23 in DMF (10 mL) was cooled to 0 °C and NaH (60% on mineral oil, 150 mg) was added. The resulting mixture was stirred at 0 °C for 30 min and BnBr (360 μL) was added dropwise. The reaction mixture was warmed to room temperature slowly and stirred for 5 h. The mixture was cooled to 0 °C and methanol (1 mL) was added to quench the reaction. The resulting mixture was diluted with CH2Cl2 (100 mL), washed by water and brine, dried over anhydrous Na2SO4, filtered, and concentrated. The residue was dissolved in CH2Cl2/methanol (10 mL/10 mL), and p-toluenesulfonic acid monohydrate (100 mg) was added. The mixture was stirred at room temperature overnight, quenched by Et3N, concentrated and purified by flash column chromatography (Hexanes/EtOAc = 4/1 → 1/1) to give compound 24 (974 mg, 1.70 mmol, 68% over three steps from 22) as light yellow syrup. = +37.4 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.50 (dd, J = 7.8, 1.7 Hz, 2H, HAr), 7.35 – 7.19 (m, 18H, HAr), 5.44 (d, J = 2.1 Hz, 1H, H-1), 4.76 – 4.65 (m, 4H, -OCHHAr), 4.52 (d, J = 11.9 Hz, 1H, -OCHHAr), 4.48 (d, J = 11.9 Hz, 1H, -OCHHAr), 4.44 (dd, J = 9.3, 1.7 Hz, 1H, H-5), 4.11 (dd, J = 3.3, 2.1 Hz, 1H, H-2), 4.02 (ddd, J = 6.9, 5.3, 1.7 Hz, 1H, H-6), 3.93 (dd, J = 8.6, 3.3 Hz, 1H, H-3), 3.85 (t, J = 9.0 Hz, 1H, H-4), 3.79 (dd, J = 10.2, 5.3 Hz, 1H, H-7a), 3.71 (dd, J = 10.2, 6.6 Hz, 1H, H-7b), 2.10 (bs, 2H, -OH). 13C{1H} NMR (150 MHz, CDCl3) δ 138.6, 138.3, 138.3, 133.9, 132.1, 129.1, 128.7, 128.5, 128.4, 128.1, 128.1, 128.0, 127.9, 127.8, 127.7, 127.6, 87.8, 78.0, 76.4, 74.4, 73.5, 72.6, 72.5, 72.2, 70.7. HRMS (ESI) m/z: [M+Na]+ Calcd for C34H36NaO6S 595.2130, found 595.2137.
Phenyl 3-O-(4-methoxybenzyl)-4,6,7-tri-O-benzyl-1-thio-α-d-glycero-d-manno-hepto-pyranoside (25).
To a solution of compound 24 (321 mg, 0.56 mmol) in anhydrous toluene (11 mL) was added Bu2SnO (153 mg, 0.61 mmol). The mixture was stirred at 110 °C for 5 h and during this period, water was trapped and removed with Dean–Stark apparatus. The mixture was cooled to room temperature, and PMBCl (91 μL) and TBAI (41 mg) were added. After the resulting mixture was stirred at 80 °C for 6 h, toluene was removed under reduced pressure. The residue was purified by flash column chromatography (Hexanes/EtOAc = 5/1) to give compound 25 (322 mg, 0.465 mmol, 83%) as light yellow syrup. = +34.3 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.52 – 7.43 (m, 2H, HAr), 7.36 – 7.18 (m, 20H, HAr), 6.93 – 6.83 (m, 2H, HAr), 5.52 (d, J = 1.8 Hz, 1H, H-1), 4.82 (d, J = 10.8 Hz, 1H, -OCHHAr), 4.73 (d, J = 11.8 Hz, 1H, -OCHHAr), 4.66 (d, J = 11.8 Hz, 1H, -OCHHAr), 4.63 – 4.59 (m, 3H, -OCHHAr), 4.51 – 4.43 (m, 3H, -OCHHAr, H-5), 4.20 (td, J = 2.9, 1.8 Hz, 1H, H-2), 4.00 (ddd, J = 6.8, 4.8, 1.5 Hz, 1H, H-6), 3.96 (dd, J = 9.8, 8.8 Hz, 1H, H-4), 3.87 (dd, J = 8.8, 3.2 Hz, 1H, H-3), 3.81 (s, 3H, -OCH3), 3.75 (dd, J = 10.4, 4.8 Hz, 1H, H-7a), 3.69 (dd, J = 10.3, 6.8 Hz, 1H, H-7b), 2.58 (d, J = 2.7 Hz, 1H, C2-OH). 13C{1H} NMR (150 MHz, CDCl3) δ 159.7, 138.8, 138.5, 138.4, 134.0, 132.0, 129.9, 129.7, 129.1, 128.5, 128.4, 128.3, 128.0, 127.9, 127.7, 127.7, 127.6, 127.6, 127.5, 114.2, 87.5, 80.5, 78.3, 74.9, 74.5, 73.3, 72.7, 72.5, 71.9, 70.9, 69.8, 55.4. HRMS (ESI) m/z: [M+Na]+ Calcd for C42H44NaO7S 715.2705, found 715.2704.
3-O-(4-Methoxybenzyl)-4,6,7-tri-O-benzyl-α/β-d-glycero-d-manno-heptopyranose (26).
To a solution of compound 25 (300 mg, 0.433 mmol) in MeCN/water (4 mL/0.4 mL) cooled at 0 °C was slowly added a solution of Br2 in CH2Cl2 (5 M, 260 μL). The mixture was stirred at 0 °C for 3 h before being quenched with 40% Na2S2O3 solution. After MeCN was removed under reduced pressure, the residue was extracted by CH2Cl2. Combined organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (Hexanes/EtOAc = 1/1 with 2% methanol) to give compound 26 (203 mg, 0.338 mmol, 78%) as colorless syrup. α/β = 1/0.16. = +2.4 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.39 – 7.12 (m, 34H, α HAr × 17, β HAr × 17), 6.89 – 6.77 (m, 2H, α HAr × 2, β HAr × 2), 5.23 (s, 1H, α H-1), 4.83 – 4.66 (m, 6H, α -OCHHAr × 3, β -OCHHAr × 3), 4.64 (d, J = 9.8 Hz, 1H, β H-1), 4.62 – 4.43 (m, 10H, α -OCHHAr × 5, β -OCHHAr × 5), 4.13 (d, J = 10.0 Hz, 1H, α H-5), 4.04 – 3.98 (m, 2H, α H-2, α H-6), 3.95 – 3.67 (m, 15H, α H-3, β -OCH3, β H-2, β H-6, β H-4, α H-4, α -OCH3, α H-7a, α H-7b, β H-7a, β H-7b), 3.58 – 3.50 (m, 2H, β H-3, β H-5), 3.28 (bs, 1H, β C1-OH), 3.23 (bs, 1H, α C1-OH), 2.43 (bs, 1H, α C2-OH), 2.37 (bs, 1H, β C2-OH). 13C{1H} NMR (150 MHz, CDCl3) δ 159.6 (β), 159.5, 138.7 (β), 138.5, 138.5, 138.4, 138.4 (β), 138.3 (β), 133.2 (β), 130.0 (β), 129.7, 128.5 (β), 128.5, 128.4, 128.4, 128.1, 128.0 (β), 127.9 (β), 127.9, 127.8 (β), 127.8, 127.7 (β), 127.7, 127.7, 127.7, 127.6, 114.1 (β), 114.0, 94.1 (β C-1), 93.9 (α C-1), 81.8 (β), 80.1 (β), 80.0, 78.2 (β), 77.5, 75.7 (β), 74.8, 74.4, 73.9 (β), 73.3, 72.8 (β), 72.4, 71.9 (β), 71.9, 71.8, 71.7 (β), 71.0 (β), 70.4, 68.6 (β), 68.5, 56.4 (β), 55.4. HRMS (ESI) m/z: [M+Na]+ Calcd for C36H40NaO8 623.2621, found 623.2620.
4-Methoxyphenyl 2,3-di-O-benzyl-4-O-[2,4,6,7-tetra-O-benzyl-3-O-(4-methoxybenzyl)-β-d-glycero-d-manno-heptopyranosyl]-α-l-rhamnopyranoside (27).
To a solution of lactol 26 (60 mg, 0.1 mmol) and nosylate 20 (126 mg, 0.30 mmol) in anhydrous 1,2-dichloroethane (1.0 mL) was added Cs2CO3 (114 mg, 0.35 mmol). The mixture was stirred at 40 °C for 24 h and filtered. The filtrate was concentrated and loaded on silica gel flash column chromatography. After unreacted nosylate 20 was quickly removed by DCM, the crude product was collected and concentrated. The residue was dissolved in acetone/water (2 mL/0.2 mL) and cooled to 0 °C, osmium tetroxide (2.5 mg, 0.0098 mmol) was added followed by addition of an aqueous solution of NMO (50 wt%, 60 μL). The mixture was stirred at 0 °C overnight and quenched with NaHSO3. After acetone was removed under reduced pressure, the residue was diluted with CH2Cl2 and washed with water and brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated. The residue was dissolved in anhydrous DMF (1.0 mL) and cooled to 0 °C. NaH (60% on mineral oil, 15 mg) was added, and the resulting mixture was stirred for 30 min. BnBr (36 μL) was added dropwise, and the reaction mixture was warmed up and stirred at room temperature for 4 h before being quenched by methanol (50 μL). The mixture was diluted by CH2Cl2, washed with brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (Hexanes/EtOAc = 5/1) to give compound 27 (55.0 mg, 0.049 mmol, 49% over three steps) as light yellow syrup. JC1′,H1′ = 153.1 Hz. = −11.3 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.46 – 7.11 (m, 32H, HAr), 7.03 – 6.92 (m, 2H, HAr), 6.87 – 6.74 (m, 4H, HAr), 5.37 (d, J = 1.8 Hz, 1H, H-1), 4.89 – 4.73 (m, 6H, -OCHHAr), 4.68 – 4.62 (m, 2H, H-1′, -OCHHAr), 4.57 – 4.52 (m, 2H, -OCHHAr), 4.49 – 4.42 (m, 2H, -OCHHAr), 4.32 – 4.22 (m, 3H, -OCHHAr), 4.04 – 3.98 (m, 2H, H-6′, H-4′), 3.93 (dd, J = 3.1, 1.9 Hz, 1H, H-2), 3.87 (dd, J = 9.3, 3.1 Hz, 1H, H-3), 3.82 (t, J = 9.3 Hz, 1H, H-4), 3.80 – 3.75 (m, 7H, -OCH3, -OCH3, H-5), 3.71 – 3.66 (m, 3H, H-7′, H-2′), 3.48 (dd, J = 9.8, 1.0 Hz, 1H, H-5′), 3.32 (dd, J = 9.4, 2.9 Hz, 1H, H-3′), 1.34 (d, J = 6.1 Hz, 3H, H-6). 13C{1H} NMR (150 MHz, CDCl3) δ 159.3, 155.1, 150.6, 139.4, 139.3, 138.7, 138.7, 138.5, 138.1, 130.5, 129.4, 128.6, 128.6, 128.4, 128.4, 128.3, 128.2, 128.2, 128.1, 128.0, 128.0, 127.8, 127.8, 127.5, 127.5, 127.4, 127.2, 117.8, 114.8, 113.9, 102.5, 97.3, 83.2, 80.1, 79.8, 78.9, 76.2, 74.8, 74.3, 74.3, 74.2, 73.8, 73.3, 72.9, 72.7, 72.0, 71.5, 71.4, 68.7, 55.8, 55.4, 18.2. HRMS (ESI) m/z: [M+Na]+ Calcd for C70H74NaO13 1145.5027, found 1145.5028.
4-Methoxyphenyl 2,3-di-O-benzyl-4-O-[2,4,6,7-tetra-O-benzyl-β-d-glycero-d-manno-hepto-pyranosyl]-α-l-rhamnopyranoside (28).
To a solution of compound 27 (203 mg, 0.181 mmol) in anhydrous CH2Cl2 (10 mL) cooled at 0 °C was added TFA (0.5 mL). The reaction mixture was stirred at 0 °C for 1.5 h and neutralized by saturated NaHCO3 solution. The organic layer was separated, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by silica gel flash column chromatography (Toluene/EtOAc = 10/1) to give compound 28 (150 mg, 0.150 mmol, 83%) as light yellow syrup. = −13.1 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.45 – 7.17 (m, 30H, HAr), 6.96 (d, J = 8.9 Hz, 2H, HAr), 6.82 (d, J = 9.0 Hz, 2H, HAr), 5.36 (s, 1H, H-1), 4.98 (d, J = 11.5 Hz, 1H, -OCHHAr), 4.89 – 4.74 (m, 5H, -OCHHAr, H-1′), 4.69 – 4.61 (m, 2H, -OCHHAr), 4.55 – 4.41 (m, 5H, -OCHHAr), 4.04 (dd, J = 7.0, 4.9 Hz, 1H, H-6′), 3.98 – 3.92 (m, 2H, H-2, H-3), 3.89 (t, J = 9.7 Hz, 1H, H-4), 3.78 (s, 3H, -OCH3), 3.76 – 3.64 (m, 4H, H-7′, H-4′, H-5), 3.62 (d, J = 3.5 Hz, 1H, H-2′), 3.54 (td, J = 9.7, 3.5 Hz, 1H, H-3′), 3.45 (d, J = 9.7 Hz, 1H, H-5′), 2.35 (d, J = 10.1 Hz, 1H, C3′-OH), 1.35 (d, J = 6.2 Hz, 3H, H-6). 13C{1H} NMR (150 MHz, CDCl3) δ 155.0, 150.6, 139.3, 138.8, 138.6, 138.6, 138.2, 138.0, 128.7, 128.6, 128.5, 128.4, 128.3, 128.2, 128.1, 128.1, 128.0, 127.9, 127.8, 127.8, 127.7, 127.6, 127.6, 127.3, 117.8, 114.7, 102.3, 97.3, 79.8, 78.5, 78.1, 76.6, 75.7, 75.0, 74.9, 74.7, 74.0, 73.3, 73.0, 72.9, 72.1, 71.4, 68.6, 55.8, 18.2. HRMS (ESI) m/z: [M+Na]+ Calcd for C62H66NaO12 1025.4452, found 1025.4414.
2,3-Di-O-benzoyl-4-O-(2-O-benzoyl-3,4,6,7-tetra-O-benzyl-β-d-glycero-d-manno-hepto-pyranosyl)-α/β-l-rhamnopyranose (29).
To compound 21 (297 mg, 0.264 mmol) in a mixture of toluene (4 mL), acetonitrile (8 mL) and water (1 mL) was added ceric ammonium nitrate (290 mg, 0.529 mmol). The reaction mixture was stirred at room temperature for 4 h before being quenched by 40% Na2S2O3 solution. After toluene and MeCN were removed under reduced pressure, the residue was extracted with CH2Cl2. The organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by silica gel flash column chromatography (Hexanes/EtOAc = 1/1 with 1% methanol) to give compound 29 (206 mg, 0.201 mmol, 76%) as light yellow syrup. α/β = 1/0.16. = +1.3 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 8.10 – 7.89 (m, 12H, α HAr × 6, β HAr × 6), 7.59 – 7.06 (m, 58H, α HAr × 29, β HAr × 29), 5.77 (d, J = 3.3 Hz, 1H, β H-2), 5.71 (dd, J = 8.8, 3.4 Hz, 1H, α H-3), 5.65 (m, 1H, α H-2), 5.56 (d, J = 2.9 Hz, 1H, α H-2′), 5.54 (d, J = 3.0 Hz, 1H, β H-2′), 5.37 (dd, J = 9.8, 3.3 Hz, 1H, β H-3), 5.31 (dd, J = 3.8, 1.8 Hz, 1H, α H-1), 5.09 – 4.96 (m, 3H, β H-1, α -OCHHAr, β -OCHHAr), 4.85 – 4.68 (m, 6H, α -OCHHAr × 2, β -OCHHAr × 2, α H-1′, β H-1′), 4.59 – 4.45 (m, 8H, α -OCHHAr × 4, β -OCHHAr × 4), 4.17 – 3.99 (m, 9H, α -OCHHAr, β -OCHHAr, α H-6′, β H-6′, α H-5, α H-4′, β H-4′, α H-4, β H-4), 3.80 – 3.71 (m, 4H, α H-7′, β H-7′), 3.61 – 3.54 (m, 3H, α H-5′, β H-5′, β H-5), 3.49 (d, J = 9.6 Hz, 1H, β C1-OH), 3.46 – 3.38 (m, 2H, α H-3′, β H-3′), 2.95 (d, J = 3.8 Hz, 1H, α C1-OH), 1.42 (d, J = 6.1 Hz, 3H, β H-6), 1.36 (d, J = 5.1 Hz, 3H, α H-6). 13C{1H} NMR (150 MHz, CDCl3) δ 166.1 (β), 165.6, 165.5 (β), 165.5, 165.0 (β), 165.0, 139.0, 138.9 (β), 138.2, 138.1 (β), 138.1, 138.0 (β), 137.3, 137.3 (β), 133.7 (β), 133.5 (β), 133.4, 133.3, 133.0 (β), 132.9, 130.1, 130.0, 129.9, 129.9, 129.8, 129.7, 129.6, 129.5, 129.4 (β), 129.2 (β), 128.7, 128.7, 128.6, 128.4, 128.4, 128.4, 128.3, 128.3, 128.2, 128.1, 128.0, 127.7, 127.6, 127.6, 127.4, 127.4, 127.2, 99.3 (α C-1′), 99.2 (β C-1′), 92.8 (β C-1), 92.3 (α C-1), 81.0, 80.9 (β), 79.6, 79.6 (β), 76.9, 76.3, 76.2 (β), 75.0, 74.4 (β), 73.8, 73.7 (β), 73.4, 73.1, 72.3, 71.7 (β), 71.3 (β), 71.2, 71.2, 71.1 (β), 71.1, 68.1, 68.1 (β), 67.2, 18.0, 17.9 (β). HRMS (ESI) m/z: [M+Na]+ Calcd for C62H60NaO14 1051.3881, found 1051.3885.
2,3-Di-O-benzoyl-4-O-(2-O-benzoyl-3,4,6,7-tetra-O-benzyl-β-d-glycero-d-manno-hepto-pyranosyl)-α-l-rhamnopyranosyl trichloroacetimidate (30).
To a solution of lactol 29 (91 mg, 0.0884 mmol) in anhydrous CH2Cl2 (3 mL) cooled at 0 °C was added Cl3CCN (27 μL) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (3 μL). The reaction mixture was stirred at 0 °C for 3 h and purified by silica gel flash column chromatography (Hexanes/EtOAc = 3/1) to give compound 30 (92 mg, 0.0787 mmol, 89%) as light yellow solids. = −14.4 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 8.71 (s, 1H, N-H), 8.07 – 7.99 (m, 4H, HAr), 7.95 – 7.86 (m, 2H, HAr), 7.59 – 7.48 (m, 3H, HAr), 7.44 – 7.23 (m, 19H, HAr), 7.21 – 7.06 (m, 7H, HAr), 6.36 (d, J = 2.1 Hz, 1H, H-1), 5.83 (dd, J = 3.5, 2.0 Hz, 1H, H-2), 5.68 (dd, J = 9.7, 3.5 Hz, 1H, H-3), 5.56 (d, J = 2.7 Hz, 1H, H-2′), 4.98 (d, J = 12.2 Hz, 1H, -OCHHAr), 4.85 – 4.74 (m, 3H, -OCHHAr, H-1′), 4.56 – 4.47 (m, 4H, -OCHHAr), 4.20 (t, J = 9.7 Hz, 1H, H-4), 4.14 – 4.09 (m, 2H, H-6′, -OCHHAr), 4.08 – 3.98 (m, 2H, H-5, H-4′), 3.80 – 3.70 (m, 2H, H-7′), 3.59 (dd, J = 9.8, 1.0 Hz, 1H, H-5′), 3.44 (dd, J = 9.3, 3.0 Hz, 1H, H-3′), 1.40 (d, J = 6.1 Hz, 3H, H-6). 13C{1H} NMR (150 MHz, CDCl3) δ 165.4, 165.2, 164.9, 160.3, 138.9, 138.2, 138.1, 137.3, 133.6, 133.4, 133.0, 130.1, 129.9, 129.9, 129.5, 129.5, 129.1, 128.7, 128.6, 128.4, 128.4, 128.3, 128.3, 128.3, 128.2, 128.0, 127.7, 127.6, 127.6, 127.6, 127.4, 127.1, 99.3, 94.9, 90.7, 81.0, 79.5, 76.3, 76.0, 75.0, 73.8, 73.4, 72.3, 72.2, 71.1, 70.2, 69.3, 68.1, 18.0. LRMS (ESI) m/z: [M+Na]+ Calcd for C64H60Cl3NNaO14 1194.3, found 1194.3.
4-Methoxyphenyl 2-O-benzoyl-3,4,6,7-tetra-O-benzyl-β-d-glycero-d-manno-hepto-pyranosyl-(1→4)-2,3-di-O-benzoyl-α-l-rhamnopyranosyl-(1→3)-3,4,6,7-tetra-O-benzyl-β-d-glycero-d-manno-heptopyranosyl-(1→4)-2,3-di-O-benzyl-α-l-rhamnopyranoside (31).
A round-bottom flask containing disaccharide acceptor 28 (32.6 mg, 0.0325 mmol), disaccharide donor 30 (42.0 mg, 0.0358 mmol) and 4 Å molecular sieves (100 mg) were dried under vacuum for 1 h. Anhydrous CH2Cl2 (3 mL) was added and the mixture was stirred at room temperature for 1 h. The mixture was cooled to −40 °C and stirred for 30 min, and TMSOTf (1.0 μL) was added. The resulting mixture was stirred at −40 °C for 30 min and then slowly warmed to 0 °C over a period of 1 h. After the reaction was quenched with Et3N (0.1 mL), molecular sieves were removed through a short pad of celite. The filtrate was concentrated and purified by silica gel flash column chromatography (Hexanes/EtOAc = 3/1) to give tetrasaccharide 31 (59.0 mg, 0.0293 mmol, 90%) as light yellow syrup. = −33.4 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 8.11 – 8.05 (m, 2H, HAr), 8.01 – 7.91 (m, 4H, HAr), 7.60 – 7.54 (m, 1H, HAr), 7.53 – 7.48 (m, 2H, HAr), 7.46 – 7.41 (m, 4H, HAr), 7.40 – 7.07 (m, 49H, HAr), 7.03 (t, J = 7.7 Hz, 2H, HAr), 6.98 – 6.90 (m, 3H, HAr), 6.82 (d, J = 9.1 Hz, 2H, HAr), 5.74 (dd, J = 10.0, 3.4 Hz, 1H, H-3′′), 5.64 (dd, J = 3.4, 1.7 Hz, 1H, H-2′′), 5.53 (d, J = 2.9 Hz, 1H, H-2′′′), 5.35 (d, J = 1.8 Hz, 1H, H-1), 5.02 (d, J = 12.2 Hz, 1H, -OCHHAr), 4.84 – 4.65 (m, 11H, -OCHHAr, H-1′, H-1′′, H-1′′′), 4.62 (d, J = 12.1 Hz, 1H, -OCHHAr), 4.59 – 4.34 (m, 9H, -OCHHAr), 4.18 – 4.08 (m, 2H, H-6′′′, H-4′′), 4.06 – 3.94 (m, 5H, H-4′′′, -OCHHAr, H-5′′, H-4′, H-6′), 3.92 (dd, J = 3.1, 1.9 Hz, 1H, H-2), 3.88 (dd, J = 9.4, 3.1 Hz, 1H, H-3), 3.83 (t, J = 9.3 Hz, 1H, H-4), 3.81 – 3.61 (m, 10H, -OCH3, H-7′′′, H-7′, H-5, H-2′, H-3′), 3.59 (d, J = 9.9 Hz, 1H, H-5′′′), 3.46 (d, J = 9.8 Hz, 1H, H-5′), 3.39 (dd, J = 9.2, 2.9 Hz, 1H, H-3′′′), 1.33 (d, J = 6.2 Hz, 3H, H-6), 1.26 (d, J = 6.1 Hz, 3H, H-6′′). 13C{1H} NMR (150 MHz, CDCl3) δ 165.5, 165.3, 164.9, 155.0, 150.6, 139.2, 139.1, 138.9, 138.7, 138.3, 138.2, 138.1, 138.1, 137.8, 137.5, 133.6, 133.4, 133.0, 130.2, 130.0, 129.9, 129.9, 129.8, 129.6, 128.8, 128.8, 128.7, 128.6, 128.5, 128.4, 128.4, 128.4, 128.4, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 127.5, 127.5, 127.4, 127.3, 127.2, 117.8, 114.7, 102.2, 99.7, 97.2, 93.4, 81.5, 79.7, 79.6, 79.6, 78.4, 78.1, 76.5, 76.1, 75.5, 75.0, 74.2, 74.0, 73.8, 73.6, 73.5, 73.3, 73.1, 73.0, 72.8, 72.7, 72.6, 72.0, 71.5, 71.4, 71.3, 71.2, 68.7, 68.1, 67.9, 55.8, 18.2, 18.1. LRMS (ESI) m/z: [M+2Na]2+ Calcd for C124H124Na2O25 1029.4, found 1029.6.
4-Methoxyphenyl β-d-glycero-d-manno-heptopyranosyl-(1→4)-α-l-rhamnopyranosyl-(1→3)-β-d-glycero-d-manno-heptopyranosyl-(1→4)-α-l-rhamnopyranoside (32).
Compound 31 (56.0 mg, 0.0278 mmol) was dissolved in CH2Cl2/methanol (1 mL/4 mL) and a solution of NaOMe in methanol (5.4 M, 0.1 mL) was added. After the mixture was stirred at 40 °C for 1 h, crude mass spectrum showed complete removal of benzoyl groups. The mixture was cooled to room temperature and neutralized by acetic acid. Solvents were removed under vacuum to give the crude product as white solids. Sodium acetate and methyl benzoate were removed by trituration using methanol (1 mL). The residual white solids were dissolved in a mixture of THF (1 mL) and methanol (4 mL), followed by addition of 10% Pd/C (24 mg). The mixture was stirred under H2 atmosphere at room temperature for 36 h. The Pd/C was removed through a short pad of celite and the filtrate was concentrated to give compound 32 (21.2 mg, 0.0265 mmol, 95% over two steps) as light yellow solids. JC1,H1 = 170.6 Hz. JC1′,H1′ = 161.4 Hz. JC1′′,H1′′ = 172.3 Hz. JC1′′′,H1′′′ = 161.4 Hz. = −143.3 (c 2.0, MeOH). 1H NMR (600 MHz, D2O) δ 7.07 (d, J = 8.5 Hz, 2H, HAr), 6.95 (d, J = 8.4 Hz, 2H, HAr), 5.38 (s, 1H, H-1), 4.95 (s, 1H, H-1′′), 4.88 – 4.79 (m, 2H, H-1′, H-1′′′, overlap with residual signal of D2O), 4.26 (s, 1H), 4.12 (s, 1H), 4.09 – 3.86 (m, 7H), 3.80 – 3.56 (m, 11H), 3.44 – 3.33 (m, 2H), 3.32 (s, 3H, -OCH3), 1.42 – 1.13 (m, 6H). 13C{1H} NMR (150 MHz, D2O) δ 154.6, 149.4, 118.9, 115.0, 100.6, 100.6, 99.2, 96.1, 79.2, 79.0, 76.9, 76.0, 75.8, 73.1, 72.3, 72.2, 70.5, 70.3, 70.2, 68.1, 68.0, 67.5, 66.2, 66.0, 62.0, 61.9, 55.7, 48.8, 16.9, 16.8. HRMS (ESI) m/z: [M+Na]+ Calcd for C33H52NaO22 823.2848, found 823.2859.
Supplementary Material
Acknowledgements
We are grateful to National Institutes of Health Common Fund Glycosciences Program (U01GM125289) and The University of Toledo for supporting this research. We would also like to thank Professor Peter Andreana and Ms. Shanika Gamage for cross validation of the synthesis of disaccharide 13 via anomeric O-alkyation of lactol 7a with triflate 8.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org
1H and 13C{1H} NMR spectra for all new compounds.
The authors declare no competing financial interest.
References
- 1.Mettu R; Chen C-Y; Wu C-Y Synthetic carbohydrate-based vaccines: challenges and opportunities. J. Biomed. Sci 2020, 27, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pakulski Z; Poly F; Dorabawila N; Guerry P; Monteiro MA 6-Deoxyheptoses in Nature, Chemistry, and Medicine. Curr. Org. Chem 2014, 18, 1818–1845. [Google Scholar]
- 3.(a) Gilbert M; Mandrell RE; Parker CT; Li J; Vinogradov E Structural Analysis of the Capsular Polysaccharide from Campylobacter jejuni RM1221. ChemBioChem 2007, 8, 625–31. [DOI] [PubMed] [Google Scholar]; (b) Guerry P; Poly F; Riddle M; Maue A; Chen Y-H; Monteiro M; Campylobacter Polysaccharide Capsules: Virulence and Vaccines. Front. Cell Infect. Microbio 2012, 2, 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Young KT; Davis LM; DiRita VJ Campylobacter jejuni: molecular biology and pathogenesis. Nat. Rev. Microbio 2007, 5, 665–679. [DOI] [PubMed] [Google Scholar]
- 4.(a) Niedziela T; Lukasiewicz J; Jachymek W; Dzieciatkowska M; Lugowski C; Kenne L Core oligosaccharides of Plesiomonas shigelloides O54:H2 (strain CNCTC 113/92): structural and serological analysis of the lipopolysaccharide core region, the O-antigen biological repeating unit, and the linkage between them. J. Biol. Chem 2002, 277, 11653–11663. [DOI] [PubMed] [Google Scholar]; (b) Czaja J; Jachymek W; Niedziela T; Lugowski C; Aldova E; Kenne L Structural studies of the O-specific polysaccharide from Plesiomonas shigelloides strain CNCTC 113/92. Eur. J. Biochem 2000, 267, 1672–1679. [DOI] [PubMed] [Google Scholar]
- 5.(a) Knirel YA; Paramonov NA; Shashkov AS; Kochetkov NK; Yarullin RG; Farber SM; Efremenko VI Structure of the polysaccharide chains of Pseudomonas pseudomallei lipopolysaccharides. Carbohydr. Res 1992, 233, 185–193. [DOI] [PubMed] [Google Scholar]; (b) Perry MB; MacLean LL; Schollaardt T; Bryan LE; Ho M Structural characterization of the lipopolysaccharide O antigens of Burkholderia pseudomallei. Infect. Immun 1995, 63, 3348–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Heiss C; Burtnick MN; Wang Z; Azadi P; Brett PJ Structural analysis of capsular polysaccharides expressed by Burkholderia mallei and Burkholderia pseudomallei. Carbohydr. Res 2012, 349, 90–94. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kosma P; Wugeditsch T; Christian R; Zayni S; Messner P Glycan structure of a heptose-containing S-layer glycoprotein of Bacillus thermoaerophilus. Glycobiology 1995, 5, 791–796. [DOI] [PubMed] [Google Scholar]; (b) Kosama P; Wugeditsh T; Christian R; Zaynl S; Messner P Glycan structure of a heptose-containing S-layer glycoprotein of Bacillus thermoaerophillus. Glycobiology 1996, 6, 5. [DOI] [PubMed] [Google Scholar]
- 7.(a) Kosma P Occurrence, Synthesis and Biosynthesis of Bacterial Heptoses. Curr. Org. Chem 2008, 12, 1021–1039. [Google Scholar]; (b) Zamyatina A; Gronow S; Puchberger M; Graziani A; Hofinger A; Kosma P Efficient chemical synthesis of both anomers of ADP L-glycero- and D-glycero-D-manno-heptopyranose. Carbohydr. Res 2003, 338, 2571–2589. [DOI] [PubMed] [Google Scholar]
- 8.For a recent report on the synthesis of 6-deoxy-D-or L-heptopyranosyl fluorides, see:; Li T; Wang J; Zhu X; Zhou X; Sun S; Wang P; Cao H; Yu G; Li M Synthesis of Rare 6-Deoxy-d-/l-Heptopyranosyl Fluorides: Assembly of a Hexasaccharide Corresponding to Campylobacter jejuni Strain CG8486 Capsular Polysaccharide. J. Am. Chem. Soc 2021, 143, 29, 11171–11179. [DOI] [PubMed] [Google Scholar]
- 9.(a) Crich D; Banerjee A Synthesis and Stereoselective Glycosylation of D- and L-glycero-β-D-manno-Heptopyranoses. Org. Lett 2005, 7, 1395–1398. [DOI] [PubMed] [Google Scholar]; (b) Crich D; Li M Block Synthesis of Tetra- and Hexasaccharides (β-d-Glycero-d-manno-Hepp-(1→4)-[α-l-Rhap-(1→3)-β-d-glycero-d-manno-Hepp-(1→4)]n-α-l-Rhap-OMe (n = 1 and 2)) Corresponding to Multiple Repeat Units of the Glycan from the Surface-Layer Glycoprotein from Bacillus thermoaerophilus . J. Org. Chem 2008, 73, 7003–7010. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jaipuri FA; Collet BYM; Pohl NL Synthesis and Quantitative Evaluation of Glycero-D-manno-heptose Binding to Concanavalin A by Fluorous-Tag Assistance. Angew Chem., Int. Ed 2008, 47, 1707–1710. [DOI] [PubMed] [Google Scholar]; (d) Dohi H; Perion R; Durka M; Bosco M; Roue Y; Moreau F; Grizot S; Ducruix A; Escaich S; Vincent SP Stereoselective Glycal Fluorophosphorylation: Synthesis of ADP-2-fluoroheptose, an Inhibitor of the LPS Biosynthesis . Chem. Eur. J 2008, 14, 9530–9539. [DOI] [PubMed] [Google Scholar]; (e) Mulani SK; Cheng K-C; Mong K-KT General Homologation Strategy for Synthesis of l-glycero- and d-glycero-Heptopyranoses. Org. Lett 2015, 17, 5536–5539. [DOI] [PubMed] [Google Scholar]; (f) Li T; Tikad A; Durka M; Pan W; Vincent SP Multigram-scale synthesis of l,d-heptoside using a Fleming-Tamao oxidation promoted by mercuric trifluoroacetate. Carbohydr. Res 2016, 432, 71–75. [DOI] [PubMed] [Google Scholar]; (g) Wang P; Huo C.-x.; Lang S; Caution K; Nick ST; Dubey P; Deora R; Huang X Chemical Synthesis and Immunological Evaluation of a Pentasaccharide Bearing Multiple Rare Sugars as a Potential Anti-pertussis Vaccine. Angew. Chem., Int. Ed 2020, 59, 6451–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Tian G; Hu J; Qin C; Li L; Zou X; Cai J; Seeberger PH; Yin J Chemical Synthesis and Immunological Evaluation of Helicobacter pylori Serotype O6 Tridecasaccharide O-Antigen Containing a dd-Heptoglycan. Angew. Chem., Int. Ed 2020, 59, 13362–13370. [DOI] [PubMed] [Google Scholar]; (i) Inuki S; Aiba T; Kawakami S; Akiyama T; Inoue J.-i.; Fujimoto Y Chemical Synthesis of d-glycero-d-manno-Heptose 1,7-Bisphosphate and Evaluation of Its Ability to Modulate NF-κB Activation. Org. Lett 2017, 19, 3079–3082. [DOI] [PubMed] [Google Scholar]
- 10.(a) Brimacombe JS; Kabir AKMS Convenient syntheses of L-glycero-D-manno-heptose and D-glycero-D-manno-heptose. Carbohydr. Res 1986, 152, 329–334. [Google Scholar]; (b) Jorgensen M; Iversen EH; Madsen R A Convenient Route to Higher Sugars by Two-Carbon Chain Elongation Using Wittig/Dihydroxylation Reactions. J. Org. Chem 2001, 66, 4625–4629. [DOI] [PubMed] [Google Scholar]; (c) Stanetty C; Walter M; Kosma P Convergent Synthesis of 4-O-Phosphorylated L-glycero-D-manno-Heptosyl Lipopolysaccharide Core Oligosaccharides Based on Regioselective Cleavage of a 6,7-O-Tetraisopropyldisiloxane-1,3-diyl Protecting Group. J. Org. Chem 2014, 79, 582–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Palmelund A; Madsen R Chain Elongation of Aldoses by Indium-Mediated Coupling with 3-Bromopropenyl Esters. J. Org. Chem 2005, 70, 8248–8251. [DOI] [PubMed] [Google Scholar]; (b) Stanetty C; Baxendale IR Large-Scale Synthesis of Crystalline 1,2,3,4,6,7-Hexa-O-acetyl-L-glycero-α-D-manno-heptopyranose. Eur. J. Org. Chem 2015, 2015, 2718–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Suster C; Baxendale IR; Mihovilovic MD; Stanetty C Straight Forward and Versatile Differentiation of the L-glycero and D-glycero-D-manno Heptose Scaffold. Front. Chem 2020, 8, 625–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Yang Y; Martin CE; Seeberger PH Total synthesis of the core tetrasaccharide of Neisseria meningitidislipopolysaccharide, a potential vaccine candidate for meningococcal diseases. Chem. Sci 2012, 3, 896–899. [Google Scholar]; (b) Ohara T; Adibekian A; Esposito D; Stallforth P; Seeberger PH Towards the synthesis of a Yersinia pestiscell wallpolysaccharide: enantioselective synthesis of an l-glycero-d-manno-heptose building block . Chem. Commun 2010, 46, 4106–4108. [DOI] [PubMed] [Google Scholar]
- 13.Wang J; Rong J; Lou Q; Zhu Y; Yang Y Synthesis of l-glycero- and d-glycero-d-manno-Heptose Building Blocks for Stereoselective Assembly of the Lipopolysaccharide Core Trisaccharide of Vibrio parahemolyticus O2. Org. Lett 2020, 22, 8018–8022. [DOI] [PubMed] [Google Scholar]
- 14.(a) Nigudkar SS; Demchenko AV Stereocontrolled 1,2-cis glycosylation as the driving force of progress in synthetic carbohydrate chemistry. Chem. Sci 2015, 6, 2687–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Demchenko AV Stereocontrolled 1,2-cis glycosylation as the driving force of progress in synthetic carbohydrate chemistry. Curr. Org. Chem 2003, 7, 35–79. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tanaka M; Nakagawa A; Nishi N; Iijima K; Sawa R; Takahashi D; Toshima K Boronic-Acid-Catalyzed Regioselective and 1,2-cis-Stereoselective Glycosylation of Unprotected Sugar Acceptors via SNi-Type Mechanism. J. Am. Chem. Soc 2018, 140, 3644–3651. [DOI] [PubMed] [Google Scholar]
- 15.(a) Sasaki K; Tohda K Recent topics in β-stereoselective mannosylation. Tetrahedron Lett 2018, 59, 496–503. [Google Scholar]; (b) Ishiwata A; Lee YJ; Ito Y Recent advances in stereoselective glycosylation through intramolecular aglycon delivery. Org. Biomol. Chem 2010, 8, 3596–3608. [DOI] [PubMed] [Google Scholar]; (c) Boltje TJ; Buskas T; Boons GJ Opportunities and challenges in synthetic oligosaccharide and glycoconjugate research. Nat. Chem 2009, 1, 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zeng Y; Kong F Synthesis of β-Mannoside and β-Mannosamine Prog . Chem 2006, 18, 907–926. [Google Scholar]; (e) Barresi F; Hindsgaul O; Khan SH; O’Neill RA (Eds.) In Modern Methods in Carbohydrate Synthesis Harwood Academic Publishers: Amsterdam, 1996, 251–276. [Google Scholar]; (f) Toshima K, Tatsuta K, Chem. Rev 1993, 93, 1503–1531. [Google Scholar]; (g) Paulsen H Advances in Selective Chemical Syntheses of Complex Oligosaccharides. Angew. Chem., Int. Ed 1982, 21, 155–173. [Google Scholar]
- 16.Crich D; Banerjee A Stereocontrolled Synthesis of the d- and l-glycero-β-d-manno-Heptopyranosides and Their 6-Deoxy Analogues. Synthesis of Methyl α-l-Rhamno-pyranosyl-(1→3)-d-glycero-β-d-manno-heptopyranosyl- (1→3)-6-deoxy-glycero-β-d-manno-heptopyranosyl-(1→4)-α-l-rhamno-pyranoside, a Tetrasaccharide Subunit of the Lipopolysaccharide from Plesimonas shigelloides J. Am. Chem. Soc 2006, 128, 8078–8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tamigney Kenfack M; Blériot Y; Gauthier C Intramolecular Aglycon Delivery Enables the Synthesis of 6-Deoxy-β-d-manno-heptosides as Fragments of Burkholderia pseudomallei and Burkholderia mallei Capsular Polysaccharide. J. Org. Chem 2014, 79, 4615–4634.: [DOI] [PubMed] [Google Scholar]
- 18.(a) Nguyen H; Zhu D; Li X; Zhu J Stereoselective Construction of β-Mannopyranosides via Anomeric O-Alkylation: Synthesis of the Trisaccharide Core of N-linked Glycans. Angew. Chem., Int. Ed 2016, 55, 4767–4771. [DOI] [PubMed] [Google Scholar]; (b) Bhetuwal BR; Woodward J Li X Zhu J Stereoselective β-Mannosylation via Anomeric O-Alkylation: Concise Synthesis of β-D-Xyl-(l→2)-β-D-Man-(1→4)-α-D-Glc-OMe, a Trisaccharide Oligomer of the Hyriopsis schlegelii Glycosphingolipid. J. Carbohydr. Chem 2017, 36, 162–172. [Google Scholar]; (c) Li X; Berry N; Saybolt K; Ahmed U; Yuan Y Stereoselective β-Mannosylation via Anomeric O-Alkylation: Formal Synthesis of Potent Calcium Signal Modulator Acremomannolipin A. Tetrahedron Lett 2017, 58, 2069–2072. [Google Scholar]; (d) Bhetuwal BR; Wu F; Meng S Zhu J Stereoselective Synthesis of 2-Azido-2-deoxy-β-D-mannosides via Cs2CO3-Mediated Anomeric O-Alkylation with Primary Triflates: Synthesis of a Tetrasaccharide Fragment of Micrococcus Luteus Teichuronic Acid. J. Org. Chem 2020, 85, 16196–16206. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Meng S Bhetuwal BR; Nguyen H; Qi X; Fang C; Saybolt K; Li X; Liu P; Zhu J β-Mannosylation via O-Alkylation of Anomeric Cesium Alkoxides: Mechanistic Studies and Synthesis of the Hexasaccharide Core of Complex Fucosylated N-Linked Glycans. Eur. J. Org. Chem 2020, 2291–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Hettiarachchi IL; Meng S; Chahine M; Li X; Zhu J Stereoselective β-Mannosylation via Anomeric O-Alkylation with L-sugar-derived electrophiles. Eur. J. Org. Chem 2021, 6682–6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.For synthesis of β-mannosides via catalytic anomeric O-alkylation, see:; Izumi S; Kobayashi Y; Takemoto Y Regio- and Stereoselective Synthesis of 1,2-cis-Glycosides by Anomeric O-Alkylation with Organoboron Catalysis. Org. Lett 2019, 21, 665–670. [DOI] [PubMed] [Google Scholar]
- 20.Meng S; Bhetuwal BR; Acharya PP; Zhu J Facile synthesis of sugar lactols via bromine-mediated oxidation of thioglycosides. J. Carbohydr. Chem 2019, 38, 109–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.See Experimental Section for details.
- 22.Crich D; Hu T; Cai F Does Neighboring Group Participation by Non-Vicinal Esters Play a Role in Glycosylation Reactions? Effective Probes for the Detection of Bridging Intermediates. J. Org. Chem 2008, 73, 8942–8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mizia JC; Bennett CS Reagent Controlled Direct Dehydrative Glycosylation with 2-Deoxy Sugars: Construction of the Saquayamycin Z Pentasaccharide. Org. Lett 2019, 21, 5922–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.