

Abstract
Rhodococcus erythropolis I-19, containing multiple copies of key dsz genes, was used to desulfurize alkylated dibenzothiophenes (Cx-DBTs) found in a hydrodesulfurized middle-distillate petroleum (MD 1850). Initial desulfurization rates of dibenzothiophene (DBT) and MD 1850 by I-19 were 5.0 and 2.5 μmol g dry cell weight−1 min−1, more than 25-fold higher than that for wild-type bacteria. According to sulfur K-edge X-ray absorption near-edge structure (XANES) analysis, thiophenic compounds accounted for >95% of the total sulfur found in MD 1850, predominantly Cx-DBTs and alkylated benzothiophenes. Extensive biodesulfurization resulted in a 67% reduction of total sulfur from 1,850 to 615 ppm S. XANES analysis of the 615-ppm material gave a sulfur distribution of 75% thiophenes, 11% sulfides, 2% sulfoxides, and 12% sulfones. I-19 preferentially desulfurized DBT and C1-DBTs, followed by the more highly alkylated Cx-DBTs. Shifting zero- to first-order (first-order) desulfurization rate kinetics were observed when MD 1850 was diluted with hexadecane. Apparent saturation rate constant (K0) and half-saturation rate constant (K1) values were calculated to be 2.8 μmol g dry cell weight−1 min−1 and 130 ppm, respectively. However, partial biocatalytic reduction of MD 1850 sulfur concentration followed by determination of initial rates with fresh biocatalyst led to a sigmoidal kinetic behavior. A competitive-substrate model suggested that the apparent K1 values for each group of Cx-DBTs increased with increasing alkylation. Overall desulfurization rate kinetics with I-19 were affected by the concentration and distribution of Cx-DBTs according to the number and/or lengths of alkyl groups attached to the basic ring structure.
Nitrogen and sulfur oxides are a subset of greenhouse gases receiving attention throughout the world with the overall agreement that reductions are required to protect the environment. Government regulations are demanding that the sulfur content of petroleum products for use in motor vehicles needs to be further reduced over the next decade, requiring refiners to increase desulfurization capacity. Depending on the sulfur content of any given crude oil supply, the sulfur concentration of the middle-distillate fraction used to make diesel fuel can range widely from <500 to >5,000 ppm (17). Current U.S. specifications for diesel fuel mandate that the sulfur concentration be less than 500 ppm, with 50 ppm anticipated for the year 2005 (17). Sulfur concentrations in crude oil supplies are increasing, resulting in upward pressure on sulfur concentrations in finished petroleum products. To achieve lower sulfur concentrations, refiners need to operate their hydrodesulfurization (HDS) units at higher temperatures and/or lower space velocities, requiring new capital investment and/or higher operating costs (14, 16). Of the different classes of sulfur compounds found in the middle-distillate fraction, alkylated benzothiophenes (Cx-BTs) and alkylated dibenzothiophenes (Cx-DBTs) are more resistant to HDS treatment than mercaptans and sulfides, with alkyl substitutions in positions 4 and 6 on the DBT ring being the most resistant (1, 7, 13). Increasing the severity of HDS also elicits undesirable effects on fuel quality as other chemical components are reduced at the higher temperatures and pressures needed to achieve low sulfur levels.
Biodesulfurization offers the potential for a more selective and cost-effective method for lowering the sulfur content of petroleum products. DBT has been used as a model polyaromatic sulfur heterocycle for the isolation and characterization of bacteria capable of transforming organosulfur compounds found in a variety of fossil fuels. There are two primary pathways for DBT desulfurization, one in which the initial attack is directed against one of the carbon atoms (the Kodama pathway) and one in which initial catalysis is directed against the sulfur center (the 4S pathway) (8, 9). Enzymatic attack at a carbon atom, typical of many aromatic hydrocarbon degradative pathways, is undesirable for a process designed to selectively remove organic sulfur compounds without oxidation of other aromatics found in petroleum products. Several bacterial species which are capable of either biotransforming DBT or growing with it as a sole sulfur source have been identified including Arthrobacter, Brevibacterium, Pseudomonas, Gordona, and Rhodococcus spp. (8, 10, 11, 15, 21–23). Consequently, development of biocatalytic desulfurization for the selective removal of polyaromatic sulfur heterocycles from petroleum products has focused on the 4S pathway.
The DBT desulfurization (dsz) operon from Rhodococcus erythropolis IGTS8, which encodes three proteins, DszC, DszA, and DszB, has been isolated, cloned, mutated, and overexpressed (2, 3, 12, 19, 20). DBT is stepwise S-oxidized by DszC, first to DBT-5-oxide (DBTO) and then to DBT-5,5′-oxide (DBTO2). DszA catalyzes the transformation of DBTO2 to 2-(2′-hydroxyphenyl)benzene sulfinate (HPBS), which opens the thiophenic ring. HPBS is then desulfinated by DszB to produce 2-hydroxybiphenyl (HBP) (Fig. 1). Each of the key Dsz pathway enzymes has been purified and characterized (4). The first two catabolic steps require oxygen, reduced flavin, and an NADPH-FMN oxidoreductase for activity (4, 19, 24). To enhance biocatalytic performance, multiple copies of dszC, dszA, and dszD were cloned back into R. erythropolis to increase enzyme production (12).
FIG. 1.

Proposed Dsz (4S) pathway for the biodesulfurization of DBT by R. erythropolis IGTS8 (4). DBT is shown with the standard numbering system. DBTO, DBT-5-oxide; DBTO2, DBT-5,5′-dioxide; HPBS, 2-(2′-hydroxyphenyl)-benzene sulfinate; HBP, 2-hydroxybiphenyl; DBT-MO, DBT mono-oxygenase; DBTO2-MO, DBTO2 mono-oxygenase (4); FMN, flavin mononucleotide.
The Dsz system (4S pathway) has been shown to transform a variety of thiophenic compounds both in synthetic mixtures and from petroleum fractions (18, 22). Recently, reductions in sulfur levels of around 30% were reported for a straight-run middle distillate derived from Oregon Basin crude oil with Rhodococcus sp. strain ECRD-1 (5). Of the 70% remaining sulfur, about 50% was transformed to oxidized forms, which remained in the oil. Although the substrate range of this organism was described in rather broad terms, little detail regarding the specific types of sulfur compounds involved and the relative rates at which they are biotransformed was given.
In this paper, we provide additional detail regarding the rate, range, and extent of microbial transformation of Cx-DBTs found in a hydrodesulfurized middle distillate. A strain of R. erythropolis with overexpression of the key desulfurization pathway enzymes was used to describe the complex kinetic behavior of this system and to provide a mechanistic basis for the observations.
MATERIALS AND METHODS
Chemicals.
A representative middle distillate was obtained from Total Raffinage S. A. following distillation and HDS to produce an oil with 1,850 mg of S kg−1 (MD 1850). DBT (Aldrich Chemical) and hexadecane (Spectrum Chemical) had >99% purity. All other chemicals were of reagent grade or better.
Microorganism, media, and culture conditions.
A genetically modified strain of R. erythropolis IGTS8, I-19 (12), was grown in either a 15- or 300-liter fermentor by using a defined basal salts medium with glucose as the carbon source and dimethylsulfoxide as the sulfur source (12). Once the culture reached an optical density at 600 nm (OD600) of 30 to 50, cells were harvested by centrifugation at 17,000 × g and stored at 4°C. Culture densities were determined by diluting the suspension with distilled water and measuring the OD in a Beckman DU 650 spectrophotometer with a 1-cm-path-length cuvette. The cell content of the harvested paste ranged from 20 to 30% solids (grams dry cell weight [DCW] per gram wet cell weight [WCW]). DCW, WCW, and percent solids were determined by placing a sample of paste on a tarred glass fiber pad, weighing the pad (WCW), and then drying it to constant weight (DCW) in a CEM 9000 microwave oven with balance (CEM Corp.). A calibration curve was generated for correlating OD to DCW.
Desulfurization rate determination.
Unless otherwise indicated, the standard protocol for desulfurization experiments was to first suspend cell paste in 150 mM phosphate buffer, pH 7.5, with 2% glucose at a concentration of 16.7 gDCW liter−1. Next, 750 ml of the suspension was transferred to a 2-liter stirred vessel (Applikon Inc.) equipped with temperature control, agitation control, pH control, and dissolved oxygen (DO) monitoring and then allowed to mix for about 60 min at 30°C. Air flow was set to 400 ml min−1 to maintain DO levels above 40% of air saturation. The pH was controlled through the automatic addition of 10% NaOH in water, as needed. To initiate desulfurization, 250 ml of oil was added and the contents were mixed at 1,000 rpm. The volumetric water-to-oil ratio (WOR) was 3:1 under these conditions. Samples were withdrawn at defined time intervals and analyzed for total sulfur content or subjected to gas chromatographic (GC) analysis for quantification of levels of individual chemical components. Withdrawn samples were centrifuged at 15,000 × g for 5 min to separate the biocatalyst from the aqueous and oil phases.
Quantification of sulfur concentration.
Between 2 and 10 ml of oil was filtered through a 0.22-μm-pore-size Millex-GP filter (Millipore, Bedford, Mass.) to remove any residual cellular particles. X-ray fluorescence (SLFA-1800H sulfur analyzer; Horiba) was used for determination of total sulfur concentration, which had a linear response from 100 to 4,000 mg of S kg−1 (ppm by weight). A calibration curve was generated by diluting a certified standard of di-n-butyl sulfide (Analytical Services, The Woodlands, Tex.) in synthetic diesel oil. Quality assurance samples were run before the first, and after the last, sample analyzed, with concentration determined by comparison to the standard curve.
The distribution of organosulfur compounds was determined with an HP 5890 II plus GC (Hewlett-Packard) equipped with a sulfur chemiluminescence detector (SCD) (Sievers 355A). For a qualitative view of the total hydrocarbon distribution and changes in total sulfur distribution, a 1-μl filtered oil sample was injected onto an 0.53-mm-interior-diameter, 15-m column with a 1.5-μm film DB1 column (J&W) operated at 50.7-cm s−1 linear velocity of helium carrier gas. The injector and detector temperatures were set to 275 and 300°C, respectively. Chromatography was accomplished over 12 min by using an oven temperature program which started at 120°C and then was ramped to 300°C at 20°C min−1 and held for 3 min. This method was not used to quantify individual components, though relative retention times have been determined.
For quantification of groups of Cx-DBTs an HP 5890 II plus GC with a 5972 mass spectrometer (MS) (Hewlett-Packard) operated in selective ion monitoring (SIM) mode was used. A 1-μl filtered oil sample was injected onto an 0.25-mm-interior-diameter, 30-m column with a 0.5-μm film RTX-1 column (Restek) operated at 50.7-cm s−1 linear velocity of helium carrier gas. The injector and detector temperatures were set to 290 and 320°C, respectively. Chromatography was accomplished over 75 min by using an oven temperature program which started at 100°C and then was ramped to 315°C at 4°C min−1 and held for 20 min. Concentrations of groups of Cx-DBTs were determined from calibration curves. While an authentic standard of DBT was used to calibrate for DBT, authentic 2-methyl DBT was used to calibrate for all C1-DBTs, and authentic 4,6-dimethyl DBT was used to calibrate for all C2-DBTs. Since no primary standards for C3- to C5-DBTs were available, the response factor for 4,6-dimethyl DBT was used to quantify these species. In each case, the calibration range was from 2.4 to 48 ppm.
RESULTS
Characterization of the middle-distillate petroleum stock.
The middle-distillate test material was collected from a refinery pilot plant following partial HDS, which lowered the total sulfur content of the straight-run middle-distillate feed stock to 1,850 ppm total sulfur. A variety of chemical and physical parameters for this material were determined (Table 1). X-ray absorption near-edge structure (XANES) analysis reported >95% of the organosulfur compounds as thiophenic. Thiophenic compounds include thiophene, benzothiophene (BT), DBT, and benzonaphthothiophene. Each of these basic aromatic structures can have a variety of alkyl substituents, thereby increasing the number of unique compounds. The sample was also subjected to GC analysis by using both SCD and MS detectors (GC-SCD and GC-MS, respectively) to compare the distributions of organosulfur compounds (Fig. 2, top curve). GC-MS was used to identify the main groups of Cx-DBT and Cx-BT compounds. A general trend of longer GC retention time with increasing degree of alkylation was observed; this was consistent with an increasing boiling point as the degree of alkylation increases. There were some sulfur-containing compounds eluting before DBT which had retention times consistent with Cx-BTs. Essentially all of the organosulfur compounds found in MD 1850 were thiophenic, with most eluting after DBT.
TABLE 1.
Characterization of middle-distillate test materials
| Test material | Boiling point (°C)
|
Density (g ml−1) | S concn (ppm)e | Amt (%) of:
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Initial | Final | Paraffin | Isoparaffin | Olefin | Naphthene | Aromatics | Sulfidic sulfur | Thiophenic sulfur | Sulfoxide | Sulfone | |||
| MD 1850 | 149 | 428 | 0.845 | 1,860 | 18.5b | 19.4b | 2.0b | 22.8b | 37.3b | <5c | >95c | NDc | NDc |
| DRMa | 615 | 11cd | 75cd | 2cd | 12cd | ||||||||
Extensively biodesulfurized MD 1850 with 615-ppm S remaining.
Hydrocarbon speciation reported as percentage of total volume by PIONA analysis (Core Laboratories, Inc., Houston, Tex.).
XANES analysis was performed at beam line X-19A of the National Synchrotron Light Source (Brookhaven National Laboratory). Estimated uncertainty in aliphatic and aromatic sulfide forms is ±5% and in oxidized forms is ±3%. ND, not detected.
DRM was chemically oxidized with a twofold molar excess of 30% (by weight) peroxyacetic acid (Aldrich) per g of oil with vigorous mixing at 22°C for 16 h. Following the reaction, the excess peroxide was removed with platinum on carbon until no peroxide was detected. The oxidized sulfur products were isolated from the oil by solid-phase extraction with a 5-g silica column. The column was first rinsed with pentane, and the oxidized sulfur products were then eluted with methylene chloride.
Total sulfur in oil was analyzed with an SLFA-1800H analyzer (Horiba).
FIG. 2.

GC profile for the time-dependent change in sulfur concentration and composition. Reaction conditions were 12.5 gDCW liter−1 I-19, 2% glucose, 3:1 WOR, 30°C, 150 mM phosphate, and pH 7.5. Each plot is for an oil sample removed from the reactor and analyzed by GC-SCD 0, 1, 3, and 6 h after initiation of the reaction. All response factors were plotted with the same scale for both x and y axes.
Time-dependent change in sulfur concentration and composition.
The rate and extent of desulfurization were determined for R. erythropolis I-19, which contained multiple copies of dszC, dszA, and dszD genes leading to overexpression of the corresponding pathway enzymes. The initial rate of DBT biotransformation was determined to be 5.0 μmol gDCW−1 min−1 with DBT (1,040 ppm S) diluted in hexadecane. The initial rate of MD 1850 desulfurization was determined to be 2.5 μmol gDCW−1 min−1, based on the change in total sulfur content in the oil phase. No significant change in sulfur concentration was observed when either DBT-hexadecane or MD 1850 was brought into contact with either cell-free buffer, killed-cell controls, or live-cell controls without active Dsz enzymes. I-19 demonstrated a higher rate of desulfurization for DBT in hexadecane than for the mixture of Cx-DBTs found in MD 1850.
The rate of MD 1850 desulfurization decreased with time over the 24-h reaction period monitored. The distribution of sulfur compounds also changed with time and the extent of desulfurization (Fig. 2). At 0, 1, 3, and 6 h, the sulfur concentrations were 1,850, 1,620, 1,314, and 949 ppm, respectively. The first 230-ppm drop in total sulfur observed after 1 h corresponded primarily to a biotransformation of DBT and mid-boiling-range sulfur compounds. Between 1 and 3 h, another 300-ppm sulfur reduction occurred, with some evidence for more highly alkylated DBTs being affected. At 3 h, most of the DBT and much of the C1-DBTs were gone. Between 3 and 6 h, desulfurization shifted to the higher-boiling-range sulfur compounds, resulting in an additional 365-ppm drop in total sulfur. The Dsz system appeared to selectively attack different groups of Cx-DBTs, leading to a change in reaction rate over time as preferred substrates were consumed.
Two procedures were used to determine the extent of biodesulfurization. First, MD 1850 was brought into contact with cells at 12.5 gDCW liter−1 and a 3:1 WOR for 24 h. Following each treatment, the oil was recovered and brought into contact with fresh cells for a total of five cycles. Second, MD 1850 was treated in one stage for 24 h with cells at 30 gDCW liter−1 and a 9:1 WOR. Both procedures produced an oil with 615 ppm sulfur which could not be further reduced following contact with fresh biocatalyst. XANES analysis of the MD 1850 biodesulfurized to 615 ppm sulfur showed the majority of the remaining sulfur to be thiophenes (75%), with 11% sulfides, 2% sulfoxides, and 12% sulfones (Table 1). GC-MS analysis of the polar material derived from the chemical oxidation of the remaining sulfur gave mass spectra consistent with a mixture of Cx-BTs and Cx-DBTs with five or more alkyl units. About 33% of the sulfur compounds found in MD 1850 were resistant to biotransformation by the Dsz system (Dsz-recalcitrant material [DRM]). These data demonstrated that the Dsz system actively desulfurized a wide range of alkylated organosulfur compounds found in middle distillate. Furthermore, these data demonstrated that reactivities for all organosulfur compounds were not equivalent.
Differential rates of desulfurization of Cx-DBTs.
To further characterize the substrate selectivity of the Dsz system, GC-MS analysis was performed. Cx-DBTs can be grouped based on the number of additional carbon residues. C1-DBTs represent four different isomers with methyl residues at either the 1, 2, 3, or 4 carbon position on the DBT ring. C2-DBTs represent dimethylated and ethylated isomers with substitution on either one or both aromatic rings. While GC-MS was not used to monitor each specific isomer, it was employed for the quantitative analysis of groups of Cx-DBTs differentiated by the level of alkyl substitution. Using SIM for the parent ions of these species, initial desulfurization rate profiles were generated for six classes of Cx-DBTs in MD 1850 (Fig. 3). DBT was the lowest-concentration compound. C2-DBTs were the highest-concentration group. However, the concentration of any individual isomer, except DBT, was not determined. The data demonstrated that DBT, C1-DBT, and C2-DBT all reacted with no apparent lag. C3-, C4-, and C5-DBTs all demonstrated an acceleration in rate over time coincident with the disappearance of the less-alkylated DBTs. Since primary standards were not available for many of the Cx-DBTs, their quantification was based on the response factors for DBT, 2-methyl DBT, and 4,6-dimethyl DBT. With this assumption, initial rates of desulfurization were calculated for each group of Cx-DBTs (Table 2). C2-DBT demonstrated the highest rate, consistent with this group having the greatest concentration. C5-DBT demonstrated the lowest rate, even though the concentration for this group was greater than that for DBT, which reacted 10 times faster. The summed reaction rates were equivalent to the rate determined by tracking total sulfur loss within experimental error. Clearly, the Cx-DBT degradation rate kinetics were dependent on both concentration and degree of alkylation. The trend showed a decrease in reaction rate with increasing alkylation.
FIG. 3.

Desulfurization of Cx-DBTs as tracked by GC-MS. Reaction conditions were 12.5 gDCW liter−1 I-19, 2% glucose, 3:1 WOR, 30°C, 15 mM phosphate, and pH 7.5. A competitive-substrate model (equation 3) was used to fit the data and plot the curve for each Cx-DBT (K0 and K1 values are listed in Table 2). Time-dependent changes in sulfur concentrations [S] for DBT (○), C1-DBT (□), C2-DBT (●), C3-DBT (▵), C4-DBT (▴), and C5-DBT (■) are shown.
TABLE 2.
Competitive-substrate model parameter estimates
| Compound | Concn (ppm) | Rate (μmol gDCW−1 min−1) | K1 (ppm) |
|---|---|---|---|
| DBT | 32 | 0.081 | 100 |
| C1-DBT | 150 | 0.381 | 100 |
| C2-DBT | 293 | 0.69 | 105 |
| C3-DBT | 230 | 0.306 | 182 |
| C4-DBT | 130 | 0.077 | 425 |
| C5-DBT | 50 | 0.008 | 1,538 |
| CR-DBTa | 379 | NAb | 2,000 |
| DRM | 615 | 0 | ∞ |
CR-DBT, C6- and higher Cx-DBTs.
NA, not applicable.
Desulfurization rate kinetic estimates.
MD 1850 was diluted with hexadecane to produce oil with intermediate levels of total sulfur (Fig. 4). The concentration of reactive sulfur compounds was plotted against initial reaction rate. The unreactive-sulfur compound component (615 ppm S in 100% MD 1850) was subtracted based on the dilution with hexadecane which contained no unreactive sulfur compounds. Desulfurization followed a shifting zero- to first-order (first-order) kinetic model (see equation 1) with an apparent half-saturation rate constant, K1, of 130 ppm and an apparent saturation rate constant, K0, of 2.8 μmol gDCW−1 min−1. This rate-versus-substrate concentration profile was similar to that determined for DBT dissolved in hexadecane, which also demonstrated a first-order kinetic profile (data not shown). All of the DBT can be transformed by the Dsz system under these conditions, with final sulfur concentrations below detection limits.
FIG. 4.

Concentration-dependent desulfurization rate profiles. Initial rates of desulfurization were determined from three independent shake flasks incubated for four different times for a total of 12 points per rate determination. Shake flasks were prepared with 40-ml total volumes, and reaction conditions were either 3 or 6 g DCW liter−1 I-19, 2% glucose, a 3:1 WOR, 30°C, 150 mM phosphate, and pH 7.5. The initial rate was calculated with a second-order polynomial (R2 > 0.98 for all of the data) and plotted as a function of initial concentration of degradable sulfur [S − DRM] for MD 1850 in hexadecane (●), partially desulfurized MD 1850 (■), and MD 1850 mixed with DRM (□). A first-order kinetic model (equation 1) (top curve) was used to fit the MD 1850 dilution data (K0 = 2.8 μmol gDCW−1 min−1; K1 = 130 ppm; 615-ppm DRM). A second-order kinetic model (equation 2) (middle curve) was used to empirically fit the partially desulfurized MD 1850 data (K0 = 2.8 μmol gDCW−1 min−1; K2 = 211,985 ppm2; 615-ppm DRM). A competitive-substrate model (equation 3) (bottom curve; K0 and K1 values as listed in Table 2; 615-ppm DRM) was also used to fit the partially desulfurized MD 1850 data. MD 1850 mixed with DRM (□) was not used for any of the fitted curves.
Desulfurization of MD 1850 was then determined by producing oil with various levels of sulfur following biodesulfurization. The oil was treated for different times with various biocatalyst concentrations to generate sulfur concentrations between those of stock oil and DRM. The oil was then recovered by centrifugation and filtered. Initial desulfurization rates were then determined for each pool of oil by using the same batch of fresh cells (Fig. 4). Desulfurization of this partially desulfurized middle distillate did not follow a simple first-order kinetic equation but could be fitted to an empirical, shifting zero- to second-order (second-order) model (see equation 2), with apparent kinetic constants of 2.8 μmol gDCW−1 min−1 and of 211,985 ppm2 for K0 and K2, respectively. This observation was consistent with the data presented in Fig. 2 and 3, which clearly demonstrated a shift in sulfur species distribution with the extent of desulfurization.
One additional rate was determined in this experiment. MD 1850 was mixed with oil which had been extensively desulfurized to lower the total sulfur concentration but not to shift the relative concentrations of the different biodegradable sulfur species. This mixture demonstrated a significantly greater rate than that of the corresponding partially desulfurized material at the same level of degradable sulfur but one that was less than that obtained by diluting MD 1850 with hexadecane (Fig. 4). Both total sulfur concentration and the distribution of Cx-DBTs contributed to changes in the overall reaction rate kinetics for the Dsz system.
DISCUSSION
A hydrodesulfurized middle-distillate petroleum used for producing diesel fuel was obtained from Total Raffinage S. A. (1,850 ppm S; 149 to 428°C boiling range petroleum fraction). The sulfur compounds eliminated by HDS treatment included most of the mercaptan and sulfidic components, leaving primarily thiophenic sulfur compounds as determined by XANES analysis (Table 1). Of the alkylated thiophenic moieties, Cx-DBTs constituted the majority of those identifiable by GC-MS, though there are Cx-BTs and alkyl-benzonaphthothiophenes present at lower concentrations. Deeper HDS treatment results in the desulfurization of Cx-BTs and less-alkylated Cx-DBTs (data not shown). As such, each group of Cx-DBTs is composed of several unique compounds. For example, C1-DBTs are a group of monomethyl compounds with four different positions of substitution. The C2-DBTs include both dimethyl- (methylation of one or both rings) and ethyl-substituted compounds. As alkylation increases, the number of unique structures also increases. Though DBT has been used extensively as a model compound for biodesulfurization research, it does not fully represent the range of thiophenic compounds found in petroleum.
The rate of DBT desulfurization by R. erythropolis I-19 was determined to be 5.0 μmol gDCW−1 min−1. Previously, rates from 0.1 to 0.2 μmol gDCW−1 min−1 were reported for wild-type R. erythropolis IGTS8 conversion of DBT to HBP (6, 19). Another desulfurizing bacterium, Gordona sp. strain CYKS1, was reported to transform DBT at a rate of 0.15 μmol gDCW−1 min−1 (22). Overexpression of key Dsz enzymes in R. erythropolis I-19 increased specific desulfurization rates by at least 25-fold over those for the naturally occurring isolates. The rate of DBT desulfurization in a model hexadecane system was greater than that observed for biotransformation of Cx-DBT from a middle-distillate stock. The lower rate of desulfurization in middle distillate can be partially attributed to the mixture of Cx-DBTs, which appear to have lower reactivities than the unalkylated parent structure, DBT.
Extensive biodesulfurization of MD 1850 led to a 67% reduction in total sulfur from 1,850 to 615 ppm. Comparable sulfur reductions have been reported previously for desulfurization of diesel oils by a Gordona species employing the 4S pathway: 70% for a middle-distillate unit feed and 50% for light gas oil (22). Rhodococcus sp. strain ECRD-1 reduced the sulfur content of a straight-run middle distillate 30% and oxidized another 35% to oil-soluble products (5). The remaining sulfur compounds, DRM, were either not substrates for the Dsz system or reacted so slowly as to not be observed under the conditions employed. XANES analysis of the DRM from MD 1850 shows the majority of the remaining sulfur to be thiophenes (75%), with 11% sulfides, 2% sulfoxides, and 12% sulfones. GC-MS analysis of the polar material derived from the chemical oxidation of the remaining sulfur gave mass spectra consistent with a mixture of Cx-BTs and Cx-DBTs with five or more alkyl units. This distribution of sulfur compounds remaining following biodesulfurization was significantly different from that reported for a straight-run middle distillate, which contained upwards of 50% oxidized sulfur compounds (5). This difference is likely due to the differences between the straight-run distillate fraction reported previously and the hydrodesulfurized material used in this study. Clearly, the Dsz system of I-19 is capable of transforming a wide range of alkylated thiophenic compounds found in petroleum distillates.
The Dsz system selectively and sequentially transformed groups of Cx-DBTs. Though DBT is a good model compound, it does not adequately represent all Cx-DBTs found in middle distillate. DBT and C1-DBT appear to be attacked preferentially, followed by the more highly alkylated DBTs. GC-MS data demonstrate that Cx-DBTs, up to at least C5-DBTs, are substrates for the Dsz system. The carbons adjacent to the sulfur center were reported to be sterically hindered, leading to lower reactivities for an Arthrobacter sp. employing a 4S pathway (11). In vivo desulfurization of 2,8-DBT and 4,6-DBT demonstrated 120 and 60%, respectively, of the rate determined for DBT by using R. erythropolis H-2 employing a 4S pathway (18). Both of these compounds are C2-DBTs and would be included in the overall rate of disappearance as quantified by the GC-MS methods used. Not only does the position of alkylation affect reactivity but also, in general, increasing alkylation decreases reactivity.
Desulfurization rate kinetics were determined by adjusting total sulfur concentration and Cx-DBT distribution independently and at the same time. First, dilution with hexadecane (i) changed the total sulfur concentration, (ii) did not shift the sulfur species distribution, (iii) decreased the DRM concentration, and (iv) shifted the composition of the bulk hydrocarbon matrix. Dilution of MD 1850 with hexadecane fitted a typical first-order kinetic, with a K1 of 130 ppm and an apparent K0 of 2.8 μmol gDCW−1 min−1 (see equation 1). Second, MD 1850 was partially biodesulfurized, generating sulfur concentrations intermediate between those for the stock oil and DRM. The oil was recovered and filtered, and then initial desulfurization rates were determined with fresh cells. Partial biodesulfurization (i) changed the total sulfur concentration, (ii) shifted the sulfur species distribution, (iii) did not change DRM concentration, and (iv) did not change the composition of the bulk hydrocarbon matrix. This rate change profile fit a second-order model with apparent kinetic constants of 2.8 μmol gDCW−1 min−1 and 211,985 ppm2 (K0 and K2, respectively; equation 2). Finally, dilution of MD 1850 with DRM (i) changed the total sulfur concentration, (ii) did not shift the sulfur species distribution, (iii) did not change the DRM concentration, and (iv) did not change the composition of the bulk hydrocarbon matrix. Desulfurization rate kinetics for the complex mixture of Cx-DBT compounds found in middle distillate were affected by both the concentration and distribution of Cx-DBTs.
Shifting order kinetics from zero to first order (Michaelis-Menten) is expressed by
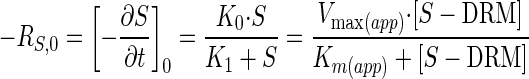 |
1 |
where RS,0 is the initial specific reaction rate for the substrate (S), [∂S/∂t]0 is the differential change in substrate concentration divided by the differential change in time (t), Vmax(app) is the Michaelis-Menten apparent saturation rate constant, Km(app) is the Michaelis-Menten apparent half saturation rate constant, and S − DRM is the total sulfur concentration (S) minus the Dsz-recalcitrant sulfur concentration (DRM).
Shifting order kinetics from zero to second order is expressed by
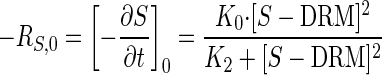 |
2 |
Use of empirical kinetic models limits our ability to estimate reaction rates for oil feed stocks with variable compositions and/or concentrations. From a theoretical standpoint, it is reasonable to assume that rates of biodesulfurization of each of the sulfur species would follow apparent first-order kinetics. In this case, the kinetic parameters K0 and K1 for each of the sulfur species would be different. A predictive model was developed based upon this assumption, breaking the sulfur species in MD 1850 into eight categories: DBT, C1-DBTs, C2-DBTs, C3-DBTs, C4-DBTs, C5-DBTs, CR-DBTs (C6- and higher Cx-DBTs), and DRM. This approach, though more complex than assuming that all Cx-DBTs are the same, is still a simplification. The GC-MS SIM method identified groups and did not attempt to distinguish individual isomers. As mentioned above, as alkylation increases, there is an increasingly complex mixture of methyl, ethyl, or longer-chain substitutions in multiple positions on one or both rings. As noted, within the C2-DBT group, 2,8-DBT and 4,6-DBT demonstrated different reactivities when rates were determined on single substrates (11). As a first approximation, it was further assumed that the K0 for each of these groups is the same but that the K1 values differ. The practical meaning of this is that the Dsz system has differential affinity for each of the species but that once the substrate is bound to the enzymes within the cell, the rate of conversion is the same. Using GC-MS data from batch biodesulfurization tests with MD 1850, parameters for each of the sulfur species were approximated. These results are presented in Table 2.
The competitive-substrate model is expressed by
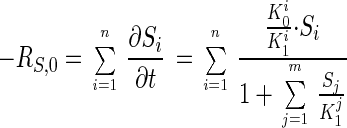 |
(3)
The competitive-substrate model closely predicts the observed change in concentration for each of the classes of Cx-DBTs as a function of time with the kinetic constants in Table 2. Figure 4 shows the difference in the predicted rates of desulfurization as a function of sulfur concentration by using an empirical second-order model (equation 2) and a mechanistically based competitive-substrate model (equation 3). These data support the conclusion that the more highly alkylated DBTs are less reactive and have higher apparent K1 values than DBT. The resulting estimates for K1 for each group of Cx-DBTs are consistent with expected changes in oil-water partitioning. The data support the view that the lower Cx-DBTs have lower K1 values and are preferentially transformed first. As this group of substrates is consumed, the biocatalyst shifts to transformation of the more highly alkylated DBTs with higher K1 values, resulting in an apparent second-order kinetic behavior. This second-order kinetic behavior appears to be consistent with a mechanistically based competitive-substrate model. Further generalization of this model would require validating the assumption that each group of Cx-DBTs has the same K0. The same type of experiment would be required to expand the database and fine-tune the kinetic constants for more than one stock oil.
ACKNOWLEDGMENTS
This work was supported by funds provided by Energy Biosystems Corp.
We thank Mark Bauer for growing and supplying the bacteria used for this work.
REFERENCES
- 1.Altgelt K H, Boduszynski M M. Composition and analysis of heavy petroleum fractions. New York, N.Y: Marcel Dekker, Inc.; 1994. pp. 417–420. [Google Scholar]
- 2.Denome S A, Olson E S, Young K D. Identification and cloning of genes involved in specific desulfurization of dibenzothiophene by Rhodococcus sp. strain IGTS8. Appl Environ Microbiol. 1993;59:2837–2843. doi: 10.1128/aem.59.9.2837-2843.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Denome S A, Oldfield C, Nash L J, Young K D. Characterization of the desulfurizing genes from Rhodococcus sp. strain IGTS8. J Bacteriol. 1994;176:6707–6716. doi: 10.1128/jb.176.21.6707-6716.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gray K A, Pogrebinsky O S, Mrachko G T, Xi L, Monticello D J, Squires C H. Molecular mechanisms of biocatalytic desulfurization of fossil fuels. Nat Biotechnol. 1996;14:1705–1709. doi: 10.1038/nbt1296-1705. [DOI] [PubMed] [Google Scholar]
- 5.Grossman M J, Lee M K, Prince R C, Garrett K K, George G N, Pickering I J. Microbiol desulfurization of a crude oil middle-distillate fraction: analysis of the extent of sulfur removal and the effect of removal on remaining sulfur. Appl Environ Microbiol. 1999;65:181–188. doi: 10.1128/aem.65.1.181-188.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Honda H, Sugiyama H, Saito I, Kobayashi T. High cell density culture of Rhodococcus rhodochrous by pH-stat feeding and dibenzothiophene degradation. J Ferment Bioeng. 1998;85:334–338. [Google Scholar]
- 7.Kabe T, Ishihara A, Tajima H. Hydrodesulfurization of sulfur-containing polyaromatic compounds in light oil. Ind Eng Chem Res. 1992;31:1577–1580. [Google Scholar]
- 8.Kilbane J J, Bielaga B A. Genetic study of biodesulfurization. In: Yunker S, Rhee K, editors. Proceedings of the 1990 First International Symposium on the Biological Processing of Coal. Palo Alto, Calif: Electric Power Research Institute, Inc.; 1990. pp. 215–232. [Google Scholar]
- 9.Kodama K, Umehara K, Shimizu K, Nakatani S, Minoda Y, Yamada K. Identification of microbial products from dibenzothiophene and its proposed oxidation pathway. Agric Biol Chem. 1973;37:45–50. [Google Scholar]
- 10.Kropp K G, Andersson J T, Fedorak P M. Bacterial transformation of 1,2,3,4-tetrahydrodibenzothiophene and dibenzothiophene. Appl Environ Microbiol. 1997;63:3032–3042. doi: 10.1128/aem.63.8.3032-3042.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee M K, Senius J D, Grossman M J. Sulfur-specific microbial desulfurization of sterically hindered analogs of dibenzothiophene. Appl Environ Microbiol. 1995;61:4362–4366. doi: 10.1128/aem.61.12.4362-4366.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li M Z, Squires C H, Monticello D J, Childs J D. Genetic analysis of the dsz promoter and associated regulatory regions of Rhodococcus erythropolis IGTS8. J Bacteriol. 1996;178:6409–6418. doi: 10.1128/jb.178.22.6409-6418.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma X, Sakanishi K, Mochita I. Hydrodesulfurization reactivities of various sulfur compounds in diesel fuel. Ind Eng Chem Res. 1994;33:218–222. [Google Scholar]
- 14.McColloch M D, Edgar M D. Higher severity diesel hydrotreating. Presented at the annual meeting of the National Petroleum Refiners Association. 1997. , San Antonio, Tex., 29 to 31 March 1987. [Google Scholar]
- 15.Monticello D J, Baker D T, Finnerty W R. Plasmid-mediated degradation of dibenzothiophene by Pseudomonas species. Appl Environ Microbiol. 1985;49:756–760. doi: 10.1128/aem.49.4.756-760.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monticello D J, Finnerty W R. Microbial desulfurization of fossil fuels. Annu Rev Microbiol. 1985;39:371–389. doi: 10.1146/annurev.mi.39.100185.002103. [DOI] [PubMed] [Google Scholar]
- 17.Monticello D J. Riding the fossil fuel biodesulfurization wave. Chemtech. 1998;28:38–45. [Google Scholar]
- 18.Ohshiro T, Hirata T, Izumi Y. Desulfurization of dibenzothiophene derivatives by whole cells of Rhodococcus erythropolis H-2. FEMS Microbiol Lett. 1996;142:65–70. [Google Scholar]
- 19.Oldfield C, Pogrebinsky O, Simmonds J, Olson E S, Kulpa C F. Elucidation of the metabolic pathway for dibenzothiophene desulfurization by Rhodococcus sp. strain IGTS8 (ATCC 53968) Microbiology. 1997;143:2961–2973. doi: 10.1099/00221287-143-9-2961. [DOI] [PubMed] [Google Scholar]
- 20.Piddington C S, Kovacevich B R, Rambosek J. Sequence and molecular characterization of a DNA region encoding the dibenzothiophene desulfurization operon of Rhodococcus sp. strain IGTS8. Appl Environ Microbiol. 1995;61:468–475. doi: 10.1128/aem.61.2.468-475.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Resnick S M, Gibson D T. Regio- and stereospecific oxidation of fluorene, dibenzofuran, and dibenzothiophene by naphthalene dioxygenase from Pseudonomas sp. strain NCIB 9816-4. Appl Environ Microbiol. 1996;62:4073–4080. doi: 10.1128/aem.62.11.4073-4080.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rhee S K, Chang J H, Chang Y K, Chang H N. Desulfurization of dibenzothiophene and diesel oils by a newly isolated Gordona strain, CYKS1. Appl Environ Microbiol. 1998;64:2327–2331. doi: 10.1128/aem.64.6.2327-2331.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Afferten M, Schacht S, Klein J, Trüper H G. Degradation of dibenzothiophene by Brevibacterium sp. DO. Arch Microbiol. 1990;153:324–328. [Google Scholar]
- 24.Xi L, Squires C H, Monticello D J, Childs J D. A flavin reductase stimulates DszA and DszC proteins of Rhodococcus erythropolis IGTS8 in vitro. Biochem Biophys Res Commun. 1997;230:73–78. doi: 10.1006/bbrc.1996.5878. [DOI] [PubMed] [Google Scholar]