

Abstract
Histone variants and the associated post-translational modifications that govern the stemness of haematopoietic stem cells (HSCs) and differentiation thereof into progenitors (HSPCs) have not been well defined. H3.3 is a replication-independent H3 histone variant in mammalian systems that is enriched at both H3K4me3- and H3K27me3-marked bivalent genes as well as H3K9me3-marked endogenous retroviral repeats. Here we show that H3.3, but not its chaperone Hira, prevents premature HSC exhaustion and differentiation into granulocyte-macrophage progenitors. H3.3-null HSPCs display reduced expression of stemness and lineage-specific genes with a predominant gain of H3K27me3 marks at their promoter regions. Concomitantly, loss of H3.3 leads to a reduction of H3K9me3 marks at endogenous retroviral repeats, opening up binding sites for the interferon regulatory factor family of transcription factors, allowing the survival of rare, persisting H3.3-null HSCs. We propose a model whereby H3.3 maintains adult HSC stemness by safeguarding the delicate interplay between H3K27me3 and H3K9me3 marks, enforcing chromatin adaptability.
The capacity of haematopoietic stem cells (HSCs) to differentiate into specific lineages at steady state or during acute stress is dependent on cell-autonomous programming and dynamic epigenetic adaptation to microenvironmental stimuli1–4. Little is known about the role of histone variants on the maintenance of the post-translational-modification (PTM) landscape in HSCs and the pathological consequences when such programmes go awry5–9. H3.3 is a replacement histone and can be incorporated into nucleosomes in a replication-independent manner. As 95% of adult long-term HSCs (LT-HSCs) reside in a G0 quiescent stage10,11, histone replacement is important for sustaining homeostasis. In mouse embryonic stem cells, H3.3 is deposited at H3K4me3- and H3K27me3-marked bivalent genes by the histone chaperone Hira (with Ubn1 and Cabin1)8,9, and at H3K9me3-marked endogenous retroviral (ERV) repeats or pericentromeric and telomeric heterochromatin regions by Daxx–Atrx5,6,12. Haematopoietic lineage commitment involves de novo establishment of lineage-specific enhancers, whose repertoire expansion precedes transcriptional changes3. We thus hypothesize that H3.3 deposition maintains and balances the self-renewal and lineage-differentiation of adult HSCs by directing the histone PTM landscape and, at a highly pliable initial differentiation stage, enforcing chromatin adaptability. We identified the two H3.3-safeguarded heterochromatin marks H3K27me3 and H3K9me3, and define a role for H3.3 in preventing the spread of H3K27me3 marks. When such spreading does occur in H3.3-null HSCs, H3K9me3 derepressed ERV repeats serve as enhancers, initiating aberrant pro-malignant myeloid transcription and inflammation signalling. By employing genetic models in which H3.3 is selectively deleted in haematopoietic stem and progenitor cells (HSPCs), we show that H3.3 regulates both cellular attributes of stemness and diverse haematopoietic lineage differentiation by sustaining HSC chromatin adaptability via the delicate interplay between H3K27me3 and H3K9me3 marks.
H3.3A and H3.3B are redundant for steady-state haematopoiesis
In mice, the histone variant H3.3 is encoded by two genes, H3.3A and H3.3B. We mapped the expression of H3.3A and H3.3B in adult haematopoietic cells (Extended Data Fig. 1a). Mice carrying the conditional H3.3A floxed allele13 under the control of Rosa26creERT2+ (AKO mice) and mice carrying the H3.3B-deletion allele (BKO mice13; Extended Data Fig. 1b) did not manifest defects in cKit+Sca1+Lin− (LKS) HSPCs or lineage compositions, suggesting compensatory roles (Extended Data Fig. 1c–l). Two exceptions were the reduced percentage of B cells in the spleen of AKO mice and reduced CD8+ T cells in the spleen of BKO mice, indicating the requirement for H3.3A and H3.3B in maintaining B-cell and CD8+ T-cell function, respectively.
We next treated BKO mice with sublethal irradiation to trigger regenerative responses14. There were no differences between the survival rates of the BKO and control mice over a period of 4 weeks (Extended Data Fig. 1m,n), with minor changes observed in the white blood cell, red blood cell (RBC) and platelet counts. Thus, H3.3B might inhibit the cell-cycle progression of lineage-differentiated cells. Next, we performed chromatin immunoprecipitation (ChIP) with sequencing (ChIP–seq) for H3K27me3 and H3K27ac in BKO and control LKS cells at steady state. Despite genome-wide changes in H3K27me3 and H3K27ac enrichment (Extended Data Fig. 1o–r), there were minimal changes in the haematopoietic phenotypes, demonstrating redundant roles of H3.3A and H3.3B in the maintenance of HSPC homeostasis.
Reduced LT-HSCs and biased myelopoiesis in H3.3-KO mice
Total H3.3 (sum of H3.3A and H3.3B) and Hira/Daxx5,7,15,16 are expressed in LT-HSCs, B cells, monocytes and dendritic cells as well as CD4+ and CD8+ T cells, but to a lesser extent (Extended Data Fig. 2a). Hira-mediated H3.3 deposition facilitates H3K27me3 enrichment at developmental genes13,17,18. Thus, we analysed the haematopoietic phenotype in Rosa26creERT2+Hirafl/fl-knockout (Hira-KO) and control Hirafl/fl mice. The Rosa26creERT2+H3.3Afl/flH3.3B−/− double-KO (DKO) mice had reduced survival rates and numbers of LT-HSCs as well as megakaryocyte erythroid progenitor cells, and increased numbers of CD150−CD48+LKS multipotent progenitor (MPP) 3 cells19–21 (Extended Data Fig. 2b–f). However, there was no change in the number of HSPCs in the Hira-KO mice (Extended Data Fig. 2c–f). Expression of the Fc fragment of the IgG receptors IIIa and IIa (Fcgr3a/2a, also known as CD16/32), a marker for granulocyte-macrophage progenitors (GMPs), increased in the HSCs of DKO mice but not the Hira-KO mice (Extended Data Fig. 2g), demonstrating bias towards the myeloid lineage. Therefore, H3.3, but not its chaperone Hira, represses the myeloid differentiation of HSCs.
In DKO mice, there was increased myelopoiesis at the expense of lymphopoiesis. The long bones of DKO, but not Hira-KO, mice had a pale appearance (Extended Data Fig. 2h); the spleen of DKO, but not Hira-KO, mice displayed splenomegaly; the thymus of both DKO and Hira-KO mice was smaller than that of the controls. The DKO mice had reduced numbers of RBCs, whereas Hira-KO mice had reduced platelet counts (Extended Data Fig. 2i and Supplementary Table 1). The percentage increase of CD11b+Gr1+ cells and reduction of B220+ B cells, CD4+ T cells and CD8+ T cells was more pronounced in DKO mice than Hira-KO mice. This indicates a more robust role of H3.3, rather than Hira, in repressing excessive myelopoiesis (Extended Data Fig. 2j–n). The erythroblast clusters in the spleen were altered in the DKO mice but not the Hira-KO mice (Extended Data Fig. 2o–q)22,23. Altogether, there was a loss of terminally differentiated erythroid lineage (cluster V) in DKO, but not Hira-KO, mice.
Histological examinations confirmed increased myelopoiesis, a reduction of the splenic white-pulp area, intra-hepatic haematopoietic infiltrations and reduced thymic medulla in DKO mice (Extended Data Fig. 3a–e). Leukocyte infiltrations were observed in the glomeruli of the DKO kidneys but not lungs (Extended Data Fig. 3f). The bone marrow (BM) of BKO mice demonstrated a balanced distribution of lymphocytes, RBCs and myeloid cells, whereas the BM of DKO mice had reduced numbers of RBCs and lymphocytes.
H3.3 is required for the survival and repopulation of HSCs
To assess the cell-intrinsic function of H3.3 in vivo, we performed a non-competitive transplantation experiment (Fig. 1a and Extended Data Fig. 4a), in which we deleted the H3.3A gene 4 weeks after the transplantation of BM mononuclear cells (BMMNCs). Several of the mice receiving DKO cell transplants died, whereas no deaths were recorded in the mice that received AKO cells (Extended Data Fig. 4b). The recipients of DKO transplants manifested splenomegaly and reduced thymus size (Fig. 1b,c) as well as an increase in the percentage of LKS cells in the lineage-negative (LinN) cell population, whereas the total numbers of LinN, cKit+Lin− (LKS) and LKS cells were reduced (Fig. 1d). In the DKO-cell recipient mice, an increased percentage of CD16/32+ LKS cells was observed, with a total reduction in the number of GMP cells (Fig. 1e and Extended Data Fig. 4c) and a trend towards a reduction in LT-HSCs (Fig. 1f). Compared with the mice that received AKO cells, the BM of the mice in the DKO-cell transplant group demonstrated hypocellularity with increased adipocytes (Extended Data Fig. 4h). Only the CD16/32+ LKS cells remained unchanged in the recipients of DKO cells, suggesting protection from apoptosis for such cell populations. Thus, H3.3 maintains adult homeostatic haematopoiesis by preventing cell death.
Fig. 1 |. H3.3 maintains adult haematopoiesis and HSC repopulation.
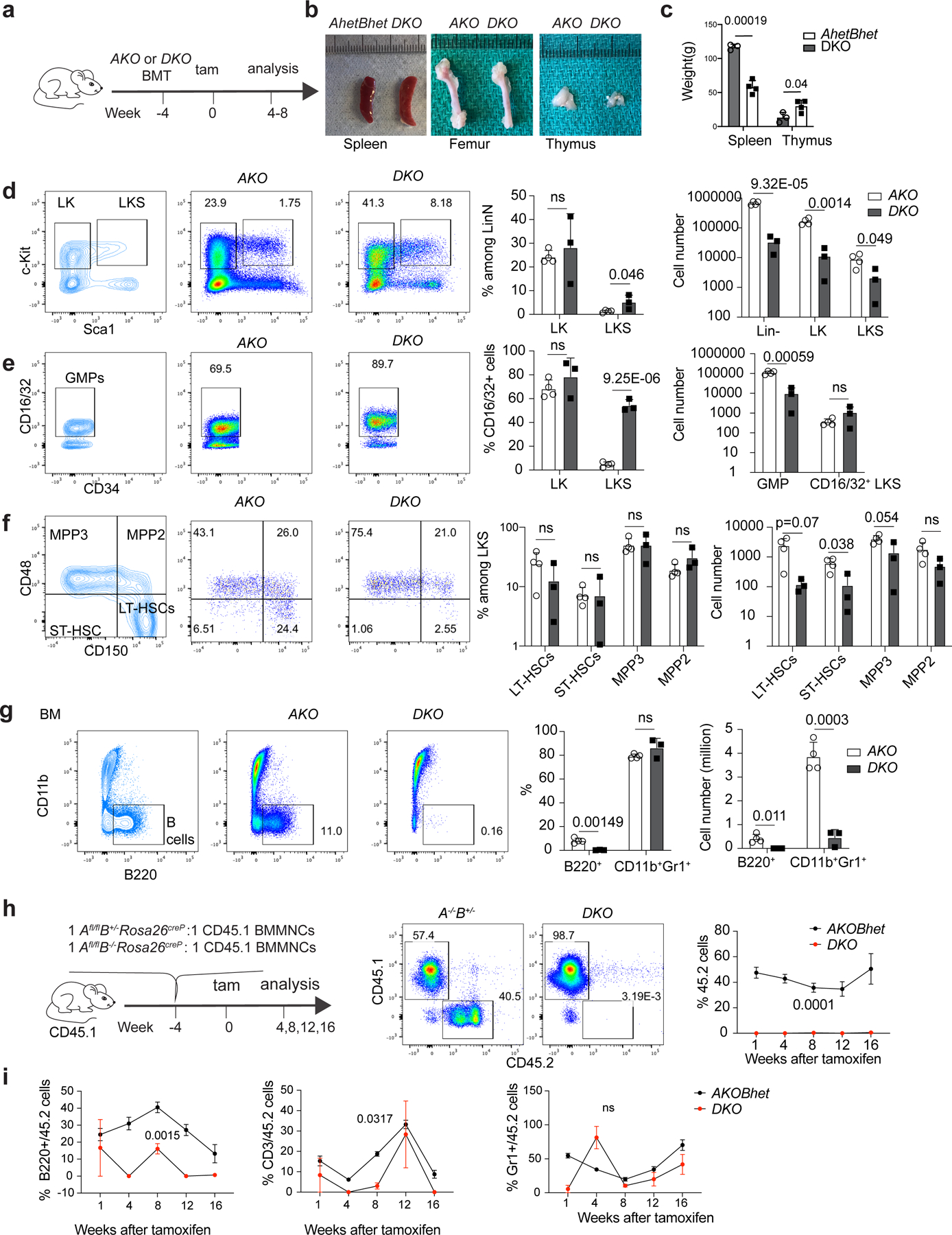
a, Schematic view of non-competitive transplantation experiments. b, Images of the spleen (left), femur (middle) and thymus (right) of the recipients transplanted with induced KO (iKO) including H3.3AiKO/iKO (AKO), H3.3AiKO/+H3.3B+/− (heterozygous H3.3A-KO and heterozygous H3.3B-KO; AhetBhet) or DKO BMMNCs at 4 weeks following tamoxifen treatment. c, Weight of the spleen and thymus of the recipients of AKO, AhetBhet or DKO BMMNCs. d, Representative flow cytometry plots (left), percentages (middle) and the total number of LKS cells (right) in the mice described in a. e, Representative flow cytometry plots (left), percentages (middle) and the total number of CD16/32+ CD34+c-Kit+Sca1−Lin− GMPs and CD16/32+ LKS cells (right) in the recipients described in a. f, Representative flow cytometry plots (left), percentages (middle) and the total number of MPP3, MPP2, LT-HSC and ST-HSC (right) cells in the recipients described in a. g, Representative flow cytometry plots (left), percentages (middle) and numbers (right) of B cells in the BM of the recipients described in a. a–g, n = 4 AKO mice and n = 3 DKO mice. h, Schematic of the competitive transplantation assay used to test the engraftment of DKO BMMNCs (left). Representative flow cytometry plots showing the percentage of CD45.2+ cells in the peripheral blood of the CD45.1 recipients (middle). Percentages of CD45.2+ cells in the peripheral blood of the CD45.1 recipients at different time points (right). i, Multilineage engraftment for the CD45.2+ BMMNCs cells from AKOBhet and DKO mice. h,i, n = 4 animals per experiment. The experiment was repeated twice. c–i, The P values were calculated using an unpaired two-tailed Student’s t-test (c–g) or a two-way analysis of variance (ANOVA; h,i) and are indicated on the graphs; NS, not significant. The error bars represent the s.e.m. d–h, The percentage of cells in the different quadrants or boxes in the flow cytometry plots are indicated. Tam, tamoxifen. Numerical source data are provided.
The percentages of B cells in the BM, spleen and peripheral blood of the recipients of the DKO-cell transplants were reduced (Fig. 1g, Extended Data Fig. 4d,e and Supplementary Table 2). The area of white pulp in the spleen was also reduced, displaying fibrosis without changes in other organs (Extended Data Fig. 4i–k). The phenotypes of the global-DKO mice and the mice that received DKO-cell transplants demonstrated that the splenomegaly, loss of lymphocytes and increased percentages of myeloid cells were due to impaired cell-intrinsic function as a result of H3.3 deletion from haematopoietic cells, except that the number of CD16/32+ LKS cells remained unchanged, demonstrating myeloid persistence and survival. Thus, H3.3 preserves the viability of subsets of haematopoietic cells at various stages of lineage-specific differentiation.
To assess the function of H3.3 in HSC engraftment and repopulation, we performed competitive transplantation of DKO cells and Rosa26creERT2+H3.3Afl/flH3.3B+/− (AKOBhet) cells (Fig. 1h). Four weeks after transplantation, after the administration of tamoxifen, we observed a reduction in donor-derived cells in the peripheral blood of mice receiving DKO BMMNC competitive transplants compared with those that received AKOBhet BMMNCs (Fig. 1h). Both B and T lymphocytes were also reduced (Fig. 1i). In the BM, the total numbers of donor-derived LinN, LK, LKS, MPP3 and MPP2 cells; LT-HSCs, ST-HSCs and differentiated lineages were reduced (Extended Data Fig. 5a–l). Therefore, H3.3 is required for the balanced survival and lineage differentiation of HSCs.
H3.3 is required for HSPC expansion and differentiation
Next, we employed a vascular niche-HSPC co-culture model to test the hypothesis that H3.3 is cell-intrinsically required for HSPCs to expand and differentiate (Fig. 2a)24. Endothelial cells maintain HSPC repopulating capacity and promote the differentiation of myeloid cells. When H3.3 was deleted from HSPCs, the total numbers of expanded, LinN and CD16/32− LKS cells were reduced (Fig. 2b). As LT-HSCs are found in small numbers in in vitro cultures, we focused our analysis on LKS cells (Extended Data Fig. 6c). The number of wild-type LKS cells expressing CD16/32 gradually increased (Extended Data Fig. 6a). When H3.3 was deleted, the total number of phenotypic LKS cells adopting the GMP marker CD16/32 increased, with an increase in G1-stage LK cells and apoptotic cells but a reduction in G0-quiescence LKS cells (Fig. 2d,e).
Fig. 2 |. H3.3 maintains HSC quiescence and blocks myeloid differentiation.
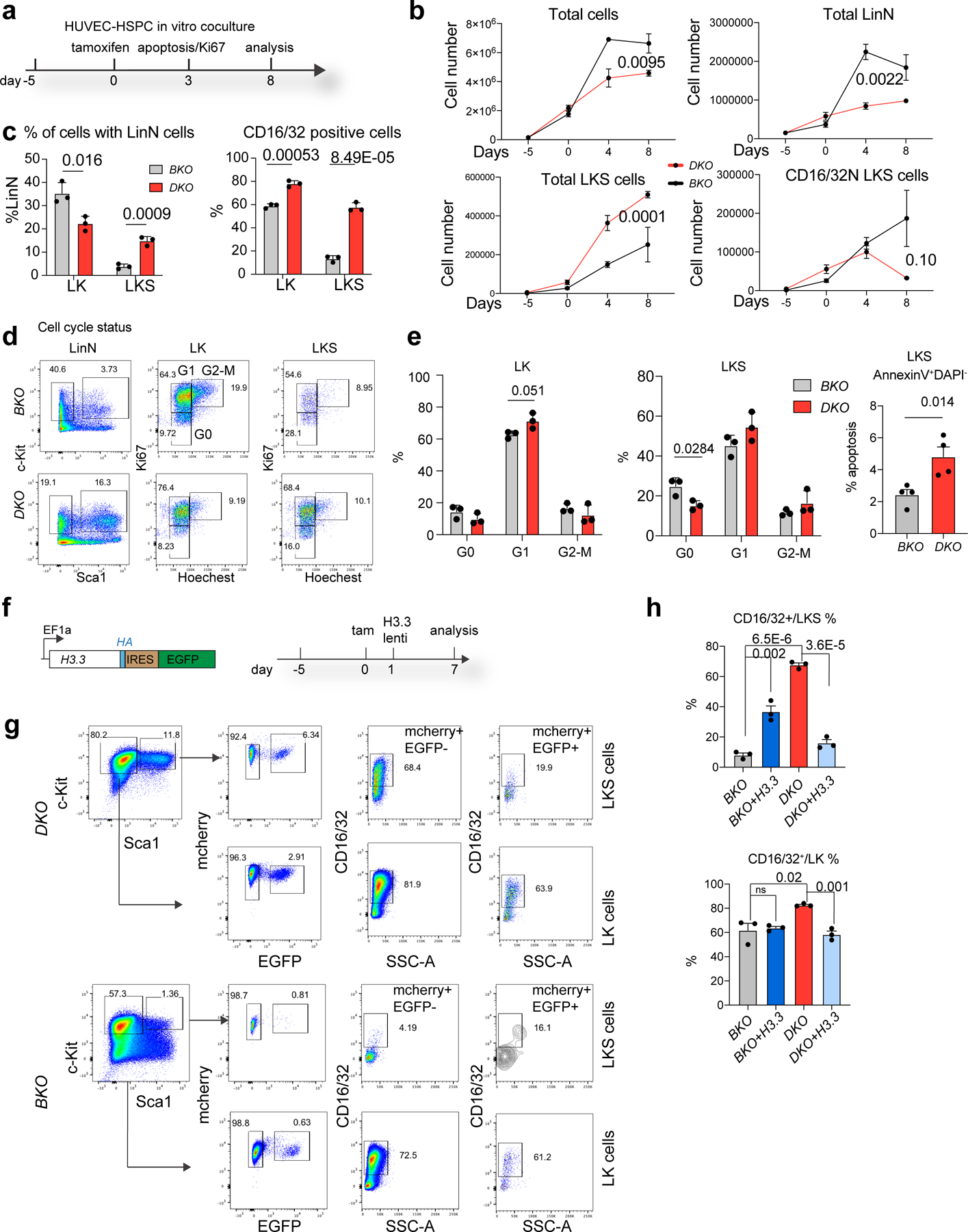
a, Schematic view of the in vitro HUVEC–HSPC co-culture experiment. The cell-cycle and apoptosis status of the cells was evaluated 3 d after tamoxifen treatment; the total number of LKS and lineage cells were quantified 8 d after the tamoxifen (Tam) treatment. b, Total number of haematopoietic, LinN, LKS and CD16/32− LKS cells at different time points during the co-culture. c, Percentages of LK and LKS cells in the LinN cell populations (left). Percentages of CD16/32+ LK and LKS cells at day 7 post tamoxifen treatment (right). d, Representative flow cytometry plots for the analysis of the cell-cycle status of LK and LKS cells on day 3 after H3.3A deletion. e, Percentages of cells in different stages of the cell cycle (LK, left; LKS, right) and the percentage of LKS cells in apoptosis. f, We carried out a rescue experiment using lentiviral-mediated overexpression of H3.3. The lentiviral construct (left) and the experimental scheme (right) are shown. Due to the limited transduction efficiency of lentivirus into LKS cells, we used H3.3BmCherry/mCherry BKO or Rosa26cre+H3.3Afl/flH3.3BmCherry/mCherry DKO cells. g, Representative flow cytometry plots for the LK and LKS cells following H3.3 overexpression. Overexpression of H3.3 (mCherry+EYFP+ cells) has reduced expression of the CD16/32+ marker in both LKS and LK DKO cells (top). Overexpression of H3.3 increased the expression of CD16/32+ in LKS BKO cells (bottom). d,g, The percentage of cells in the boxed regions in the flow cytometry plots are indicated. h, Percentages of CD16/32+ BKO, BKO + H3.3, DKO and DKO + H3.3 LKS (top) and LK (bottom) cells. Error bars represent the s.e.m.; n = 3 independent biological samples for all panels, except the apoptosis assay in e, where n = 4. The P values were calculated using an unpaired two-tailed Student’s t-test (c,e,h), two-way ANOVA (b, total, LinN and LKS cells) or a two-tailed Student’s t-test (b, CD16/32− LKS cells on day 8) and are indicated on the graphs; NS, not significant. Numerical source data are provided.
We utilized lentivirus-mediated H3.3 overexpression for rescue experiments to examine whether the changes in the growth kinetics of DKO LKS cells is H3.3 dependent (Fig. 2f,g). When H3.3 was overexpressed, the percentages of CD16/32+ cells were reduced in the DKO LK and LKS cell populations (Fig. 2h). H3.3 overexpression repressed CD11b+Gr1+ cells but did not result in an increase in B220+ cells (Extended Data Fig. 6d). Hence, H3.3 preserves LKS identity by maintaining their quiescence and inhibiting excessive myeloid-marker expression. When Hira was deleted, there was a reduction in the total number of expanded cells and CD16/32− LKS cells; however, the percentages of CD16/32+ cells were increased (Extended Data Fig. 6e–k). For lineage-differentiated cells, the percentages (but not the total number) of B cells were increased, whereas CD11b+Gr1− cells were decreased. Therefore, Hira represses CD16/32+ cells ex vivo but is dispensable for B-cell differentiation.
H3.3 deposition colocalizes with heterochromatin
We used H3.3B–haemagglutinin (HA) knock-in reporter mice22 and performed ChIP–seq on the HA epitope tag to detect H3.3 localization (Extended Data Fig. 7a). Genome-wide, 30,651 H3.3 peaks localized at intron, intergenic, SINE and promoter regions (Extended Data Fig. 7b). We also performed ChIP–seq for the active histone modifications H3K4me3 and repressive histone marks H3K27me3 and H3K9me3 (refs. 12,13,23,25,26). We show that 14,720 regions with H3.3 deposition overlapped with H3K4me3-enriched regions, which were associated with 9,031 genes (Extended Data Fig. 7c,k). We identified 5,142 bivalent regions, enriched with both H3K4me3 and H3K27me3 modifications (Extended Data Fig. 7d,e,h,k)27,28; a significant proportion of which (2,012 of 5,142 peaks, 1,722 of 4,111 associated genes) colocalized with H3.3 deposition. Thus, H3.3 can potentially maintain the bivalency of developmental genes in HSPCs.
H3K9me3 is a marker for poised enhancers that can control gene regulation in a cell-specific manner29–31. We hypothesize that H3K9me3-marked enhancers are dependent on H3.3 deposition and control the expression of lineage-specific genes. H3.3-dependent H3K9me3 peaks localized at long terminal repeat (LTR), intergenic, intron and SINE regions (Extended Data Fig. 7f,g,k), and were near the genes involved in intracellular signal transduction, immune-system process, regulation of the apoptotic process and positive regulation of NF-κB transcription factor activity (Extended Data Fig. 7i). Genes with H3K4me3 enrichment were more abundantly expressed than genes with the repressive histone marks H3K9me3 or H3K27me3 (Extended Data Fig. 7j). Notably, genes near the H3.3-dependent H3K9me3 marks had increased levels of messenger RNA than genes with H3K27me3 marks, suggesting an intermediate stage of transcription status that is between active and repressed. It remains to be determined whether H3.3-dependent deposition maintains the integrity of histone PTMs and safeguards transcription.
H3.3-null HSPCs transcriptionally resemble GMPs
Loss of H3.3 forces HSPCs to assume a GMP-like transcriptomic signature. To define the transcriptome and epigenetic changes in DKO LKS cells, we sorted out HSPCs on days 15, 21 and 56 after H3.3A deletion as well as on day 15 after Hira deletion. The differentially expressed genes (DEGs) at these time points were analysed (Fig. 3a,b and Supplementary Tables 3,4). The early–mid upregulated genes were enriched in biological pathways including apoptosis, T-cell cytotoxicity and TNF-α activities (Fig. 3c and Extended Data Fig. 8a,b). The mid–late response genes are involved in the cell cycle, chromosome segregation and immune system (Fig. 3c and Extended Data Fig. 8c,d). While BKO and DKO LKS cells showed diverse clustering (Fig. 3d), Hira-KO LKS cells demonstrated similar transcriptomic profiles to Hirafl/fl and BKO LKS cells, matching the minimum changes in LT-HSC and LKS cell numbers following Hira deletion. Loss of H3.3, but not Hira, brings HSPCs transcriptionally closer to GMPs in vivo. H3.3-null HSPCs facilitated the transition from LKS cells to GMP cells, suggesting that an H3.3/Daxx-dependent pathway regulates GMP commitment. H3.3-null HSPCs have reduced stemness markers, including Meis1, Procr, Nkx2.3, Mycn, Mpl, Egr1, Gfi1b and Mllt3 (Fig. 3e). Lineage-differentiation genes were also reduced (Fig. 3f), matching the in vivo haematopoietic phenotypes following H3.3 deletion.
Fig. 3 |. H3.3-null HSPCs demonstrated a GMP-like transcriptomic signature and a predominant gain of H3K27me3 marks.
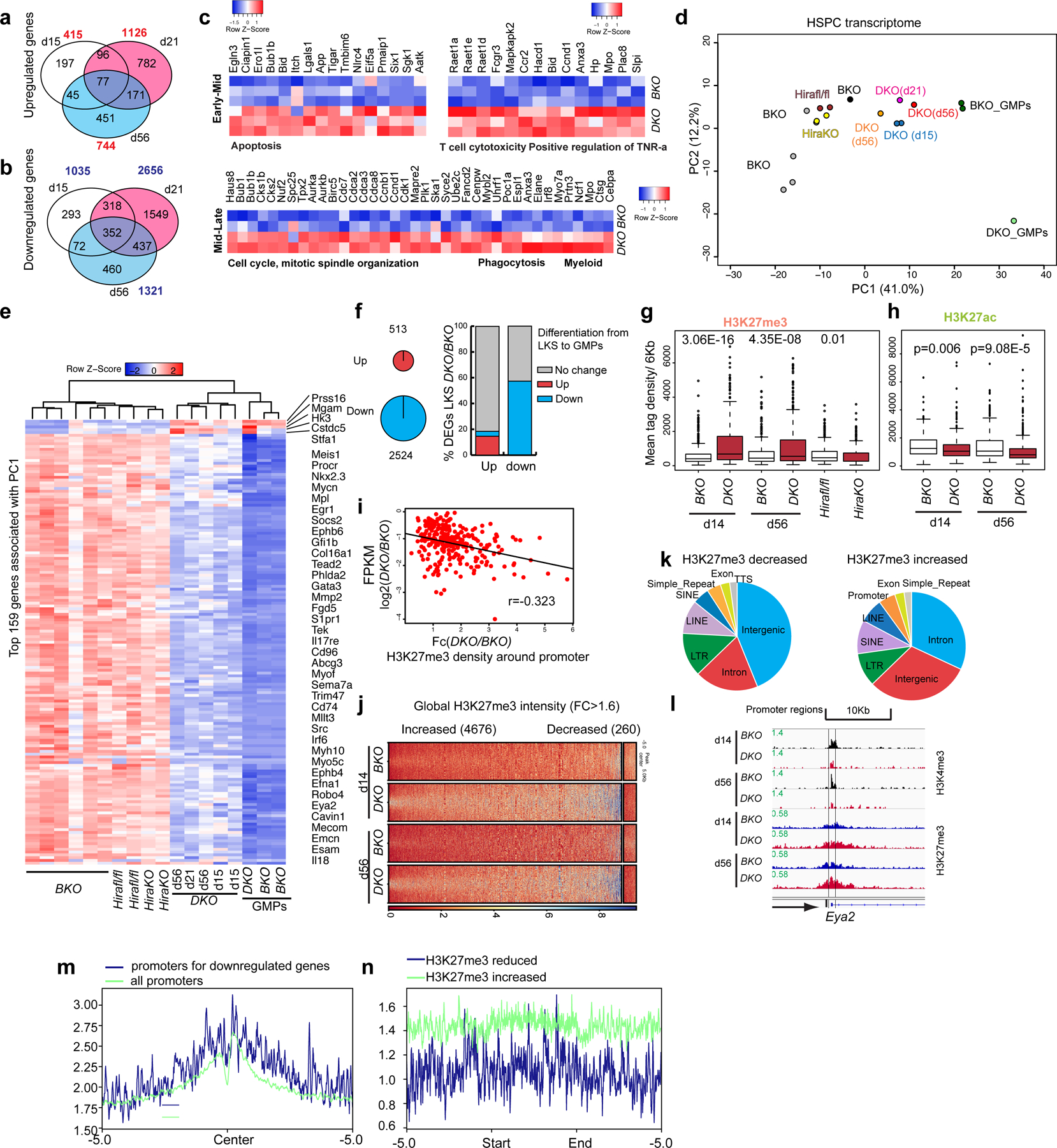
a,b, Number of upregulated (a) and downregulated (b) DEGs on days 15, 21 and 56 after H3.3A deletion (P < 0.05; FC > 1.8). The P values were calculated using a two-tailed Student’s t-test. The total numbers of genes that are upregulated or downregulated at the different time points are shown in red (a) and blue (b), respectively. c, We arbitrarily divided the time following H3.3 deletion into early–mid and mid–late stages. The early–mid DEGs are enriched for apoptosis, T-cell cytotoxicity and positive regulation of TNF-ɑ. The mid–late stage DEGs are enriched for the cell-cycle, mitotic-spindle organization, phagocytosis and myeloid cell biological pathways. d, HSPC transcriptome. Principal component (PC) analysis of FPKM values in different samples, using the most variably expressed genes including the DEGs at early (n = 1,450) and late (n = 329) time points after H3.3 deletion, and the DEGs between Hirafl/fl and Hira-KO HSPCs (n = 313). e, Heatmap showing the expression of the top 159 genes associated with PC1 in LKS cells from BKO, Hirafl/fl, Hira-KO and DKO mice as well as GMPs from BKO and DKO mice. Among the top 159 genes associated with PC1, five are increased in DKO LKS cells; the remaining 154 genes are downregulated. f, In the differentiation of LKS cells towards GMPs, there are 513 and 2,524 genes that are significantly upregulated and downregulated, respectively (left). Percentage of overlap between the up- and downregulated genes in DKO cells relative to BKO cells (y axis) and the up and downregulated genes from LKS-to-GMP differentiation (x axis; right). g, Levels of H3K27me3 enrichment at the promoter regions (TSS ± 3 kb) of the top 385 downregulated genes associated with PC1. The experiment was repeated twice independently for the BKO and DKO cells at each time point; the experiment was performed once for the Hirafl/fl and Hira-KO cells. h, H3K27ac profiles at the promoter regions of the top 385 downregulated genes associated with PC1. The experiment was repeated twice independently for each column. g,h, The P values were calculated using an unpaired two-tailed Student’s t-test using one biological sample for each genotype and are indicated on the graphs. In the box-and-whisker plots, the boundaries of the box indicate the 25th and 75th percentiles, the centre line indicates the median and the whiskers (dashed lines) indicate 1.5× the interquartile range. i, Correlation between the mRNA level changes of the 385 downregulated genes (FPKM) and the density of H3K27me3 around the promoter (DKO/BKO FC); r, coefficient of multiple correlation. j, Globally, a larger number of regions had increased H3K27me3-peak intensity than decreased H3K27me3-peak intensity in DKO LKS cells compared with BKO LKS cells. k, Genome-wide distribution of the H3K27me3-decreased (left) and H3K27me3-increased (right) peaks. l, Representative genome browser track showing the reduced H3K4me3 and increased H3K27me3 at the Eya2 promoter locus on days 14 and 56 after H3.3 deletion in vivo. m, H3.3 enrichment at the 385 downregulated genes associated with PC1 compared with all promoters. n, H3.3 enrichment at the H3K27me3-reduced regions and H3K27me3-increased regions. There is more H3.3 enrichment at the H3K27me3-increased regions compared with the H3K27me3-reduced regions.
The promoter regions of downregulated genes in DKO HSPCs manifested a reduction in H3K4me3, an increase in H3K27me3 and a decrease in H3K27ac (Fig. 3g,h and Extended Data Fig. 8e,f). For the upregulated genes, there was an increase in H3K4me3 but no change in H3K27me3 at their promoter regions (Extended Data Fig. 8h–k). Consistent with the predominant reduced gene expression in DKO LKS cells, genome-wide, there were 260 regions with decreased H3K27me3 enrichment and 4,676 regions with increased H3K27me3 enrichment (fold change (FC) > 1.6; Fig. 3i–l and Extended Data Fig. 8g). We next investigated whether H3.3 enrichment directly prevents H3K27me3 density. H3.3 is enriched at the promoters of the downregulated genes (Fig. 3m); H3.3 deposition is more enriched in the regions with increased H3K27me3 marks compared with regions with reduced H3K27me3 (Fig. 3n), suggesting a direct role of H3.3 in competing with H3K27me3 enrichment in HSPCs. The global gain of H3K27me3 in DKO HSPCs predisposes their differentiation path towards GMPs. The erythroid-lineage differentiation programme in DKO LKS cells is downregulated (Extended Data Fig. 8l), with an increase in H3K27me3 enrichment. Therefore, H3.3 is required for the maintenance of lineage-specific active gene transcription by preventing the spread of H3K27me3 marks.
We also compared H3K27me3-altered regions in DKO and Hira-KO HSPCs (Fig. 4a and Extended Data Fig. 8m–p), and identified genes with increased mRNA expression and reduced H3K27me3 enrichment, such as inhibitor of differentiation 1 (Id1; Fig. 4b–d). To examine whether Id1 increases in DKO HSPCs promoted myelopoiesis or was a passive result of H3.3 loss, we crossed Id1-mutant mice with DKO mice and quantified the haematopoietic cells in the BM, spleen and peripheral blood of the triple-KO (TKO) mice (Fig. 4e–l and Supplementary Table 5). Id1 deletion did not rescue the reduction in LT-HSC numbers and worsened the myeloid skewing in DKO HSPCs. Therefore, Id1 overexpression did not contribute to the overt myelopoiesis phenotype of DKO HSPCs.
Fig. 4 |. Id1 deletion does not rescue the increased myelopoiesis in DKO mice.
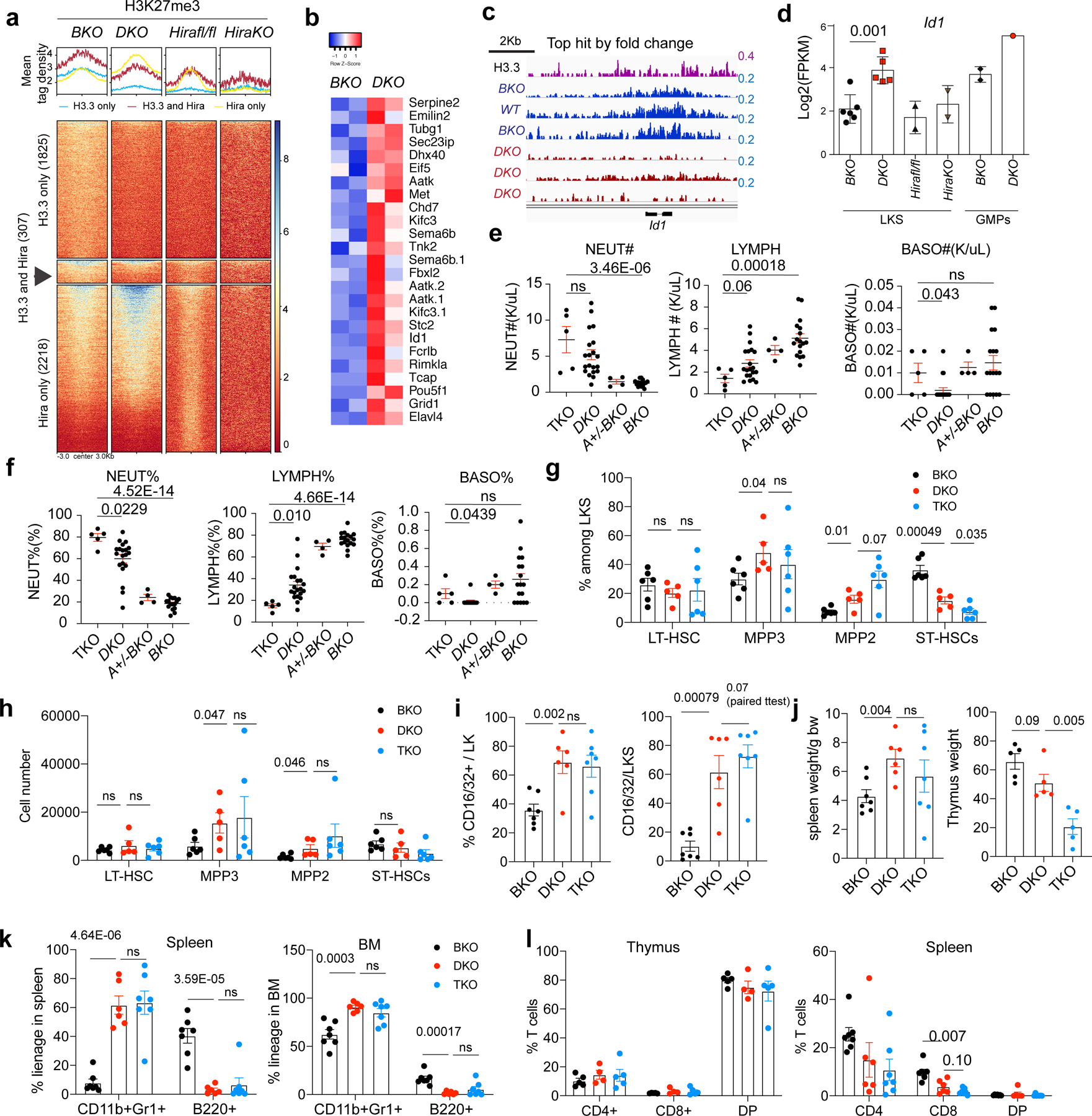
a, Histograms (top) and heatmap (bottom) showing the H3K27me3 mean tag density at H3K27me3-reduced regions in BKO, DKO, Hirafl/fl and Hira-KO LKS cells. The heatmap shows the H3K27me3-reduced regions for the DKO only, DKO and Hira-KO shared and Hira-KO only cells. b, FPKM levels of genes with concomitant promoter-region H3K27me3 reduction and increased mRNA expression levels in DKO HSPCs. Aatk.1 and Aatk.2 are two isoforms of Aatk. c, The gene with the highest H3K27me3 reduction, ranked according to the FC, is Id1. d, Expression levels of Id1 mRNA in BKO, DKO, Hirafl/fl and Hira-KO LKS cells as well as BKO and DKO GMPs. e,f, We crossed DKO mice with Id1-mutant mice35 and generated Rosa26creERT2+H3.3Afl/flH3.3B−/−Id1−/− mice. After tamoxifen-induced H3.3A deletion, the resulting mice were termed TKO mice. The total number (e) and percentages (f) of neutrophils, lymphocytes and basophils in the peripheral blood of the indicated mice were determined. g,h, Percentage (g) and total number (h) of LT-HSCs, ST-HSCs, and MPP3 and MPP2 cells in BKO, DKO and TKO mice. i, Percentage of GMPs in the LK cell populations (left) and of CD16/32+ cells in the LKS cell populations (right). j, Spleen and thymus weight (normalized to the body weight of the mouse) for the BKO, DKO and TKO mice. k, Percentage of lineage cells in the spleen (left) and BM (right) of the TKO mice. l, Percentage of T cells in the thymus (left) and spleen (right) of the TKO mice. DP, CD4+CD8+ double-positive cells, k,l, Each dot represents an individual mouse. Error bars represent the s.e.m. P values were calculated using an unpaired two-tailed Student’s t-test, except for the panel in Fig. 4i (P = 0.07), and are indicated on the graphs. NS, not significant. Numerical source data are provided.
Reduction of H3K9me3 at ERVs in DKO HSPCs
The mouse genome harbours ERVs that can be classified into different subtypes, termed repfamilies. H3.3 is deposited at ERVs in mouse embryonic stem cells (ESCs)12 and might repress class I and II ERVs by maintaining H3K9me3 enrichment in adult HSPCs. Following H3.3 deletion, H3K9me3 was decreased at class II and I ERVs (ERV1) but not at class III ERV or LINE1 elements, suggesting specific regulation of class I and II ERVs by H3.3 (Fig. 5a and Extended Data Fig. 9a). We also quantified the mRNA expression of ERV repfamilies after H3.3 deletion (Fig. 5b and Extended Data Fig. 9b,c). Although the degree of H3K9me3 reduction was most severe for class II ERVs, which include MMERVK10C-int and IAPEz-int, the most apparent mRNA changes happened in class I ERVs, including MMVL30-int and RLTR6-int (Fig. 5b). The kinetics of ERV mRNA expression has unique features in vitro versus in vivo (Extended Data Fig. 9b). In vitro, the degree of mRNA derepression of MMVL30-int and MMERVK10C-int on day 11 following H3.3 deletion was more than the degree of derepression on day 4; in vivo, the increase in ERVs on day 21 resolved on day 56, suggesting that the microenvironment could play a role in counterbalancing ERV expression (Supplementary Table 6). Notably, the expression levels of RLTR6-int mRNA were higher on day 56 compared with day 21 post H3.3 deletion, highlighting repfamily-specific expression patterns.
Fig. 5 |. Reduction of H3K9me3 and dysregulated ERV expression in DKO HSPCs.
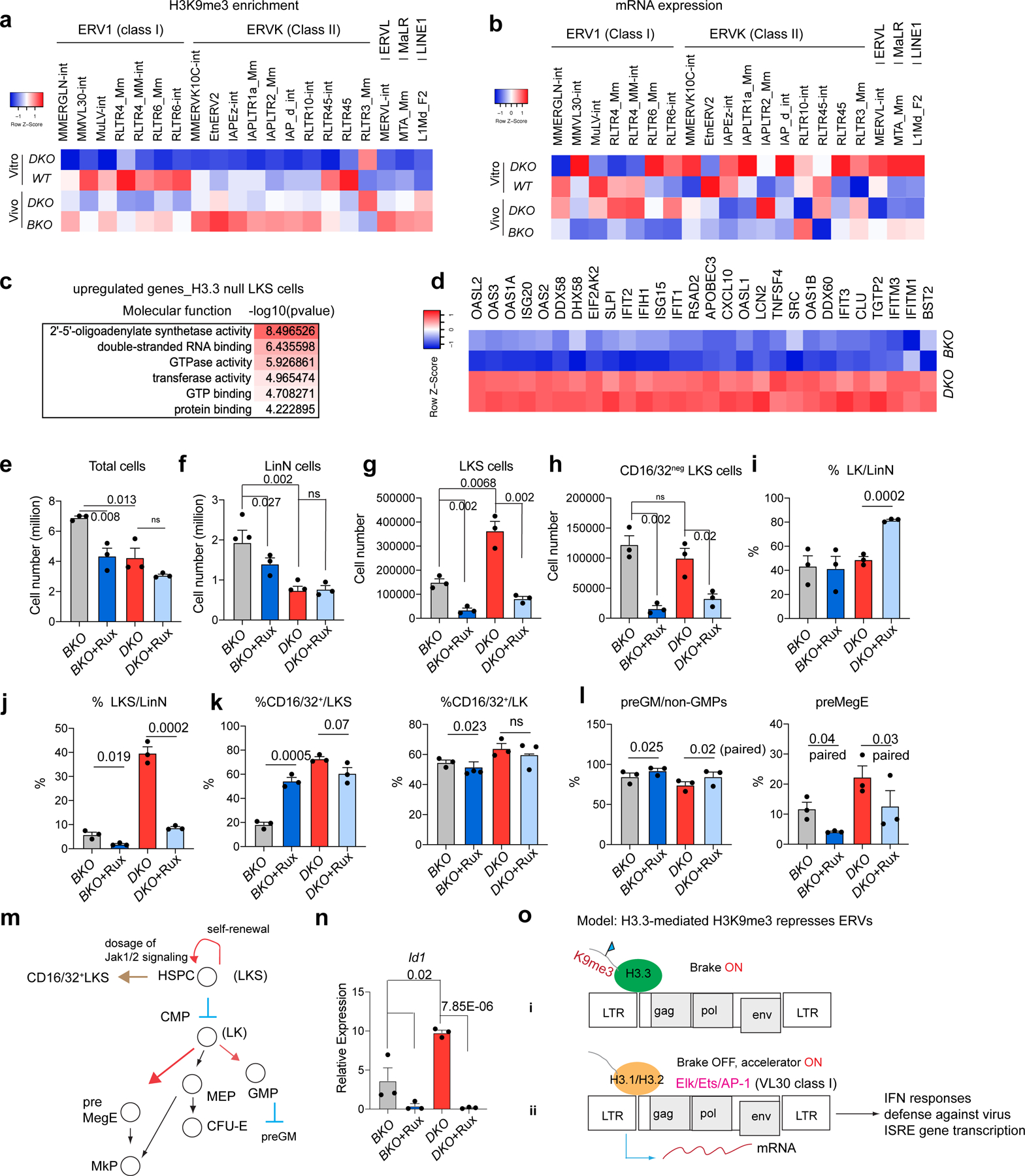
a, Heatmap showing the levels of H3K9me3 enrichment at representative ERV repfamilies in wild-type and DKO LKS cells (in vitro), and BKO and DKO LKS cells (in vivo). b, Levels of mRNA expression of representative ERV repfamilies. c, Molecular functions associated with upregulated DEGs in H3.3-null LKS cells. d, Levels of RNA expression of genes involved in the cellular response to interferon-β. e, Total number of BKO and DKO LKS cells following treatment with Rux or vehicle. f,g, Total number of LinN (f) and LKS (g) cells in the BKO and DKO cells following treatment with Rux or vehicle. h, Total number of CD16/32− LKS cells for the indicated treatment groups. i,j, Percentage of LK (i) and LKS (j) cells in the LinN cell populations during the co-culture. k, Percentage of CD16/32+ cells in the LK (right) and LKS (left) cell populations. l, Percentages of progenitors to granulocytes and macrophages (preGM; left) and pre-megakaryocyte-erythrocyte progenitors (preMegE; right) subpopulations in the CD16/32− LK (non-GMPs) cells. m, Cartoon showing the regulation of HSC proliferation and differentiation by Jak1–Jak2 signalling. MEP, megakaryocyte erythroid progenitor. n, Expression levels of Id1 in LKS cells for the indicated treatment groups. o, Working model. The expression of ERV is regulated by a ‘brake and accelerator’ mechanism. H3.3-mediated H3K9me3 deposition serves as a repressive histone modification brake (i); when H3.3 is deleted, the brake is released (ii). The expression levels of ERV mRNA also depend on the signalling context. The ERV families with the binding sites for Elk/Ets/AP1 are expressed at higher levels in the presence of Elk/Ets/AP1, similar to RLTR6-int. e–l,n, Each dot indicates an independent biological sample. The error bars indicate the s.e.m. The P values were calculated using an unpaired two-tailed Student’s t-test, except for the two of the panels in Fig. 5l (P = 0.02 and 0.03), and have been indicated on the graphs. NS, not significant; WT, wild type. Numerical source data are provided.
We confirmed the dysregulation of ERV mRNAs in the LKS, LK and lineage-positive cells from DKO mice by quantitative PCR with reverse transcription (Fig. 5b and Extended Data Fig. 9d)32–35. Along with the derepression of ERV repfamilies, the expression levels of other ERVs were reduced, suggesting ERV-dysregulation rather than global ERV derepression following the loss of H3.3 (Extended Data Fig. 10a,b).
Derepressed ERV elicits antiviral responses
We examined the molecular functions of upregulated genes in HSPCs after H3.3 deletion in vitro. The top terms were double-stranded RNA binding and 2′−5′-oligoadenylate synthetase activity (Fig. 5c), which is an antiviral enzyme that counteracts viral attack by degrading viral mRNA; such as ERV life cycles36. There is a link between histone variants or histone modifying enzymes in suppressing ERVs and interferon signalling pathways37–39. The upregulated genes in DKO HSPCs include genes involved in the defence response to virus and interferon-β (Fig. 5d, Extended Data Fig. 9e and Supplementary Table 7) as well as Apobec3, a cytidine deaminase that plays a dominant role in retroviral-element restriction in host cells40, which was upregulated on both day 4 and day 11.
For the enhancer landscape in DKO HSPCs, (Extended Data Fig. 9f), the 15,484 regions with increased H3K27ac were enriched with binding sites for transcription factors including erythroblast transformation-specific (ETS) family members (Ets1, Fli1 and Etv2), Irf4 and the bZIP family members Jun:AP1. ERV derepression involves two mechanisms, a ‘brake’ (H3.3-mediated H3K9me3 deposition) and an ‘accelerator’ (transcription factor binding, Jun:AP1 in the case of DKO HSPCs41; Fig. 5o). A consequence of dysregulated ERV expression is aberrant interferon signalling in DKO HSPCs, including Fcgr2b/Frgr3 (CD16/32; Extended Data Fig. 10h,i and Supplementary Tables 8,9)42, which is a marker for GMPs. The levels of Fcgr2b/Fcgr3 in LT-HSCs and HSPCs were increased following loss of H3.3 (Extended Data Fig. 2g), suggesting a myeloid bias at the LT-HSC and HSPC levels. Therefore, we hypothesize that H3.3 regulates ERV repression in HSPCs, preventing myeloid skewing by suppressing interferon signalling, possibly mediated by Jak1–Jak2 (refs. 43–46). When BKO and DKO LKS cells were treated with Ruxolitinib (Rux), a Jak1/Jak2/Jak2V617 inhibitor41,47, there was a reduction in the total number of expanded, LinN and LKS cells, and an increase in CD16/32+ LKS cells in the BKO, but not DKO, cells (Extended Data Fig. 9g,h and Fig. 5e–m). Treatment with Rux reduced the percentage of B220+ cells in the BKO cultures and increased the B220+ cell percentages in the DKO cells ex vivo (Extended Data Fig. 9i,j)48. Hence, inhibition of Jak1–Jak2 partially restores the myeloid bias in DKO cells by promoting B-cell differentiation.
The levels of H3.3A and Daxx mRNA were reduced in DKO LKS cells, whereas Hira was unchanged (Extended Data Fig. 9k–r). The levels of Daxx in GMPs was also reduced in DKO mice (Extended Data Fig. 9n). As Daxx represses ERVs49, the dysregulated ERV phenotype could be due to Daxx downregulation. Notably, the increased expression of Id1 in DKO cells, which had the biggest reduction in H3K27me3, was dependent on Jak1–Jak2 signalling (Fig. 5n). Therefore, H3.3 might mediate the crosstalk between of H3K9me3 and H3K27me3 enrichment through the modulation of interferon signalling.
H3K9me3-reduced ERV regions as enhancers, regulating survival
H3K9me3 is a marker of repressed enhancers29; thus, reduced H3K9me3 in H3.3-null HSPCs may be associated with enhanced gene transcription. There were 1,361 regions with decreased H3K9me3 enrichment and 484 regions with increased H3K9me3 enrichment (FC > 1.6) in DKO LKS cells (Fig. 6a–d). The H3K9me3 density50 was reduced at H3K9me3 mountain regions in DKO HSPCs but not in Hira-KO HSPCs (Fig. 6e, f and Extended Data Fig. 10c,d); a portion of thee H3K9me3-reduced mountains was located at sub-telomeric regions (Extended Data Fig. 10e,f). The H3K9me3-reduced mountains in H3.3-null HSPCs suggested premature ageing of HSCs, consistent with the myeloid skewing51. We hypothesized that H3K9me3-reduced ERV regions serve as enhancers to promote myeloid gene expression, fuelling the adaptive evolution of H3.3-null cells. Notably, 38.2% of the total 3,079 H3K9me3-reduced regions were at LTR regions (Fig. 6c); LTR/ERV are exclusively enriched at the distal H3K9me3 regions (Fig. 6g). Motif analysis identified the enrichment of key transcription-factor binding sites, including Spi1, Myc and Relb within the H3K9me3-reduced ERV regions (Fig. 6h). We quantified the fold change in mRNA levels in DKO LKS cells compared with BKO LKS cells and plotted this against the distance of the ERV to its trancription start site (TSS; Fig. 6i). As the distance to the TSS increased, the influence of the ERV on gene expression became smaller. There was both increased and decreased gene expression near the H3K9me3-reduced ERV sites. One example of a gene with increased expression was Il3ra, whose enhancer has a putative binding site for Myc and Irf4 (Fig. 6j). H3K9me3 marks were reduced at its enhancer region, which overlaps with an IAPEz-int ERV repeat and an increase of H3K27ac enrichment at its promoter region. The complete list of altered genes with H3K9me3 reduction (TSS ± 10 kb) and increased mRNA expression include apoptotic or cell proliferation, C-type-lectin receptors, myeloid receptors, epigenetic modifiers, mitochondrial and peroxisomal and extracellular matrix-related genes (Extended Data Fig. 10g). Representative genes with reduced H3K9me3 around promoters and reduced expression are Zfp84 and Zfp30, suggesting context-specific transcriptional regulations (Extended Data Fig. 10i and Supplementary Table 9).
Fig. 6 |. ERVs in the H3K9me3-reduced regions as potential enhancers to regulate survival genes.
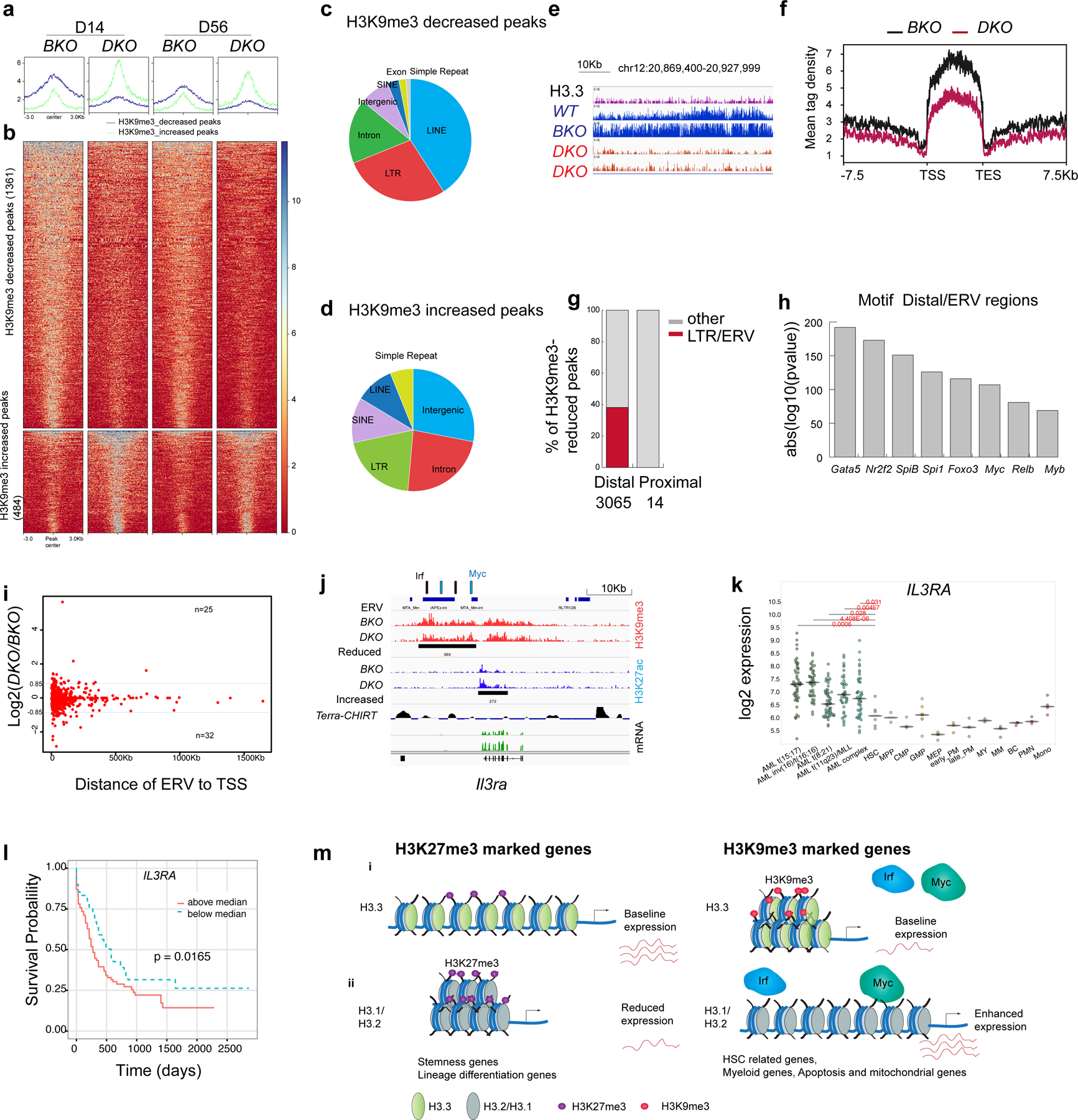
a,b, Histograms (a) and heatmaps (b) of the H3K9me3 densities for H3K9me3-decreased and -increased regions in BKO and DKO HSPCs (false-detection rate (FDR) < 0.01, FC > 1.6). c,d, Genome-wide distribution of H3K9me3-decreased (c) and -increased (d) peaks. e, Example of H3K9me3-reduced mountains. f, Histogram of the H3K9me3-reduced mountains in BKO and DKO HSPCs on day 14 after H3.3A deletion. TES, transcription end site. g, Percentage of ERVs at the distal and proximal loci within the H3K9me3-reduced regions. Distal peaks, distance to TSS of >1,000 bp. Proximal regions, distance to TSS of <1,000 bp. h, Motif analysis of ERV-overlapped distal H3K9me3-reduced regions. i, Fold change in mRNA levels in DKO LKS cells compared with BKO LKS cells plotted against the distance of the ERV to its TSS. Blue line, Log2(DKO/BKO) = 0.85 or −0.85. j, Integrative Genomics Viewer view of the promoter and enhancer regions for Il3ra showing the reduced H3K9me3 enrichment upstream of its TSS, the ERV elements and the putative binding sites for the transcription factors Irf4 and Myc near the H3K9me3-reduced region. Terra, telomeric repeat-containing RNA. Terra has been shown to serve as an epigenomic modulator in trans and regulate the telomerase function in cis1. The Terra binding site is shown here as a reference. CHIRT, chromatin isolation of RNA targets. k, Expression levels of IL3RA in AML, normal haematopoietic progenitor and normal differentiated lineage cells2. Each dot represents an independent biological sample. Mono, monocyte; MEP, megakaryocyte erythroid progenitor; MY, myeloid; BC, B cell. The P values were calculated using an unpaired two-tailed Student’s t-test; *P < 0.05, **P < 0.01 and ***P < 0.001. l, Kaplan–Meier survival curve of patients with AML stratified into two groups—that is, individuals with IL3RA expression levels above or below the median51. m, Working model. (i) H3.3 maintains adult HSC homeostasis and prevents premature myeloid differentiation by regulating H3K27me3 and H3K9me3 enrichment. After H3.3A deletion, the reduction in H3K9me3 promotes transcription-factor binding and enhanced mRNA expression of target genes, conferring adapted survival for H3.3-null cells (right). (ii) For stemness genes and lineage-differentiation genes (RBCs), H3.3 prevents H3K27me3 enrichment at their promoter regions. H3.3 represses gene expression via H3K9me3 enrichment. After H3.3 deletion, the deposition of canonical histones H3.1 and H3.2 is unaffected (right). H3.2 and H3.1 are most probably deposited into the H3.3-null nucleosomes. Numerical source data are provided.
Gene sets that convey stemness can predict the prognosis of acute myeloid leukaemia (AML)52,53. Hence, aberrantly upregulated genes could enhance the survival of H3.3-null HSPCs and partially overlap with leukaemic programmes. Indeed, IL3RA is upregulated in AML compared with normal HSCs (Fig. 6k)54. Increased IL3RA and RELB expression reduced the survival probability of patients with AML (Fig. 6l and Extended Data Fig. 10k). Increased expression of HUSH complex components, SETDB1 and TRIM28, correlated with an increased probability of survival (Extended Data Fig. 10l,m). Altogether, H3.3 maintains H3K9me3 at repressed enhancers; following the loss of H3.3, the reduction of H3K9me3 augments aberrant myeloid gene expression and altered cell-cycle programme (Fig. 6m). Therefore, H3.3 sustains homeostasis, silences inflammatory signalling and prevents maladaptation of HSPCs to the environmental stressors by safeguarding heterochromatin H3K27me3 and H3K9me3 enrichment.
Discussion
Deciphering the molecular underpinning of how HSCs decide to undergo coordinated self-renewal or commit to differentiation is a central question of adult stem cell biology. Here we focused on the role of the replacement histone variant H3.3, a critical regulator of dynamic chromatin55–58. Deposition of H3.3 colocalizes with active PTM H3K4me3 marks59, bivalent H3K4me3–H3K27me3 marks at developmental genes13 and H3K9me3 repressive marks at LTR ERV regions (Extended Data Fig. 7)12. As such, mouse models with adult-inducible deletion of H3.3 serve as unique tools to test the hypothesis that H3.3-dependent PTMs are integral for stem cell maintenance. By comparing the haematopoietic phenotype and epigenetic changes of DKO mice with Hira-KO and Daxx-KO mice, we uncovered a role for heterochromatin in modulating HSC function. H3.3 maintains HSC stemness and prevents the differentiation of HSCs towards GMPs by repressing global spreading of H3K27me3 and silencing ERV-induced inflammation through deposition at H3K9me3 marks. In H3.3-null HSCs, H3K9me3 is reduced at key genes in the surviving cells, thereby enhancing their adaptive survival.
Hira has been shown to confer chromatin accessibility in adult HSCs60. The differences between the previously reported Hira-KO phenotypes and those observed in this study could be a result of the different models used, including developmental haematopoietic Vav1–cre versus the global adult-inducible Rosa26creERT2+ and the different Hira floxed alleles in the transgenic mice. Nonetheless, both of these studies highlight the role of H3.3A and H3.3B as epigenetic regulators in developmental and adult haematopoietic homeostasis.
H3.3 deposition safeguards active transcription.
Constant replacement of H3.3 maintains accessible chromatin configuration at active genes25,59,61,62. Our in vitro expansion and in vivo transplantation assays demonstrated that H3.3 is cell-autonomously required for sustaining the number and stemness of LT-HSCs and B-cell differentiation. H3.3-null HSPCs mimic the GMP phenotype, with both surface expression of the GMP marker CD16/32 and GMP-like transcriptomic signatures. Chromatin-state dynamics have demonstrated the smallest ratio of active/inactive enhancers in GMPs2. In the differentiation of wild-type LKS cells to GMPs, 513 genes were upregulated compared with 2,523 genes that were downregulated. The downregulated genes in DKO HSPCs largely overlapped with the genes that were downregulated in the LKS-to-GMP differentiation (Fig. 3f), including the genes involved in stem cell self-renewal and lineage differentiation (Fig. 3e). Epigenetically, H3K27me3 was enriched at the promoter regions of the downregulated regions (Fig. 3g,i). Thus, in the adult HSPCs, through bookmarking the actively transcribed key stemness genes and preventing the spreading of repressive H3K27me3 marks (Fig. 3j), H3.3 supports the self-renewal and balanced differentiation of HSPCs into GMPs. The target genes regulated by H3.3 are primarily stemness and inflammatory genes, such as Il3ra and Spib, which have been to shown to regulate HSPC homeostasis. Increased Id1 expression in the DKO LKS cells reflects the adaptive responses of the cells to strengthen survival in the event of global epigenetic changes, rather than being responsible for the increased myelopoiesis and reduced HSC repopulating function.
H3.3 maintains constitutive heterochromatin.
H3.3 deposition into constitutive heterochromatin is dependent on Daxx7. The Daxx–H3.3 complex functionally represses retrotransposon elements/ERVs, maintains telomere structure and represses gene expression6,7. How heterochromatin is maintained in adult HSPCs is unknown. When H3.3 was deleted from HSPCs, there was a reduction in H3K9me3 mountains, a proportion of which were located near the telomeres. In HSPCs, the deletion of H3.3 led to a reduction of H3K9me3 at class I and II ERVs (Fig. 5a), dysregulated ERV expression and increased gene expression associated with the defence response to viruses, interferon-β and the immune-system responses. The dysregulated ERVs correlated with heightened interferon response and elevated expression of CD16/32 mRNA and protein, promoting the myeloid bias of DKO HSPCs (Extended Data Fig. 10i). When the Jak1–Jak2 signalling downstream of interferon signalling pathway was blocked, there was not only a reduction in DKO LKS cells but also a reduction in CD16/32+ LKS cells, suggesting the requirement of active Jak1–Jak2 signalling to drive myeloid bias. Notably, Jak1–Jak2 signalling enhances the expression of Hira and Id1 but antagonizes the expression of Daxx. Therefore, crosstalk might exist between signalling pathways and histone deposition pathways (Hira and Daxx), collectively contributing to dynamics of epigenetic landscape in DKO HSPCs.
H3.3-dependent H3K9me3 regulates transcription.
H3.3-mediated H3K9me3 deposition represses gene expression in HSPCs. In H3.3-null HSPCs, H3K9me3-reduced ERV regions could serve as enhancers. The ERV regions with H3K9me3 reduction are enriched in transcription factor binding sites including Nr2f2, Spi1, Myc and Relb. Notably, the IRF motif is prevalent at the ERV regions near to the upregulated genes (Il3ra, Fcgr2b and Fcgr3), suggesting the co-option of innate immune-gene upregulation with ERV derepression via the interferon signalling pathway. This list of genes with increased mRNA expression and reduced H3K9me3 near the distal enhancer regions includes myeloid genes and genes involved in apoptosis and proliferation kinetics. Thus, H3.3 deletion and reduced H3K9me3 at telomere regions and ERVs may initially lead to increased apoptosis and elevated inflammatory signalling; ultimately, H3K9me3-reduced ERV regions serve as a mechanism to increase the adaptive fitness of persisting H3.3-null HSPCs.
We identified a role for H3.3 in maintaining adult HSC homeostasis and balanced lineage output. Maintenance of HSPC stemness and lineage differentiation is epigenetically scripted; loss of H3.3 leads to a predominant gain of H3K27me3 marks, enforcing GMP differentiation. Meanwhile, H3.3 deletion opens up ERV regions, which potentially function as enhancers to induce myeloid genes and proliferation-related genes, allowing H3.3-null HSPCs to persist via H3K9me3-mediated mechanisms. The changes of dynamic chromatin configuration in HSCs, as influenced by a unique histone variant H3.3, can serve as a paradigm for our understanding of the fine balance between normal and pathological haematopoiesis.
Note added in proof:
The Salomoni group has recently published a paper63 which demonstrates similar findings in their Mx1-cre, Daxx-KO and our DKO mice, which include increased percentages of neutrophils in the spleen and BM, increased percentages and numbers of GMPs, an altered transcriptomic signature of Daxx-KO and DKO HSPCs towards GMPs, derepression of ERVs in HSPCs and an increased distal enhancer motif marked with ETS transcription factors such as Pu.1. Due to its well-established role as a H3.3 chaperone in the heterochromatin regions including ERV repeats, telomeric regions and repressive enhancer marks, it is highly probable that Daxx mediated the shared phenotypic changes in DKO mice.
Methods
Study approval.
All breeding of animals and animal experiments were performed under the approval of Weill Cornell Medicine Institutional Animal Care and Use Committee. The animal protocol numbers are 2009–0061 and 2015–0008. To compare the phenotypes between different mouse genotypes, sex-and weight-matched control and H3.3 DKO mice were used. The mice used in experiments had a body weight of 20–28 g.
Animals.
The parental ESC line used to generate both H3.3A-and H3.3B-KO mice was derived from F1 hybrid embryos crossed with 129SV and C57BL/6J mice. The H3.3A-floxed ESC line was generated through knock-in of loxP sites to exon 3 of the h3f3a gene via a conventional recombinational approach. The H3.3B−/− ESC lines were generated through knock-in of a fluorescence (EYFP/mCherry) reporter and a stop codon to replace the three exons of the h3f3b gene via zinc finger nucleases. The mutant mice were produced through generation of chimaeras with the targeted ESCs, which were intercrossed with the germline transmission mice to obtain homozygous mutant mice. The CD45.1 NSG mice used for the BM transplantations were purchased from The Jackson Laboratory (strain 005557).
Rosa26creERT2+ mice were purchased from The Jackson Laboratory (https://www.jax.org/strain/008463). The Rosa26creERT2+; H3.3Afl/fl, H3.3B−/− breeder and offspring mice were maintained in a B6/129 background and backcrossed to C57BL/6J mice more than five times.
Genotyping.
Tail DNA was used for genotyping using a KAPA HS mouse genotyping kit (KAPA Biosystems, KK7352). The PCR reaction was carried out using the KAPA2G fast HotStart genotyping mix with dye and the following protocol: 95.0 °C for 180 s; 35 cycles of 95 °C for 15 s, 64 °C for 15 s and 72 °C for 30 s; and 72 °C for another 5 min. The primers used are provided in Supplementary Table 11.
Tamoxifen administration.
Tamoxifen (4-hydroxyl tamoxifen; Sigma, T5648–5G) was dissolved in sunflower oil (Sigma, S5007) at a concentration of 20 mg ml−1. The mice (8–12 weeks old) were intraperitoneally injected with tamoxifen at a dose of 100 mg kg−1 for 3 d, rested for 3 d, and then administered another three doses of tamoxifen for another 3 d. The haematopoietic phenotypes were analysed at 2, 3, 8 and 16 weeks after the tamoxifen treatment.
Complete blood counts.
The mice were anaesthetized in an anaesthetic vaporizer delivery system delivering vaporized isoflurane (Henry Schein Animal Health, 1169567762) into the chamber at constant flow rate of 2.5 l min−1 at 3–5% for the initial induction and kept at 1–2% after the first 5 min; the oxygen (Tech Air) flow rate was adjusted to 1.0 l min−1. Blood was drawn from the retro-orbital plexus using capillary tubes (1.1 mm × 75 mm, colour-coded, red; Kimble Chase, 41B2501). A tube of blood (about 66 μl) was added into 200 μl of 10 mM EDTA in PBS buffer. After brief vortexing, the blood was run on an ADVIA120 haematology analyzer (Siemens). The white blood cell, RBC, platelet, haemoglobin and haematocrit counts as well as the differential count of neutrophils versus lymphocytes were obtained.
Flow cytometry analysis of cells in the peripheral blood, BM and spleen.
The mice were anaesthetized using isofluorane according to the protocol described earlier. Two calibre tubes of blood were drawn from the retro-orbital veins and added into 200 μl of 10 mM EDTA in PBS buffer. After brief vortexing, the sample was put on ice until all of the blood samples had been collected. The RBCs were lysed on ice using 5 ml of 1×RBC lysis buffer (BioLegend, 420301) for 10 min. The cells were then transferred into an Eppendorf tube for staining. The cells were incubated with mouse FcR block in 50 μl MACS buffer for 10 min. Antibodies to Ter119, CD3, B220 and Gr1/CD11b were then mixed to generate a master mix and added to each tube. For analysis of the multilineage components in the BM and spleen, single-cell suspensions of haematopoietic cells were obtained and then stained with antibodies to Ter119, CD3, Gr1, CD11b and B220. For all of the experiments, unstained sample or the fluorescence-minus-one rule was used for the gating of positive populations. Unstained control and single-stained cells or ultracompensation beads (eBiosciences, 01–2222-42) were used to calculate the fluorescence spillover from all channels. The cells were analysed using the flow cytometry analyzer or sorter on the BD Diva software, V8.02. For the subsequent gating and analysis of cell populations, FlowJo 10.4.0 was used. The full list of antibodies and their dilutions are provided in Supplementary Table 12.
Quantification of HSPCs in DKO mice.
The mice were euthanized at the indicated time points following the tamoxifen injections. Their femurs and tibias were dissected. Following muscle clean-up, two femurs and two tibias per mouse were ground in 5 ml MACS buffer (PBS + 2 mM EDTA + 0.5% BSA + 1% Penicillin/Streptomycin/Amphotericin B) using a mortar and pestle. The cells were filtered through a 40 μm cell strainer. The remaining bones were ground twice more following the same procedure until the bones became white. The total number of filtered cells was counted as the total haematopoietic cells (excluding mature RBCs) in the BM. Lineage depletion was carried out for the haematopoietic cells using a direct lineage depletion kit (Miltenyi, 130–110-470). Lineage antibody cocktail (50 μl) was added to the cells obtained from the two femurs and two tibias. The numbers of LinN cells were recorded. The LinN cells were blocked with rat IgGs for 5 min on ice and then stained with the HSC and progenitor cell markers c-Kit, Sca1, CD150, CD48, CD34, CD16/32 and CD105 to identify the different cell populations—LT-HSC, GMP, CMP, megakaryocyte erythroid progenitor, and so on. The total number of HSCs per femur and tibia was calculated using the total number of LinN cells and the frequencies in the CD150+CD48−c-Kit+Sca1+ gating. Similarly, the total number of GMPs, or any other progenitor cell type, was calculated using the total number of Lin−cKit+ cells and frequency of the respective cell type.
To determine the total number of spleen cells, the weight of the whole spleen was recorded. A small fraction of the spleen was cut out and weighed. We could then infer the total number of mononuclear cells in the spleen or thymus by determining the total cell number in the spleen fraction. For the quantification of BM and spleen lineage cell types, the cells were blocked with mouse FcR block for 5 min on ice. The cells were stained with an antibody cocktail (anti-Ter119, -CD11b, -Gr1 and -B220). The percentages of myeloid and B cells were obtained. The total numbers of lineage cell types were calculated by multiplying the total cell number in the spleen and the percentage of lineage-positive cells.
In vitro E4-HUVEC and HSPC co-culture.
Human umbilical endothelial cells (HUVECs) were enzymatically digested from umbilical cord. Passage 1 or 2 HUVECs were transduced with lentivirus encoding adenoviral E4ORF1 (E4-HUVECs). Positively transduced E4-HUVECs were selected using the selection medium X-VIVO-20 (Lonza, 04–448Q) for 4 d. The E4-HUVECs were then cultured in endothelial cell growth medium for expansion until the co-culture assays were set up. The E4-HUVECs were split into 12-well plates and cultured to confluency. Lineage depletion was carried out to enrich the LinN cells from the femurs of Rosa26creERT2−H3f3afl/flH3f3b−/− and Rosa26creERT2+H3f3afl/flH3f3b−/− mice. The LinN cells (0.1 × 106) were plated into one well of a 12-well plate and cultured in StemSpan medium (Stemcell Technologies, 09650) supplemented with knockout serum replacement (Thermo Fisher Scientific, 10828028) and cytokines (10 ng ml−1 Flt3, 20 ng ml−1 sKitL and 10 ng ml−1 TPO). Fresh medium (1 ml) was added to the co-culture on day 2. On day 4, the culture was further expanded by collecting the floating haematopoietic cells and culturing them on a fresh layer of E4-HUVECs. Tamoxifen was added to the medium on day 5 at a final concentration of 1 μg ml−1. The expansion results were analysed on days 2, 4, 8, 11 and 14 post tamoxifen treatment. Suspension and attached cells from the wells were detached using accutase cell detachment solution (Corning). The majority (90%) of the cells were blocked with rat IgG and stained with lineage antibodies (antibodies to c-Kit, Sca1, CD34 and CD16/32). The remaining cells (10%) were blocked with mouse FcR block and stained for CD11b, Gr1 and B220 to examine the lineage-differentiation potential. Cell counting beads were used to quantify the total cell number as well as the total number of LinN and LKS cells. The HSCs and haematopoietic progenitor cKit+Lin− cells were sorted on a BD FACS Aria II system. The HSCs were split into two samples, one portion was used for RNA extraction using a PicoPure RNA extraction kit (Thermo Fisher Scientific) and the other for the native ChIP–seq experiment, which were performed on the same day of the cell harvest.
Treatment with Rux in vitro.
LinN cells were co-cultured with HUVECs for 5 d before H3.3A deletion was induced with tamoxifen. At the same time of the tamoxifen treatment, Rux (Sigma-Aldrich, G-6185) or vehicle was added to the culture at a concentration of 10 μM according to the manufacturer’s instructions. After 3 d, all of the cells were detached using accutase cell detachment medium (Corning), stained with antibodies and subjected to flow cytometry. The cells were quantified using an automatic cell counter (Nexcelom) and the number of LKS cells was determined using the total expanded cells times the percentages of LKS cells obtained from the flow cytometry analysis.
Cell-cycle and apoptosis assay.
Equal numbers of total expanded cells from each condition were used. Following cell-surface staining of c-Kit, Sca1 and lineage antibodies, the cells were fixed with 1% paraformaldehyde (PFA) at room temperature for 3 min and the PFA was washed off. The cell pellet was then permeabilized with BD cytoperm/cytofix buffer (BD) and stained with anti-Ki67 (BD, 561165) for 30 min in BD cytoperm buffer according to the manufacturer’s protocol. After washing once with cytoperm buffer, the cells were stained overnight with Hoechst 33342 (Invitrogen) at a final concentration of 20 μg ml−1. For apoptosis analysis, equal numbers of total expanded cells from each condition were used. The cells were washed in MACS buffer and resuspended in Annexin V binding buffer (BioLegend, 422201). Annexin V antibody (5 μl) was added to each sample. DAPI (BioLegend, 422801) was added at a final concentration of 1 μg ml−1. The samples were run on a BD LSRII system on the same day of the sample preparation without additional washing.
Quantitative real-time PCR.
For whole-organ mRNA expression analysis, a sample of tissue from the lung, liver or BM was homogenized in RNA lysis buffer, RLT buffer (Qiagen) with 1% β-mercaptoethanol. The RLT solution was cleared of undigested tissue using a QIAthredder column (Qiagen). The RNA was prepared following the RNeasy protocol (Qiagen). The DNA was digested using DNase I (Qiagen) during the RNA extraction process. The concentration of the extracted RNA was determined using a NanoDrop machine (Thermo Fisher Scientific). The RNA (200 ng) was converted to complementary DNA using qScript Super Mix (Quanta Biosciences). After cDNA dilution with double-distilled H2O at a 1:5 ratio, the quantitative PCR reaction was set up by mixing the gene-specific primers and SYBR Green qPCR reaction mix (KAPA Biosystems). The quantitative PCR was run and detected using a ViiA-7 qPCR machine (Life Technologies, Thermo Fisher Scientific). The primer sequence(s) for the different genes are provided in Supplementary Table 11. For RNA extraction of abundant cells, a Qiagen RNeasy kit was used. For RNA extraction of low numbers of cells, a PicoPure RNA isolation kit (Life Technologies, Thermo Fisher Scientific) was used.
RNA-seq and DEG analysis.
HSPCs were sorted from the in vitro co-culture system at the indicated time points following tamoxifen treatment. For the in vivo experiments, HSPCs from BKO or DKO mice were sorted on days 14, 21 and 56 following H3.3 deletion. The total RNA was extracted. The RNA concentration was measured and a quality check was performed using an Agilent 2100 Bioanalyzer (Agilent). The integrity of the RNA is indicated by the RNA integrity number. RNA samples with sufficient concentration and an RNA integrity number greater than 8.0 were used for cDNA library preparation using poly-A selection and unstranded library preparation using a Truseq library preparation kit (Illumina). The DNA library was then sequenced using an Illumina Hiseq 4000 system (single read, 50 cycles). The reads from the RNA sequencing (RNA-seq) were aligned to mouse genome version mm9 using TopHat64. The fragments per kilobase of transcript per million fragments mapped (FPKM) was determined using Cufflinks65 with upper-quartile and GC normalization. After log2 transformation of the FPKM values, DEGs were identified using the limma packaged in R. A cutoff of P < 0.05 and FC < −log2(1.8) was used to identify downregulated genes. A cutoff of P < 0.05 and FC > log2(1.8) was used to identify upregulated genes. The gene ontology function was revealed using the DAVID Gene Ontology website (https://david.ncifcrf.gov/), based on biological process and molecular functions.
ChIP of histone PTMs.
For the detection of the H3.3 binding sites in HSPCs, LinN cells from H3.3B–HA-IRES-mCherry mice were first co-cultured with E4-HUVECs for 7 d to obtain sufficient cells. The LKS cells were sorted on day 7. A total of 3 × 106 cells were collected and fixed in 1% PFA for 10 min at 37 °C. The PFA was washed off using 125 mM glycine and the pellet was snap-frozen using liquid nitrogen. The nucleosome was extracted using a TruChip chromatin shearing reagent kit (Covaris, 520127). The DNA was sheared using a Covaris E220/S220 system for 20 min in cold circulating H2O. ChIP was performed using a ChIP-IT high sensitivity kit (Active Motif, 53040). The sonicated nucleosomes were incubated overnight with antibodies to HA (Thermo Fisher Scientific, clone 12CA5), H3K4me3 (Abcam, ab8580), H3K27me3 (Millipore, 07–449) and H3K27ac (Abcam, ab4729). The DNA was eluted and a library was prepared using ThruPLEX DNA-seq kit dual index (Rubicon Genomics, R400523).
For ChIP–seq of LKS cells cultured in vitro or freshly isolated LKS cells from mouse BM, between 5,000 and 20,000 sorted LKS cells were used for two or three ChIP reactions. The nuclei were extracted using NP40 buffer and digested into single nucleosomes using diluted MNase (NEB, M02475), followed by overnight incubation with antibodies to: H3K4me3 (Cell Signaling Technology, 9727), H3K9me3 (Active Motif, 39161), H3K27me3 (Diagnode, C15410069, pAb-069–050) and H3K27ac (Active Motif, 39133). The nucleosomes were washed twice with low-salt wash buffer and twice with high-salt wash buffer at 4 °C in a rotator. The DNA was eluted using SDS–NaHCO3 buffer at 65 °C for 1.5 h, extracted using phenol:chloroform:isoamyl alcohol (Invitrogen, 15593–031) and precipitated using 2-isopropanol (Sigma, 190764) at −80 °C overnight or longer. The DNA library was prepared using a KAPA hyper kit (KK8504) according to the product manual. The adaptor was from the KAPA single-indexed adaptor kit (KAPA, 8700 08005699001). The DNA was purified using the DNA purification kit AMPure XP (60 ml; Beckman Coulter, A63800). The DNA library was sequenced on the Illumina platform, using Hiseq 4000 (single read, 50 cycles). Two or three biological experiments were performed for each histone mark examined in the BKO or DKO LKS cells.
Identification of significantly changed peaks using SICER.
Raw reads (fastq files) from ChIP–seq were aligned to the mouse genome mm9 using bwa. Duplicate reads and reads aligning to more than one location were excluded. After quality control and alignment, bam files were obtained. The bam files were then converted to bed files. SICER66 was used to identify the peaks for H3.3 (HA) by comparison to the input control sample using: FDR < 0.01; fragment size, 150 base pairs (bp); window size, 200 bp; and gap size, 600 bp. For the identification of significantly decreased or increased H3K9me3, H3K27me3 and H3K27ac peak regions, bed files for BKO H3K9me3, BKO input, DKO H3K9me3 and DKO input were used. The parameters used were as follows: FDR < 0.01; fragment size, 150 bp; window size, 200 bp; and gap size, 600 bp. For H3K4me3 peak calling, a gap size of 200 bp was used. The peak-region bed files were annotated to the nearest TSS using the HOMER annotatepeaks.pl function. To visualize the changes in H3K9me3 at a given locus, the ChIP–seq bam files were converted to tdf files and then visualized using the Integrative Genomics Viewer browser (Broad Institute). Normalization was selected for visualization. Annotations of RepeatMasker were loaded from the UCSC Table Browser by selecting the desired class of repeats.
To calculate the changes in histone PTM profiles between wild-type and BKO or DKO samples, deepTools was used. Bam files were first converted to bw files using the bamcoverage function with a binsize of 10 bp and the normalization method of RPGC (the mm9 genome size is 2,620,345,972 bp). To compare the H3K9me3 marks at the significantly decreased or increased H3K9me3 regions in wild-type and DKO HSPCs, the computematrix scale region function was used. The normalized tag density at these peak regions was reported by the ‘outFileNameMatrix’ function, with each row representing the rows from the original bed files and the column representing the tag density within the binned chromosome regions. The mean tag density of wild-type and DKO H3K9me3 were generated using the plotProfile function. The heatmap and k-means clusters or Hclusters were generated using the plotHeatmap function.
Histone modification changes and motif analysis of the promoter regions of DEGs.
To examine how the histone modification changes influence gene expression, after identifying the DEGs, the promoter regions of the up- or downregulated genes were extracted to form a bed file. The Bedtools ‘intersectBed’ function was used to find the overlapping regions between the H3K9me3-decreased peaks and the promoter regions of upregulated genes. To identify the transcription factors that influence of the DEGs, we carried out motif analysis for H3K27ac-marked enhancer regions or the promoter regions of the DEGs. Motif discovery was carried out using HOMER, using the 400 bp size option. The transcription factors identified were defined by HOMER. The reported transcription factors were selected based on the P value.
Quantification of H3K9me3 changes at ERVs.
The genome coordinates of different repfamilies of repeats were downloaded from the UCSC genome browser. The deepTools compute matrix (reference point) function was used to calculate the tag density (H3K9me3) within each repeat region for wild-type and DKO HSCs. The matrix was exported using the outfileNameMatrix function; the boxplots of the tag density at specified regions were plotted using R. A Student’s t-test was used to test whether the resulting wild-type and DKO H3K9me3 tags were significantly different from each other.
Quantification of changes in ERV mRNA expression.
Two methods were used to quantify the changes in mRNA expression of ERVs. The first was quantification using RNA-seq methods, followed by the analyzerepeat function of HOMER. In detail, RNA-seq raw reads were mapped to the mouse genome mm9 using TopHat tools. The resulting bam files were converted to bed files. The make tag directory and analyzerepeat function were then used to generate the read count within each repeat region. The read counts were normalized to ten million mapped reads. The resulting matrix loaded in R; differentially expressed ERVs were calculated using the package limma. A cutoff of P < 0.05 was used to find the significantly changed ERVs. The genome coordinate bed or gtf files for a specific repfamily, such as MMERVK10C-int and IAPEz-int, were downloaded from the UCSC table browser. To unbiasedly identify the significantly changed ERVs across the genome, gtf files for LTR class of repeats from chromosome 1 were downloaded from the UCSC table browser. The name and locus of the significantly changed ERV were identified according to the above method. The coding genes near the up-or downregulated ERVs were annotated using HOMER. The second method to quantify the changes of a repfamily of ERVs was via quantitative real-time PCR. The primer sequences and their original descriptions are summarized in Supplementary Table 11.
Statistics and reproducibility.
Differences in the phenotypic analysis of the experimental parameters were statistically compared using unpaired two-tailed t-tests, paired t-tests or a two-way ANOVA. A P value of less than 0.05 was considered to be statistically significant. Experiments were repeated at least two or three times. For Fig. 3g, n = 2 independent biological samples for BKO and DKO at each time point and n = 1 biological sample for Hirafl/fl and Hira-KO. For Fig. 3h, n = 2 biological samples for each column. For Extended Data Fig. 7a, n = 1 biological sample. For Extended Data Fig. 8e,f, n = 2 biological samples for d14; and n = 1 biological sample for d56, Hirafl/fl and Hira-KO. For Extended Data Fig. 8l, n = 2 biological samples for each genotype. For Extended Data Fig. 9a, n = 2 independent biological samples. For Extended Data Fig. 9b, n = 1 biological sample for each column. The variance was similar between the groups being statistically compared. If higher variability was found, we increased the sample size to fully confirm statistical significance. No statistical method was used to pre-determine the sample size for a given effect size. No data were excluded from the analyses. The investigators were blinded to the genotypes of the animals.
Extended Data
Extended Data Fig. 1 |. H3.3A and H3.3B are redundant during steady state hematopoiesis.
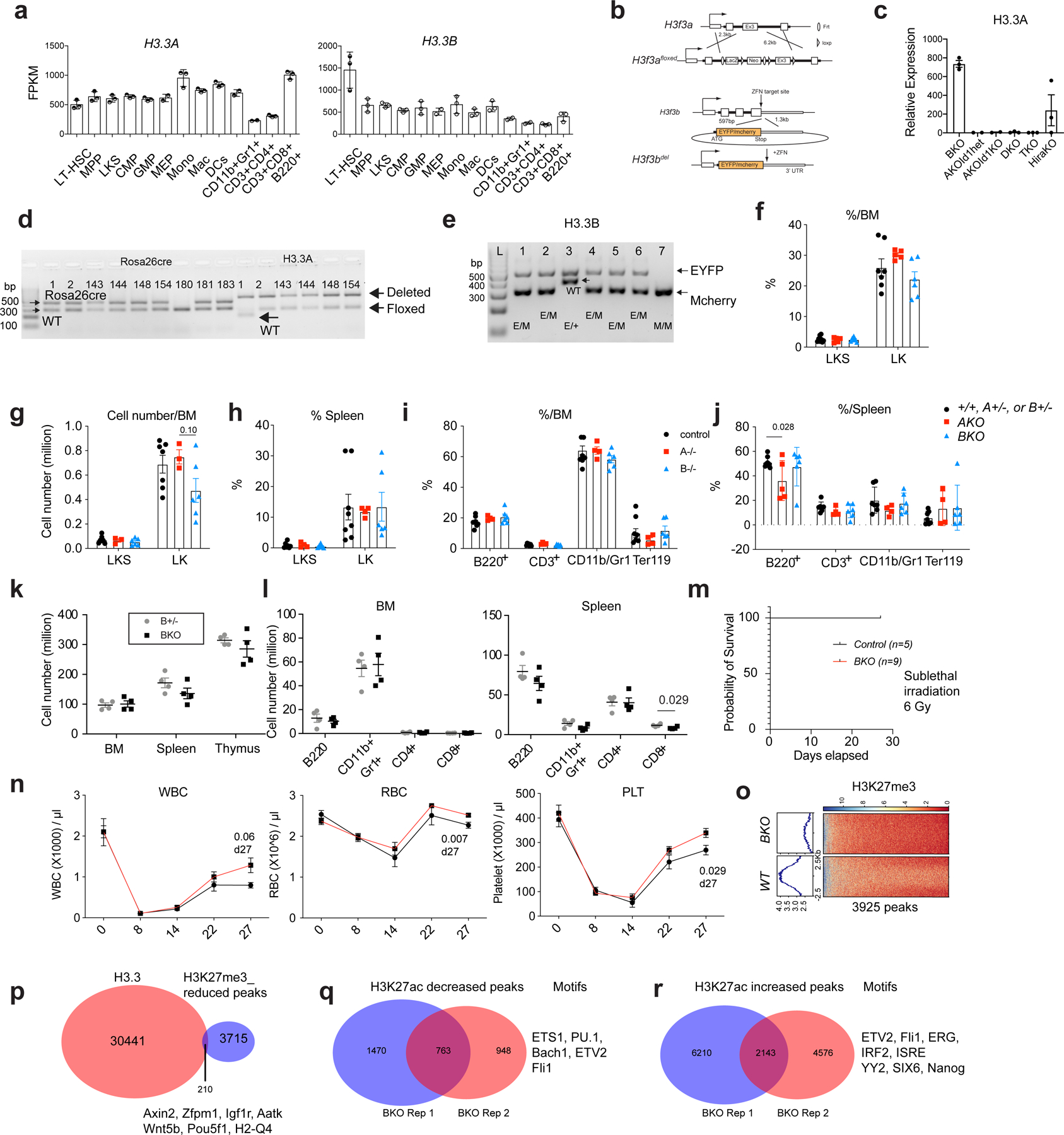
a. the Fragments Per Kilobase of transcript per Million mapped reads (FPKM) of H3.3A and H3.3B in hematopoietic cells. LT-HSC, long-term hematopoietic stem cells; MPP, multipotent progenitor cells; CMP, common myeloid progenitor cells; GMP, granulocyte macrophage progenitor cells; MEPs, megakaryocyte erythroid progenitor cells. Mono, Monocytes; Mac, macrophages; DCs, dendritic cells; CD11b+Gr1+ cells, granulocytes. The number of dots indicated the number of biological replicates. b. The design of the H3.3Afl/fl allele and the H3.3BEYFP/mcherry knockin knockout allele. c. qRT-PCR quantification of H3.3A mRNA expression within LKS cells after tamoxifen treatment. TKO is Id1mut/mut; DKO mice. d-e, genotyping of Rosa26creERT2, H3.3A floxed, WT, and deletion allele; H3.3B-EYFP or H3,3-mcherry allele (n>3 biological replicates were used). f-g. Percentages and numbers of c-Kit+Sca1+Lin− (LKS) or c-Kit+Sca1−Lin− LK cells within control (+/+, H3.3A+/−, or H3.3B+/−), H3.3AiKO/iKO (AKO), and H3.3B−/− (BKO) mice. h. Percentages of LKS and LK cells among LinN cells of control, AKO, and BKO mice. i. percentages of lineage cells within BM or spleen of control, AKO, and BKO mice. j. The total number of cells within BM, spleen, and thymus of H3.3B+/− (Bhet) or BKO mice. k-l, The total number of lineage cells with BM and spleen or Bhet or BKO mice. m-n, Recovery after myelosuppression with sublethal total body irradiation at 600 cGy. m, Survival probability after total body irradiation. n. white blood cells (WBC), red blood cells (RBC) and platelets (PLT) in the PB of control or BKO mice. o. Trimethylation at histone 3 lysine 27 (H3K27me3) profiling at WT or BKO LKS cells. Genome-wide, there were 3925 regions with reduced H3K27me3 enrichment. p. 210 of the H3K27me3 reduced regions in BKO LKS cells colocalize with H3.3 enrichment. q-r. We also profiled the enhancer mark, H3K27ac in BKO or WT cells, and identified the regions with increased or decreased H3K27ac enrichment. Motifs within those regions are also shown. For panels a, c, f-l, the number of dots indicate the number of animals. For m, n, n=5 mice for control group and n=9 for BKO group. Error bars indicate standard error of mean. p-value is calculated using unpaired, 2-tailed t-test. For h, p-value for the data points at day 27 was calculated using unpaired, 2-tailed t-test. Numerical source data are provided in Source Data.
Extended Data Fig. 2 |. Adult inducible deletion of H3.3A, but not its chaperone Hira resulted in myeloid bias.
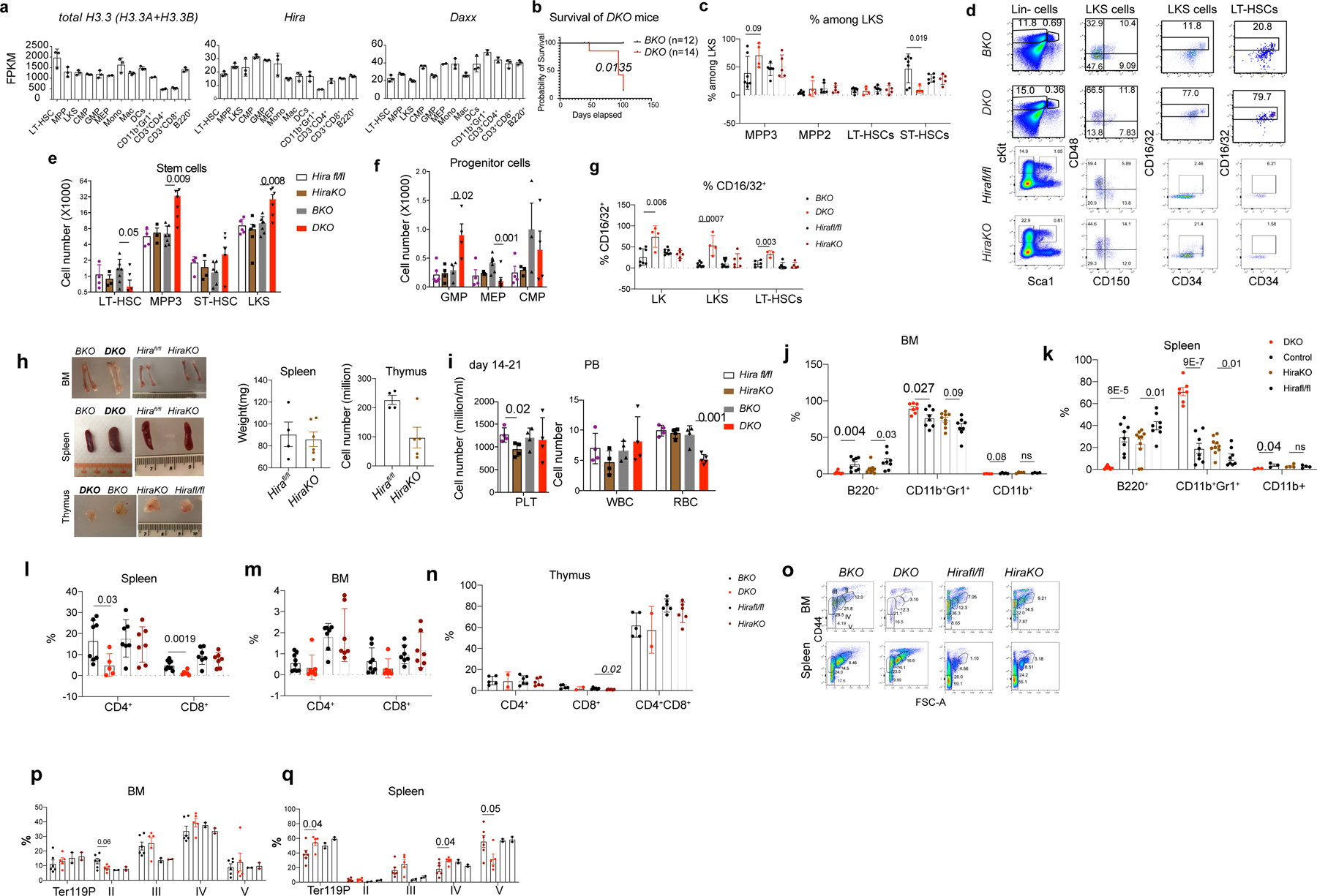
a. FPKM of total H3.3, calculated as the sum of H3.3A and H3.3B, Hira, and Daxx in hematopoietic cells. b. Survival probability of DKO or BKO mice following tamoxifen treatment. c. Representative flow cytometric plots for the gating of LKS cells, LT-HSCs (CD150+CD48− LKS), MPP3 (CD150−CD48+ LKS), MPP2 (CD150+CD48+ LKS), and ST-HSCs (CD150−CD48− LKS) for BKO, DKO, Hirafl/fl or HiraKO mice. d-e. The percentages and the total number of LT-HSCs, MPP3, ST-HSCs, and LKS cells. f. The total number of progenitor cells at 2–3 weeks after H3.3 or Hira deletion. GMP, CD16/32+CD34+LK; MEPs, CD16/32−CD34−LK-cells; CMP, CD16/32−CD34+LK. g. At week 2, 3, 4, or 7 after tamoxifen injection, the upregulation of GMP marker CD16/32 on LKS and LT-HSCs in DKO mice. h. BM, spleen, and thymus of BKO, DKO, Hirafl/fl or HiraKO mice. Note the BM anemia phenotype and enlarged spleen for DKO mice. On the right, the spleen weight and thymocytes numbers are shown for HiraKO mice. i. CBC for BKO, DKO, Hirafl/fl or HiraKO mice. j-k. the percentages of granulocytes or B cells in the spleen or BM. l-n, the percentages of CD4+, CD8+, and CD4+CD8+ cells within BM, spleen, and thymus of the indicated mice. o. Representative flow cytometric plots of the erythroid cell clusters II-V. p-q. the percentages and numbers of Ter119+, erythroid cells II, III, IV, and V within the BM and spleen of BKO, DKO, Hirafl/fl, and HiraKO mice. For panels 2a, c, e-n, p-q, the number of dots indicates the number of animals Error bars indicate standard error of mean (SEM). P-values are calculated using unpaired, two-tailed, t-test. ‘ns’, non-significant. Numerical source data and unprocessed gels are provided in Source Data.
Extended Data Fig. 3 |. Histology of hematopoietic organs demonstrated myeloid hyperplasia in H3.3DKO mice.
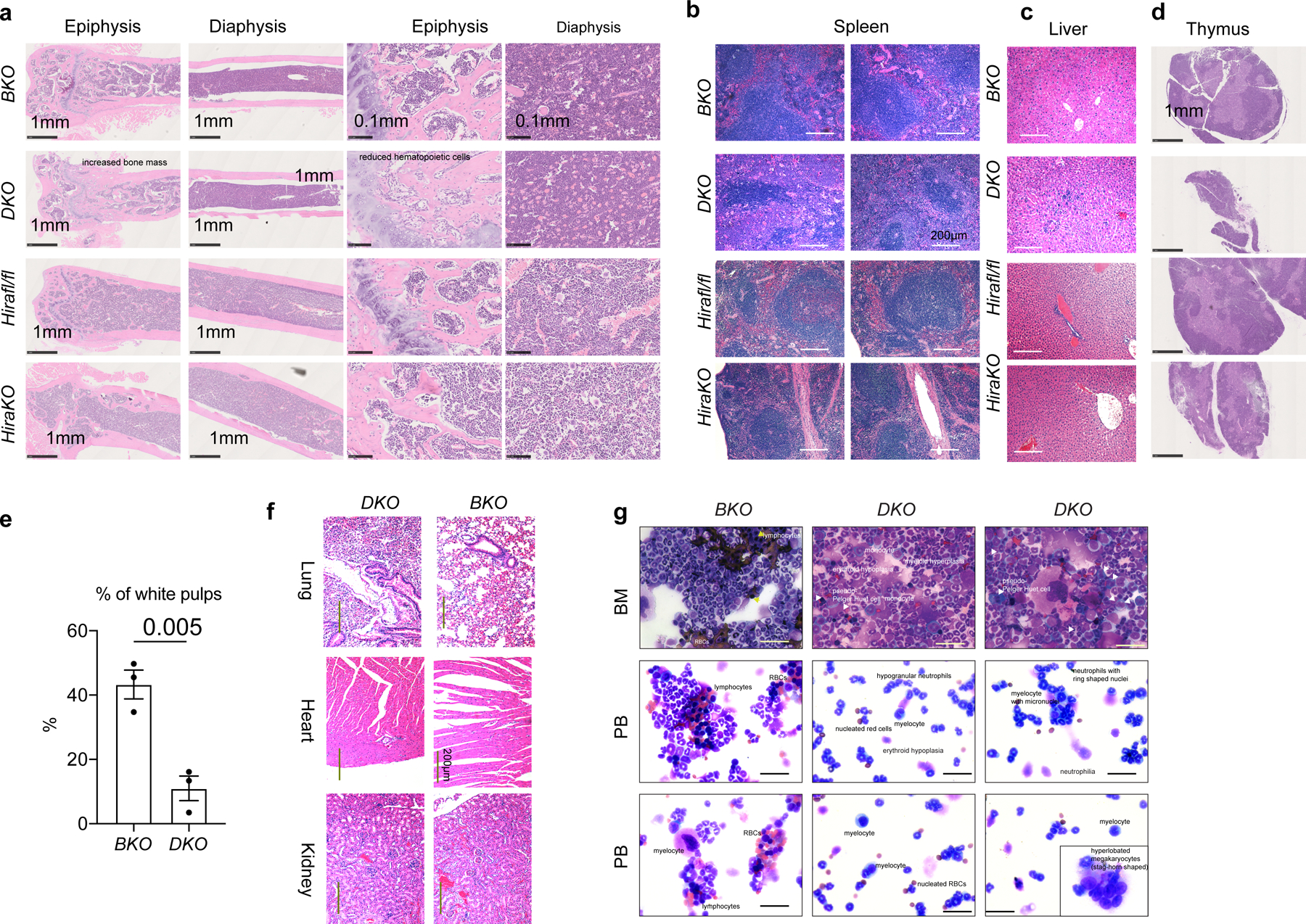
a. Hematoxylin and eosin (HE) staining of the BM epiphysis and diaphysis of BKO, DKO, Hirafl/fl, and HiraKO mice. Note the increased bone mass and reduced hematopoietic cells in the epiphysis region of DKO mice, compared with BKO mice. b-d. HE staining of spleen (b), liver (c), and thymus (d) for BKO, DKO, Hirafl/fl, and HiraKO mice. Note the significantly smaller white pup in the spleen of DKO, but not that of HiraKO mice; the infiltration of hematopoietic cells in the liver of DKO mice, but not that of HiraKO mice. For a-d, n=4 independent animals for each genotype. e. Quantification of the percentages of the white pulp area in the spleens of BKO and DKO mice. The number of dots indicates the number of independent animals per experiment. The experiment in a-e were repeated 2–3 times. Error bar indicates SEM. P-value was calculated using two-tailed t-test. f. HE staining of lung, heart, and kidney from BKO and DKO mice. There were leukocyte infiltrations in the heart and kidney of DKO mice. g. Qiff-quik staining of BMMNCs and PB of BKO and DKO mice. Upper panel, BMMNCs, note the presence of red blood cells and lymphocytes in the BKO mice; however, DKO mice demonstrated myeloid hyperplasia, erythroid hypoplasia, and Peudo-Pelger huet cells (white arrows). Middle and lower panels, PB. In BKO PB, there were lymphocytes, RBCs, and neutrophils; in DKO mice, the PB contains predominantly myeloid cells (myeloid hyperplasia), including nucleated red cells, hypogranular neutrophils, ring-shaped neutrophils, myelocytes, myelocyte with micronuclei, and hyperlobated megakaryocytes (stag-horn shaped). For g, scale bar is 50 μm for each figure panel. For f-g, n=3 biological samples were used. Numerical source data are provided in Source Data.
Extended Data Fig. 4 |. H3.3 maintains hematopoietic homeostasis and HSC repopulation.
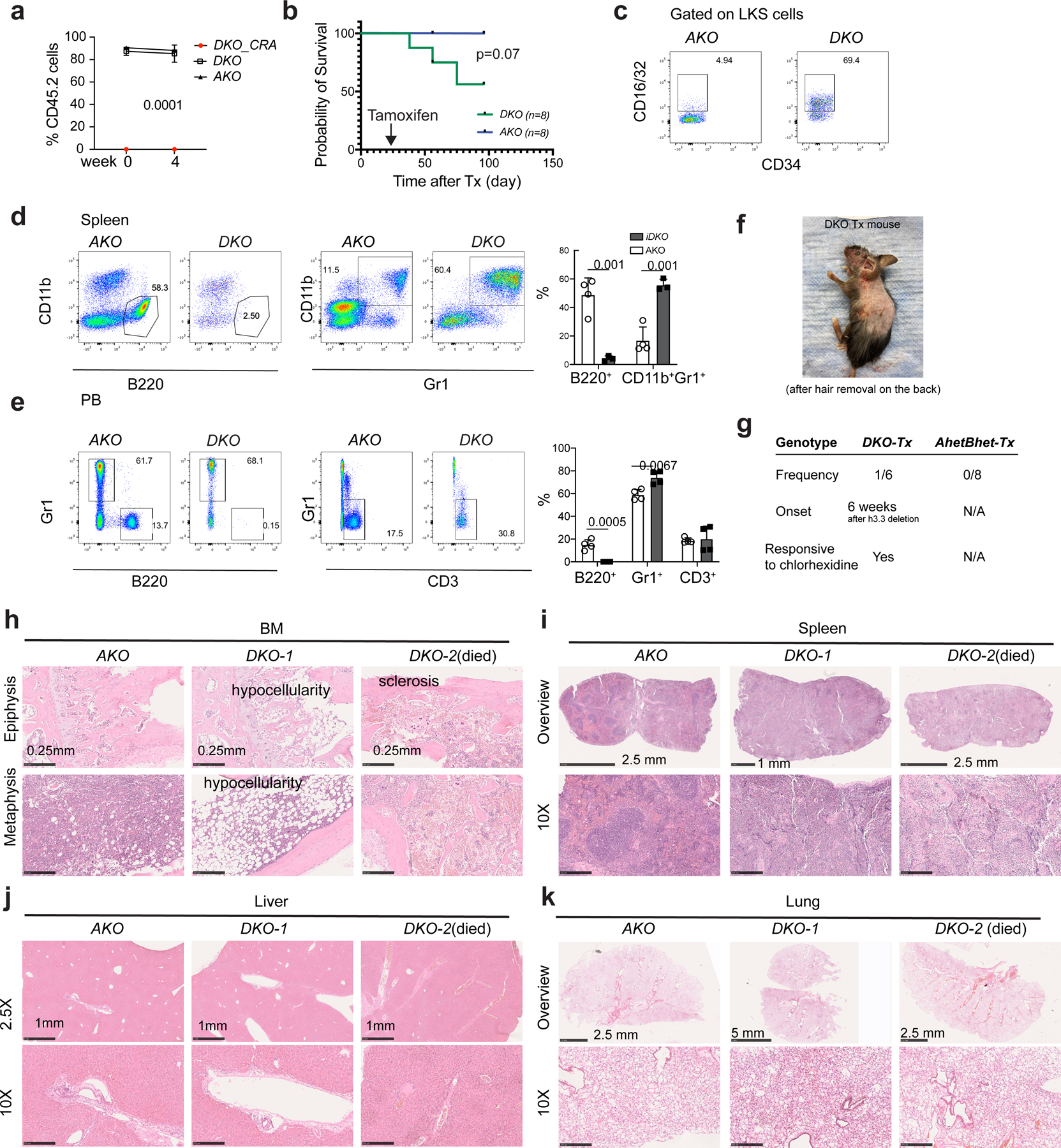
a. Percentages of CD45.2+ cells at the week 0 and week 4 after tamoxifen injection; note the absence of DKO-CRA cells in the PB of recipients CD45.1+ recipients before tamoxifen induced deletion of H3.3A gene, suggesting a disadvantage of BKO HSPCs to repopulate the wild type BM in a competitive setting. Experiments were repeated twice; each with n=4 animals for each group. b. Survival probability of mice with different transplantation schemes. c. Representative flow cytometric plots for CD16/32+ LKS cells in the recipients transplanted with DKO or AKO BMMNCs. d. Percentages of B220+ B cells and CD11b+Gr1+ granulocytes in the spleen of recipients transplanted with DKO or AKO BMMNCs. e. Percentages of B220+ B cells, Gr1+ myeloid cells, and CD3+ T cells in the PB of recipients transplanted with DKO or AKO BMMNCs. f-g. Images showing the skin lesions of the DKO BMMNCs-transplanted recipient animals, at week 7 after H3.3A deletion. The frequency, onset, and treatment for such skin lesion is listed in g. h-k. Hematoxylin and eosin (H and E) staining of BM, spleen, liver, and lung of recipients transplanted with DKO or AKO BMMNCs (n=3 for DKO, n=4 for AKO). For the BM, note the hypocellularity and sclerosis at the epiphysis and metaphysis regions. For the spleen, note the purple pro-fibrotic fibrous structures and disappearances of white pups in the DKO mice. There was no apparent hematopoietic cell infiltration into the liver for DKO BMMNC cells transplanted mice. For a, p-value is calculated using two-way ANOVA. For panels d, e, the number of dots indicates the number of independent animals. Error bars indicate standard error of mean. For d, e, p-value is calculated using unpaired, two-tailed t-test; the number of dots equals to the number of biological replicates in d, e. Numerical source data are provided in Source Data.
Extended Data Fig. 5 |. H3.3 maintains HSC repopulation in a competitive setting.
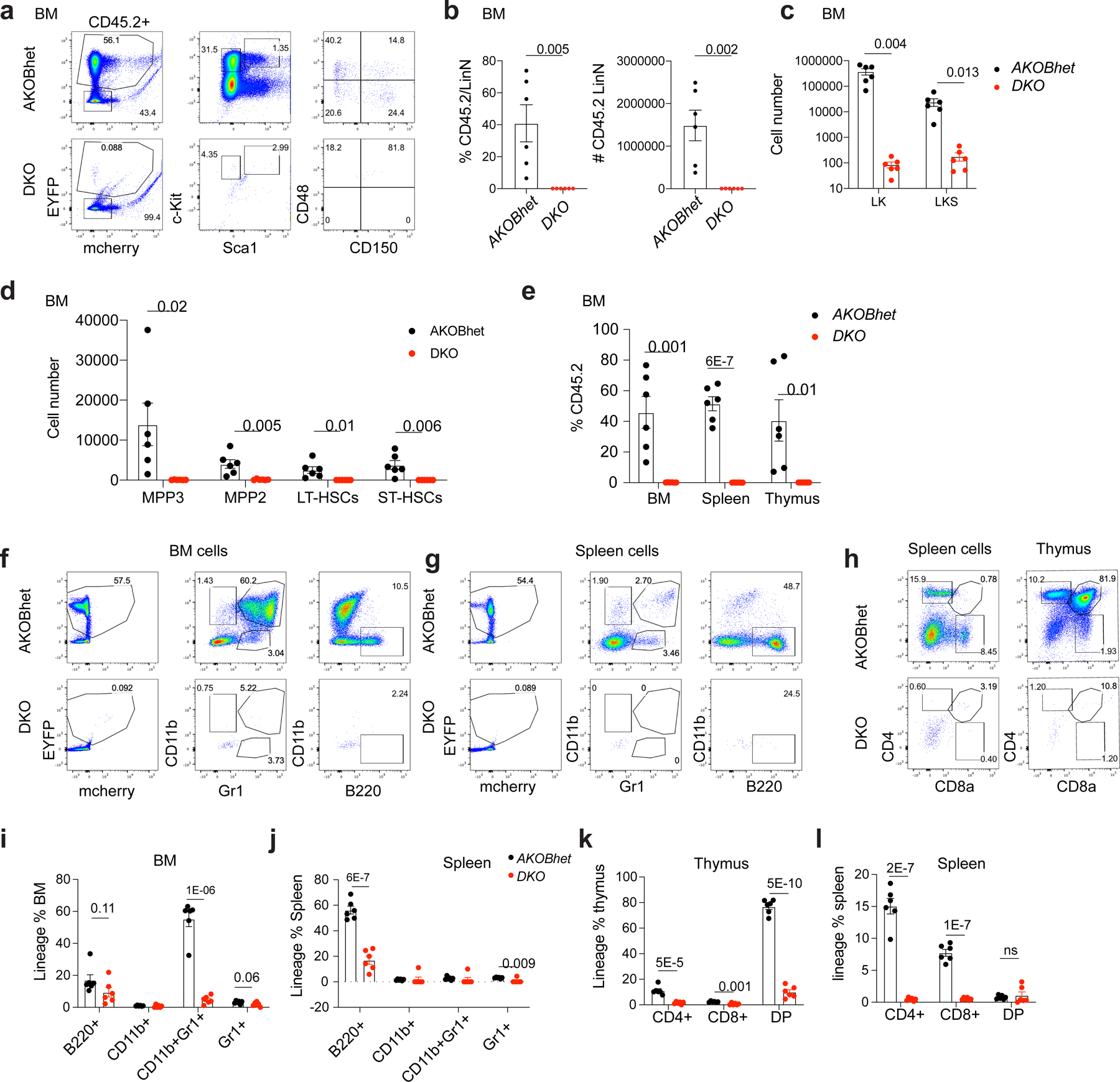
a. Representative flow cytometric plots for the CD45.2+ LinN cells and LT-HSC at week 16 following tamoxifen injection. b. The percentage and total number of CD45.2+ LinN cells within the BM of recipient mice. c. The percentages of CD45.2+ cells within the BM, spleen, and thymus of recipient mice. d-e. The total numbers of stem and progenitor cells, and lineage positive cells within the BM of recipient mice. f-g. Representative flow cytometric plots for the lineage cells (CD11b+, CD11b+Gr1+, Gr1+, or B220+ cells) within the BM or spleen of recipient mice. h. Representative flow cytometric plots for lineage cells (CD4+, CD8+, or CD4+CD8+ double positive cells) within thymus or spleen. i-j. The percentages of lineage-specific cells within BM or spleen of the recipients. k-l, The percentages of lineage cells within thymus or spleen of the recipients. For all the panels, n=6 animals for AKOBhet group; n=6 animals for DKO group. Error bars represent standard error of the mean. p-value is calculated using unpaired, 2-tailed t-test.
Extended Data Fig. 6 |. H3.3 prevents HSC myeloid differentiation.
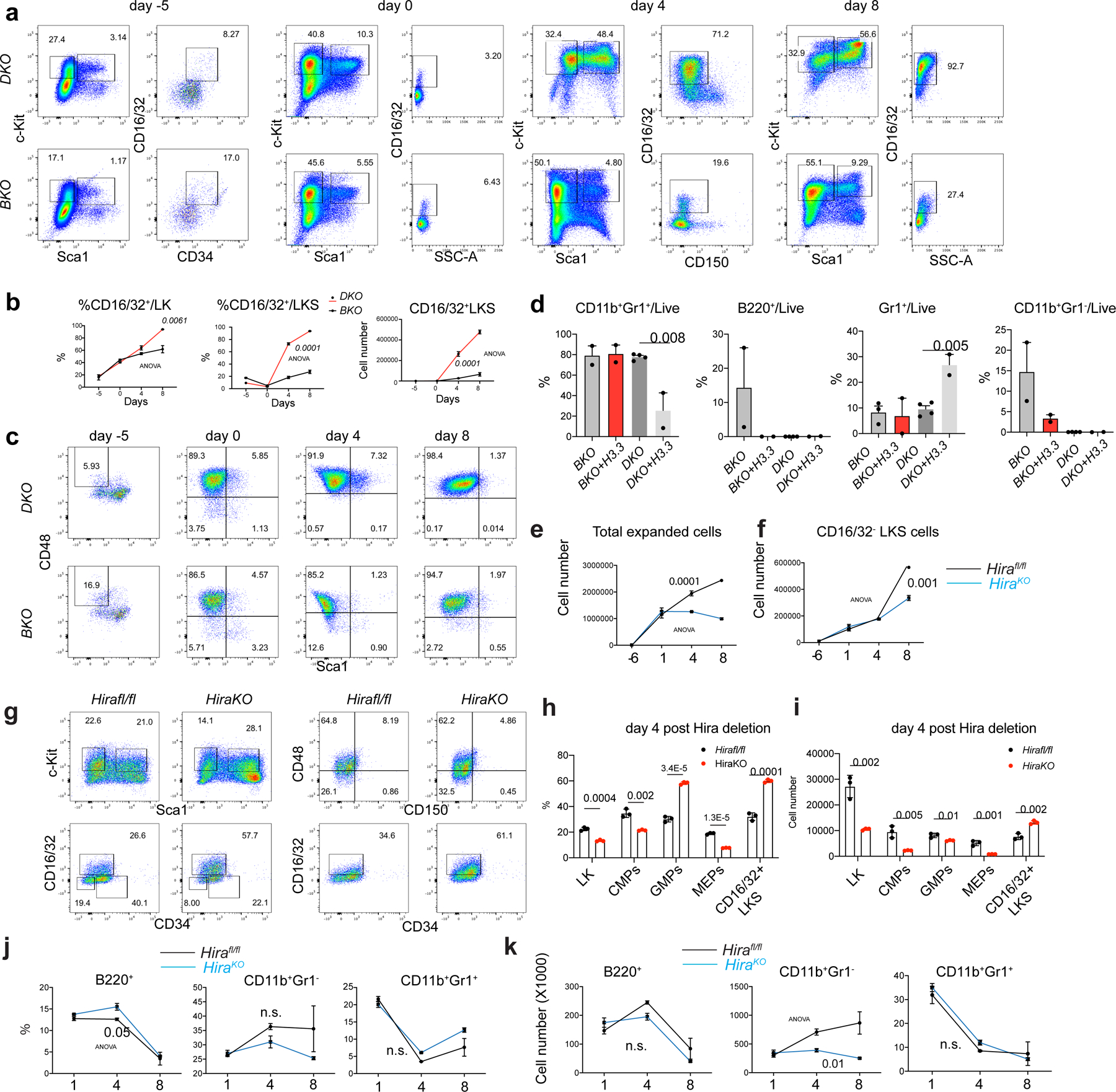
a. Representative flow cytometric plots for the kinetics of LKS cell at different time points after tamoxifen treatment. Note the emergence of CD16/32+ LKS cells with the increase of culture time, and the elevated percentages of CD16/32+ cells in DKO LKS cells, compared with BKO LKS cells. b. percentages of the CD16/32+ among LK or LKS cells. c. Representative flow cytometric plots for the LT-HSCs during the co-culture. d. Lineage differentiation potential of BKO and DKO cells. There is reduced capacity for DKO to differentiate into B220+ B cells; overexpressing H3.3 did not rescue this defect. e. We set up in vitro culture assays for Hirafl/fl and Rosa26creERT2+ ;Hirafl/fl (HiraKO) HSCs. The growth curve of total hematopoietic cells are shown. f. the growth kinetics of total number of CD16/32+LKS cells. g. Representative flow cytometric plots for the LKS cells, LT-HSCs, GMPs, and CD16/32+ LKS cells in Hirafl/fl or HiraKO cultures. h-i, at day 4 after Hira deletion, the percentages and total numbers of LK, CMPs, GMPs, MEPs, and CD16/32+ LKS cells. j-k. the percentages and the total numbers of lineage cells, including B220+ B cells, CD11b+Gr1+ early myeloid cells, and CD11b+Gr1+ granulocytes. For panels 6b, d, e-f, h-k, the number of dots indicate the number of independent biological samples per experiment. Error bars indicate standard error of mean (SEM). For panels d, h, and i, P-values were calculated using unpaired, two-tailed, t-test; for panels b, e, f, j, and k, p-value was calculated using two-way ANOVA. Numerical source data are provided in Source Data.
Extended Data Fig. 7 |. H3.3 enrichment colocalizes with active histone marks H3K4me3 and repressive histone mark H3K27me3 or H3K9me3 within HSPCs.
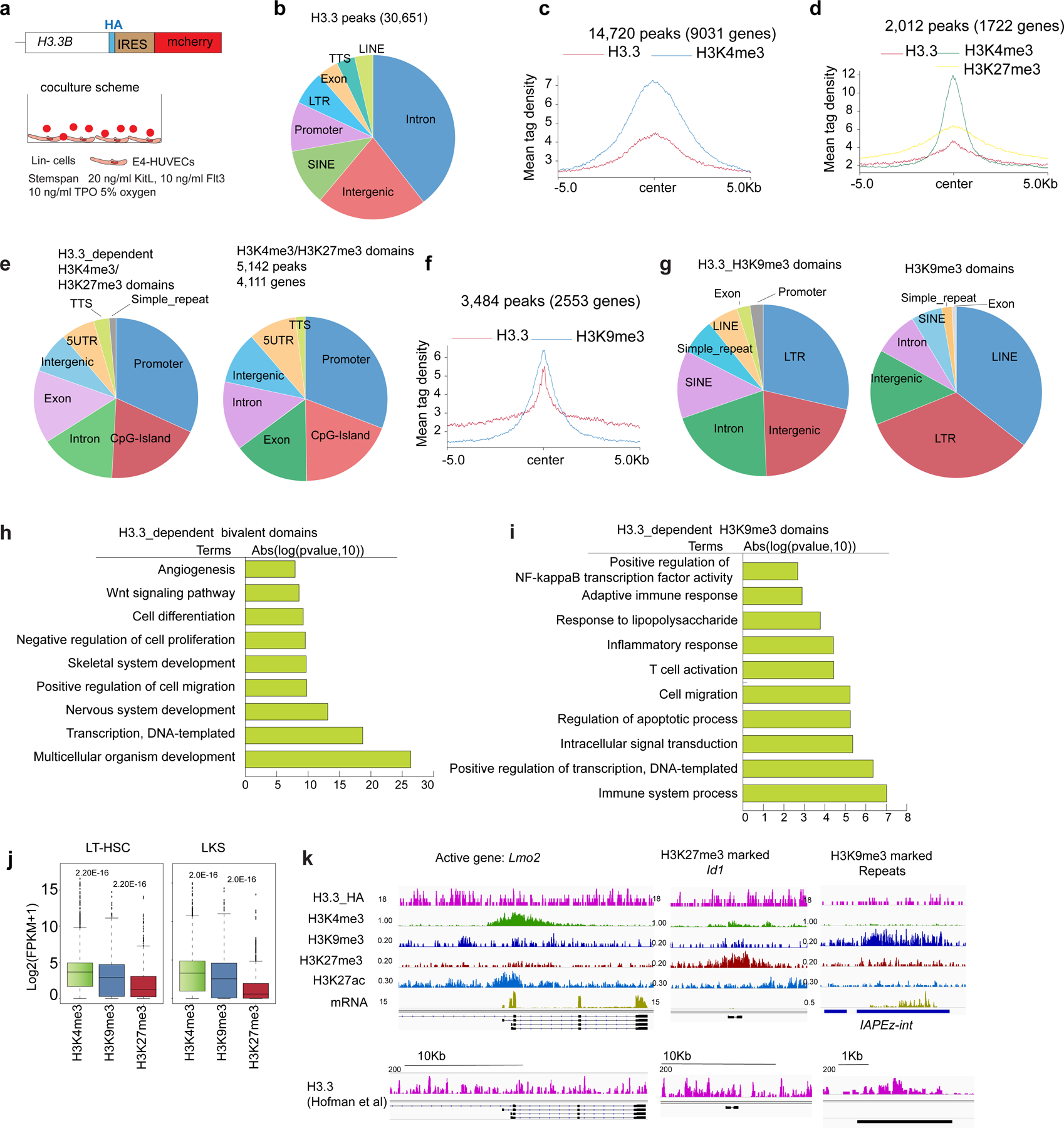
a. Schematic view of the H3.3B-HA-IRES-mcherry knockin mice22 used to map the H3.3 binding site within HSPCs. Lin− cells were co-cultured with E4-HUVECs for 1 week to expand LKS cells; ChIP-seq was performed using HA antibody. n=1 biological sample. b. Genome-wide distribution of 30,651 H3.3 peaks in LKS cells. Across the genome, there are 14,720 H3K4me3_H3.3 overlapped peaks (c), 2,012 H3.3 dependent H3K4me3/H3K27me3 bivalent peaks (d), and 3,484 H3K9me3_H3.3 overlapped peaks (f). e. The genome-wide localization of the H3.3 dependent H3K4me3/H3K27me3 regions. g. The genome-wide localization of the H3.3 dependent H3K9me3 marked regions. For panels c, d, e, f, ChIP-seq for histone modifications H3Km4e3, H3K9me3, H3K27me3 has been performed on n=3 biological samples. h. Genes at or near the H3.3-dependent H3K4me3/H3K27me3 bivalent regions. i. Genes at or near the H3.3 dependent H3K9me3 marked regions. j. There are a total of 9,031 genes with H3.3-dependent H3K4me3 enrichment, 2,533 genes with H3.3-dependent H3K9me3 enrichment, and 1,722 genes with H3.3-dependent H3K4me3/H3K27me3 enrichment. The expression levels of these three categories of genes are shown for both LT-HSCs and LKS cells. p-values were calculated using two-tailed t-test using one biological sample for each cell type. The experiment was repeated three times. The boundaries of box indicate the 25 and 75 percentiles, center indicate median number, whiskers indicate 1.5X interquartile range. k. Representative genome browser view of enrichment of H3.3, H3K4me3, H3K9me3, H3K27me3, H3K27ac and mRNA expression levels for active genes, bivalent genes and repetitive elements. Also shown here is the genome browser view of H3.3 enrichment using anti-H3.3 antibody, kindly shared by another group. P-value is labelled on the top of the box plot in panel j.
Extended Data Fig. 8 |. Dynamic changes of transcriptomic landscape within HSPCs following H3.3 deletion in vitro and in vivo.
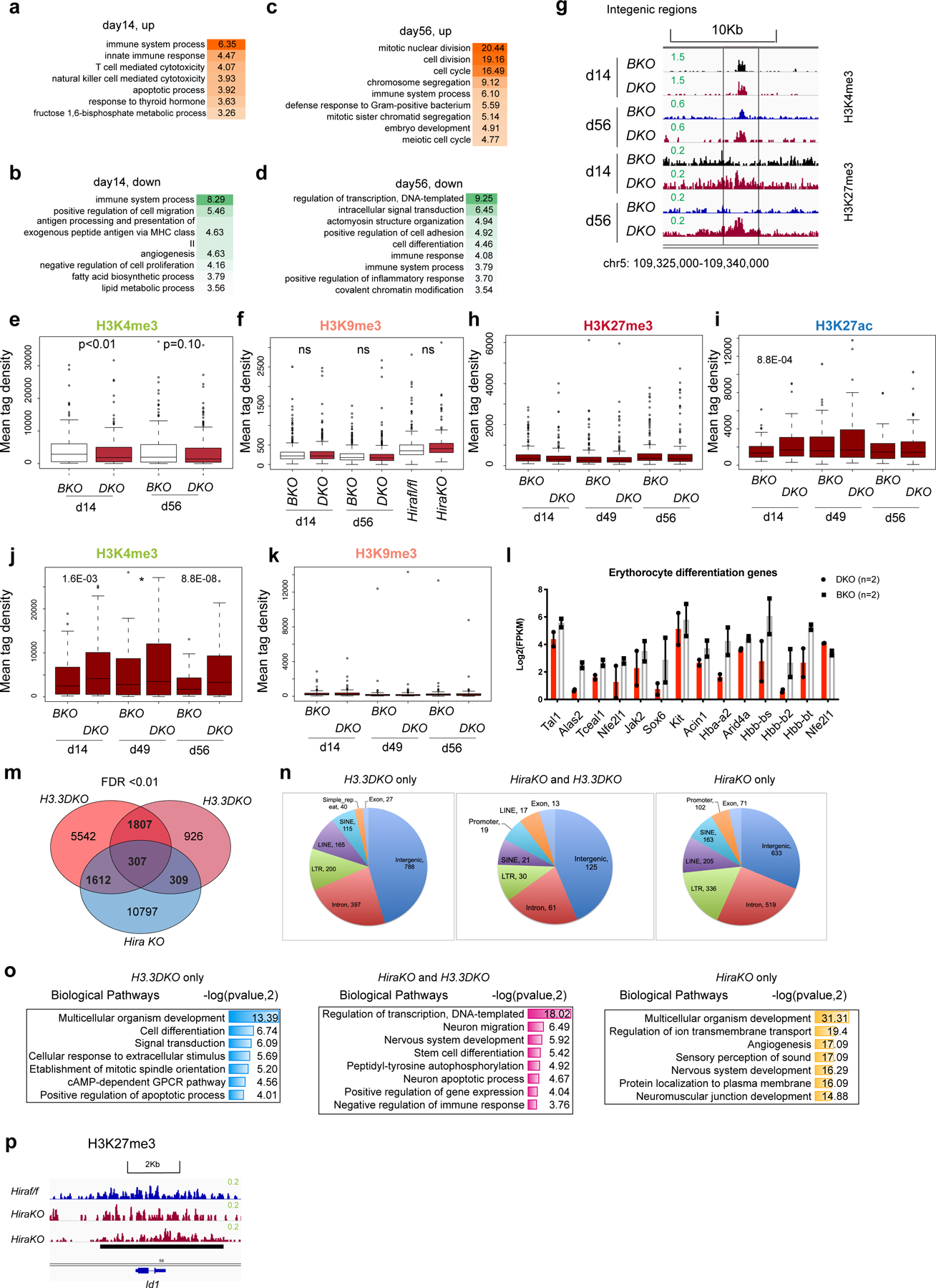
a-b, DAVID gene ontology biological pathway of differentially expressed genes (DEGs) at day 14-post H3.3 deletion in DKO LKS cells, compared with BKO LKS cells (in vivo). c-d, DAVID gene ontology biological pathway analysis of DEGs at day 56 post H3.3 deletion in DKO LKS cells, compared with BKO LKS cells (in vivo). e-f, the mean tag density of H3K4me3 and H3K9me3 at the promoter regions of the 385 downregulated genes associated with PC1. P-values were calculated using unpaired two-tailed t-test using one biological sample of each genotype. The experiment was performed twice independently for d14, once for d49, d56, and once for Hirafl/fl and HiraKO. g. Representative genome browser view of H3K4me3 and H3K27me3 intensity at intergenic region, at day 14 and day 56 post H3.3A deletion. h-k. For the commonly upregulated genes at d14 and d56 (171 genes), the enrichment of histone modifications H3K27me3, H3K27ac, H3K4me3, and H3K9me3 at the promoter regions is shown. For panels h-k, P-values were calculated using unpaired two-tailed t-test using one biological sample of each genotype. The experiment was performed twice independently for d14, once for d49, d56, and once for Hirafl/fl and HiraKO. l. Downregulation of key genes involved in erythrocyte differentiation within DKO LKS cells, compared with BKO LKS cells (n=2 biological samples). m, Venn diagram showing the overlap between the H3K27me3-reduced peaks within H3.3DKO cells and HiraKO LKS cells. n. The genome-wide distribution of DKO only, HiraKO and H3.3DKO shared, and HiraKO only H3K27me3-reduced peaks. o. The genes nearby the H3K27me3_reduced peaks are enriched in distinct biological pathways, as shown in the table. p. The H3K27me3 enrichment around the promoter regions of Id1 gene in Hirafl/fl or HiraKO LKS cells. For 8l, error bar indicates standard error of the mean. For the box plots in panels e-f, h-k, the boundaries of box indicate the 25 and 75 percentiles, center indicate median number, whiskers indicate 1.5X interquartile range. For e,-f, h-k, and o, p-values were calculated using two-tailed t-test. P-value is indicated on top of the box plot in panels e and f.
Extended Data Fig. 9 |. Dysregulated ERV expressions in H3.3DKO LKS cells triggered interferon responses, responsible for the myeloid bias.
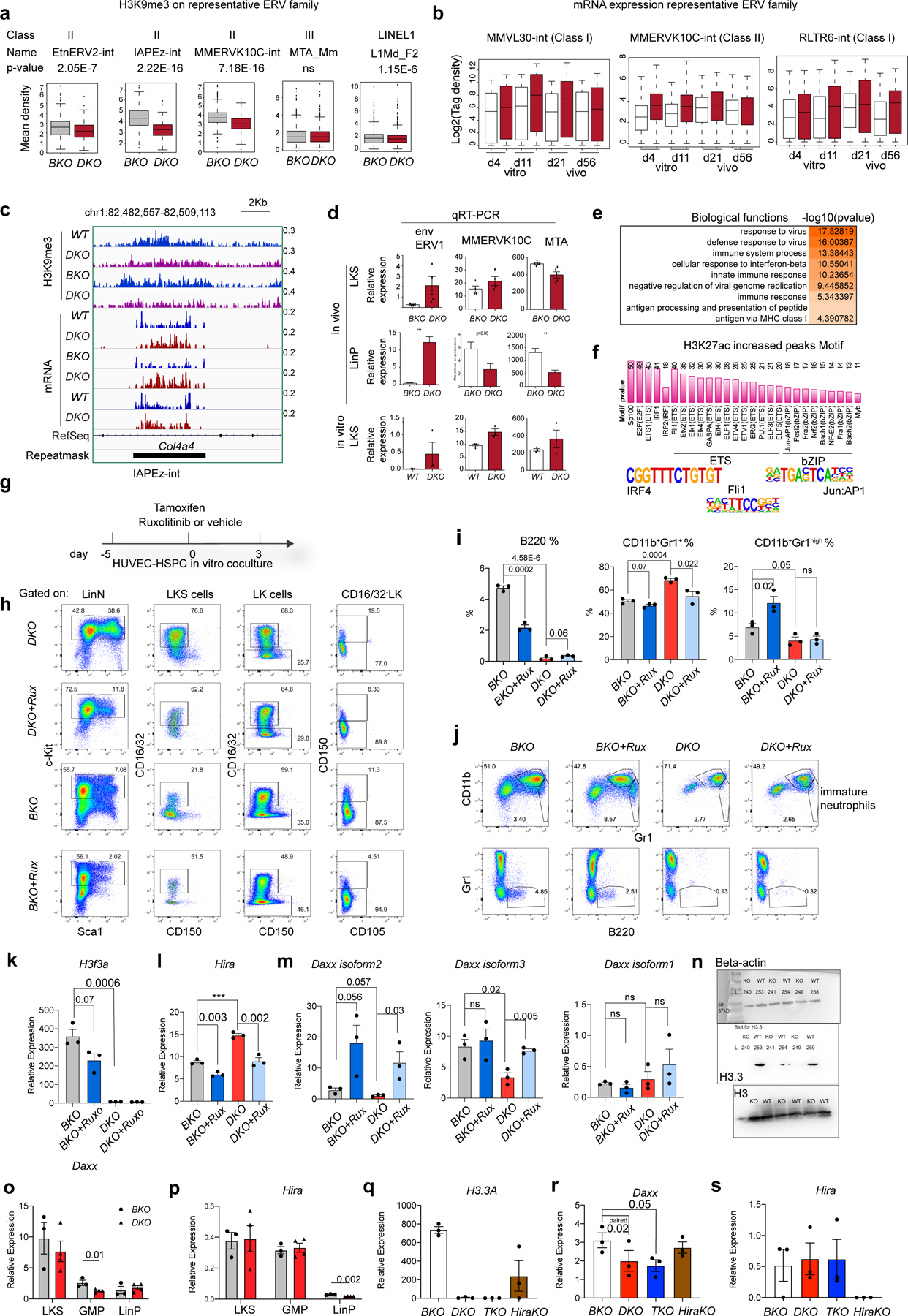
a. H3K9me3 enrichment at representative ERV family. p-value was calculated using unpaired two-tailed t-test using one biological sample for each genotype. The experiment was repeated twice independently. b. The dynamic changes of ERV mRNA expression at in vitro or in vivo settings with DKO and BKO LKS cells (n=1 biological sample for each column). For the box plots in a, b, the boundaries of box indicate the 25 and 75 percentiles, center indicate median number, whiskers indicate 1.5X interquartile range. c. Genome browser plots showing the reduction of H3K9me3 and increased mRNA expression at a IAPEz-int ERV region. d. qRT-PCR quantifications of ERV expression at in vivo or in vitro LKS cells. env encodes for the envelope protein of ERV1. The number of dots indicate the number of biological samples]. e. At day 4 after in vitro deletion of H3.3A gene, the biological pathways associated with upregulated DEGs. f. We performed ChIP-seq for H3K27ac, an enhancer mark. The motifs within the H3K27ac increased regions include interferon pathway downstream transcription factor, Irf. g. The experimental scheme to test the effect of Jak2 inhibitor Ruxolitinib on the expansion and differentiation of HSPCs. h. Representative flow cytometric plots for the LKS, CD16/32+ LKS cells, pre-MegE, and pre-GM populations within the cocultures. i. The percentages of lineage positive cells within the total cultured cells. CD11b+Gr1+, immature neutrophils; CD11b+Gr1high, mature neutrophils. j. Representative flow cytometric plots for the lineage cells within the cocultures. k-m, at in vitro cultures, the mRNA expression of H3.3A, Hira, and Daxx within LKS cells. n-o, Daxx or Hira mRNA expression within LKS cells, GMPs, and LinP cells at in vivo scenarios. p-r, another batch of in vivo experiments, the mRNA expression of H3.3A, Hira, and Daxx within LKS cells was quantified. For panels I, k-m, o-s, the number of dots indicate the number of independent biological samples. Error bars indicate SEM. p-value is calculated using unpaired, two-tailed t-test unless otherwise indicated on the plot. P-value for panel a is labelled in the legend; p-value for panel b is not shown. Numerical source data are provided in Source Data.
Extended Data Fig. 10 |. Reduction of H3K9me3 opens up the accessibility of transcription factor binding sites.
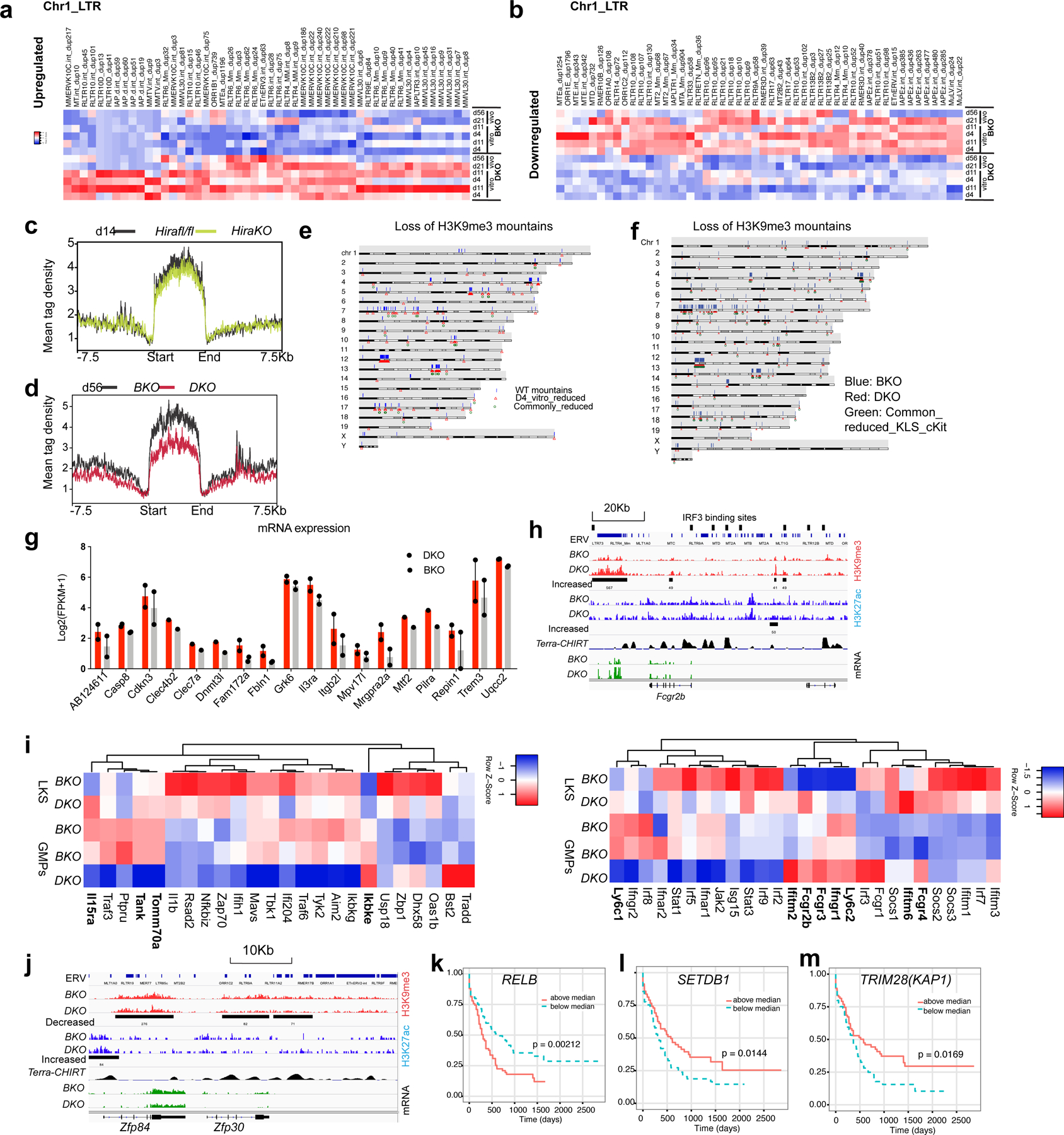
a. Heatmap showing the increased ERV RNA expression within DKO LKS cells, compared with BKO LKS cells in chromosome 1. b. Heatmap showing the decreased ERV RNA expression within DKO cells, compared with BKO LKS cells. c. There is no H3K9me3 reduction mountain in HiraKO LKS cells. d. The H3K9me3 mountain in DKO cells persist into later stage d56 after H3.3A deletion. e. Karyotype plot showing the localization of H3K9me3 mountain in BKO LKS cells (blue bars), reduced H3K9me3 mountain in D4 DKO LKS cells (in vitro) (red triangle), and commonly reduced H3K9me3 mountains within three biological replicates. f. Karyotype plot showing the localization of H3K9me3 mountain in BKO LKS cells (blue bars), reduced H3K9me3 mountain in DKO LKS cells (red triangle), and commonly reduced H3K9me3 mountains within LKS and LK cells. g. The increased mRNA expression near decreased H3K9me3 peaks (distance from TSS < 10Kb) (n=2 biological samples). h. GMP marker Fcgr2b mRNA is upregulated in DKO HSPCs. Near Fcgr2b gene, there is increased H3K9me3 enrichment at ERV site, putative IRF binding site and Terra binding site67. i. Heatmap showing the expression of interferon target genes in BKO or DKO LKS cells, BKO or DKO GMPs. The gene with increased mRNA expression in DKO LKS cells compared with BKO LKS cells were labelled in bold. j, example of reduced mRNA expression near the H3K9me3_reduced_ERV regions. k-m. Kaplan Meyer survival curve for acute myeloid leukaemia patients striated according to the mRNA expression level of RELB (k), SETDB1 (k), and TRIM28 (l)54. For k and m, p-values were calculated using log-rank test. Error bars represent SEM. Numerical source data and unprocessed blots are provided in Source Data.
Supplementary Material
Acknowledgements
We thank M. Ramalho-Santos at the Lunenfeld–Tanenbaum Research Institute, Mount Sinai Hospital for providing the Hirafl/fl mice. We thank L. Banaszynski for generating the plasmid containing the H3.3A floxed allele. We thank B. Kunar for consultation in the bioinformatics analyses. Y.L. is a New York Stem Cell Foundation Druckenmiller Fellow. We thank the Starr Foundation Stem Cell Derivation laboratory at Weill Cornell Medicine for the use of the FACS Aria II/III and BD LRSII systems. We thank the Mount Sinai Dean CoREs Flow Cytometry for their timely help and discussions with flow cytometry analysis and FACS. We thank the Mount Sinai CCMS for help with the complete blood count measurements of mouse peripheral blood. This work was funded by the New York State Stem Cell Science Program (NYSTEM; contract C32581GG to D.W. and S.R.) and grant no. R01 GM129380-01 from the National Institutes of Health (to D.W.). S.R. was funded by the Ansary Stem Cell Institute, National Institute of Health (grant nos R35 HL150809, RC2 DK114777 and U01AI138329), Empire State Stem Cell Board NYSTEM (grant no. C030160), Daedalus Fund for Innovation and Selma and Lawrence Ruben Science to Industry Bridge Fund from Weill Cornell Medicine, Starr Foundation Stem Cell Core Project and initiatives TRI-SCI 2019-029. R.S. was funded by NYSTEM contract C32596GG. B.S.D. was supported by the National Heart Lung and Blood Institute (grant no. R01HL130826) and NYSTEM (grant no. C34052GG).
Footnotes
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
All the code will be available on request, including but not limited to the following: identification of the DEGs, identification of significantly increased or decreased peaks for H3K27me3 and H3K9me3 enrichment, calculation of H3K9me3 at the ERV sites, calculation of ERV mRNA expression and motif analysis.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41556-021-00795-7.
Competing interests
S.R. is the non-paid co-founder of Angiocrine Bioscience (San Diego). The other authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41556-021-00795-7.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41556-021-00795-7.
Peer review information Nature Cell Biology thanks Gerald de Haan, Yuin-Han Loh and the other, anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
The ChIP–seq data, including the fastq data and processed wig files, have been uploaded to the Gene Expression Omnibus website. The accession number is GSE148750. Please refer to Supplementary Table 10 for detailed information. Source data are provided with this paper.
References
- 1.Yu VWC et al. Epigenetic memory underlies cell-autonomous heterogeneous behavior of hematopoietic stem cells. Cell 167, 1310–1322 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Yu X et al. Chromatin dynamics during the differentiation of long-term hematopoietic stem cells to multipotent progenitors. Blood Adv 1, 887–898 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lara-Astiaso D et al. Chromatin state dynamics during blood formation. Science 345, 943–949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corces MR et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet 48, 1193–1203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldberg AD et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140, 678–691 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoelper D, Huang H, Patel DJ & Lewis PW Structural and mechanistic insights into ATRX-dependent and -independent functions of the histone chaperone DAXX. Nat. Commun 8, 1193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewis PW, Elsaesser SJ, Noh K-M, Stadler SC & Allis CD Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl Acad. Sci. USA 107, 14075–14080 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ricketts MD et al. Ubinuclein-1 confers histone H3.3-specific-binding by the HIRA histone chaperone complex. Nat. Commun 6, 7711 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rai TS et al. Human CABIN1 is a functional member of the human HIRA/UBN1/ASF1a histone H3.3 chaperone complex. Mol. Cell. Biol 31, 4107–4118 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pietras EM, Warr MR & Passegué E Cell cycle regulation in hematopoietic stem cells. J. Cell Biol 195, 709–720 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakamura-Ishizu A, Takizawa H & Suda T The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development 141, 4656–4666 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Elsässer SJ, Noh K-M, Diaz N, Allis CD & Banaszynski LA Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 522, 240–244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banaszynski LA et al. Hira-dependent histone H3.3 deposition facilitates PRC2 recruitment at developmental loci in ES cells. Cell 155, 107–120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo P et al. Endothelial jagged-2 sustains hematopoietic stem and progenitor reconstitution after myelosuppression. J. Clin. Invest 127, 4242–4256 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elsaesser SJ & Allis CD HIRA and Daxx constitute two independent histone H3.3-containing predeposition complexes. Cold Spring Harb. Sym. Quant. Biol 75, 27–34 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Wong LH et al. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res 20, 351–360 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui K et al. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 4, 80–93 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adli M, Zhu J & Bernstein BE Genome-wide chromatin maps derived from limited numbers of hematopoietic progenitors. Nat. Methods 7, 615–618 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schepers K, Hsiao EC, Garg T, Scott MJ & Passegué E Activated Gs signaling in osteoblastic cells altersthe hematopoietic stem cell niche in mice. Blood 120, 3425–3435 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pietras EM et al. Functionally distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell 17, 35–46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grinenko T et al. Hematopoietic stem cells can differentiate into restricted myeloid progenitors before cell division in mice. Nat. Commun 9, 1898 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wen D et al. Genome editing a mouse locus encoding a variant histone, H3.3B, to report on its expression in live animals. Genesis 52, 959–966 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pradhan SK et al. EP400 deposits H3.3 into promoters and enhancers during gene activation. Mol. Cell 61, 27–38 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Butler JM et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell 6, 251–264 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen P et al. H3.3 actively marks enhancers and primes gene transcription via opening higher-ordered chromatin. Gene Dev 27, 2109–2124 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martire S et al. Phosphorylation of histone H3.3 at serine 31 promotes p300 activity and enhancer acetylation. Nat. Genet 51, 941–946 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernstein BE et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Sachs M et al. Bivalent chromatin marks developmental regulatory genes in the mouse embryonic germline in vivo. Cell Rep 3, 1777–1784 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zentner GE, Tesar PJ & Scacheri PC Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res 21, 1273–1283 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsumura Y et al. H3K4/H3K9me3 bivalent chromatin domains targeted by lineage-specific DNA methylation pauses adipocyte differentiation. Mol. Cell 60, 584–596 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Zhu Y, van Essen D & Saccani S Cell-type-specific control of enhancer activity by H3K9 trimethylation. Mol. Cell 46, 408–423 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Young GR et al. Resurrection of endogenous retroviruses in antibody-deficient mice. Nature 491, 774–778 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshinobu K et al. Selective up-regulation of intact, but not defective env RNAs of endogenous modified polytropic retrovirus by the Sgp3 locus of lupus-prone mice. J. Immunol 182, 8094–8103 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karimi MM et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 8, 676–687 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheng W et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 174, 549–563 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiappinelli KB et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162, 974–986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Izquierdo-Bouldstridge A et al. Histone H1 depletion triggers an interferon response in cancer cells via activation of heterochromatic repeats. Nucleic Acids Res 45, 11622–11642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cuellar TL et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemiaSETDB1 regulates the antiviral response in AML. J. Cell Biol 216, 3535–3549 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collins PL, Kyle, Shinkai Y & Oltz EM The histone methyltransferase SETDB1 represses endogenous and exogenous retroviruses in B lymphocytes. Proc. Natl Acad. Sci. USA 112, 8367–8372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stavrou S & Ross SR APOBEC3 proteins in viral immunity. J. Immunol 195, 4565–4570 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elli EM, Baratè C, Mendicino F, Palandri F & Palumbo GA Mechanisms underlying the anti-inflammatory and immunosuppressive activity of ruxolitinib. Front. Oncol 9, 1186 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li W et al. Type I interferon-regulated gene expression and signaling in murine mixed glial cells lacking signal transducers and activators of transcription 1 or 2 or interferon regulatory factor 9. J. Biol. Chem 292, 5845–5859 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Platanias LC Mechanisms of type-I-and type-II-interferon-mediated signalling. Nat. Rev. Immunol 5, 375–386 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Passegué E & Ernst P IFN-α wakes up sleeping hematopoietic stem cells. Nat. Med 15, 612–613 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baldridge MT, King KY & Goodell MA Inflammatory signals regulate hematopoietic stem cells. Trends Immunol 32, 57–65 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.King KY & Goodell MA Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat. Rev. Immunol 11, 685–692 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mascarenhas J & Hoffman R Ruxolitinib: the first FDA approved therapy for the treatment of myelofibrosis. Clin. Cancer Res 18, 3008–3014 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Ueda Y, Kondo M & Kelsoe G Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J. Exp. Med 201, 1771–1780 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He Q et al. The Daxx/Atrx complex protects tandem repetitive elements during DNA hypomethylation by promoting H3K9 trimethylation. Cell Stem Cell 17, 273–286 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang W et al. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 348, 1160–1163 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Challen GA, Boles NC, Chambers SM & Goodell MA Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-β1. Cell Stem Cell 6, 265–278 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ng SWK et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 540, 433–437 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Shlush LI et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 547, 104–108 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Bagger FO, Kinalis S & Rapin N BloodSpot: a database of healthy and malignant haematopoiesis updated with purified and single cell mRNA sequencing profiles. Nucleic Acids Res 47, D881–D885 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi L, Wen H & Shi X The histone variant H3.3 in transcriptional regulation and human disease. J. Mol. Biol 429, 1934–1945 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buschbeck M & Hake SB Variants of core histones and their roles in cell fate decisions, development and cancer. Nat. Rev. Mol. Cell Biol 18, 299–314 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Henikoff S & Smith MM Histone variants and epigenetics. Cold Spring Harb. Perspect. Biol 7, a019364 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Szenker E, Ray-Gallet D & Almouzni G The double face of the histone variant H3.3. Cell Res 21, 421–434 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McKittrick E, Gafhen PR, Ahmad K & Henikoff S Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc. Natl Acad. Sci. USA 101, 1525–1530 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Farrell MJ et al. HIRA, a digeorge syndrome candidate gene, is required for cardiac outflow tract septation. Circ. Res 84, 127–135 (1999). [DOI] [PubMed] [Google Scholar]
- 61.Schwartz BE & Ahmad K Transcriptional activation triggers deposition and removal of the histone variant H3.3. Gene Dev 19, 804–814 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wen D et al. Histone variant H3.3 is an essential maternal factor for oocyte reprogramming. Proc. Natl Acad. Sci. USA 111, 7325–7330 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gerber JP et al. Aberrant chromatin landscape following loss of the H3.3 chaperone Daxx in haematopoietic precursors leads to Pu.1-mediated neutrophilia and inflammation. Nat. Cell Biol 10.1038/s41556-021-00774-y (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Garber M, Grabherr MG, Guttman M & Trapnell C Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 8, 469–477 (2011). [DOI] [PubMed] [Google Scholar]
- 65.Trapnell C et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc 7, 562–578 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zang C et al. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25, 1952–1958 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chu H-P et al. TERRA RNA antagonizes ATRX and protects telomeres. Cell 170, 86–101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ChIP–seq data, including the fastq data and processed wig files, have been uploaded to the Gene Expression Omnibus website. The accession number is GSE148750. Please refer to Supplementary Table 10 for detailed information. Source data are provided with this paper.