

Abstract
Endothelial cell (EC) aging plays a vital role in the pathogenesis of cardiovascular disease (CVD). MicroRNAs have emerged as crucial regulators of target gene expression by inhibiting mRNA translation and/or promoting mRNA degradation. We identify an aging-related and oxidative stress-responsive microRNA, miR-181b, that inhibits endothelial cell apoptosis and senescence. In gain-or-loss function studies, miR-181b regulated the expression of key apoptosis markers (Bcl2, Bax, cleaved-Caspase3) and senescence markers (p16, p21, γH2AX) and the ratio of apoptotic cells (TUNEL positive) and senescent cells (SA-βgal positive) in H2O2-induced ECs. Mechanistically, miR-181b targets MAP3K3 and modulates a MAP3K3/MKK/MAPK signaling pathway. MAP3K3 knockdown recapitulated the phenotype of miR-181b overexpression and miR-181b was dependent on MAP3K3 for regulating EC apoptosis and senescence. In vivo, miR-181b expression showed a negative correlation with increasing age in the mouse aorta. Endothelial-specific deficiency of miR-181a2b2 increased the target MAP3K3, markers of vascular senescence (p16, p21), and DNA double-strand breaks (γH2AX) in the aorta of aged mice. Collectively, this study unveils an important role of miR-181b in regulating vascular endothelial aging via a MAP3K3-MAPK signaling pathway, providing new potential therapeutic targets for anti-aging therapy in CVD.
Keywords: atherosclerosis, endothelium, microRNA, apoptosis, senescence
Introduction
Aging is a major risk factor for the development of cardiovascular disease, and people >65 years of age (especially >80 years of age) have a higher prevalence for all CVD1. Manifestation of disease is the culmination of the interaction between inherent genotypes and extrinsic factors, and aging is no exception2. Extrinsic factors can potently promote or postpone cellular and molecular cardiovascular aging and are regulated by therapeutic intervention, while genotype is a challenge to modify by current medical treatments3. Therefore, unveiling the mechanisms of extrinsic factors affecting aging and uncovering key nodes is valuable for anti-aging therapies. Oxidative stress, as an aging-accelerated extrinsic factor, plays an important role in the pathological process of vascular aging by producing excessive amounts of oxygen free radicals, which trigger senescence, and activate apoptotic signals in endothelial cells4–6. Apoptosis and senescence, as the key cellular response to environmental damages, are hallmarks of aging and are increasingly implicated in the aging process7,8. Oxidative stress induces cellular apoptosis in the intrinsic pathway by triggering the mitochondrial outer membrane permeabilization (MOMP), a tightly regulated event controlled by the Bcl2 family, including pro-apoptotic effector proteins, e.g. Bax and the antiapoptotic proteins, e.g. Bcl29,10. Senescence includes replicative senescence (RS) and stress-induced premature senescence (SIPS). While the former naturally progresses over time, the latter is induced by extrinsic factors, e.g. oxidative stress11. Senescent cells can be identified by elevated senescence-associated β-galactosidase (SA-β-gal) activity, which is the most widely and easily used gold standard12,13. As terminally growth arrest appears in senescent cells, cell cycle regulators, e.g. p16 and p21, are also broadly employed biomarkers14. As a marker of DNA double-strand breaks and genomic instability in human population studies, γH2AX is also an important senescent biomarker15.
MicroRNAs (miRNAs), are an emerging class of biomarkers and potential therapeutic targets for cardiovascular disease that negatively regulate gene expression by targeting specific mRNAs for cleavage or translational repression16–18. Accumulating studies highlight that miRNAs serve as key regulators in endothelial aging, often with an amplifying effect from the onset and progression of aging-related diseases. For example, miR‐217, miR‐34a, and miR‐21 regulate endothelial aging by targeting SIRT1, a crucial controller in vascular aging19–21. miR-200c is induced by oxidative stress and regulates endothelial apoptosis and senescence via targeting ZEB122. miR‐181 family plays diverse roles in regulating key aspects of physiological and pathological processes in endothelial cells23. In our previous studies, systemic delivery of miR‐181b demonstrated strong anti-atherosclerosis effects in mice24. Increasing aging is an independent risk factor for atherogenesis, and atherosclerosis per se is a disease of aging25. Therefore, identifying the potential effect of miR‐181b in endothelial aging could open a new strategy to anti-aging therapies in CVD.
MAPK signal transduction pathways play a crucial role in regulating aging-related senescence and apoptosis26. MAPK cascades propel signal amplification and maintain signaling fidelity depending on a series of kinase proteins from MKKK (MAPK kinase kinase) through MKK (MAPK kinase) to a terminal MAPK such as p38, JNK, ERK1/227. As a result of MAPK signal transduction, the expressions of aging markers (e.g. Bcl2, Bax, p21) are regulated during the process of aging28–30. MAP3K3 (Mitogen-activated protein kinase kinase kinase 3), a key upstream regulator in the MAPK cascade, directly modulates phosphorylation of MKK3/MKK6 or MKK4, which then regulates substrates e.g. p38, JNK31,32. Some studies demonstrate key effects of MAP3K3 in apoptosis and senescence in diverse cell types14,33–36.
In this study, we identify that miR-181b regulates endothelial aging by modulating the MAPK signaling pathway. We explore the mechanistic basis for miR-181b regulation of apoptosis and senescence in ECs that is dependent on a MAP3K3/MAPK signaling cascade. Finally, using EC-specific miR-181a2b2 knockout mice, we explore pre-mature aging markers for both senescence and apoptosis to reveal that this miR-181b-MAP3K3/MAPK signaling pathway may figure prominently in anti-aging strategies in CVD.
Materials and Methods
Animal Studies
MiR-181a2b2flox/flox (C57BL/6J) and miR-181a2b2 systemic knockout (miR-181a2b2−/−) (C57BL/6J) mice were used as previously described37 and tamoxifen-inducible endothelial-specific VE-cadherin (VECad-Cre-ERT2) (C57BL/6J) mice was kindly provided by R. Adams38. Inducible endothelial cell-specific miR-a2b2 deficient mice (MiR-181a2b2flox/flox; VECad-Cre-ERT2) was generated by crossbreeding miR-181a2b2flox/flox and VECad-Cre-ERT2 mice. For induction of Cre recombination, 4 weeks old male MiR-181a2b2flox/flox; VECad-Cre-ERT2 mice were treated with either 4-hydroxytamoxifen (H6278, Sigma) (10 mg/kg, i.p.) or same volume of corn oil vehicle for five consecutive days to generate ECs-specific miR-181a2b2 deficient mice (miR-181a2b2iECKO) and control mice (Ctrl). Male C57BL/6J mice at 20 weeks and 80 weeks old were purchased from Charles River Laboratory. Age- and cage-matched male littermates were used for experiments. All mice were maintained under SPF conditions at an American Association for the Accreditation of Laboratory Animal Care-accredited animal facility at the Brigham and Women’s Hospital. All animal protocols were approved by the Institutional Animal Care and Use Committee at Brigham and Women’s Hospital, Boston, MA and conducted in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals.
Cell culture and transfection
HUVECs were purchased from Lonza (cc-2159) and cultured in EC growth medium EGM-2 (cc-3162, Lonza). HUVECs were plated on 12-well plates at 60, 000/well or 150, 000/well and allowed to grow to 80%−90% confluence under growing conditions or 70%−80% for transfection. Cells were stimulated with or without H2O2 (Sigma) at indicated dosage or camptothecin (CPT, 10 μM, C9911, Sigma) for various times, according to the experiment: Western blot, 2, 4, 8 or 16 hours; Real-time PCR, 1, 4 or 12 hours; Tunnel staining, 4 hours; senescence β-Galactosidase staining; 1 hour. Lipofectamine 2000 transfection reagent (11668019, Invitrogen) was used for transfection, following the manufacturer’s instructions. MiRNA negative control (NS-m) (AM17110, Ambion) or pre-miR-181b (181b-m) (PM12442, Ambion) were transfected at 10 nM, and miRNA inhibitor negative control (NS-i) (AM17010, Ambion) or miR-181b inhibitor (181b-i) (AM12442, Ambion) were transfected at 100 nM. SiRNA control (Ctrl si) (AM4636) and validated MEKK3 siRNA (MEKK3 si) (AM51331) from Invitrogen were transfected at 20 nM. For co-transfection studies, NS-i (100 nM), 181b-i (100 nM), Ctrl si (20 nM), or MEKK3 si (20 nM) were transfected as indicated in respective experiments.
Luciferase activity assay
Putative miR-181b binding sites in the MEKK3 gene 3’UTR were predicted by the TargetScan algorithm. Individual wild-type or mutant binding site sequence was generated by annealing the forward and reverse oligonucleotides containing Xhol and Xbal sticky ends. The double-stranded oligonucleotides were ligated into the pcDNA3.1 (+) Luciferase vector, by using T4 DNA ligase. HUVECs were plated on 12-well plates at 60, 000/well up to 70%−80% confluency and transfected with 100 ng of wild-type or mutant MEKK3 reporter constructs and 10 ng Renilla plasmid (E2231, Promega). NS-m or 181b-m was co-transfected at 10 nM final concentration for 12 hours. Transfected cells were collected in 200 μl reporter lysis buffer (E1910, Promega) and luciferase activity was measured using a Dual-Luciferase reporter assay system (E1910, Promega). Each reading of luciferase activity was normalized to the Renilla activity.
β-Galactosidase staining
β-Galactosidase activity assay was performed as previously described39. Briefly, HUVECs were transfected with and 24 hours post-transfection treated for 1 hours with H2O2 (30 μM) and cultured for another 3 days in normal growth medium before cells were fixed. β-Galactosidase activity at pH 6 was measured using X-Gal staining kit (9860, Cell Signaling).
Immunofluorescence and Tunnel staining assay
For Immunofluorescence, 6 μm frozen sections of aortic roots were fixed and permeabilized with cold-acetone for 5 min and blocked in PBS containing 3% BSA for 1 hour at room temperature. Sections were stained with rat anti-CD31 (BD Pharmingen, 550274, 1:30), rabbit anti-p16 (1:100, ab51243, Abcam), rabbit anti-p21 (1:400, 2947, Cell Signaling), rabbit anti-γH2AX (2577, 1:800, Cell Signaling), cleaved caspase3 (1:400, 9661, Cell Signaling), Importin-α3 (1:100, ab84706, Abcam) and rabbit anti-MEKK3 (Cell Signaling) in PBS at 4 oC overnight. After three washes with PBS, the sections were further incubated with Alexa Flour 488-conjugated donkey anti-Rat IgG (Invitrogen, A21208, 1:300) and Alexa Flour 555-conjugated goat anti Rabbit IgG (Invitrogen, 1:300) in PBS for 1 hour at room temperature. Nuclei were stained for 5min at room temperature in PBS containing DAPI (Cell signaling, 4083, 0.5μg/ml). Coverslips were mounted with ProLong Gold antifade reagent (Invitrogen). Images were acquired on an upright Carl Zeiss LSM 510 confocal. The data were calculated from at least 40 cells for each group with 6–10 mice. For each mouse, 2–5 plaques were selected and used for quantification. Tunnel protocol was performed as described by manufacturer’s protocol (Millipore, ApopTaq Peroxidase Detection kit).
RNA isolation and Real-time qPCR
Tissues were homogenized using TissueLyser II (Qiagen) according to manufacturer/s instructions. RNA isolation was using TRIzol Reagent according to the manufacturer’s instructions. Subsequent RT-qPCR was performed using a High-Capacity cDNA Reverse Transcription Kit (4368813, Applied Biosystems). For SyberGreen-based assay GoTaq qPCR Master Mix (A6001, Promega) was used, and for TaqMan Universal Master Mix II, UNG (4440038, Life Technologies) was used. Expression of mRNAs and miRNA expression were normalized to β-actin or U6 (Aglient, AriaMx Real Time PCR System). Specific primers including miR-181b-5p (#001098), U6 (#001973), human-HPRT (Hs05647472_cn), human-primary-miR-181b1 (Hs03302966_pri), human-primary-miR-181b2 (Hs03302963_pri), mouse-HPRT (Mm00522878_cn), mouse-primary-miR-181b1 (Mm03307120_pri) and mouse-primary-miR-181b2 (Mm03307414_pri) were purchased from Life Technologies. Changes in expression were calculated using delta delta Ct method.
Western blot
Proteins were isolated using RIPA buffer (Boston BIoProductus, BP-115) with protease inhibitor and phosphatase inhibitor. Protein concentrations were determined using Pierce BCA kit (Thermo Scientific). 20 μg protein were loaded per lane on a 4–20% Mini-PROTEAN TGX Gel (Bio-Rad, 456–1096). Separated proteins were transferred to PVDF membranes using the Transfer Turbo Blot system (Bio-Rad) and Trans-Blot Turbo RTA Transfer Kit (Bio-Rad, 170–4272). The membrane was blocked with 5% nonfat milk in TBST for 1 hour at room temperature. After blocking, the membrane was incubated overnight at 4°C with antibodies against Bax (1:1000, 2772, Cell Signaling), Bcl-2 (1:1000, 3498, Cell Signaling), cleaved caspase-3 (1:1000, 9661, Cell Signaling), MEKK3 (1:1000, 5727, Cell Signaling), p16 (1:1000, ab51243, Abcam), p21(1:1000, ab109520, Abcam), p-γH2AX (1:1000, 9718, Cell Signaling), p-MKK3/6 (1:1000, 12280, Cell Signaling), p-SEK1/MKK4 (1:1000, 4514, Cell Signaling), p-JNK (1:1000, 9255, Cell Signaling), p-p38 (1:1000, 8690, Cell Signaling), p-ERK1/2 (1:1000, 8544, Cell Signaling), p-IKBα (1:1000, 2859, Cell Signaling), JNK (1:1000, 9252, Cell Signaling), p38 (1:1000, 8690, Cell Signaling), ERK1/2 (1:1000, 4696, Cell Signaling), IKBα (1:1000, 4814, Cell Signaling), TAOK1 (1:1000, ab197891, Abcam) or β-actin (1:4000, 4970, Cell Signaling). Quantification of protein bands were performed using a luminescent image analyzer (BioRad, Chemidoc).
Statistical analysis
GraphPad 7.0 software package (GraphPad Software, Inc) was used for statistical analysis. Unpaired two-tailed Student’s t test was used to determine statistical significance between two groups for normally distributed continuous variables. For data without normal distribution, non-parametric Mann-Whitney U test was used. All data were expressed as mean ± SEM. P < 0.05 was considered significant for all tests.
Results
miR-181b is an oxidative stress-responsive microRNA reduced by aging.
miR-181b is a key regulator in multiple processes of pathology and physiology. In our previous studies, we found miR-181b exerted vasculo-protective effects in atherosclerosis, insulin resistance, and thrombosis24,40,41. Aging, as an independent risk factor, threatens vascular health and induces cardiovascular disease42. In order to investigate the role of miR-181b in vascular aging, we isolated RNA from the aorta of young (10 weeks old) and aged (80 weeks old) C57BL/6 mice for RT-qPCR (Fig.1 A). Compared to young mice, miR-181b expression of the aorta decreased by 40% in the aged mice. This decrease is in line with other stimuli known to reduce miR-181b expression including hyperlipidemia.24,43 We further assessed the expression changes of miR-181b, the dominantly expressed miR-181 family member in endothelial cells40, in H2O2-induced human umbilical vein endothelial cells (HUVECs). Expression of miR-181b was reduced in response to H2O2 in a dose- and time-dependent manner (Fig.1 B–C).
Fig.1. miR-181b expression is regulated by aging.
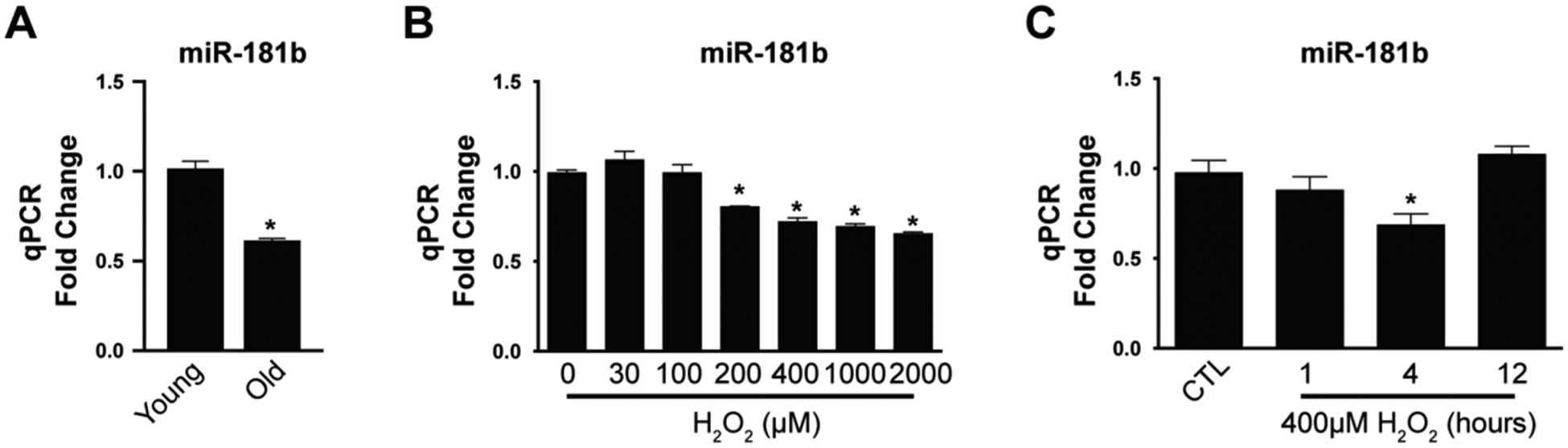
(A) miR-181b expression of the aorta between young (10w) and old mice (80w) (n=4). (B) Expression of miR-181b in response to different concentrations of H2O2 (n=4). (C) Kinetics of miR-181b in ECs stimulated with H2O2 (n=4).
miR-181b regulates apoptosis and senescence in ECs.
To assess the potential role of miR-181b in endothelial apoptosis and senescence, we examined the effect of miR-181b on aging-relevant phenotypes in H2O2–induced HUVECs by using gain- and loss-of-function experiments. Gain-of-function was induced by miR-181b mimics for miR-181b overexpression. Loss-of-function was induced by use of the complementary antagonist of miR-181b for miR-181b inhibition. We analyzed apoptosis markers, including Bax, Bcl-2, cleaved-Caspase3, and TUNEL staining, in H2O2–induced HUVECs. miR-181b overexpression reduced Bax, cleaved-Caspase3 protein expression by 17% and 54%, respectively, and increased Bcl-2 protein expression by 210% (Fig.2 A), while miR-181b inhibitors increased Bax and cleaved-Caspase3 protein expression by 129% and 127%, respectively, and reduced Bcl-2 protein expression by 45% (Fig.2 B). In TUNEL assays, miR-181b overexpression decreased apoptotic cells (TUNEL+) by 25%, while miR-181b inhibitors increased it by 30% (Fig.2 C). For induction of senescence, H2O2 and CPT, an inhibitor of DNA topoisomerase I, can trigger DNA strand breaks44. In HUVECs stimulated by H2O2 or CPT, miR-181b overexpression reduced senescence markers, p16, p21, and γH2AX (a specific marker of DNA double-strand breaks) protein expression by 42%, 30%, and 41%, respectively (Fig.3 A). Using senescence-associated βgal (SA-βgal) as a marker of senescence in H2O2–induced HUVECs, we find that miR-181b overexpression decreased senescence cells (SA-βgal +) by 50%, while miR-181b inhibitors increased it by 30% (Fig.3 B). Taken together, these findings indicate that miR-181b can inhibit apoptosis and senescence in H2O2–induced HUVECs.
Fig.2. miR-181b regulation of endothelial apoptosis.
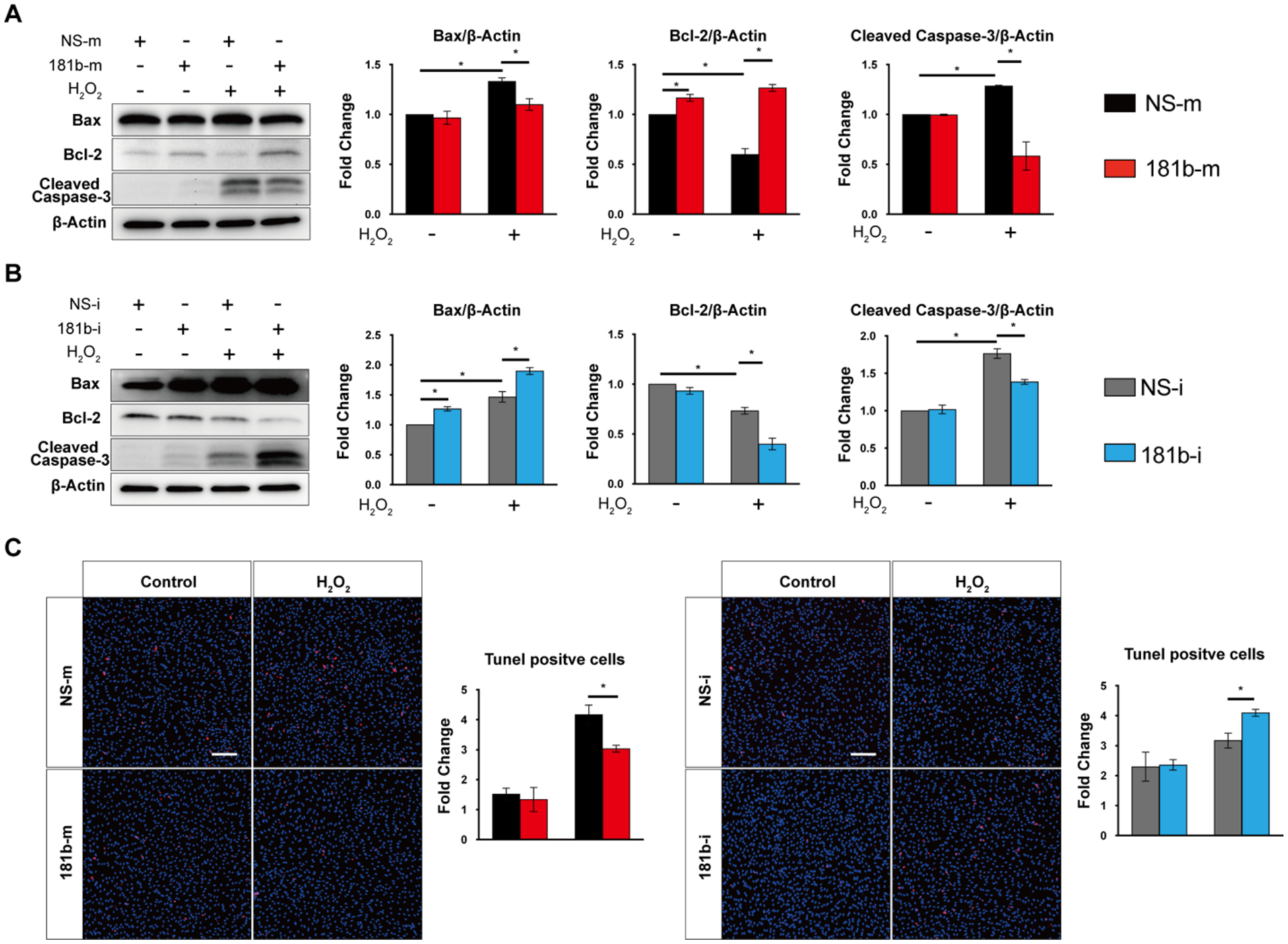
miR-181b mimics (A) and inhibitors (B) regulate apoptotic markers (n=3). (C) TUNEL staining of HUVEC transfected with miR-181b mimics (n=4–5) or miR-181b inhibitors (n=4–5).
Fig.3. miR-181b regulation of endothelial senescence.
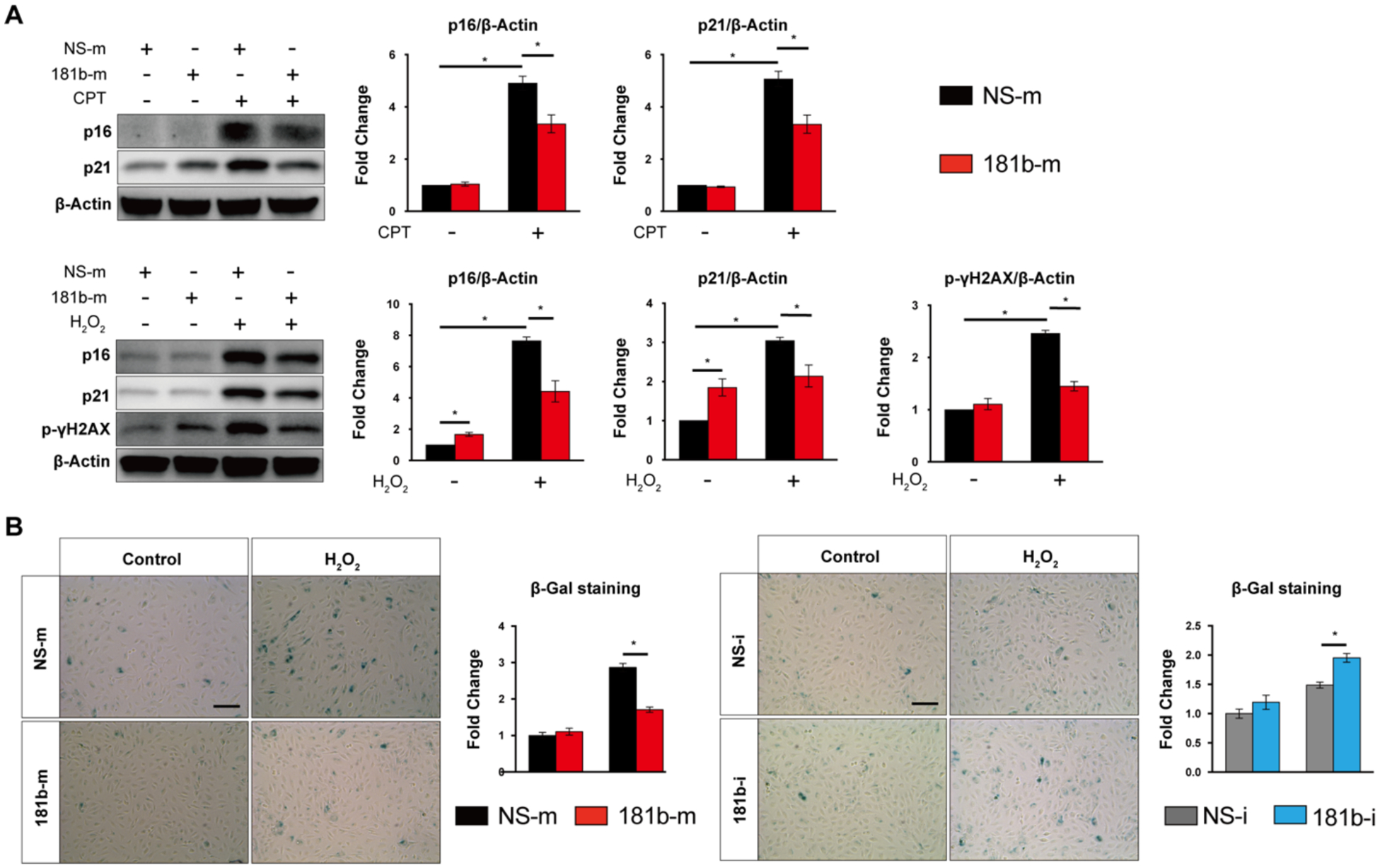
(A) miR-181b mimics regulate senescence markers with stimuli of CPT or H2O2 (n=3). (B) SA-βgal staining of HUVEC transfected with miR-181b mimics (n=4–6) or miR-181b inhibitors (n=4–6).
miR-181b regulates a MAPK signaling pathway.
To identify potential signaling pathways subject to miR-181b regulation of apoptosis and senescence, we overexpressed miR-181b using mimics. HUVECs were treated with H2O2 for 5, 15, 30, and 60 minutes, and we evaluated the phosphorylation of related signaling pathways. We found that miR-181b overexpression significantly reduced the phosphorylation of MKK3/MKK6 by 29%, and MKK4 by 36 % after 5, 15 minutes, respectively (Fig.4 A). Furthermore, we investigated the phosphorylation of JNK or p38, which are the downstream substrates of MKK3/MKK6 or MKK4, that directly regulate transcription factors to affect gene expression45. In H2O2-induced HUVECs, miR-181b overexpression significantly reduced the phosphorylation of p38 by 48%, while JNK, ERK1/2 was not impacted (Fig.4 A, Supplementary Figure 1). Interestingly, in H2O2-induced HUVECs, miR-181b had no effect on the NF-κB signaling, a pathway that miR-181b markedly repressed in response to TNF-α or LPS46 (Supplementary Figure 1). Collectively, these data indicated that in response to H2O2, miR-181b inhibited the MAPK signaling pathway.
Fig. 4. miR-181b regulates MKKs/MAPK signaling pathway.
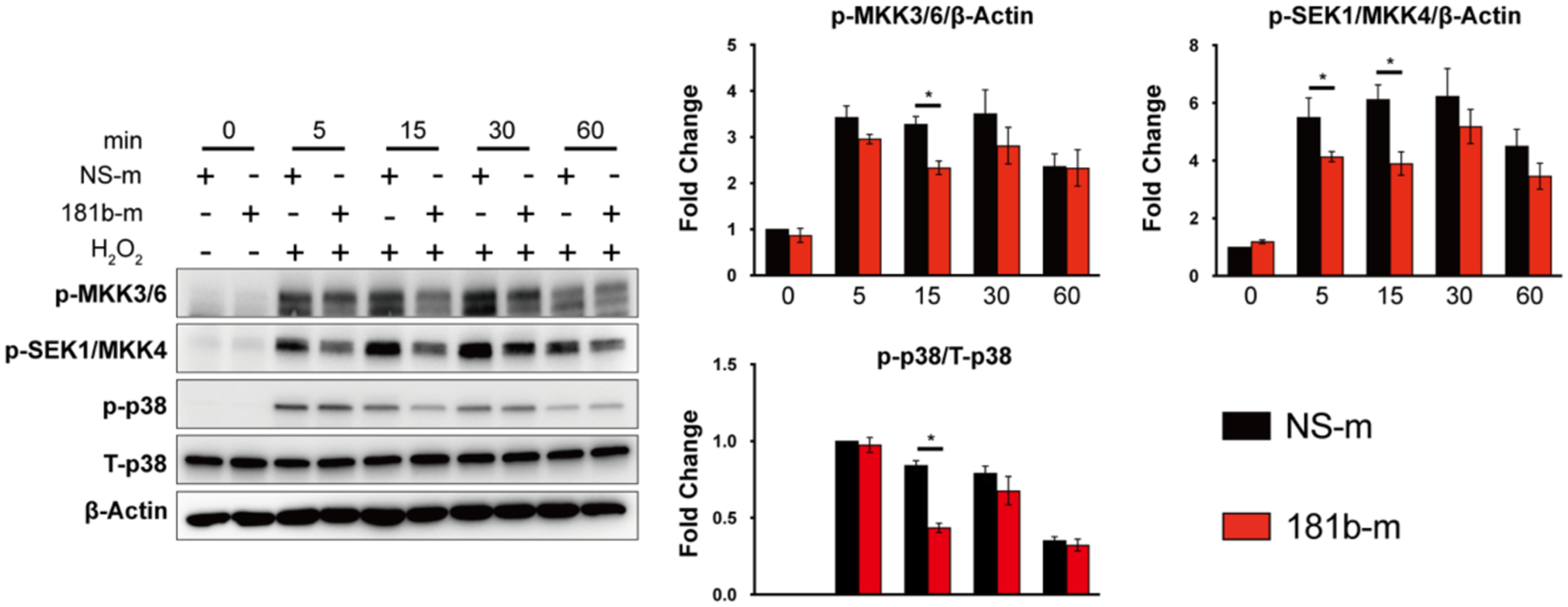
(A) miR-181b overexpression reduces the phosphorylation of MKK3/6, SEK1/MKK, p38 (n=3).
miR-181b targets the expression of MAP3K3.
Using algorithms of TargetScan, miRDB, and miRWalk, predicted targets of miR-181b were identified (Fig.5 A). Aligning these potential targets with those known to be involved in MAPK signaling pathways revealed two targets of interest, MAP3K3 and TAOK1. Overexpression of miR-181b did not affect the protein expression of TAOK1 or phosphorylated TAOK1 (Supplementary Figure 2). However, in miR-181b–overexpressing HUVECs, mRNA expression of MAP3K3 was reduced by 40% (Fig.5 B), and protein expression of MAP3K3 was reduced by 50% (Fig.5 C). In contrast, miR-181b inhibition in HUVECs increased MAP3K3 mRNA expression by 50% (Fig.5 D) and protein expression by 40% (Fig.5 E), suggesting that miR-181b is a negative regulator of MAP3K3. Analysis of miR-181b–binding sites in the 3’-UTR of MAP3K3 by TargetScan revealed conserved sites across species (Fig.5 F). To investigate whether the miR-181b binding site is functionally active in the MAP3K3 3’-UTR, we generated a luciferase-reporter construct harboring wildtype and mutant miR-181b binding sites (Fig.5 G). MiR-181b overexpression decreased luciferase activity by 25% in luciferase-reporter constructs containing wildtype 3’UTR of MAP3K3, while constructs that contained the mutated 3’-UTR was not affected (Fig.5 H). Taken together, these findings indicate that miR-181b targets the 3’UTR of MAP3K3 to repress its mRNA and protein expression. Because MAP3K3, is known as a key upstream regulator in the MAPK signaling cascade, and modulates signal transduction by directly regulating phosphorylation of MKK3/MKK6, MKK431,32, these findings are in accordance with the signaling pathway modulation observed by miR-181b (Fig.4 A).
Fig. 5. miR-181b targets MAP3K3.
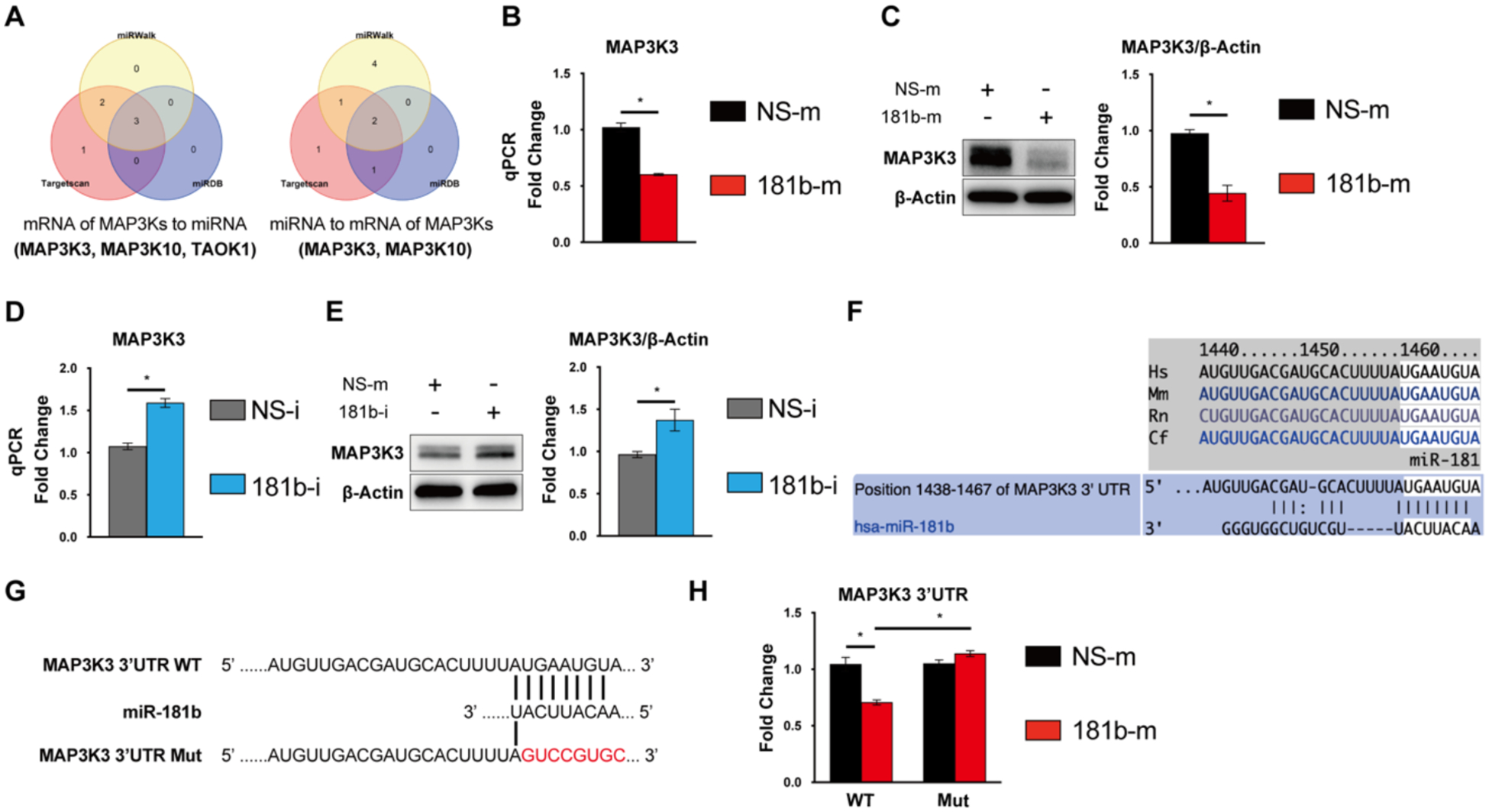
(A) The genes of MAPK signaling pathway targeted by miR-181b were predicted by algorithms miRWalk, TargetScan, or miRDB. miR-181b mimics reduce mRNA expression (n=3) (B) and protein expression (n=3). (C) of MAP3K3. miR-181b inhibition increases mRNA expression (n=3). (D) and protein expression (E) of MAP3K3. (F) The binding site in MAP3K3 3’UTR for miR-181b was conserved across species and predicted by TargetScan3.1. (G) Schematic diagram of luciferase-reporter constructs containing wild-type or mutated 3’UTR of MAP3K3. (H) miR-181b inhibits MAP3K3 with WT 3’-UTR but not the MAP3K3 with the 3’-UTR mutation (n=4).
MAP3K3 regulates endothelial apoptosis and senescence by modulating the MAPK signaling pathway.
To investigate whether MAP3K3 deficiency can reproduce the inhibitory effects of miR-181b by modulating the MAPK signaling pathway, we performed siRNA-mediated knockdown studies in HUVECs47. For apoptosis, MAP3K3 knockdown reduced protein expression of Bax and cleaved-Caspase3 by 16% and 21%, respectively, and increased Bcl-2 by 174% (Fig.6 A). MAP3K3 knockdown also decreased apoptotic cells (TUNEL+) by 25% in H2O2–induced HUVECs (Fig.6 B) and senescence cells (SA-βgal +) by 50% (Fig.6 C). Moreover, we evaluated if MAP3K3, as a key upstream regulator in the MAPK signaling cascade, modulated the MAPK signaling pathway in H2O2–induced HUVECs. Indeed, MAP3K3 knockdown significantly reduced the phosphorylation of MAP2Ks, (e.g., MKK3/MKK6, MKK4), and MAPKs, (e.g., ERK, p38) (Fig.6 D). Taken together, siRNA-mediated knockdown of MAP3K3 recapitulates the effects of miR-181b on apoptosis and senescence by modulating the MAPK signaling pathway in H2O2–induced HUVECs.
Fig. 6. MAP3K3 regulates endothelial apoptosis and senescence via a MAPK signaling pathway.
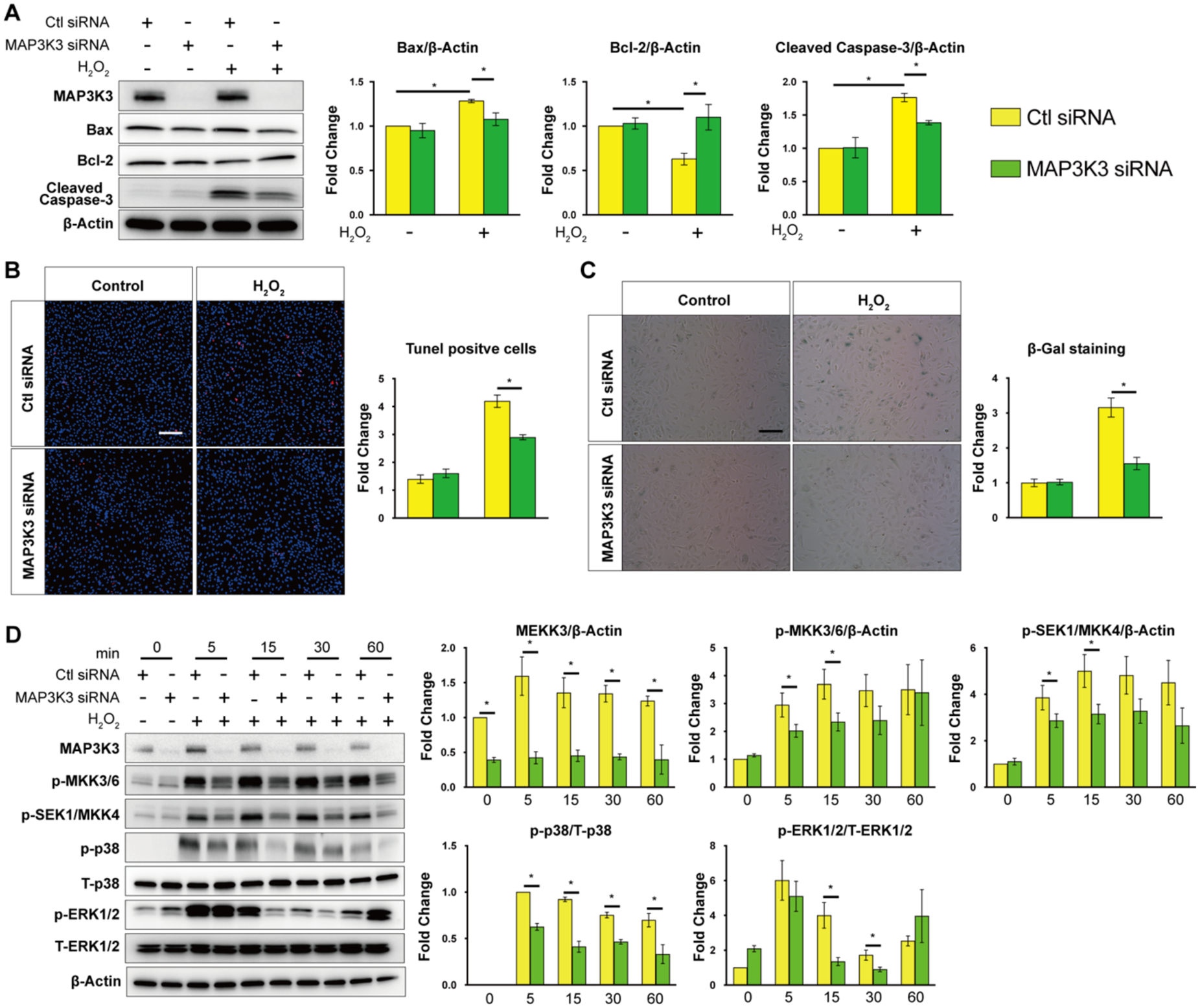
(A) MAP3K3 knockdown regulates protein expression of Bax, Bcl-2, cleaved-Caspase3 in ECs (n=3). (B) Quantification of TUNEL staining in siRNA-transfected HUVECs stimulated with H2O2 (1hr, 30μM) (n=4–5). (C) Quantification of SA-βgal staining in siRNA-transfected HUVECs stimulated with H2O2 (1hr, 30μM) (n=4–6). (D) MAP3K3 knockdown modulates the phosphorylation of MKK3/6, SEK1/MKK, p38, and ERK1/2 in ECs (n=3).
miR-181b is dependent on MAP3K3 for regulating apoptosis and senescence in ECs.
To test whether the miR-181b-mediated effect on apoptosis and senescence is dependent on MAP3K3, we used miR-181b inhibitors or mimics with simultaneous MAP3K3 siRNA knockdown. In HUVECs stimulated with H2O2, MAP3K3 knockdown reversed the increased expression of Bax or cleaved-Caspase3 and the reduction of Bcl-2 mediated by miR-181b inhibitors at the protein level (Fig.7 A). Furthermore, MAP3K3 silencing neutralized the increase of apoptotic cells (TUNEL+) induced by miR-181b inhibitors (Fig.7 B). In SA-βgal staining of H2O2–induced HUVECs, MAP3K3 knockdown counteracted the increase in senescence cells (SA-βgal +) induced by miR-181b inhibitors (Fig.7 C). Finally, silencing of MAP3K3 reinforced senescence induced by miR-181b overexpression (Fig.7 D).
Fig. 7. miR-181b is dependent on MAP3K3 for regulating EC apoptosis and senescence.
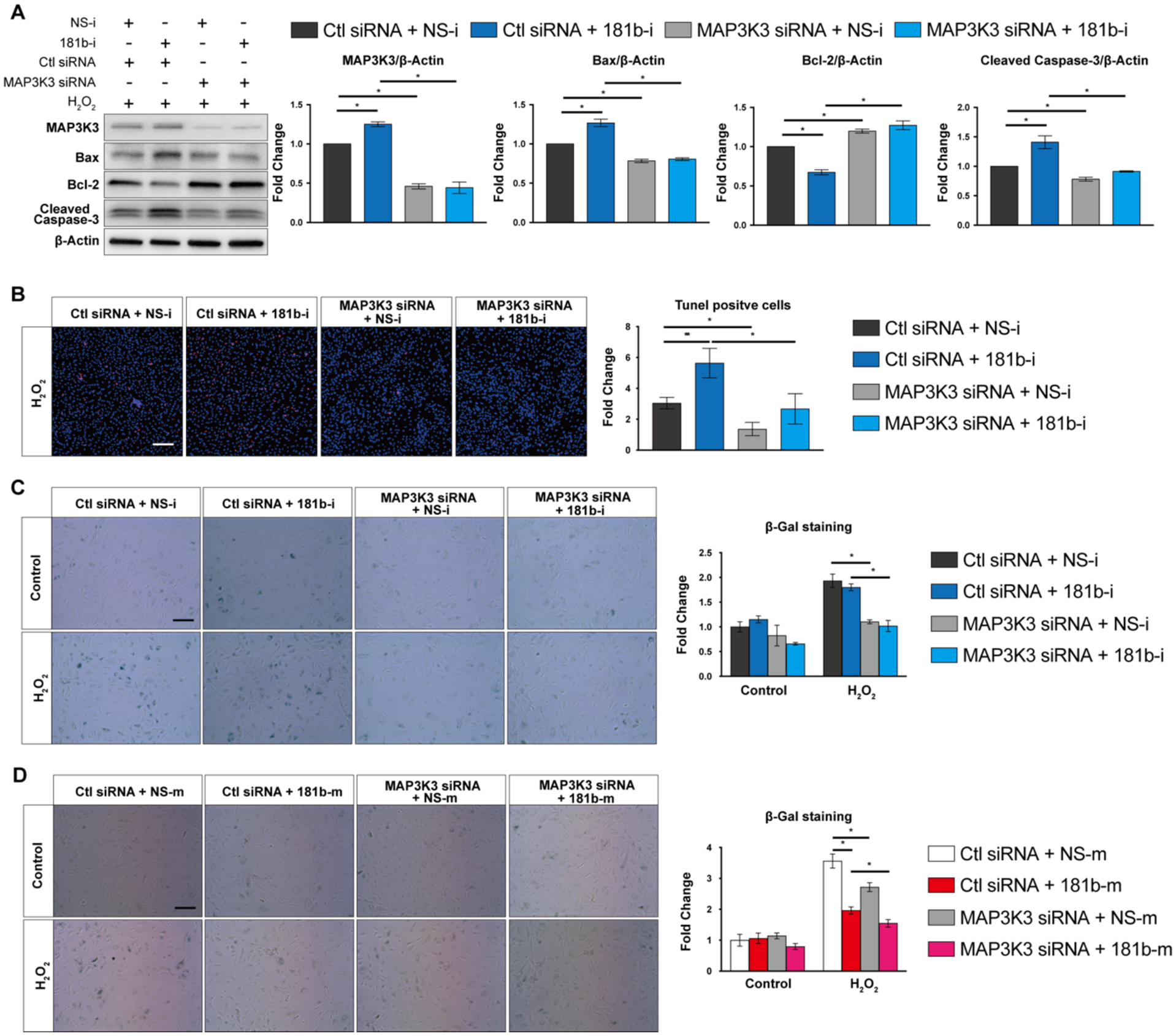
(A) Dependency study of miR-181b inhibitor and MAP3K3 siRNA on protein expression of Bax, Bcl-2, and cleaved-Caspase-3 (n=3). (B) Effect of miR-181b inhibitor in the presence of MAP3K3 knockdown by siRNA on TUNEL staining in ECs (n=4–5). Effect of miR-181b inhibitor (n=4–6) (C) or mimic (n=4–6) (D) in the presence of MAP3K3 knockdown on SA-β-gal staining in ECs.
miR-181b deficiency induces aging in vivo.
To investigate the effect of miR-181b on aging in vivo, we first isolated the RNA of different organs from systemic miR-181a2b2 knockout mice to examine the global effect of miR-181b on apoptosis and senescence. We found the mRNA expression of p21 was induced by systemic miR-181a2b2 knockout in several organs, while other markers of apoptosis or senescence were unchanged (Supplementary Figure 3). To assess the effects on apoptosis and senescence in ECs, we recently generated the endothelial-specific miR-181a2b2 knockout (EC-miR-181a2b2 KO) mice to further explore the in vivo phenotype of miR-181b in the vascular endothelium.43 These EC-miR-181a2b2 KO mice harbor a marked reduction in miR-181b expression in the aortic intima, whereas there was minimal change in miR-181a likely due to its much lower relative expression.43 Immunofluorescence staining results showed markedly increased positive cells of p21, p16, γH2AX, and MAP3K3 in the aorta sinus of endothelial-specific miR-181a2b2 knockout mice by 18 months (Fig.8) compared to controls. Taken together, these findings reveal that deficiency of miR-181a2b2 triggers hallmarks of vascular endothelial senescence in vivo.
Fig. 8. Endothelial-specific deficiency of miR-181a2b2 increases senescence markers in the aorta.
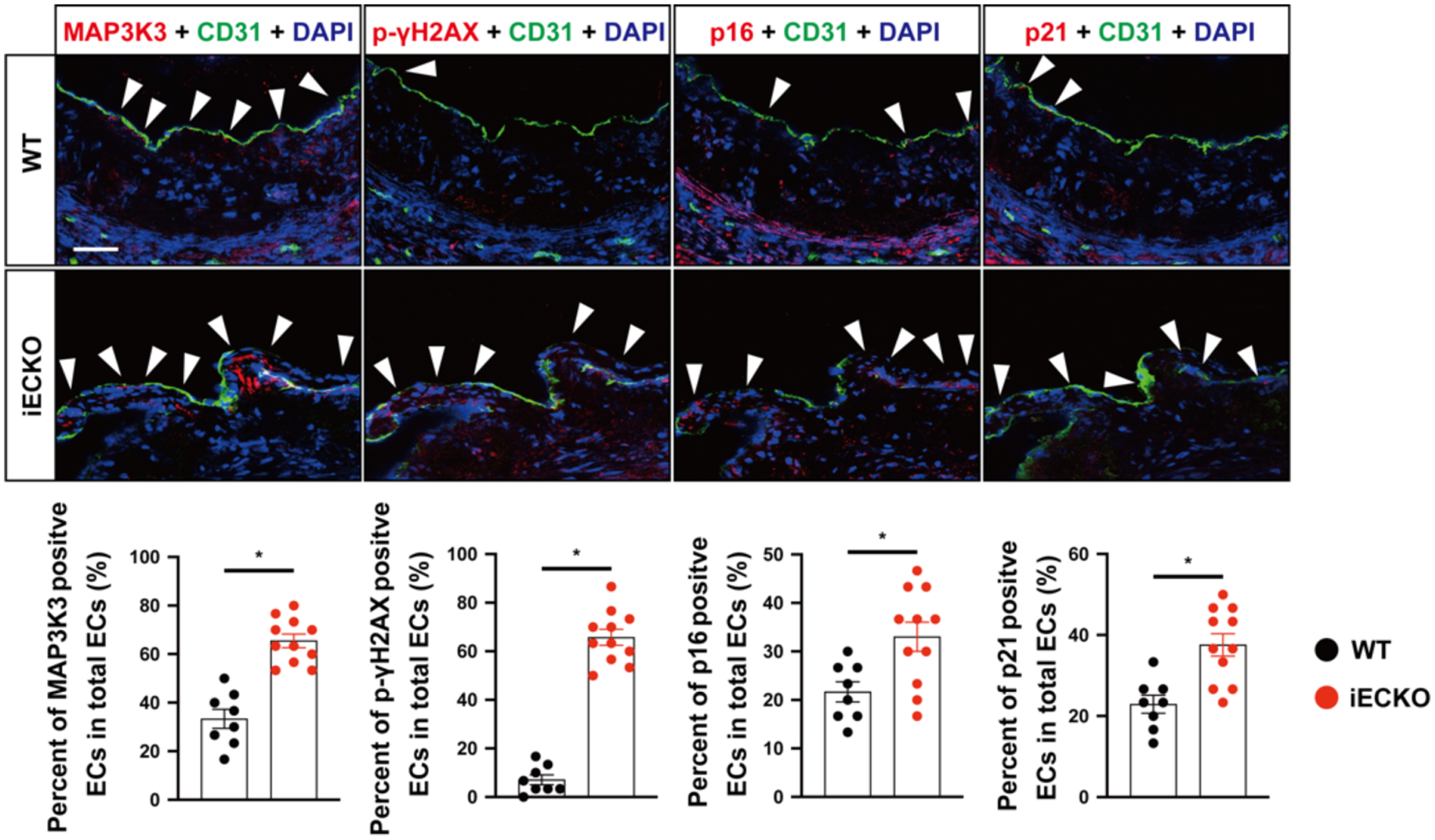
Immunofluorescence staining of p21, p16, γ-H2AX, MAP3K3 in the aorta sinus of miR-181a2b2 iEC KO old (n=11) or control mice (18 months) (n=8): representative images are shown on the left, statistical diagrams are shown on the right.
Discussion
Aging is among the strongest risk factors that threatens health and contributes to organ dysfunction. Cellular aging ultimately leads to organismal aging48. In the cardiovascular system, endothelial aging alters endothelial homeostasis triggering atherosclerosis, coronary heart disease, stroke, and a range of other ischemic cardiovascular diseases42,49. A better understanding of key drivers of endothelial aging may provide insights to promote reparative cell and organismal function. In our study, we identified miR-181b as a key regulator in endothelial aging by targeting MAP3K3, an upstream regulator of MAPK signaling. In mice, miR-181b expression was inversely correlated with increasing age (Fig.1 A).
In the biological process of cells, miR-181b regulates a diverse array of cellular functions and maintains cellular homeostasis by controlling cell death and cellular senescence, which are two closely related pathways in aging. In our previous studies, miR-181b, as an inflammation-responsive microRNA, inhibited ECs inflammation, which protected vessels from atherosclerosis in ApoE–deficient mice24. Apoptosis is an aging-related programmed cell death. In different cell types, miR-181b broadly exerts inhibition on apoptosis, such as in hypoxia-evoked H9c2 cells50, hypertrophic scar fibroblasts51, AML cells52, and SW480 cells53. However, to date there has not been a definitive study to illustrate the effect of miR-181b on endothelial apoptosis. In this study, we identified the anti-apoptosis phenotype of miR-181b in H2O2–induced ECs by gain- or loss-of-function. The intrinsic pathway takes a dominant role in oxidative stress-induced apoptosis. Indeed, miR-181b decreased Bax, a proapoptotic effector protein, and increased Bcl2, an antiapoptotic protein, in response to H2O2-mediated oxidative stress (Fig.2 A, B). As the shared key mediator of apoptosis by both intrinsic and extrinsic pathways, caspase3 is cleaved at an aspartate residue to yield cleaved caspase-3, which then initiates cellular apoptosis. MiR-181b overexpression decreased, while miR-181b inhibition increased the protein expression of cleaved caspase3 in H2O2–induced ECs (Fig.2 A, B). Moreover, TUNEL positive cells were also significantly reduced by miR-181b mimics, while increased by miR-181b inhibitor (Fig.2 C). Collectively, these findings suggest that miR-181b plays a vital role in the regulation of endothelial apoptosis.
In cancer biology, the effect of miR-181b on apoptosis is complicated54. In human gastric adenocarcinoma cells and human lung cancer cells, miR-181b induced Bcl-2 expression and protected cells from apoptosis55. miR-181b overexpression induced Bcl-2 and reduced Bax and cleaved caspase3 expression, which decreased the proportion of apoptotic human colon cancer cells56. In human cervical cancer cells, prostate cancer cells, and thyroid papillary cancer cells, miR-181b also showed the anti-apoptotic effect57–59. However, in chronic lymphocytic leukemia and astrocytoma, miR-181b induced apoptosis60,61. In different non-tumor cell types, miR-181b usually exerts inhibition on apoptosis. miR-181b-5p inhibited fibroblast apoptosis through MEK/ERK/p21 pathway62. Cardiomyocyte apoptosis is inhibited by miR-181b via directly targeting HMGB163. miR-181b suppressed mesangial apoptosis by regulating TIMP364. In human renal proximal tubule cells, miR-181b inhibitor promoted apoptosis and miR‐181b overexpression attenuated apoptosis induced by MEG3 overexpression65. Finally, by interacting with NEAT1, inhibition of miR-181b induced apoptosis in hypoxia-evoked cardiomyoblasts via PI3K/AKT/mTOR and JAK1/STAT3 pathways50. Taken together, these data indicate that miR-181b can exhibit either anti-apoptotic or pro-apoptotic effects in different cancer cell types, whereas in primary cells miR-181b generally exerts anti-apoptotic effects.
Cellular senescence, a stable form of cell cycle arrest, is typically induced in response to diverse stressors66,67. Hallmarks of senescent cells include DNA damage, expression of cyclin-dependent kinase inhibitors and senescence-associated β-galactosidase (SA-β-gal), induction of the senescence-associated secretory phenotype, alterations in chromatin remodeling, and metabolism68,69. In support of a role of miR-181b in endothelial senescence, we found that miR-181b expression decreased in the aorta of aged mice compared to young mice (Fig.1 A), and fell in response to H2O2 treatment in endothelial cells (Fig.1 B, C), which hinted at its potential regulation in the endothelial senescence. In ECs stimulated by H2O2 or CPT (which induces DNA double-strand breaks), miR-181b overexpression reduced the cyclin dependent kinase inhibitor senescent markers, p16, p21 and γH2AX, a definitive marker for DNA double-strand breaks70 (Fig.3 A). In the SA-βgal staining of H2O2–induced ECs, miR-181b overexpression potently reduced the proportion of senescent ECs, while miR-181b inhibition induced it (Fig.3 B). To unveil the regulation of miR-181b on apoptosis and senescence in the vascular endothelium in vivo, we utilized endothelial-specific miR-181a2b2 knockout mice, which showed robust expression of senescent markers p16, p21, and γH2AX in aortic endothelium (Fig.8). Because there was a potent decrease in p21 expression in aortas and several other organs in the systemic miR-181a2b2 knockout (Supplementary Figure 3), it is likely that other cell types in addition to endothelial cells contribute to cellular senescence in this mouse model. We recently found that these mice develop accelerated atherosclerotic lesion formation in the aorta when placed on a Western diet in the presence of AAV-PCSK9 gain-of-function mutation overexpression.43 While we have not performed formal longitudinal studies that would be required to address the life span of these mice, the constellation of aging markers in the vascular endothelium (Fig. 8) including γH2AX for DNA double strand breaks, p16 and p21 for cellular senescence, and MAP3K3 for inflammatory signaling constitute some of the key features found in several aging mouse models.71 Collectively, the miR-181b-mediated regulation of apoptosis and senescence indicates that miR-181b plays an important role in endothelial aging.
As an essential upstream regulator in the MAPK cascade, MAP3K3 regulates apoptosis and senescence. MAP3K3 knockdown blocked mitochondrial impairment by inhibiting the loss of mitochondrial membrane potential and cytochrome C expression as well as promoting ATP synthesis and reduced apoptosis and mitochondrial damage in cardiomyocytes in response to hypoxia/reoxygenation (H/R)34. MAP3K3 deficiency also increased the lifespan in C.elegans and decreased the levels of ROS72. In the current study, we also found that MAP3K3 knockdown reduced apoptosis and senescence markers in ECs responding to H2O2 through the MAPK signaling pathway (Fig.6 A–D). Using a combination of prediction algorithms and alignment with potential MAPK-associated targets, we verified that MAP3K3 is a target of miR-181b in ECs. Importantly, miR-181b was dependent on MAP3K3 to regulate EC apoptosis and senescence (Fig.7 A–D). Given the known cross-talk between NF-kB and MAPK signaling pathways, it is possible that the reduction in miR-181b expression in response to senescent stimuli may be mediated in part by NF-κB site. In support, we identified several putative NF-κB binding sites in the promoter region of MIR181B2 (Supplementary Figure 4), the dominantly expressed miR-181b isoform in endothelial cells.24,40 Future studies will be of interest to further delineate this potential feedback loop. Taken together, these findings underscore a prominent role for miR-181b in regulating apoptosis and senescence in ECs via a MAP3K3/MAPK pathway.
In summary, we identified miR-181b as an aging-related and oxidative stress-responsive microRNA that inhibits endothelial cell apoptosis and senescence. MiR-181b expression inversely correlated with increasing age in the aorta and was repressed by H2O2-treated ECs. Gain or loss-of-function studies revealed that miR-181b regulated the expression of key apoptosis (Bcl2, Bax, cleaved-Caspase3) and senescence markers (p16, p21, γH2AX) in H2O2-induced ECs. On the molecular level, miR-181b targets MAP3K3, a known upstream regulator of MAPK signaling. MiR-181b was dependent on MAP3K3 for regulating EC apoptosis and senescence, and MAP3K3 knockdown recapitulated the phenotype of miR-181b overexpression. EC-miR-181a2b2 KO mice showed increased expression of the target MAP3K3, markers of vascular senescence (p16, p21), and DNA double-strand breaks (γH2AX) in the aorta of aged mice. These findings establish an important role of miR-181b in regulating vascular endothelial aging via a MAP3K3-MAPK signaling pathway, providing new insights for anti-aging therapies in the vascular endothelium.
Supplementary Material
Acknowledgments
The authors would like to thank Ana Lay-Hong for their assistance with immunofluorescence imaging (Harvard Digestive Disease Center, NIH P30DK034854).
Funding
This work was supported by the National Institutes of Health (HL115141, HL134849, HL148207, HL148355, HL153356 to M.W.F.), and the American Heart Association (18SFRN33900144 and 20SFRN35200163 to M.W.F.).
Non-standard Abbreviations and Acronyms
- BMDM
Bone marrow derived macrophages
- CVD
Cardiovascular disease
- EC
Endothelial cell
- HCD
High cholesterol diet
- IHC
Immunohistochemistry
- MAPK
Mitogen-activated protein kinases
- PBMC
Peripheral blood mononuclear cell
- RT-qPCR
Real-time polymerase chain reaction
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
Footnotes
Competing interests
Authors declare that they have no competing interests.
Data and materials availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
References
- 1.Heidenreich PA et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation 123, 933–944, doi: 10.1161/CIR.0b013e31820a55f5 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Anton B, Vitetta L, Cortizo F & Sali A Can we delay aging? The biology and science of aging. Ann N Y Acad Sci 1057, 525–535 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Gude NA, Broughton KM, Firouzi F & Sussman MA Cardiac ageing: extrinsic and intrinsic factors in cellular renewal and senescence. Nat Rev Cardiol 15, 523–542, doi: 10.1038/s41569-018-0061-5 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Munzel T et al. Impact of Oxidative Stress on the Heart and Vasculature: Part 2 of a 3-Part Series. J Am Coll Cardiol 70, 212–229, doi: 10.1016/j.jacc.2017.05.035 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donato AJ, Machin DR & Lesniewski LA Mechanisms of Dysfunction in the Aging Vasculature and Role in Age-Related Disease. Circ Res 123, 825–848, doi: 10.1161/CIRCRESAHA.118.312563 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aoki M et al. Endothelial apoptosis induced by oxidative stress through activation of NF-kappaB: antiapoptotic effect of antioxidant agents on endothelial cells. Hypertension 38, 48–55 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Tower J Programmed cell death in aging. Ageing Res Rev 23, doi: 10.1016/j.arr.2015.04.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McHugh D & Gil J Senescence and aging: Causes, consequences, and therapeutic avenues. J Cell Biol 217, 65–77, doi: 10.1083/jcb.201708092 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green DR & Reed JC Mitochondria and apoptosis. Science 281, 1309–1312, doi: 10.1126/science.281.5381.1309 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Green DR & Llambi F Cell Death Signaling. Cold Spring Harb Perspect Biol 7, doi: 10.1101/cshperspect.a006080 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sikora E, Arendt T, Bennett M & Narita M Impact of cellular senescence signature on ageing research. Ageing Res Rev 10, 146–152, doi: 10.1016/j.arr.2010.10.002 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Debacq-Chainiaux F, Erusalimsky JD, Campisi J & Toussaint O Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 4, 1798–1806, doi: 10.1038/nprot.2009.191 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Dimri GP et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 92, 9363–9367 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collado M & Serrano M Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 10, 51–57, doi: 10.1038/nrc2772 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valdiglesias V, Giunta S, Fenech M, Neri M & Bonassi S gammaH2AX as a marker of DNA double strand breaks and genomic instability in human population studies. Mutat Res 753, 24–40, doi: 10.1016/j.mrrev.2013.02.001 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Condorelli G, Latronico MV & Cavarretta E microRNAs in cardiovascular diseases: current knowledge and the road ahead. J Am Coll Cardiol 63, 2177–2187, doi: 10.1016/j.jacc.2014.01.050 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Dangwal S & Thum T microRNA therapeutics in cardiovascular disease models. Annu Rev Pharmacol Toxicol 54, 185–203, doi: 10.1146/annurev-pharmtox-011613-135957 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Bartel DP MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233, doi: 10.1016/j.cell.2009.01.002 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menghini R et al. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation 120, 1524–1532, doi: 10.1161/CIRCULATIONAHA.109.864629 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Ito T, Yagi S & Yamakuchi M MicroRNA-34a regulation of endothelial senescence. Biochem Biophys Res Commun 398, 735–740, doi: 10.1016/j.bbrc.2010.07.012 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Guo Y et al. Kallistatin reduces vascular senescence and aging by regulating microRNA-34a-SIRT1 pathway. Aging Cell 16, 837–846, doi: 10.1111/acel.12615 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Magenta A et al. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ 18, 1628–1639, doi: 10.1038/cdd.2011.42 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun X, Sit A & Feinberg MW Role of miR-181 family in regulating vascular inflammation and immunity. Trends Cardiovasc Med 24, 105–112, doi: 10.1016/j.tcm.2013.09.002 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun X et al. Systemic delivery of microRNA-181b inhibits nuclear factor-kappaB activation, vascular inflammation, and atherosclerosis in apolipoprotein E-deficient mice. Circ Res 114, 32–40, doi: 10.1161/CIRCRESAHA.113.302089 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang JC & Bennett M Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res 111, 245–259, doi: 10.1161/CIRCRESAHA.111.261388 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Pearson G et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22, 153–183, doi: 10.1210/edrv.22.2.0428 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Raman M, Chen W & Cobb MH Differential regulation and properties of MAPKs. Oncogene 26, 3100–3112, doi: 10.1038/sj.onc.1210392 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Liu YZ, Boxer LM & Latchman DS Activation of the Bcl-2 promoter by nerve growth factor is mediated by the p42/p44 MAPK cascade. Nucleic Acids Res 27, 2086–2090, doi: 10.1093/nar/27.10.2086 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Papadakis ES et al. The regulation of Bax by c-Jun N-terminal protein kinase (JNK) is a prerequisite to the mitochondrial-induced apoptotic pathway. FEBS Lett 580, 1320–1326, doi: 10.1016/j.febslet.2006.01.053 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Beier F, Taylor AC & LuValle P The Raf-1/MEK/ERK pathway regulates the expression of the p21(Cip1/Waf1) gene in chondrocytes. J Biol Chem 274, 30273–30279, doi: 10.1074/jbc.274.42.30273 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Deacon K & Blank JL MEK kinase 3 directly activates MKK6 and MKK7, specific activators of the p38 and c-Jun NH2-terminal kinases. The Journal of biological chemistry 274, 16604–16610 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Deacon K & Blank JL Characterization of the mitogen-activated protein kinase kinase 4 (MKK4)/c-Jun NH2-terminal kinase 1 and MKK3/p38 pathways regulated by MEK kinases 2 and 3. MEK kinase 3 activates MKK3 but does not cause activation of p38 kinase in vivo. The Journal of biological chemistry 272, 14489–14496 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Guo SY, Liu SG, Liu L, Zhou XJ & Gu Y RNAi silencing of the MEKK3 gene promotes TRAIL-induced apoptosis in MCF-7 cells and suppresses the transcriptional activity of NF-kappaB. Oncol Rep 27, 441–446, doi: 10.3892/or.2011.1509 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Wang L, Yan W & Wang J Silencing MEKK3 attenuates cardiomyocyte injury caused by hypoxia/reoxygenation via the sonic hedgehog pathway. J Cell Physiol, doi: 10.1002/jcp.28162 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Xu LG, Li LY & Shu HB TRAF7 potentiates MEKK3-induced AP1 and CHOP activation and induces apoptosis. J Biol Chem 279, 17278–17282, doi: 10.1074/jbc.C400063200 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Ellinger-Ziegelbauer H, Kelly K & Siebenlist U Cell cycle arrest and reversion of Ras-induced transformation by a conditionally activated form of mitogen-activated protein kinase kinase kinase 3. Mol Cell Biol 19, 3857–3868 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henao-Mejia J et al. The microRNA miR-181 is a critical cellular metabolic rheostat essential for NKT cell ontogenesis and lymphocyte development and homeostasis. Immunity 38, 984–997, doi: 10.1016/j.immuni.2013.02.021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486, doi: 10.1038/nature09002 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Haemmig S et al. Long noncoding RNA SNHG12 integrates a DNA-PK-mediated DNA damage response and vascular senescence. Science translational medicine 12, doi: 10.1126/scitranslmed.aaw1868 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Sun X et al. MicroRNA-181b regulates NF-κB-mediated vascular inflammation. J Clin Invest 122, 1973–1990, doi: 10.1172/JCI61495 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin J et al. MicroRNA-181b inhibits thrombin-mediated endothelial activation and arterial thrombosis by targeting caspase recruitment domain family member 10. FASEB J 30, 3216–3226, doi: 10.1096/fj.201500163R (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paneni F, Diaz Canestro C, Libby P, Luscher TF & Camici GG The Aging Cardiovascular System: Understanding It at the Cellular and Clinical Levels. J Am Coll Cardiol 69, 1952–1967, doi: 10.1016/j.jacc.2017.01.064 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Yang DH,S; Chen J; McCoy MG; Cheng HS; Zhou H; Pérez-Cremades D; Cheng X; Sun X; Haneo-Mejia J; Vellarikkal SK; Gupta RM; Barrera V; Feinberg MW Endothelial cell-specific deletion of a microRNA accelerates atherosclerosis. Atherosclerosis, doi:DOI: 10.1016/j.atherosclerosis.2022.04.010 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pommier Y Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer 6, 789–802, doi: 10.1038/nrc1977 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Dérijard B et al. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science (New York, N.Y.) 267, 682–685 (1995). [DOI] [PubMed] [Google Scholar]
- 46.Sun X et al. MicroRNA-181b regulates NF-kappaB-mediated vascular inflammation. J Clin Invest 122, 1973–1990, doi: 10.1172/JCI61495 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lam JK, Chow MY, Zhang Y & Leung SW siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol Ther Nucleic Acids 4, e252, doi: 10.1038/mtna.2015.23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jeyapalan JC & Sedivy JM Cellular senescence and organismal aging. Mech Ageing Dev 129, 467–474, doi: 10.1016/j.mad.2008.04.001 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ungvari Z et al. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat Rev Cardiol 15, 555–565, doi: 10.1038/s41569-018-0030-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lv Y et al. LncRNA nuclear-enriched abundant transcript 1 regulates hypoxia-evoked apoptosis and autophagy via mediation of microRNA-181b. Mol Cell Biochem 464, 193–203, doi: 10.1007/s11010-019-03660-2 (2020). [DOI] [PubMed] [Google Scholar]
- 51.Liu B, Guo Z & Gao W miR-181b-5p promotes proliferation and inhibits apoptosis of hypertrophic scar fibroblasts through regulating the MEK/ERK/p21 pathway. Exp Ther Med 17, 1537–1544, doi: 10.3892/etm.2019.7159 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen H, Chen Q, Fang M & Mi Y microRNA-181b targets MLK2 in HL-60 cells. Sci China Life Sci 53, 101–106, doi: 10.1007/s11427-010-0002-y (2010). [DOI] [PubMed] [Google Scholar]
- 53.Liu Y et al. miR-181b functions as an oncomiR in colorectal cancer by targeting PDCD4. Protein Cell 7, 722–734, doi: 10.1007/s13238-016-0313-2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu J, Shi W, Wu C, Ju J & Jiang J miR-181b as a key regulator of the oncogenic process and its clinical implications in cancer (Review). Biomed Rep 2, 7–11, doi: 10.3892/br.2013.199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu W, Shan X, Wang T, Shu Y & Liu P miR-181b modulates multidrug resistance by targeting BCL2 in human cancer cell lines. Int J Cancer 127, 2520–2529, doi: 10.1002/ijc.25260 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Yang X, Sun Y, Zhang Y & Han S Downregulation of miR181b inhibits human colon cancer cell proliferation by targeting CYLD and inhibiting the NFkappaB signaling pathway. Int J Mol Med 46, 1755–1764, doi: 10.3892/ijmm.2020.4720 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang L et al. miR-181b promotes cell proliferation and reduces apoptosis by repressing the expression of adenylyl cyclase 9 (AC9) in cervical cancer cells. FEBS Lett 588, 124–130, doi: 10.1016/j.febslet.2013.11.019 (2014). [DOI] [PubMed] [Google Scholar]
- 58.He L et al. MicroRNA-181b expression in prostate cancer tissues and its influence on the biological behavior of the prostate cancer cell line PC-3. Genet Mol Res 12, 1012–1021, doi: 10.4238/2013.April.2.17 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Li D et al. Down-regulation of miR-181b promotes apoptosis by targeting CYLD in thyroid papillary cancer. Int J Clin Exp Pathol 7, 7672–7680 (2014). [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu DX et al. miR-181a/b significantly enhances drug sensitivity in chronic lymphocytic leukemia cells via targeting multiple anti-apoptosis genes. Carcinogenesis 33, 1294–1301, doi: 10.1093/carcin/bgs179 (2012). [DOI] [PubMed] [Google Scholar]
- 61.Zhi F et al. MiR-181b-5p downregulates NOVA1 to suppress proliferation, migration and invasion and promote apoptosis in astrocytoma. PLoS One 9, e109124, doi: 10.1371/journal.pone.0109124 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu B, Guo Z & Gao W miR-181b-5p promotes proliferation and inhibits apoptosis of hypertrophic scar fibroblasts through regulating the MEK/ERK/p21 pathway. Exp Ther Med 17, 1537–1544, doi: 10.3892/etm.2019.7159 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ling L, Zhi L, Wang H, Deng Y & Gu C MicroRNA-181b Inhibits Inflammatory Response and Reduces Myocardial Injury in Sepsis by Downregulating HMGB1. Inflammation 44, 1263–1273, doi: 10.1007/s10753-020-01411-w (2021). [DOI] [PubMed] [Google Scholar]
- 64.Zhu FX, Wu HL, Chen JX, Han B & Guo YF Dysregulation of microRNA-181b and TIMP3 is functionally involved in the pathogenesis of diabetic nephropathy. J Cell Physiol 234, 18963–18969, doi: 10.1002/jcp.28536 (2019). [DOI] [PubMed] [Google Scholar]
- 65.Pang X et al. Inhibition of lncRNA MEG3 protects renal tubular from hypoxia-induced kidney injury in acute renal allografts by regulating miR-181b/TNF-alpha signaling pathway. J Cell Biochem 120, 12822–12831, doi: 10.1002/jcb.28553 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Hernandez-Segura A, Nehme J & Demaria M Hallmarks of Cellular Senescence. Trends Cell Biol 28, 436–453, doi: 10.1016/j.tcb.2018.02.001 (2018). [DOI] [PubMed] [Google Scholar]
- 67.Munoz-Espin D & Serrano M Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15, 482–496, doi: 10.1038/nrm3823 (2014). [DOI] [PubMed] [Google Scholar]
- 68.Herranz N & Gil J Mechanisms and functions of cellular senescence. J Clin Invest 128, 1238–1246, doi: 10.1172/JCI95148 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gorgoulis V et al. Cellular Senescence: Defining a Path Forward. Cell 179, 813–827, doi: 10.1016/j.cell.2019.10.005 (2019). [DOI] [PubMed] [Google Scholar]
- 70.Kuo LJ & Yang LX Gamma-H2AX - a novel biomarker for DNA double-strand breaks. In Vivo 22, 305–309 (2008). [PubMed] [Google Scholar]
- 71.Koks S et al. Mouse models of ageing and their relevance to disease. Mech Ageing Dev 160, 41–53, doi: 10.1016/j.mad.2016.10.001 (2016). [DOI] [PubMed] [Google Scholar]
- 72.Chamoli M, Singh A, Malik Y & Mukhopadhyay A A novel kinase regulates dietary restriction-mediated longevity in Caenorhabditis elegans. Aging Cell 13, 641–655, doi: 10.1111/acel.12218 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.