

Introduction
The mismatches between RNA-Seq reads and reference genome come from many sources. Two major biological sources are SNPs (replication errors) and RNA editing events. These two mutation sources are generally distinguishable based on their own unique features. Several bioinformatic tools are invented to faithfully identify RNA editing sites (Porath et al. 2014; Zhang et al. 2017; Zhang and Xiao 2015) and the pipelines of which are different from (and even beyond) the traditional SNP-calling pipeline. Here, we would discuss that a previous work by Dr. Conticello’s group (Di Giorgio et al. 2020) utilized the SNP-calling pipeline to identify RNA editing in SARS-CoV-2, leading to many false-positive sites as we and other scientists revealed (Picardi et al. 2021; Song et al. 2022; Zong et al. 2022). Without deep thinking on how to distinguish RNA editing and SNPs, one should not automatically regard all the A>G mismatches (SNVs) as A-to-I RNA editing sites. We emphasize that this “take-for-granted” logic needs to be reconsidered.
Traditional SNV identification pipeline for SNP calling
The identification of single-nucleotide variant (SNV) is a basic bioinformatic skill in nearly all the genome-wide mutation analyses and comparative genomics studies (Jiang et al. 2021; Jiang et al. 2022; Li et al. 2021). For example, in the 1000-genome project (Kuehn 2008), the ultimate goal of the SNP (single-nucleotide polymorphism)-calling step is to find out the difference between the reference genome and the genome of a given individual. The mapping of DNA-resequencing data to the reference genome is required before SNP calling. We will not discuss the tricks in the mapping step and only focus on the motivation of the downstream SN calling. Although different software was developed for SNP calling (Li et al. 2009; McKenna et al. 2010), the conception is almost identical, intuitive, and simple, which is called the “pile-up” algorithm. On each genomic position, it counts the number of reads supporting the reference genome versus the number of reads supporting an alternative allele. With a few additional necessary steps of quality control like considering the mapping quality and potential sequencing errors, the SNVs in the DNA-resequencing data should be reliable SNPs in the tested samples.
Traditional RNA editing detection when DNA resequencing is available
The detection of RNA editing sites requires RNA-sequencing data. Obviously, an SNV between the RNA and the reference genome is not an evidence of RNA editing. In most cases, the SNV between RNA and reference genome reflects a SNP in the individual. To exclude the SNPs, one needs to look for the real RNA-DNA difference (RDD). Accordingly, REDItools_DnaRNA.py was developed (Picardi and Pesole 2013). When both RNA-sequencing and DNA-resequencing data are available, the software directly compares the differences between the RNA and DNA of the same individual (Fig. 1). By this way, the detected RDDs should be real RNA editing events. As a software, REDItools is user-friendly and widely accepted by the RNA editing community (Liscovitch-Brauer et al. 2017). In animals, where A-to-I RNA editing is prevalent, one could always find a dominant peak at A-to-G among all SNVs after applying the RDD method (Alon et al. 2015; Li et al. 2014), which is a strong evidence for genuine A-to-I editing.
Fig. 1.
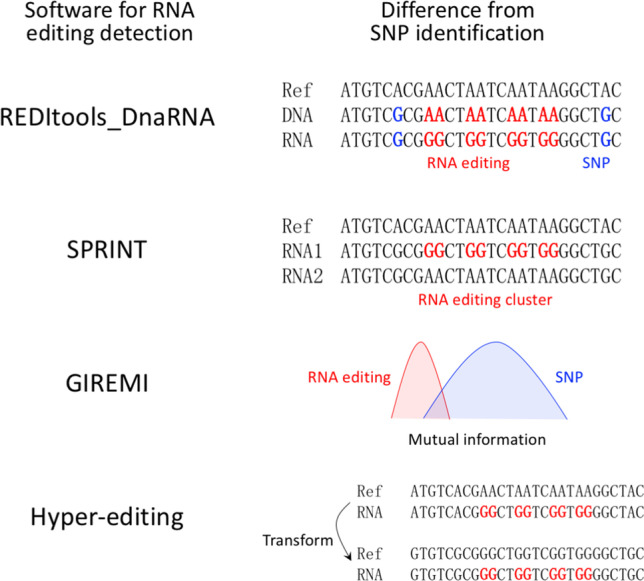
The bioinformatic software for RNA editing detection and how they differ from the traditional SNP-calling pipeline
Traditional RNA editing detection when DNA resequencing is absent
The RDD method (Picardi and Pesole 2013) is the golden standard for RNA editing detection. However, the DNA-resequencing data are not always available. Without the matched DNA sequence of a sample, one could hardly determine whether the SNV between RNA and reference genome is SNP or RNA editing (Li et al. 2020b; Li et al. 2020d). Nevertheless, researchers have managed to find out multiple approaches to distinguish RNA editing events and SNPs by using RNA-sequencing data alone (Porath et al. 2014; Zhang et al. 2017; Zhang and Xiao 2015).
Software SPRINT (Zhang et al. 2017) hold an idea based on the interaction between editing enzyme ADAR and the target RNAs. Nearby adenosine sites are usually targeted by ADAR at the same time so that the A-to-I editing sites should appear in clusters. In contrast, SNPs in the genomes are largely randomly distributed. This is how SPRINT distinguish RNA editing sites from SNPs (Fig. 1); an additional clustering is performed after the traditional “pile-up” step.
Software GIREMI (Zhang and Xiao 2015) considers that SNPs should be tightly linked in the RNA-sequencing data, while RNA editing events do not have such strong linkage. Using a mutual information methodology (Fig. 1), GIREMI successfully distinguishes RNA editing events from SNPs by using RNA-sequencing data alone. It is also essentially different from the traditional SNV detection pipeline where an additional mutual information analysis is performed.
The hyper-editing method (Porath et al. 2014) concerns a situation where an RNA read contains multiple RNA editing events. Under this circumstance, the hyper-edited reads could not be mapped to the reference genome. Therefore, an A>G transformation is done for both RNA reads and reference sequence to retrieve the heavily edited RNAs (Fig. 1). SNPs would not show such clusters within a sequencing read so that this methodology identifies RNA editing events without the need of DNA-resequencing data. Again, an additional transformation step is required beyond the simple “pile-up” strategy.
In a word: RNA editing detection has additional steps beyond SNP-calling pipeline
Apart from the basic quality controls like requiring mapping quality and removing some potential sequencing errors, those aforementioned highly acknowledged RNA editing detection pipelines have additional steps compared to the SNP-calling pipeline. The additional steps are strictly necessary especially when DNA-resequencing data is absent (Porath et al. 2014; Zhang et al. 2017; Zhang and Xiao 2015). The purpose is to remove the SNPs and “enrich” the RNA editing sites.
SARS-CoV-2 is an RNA virus. Conceptually, it does not have DNA resequencing at all. Therefore, the RDD method (Picardi and Pesole 2013) is not applicable. To reliably identify real RNA editing sites in the viral transcriptome, one has to utilize at least one of the “RNA-alone” methods (Porath et al. 2014; Zhang et al. 2017; Zhang and Xiao 2015) beyond the traditional SNP-calling pipeline. This idea agrees with the methodology of some recent studies (Picardi et al. 2021; Song et al. 2022) who successfully enriched the A-to-G variations from a mixture of SNVs in SARS-CoV-2 transcriptome.
Di Giorgio et al. used the SNP-calling pipeline to identify RNA editing sites
In sharp contrast, the Di Giorgio et al. (2020) paper merely used the traditional SNP-calling pipeline to identify the A-to-I RNA editing sites in SARS-CoV-2 transcriptome. They indeed performed some seemingly professional filters on, e.g., base quality (Q), mapping quality (q), and reads trimming (Di Giorgio et al. 2020). However, as we have explained, these basic procedures still belong to the SNP-calling pipeline because if you want to identify the SNPs in SARS-CoV-2, you will still use exactly the same pipeline. What makes RNA editing different from SNPs? Their original article title is “Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2,” and then, one could envision a paradox that they could write a new paper entitled “Evidence for replication error-induced SNPs in the transcriptome of SARS-CoV-2” by using exactly the same pipeline.
Our point here is that SNPs and RNA editing sites have many features in common, but they also have some differences. In order to distinguish the two sets of sites, one needs to know their differences instead of using the common pipeline and then “automatically” regard the results as RNA editing sites. The purpose of the additional steps like clustering (Zhang et al. 2017), mutual information (Zhang and Xiao 2015), or transformation (Porath et al. 2014) is to remove the SNPs and enrich the RNA editing sites. Without those additional steps, one could not “take it for granted” to regard all the A-to-G SNVs as A-to-I RNA editing sites because no enrichment on A-to-G is observed.
Here, we must emphasize that although their logic remains questionable, we do respect Dr. Conticello and colleagues for the works done in many other fields (Saraconi et al. 2014; Severi et al. 2011; Severi and Conticello 2015). Our criticism is impersonal. We only claim that the (Di Giorgio et al. 2020) SARS-CoV-2 paper was imperfect in logic (as it is so conspicuous that this logic does not meet the criteria of an RNA editing literature). As we will explain in the following sections, tolerating this paper would jeopardize the golden standard of the RNA editing community. This is the reason why we should keep trying our best to correct this fallacy. However, imperfection of a single paper does not reflect the unreliability of their other works. We try to provide the community with an objective judgement on this SARS-CoV-2 RNA editing paper.
How to define enrichment and false-positive rate?
Without observing the enrichment of a particular SNV type, one could not automatically regard A>G sites as A-to-I editing events (Wei 2022). Question comes that how to define “enrichment”? Even if the SNV between RNA versus reference genome is the typical symmetric distribution (Fig. 2A), one could still argue (claim) that “there must be RNA editing sites contained in these SNVs” (Di Giorgio et al. 2020; Martignano et al. 2022). Of course, this statement is absolutely true and undeniable as the total SNVs are “necessary but insufficient” for RNA editing sites. However, let us imagine that one may also potentially show the nucleotide composition of the whole reference genome and claim that “there must be RNA editing sites contained in these sites.” This logic obviously violates the motivation of RNA editing detection. Conceptually, this is a matter of false-positive rate. Only when the fraction of true positive A-to-G sites is sufficiently high (Fig. 2B) could one be certain that “most of the A-to-G SNVs are RNA editing sites.” In a classic A-to-I study in animals (Li et al. 2014), > 96% of the SNVs were A-to-G, suggesting that the false-positive rate was less than 4%. One may define false-positive rate = fraction of non-A>G sites.
Fig. 2.

Schematic diagrams. A The SNP profile which is symmetric. B The RNA editing profile which shows a striking peak at A-to-G
In contrast, in the Di Giorgio et al. study (Di Giorgio et al. 2020), even if they regard both A>G and T>C SNVs as A-to-I RNA editing sites (Martignano et al. 2022), the total fraction of (A>G + T>C) is not high enough to be confident that most of them are A-to-I RNA editing sites (Di Giorgio et al. 2020). Let alone the “A>G + T>C” assumption is not widely accepted. We will discuss the “A>G + T>C” issue in the following section.
Treating both A>G and T>C as A-to-I editing sites is not widely accepted
Here, we will show that treating both A>G and T>C SNVs as A-to-I RNA editing sites is not a widely accepted logic. For example, the Picardi et al. (2021) paper used strict pipeline and successfully reached a high A>G peak (but not two peaks at A>G and T>C). Picardi et al. did not include the T>C sites at all. If one wants to regard T>C as A-to-I editing, it must come from the antisense editing during RNA replication by RDRP. As we have previously explained (Zong et al. 2022), (1) this intermolecular dsRNA status is transient, and (2) RDRP may prevent ADAR binding due to steric effect. How could one expect so many antisense editing events (T>C) come from this transient status? We believe that RNA editing needs time to take place. If the T>C sites were as abundant as the A>G sites (Di Giorgio et al. 2020), does this mean that the whole life cycle of SARS-CoV-2 RNA belong to the replication state (intermolecular dsRNA state)? When replication is done, positive ssRNA is released (for translation or other biological processes) so that the positive ssRNA itself could be targeted by ADARs (by forming intramolecular hairpin structures). Importantly, this process on positive ssRNA only produces A>G sites but not T>C sites. Therefore, the final A>G sites must outnumber T>C sites (if one admits that SARS-CoV-2 is not always in the replication state). Thus, to prove reliable RNA editing events in SARS-CoV-2, the symmetric distribution shown in Di Giorgio et al. (2020) paper is not as convincing as the asymmetric plot shown by Picardi et al. (2021).
In fact, in a well-acknowledged study which performed systematic in vivo/vitro measurement of SARS-CoV-2 RNA structure (Sun et al. 2021), researchers only mentioned intramolecular dsRNA and did not consider the intermolecular dsRNA at all, suggesting that the transient replication status is really inconsequential if one intends to study the dsRNA structure. In most conditions during SARS-CoV-2 life cycle, the dsRNA should be formed by a single positive strand RNA molecule so that ADARs only produce A>G variations in the RNA sequencing reads.
Moreover, during RNA replication, the RDRP first binds the 3′ of positive strand RNA and starts to synthesize the antisense strand RNA. This means that the movement of RDRP is from 3′ to 5′ (we refer to the coordinates labeled on positive strand). As a consequence, the 3′ part of positive strand RNA has “more time” to be in the “dsRNA state.” Therefore, antisense editing (which requires the existence of both strands) has higher chance to take place at the 3′ region of the reference sequence.
Another critical issue is the layout of the RNA-sequencing library. Strand-specific RNA-sequencing reads capture all variations taken place in RNAs, while for non-strand-specific RNA reads, an A>G (editing event) on positive strand would also produce a T>C variation on antisense strand (due to the experimental procedure of library construction). Then, the antisense editing could not be confirmed by non-strand-specific libraries at all. This concern remains unanswered in Martignano et al. (2022). Altogether, we propose that without deep thinking on these detailed biological or technical issues, automatically regarding T>C variations as antisense editing is questionable.
A-to-I plus C-to-U editing is not an explanation for the SNP-like profile
The original Di Giorgio et al. (2020) paper provided a SNP-like profile, where the A>G (even T>C was added as they argued) was not dominant and contained many potential false-positive sites. Then they (Martignano et al. 2022) argued that they intended to regard A>G + T>C as A-to-I editing and C>T + G>A as C-to-U editing. The “A-to-I editing plus C-to-U editing” would produce the profile similar to the SNP profile.
Unfortunately, many following researchers have misunderstood this point. For example, some following papers (Picardi et al. 2021; Song et al. 2022) thought (Di Giorgio et al. 2020) was highlighting A-to-I editing, while some other papers (Rice et al. 2021; van Dorp et al. 2020) thought (misunderstood) that the result of Di Giorgio et al. (2020) was “considered to be consistent with APOBEC editing.” Actually, Di Giorgio et al. (2020) intended to highlight both editing types (if we understood correctly). This controversy already sheds doubt on the interpretation and logic of the Di Giorgio et al. (2020) paper. Besides, a recent paper has shown that the occurrence of C-to-U editing (leading to C>T mismatches) is dominant among all 12 variation types in SARS-CoV-2 as shown by both fixed and polymorphic mutations (Liu et al. 2022). This result again contradicts with the profile shown by Di Giorgio et al. (2020) where C-to-U editing is no more than A-to-I editing in many samples.
We propose that the existence of both A-to-I and C-to-U editing is not a reason for displaying an SNP-like profile (Fig. 2A) as Di Giorgio et al. (2020) did. Frankly speaking, if this logic is allowed, then the 30-year RNA editing field would be “overturned.” Please do not take our intention badly. Let us objectively retrospect the scientific progresses in the past years; bioinformatic researchers spent so many efforts to remove the false-positive sites and try to elevate the A>G percentage just to prove that the A>G SNVs are reliable A-to-I editing sites (Adetula et al. 2021; Alon et al. 2015; Huang et al. 2021; Levanon et al. 2004; Li et al. 2014; Porath et al. 2014; Ramaswami et al. 2013). However, now there comes a paper (Di Giorgio et al. 2020) that, for the first time in history, shows a symmetric profile and claims that these SNVs consists of A-to-I editing plus C-to-U editing. Then, why should others work so hard to elevate the A>G percentage? Even if one does not make any efforts to filter the SNVs, one would still get a symmetric SNV profile so one could claim that these SNVs contain both A-to-I and C-to-U editing sites. The golden standard of the RNA editing community would be infringed if the symmetric SNV profile is allowed to be an evidence for RNA editing (Di Giorgio et al. 2020).
What if one intends to find out both A-to-I and C-to-U editing sites from a single dataset?
Note that we have never denied the existence of any types of RNA editing. Indeed, it is true that both A-to-I and C-to-U RNA editing exist. As far as we know, identification of both types of RNA editing at the same time (within the same dataset) has not been performed until the Di Giorgio et al. (2020) paper. Let us propose a set of reasonable criteria if one intends to find out both A-to-I and C-to-U sites simultaneously (Fig. 3):
Step no. 1, use a pipeline (denoted as pipeline 1) to obtain a sharp peak at A>G (> 80%) in the SNV profile to prove that these A>G sites are reliable A-to-I editing sites (Picardi et al. 2021). Or alternative, as one argued (Martignano et al. 2022), one may obtain two peaks at both A>G and T>C and let the sum of A>G + T>C be higher than 80% (Fig. 3). Note that 80% is not an extravagant cutoff since many RNA editing studies were required (or obligated) to achieve that.
Step no. 2, use a different pipeline (denoted as pipeline 2) to obtain a sharp peak at C>T (> 80%) in the SNV profile to prove that these C>T sites are reliable C-to-U editing sites (Chu and Wei 2019, 2020; Li et al. 2020a; Li et al. 2020c). Or alternative, as one argued (Martignano et al. 2022), one may obtain two peaks at both C>T and G>A and let the sum of C>T + G>A be higher than 80% (Fig. 3).
Step no. 3, combine the A-to-I and C-to-U editing sites (Fig. 3) and claim that “We found both ADAR and APOBEC target sites. Although it resembles SNP profile, it clearly comes from two origins.”
Fig. 3.

If one intends to identify A-to-I and C-to-U editing sites from the same dataset, then one needs to follow the stringent pipelines. Theoretically, one should only regard A > G sites as A-to-I editing. However, this golden standard might be “compromised” if one argues that there is antisense editing on viral RNAs. The same logic goes for C-to-U editing
Although the final results of steps 1 to 3 look similar to the pure symmetric SNP profile (Di Giorgio et al. 2020), the logic chains of the two strategies are essentially different. If one omits steps 1 and 2 and directly obtains the SNP profile (symmetric distribution) in step 3, common readers would naturally suspect that those SNVs are actually SNPs. As we have discussed, the symmetric distribution could be easily obtained even without any efforts or filters. The pertinent way is to separately identify A-to-I and C-to-U sites and then combine them. Again, we think this idea is intuitive. Indeed, successfully finding the two sets of editing sites (within the same sample) might not be realistic in some cases. It is possible that the sequence context (motif) around editing sites might help identify both sets of sites. However, in most cases, A-to-I editing sites would better be identified in normal versus ADAR-KO samples, while C-to-U editing sites would better be identified in normal versus APOBEC-KO samples. Simultaneously, finding two editing types within the same sample is highly challenging. As far as we know, only the Di Giorgio et al. (2020) paper has intended to do so. Common papers only try to identify one of the two types of RNA editing sites.
Several literatures citing the Di Giorgio et al. paper questioned its conclusions
Again, we fully respect Dr. Conticello for the works done in many other fields (Saraconi et al. 2014; Severi et al. 2011; Severi and Conticello 2015). However, we have to say that the logic presented in the Di Giorgio et al. (2020) paper was not the way that many RNA editing papers do. In their response to our concerns (Martignano et al. 2022), they tried to provide a bunch of literatures that have cited their original paper (Di Giorgio et al. 2020) in order to prove that their work was “highly acknowledged.”
Unfortunately, just like the famous case where many literatures (Kleinman and Majewski 2012; Lin et al. 2012; Pickrell et al. 2012) cited the Li-MY et al. paper (Li et al. 2011) to disprove its results, this time, many literatures citing the Di Giorgio et al. (2020) paper were also trying to question their original interpretation. For the literatures mentioned in Conticello’s commentary (Martignano et al. 2022) which they thought were supporting their original conclusion (Di Giorgio et al. 2020), we found that (1) at least two papers (Picardi et al. 2021; Song et al. 2022) have disproved the conclusion of Di Giorgio et al. (2020); (2) at least two papers (Rice et al. 2021; van Dorp et al. 2020) have misunderstood what Di Giorgio et al. (2020) were trying to highlight (abundant A-to-I events or abundant C-to-U events); and (3) some papers (Popa et al. 2020) showed excessive C-to-U editing events instead of A-to-I editing, which contradicts with the main intention of Di Giorgio et al. (2020) paper. These facts indicate that the flaws we found in the Di Giorgio et al. (2020) paper were not caused by our own misunderstanding. Instead, our concerns are highly reasonable and have been independently found by other scientists. Since the interpretation of the variants in SARS-CoV-2 directly determines our understanding of virus evolution and is connected to the control of the pandemic (Yu et al. 2021; Zhang et al. 2022; Zhang et al. 2021), one should be cautious when coming to a conclusion.
Summary points
In summary, our points are as follows:
The detection of RNA editing sites in the SARS-CoV-2 transcriptome should require additional bioinformatic steps compared to the traditional SNP-calling pipeline (in order to enrich the RNA editing sites) (Porath et al. 2014; Zhang et al. 2017; Zhang and Xiao 2015).
The seemingly professional filters performed by Di Giorgio et al. (2020) essentially belong to the traditional SNP-calling pipeline. The lack of A>G enrichment in their mutation profile suggests many false-positive sites, which is not strong evidence for A-to-I editing (Liu et al. 2022; Picardi et al. 2021; Song et al. 2022; Wei 2022).
Regarding T>C sites as antisense editing by ADAR is still questionable. Thus, a sharp peak at A>G is still needed in order to prove a reliable set of A-to-I editing sites (Picardi et al. 2021).
The claim of A-to-I plus C-to-U editing is not a reason for producing a SNP-like profile. The pertinent way is to separately identify A-to-I and C-to-U sites and then combine them.
Literatures focusing on A-to-I editing (Picardi et al. 2021; Song et al. 2022) are citing the Di Giorgio et al. (2020) paper because they thought Di Giorgio et al. (2020) were highlighting A-to-I editing. Literatures focusing on C-to-U editing (Rice et al. 2021; van Dorp et al. 2020) are also citing the Di Giorgio et al. (2020) paper because they thought Di Giorgio et al. (2020) were highlighting C-to-U editing. This dilemma might indicate the ambiguous interpretation and unrecognized logic of the original paper (Di Giorgio et al. 2020).
Finally, we re-emphasize that we fully respect Dr. Conticello for the works done in many other fields. We only think that the Di Giorgio et al. (2020) paper lacks sufficient skills to justify their paper’s title “Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2.” When many false-positive sites could not be excluded, the evidence was very weak.
Acknowledgements
We thank all the colleagues that have given suggestions to this project.
Author contribution
All authors approved the submission and publication of this manuscript.
Data Availability
This is not a research article and does not contain original data and materials.
Declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Adetula AA, Fan X, Zhang Y, Yao Y, Yan J, Chen M, Tang Y, Liu Y, Yi G, Li K, et al. Landscape of tissue-specific RNA editome provides insight into co-regulated and altered gene expression in pigs (Sus-scrofa) RNA Biol. 2021;18:439–450. doi: 10.1080/15476286.2021.1954380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon S, Garrett SC, Levanon EY, Olson S, Graveley BR, Rosenthal JJ, Eisenberg E (2015) The majority of transcripts in the squid nervous system are extensively recoded by A-to-I RNA editing. Elife:4 [DOI] [PMC free article] [PubMed]
- Chu D, Wei L. The chloroplast and mitochondrial C-to-U RNA editing in Arabidopsis thaliana shows signals of adaptation. Plant Direct. 2019;3:e00169. doi: 10.1002/pld3.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu D, Wei L. Systematic analysis reveals cis and trans determinants affecting C-to-U RNA editing in Arabidopsis thaliana. BMC Genet. 2020;21:98. doi: 10.1186/s12863-020-00907-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio S, Martignano F, Torcia MG, Mattiuz G, Conticello SG. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci Adv. 2020;6:eabb5813. doi: 10.1126/sciadv.abb5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Lin L, Dong Z, Yang L, Zheng T, Gu W, Zhang Y, Yin T, Sjostedt E, Mulder J, et al. A porcine brain-wide RNA editing landscape. Commun Biol. 2021;4:717. doi: 10.1038/s42003-021-02238-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Cao X, Wang H. Mutation profiling of a limbless pig reveals genome-wide regulation of RNA processing related to bone development. J Appl Genet. 2021;62:643–653. doi: 10.1007/s13353-021-00653-0. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Ge F, Sun G, Wang H (2022) An evolutionarily conserved mechanism that amplifies the effect of deleterious mutations in osteosarcoma. Mol Genet Genomics. [DOI] [PubMed]
- Kleinman CL, Majewski J. Comment on "Widespread RNA and DNA sequence differences in the human transcriptome". Science. 2012;335:1302. doi: 10.1126/science.1209658. [DOI] [PubMed] [Google Scholar]
- Kuehn BM. 1000 genomes project promises closer look at variation in human genome. JAMA. 2008;300:2715. doi: 10.1001/jama.2008.823. [DOI] [PubMed] [Google Scholar]
- Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol. 2004;22:1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Proc GPD. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Wang IX, Li Y, Bruzel A, Richards AL, Toung JM, Cheung VG. Widespread RNA and DNA sequence differences in the human transcriptome. Sci. 2011;333:53–58. doi: 10.1126/science.1207018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Wang Z, Lian J, Schiott M, Jin L, Zhang P, Zhang Y, Nygaard S, Peng Z, Zhou Y, et al. Caste-specific RNA editomes in the leaf-cutting ant Acromyrmex echinatior. Nat Commun. 2014;5:4943. doi: 10.1038/ncomms5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Yang X, Wang N, Wang H, Yin B, Yang X, Jiang W. Mutation profile of over 4500 SARS-CoV-2 isolations reveals prevalent cytosine-to-uridine deamination on viral RNAs. Future Microbiol. 2020;15:1343–1352. doi: 10.2217/fmb-2020-0149. [DOI] [PubMed] [Google Scholar]
- Li Y, Yang X, Wang N, Wang H, Yin B, Yang X, Jiang W. SNPs or RNA modifications? Concerns on mutation-based evolutionary studies of SARS-CoV-2. PLoS ONE. 2020;15:e0238490. doi: 10.1371/journal.pone.0238490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Yang XN, Wang N, Wang HY, Yin B, Yang XP, Jiang WQ. The divergence between SARS-CoV-2 and RaTG13 might be overestimated due to the extensive RNA modification. Future Virol. 2020;15:341–347. doi: 10.2217/fvl-2020-0066. [DOI] [Google Scholar]
- Li Y, Yang XN, Wang N, Wang HY, Yin B, Yang XP, Jiang WQ. Pros and cons of the application of evolutionary theories to the evolution of SARS-CoV-2. Future Virol. 2020;15:369–372. doi: 10.2217/fvl-2020-0048. [DOI] [Google Scholar]
- Li Q, Li J, Yu CP, Chang S, Xie LL, Wang S. Synonymous mutations that regulate translation speed might play a non-negligible role in liver cancer development. BMC Cancer. 2021;21:388. doi: 10.1186/s12885-021-08131-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Piskol R, Tan MH, Li JB. Comment on "Widespread RNA and DNA sequence differences in the human transcriptome". Sci. 2012;335:1302. doi: 10.1126/science.1210624. [DOI] [PubMed] [Google Scholar]
- Liscovitch-Brauer N, Alon S, Porath HT, Elstein B, Unger R, Ziv T, Admon A, Levanon EY, Rosenthal JJC, Eisenberg E. Trade-off between transcriptome plasticity and genome evolution in cephalopods. Cell. 2017;169(191-202):e111. doi: 10.1016/j.cell.2017.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Liu X, Zhou J, Dong Y, Jiang W, Jiang W (2022) Rampant C-to-U deamination accounts for the intrinsically high mutation rate in SARS-CoV-2 spike gene. RNA. [DOI] [PMC free article] [PubMed]
- Martignano F, Di Giorgio S, Mattiuz G, Conticello SG (2022) Commentary on "Poor evidence for host-dependent regular RNA editing in the transcriptome of SARS-CoV-2". J Appl Genet. [DOI] [PMC free article] [PubMed]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardi E, Pesole G. REDItools: high-throughput RNA editing detection made easy. Bioinformatics. 2013;29:1813–1814. doi: 10.1093/bioinformatics/btt287. [DOI] [PubMed] [Google Scholar]
- Picardi E, Mansi L, Pesole G (2021) Detection of A-to-I RNA editing in SARS-COV-2. Genes (Basel) 13 [DOI] [PMC free article] [PubMed]
- Pickrell JK, Gilad Y, Pritchard JK. Comment on "Widespread RNA and DNA sequence differences in the human transcriptome". Sci. 2012;335:1302. doi: 10.1126/science.1210484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa A, Genger JW, Nicholson MD, Penz T, Schmid D, Aberle SW, Agerer B, Lercher A, Endler L, Colaco H et al (2020) Genomic epidemiology of superspreading events in Austria reveals mutational dynamics and transmission properties of SARS-CoV-2. Sci Transl Med 12 [DOI] [PMC free article] [PubMed]
- Porath HT, Carmi S, Levanon EY. A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat Commun. 2014;5:4726. doi: 10.1038/ncomms5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, O'Connell MA, Li JB. Identifying RNA editing sites using RNA sequencing data alone. Nat Methods. 2013;10:128–132. doi: 10.1038/nmeth.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice AM, Castillo Morales A, Ho AT, Mordstein C, Muhlhausen S, Watson S, Cano L, Young B, Kudla G, Hurst LD. Evidence for strong mutation bias toward, and selection against, U content in SARS-CoV-2: implications for vaccine design. Mol Biol Evol. 2021;38:67–83. doi: 10.1093/molbev/msaa188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraconi G, Severi F, Sala C, Mattiuz G, Conticello SG. The RNA editing enzyme APOBEC1 induces somatic mutations and a compatible mutational signature is present in esophageal adenocarcinomas. Genome Biol. 2014;15:417. doi: 10.1186/s13059-014-0417-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severi F, Conticello SG. Flow-cytometric visualization of C>U mRNA editing reveals the dynamics of the process in live cells. RNA Biol. 2015;12:389–397. doi: 10.1080/15476286.2015.1026033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severi F, Chicca A, Conticello SG. Analysis of reptilian APOBEC1 suggests that RNA editing may not be its ancestral function. Mol Biol Evol. 2011;28:1125–1129. doi: 10.1093/molbev/msq338. [DOI] [PubMed] [Google Scholar]
- Song Y, He X, Yang W, Wu Y, Cui J, Tang T, Zhang R. Virus-specific editing identification approach reveals the landscape of A-to-I editing and its impacts on SARS-CoV-2 characteristics and evolution. Nucleic Acids Res. 2022;50:2509–2521. doi: 10.1093/nar/gkac120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Li P, Ju X, Rao J, Huang W, Ren L, Zhang S, Xiong T, Xu K, Zhou X, et al. In vivo structural characterization of the SARS-CoV-2 RNA genome identifies host proteins vulnerable to repurposed drugs. Cell. 2021;184(1865-1883):e1820. doi: 10.1016/j.cell.2021.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dorp L, Richard D, Tan CCS, Shaw LP, Acman M, Balloux F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat Commun. 2020;11:5986. doi: 10.1038/s41467-020-19818-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L (2022) Reconciling the debate on deamination on viral RNA. J Appl Genet. [DOI] [PMC free article] [PubMed]
- Yu YY, Li Y, Dong Y, Wang XK, Li CX, Jiang WQ. Natural selection on synonymous mutations in SARS-CoV-2 and the impact on estimating divergence time. Future Virol. 2021;16:447–450. doi: 10.2217/fvl-2021-0078. [DOI] [Google Scholar]
- Zhang Q, Xiao X. Genome sequence-independent identification of RNA editing sites. Nat Methods. 2015;12:347–350. doi: 10.1038/nmeth.3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Lu Y, Yan S, Xing Q, Tian W. SPRINT: an SNP-free toolkit for identifying RNA editing sites. Bioinformatics. 2017;33:3538–3548. doi: 10.1093/bioinformatics/btx473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YP, Jiang W, Li Y, Jin XJ, Yang XP, Zhang PR, Jiang WQ, Yin B. Fast evolution of SARS-CoV-2 driven by deamination systems in hosts. Future Virol. 2021;16:587–590. doi: 10.2217/fvl-2021-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Jin X, Wang H, Miao Y, Yang X, Jiang W, Yin B. SARS-CoV-2 competes with host mRNAs for efficient translation by maintaining the mutations favorable for translation initiation. J Appl Genet. 2022;63:159–167. doi: 10.1007/s13353-021-00665-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong J, Zhang Y, Guo F, Wang C, Li H, Lin G, Jiang W, Song X, Zhang X, Huang F, et al. Poor evidence for host-dependent regular RNA editing in the transcriptome of SARS-CoV-2. J Appl Genet. 2022;63:413–421. doi: 10.1007/s13353-022-00687-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This is not a research article and does not contain original data and materials.