

Abstract
Several CSF and blood biomarkers for genetic frontotemporal dementia have been proposed, including those reflecting neuroaxonal loss (neurofilament light chain and phosphorylated neurofilament heavy chain), synapse dysfunction [neuronal pentraxin 2 (NPTX2)], astrogliosis (glial fibrillary acidic protein) and complement activation (C1q, C3b). Determining the sequence in which biomarkers become abnormal over the course of disease could facilitate disease staging and help identify mutation carriers with prodromal or early-stage frontotemporal dementia, which is especially important as pharmaceutical trials emerge. We aimed to model the sequence of biomarker abnormalities in presymptomatic and symptomatic genetic frontotemporal dementia using cross-sectional data from the Genetic Frontotemporal dementia Initiative (GENFI), a longitudinal cohort study.
Two-hundred and seventy-five presymptomatic and 127 symptomatic carriers of mutations in GRN, C9orf72 or MAPT, as well as 247 non-carriers, were selected from the GENFI cohort based on availability of one or more of the aforementioned biomarkers. Nine presymptomatic carriers developed symptoms within 18 months of sample collection (‘converters’). Sequences of biomarker abnormalities were modelled for the entire group using discriminative event-based modelling (DEBM) and for each genetic subgroup using co-initialized DEBM. These models estimate probabilistic biomarker abnormalities in a data-driven way and do not rely on previous diagnostic information or biomarker cut-off points. Using cross-validation, subjects were subsequently assigned a disease stage based on their position along the disease progression timeline.
CSF NPTX2 was the first biomarker to become abnormal, followed by blood and CSF neurofilament light chain, blood phosphorylated neurofilament heavy chain, blood glial fibrillary acidic protein and finally CSF C3b and C1q. Biomarker orderings did not differ significantly between genetic subgroups, but more uncertainty was noted in the C9orf72 and MAPT groups than for GRN. Estimated disease stages could distinguish symptomatic from presymptomatic carriers and non-carriers with areas under the curve of 0.84 (95% confidence interval 0.80–0.89) and 0.90 (0.86–0.94) respectively. The areas under the curve to distinguish converters from non-converting presymptomatic carriers was 0.85 (0.75–0.95).
Our data-driven model of genetic frontotemporal dementia revealed that NPTX2 and neurofilament light chain are the earliest to change among the selected biomarkers. Further research should investigate their utility as candidate selection tools for pharmaceutical trials. The model’s ability to accurately estimate individual disease stages could improve patient stratification and track the efficacy of therapeutic interventions.
Keywords: biomarker, disease progression model, event-based modelling, frontotemporal dementia, neurofilament light chain
Van der Ende et al. construct a disease progression model of fluid biomarkers in genetic frontotemporal dementia. Neuronal pentraxin 2 and neurofilament light chain are the first biomarkers to become abnormal, suggesting that they may be the most suitable for detecting early-stage disease.
Introduction
Frontotemporal dementia (FTD), a form of early-onset dementia characterized by prominent behavioural and/or language impairments, is frequently caused by autosomal dominant mutations in granulin (GRN), chromosome 9 open reading frame 72 (C9orf72) or microtubule-associated protein tau (MAPT).1,2 Upcoming therapeutic trials may be most effective in early-stage FTD, when neuronal damage is minimal. In contrast to genetic Alzheimer’s disease, disease onset in FTD mutation carriers cannot be predicted based on familial onset age,3 highlighting the need for biomarkers that can identify early disease activity. Furthermore, as drug efficacy may vary depending on disease severity, objective tools to stratify patients according to their disease stage are needed.4,5
Several promising CSF and blood biomarkers of FTD have been investigated, including neurofilament light chain (NfL) and phosphorylated neurofilament heavy chain (pNfH), which reflect neuroaxonal degeneration6-9; neuronal pentraxin 2 (NPTX2), a marker of synapse integrity10,11; glial fibrillary acidic protein (GFAP), a marker of astrogliosis12–14 and complement factors C1q and C3b, which reflect activation of the complement system.15,16 As of yet, it is unclear when these biomarkers become abnormal and what temporal relationship exists between them. Determining the sequence in which biomarkers change could facilitate disease staging and elucidate which biomarker is the most sensitive to detect early disease activity in presymptomatic mutation carriers.
Discriminative event-based modelling (DEBM) is a class of disease progression modelling that uses cross-sectional data to estimate the most probable order of events, in this case abnormality of biomarkers, over the course of disease. Individuals are subsequently assigned a disease stage within this sequence based on their biomarker values. These models are robust to missing data and do not rely on predetermined clinical diagnoses or biomarker cut-off points.17 We previously used DEBM to study the sequence of mostly cognitive and neuroimaging biomarker changes in GRN-associated FTD,18 and similar event-based models have been applied to various other neurological disorders, including Alzheimer’s disease,17,19–22 Parkinson’s disease,23 amyotrophic lateral sclerosis24 and multiple sclerosis.25 The encouraging results of these studies indicate that DEBM may be a promising strategy to model fluid biomarker changes.
The current study aimed to estimate the sequence in which the aforementioned fluid biomarkers become abnormal over the course of genetic FTD, by applying DEBM to data from the Genetic Frontotemporal dementia Initiative (GENFI).26
Materials and methods
Subjects
Subjects were included from 21 centres across Europe and Canada participating in GENFI, an ongoing longitudinal cohort study since 2012 of patients with FTD due to a pathogenic mutation in GRN, C9orf72 or MAPT and healthy 50% at-risk relatives (either presymptomatic mutation carriers or non-carriers). Participants underwent an annual assessment as previously described,26 including neurological and neuropsychological examination, MRI of the brain and collection of blood and CSF. Knowledgeable informants completed questionnaires about potential changes in cognition or behaviour.
For the present study, participants were selected based on the availability of one or more of the following biomarker measurements: CSF or serum NfL, serum pNfH, CSF NPTX2, plasma GFAP, CSF C1q and CSF C3b (Table 1). The final cohort consisted of 127 symptomatic mutation carriers (49 GRN, 54 C9orf72, 24 MAPT), 275 presymptomatic mutation carriers (128 GRN, 102 C9orf72, 45 MAPT) and 247 non-carriers. In the case of multiple available biomarkers at different time points, the time point with the most available biomarkers was selected. The follow-up duration after sample collection was at least 18 months for all subjects.
Table 1.
Selected fluid biomarkers and their biological significance
| Biomarker | Primary source in the nervous system | Primary function in the nervous system | Direction of biomarker change in FTD | Process implicated in biomarker change |
|---|---|---|---|---|
| CSF and serum NfL6–9,27 | Axonal cytoskeleton | Axon stability and transport | Increase | Neuroaxonal breakdown |
| Serum pNfH8,27,28 | Axonal cytoskeleton | Axon stability and transport | Increase | Neuroaxonal breakdown |
| CSF NPTX210,11,29–31 | Excitatory synapses on GABAergic interneurons | Glutamate-receptor recruitment, synaptic plasticity | Decrease | Loss and/or dysfunction of synaptic connectivity |
| Plasma GFAP12–14 | Astrocytes | Structural integrity, movement and shape change | Increase | Astrocytosis |
| CSF C1q15,16 | Neurons and microglia | Classical complement pathway activation | Increase | Activation of the complement system |
| CSF C3b15,16 | Neurons and microglia | Opsonization and downstream activation of the complement system | Increase | Activation of the complement system |
Mutation carriers were considered symptomatic if they fulfilled international consensus criteria for behavioural variant FTD (bvFTD) or primary progressive aphasia (PPA).32,33 Subjects with isolated or concomitant amyotrophic lateral sclerosis were excluded from the current study, as biomarker trajectories in this clinically distinct phenotype differ from those in other FTD subtypes,34–36 which could affect the overall model. We calculated disease duration based on a caregiver’s estimation of the emergence of first symptoms. The Mini-Mental State Examination (MMSE) and the Clinical Dementia Rating scale plus NACC FTLD-sum of boxes (CDR® + NACC FTLD-SB) were used as measures of global cognition.37
T1-weighted MRI on 3-T scanners was obtained within 6 months of sample collection using a standardized GENFI protocol. T1-weighted volumetric MRI scans were parcellated into brain regions as previously described,26 using an atlas propagation and fusion strategy to generate grey matter volumes of the whole brain and frontal, temporal, parietal and occipital lobes. Brain volumes were expressed as a percentage of total intracranial volume, computed with SPM12 running under MATLAB R2014b (MathWorks, Natick, MA, USA).38
Sample collection and laboratory methods
Serum and plasma were collected by venipuncture in serum-separating tubes and EDTA tubes, respectively, and CSF was collected in polypropylene tubes. Samples were centrifuged and stored at −80°C until use according to a standardized GENFI protocol.
All biomarker measurements were performed as part of previous or ongoing GENFI studies,7,10,12 leading to minor variations in the sample set per biomarker (Supplementary Table 1). Measurements for each biomarker were performed in duplicate, and samples with a duplicate coefficient of variation >20% were remeasured or excluded from the analyses. Laboratory technicians were blinded to clinical and genetic status.
CSF and serum NfL and serum pNfH were measured using the Simoa NF-light Advantage kit and pNfH Discovery kit from Quanterix (Billerica, MA, USA) on a Simoa HD-1 Analyzer.7,8,10 An in-house ELISA was used to measure CSF NPTX2.10,11 Plasma GFAP was measured using the multiplex Neurology 4-plex A kit from Quanterix on a Simoa HD-1 Analyzer.12 CSF C1q and C3b were measured using the ELISA kits Human Complement C1q (ab170246) and Human Complement C3b (ab195461) from Abcam (Boston, MA, USA).15
Standard protocols and patient consents
Local ethics committees at each site approved the study, and written informed consent was obtained from all participants according to the declaration of Helsinki. Clinical researchers were blinded to the genetic status of at-risk individuals unless they had undergone predictive testing.
Statistical analysis
Demographic and clinical variables were compared between groups (symptomatic, presymptomatic, non-carrier) using Kruskal–Wallis tests for continuous variables and a Chi-square test for sex. Biomarker levels were correlated with MMSE and CDR® + NACC FTLD scores as well as with grey matter volumes using Spearman’s rho.
All biomarkers were non-normally distributed. Normal distributions for biomarkers were achieved after log-transformation, and log-transformed data was used for subsequent analyses. DEBM requires sufficient separation of biomarker distributions between symptomatic mutation carriers and non-carriers, and we used independent sample t-tests to ensure the presence of statistically significant differences in biomarker distributions between these groups. All models were corrected for age, sex and study site.
Estimating biomarker ordering using discriminative event-based modelling
To estimate the most likely biomarker ordering, DEBM follows a three-step process.17 First, it estimates the distribution of normal and abnormal values for each biomarker using Gaussian mixture modelling (GMM) and computes the probability for each subject that the biomarker is abnormal. In the present study, normal Gaussians were fixed to the mean and standard deviation of biomarker values from the non-carriers, and GMM was subsequently used to estimate the abnormal Gaussians and the mixing parameter.18 Next, based on the probability distributions of the biomarkers, an approximate sequence of biomarker abnormality is calculated for each subject. Finally, these individual sequences are combined to create a robust biomarker ordering for the whole population. To estimate the uncertainty of this ordering, we performed bootstrap resampling with 100 different random seeds from the same cohort and estimated biomarker ordering for each of those randomly sampled datasets.
The number of subjects for whom CSF biomarkers were available was smaller than for blood biomarkers due to the relative difficulty of obtaining CSF. As these differences in sample size could potentially affect biomarker ordering, we additionally constructed models that included only subjects for whom all CSF biomarkers were available (‘CSF only model’) and for whom all blood biomarkers were available (‘blood only model’); in these models, the sample size was equal for all biomarkers.
To detect potential gene-specific biomarker orderings, we built separate models for each genetic subgroup (GRN, C9orf72 and MAPT) using co-initialized DEBM, a modified version of DEBM.21 Briefly, co-initialized DEBM splits the different steps of DEBM into group-unspecific and group-specific parts. The entire dataset is used to train the group-unspecific parts and data from genetic subgroups is used to train the group-specific parts, resulting in more accurate orderings than the default approach of independently training a DEBM model in each group. To test for differences in biomarker ordering between genetic subgroups, we estimated the distribution of the Kendall’s Tau distance under the null hypothesis using 10 000 random permutations of the three subgroups. One-sided P-values were computed for the actual Kendall’s Tau distances between the orderings of the groups based on the proportion of the sampled permutations where the distance was greater than or equal to the actual distance.
Model validation and estimating disease stages
Using 10-fold cross-validation, each subject was assigned a disease stage on a continuous scale from zero to one. These disease stages were solely based on individual biomarker profiles and their position along the disease progression timeline (based on the estimated order of biomarker changes), without the use of clinical labels (e.g. presymptomatic or symptomatic) or conventional clinical severity measures such as grey matter atrophy or clinical disease severity. We calculated areas under the curve (AUCs) to discriminate between symptomatic and presymptomatic carriers, as well as between symptomatic carriers and non-carriers. Presymptomatic carriers were subsequently split into two groups: those who became symptomatic within 18 months of follow-up (‘converters’) and those who remained presymptomatic (‘non-converters’), and we calculated the AUC to discriminate between these two groups as well. Using Spearman’s rank correlations, we examined whether estimated disease stages correlated with (i) MMSE score and CDR® + NACC FTLD-SB score in mutation carriers, (ii) grey matter volume of the whole brain and frontal, temporal, parietal and occipital lobes in mutation carriers (iii) disease duration in symptomatic carriers and (iv) time to symptom onset in converters.
Data availability
The raw data of this project is part of GENFI and de-identified participant data can be accessed on reasonable request to h.seelaar@erasmusmc.nl and genfi@ucl.ac.uk.
Results
Subjects
Subject characteristics are shown in Table 2. Symptomatic mutation carriers were older and had lower MMSE and higher CDR® + NACC FTLD-SB scores than presymptomatic carriers and non-carriers. Furthermore, symptomatic carriers had significantly higher levels of serum and CSF NfL, serum pNfH, plasma GFAP, CSF C1q and C3b, as well as lower levels of CSF NPTX2, compared to presymptomatic carriers and non-carriers. Nine presymptomatic carriers converted to the symptomatic stage during follow-up; the median time interval between sample collection and symptom onset was 6 months (range 2–13 months). For all biomarkers, we observed correlations with MMSE and CDR® + NACC FTLD-SB scores as well as grey matter volume among mutation carriers (Supplementary Table 2).
Table 2.
Subject characteristics
| Non-carriers | Presymptomatic carriers | Symptomatic carriersa | P | |
|---|---|---|---|---|
| n | 247 | 275 | 127 | – |
| GRN | – | 128 | 49 | – |
| C9orf72 | – | 102 | 54 | – |
| APT | – | 45 | 24 | – |
| Age | 45 (37–58) | 44 (35–54) | 63 (56–69) | <0.001e |
| Sex, male (%) | 109 (44%) | 105 (38%) | 75 (59%) | <0.001 |
| MMSEb | 30 (29–30) | 30 (29–30) | 25 (20–27) | <0.001e |
| CDR® + NACC FTLD-SBc | 0 (0–0) | 0 (0–0) | 9.5 (3.5–14) | <0.001e |
| Whole brain volumed | 80.4 (78.8–82.4) | 80.2 (77.7–81.9) | 71.6 (69.3–75.0) | <0.001e |
| Frontal lobe volumed | 12.5 (12.0–12.9) | 12.5 (11.9–13.0) | 10.6 (9.8–11.3) | <0.001e |
| Temporal lobe volumed | 8.5 (8.2–8.8) | 8.4 (8.1–8.7) | 7.5 (6.9–7.9) | <0.001e |
| Parietal lobe volumed | 6.6 (6.3–6.9) | 6.5 (6.2–6.9) | 5.7 (5.4–6.1) | <0.001e |
| Occipital lobe volumed | 5.1 (4.8–5.5) | 5.1 (4.8–5.3) | 4.8 (4.4–5.0) | <0.001e |
| CSF NfL, pg/ml | 422 (298–555) | 470 (315–731) | 2393 (951–4023) | <0.001e |
| Serum NfL, pg/ml | 7 (5–11) | 8 (5–8) | 40 (23–62) | <0.001e |
| Serum pNfH, pg/ml | 48 (21–101) | 42 (20–94) | 139 (80–326) | <0.001e |
| CSF NPTX2, pg/ml | 990 (604–1373) | 988 (633–1274) | 624 (291–872) | <0.001e |
| Plasma GFAP, pg/ml | 105 (80–144) | 109 (82–156) | 212 (131–310) | <0.001e |
| CSF C1q, ng/ml | 295 (208–397) | 265 (207–350) | 339 (279–464) | 0.002f |
| CSF C3b, ng/ml | 2634 (1730–3556) | 2456 (1786–3285) | 3296 (2612–4737) | 0.001g |
Continuous variables are shown as medians (interquartile range, IQR). All continuous variables were compared between groups using Kruskal–Wallis tests, and, in the case of statistically significant differences, post hoc tests with Bonferroni correction were applied. Sex distributions were compared using a Chi-square test. Regional grey matter volumes are expressed as a percentage of total intracranial volume. The sample size for each of the fluid biomarkers is reported in Supplementary Table 1.
Phenotypes: behavioural variant FTD (n = 93), primary progressive aphasia (PPA) not otherwise specified (n = 13), non-fluent variant PPA (n = 12), memory-predominant FTD (n = 3), corticobasal syndrome (n = 2), dementia not otherwise specified (n = 2), semantic variant PPA (n = 1), progressive supranuclear palsy (n = 1).
MMSE score available in 231 non-carriers, 263 presymptomatic and 111 symptomatic carriers.
CDR® + NACC FTLD-SB score available in 173 non-carriers, 205 presymptomatic and 85 symptomatic carriers.
Neuroimaging data available in 225 non-carriers, 255 presymptomatic and 79 symptomatic carriers.
Symptomatic versus presymptomatic carriers: P < 0.001; symptomatic versus non-carriers: P < 0.001.
Symptomatic versus presymptomatic carriers: P = 0.001; symptomatic versus non-carriers: P = 0.019.
Symptomatic versus presymptomatic carriers: P = 0.001; symptomatic versus non-carriers: P = 0.002.
Sequence of biomarker abnormalities
Figure 1 shows GMM estimations with normal and abnormal Gaussian distributions for each biomarker. Overall, estimated Gaussians fitted the observed histograms well.
Figure 1.

GMM distributions for each biomarker. Histogram bins are shown for non-carriers (blue), presymptomatic carriers (orange) and symptomatic carriers (dark pink). The blue Gaussian represents the distribution of normal biomarker values based on non-carriers, whereas the dark pink Gaussian shows the distribution for abnormal biomarker values, as estimated by GMM. The amplitudes of these Gaussians are based on an estimated mixing parameter. Black curves show the total estimated biomarker distribution, i.e. the summation of blue and dark pink Gaussians, and indicate the overall fit of the estimated Gaussians to the observed data. All biomarker values were log-transformed.
The estimated sequence of biomarker abnormalities and associated uncertainty is shown in Fig. 2. CSF NPTX2 was ordered first, followed by serum and CSF NfL, serum pNfH, plasma GFAP and finally CSF C3b and C1q. Two clusters of relatively large uncertainty were noted: the first for NfL measurements in serum and CSF, and the second for biomarkers that became abnormal at later disease stages (pNfH, GFAP, C3b and C1q).
Figure 2.
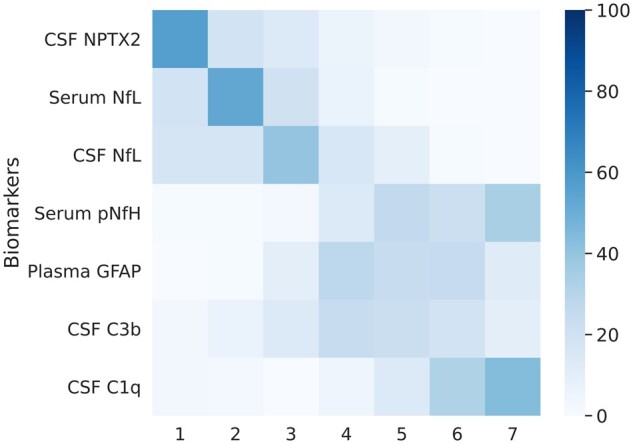
Positional variance diagram showing the sequence of biomarker abnormalities. The colour intensity of each of the squares represents the number of bootstrap resampling iterations in which the biomarker was placed at a certain position. The darkest square for each biomarker therefore signifies the mode, i.e. the position where the biomarker was placed most frequently. The spread obtained from bootstrap resampling represents the standard error of the distribution and signifies uncertainty in the estimation of the ordering. The ordering of biomarkers is based on their position in the entire dataset (without bootstrap resampling), which is akin to mean position.
The models including only subjects with all CSF biomarkers (n = 225) or all blood biomarkers (n = 342) estimated the same ordering as the full model (Supplementary Fig. 1).
Estimation of disease stage
Overall, most non-carriers and presymptomatic carriers were assigned low disease stages, with little or no biomarker abnormality, whereas most symptomatic carriers were assigned later stages of disease. Furthermore, converters (n = 9) were assigned higher disease stages than non-converting presymptomatic carriers (n = 267) (Fig. 3). Estimated disease stages could discriminate symptomatic from presymptomatic carriers with an AUC of 0.84 [95% confidence interval (CI) 0.80–0.89], and symptomatic carriers from non-carriers with an AUC of 0.90 (95% CI 0.86–0.94). The AUC to discriminate converters from non-converters was 0.85 (95% CI 0.75–0.95).
Figure 3.

Estimated disease stages per clinical group. Disease stages were obtained using 10-fold cross-validation. (A) Histogram showing the frequency of occurrence of each of the disease stages per clinical group, normalized for each clinical group. Estimated disease stages are a continuous measure and were discretized for visualization purposes only. (B) Box plots of estimated disease stages for each clinical group. Box plots indicate median ± IQR; whiskers indicate median ±1.5× IQR. Symptomatic carriers and converters had higher estimated disease stages than presymptomatic non-converters (P < 0.001 and P = 0.004 respectively), but no difference was found between symptomatic carriers and converters (P = 0.712) (by Kruskal–Wallis tests).
Estimated disease stages in mutation carriers correlated with MMSE (rs = −0.467, P < 0.001) and CDR® + NACC FTLD-SB scores (rs = 0.530, P < 0.001), but not with disease duration (rs = 0.124, P = 0.127) (Fig. 4). Correlations were additionally found with whole brain volume (rs = −0.392), frontal (rs = −0.401), temporal (rs = −0.334), parietal (rs = −0.298) and occipital (rs = −0.226) lobe volume (all correlations: P < 0.001). In converters, estimated disease stages correlated with the time to symptom onset (rs = −0.678, P = 0.045).
Figure 4.

Relationship between estimated disease stage and disease severity measures in mutation carriers. (A) MMSE score (rs = −0.467, P < 0.001); (B) CDR® + NACC FTLD-SB score (rs = 0.530, P < 0.001); (C) disease duration in years (rs = −0.124, P = 0.127) and (D) whole brain volume (rs = −0.392, P < 0.001). Whole brain volume was expressed as a percentage of total intracranial volume. Presymptomatic carriers are shown in orange, and symptomatic carriers in dark pink. The regression lines were fit using splines; dotted lines indicate 95% prediction intervals.
A small number of symptomatic carriers was assigned relatively low disease stages. To further investigate this, we divided symptomatic carriers into three equal-sized groups with low, moderate and high disease stages as estimated by the model. Subjects with low disease stages were more commonly C9orf72 or MAPT mutation carriers, more frequently suffered from bvFTD, had higher MMSE scores and a trend towards longer disease duration than those with higher disease stages. Furthermore, these subjects had significantly lower levels of CSF NfL, serum NfL and plasma GFAP than symptomatic carriers with moderate or high disease stages (Table 3).
Table 3.
Clinical characteristics of symptomatic carriers with low, moderate and high disease stages
| Estimated disease stage | P | |||
|---|---|---|---|---|
| Low (≤0.55) | Moderate (0.56–0.78) | High (≥0.79) | ||
| n | 43 | 42 | 42 | – |
| GRN | 9 | 18 | 22 | 0.038 |
| C9orf72 | 22 | 18 | 14 | |
| MAPT | 12 | 6 | 6 | |
| Sex, male | 29 | 27 | 19 | 0.080 |
| Age at sample, years | 62 (56–68) | 62 (57–69) | 64 (54–69) | 0.741 |
| MMSE | 26 (24–29) | 24 (19–27) | 23 (16–27) | 0.004a |
| CDR® + NACC FTLD-SB | 8 (4–10) | 10 (4–16) | 11 (4–15) | 0.430 |
| Disease duration, years | 4.4 (2.5–8.0) | 2.6 (1.9–5.0) | 3.9 (2.0–6.3) | 0.052 |
| Phenotype, n | ||||
| bvFTD | 37 | 28 | 28 | 0.038 |
| PPA | 3 | 12 | 11 | |
| Memory-predominant FTD | 2 | 1 | 0 | |
| Dementia not otherwise specified | 1 | 1 | 0 | |
| Corticobasal syndrome or progressive supranuclear palsy | 0 | 0 | 3 | |
| CSF NfL, pg/ml | 933 (722–1750) | 3489 (1870–6073) | 2867 (2480–5278) | <0.001b |
| Serum NfL, pg/ml | 15 (12–22) | 48 (37–89) | 58 (41–85) | <0.001c |
| Serum pNfH, pg/ml | 110 (56–235) | 122 (84–206) | 206 (76–464) | 0.135 |
| CSF NPTX2, pg/ml | 741 (483–873) | 399 (237–619) | 780 (243–1111) | 0.116 |
| Plasma GFAP, pg/ml | 127 (93–207) | 222 (169–329) | 294 (186–472) | <0.001d |
| CSF C1q, ng/ml | 319 (267–464) | 308 (215–369) | 373 (318–598) | 0.078 |
| CSF C3b, ng/ml | 2974 (2546–4501) | 3162 (2657–3668) | 4568 (2596–5819) | 0.418 |
Continuous variables are shown as medians (IQR). Categorical variables were compared using Chi-square tests. Continuous variables were compared using Kruskal–Wallis tests, and in the case of statistically significant differences, post hoc tests with Bonferroni correction were performed. bvFTD = behavioural variant FTD.
High versus low disease stage: P = 0.005; moderate versus low disease stage: P = 0.045.
High versus low disease stage: P < 0.001; moderate versus low disease stage: P = 0.003.
High versus low disease stage: P < 0.001; moderate versus low disease stage: P < 0.001.
High versus low disease stage: P < 0.001; moderate versus low disease stage: P = 0.010.
Genetic subgroup analyses
Biomarker levels for genetic subgroups are reported in Supplementary Table 3. Supplementary Fig. 2 shows the gene-specific GMM estimations of normal and abnormal biomarker distributions.
Estimated sequences of biomarker abnormality did not differ significantly between each of the genetic subgroups or compared to the full model. More uncertainty was observed than in the full model, especially for C9orf72 and MAPT mutation carriers (Supplementary Fig. 3).
In each genetic subgroup, symptomatic carriers were assigned higher disease stages than presymptomatic carriers (Fig. 5). However, separation of symptomatic and presymptomatic carriers was much clearer for GRN than for C9orf72 and MAPT mutation carriers, which was reflected in the AUCs [GRN: 0.91 (95% CI 0.87–0.95); C9orf72: 0.75 (0.66–0.84); MAPT: 0.68 (0.52–0.84)].
Figure 5.
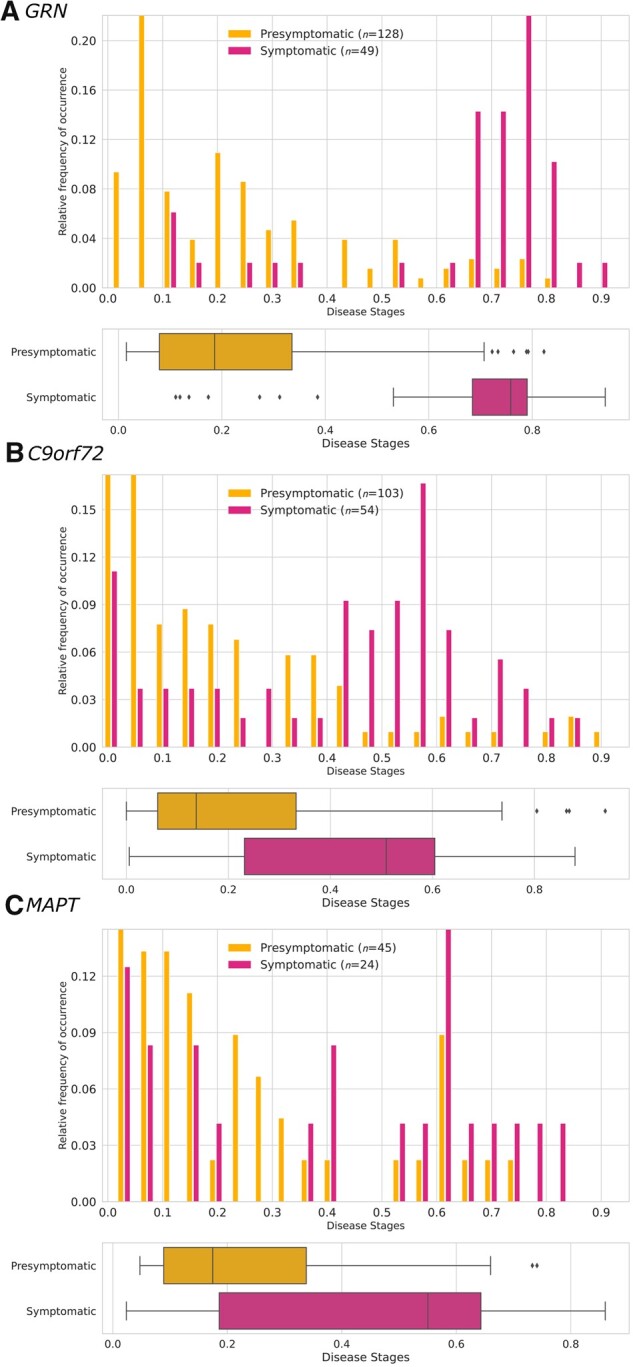
Estimated disease stages for each genetic subgroup. Disease stages were obtained using co-initialized DEBM with 10-fold cross-validation. Histograms show the relative frequency of occurrence of each disease stage and box plots show the estimated disease stages per clinical group in (A) GRN, (B) C9orf72 and (C) MAPT mutation carriers. Estimated disease stages are a continuous measure and were discretized for visualization purposes only. Box plots indicate median ± IQR; whiskers indicate median ±1.5× IQR. Symptomatic carriers had significantly higher disease stages than presymptomatic carriers in all genetic subgroups (GRN and C9orf72: P < 0.001; MAPT: P = 0.004 by Mann–Whitney U-tests).
Discussion
In this large, international study, we constructed a disease progression timeline of genetic FTD using a broad selection of fluid biomarkers. DEBM revealed that CSF NPTX2 was the first biomarker to become detectably abnormal, followed by NfL in serum and CSF, whereas pNfH, GFAP, C3b and C1q abnormality was estimated to occur at later disease stages. These findings provide novel insights into biomarker trajectories in genetic FTD and have important consequences for clinical trial design.
Our model indicates that NPTX2 reductions, which are thought to reflect a loss or dysfunction of certain excitatory synapses,11,29–31 occur at a relatively early stage in FTD. This finding is in line with the NPTX2 decreases observed before and around symptom onset in a small number of longitudinal CSF samples10 in genetic FTD, and corroborates extensive evidence for synapse pathology as an early event in the neurodegenerative process.39–41 The early changes in serum and CSF NfL similarly correspond with previous studies,6,7 including our DEBM study of GRN mutation carriers,18 and support the use of these biomarkers as tools to identify early FTD in mutation carriers.4 Within-individual changes in NPTX2 and NfL might be even more sensitive to disease activity than single measurements.7,10,27,28 Using CSF biomarkers to screen mutation carriers for clinical trial enrolment is inevitably hampered by the invasive nature of lumbar punctures, especially for repeated measurements, and blood is the preferred medium. Therefore, it is promising that NPTX2 also appears to be measurable in blood,42 and future studies should aim to determine blood NPTX2 levels in FTD. Furthermore, it is reassuring that our model ordered serum NfL change before CSF NfL. While this finding could partly be influenced by the larger sample size for serum than CSF, and therefore greater statistical power to detect biomarker abnormality, it suggests that serum NfL is not inferior as an early disease marker.
Serum pNfH was estimated to become abnormal later than CSF or serum NfL. This suggests that early-stage FTD might be characterized by elevated levels of serum NfL, but not pNfH, in which case the simultaneous measurement of both neurofilaments could facilitate disease staging. pNfH and NfL are both major structural components of axons normally present in a ratio of 4:1 (NfL:pNfH) and are released on neuroaxonal damage.43 While pNfH has not been extensively studied in FTD, a longitudinal study of familial amyotrophic lateral sclerosis showed that both serum NfL and pNfH levels increased before symptom onset, but only NfL exceeded absolute thresholds in the presymptomatic stage.28 A possible explanation for this discrepancy in NfL and pNfH dynamics lies in the ratio of NfL to pNfH expression, which increases during neurodegeneration as an energy-saving mechanism,44,45 leading to relatively more NfL and less pNfH release. Consequently, only measuring pNfH could underestimate the degree of axonal breakdown. Furthermore, in contrast to NfL, CSF and serum pNfH are only moderately correlated8,46,47; whether this reflects differences in clearance dynamics,8,28,48 or difficulties in detecting serum pNfH, e.g. due to aggregate formation or dephosphorylation,43,49–51 is unclear. Including CSF pNfH measurements in similar models might help elucidate these complex relationships.
The relatively late ordering of GFAP in our model contrasts somewhat with a previous study that indicated GFAP levels increase in the presymptomatic stage in conjunction with early atrophy.12 This discrepancy might be explained by the overlapping GFAP levels between presymptomatic and symptomatic mutation carriers, especially in C9orf72 and MAPT carriers. The ensuing uncertainty could lead the model to estimate detectable abnormality at a later stage. Similarly, C1q and C3b are relatively weak biomarkers, as is seen in the GMM distribution figures, possibly explaining their late position on the disease progression timeline. These considerations underline that one cannot comment on the order of underlying pathological processes, as the estimated sequence may be affected by the strength of the biomarkers.17,19 The reported biomarker changes provide valuable insights into the pathological processes occurring during FTD; animal and cellular models might elucidate their role in disease pathogenesis (i.e. whether these processes are causal or secondary and whether they are protective or detrimental).
We validated the model by showing that the estimated disease stages could accurately distinguish presymptomatic from symptomatic mutation carriers and correlated with MMSE and CDR® + NACC FTLD-SB scores. Interestingly, converters had similar disease stages to symptomatic carriers, providing evidence that at least some of the included biomarkers were already abnormal before symptom onset. The high accuracy to distinguish converters from non-converting presymptomatic carriers and the inverse correlation between estimated disease stages and time to symptom onset tentatively suggest that the model might be able to predict conversion. Replication of our findings with more converters is needed to confirm this. The small number of presymptomatic non-converters with high disease stages raises the question whether these subjects may be approaching conversion, or whether unrecognized factors might be either delaying symptom onset or affecting biomarker levels. Further follow-up as part of the GENFI study might clarify this.
Notably, we observed several mutation carriers, both presymptomatic and symptomatic, with high estimated disease stages but near-normal MMSE and CDR® + NACC FTLD-SB scores, suggesting that these clinical measures might underestimate the ongoing pathological process. It would be interesting to examine the relationship between estimated disease stages and more sensitive cognitive and behavioural measures of FTD, such as those specifically reflecting executive functioning, language and social cognition.52,53
Some symptomatic mutation carriers were assigned unexpectedly low disease stages. These subjects had less severe disease, as measured by the MMSE and CDR® + NACC FTLD-SB, and a trend towards a longer disease duration than those with higher disease stages. The low disease stages in these subjects are probably driven by the low levels of CSF and serum NfL as well as plasma GFAP. These biomarkers have previously been shown to predict the rate of subsequent decline in clinical and/or neuroimaging parameters, with low levels predicting a slow disease course.6,7,12,14 Taken together, these subjects might have a relatively indolent disease course. The high frequency of C9orf72 mutation carriers with low disease stages further supports this: progression is often slow54 and can even span several decades.55,56 An alternative explanation for the low disease stages lies in the fact that disease stages are computed based on the average estimated sequence of biomarker changes in the study population, and subjects who do not follow this sequence might wrongly be assigned low disease stages. Future studies using data-driven subtyping methods, such as SuStaIn,57 could potentially identify clusters of subjects with a differential sequence of events.
Using co-initialized DEBM, a recently proposed approach to compute biomarker orderings in predefined subgroups,21 we demonstrated that GRN, C9orf72 and MAPT mutation carriers followed the same biomarker ordering. This is of particular importance for clinical trials, which will most likely target genetic subgroups.4 However, much more uncertainty was noted for C9orf72 and MAPT, with poorer ability to discriminate symptomatic from presymptomatic carriers than for GRN. Several possible explanations for this uncertainty exist. First, CSF and blood NfL levels are less markedly elevated in C9orf72 and MAPT mutation carriers (with the exception of patients with amyotrophic lateral sclerosis58) than in GRN,6,7 which could reduce the model’s power to detect abnormal levels. Second, especially in C9orf72 mutation carriers, the striking clinical heterogeneity might translate into more variable biomarker levels.56 Third, previous neuroimaging and cognitive studies have suggested that C9orf72 mutation carriers may have a more protracted disease onset compared to the relatively abrupt onset presumed to occur in GRN mutation carriers.53,59–62 This might be reflected in a more gradual change of fluid biomarkers,63 which would explain the relatively large number of presymptomatic C9orf72 mutation carriers with high disease stages and complicates discriminating presymptomatic from symptomatic carriers based on biomarker data. Finally, especially for MAPT, the smaller sample size compared to other genetic subgroups may have contributed to the observed uncertainty, underlining the necessity to validate our findings in larger cohorts.
Strengths of this study include the well-characterized cohort of presymptomatic and symptomatic mutation carriers and the availability of several fluid biomarkers of different FTD-related processes. These biomarkers are not currently used for the diagnosis of FTD, enabling us to investigate the relationship between estimated disease stage and clinical diagnosis (presymptomatic or symptomatic) in a non-circular way. Using non-carriers as our control group ensured that they were ‘true controls’, in contrast to similar models of sporadic diseases, which run the risk of controls in fact being asymptomatic cases.22 The ability of DEBM to compute a disease progression timeline using cross-sectional rather than longitudinal data enabled relatively large sample sizes despite the rarity of the disease. Our model can be extended to include other biomarkers; of note, the model assumes monotonic changes over the course of disease.17 Serum NfL is highly stable in the presymptomatic stage7; for the remaining biomarkers, obtaining longitudinal data will improve our understanding of their natural dynamics. In the current model, which combines multiple markers of different pathological processes, presymptomatic fluctuations in individual biomarkers are not expected to affect the obtained disease stage, and hence should not affect inclusion or exclusion in clinical trials. This is because the model derives disease stage by comparing the abnormality of biomarkers with the estimated sequence of abnormalities in genetic FTD progression, and fluctuations in individual biomarkers (e.g. due to unrelated neurological disease) would most probably not align well with the estimated sequence of changes.
Our findings must be viewed in light of some limitations. First, the model estimates the ordering of biomarker events relative to one another, but not whether biomarkers become abnormal before or after symptom onset, highlighting the need for longitudinal studies. Second, biomarker studies of neurodegenerative diseases are inevitably hampered by a relative under-representation of end-stage disease, which could bias the model towards earlier disease stages.64
In conclusion, the present study provides an insightful event ordering of a range of fluid biomarkers in genetic FTD. Future research should aim to validate our findings in independent cohorts and using longitudinal data. The accurate estimation of disease stages demonstrates the model’s potential as a tool for patient stratification, which could in turn reduce heterogeneity in clinical trials.
Supplementary Material
Acknowledgements
We thank all participants and their family members for taking part in this study. Several authors of this publication are members of the European Reference Network for Rare Neurological Diseases—Project ID no. 739510.
Funding
This study was supported in the Netherlands by two Memorabel grants from Deltaplan Dementie (The Netherlands Organisation for Health Research and Development and Alzheimer Nederland; grant numbers 733050813,733050103 and 733050513), the Bluefield Project to Cure Frontotemporal Dementia, the Dioraphte foundation (grant number 1402 1300), the European Joint Programme—Neurodegenerative Disease Research and the Netherlands Organisation for Health Research and Development (PreFrontALS: 733051042, RiMod-FTD: 733051024); V.V. and S.K. have received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 666992 (EuroPOND). E.B. was supported by the Hartstichting (PPP Allowance, 2018B011); in Belgium by the Mady Browaeys Fonds voor Onderzoek naar Frontotemporale Degeneratie; in the UK by the MRC UK GENFI grant (MR/M023664/1); J.D.R. is supported by an MRC Clinician Scientist Fellowship (MR/M008525/1) and has received funding from the NIHR Rare Disease Translational Research Collaboration (BRC149/NS/MH); I.J.S. is supported by the Alzheimer’s Association; J.B.R. is supported by the Wellcome Trust (103838); in Spain by the Fundació Marató de TV3 (20143810 to R.S.V.); in Germany by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy—ID 390857198) and by grant 779357 ‘Solve-RD’ from the Horizon 2020 Research and Innovation Programme (to MS); in Sweden by grants from the Swedish FTD Initiative funded by the Schörling Foundation, grants from JPND PreFrontALS Swedish Research Council (VR) 529–2014-7504, Swedish Research Council (VR) 2015–02926, Swedish Research Council (VR) 2018–02754, Swedish Brain Foundation, Swedish Alzheimer Foundation, Stockholm County Council ALF, Swedish Demensfonden, Stohnes foundation, Gamla Tjänarinnor, Karolinska Institutet Doctoral Funding and StratNeuro. H.Z. is a Wallenberg Scholar.
Competing interests
J.L. reports speaker fees from Bayer Vital and Roche, consulting fees from Axon Neuroscience, author fees from Thieme medical publishers and W. Kohlhammer GmbH medical publishers, non-financial support from Abbvie and compensation for duty as part-time CMO from MODAG, outside the submitted work. H.Z. has served at scientific advisory boards for Alector, Eisai, Denali, Roche Diagnostics, Wave, Samumed, Siemens Healthineers, Pinteon Therapeutics, Nervgen, AZTherapies and CogRx, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure and Biogen, and is a cofounder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. The other authors report no competing interests relevant to this study.
Glossary
Abbreviations
- AUC
area under the curve
- CDR® + NACC FTLD-SB
Clinical Dementia Rating scale plus NACC FTLD-sum of boxes
- DEBM
discriminative event-based modelling
- FTD
frontotemporal dementia
- GENFI
Genetic Frontotemporal dementia Initiative
- GMM
Gaussian Mixture Modelling
- MMSE
Mini Mental State Examination
- NfL
neurofilament light chain
- pNfH
phosphorylated neurofilament heavy chain
- PPA
primary progressive aphasia
Appendix I
GENFI consortium members
Full details are available in the Supplementary material.
Sónia Afonso, Maria Rosario Almeida, Sarah Anderl-Straub, Christin Andersson, Anna Antonell, Silvana Archetti, Andrea Arighi, Mircea Balasa, Myriam Barandiaran, Nuria Bargalló, Robart Bartha, Benjamin Bender, Alberto Benussi, Luisa Benussi, Valentina Bessi, Giuliano Binetti, Sandra Black, Martina Bocchetta, Sergi Borrego-Ecija, Jose Bras, Rose Bruffaerts, Marta Cañada, Valentina Cantoni, Paola Caroppo, David Cash, Miguel Castelo-Branco, Rhian Convery, Thomas Cope, Giuseppe Di Fede, Alina Díez, Diana Duro, Chiara Fenoglio, Camilla Ferrari, Catarina B. Ferreira, Nick Fox, Morris Freedman, Giorgio Fumagalli, Alazne Gabilondo, Roberto Gasparotti, Serge Gauthier, Stefano Gazzina, Giorgio Giaccone, Ana Gorostidi, Caroline Greaves, Rita Guerreiro, Tobias Hoegen, Begoña Indakoetxea, Vesna Jelic, Hans-Otto Karnath, Ron Keren, Tobias Langheinrich, Maria João Leitão, Albert Lladó, Gemma Lombardi, Sandra Loosli, Carolina Maruta, Simon Mead, Gabriel Miltenberger, Rick van Minkelen, Sara Mitchell, Katrina Moore, Benedetta Nacmias, Jennifer Nicholas, Linn Öijerstedt, Jaume Olives, Sebastien Ourselin, Alessandro Padovani, Georgia Peakman, Michela Pievani, Yolande Pijnenburg, Cristina Polito, Enrico Premi, Sara Prioni, Catharina Prix, Rosa Rademakers, Veronica Redaelli, Tim Rittman, Ekaterina Rogaeva, Pedro Rosa-Neto, Giacomina Rossi, Martin Rosser, Beatriz Santiago, Elio Scarpini, Sonja Schönecker, Elisa Semler, Rachelle Shafei, Christen Shoesmith, Miguel Tábuas-Pereira, Mikel Tainta, Ricardo Taipa, David Tang-Wai, David L Thomas, Paul Thompson, Hakan Thonberg, Carolyn Timberlake, Pietro Tiraboschi, Emily Todd, Philip Van Damme, Mathieu Vandenbulcke, Michele Veldsman, Ana Verdelho, Jorge Villanua, Jason Warren, Ione Woollacott, Elisabeth Wlasich, Miren Zulaica.
Contributor Information
GENFI consortium:
Sónia Afonso, Maria Rosario Almeida, Sarah Anderl-Straub, Christin Andersson, Anna Antonell, Silvana Archetti, Andrea Arighi, Mircea Balasa, Myriam Barandiaran, Nuria Bargalló, Robart Bartha, Benjamin Bender, Alberto Benussi, Luisa Benussi, Valentina Bessi, Giuliano Binetti, Sandra Black, Martina Bocchetta, Sergi Borrego-Ecija, Jose Bras, Rose Bruffaerts, Marta Cañada, Valentina Cantoni, Paola Caroppo, David Cash, Miguel Castelo-Branco, Rhian Convery, Thomas Cope, Giuseppe Di Fede, Alina Díez, Diana Duro, Chiara Fenoglio, Camilla Ferrari, Catarina B Ferreira, Nick Fox, Morris Freedman, Giorgio Fumagalli, Alazne Gabilondo, Roberto Gasparotti, Serge Gauthier, Stefano Gazzina, Giorgio Giaccone, Ana Gorostidi, Caroline Greaves, Rita Guerreiro, Tobias Hoegen, Begoña Indakoetxea, Vesna Jelic, Hans-Otto Karnath, Ron Keren, Tobias Langheinrich, Maria João Leitão, Albert Lladó, Gemma Lombardi, Sandra Loosli, Carolina Maruta, Simon Mead, Gabriel Miltenberger, Rick van Minkelen, Sara Mitchell, Katrina Moore, Benedetta Nacmias, Jennifer Nicholas, Linn Öijerstedt, Jaume Olives, Sebastien Ourselin, Alessandro Padovani, Georgia Peakman, Michela Pievani, Yolande Pijnenburg, Cristina Polito, Enrico Premi, Sara Prioni, Catharina Prix, Rosa Rademakers, Veronica Redaelli, Tim Rittman, Ekaterina Rogaeva, Pedro Rosa-Neto, Giacomina Rossi, Martin Rosser, Beatriz Santiago, Elio Scarpini, Sonja Schönecker, Elisa Semler, Rachelle Shafei, Christen Shoesmith, Miguel Tábuas-Pereira, Mikel Tainta, Ricardo Taipa, David Tang-Wai, David L Thomas, Paul Thompson, Hakan Thonberg, Carolyn Timberlake, Pietro Tiraboschi, Emily Todd, Philip Van Damme, Mathieu Vandenbulcke, Michele Veldsman, Ana Verdelho, Jorge Villanua, Jason Warren, Ione Woollacott, Elisabeth Wlasich, and Miren Zulaica
Supplementary material
Supplementary material is available at Brain online.
References
- 1. Lashley T, Rohrer JD, Mead S, Revesz T.. Review: An update on clinical, genetic and pathological aspects of frontotemporal lobar degenerations. Neuropathol Appl Neurobiol. 2015;41(7):858–881. [DOI] [PubMed] [Google Scholar]
- 2. Seelaar H, Rohrer JD, Pijnenburg YA, Fox NC, van Swieten JC.. Clinical, genetic and pathological heterogeneity of frontotemporal dementia: A review. J Neurol Neurosurg Psychiatry. 2011;82(5):476–486. [DOI] [PubMed] [Google Scholar]
- 3. Moore KM, Nicholas J, Grossman M, et al. . Age at symptom onset and death and disease duration in genetic frontotemporal dementia: An international retrospective cohort study. Lancet Neurol. 2020;19(2):145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boxer AL, Gold M, Feldman H, et al. . New directions in clinical trials for frontotemporal lobar degeneration: Methods and outcome measures. Alzheimers Dement. 2020;16(1):131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Desmarais P, Rohrer JD, Nguyen QD, et al. . Therapeutic trial design for frontotemporal dementia and related disorders. J Neurol Neurosurg Psychiatry. 2019;90(4):412–423. [DOI] [PubMed] [Google Scholar]
- 6. Meeter LH, Dopper EG, Jiskoot LC, et al. . Neurofilament light chain: A biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol. 2016;3(8):623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van der Ende EL, Meeter LH, Poos JM, et al. . Serum neurofilament light chain in genetic frontotemporal dementia: A longitudinal, multicentre cohort study. Lancet Neurology. 2019;18(12):1103–1111. [DOI] [PubMed] [Google Scholar]
- 8. Wilke C, Pujol-Calderón F, Barro C, et al. . Correlations between serum and CSF pNfH levels in ALS, FTD and controls: A comparison of three analytical approaches. Clin Chem Lab Med. 2019;57(10):1556–1564. [DOI] [PubMed] [Google Scholar]
- 9. Rohrer JD, Woollacott IO, Dick KM, et al. . Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology. 2016;87(13):1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van der Ende EL, Xiao M, Xu D, et al. . Neuronal pentraxin 2: A synapse-derived CSF biomarker in genetic frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2020;91(6):612–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xiao MF, Xu D, Craig MT, et al. . NPTX2 and cognitive dysfunction in Alzheimer's Disease. eLife. 2017;6:e23798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heller C, Foiani MS, Moore K, et al. . Plasma glial fibrillary acidic protein is raised in progranulin-associated frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2020;91(3):263–270. [DOI] [PubMed] [Google Scholar]
- 13. Eng LF, Ghirnikar RS.. GFAP and astrogliosis. Brain Pathol. 1994;4(3):229–237. [DOI] [PubMed] [Google Scholar]
- 14. Benussi A, Ashton NJ, Karikari TK, et al. . Serum glial fibrillary acidic protein (GFAP) is a marker of disease severity in frontotemporal lobar degeneration. J Alzheimers Dis. 2020;77(3):1129–1141. [DOI] [PubMed] [Google Scholar]
- 15. Lui H, Zhang J, Makinson SR, et al. . Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell. 2016;165(4):921–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rutkowski MJ, Sughrue ME, Kane AJ, Mills SA, Fang S, Parsa AT.. Complement and the central nervous system: Emerging roles in development, protection and regeneration. Immunol Cell Biol. 2010;88(8):781–786. [DOI] [PubMed] [Google Scholar]
- 17. Venkatraghavan V, Bron EE, Niessen WJ, Klein S; Alzheimer's Disease Neuroimaging Initiative . Disease progression timeline estimation for Alzheimer's disease using discriminative event based modeling. Neuroimage. 2019;186:518–532. [DOI] [PubMed] [Google Scholar]
- 18. Panman JL, Venkatraghavan V, van der Ende EL, et al. . Modelling the cascade of biomarker changes in GRN-related frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2021;92(5):494–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oxtoby NP, Young AL, Cash DM, et al. . Data-driven models of dominantly-inherited Alzheimer's disease progression. Brain. 2018;141(5):1529–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Archetti D, Ingala S, Venkatraghavan V, et al. ; for EuroPOND Consortium . Multi-study validation of data-driven disease progression models to characterize evolution of biomarkers in Alzheimer's disease. Neuroimage Clin. 2019;24:101954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Venkatraghavan V, Klein S, Fani L, et al. ; Alzheimer’s Disease Neuroimaging Initiative . Analyzing the effect of APOE on Alzheimer's disease progression using an event-based model for stratified populations. Neuroimage. 2021;227:117646. [DOI] [PubMed] [Google Scholar]
- 22. Young AL, Oxtoby NP, Daga P, et al. . A data-driven model of biomarker changes in sporadic Alzheimer's disease. Brain. 2014;137(9):2564–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oxtoby NP, Leyland LA, Aksman LM, et al. . Sequence of clinical and neurodegeneration events in Parkinson's disease progression. Brain. 2021;144(3):975–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gabel MC, Broad RJ, Young AL, et al. . Evolution of white matter damage in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2020;7(5):722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dekker I, Schoonheim MM, Venkatraghavan V, et al. . The sequence of structural, functional and cognitive changes in multiple sclerosis. Neuroimage Clin. 2021;29:102550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rohrer JD, Nicholas JM, Cash DM, et al. . Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: A cross-sectional analysis. Lancet Neurol. 2015;14(3):253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Preische O, Schultz SA, Apel A, et al. ; Dominantly Inherited Alzheimer Network . Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer's disease. Nat Med. 2019;25(2):277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Benatar M, Wuu J, Lombardi V, et al. . Neurofilaments in pre-symptomatic ALS and the impact of genotype. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20(7-8):538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pelkey KA, Barksdale E, Craig MT, et al. . Pentraxins coordinate excitatory synapse maturation and circuit integration of parvalbumin interneurons. Neuron. 2015;85(6):1257–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu D, Hopf C, Reddy R, et al. . Narp and NP1 form heterocomplexes that function in developmental and activity-dependent synaptic plasticity. Neuron. 2003;39(3):513–528. [DOI] [PubMed] [Google Scholar]
- 31. Galasko D, Xiao M, Xu D, et al. ; Alzheimer's Disease Neuroimaging Initiative (ADNI) . Synaptic biomarkers in CSF aid in diagnosis, correlate with cognition and predict progression in MCI and Alzheimer's disease. Alzheimers Dement (N Y). 2019;5(1):871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rascovsky K, Hodges JR, Knopman D, et al. . Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gorno-Tempini ML, Hillis AE, Weintraub S, et al. . Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barschke P, Oeckl P, Steinacker P, et al. . Different CSF protein profiles in amyotrophic lateral sclerosis and frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. J Neurol Neurosurg Psychiatry. 2020;91(5):503–511. [DOI] [PubMed] [Google Scholar]
- 35. Bridel C, van Wieringen WN, Zetterberg H, et al. ; and the NFL Group . Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: A systematic review and meta-analysis. JAMA Neurol. 2019;76(9):1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Oeckl P, Weydt P, Steinacker P, et al. ; German Consortium for Frontotemporal Lobar Degeneration . Different neuroinflammatory profile in amyotrophic lateral sclerosis and frontotemporal dementia is linked to the clinical phase. J Neurol Neurosurg Psychiatry. 2019;90(1):4–10. [DOI] [PubMed] [Google Scholar]
- 37. Miyagawa T, Brushaber D, Syrjanen J, et al. . Use of the CDR(R) plus NACC FTLD in mild FTLD: Data from the ARTFL/LEFFTDS consortium. Alzheimers Dement. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Malone IB, Leung KK, Clegg S, et al. . Accurate automatic estimation of total intracranial volume: A nuisance variable with less nuisance. Neuroimage. 2015;104:366–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ling SC. Synaptic paths to neurodegeneration: The emerging role of TDP-43 and FUS in synaptic functions. Neural Plast. 2018;2018:8413496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marttinen M, Kurkinen KM, Soininen H, Haapasalo A, Hiltunen M.. Synaptic dysfunction and septin protein family members in neurodegenerative diseases. Mol Neurodegener. 2015;10:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Terry RD, Masliah E, Salmon DP, et al. . Physical basis of cognitive alterations in Alzheimer's disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30(4):572–580. [DOI] [PubMed] [Google Scholar]
- 42. Shao K, Shan S, Ru W, Ma C.. Association between serum NPTX2 and cognitive function in patients with vascular dementia. Brain Behav. 2020;10(10):e01779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Petzold A. Neurofilament phosphoforms: Surrogate markers for axonal injury, degeneration and loss. J Neurol Sci. 2005;233(1-2):183–198. [DOI] [PubMed] [Google Scholar]
- 44. Petzold A, Thompson EJ, Keir G, et al. . Longitudinal one-year study of levels and stoichiometry of neurofilament heavy and light chain concentrations in CSF in patients with multiple system atrophy. J Neurol Sci. 2009;279(1-2):76–79. [DOI] [PubMed] [Google Scholar]
- 45. Zucchi E, Lu CH, Cho Y, et al. . A motor neuron strategy to save time and energy in neurodegeneration: Adaptive protein stoichiometry. J Neurochem. 2018;146(5):631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kušnierová P, Zeman D, Hradílek P, Čábal M, Zapletalová O.. Neurofilament levels in patients with neurological diseases: A comparison of neurofilament light and heavy chain levels. J Clin Lab Anal. 2019;33(7):e22948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Benatar M, Zhang L, Wang L, et al. ; CReATe Consortium . Validation of serum neurofilaments as prognostic and potential pharmacodynamic biomarkers for ALS. Neurology. 2020;95(1):e59–e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lu CH, Petzold A, Topping J, et al. . Plasma neurofilament heavy chain levels and disease progression in amyotrophic lateral sclerosis: Insights from a longitudinal study. J Neurol Neurosurg Psychiatry. 2015;86(5):565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Adiutori R, Aarum J, Zubiri I, et al. . The proteome of neurofilament-containing protein aggregates in blood. Biochem Biophys Rep. 2018;14:168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lu CH, Kalmar B, Malaspina A, Greensmith L, Petzold A.. A method to solubilise protein aggregates for immunoassay quantification which overcomes the neurofilament "hook" effect. J Neurosci Methods. 2011;195(2):143–150. [DOI] [PubMed] [Google Scholar]
- 51. Petzold A, Keir G, Green AJ, Giovannoni G, Thompson EJ.. A specific ELISA for measuring neurofilament heavy chain phosphoforms. J Immunol Methods. 2003;278(1-2):179–190. [DOI] [PubMed] [Google Scholar]
- 52. Jiskoot LC, Dopper EG, Heijer T, et al. . Presymptomatic cognitive decline in familial frontotemporal dementia: A longitudinal study. Neurology. 2016;87(4):384–391. [DOI] [PubMed] [Google Scholar]
- 53. Jiskoot LC, Panman JL, van Asseldonk L, et al. . Longitudinal cognitive biomarkers predicting symptom onset in presymptomatic frontotemporal dementia. J Neurol. 2018;265(6):1381–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Devenney E, Hornberger M, Irish M, et al. . Frontotemporal dementia associated with the C9ORF72 mutation: A unique clinical profile. JAMA Neurol. 2014;71(3):331–339. [DOI] [PubMed] [Google Scholar]
- 55. Valente ES, Caramelli P, Gambogi LB, et al. . Phenocopy syndrome of behavioral variant frontotemporal dementia: A systematic review. Alzheimers Res Ther. 2019;11(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. van der Ende EL, Jackson JL, White A, Seelaar H, van Blitterswijk M, Van Swieten JC.. Unravelling the clinical spectrum and the role of repeat length in C9ORF72 repeat expansions. J Neurol Neurosurg Psychiatry. 2021;92(5):502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Young AL, Marinescu RV, Oxtoby NP, et al. ; Alzheimer’s Disease Neuroimaging Initiative (ADNI) . Uncovering the heterogeneity and temporal complexity of neurodegenerative diseases with Subtype and Stage Inference. Nat Commun. 2018;9(1):4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Scherling CS, Hall T, Berisha F, et al. . Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol. 2014;75(1):116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bertrand A, Wen J, Rinaldi D, et al. ; Predict to Prevent Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis (PREV-DEMALS) Study Group . Early cognitive, structural, and microstructural changes in presymptomatic C9orf72 carriers younger than 40 years. JAMA Neurol. 2018;75(2):236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Staffaroni AM, Goh SM, Cobigo Y, et al. ; ARTFL-LEFFTDS Longitudinal Frontotemporal Lobar Degeneration Consortium . Rates of brain atrophy across disease stages in familial frontotemporal dementia associated with MAPT, GRN, and C9orf72 pathogenic variants. JAMA Netw Open. 2020;3(10):e2022847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cash DM, Bocchetta M, Thomas DL, et al. ; Genetic FTD Initiative, GENFI . Patterns of gray matter atrophy in genetic frontotemporal dementia: Results from the GENFI study. Neurobiol Aging. 2018;62:191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jiskoot LC, Panman JL, Meeter LH, et al. . Longitudinal multimodal MRI as prognostic and diagnostic biomarker in presymptomatic familial frontotemporal dementia. Brain. 2019;142(1):193–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rojas JC, Wang P, Staffaroni AM, et al. ; on behalf of the ALLFTD and GENFI consortia. Plasma neurofilament light for prediction of disease progression in familial frontotemporal lobar degeneration. Neurology. 2021;96(18):e2296–e2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Oxtoby NP, Garbarino S, Firth NC, et al. ; Alzheimer’s Disease Neuroimaging Initiative . Data-driven sequence of changes to anatomical brain connectivity in sporadic Alzheimer's disease. Front Neurol. 2017;8:580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data of this project is part of GENFI and de-identified participant data can be accessed on reasonable request to h.seelaar@erasmusmc.nl and genfi@ucl.ac.uk.