

Abstract
Background:
While there is high comorbidity of stress-related disorders and alcohol use disorder (AUD), few effective treatments are available and elucidating underlying neurobiological mechanisms has been hampered by a general lack of reliable animal models. Here we utilize a novel mouse model demonstrating robust and reproducible stress-enhanced alcohol drinking to examine the role of dynorphin/kappa opioid receptor (DYN/KOR) activity within the extended amygdala in mediating this stress-alcohol interaction.
Methods:
Mice received repeated weekly cycles of chronic intermittent ethanol (CIE) exposure alternating with weekly drinking sessions ± forced swim stress (FSS) exposure. Pdyn mRNA expression was measured in the central amygdala (CeA) and DYN-containing CeA neurons (CeADYN) were then targeted for chemogenetic inhibition. Lastly, a KOR antagonist was microinjected into the CeA or bed nucleus stria terminalis (BNST) to examine the role of KOR signaling in promoting stress-enhanced drinking.
Results:
Stress (FSS) selectively increased alcohol drinking in mice with a history of CIE exposure, and this was accompanied by elevated Pdyn mRNA levels in CeA. Targeted chemogenetic silencing of CeADYN neurons blocked stress-enhanced drinking and KOR antagonism in the CeA or BNST significantly reduced stress-induced elevated alcohol consumption without altering moderate intake in controls.
Conclusions:
Utilizing a novel and robust model of stress-enhanced alcohol drinking, a significant role for DYN/KOR activity within extended amygdala circuitry in mediating this effect was demonstrated, thereby providing further evidence that the DYN/KOR system may be a valuable target in the development of more effective treatments for individuals presenting with comorbidity of stress-related disorders and AUD.
Keywords: Stress, Alcohol, Dynorphin, Kappa opioid receptor, Amygdala, Bed nucleus stria terminalis
INTRODUCTION
Stress is commonly regarded as a potent trigger for relapse and a significant factor in promoting excessive alcohol (ethanol) drinking (1–3). There is a high comorbidity between stress-related disorders (including depression, anxiety disorders, and post-traumatic stress disorder; PTSD) and alcohol use disorder (AUD) (4–8). The prevalence and magnitude of the problem underscores the importance of understanding mechanisms underlying the influence of stress on alcohol use, which is essential for developing new and more effective treatment strategies for individuals suffering with co-existing stress and alcohol use disorders.
Stress and alcohol exposure influence overlapping systems and neural circuits in the brain, producing adaptations that compromise the ability of individuals to engage in behavioral flexibility that would enhance control over alcohol consumption, as well as appropriately respond to stressful events that may provoke return to excessive drinking (9,10). Despite significant advances in our understanding about how stress and alcohol alter brain function, the mechanisms and neurocircuits underlying the complex interactions between stress and alcohol consumption are not fully understood (11). Indeed, demonstrating reliable and consistent effects related to the interaction between stress and alcohol drinking in animal models has been challenging (12–14). The general lack of reliable preclinical models has, at least in part, impeded progress toward developing effective therapeutics that especially target stress-related excessive drinking.
To address this shortcoming, we developed a mouse model wherein repeated brief forced swim stress (FSS) exposure interacts with chronic intermittent ethanol (CIE) exposure to enhance alcohol drinking in dependent (CIE-exposed) mice but not alter more moderate stable intake in nondependent mice (15,16). Corroborating an earlier study in rats (17), this stress-enhanced drinking in CIE-exposed mice is robust and reproducible, having been demonstrated by other research groups as well (18–20). Increased drinking in mice with a history of both chronic alcohol exposure and stress experience provides an ideal opportunity to utilize this CIE-FSS Drinking model to probe mechanisms and potential targets relevant to the problem of stress-related excessive alcohol drinking.
Because both stress and chronic alcohol engage the dynorphin/kappa opioid receptor system, the role of this neuropeptide system in chronic alcohol-related dysphoria and elevated drinking has gained increasing attention (21–23). Dynorphins (DYN) are peptides derived from the precursor prodynorphin (Pdyn) that preferentially bind to kappa opioid receptors (KOR), producing physiological and behavioral effects via inhibitory G-protein (Gi) coupling and other signaling cascades (24–27). KOR activation has been shown to produce aversive/dysphoric effects as indicated by measures of conditioned avoidance, anxiety-like, and depression-like behavior (28,29). Stress exposure activates the DYN/KOR system, eliciting dysphoria- and anxiety-like behaviors (30) along with elevated DYN immunoreactivity in brain regions that are integral to reward and stress circuitries involved in alcohol/drug addiction (31).
Pharmacological manipulation of DYN/KOR activity has been shown to alter behavioral responses to stress and motivational effects of alcohol in a variety of experimental conditions (22,32). Systemic administration of KOR antagonists has been shown to reduce high levels of alcohol consumption associated with dependence and binge-drinking models in rats (33,34) and mice (20,35,36). Evidence points to involvement of the extended amygdala in mediating these effects. Interconnected brain structures comprising extended amygdala circuitry including the nucleus accumbens, central amygdala (CeA), and the bed nucleus of the stria terminalis (BNST) are rich in DYN and KOR and highly responsive to stress and chronic alcohol exposure (37–41). Direct infusion of the KOR antagonist norbinaltorphimine (norBNI) into these structures reduces excessive alcohol drinking in models of dependence (42–44) and binge-like drinking (35,45). While we recently demonstrated that systemic administration of a KOR antagonist can block the ability of stress to enhance alcohol consumption (36), the site of action mediating this effect is not known.
The present series of studies were designed to probe the role of DYN/KOR activity in two prominent structures within extended amygdala circuitry (CeA and BNST) as it relates to stress-enhanced alcohol drinking. Specifically, our CIE-FSS Drinking model was employed to: 1) examine how a history of CIE and FSS exposure, alone and in combination, affect expression of Pdyn mRNA within the CeA; 2) determine the effect of targeted chemogenetic silencing of DYN-expressing neurons within the CeA on stress-enhanced alcohol drinking; and 3) determine the contribution of KOR signaling within the CeA and BNST in mediating the ability of stress to further enhance elevated alcohol consumption associated with dependence. Results from these studies provide critical evidence indicating that a history of chronic alcohol exposure and stress engage the DYN/KOR system within the extended amygdala in mediating stress-enhanced drinking. As such, these findings support recent clinical efforts devoted to evaluating the therapeutic value of targeting this neuropeptide system in reducing heavy drinking in individuals comorbid with stress-related disorders and AUD (46).
MATERIALS and METHODS
A detailed description of all experimental procedures, including the CIE-FSS Drinking model, assays for Pdyn mRNA measurement, stereotaxic surgery, virus and drug infusions, histology, and drug preparations are provided in Supplemental Materials.
Chronic Intermittent Ethanol (CIE) and Forced Swim Stress (FSS) Drinking Model
All studies involved use of adult male C57BL/6J mice and Pdyn-IRES-Cre mice (35,47) treated in the CIE-FSS Drinking model, as previously described (15,16,36). Briefly, after establishing stable 1-hr daily ethanol (15%; v/v) intake, mice were separated into four groups: control (CTL), CIE-alone, FSS-alone, and CIE+FSS. Mice received chronic intermittent ethanol (CIE) vapor or air exposure in inhalation chambers followed by test drinking sessions for 5 consecutive days. This pattern of weekly CIE (or air) exposure alternating with weekly test drinking sessions was repeated for 3 or 4 cycles (Figure 1). Mice in the FSS-alone and CIE+FSS groups experienced brief (10-min) FSS exposure 4-hr prior to each of the test drinking sessions. The remaining non-stressed mice (CTL and CIE groups) were left in their home cage undisturbed.
Figure 1: CIE-FSS Drinking Model.
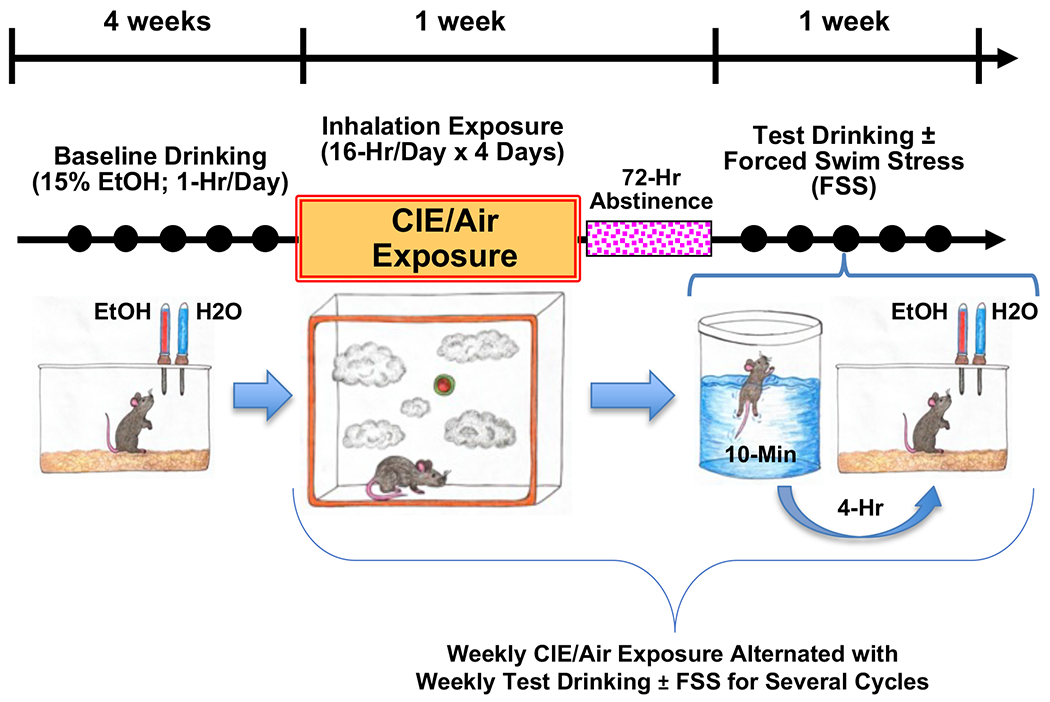
Mice were treated in the CIE-FSS Drinking model involving weekly CIE or Air exposure cycles alternating with weekly Test drinking sessions (1-hr) with or without FSS exposure (10-min; 4-hr prior to each drinking session). Solid circles represent daily 1-hr drinking sessions. After establishing stable Baseline drinking over 4-weeks, mice were separated into 4 groups: CTL, CIE-alone, FSS-alone, CIE+FSS. Weekly CIE/Air exposures alternated with weekly (5-Day) Test drinking sessions for several cycles.
Study Procedures
Effects of CIE and FSS exposure on Pdyn mRNA expression in the CeA:
C57BL/6J mice treated in the CIE-FSS Drinking model were sacrificed on the final day of Test 4 at 30-min, 4-hr, or 24-hr after FSS exposure (or at equivalent times for no-stress groups) (N= 6-10/group/time point). Collection of CeA samples, RNA extraction, and TaqMan quantitative reverse transcription-polymerase chain reaction (qRT-PCR) assays were performed as previously described (48,49).
Effect of chemogenetic inhibition of Pdyn-expressing neurons in CeA on alcohol drinking in the CIE-FSS model:
Adult male Pdyn-IRES-Cre mice received bilateral infusions of a Cre-dependent virus containing an inhibitory DREADD (AAV8-hSyn-DIO-hM4Di-mCherry) (N= 10-11/group) or control virus (AAV8-hSyn-DIO-mCherry) (N= 6-7/group) into the CeA to target Pdyn-expressing CeA neurons (CeADYN), as previously described (50). After at least 2 weeks of recovery, the Baseline phase of the study commenced. On Day-2 and Day-4 of Test 3, mice were injected (ip.) with vehicle (0.9% saline) or clozapine-N-oxide (CNO; 3 mg/kg) to activate the DREADD. The order of drug administration was counter-balanced for each group.
Effect of KOR antagonism in CeA or BNST on alcohol drinking in the CIE-FSS model:
C57BL/6J mice received bilateral guide cannula positioned above the CeA (N= 7-9/group) or BNST (N= 8-10/group) (45,50). After at least 2 weeks recovery, Baseline drinking commenced. At 16-hr prior to the start of the drinking session on Day-3 of Test 3, separate groups of mice in each experimental condition received microinjection of the KOR antagonist norbinaltorphimine (norBNI; 2.5 μg/side) or vehicle (1X PBS) into the CeA or BNST.
Statistical Analysis
Alcohol intake (g/kg) was analyzed by ANOVA, with Group (CTL, CIE, FSS, CIE+FSS) as a between-subjects factor and Test Cycle (average weekly intake for Baseline and Tests 1-4) as a repeated measure. Pdyn mRNA expression in the CeA was expressed as a fold-change from the CTL condition and analyzed at each timepoint by ANOVA, with Group as the main factor. For the chemogenetic study, alcohol intake and change in intake relative to vehicle was analyzed by ANOVA, with Group and Virus as a between-subject variables and Drug (CNO vs. saline) as a repeated measure. For microinjection studies, ANOVAs included Group and Drug (norBNI vs. vehicle) as between-subject variables and Day as a repeated measure in analyses of alcohol consumption (average of Days 3 and 4) and difference in intake from respective vehicle group. When appropriate, significant main effects and interactions were further analyzed using Newman—Keuls post-hoc comparisons. In chemogenetic and pharmacological studies, only data from subjects in which targeted viral expression or placement of microinjectors were verified as correct were used in analyses.
RESULTS
Combined Chronic Intermittent Ethanol (CIE) and Stress (FSS) Exposure Increases Pdyn mRNA Expression in the CeA.
While alcohol consumption remained relatively stable over successive Test cycles in the in the FSS-alone and CTL groups, intake increased above baseline levels in CIE-exposed groups, with this effect most robust in the CIE+FSS group (Figures 2A, 2B). This was supported by a significant Group x Test Cycle interaction [F(12,352)= 5.942, p< 0.001], which revealed a modest increase in alcohol consumption in the CIE-alone group, with intake during Test 2 and Test 4 greater than Baseline (ps< 0.05). Alcohol intake in the CIE+FSS group was greater during all Test weeks compared to their Baseline level of intake (ps< 0.001), and drinking was greater during each Test week in the CIE+FSS condition compared to the CIE-alone group (ps< 0.05). Separate analysis of alcohol intake during the final test period prior to sacrifice (Test 4) revealed a main effect of Group [F(3,88)= 13.025, p< 0.001]. While alcohol intake for CIE-alone and FSS-alone groups did not significantly differ from the CTL group, CIE+FSS mice exhibited greater alcohol intake compared to all other groups (ps< 0.001) (Figure 2C). These results confirm our previous findings showing that stress (FSS) experience selectively enhances alcohol drinking in dependent (CIE-exposed) mice while not altering intake in nondependent (FSS-alone) mice (15,16,36).
Figure 2: A History of CIE+FSS Exposure Increases Pdyn mRNA Expression in the CeA.
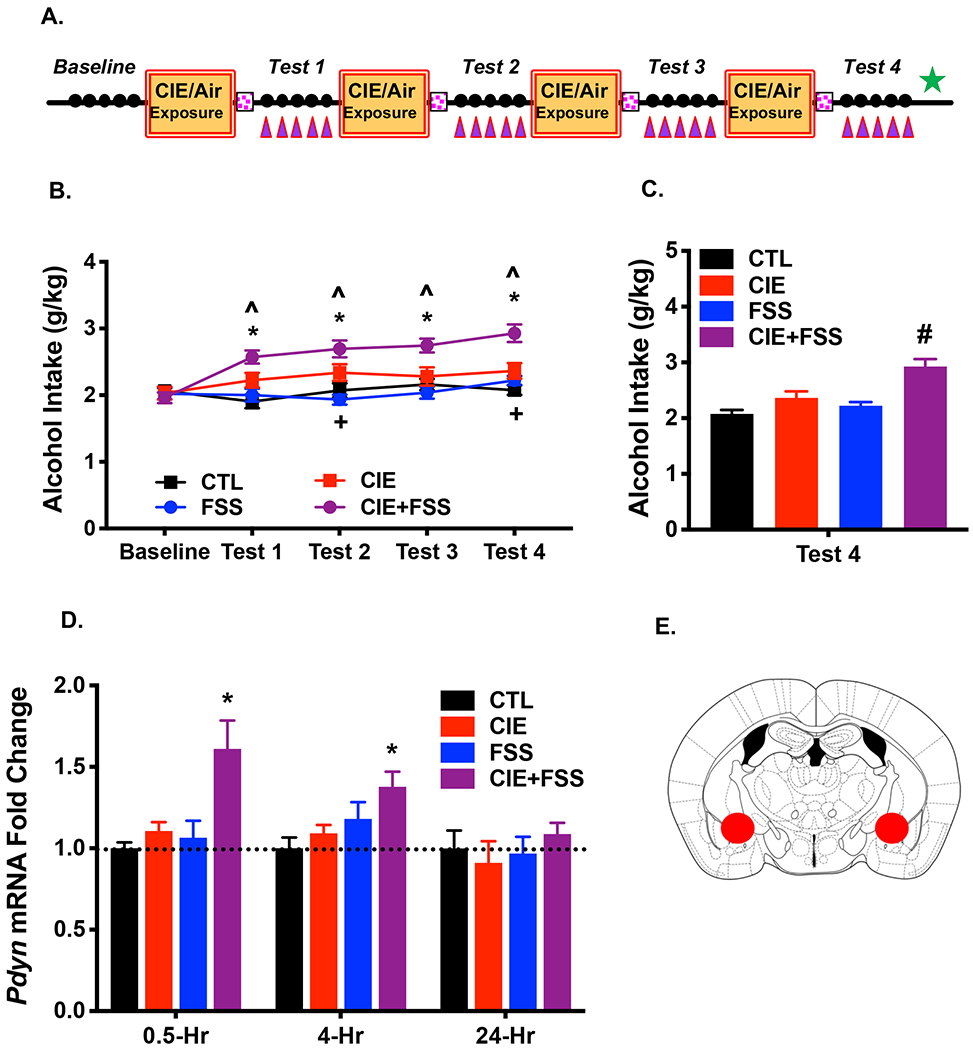
(A) Mice were sacrificed, and brain tissue was collected at 30-min, 4-hr, or 24-hr following the last FSS exposure during Test 4 (denoted by green star) to assess Pdyn mRNA expression in CeA (purple triangles denote FSS exposures) (N= 6-10/group/time point). (B) Average weekly alcohol consumption across each phase of the study. Alcohol intake increased compared to Baseline in the CIE-alone group during Test 2 and Test 4 (+ ps< 0.05); Alcohol drinking in the CIE+FSS group was significantly greater during each of the Test cycles compared to Baseline (* ps< 0.05) and compared to the CIE-alone group (^ ps< 0.05); Alcohol intake in FSS-alone and CTL groups during Test cycles did not differ from Baseline levels. (C) Average alcohol consumption during Test 4. Alcohol intake was greater in the CIE+FSS group compared to all other groups which did not significantly differ from each other (# ps< 0.001). (D) Pdyn mRNA expression in the CeA was significantly elevated in CIE+FSS compared to CTL mice at 30-min (* p< 0.01) and 4-hr (* p< 0.05) post-FSS exposure at the end of Test 4, returning to CTL levels at 24-hr. All values are mean ± s.e.m. (E) Schematic representation of tissue punches collected from the CeA.
Analysis of Pdyn mRNA expression in the CeA at the end of Test 4 revealed main effects of Group at 30-min [F(3,30)= 7.055, p< 0.001] and 4-hr [F(3,23)= 3.33, p= 0.037] following the last FSS exposure but not at 24-hr post-FSS [F(3,27)= 0.415, p= 0.744]. Post-hoc analyses showed that Pdyn mRNA expression was selectively elevated in the CIE+FSS group relative to the CTL group at 30-min and 4-hr after FSS exposure, with values normalizing at the 24-hr time point (Figure 2D). Neither CIE-alone nor FSS-alone treatments produced an increase in Pdyn mRNA levels in the CeA. Schematic depiction of bilateral tissue punches collected from the CeA is shown in Figure 2E.
Chemogenetic Inhibition of CeADYN Neurons Reduces Stress (FSS)-Enhanced Alcohol Drinking in CIE-Exposed Pdyn-IRES-Cre Mice.
Pdyn-IRES-Cre mice were used to target expression of an inhibitory (hM4Di) DREADD or control virus in CeADYN neurons (Figure 3A). Using this same strategy, we previously demonstrated fidelity of expression and functionality of this viral vector (35). In the present study, expression of the mCherry marker was confined to the CeA (Figure 3B).
Figure 3: Chemogenetic Inhibition of CeADYN Attenuates Stress-Enhanced Alcohol Drinking.
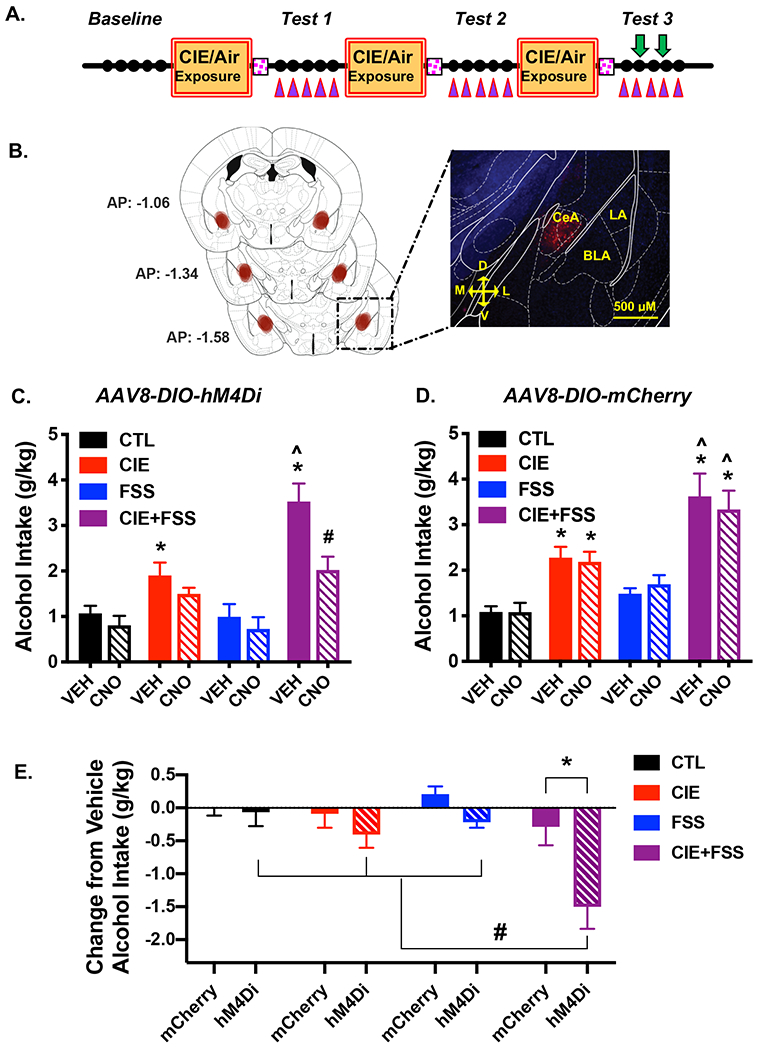
(A) For targeted expression of an inhibitory DREADD, Pdyn-IRES-Cre mice were bilaterally infused with AAV8-hSyn-DIO-hM4Di-mCherry or AAV8-hSyn-DIO-mCherry into the CeA 2-weeks prior to start of Baseline drinking; vehicle or CNO (3 mg/kg) was injected 30-min prior to the 2nd and 4th drinking sessions during Test 3 in a balanced cross-over design (denoted by green arrows). (B) Viral expression was localized to the CeA as indicated by visualization of mCherry fluorescence. (C) Average alcohol intake in mice expressing hM4Di-contaning virus (N= 10-11/group). In vehicle-treated mice, alcohol consumption was greater in CIE-alone and CIE+FSS groups compared to CTL and FSS-alone groups which did not differ (* ps< 0.05). Also, intake following vehicle injection was greater in CIE+FSS mice compared to CIE-alone mice (^ p< 0.005). Activation of the inhibitory DREADD expressed in CeADYN neurons via CNO injection resulted in a significant reduction in alcohol intake in the CIE+FSS group compared to those mice receiving vehicle (# p< 0.001). (D) Average alcohol intake in mice treated with control virus (N= 6-7/group). Vehicle injection resulted in the expected greater alcohol intake in CIE-alone and CIE+FSS groups compared to the other groups (* ps< 0.05), and intake was greater in the CIE+FSS mice compared to CIE-alone mice (^ p< 0.005). CNO injection did not alter alcohol drinking in any group relative to when those mice received vehicle injection. (E) Change from respective vehicle alcohol intake across treatment and virus groups. CNO injection significantly reduced alcohol intake (relative to vehicle) in CIE+FSS mice that harbored active vs. control virus (* p< 0.001) and this reduction was greater in CIE+FSS mice compared to when CNO was injected in CIE-alone, FSS-alone, and CTL groups that were treated with active virus (# ps< 0.01). All values are mean ± s.e.m.
Average weekly alcohol intake prior to administering CNO during Test 3 is shown in Table 1. ANOVA revealed a main effect of Group [F(3,60)= 8.397, p< 0.001] and Test Cycle [F(2,120)= 22.632, p< 0.001] but no effect of Virus [F(1,60)= 1.895, p= 0.174], indicating that viral expression did not influence alcohol drinking prior to testing. A Group x Test Cycle interaction [F(6,120)= 6.258, p< 0.001] was observed and post-hoc analyses revealed increased alcohol intake in the CIE-alone and CIE+FSS groups during Test 1 and Test 2 compared to their respective Baseline levels of intake (ps< 0.05).
Table 1: Average Weekly Alcohol Intake Prior to Chemogenetic Inhibition of CeADYN Neurons.
Alcohol intake (g/kg) in mice expressing hM4Di or mCherry in CeADYN neurons during Baseline and Test weeks prior to challenge with vehicle or CNO (3 mg/kg) during Test 3. Values are mean ± s.e.m.
| AAV | Group | Baseline | Test 1 | Test 2 |
|---|---|---|---|---|
| hM4Di | CTL | 1.48 ± 0.12 | 1.44 ± 0.14 | 1.49 ± 0.16 |
| FSS | 1.57 ± 0.13 | 1.78 ± 0.12 | 1.94 ± 0.17 | |
| CIE | 1.49 ± 0.18 | 1.95 ± 0.26* | 1.84 ± 0.20* | |
| CIE+FSS | 1.54 ± 0.15 | 2.13 ± 0.25* | 2.54 ± 0.22* | |
|
| ||||
| mCherry | CTL | 1.62 ± 0.17 | 1.72 ± 0.17 | 1.33 ± 0.13 |
| FSS | 1.49 ± 0.11 | 1.56 ± 0.19 | 1.65 ± 0.06 | |
| CIE | 1.61 ± 0.11 | 2.53 ± 0.19* | 2.37 ± 0.20* | |
| CIE+FSS | 1.73 ± 0.13 | 2.83 ± 0.34* | 2.57 ± 0.32* | |
differs from respective Baseline levels (p< 0.05).
Analysis of alcohol intake during Test 3 indicated a marginally significant Group x Virus x Drug interaction [F(3,60)= 2.340, p= 0.082]. Separate analysis of alcohol intake in mice expressing hM4Di in CeADYN neurons revealed a significant Group x Drug interaction [F(3,38)= 8.521, p< 0.001]. Vehicle (saline) administration in mice with a history of CIE-alone and CIE+FSS consumed more alcohol than CTL mice that received vehicle (ps< 0.05). Further, alcohol intake was significantly greater in the CIE+FSS group compared to the CIE-alone condition (p< 0.005) whereas FSS-alone did not significantly alter alcohol consumption Selective silencing of CeADYN neurons following CNO injection resulted in significant reduction in alcohol intake in the CIE+FSS group (p< 0.001). CNO injection produced a marginal reduction in drinking in the CIE-alone group (p= 0.082) but did not alter alcohol intake in the FSS-alone or CTL groups (Figure 3C). Similar analysis of alcohol consumption in mice treated with control virus indicated a main effect of Group [F(3,22)= 16.14, p< 0.001], but no Group x Drug interaction [F(3,22)= 0.128, p= 0.381]. As expected, alcohol intake was significantly greater in CIE-alone mice compared to CTL mice (p< 0.05) and stress further enhanced this elevated drinking (CIE+FSS > CIE-alone) (p< 0.01). Alcohol intake in the FSS-alone group did not differ from the CTL group. CNO injection in mice that harbored the control virus did not alter alcohol drinking in any of the groups (Figure 3D).
To further examine the apparent “selective” effect of CNO in mice treated with the active (hM4Di-containing) virus, data expressed as a change from the vehicle (saline) condition for each subject was analyzed (Figure 3E). Post-hoc analysis of the Group x Virus interaction [F3,60)= 2.34, p< 0.05] indicated that CNO injection significantly reduced alcohol intake (relative to vehicle) only in CIE+FSS mice that harbored active vs. control virus (p< 0.001) and this reduction was greater in CIE+FSS mice compared to all other groups that were treated with active virus (ps< 0.01). Thus, inactivation of CeADYN neurons following CNO injection significantly attenuated the ability of stress (FSS) to further enhance alcohol consumption in CIE-exposed mice, and this effect was only observed in mice treated with the inhibitory (hM4Di) DREADD-containing virus.
Intra-CeA Injection of a KOR Antagonist Blocks Stress Enhancement of Alcohol Drinking in CIE-Exposed Mice.
Alcohol intake prior to microinjection of the KOR antagonist during Test 3 is shown in Table 2. ANOVA indicated significant main effects of Group [F(3,64)= 14.823, p< 0.001] and Test Cycle [F(2,128)= 75.339, p< 0.001], and a Group x Test Cycle interaction [F(6,128)= 12.882, p< 0.001]. Post-hoc analyses indicated that alcohol intake in the CIE-alone and CIE+FSS groups during Test 1 and Test 2 was greater than their respective Baseline levels of intake (ps< 0.05). In contrast, FSS-alone did not alter alcohol drinking in nondependent mice.
Table 2: Average Weekly Alcohol Intake Prior to Injection of KOR Antagonist into CeA.
Alcohol intake (g/kg) in mice during Baseline and Test weeks prior to bilateral microinjection of vehicle of norBNI (2.5 μg/side) into the CeA during Test 3. Values are mean ± s.e.m.
| Group | Baseline | Test 1 | Test 2 |
|---|---|---|---|
| CTL | 1.88 ± 0.07 | 2.19 ± 0.09 | 1.89 ± 0.08 |
| FSS | 1.90 ± 0.08 | 2.37 ± 0.15 | 2.03 ± 0.12 |
| CIE | 1.88 ± 0.08 | 2.82 ± 0.11* | 2.94 ± 0.16* |
| CIE+FSS | 1.82 ± 0.08 | 3.04 ± 0.14* | 3.08 ± 0.14* |
differs from respective Baseline levels (p< 0.05).
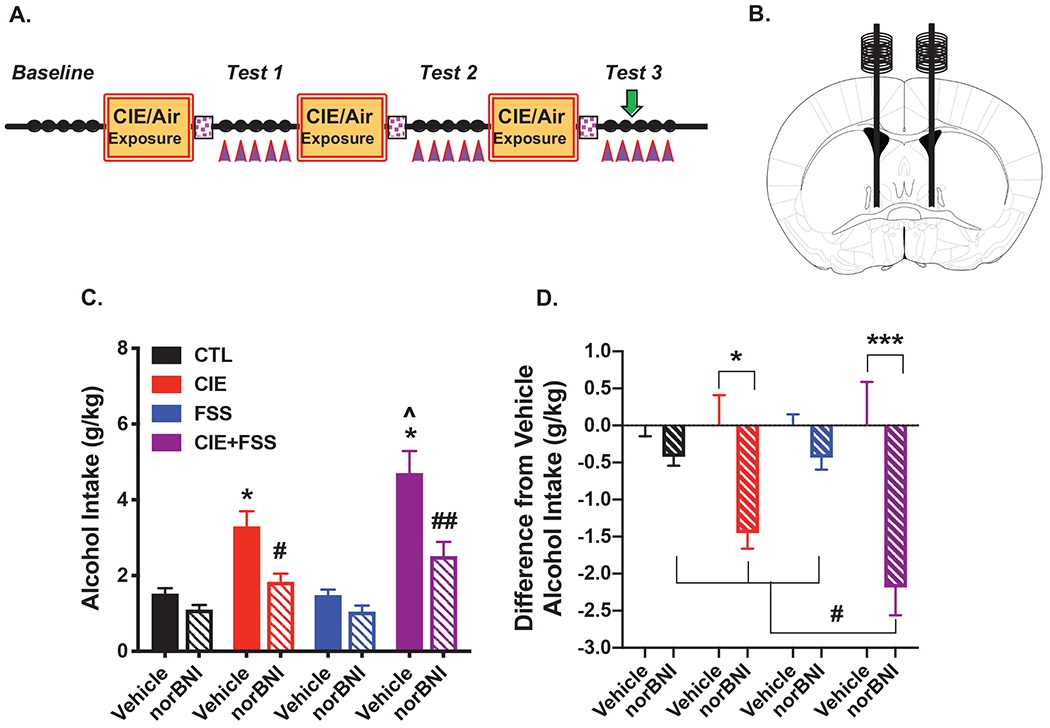
Microinjection of norBNI into the CeA during Test 3 (Figures 4A, 4B) selectively attenuated elevated drinking in mice with a history of CIE-alone and CIE+FSS without affecting more moderate levels of consumption in FSS-alone and CTL groups (Figure 4C). This finding is supported by analysis of average alcohol intake during the two days following vehicle or norBNI microinjection, which revealed a significant Group x Drug interaction [F(3,60)= 6.742; p< 0.001]. Post-hoc analyses showed that vehicle-treated mice with a history of CIE-alone and CIE+FSS exposure consumed more alcohol than CTL mice (ps< 0.005) and, replicating our earlier finding, alcohol intake was significantly greater in CIE+FSS compared to CIE-alone mice (p< 0.05). Microinjection of norBNI reversed elevated alcohol intake in both CIE-alone and CIE+FSS groups (ps< 0.01). Analysis of daily alcohol intake during Test 3 revealed a similar profile of results (Figures S2A and S2B). To evaluate the relative magnitude of the norBNI effect across all groups, data were also analyzed as a difference from the average vehicle intake for each group. ANOVA revealed a significant Group x Drug interaction [F(3,60)= 6.742, p< 0.001], and post-hoc analysis indicated that while a trend was apparent in the CIE-alone group (p= 0.125), norBNI significantly reduced alcohol intake relative to the respective vehicle condition only in the CIE+FSS group (p< 0.001). Further, the reduction in alcohol drinking following intra-CeA norBNI injection (relative to vehicle) was significantly greater in the CIE+FSS group compared to all other groups, which did not differ from each other (ps< 0.01) (Figure 4D). Collectively, these results suggest that blocking KOR signaling in the CeA was especially effective in reducing stress-enhanced drinking in the model. Schematic representation of injection sites for mice that received vehicle or norBNI into the CeA is shown in Figures S3A and S3B, respectively.
Figure 4: Microinjection of norBNI into the CeA Attenuates Stress-Enhanced Alcohol Drinking.
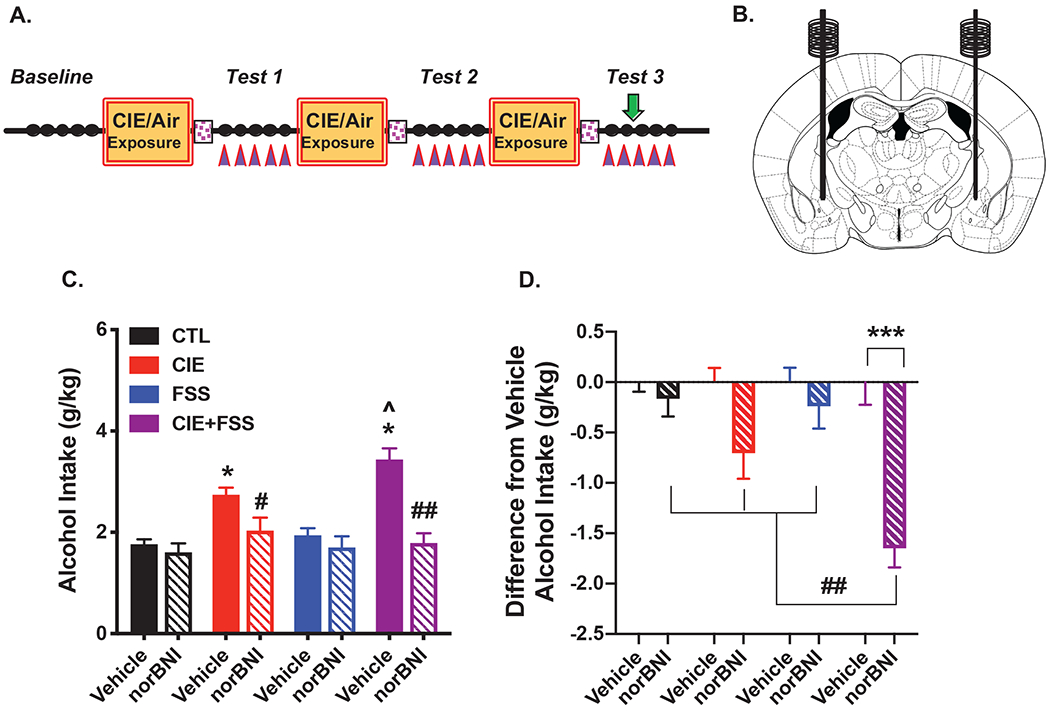
(A) Guide cannulae were implanted over the CeA 2-weeks prior to the start of Baseline drinking; vehicle or norBNI (2.5 μg/side) was infused 16-hr prior to the 3rd drinking session during Test 3 (denoted by green arrow). (B) Schematic representation of stereotaxic bilateral placement of guide cannulae over the CeA. (C) Average alcohol intake (over Day 3 and Day 4) in mice receiving vehicle (N= 7-9/group) or norBNI (N= 8-9/group) injection in the CeA. Vehicle-treated CIE-alone and CIE+FSS groups consumed significantly more alcohol than CTL and FSS-alone groups (which did not differ) (* ps< 0.05) and alcohol intake was greater in CIE+FSS compared to CIE-alone mice (^ p< 0.05); norBNI treatment blocked elevated drinking in CIE-alone (# p< 0.01) and CIE+FSS (## p< 0.001) groups without altering intake in the FSS-alone or CTL groups. (D) Difference from respective vehicle alcohol intake across treatment groups. Intra-CeA norBNI injection significantly reduced alcohol intake (relative to vehicle) only in the CIE+FSS group (*** p< 0.001), and this effect was significantly greater in the CIE+FSS group compared to all other groups, which did not differ from each other (## ps< 0.01).
Intra-BNST Injection of a KOR Antagonist Blocks Stress Enhancement of Alcohol Drinking in CIE-Exposed Mice.
Given our previous work showing that KOR antagonism in the BNST reduced heavy (binge-like) alcohol drinking (45) and that dynorphinergic neurons in the CeA project to the BNST (40,51), this study was conducted to examine whether blocking KOR signaling in the BNST attenuates the ability of stress to further enhance elevated drinking in CIE-exposed mice. As in previous experiments, alcohol consumption increased over Baseline levels during Test 1 and Test 2 in CIE-alone and CIE+FSS groups while intake remained relatively stable for FSS-alone and CTL groups (Table 3). This was supported by significant main effects of Group [F(3,68)= 13.527, p< 0.001] and Test Cycle [F(2,136)= 21.644, p< 0.001], and the Group x Test Cycle interaction [F(6,136)= 9.009, p< 0.001].
Table 3: Average Weekly Alcohol Intake Prior to Injection of KOR Antagonist into BNST.
Alcohol intake (g/kg) in mice during Baseline and Test weeks prior to bilateral microinjection of vehicle of norBNI (2.5 μg/side) into the BNST during Test 3. Values are mean ± s.e.m.
| Group | Baseline | Test 1 | Test 2 |
|---|---|---|---|
| CTL | 1.97 ± 0.12 | 1.85 ± 0.12 | 1.84 ± 0.15 |
| FSS | 2.02 ± 0.19 | 2.18 ± 0.12 | 2.02 ± 0.16 |
| CIE | 2.13 ± 0.13 | 2.84 ± 0.13* | 2.99 ± 0.16* |
| CIE+FSS | 1.99 ± 0.12 | 2.88 ± 0.12* | 2.93 ± 0.15* |
differs from respective Baseline levels (p< 0.05).
Intra-BNST norBNI administration during Test 3 (Figures 5A, 5B) reduced elevated alcohol intake in CIE-alone and CIE+FSS groups without altering moderate intake in FSS-alone and CTL groups (Figure 5C). Analysis of average intake during the two days after microinjection revealed a Group x Drug interaction [F(3,64)= 3.975, p= 0.012]. Post-hoc analyses showed that alcohol intake following vehicle administration was significantly greater in CIE+FSS mice compared to the CIE-alone group (p< 0.01), and both groups consumed more alcohol than FSS-alone and CTL mice (ps< 0.001), which did not significantly differ from each other. Microinjection of norBNI into the BNST significantly reduced elevated alcohol consumption in the CIE-alone (p< 0.005) and CIE+FSS (p< 0.001) groups. Analysis of daily alcohol intake during Test 3 produced a similar profile of results (Figures S4A and S4B). Analysis of the difference in alcohol intake following intra-BNST norBNI injection relative to the appropriate average vehicle intake for each group revealed a significant Group x Drug interaction [F(3,64)= 3.975, p< 0.01]. While intra-BNST norBNI reduced alcohol drinking (relative to vehicle levels) in the CIE-alone group (p= 0.024) and CIE+FSS mice (p< 0.001), this effect was significantly more robust in the CIE+FSS group compared to all other groups, including CIE-alone mice (ps< 0.05) (Figure 5D). In contrast, norBNI injection into the BNST did not alter alcohol intake in FSS-alone or CTL groups. Schematic representation of injection sites for mice that received vehicle or norBNI into the BNST is shown in Figure S5A and S5B, respectively.
Figure 5: Microinjection of norBNI into the BNST Attenuates Stress-Enhanced Alcohol Drinking.
(A) Guide cannulae were implanted over the BNST 2-weeks prior to the start of Baseline drinking; vehicle or norBNI (2.5 μg/side) was infused 16-hr prior to the 3rd drinking session during Test 3 (denoted by green arrow). (B) Schematic representation of stereotaxic bilateral placement of guide cannulae over the BNST. (C) Average alcohol intake (over Day 3 and Day 4) in mice receiving vehicle (N= 8-10/group) or norBNI (N= 8-10/group) injection in the BNST. Following vehicle injection, CIE+FSS mice consumed significantly more alcohol than the CIE-alone group (^ p< 0.05), and both CIE+FSS and CIE-alone groups consumed more alcohol than CTL and FSS-alone groups (which did not differ) (* ps< 0.05); norBNI treatment blocked elevated drinking in CIE-alone (# p< 0.005) and CIE+FSS (## p< 0.001) groups without altering intake in the FSS-alone or CTL groups. (D) Difference from respective vehicle alcohol intake across treatment groups. Intra-BNST norBNI injection significantly reduced alcohol intake (relative to vehicle) in the CIE-alone group (* p< 0.05) and the CIE+FSS group (*** p< 0.001), and this effect was significantly greater in the CIE+FSS group compared to all other groups, which did not differ from each other (# ps< 0.05).
DISCUSSION
The present studies validate the CIE-FSS Drinking paradigm as a framework for modeling stress-enhanced alcohol drinking and demonstrate the significant contribution of DYN/KOR activity within the extended amygdala in mediating this behavior. A history of repeated FSS in CIE-exposed mice resulted in escalation of voluntary alcohol consumption and this was accompanied by elevated Pdyn mRNA expression in the CeA. Targeted chemogenetic silencing of DYN-containing neurons in the CeA completely blocked the ability of stress to enhance alcohol drinking in the model. Further, pharmacological blockade of KOR within the CeA or BNST normalized drinking in mice with a history of both stress and CIE exposure. Together, these data suggest that CeADYN neurons are uniquely responsive to a history of chronic alcohol exposure and stress, and KOR signaling within the CeA and BNST plays an important role in mediating stress-enhanced alcohol drinking.
While chronic alcohol exposure and stress are known to produce dynamic alterations in brain gene expression (52–56), only recently have genomic changes in relation to stress-alcohol interactions been explored (18). This latter study revealed both unique transitory and long-lasting changes in gene expression in prefrontal cortex associated with stress-enhanced alcohol consumption in the CIE-FSS Drinking model. In the present study, we show that Pdyn mRNA levels in the CeA are elevated at 30-min and 4-hr after FSS exposure only in mice with a history of both FSS and CIE exposure. This change was not observed in other groups (CIE-alone and FSS-alone conditions) that did not exhibit increased alcohol intake relative to the CTL condition. Studies in rats have shown an upregulation in Pdyn mRNA expression in CeA following chronic alcohol drinking (57) and acute withdrawal from CIE exposure (43). Forced swim stress experience also has been reported to increase Pdyn mRNA levels in extended amygdala structures (58,59). Together these findings indicate that the CeA is highly responsive to stress and chronic alcohol exposure and changes in Pdyn transcriptional activity may contribute to enhanced motivation to drink following combined stress and chronic alcohol exposure.
The CeA is a key structure within extended amygdala circuitry with rich expression of both DYN and KORs (38–40). In the present study, using a validated transgenic mouse model (47) along with a validated DREADD-containing viral construct (35), targeted chemogenetic inactivation of CeADYN neurons blocked stress-enhanced alcohol drinking. Vehicle injections in mice expressing the inhibitory (hM4Di) DREADD did not alter alcohol drinking and reduced alcohol intake in mice injected with CNO is not likely to be attributed to off-target effects of CNO since the ligand did not alter alcohol consumption in mice that received control virus treatment. Using a similar experimental strategy, silencing CeADYN neurons was shown to significantly reduce alcohol consumption in a binge-drinking model (35). Likewise, genetic deletion of Pdyn in the CeA reduced alcohol drinking in models of high intake (38). Together, these data indicate that dynorphinergic activity in the CeA plays a significant role in regulating alcohol consumption, including elevated drinking associated with stress.
CeADYN neurons produce effects through signaling at KOR both locally within the CeA as well as in several projection regions. Studies have shown KOR antagonism within the CeA and other extended amygdala structures (e.g., BNST) reduces alcohol drinking (35,42,43,45), and this is congruent with findings showing that systemic administration of KOR antagonists reduce alcohol consumption in a variety of models (20,22,33–36). Results from the present study indicate that direct injection of the KOR antagonist norBNI into the CeA or BNST blocked the ability of stress to enhance voluntary alcohol drinking in the CIE+FSS group. Thus, these are the first data to directly implicate a role for DYN/KOR activity within extended amygdala circuitry in contributing to stress-induced excessive drinking. While there is evidence for dynorphinergic projections from the CeA to the BNST (40,51), the extent to which these results are mediated by KOR signaling within the CeA and other projection sites, including the BNST, will require more direct circuitry-based examination.
DYN-containing neurons in the CeA are primarily GABAergic and known to co-express other neuropeptides that influence alcohol drinking, such as corticotropin release factor, and neurotensin (60,61). Thus, co-release of GABA and other peptides may contribute to stress-enhanced drinking in this model, and this possibility cannot be ruled out. However, results from our chemogenetic and pharmacological studies strongly implicate a significant role for engagement of DYN/KOR activity within extended amygdala circuitry in the ability of stress to further elevate alcohol consumption in subjects with a history of chronic alcohol exposure.
While stress-enhanced drinking in the CIE-FSS model has been demonstrated in both female and male mice (62,63), one limitation of this study is that only males were used to examine the role of DYN/KOR activity. Sex-related differences in sensitivity to DYN/KOR function have been noted, especially regarding reward and aversion/dysphoria related behaviors (64–66). Thus, it will be important in future studies to examine whether manipulation of DYN/KOR activity within extended amygdala circuitry produces sex-related differences in stress-enhanced alcohol drinking. This is especially relevant given the high prevalence of co-occurring stress-related disorders and alcohol use disorder in women (5,8).
In summary, despite stress being a significant contributing factor in heavy drinking, as reflected by high comorbidity of stress-related disorders and AUD, few effective treatments are available and the lack of preclinical models that reliably demonstrate stress-enhanced drinking has hindered efforts to address the problem. We have established a model demonstrating robust and highly reproducible stress-induced elevation of alcohol consumption. Using chemogenetic and pharmacological approaches, we show that DYN/KOR activity within extended amygdala circuitry plays a significant role in mediating the ability of stress to increase drinking in mice with a history of chronic alcohol exposure. These findings align with other preclinical studies showing that long-acting (20,33–35) and short-acting (36,67,68) KOR antagonists reduce high levels of alcohol consumption and relapse-like behavior provoked by stress. Together, these results support clinical studies that target the DYN/KOR system in the development of more effective treatments for individuals presenting with comorbidity of stress-related disorders and AUD (46,69).
Supplementary Material
KEY RESOURCES TABLE
| Resource Type | Specific Reagent or Resource | Source or Reference | Identifiers | Additional Information |
|---|---|---|---|---|
| Add additional rows as needed for each resource type | Include species and sex when applicable. | Include name of manufacturer, company, repository, individual, or research lab. Include PMID or DOI for references; use “this paper” if new. | Include catalog numbers, stock numbers, database IDs or accession numbers, and/or RRIDs. RRIDs are highly encouraged; search for RRIDs at https://scicrunch.org/resources. | Include any additional information or notes if necessary. |
| Antibody | Rat, anti-mCherry | Millipore | # M11217 | |
| Antibody | Goat anti-Rat conjugated AlexaFluor 555 | Invitrogen | # P36961 | |
| Bacterial or Viral Strain | AAV8-hSyn-DIO-hM4Di-mCherry | Addgene | # 50459-AAV8 | |
| Bacterial or Viral Strain | AAV8-hSyn-DIO-mCherry | Addgene | # 44362-AAV8 | |
| Biological Sample | Mouse brain | This study | N/A | |
| Cell line | N/A | |||
| Chemical Compound or Drug | clozapine-N-oxide hydrochloride | Tocris | # 6329 | |
| Chemical Compound or Drug | nor-binaltorphiminie dihydrochloride | Tocris | #0347 | |
| Commercial Assay Or Kit | mirVana miRNA Extraction kit | Invitrogen | # AM1560 | |
| Commercial Assay Or Kit | QuantiTect Reverse Transcription kit | Quagen | # 205314 | |
| Deposited Data; Public Database | N/A | |||
| Genetic Reagent | N/A | |||
| Organism/Strain | Mouse, C57BL/6J, male | Jackson Laboratories | #000664 | |
| Organism/Strain | Mouse, Pdyn-IRES-Cre, male | PMID:30555162; PMID: 24487620 | N/A | |
| Peptide, Recombinant Protein | N/A | |||
| Recombinant DNA | N/A | |||
| Sequence-Based Reagent | TaqMan qRT-PCR primers (Pdyn, Ppia) | ThermoFisher Scientific | Pdyn (# Mm00457573_ml) Ppia (# Mm02342430_gl) | |
| Software; Algorithm | N/A | |||
| Transfected Construct | N/A | |||
| Other |
ACKNOWLEDGEMENTS
This work was supported by NIH grants P50 AA010761 (HCB), U01 AA 014095 (HCB), U24 AA020929 (MFL), R01 AA026536 (HCB), F31 AA027420 (HLH) and a grant from the Department of Veterans Affairs (BLRD BX000813) (HCB).
We thank Ms. Olivia C. Sweatt for creating study design schematics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
The authors report no biomedical financial interests or potential conflicts of interest.
REFERENCES
- 1.Blaine SK & Sinha R (2017). Alcohol, stress, and glucocorticoids: From risk to dependence and relapse in alcohol use disorders. Neuropharmacology 122:136–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sinha R (2012). How does stress lead to risk of alcohol relapse? Alcohol research : current reviews 34(4):432–440. [PMC free article] [PubMed] [Google Scholar]
- 3.Uhart M & Wand GS (2009). Stress, alcohol and drug interaction: an update of human research. Addiction biology 14(1):43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grant BF, Goldstein RB, Saha TD, et al. (2015). Epidemiology of DSM-5 Alcohol Use Disorder: Results From the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA Psychiatry 72(8):757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peltier MR, Verplaetse TL, Mineur YS, et al. (2019). Sex differences in stress-related alcohol use. Neurobiol Stress 10:100149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petrakis IL & Simpson TL (2017). Posttraumatic Stress Disorder and Alcohol Use Disorder: A Critical Review of Pharmacologic Treatments. Alcohol Clin Exp Res 41(2):226–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schellekens AF, de Jong CA, Buitelaar JK, et al. (2015). Co-morbid anxiety disorders predict early relapse after inpatient alcohol treatment. Eur Psychiatry 30(1):128–136. [DOI] [PubMed] [Google Scholar]
- 8.Guinle MIB & Sinha R (2020). The Role of Stress, Trauma, and Negative Affect in Alcohol Misuse and Alcohol Use Disorder in Women. Alcohol research : current reviews 40(2):05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koob GF (2021). Drug Addiction: Hyperkatifeia/Negative Reinforcement as a Framework for Medications Development. Pharmacol Rev 73(1):163–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koob GF & Volkow ND (2016). Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry 3(8):760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Becker HC (2017). Influence of stress associated with chronic alcohol exposure on drinking. Neuropharmacology 122:115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Becker HC, Lopez MF & Doremus-Fitzwater TL (2011). Effects of stress on alcohol drinking: a review of animal studies. Psychopharmacology 218(1):131–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noori HR, Helinski S & Spanagel R (2014). Cluster and meta-analyses on factors influencing stress-induced alcohol drinking and relapse in rodents. Addiction biology 19(2):225–232. [DOI] [PubMed] [Google Scholar]
- 14.Spanagel R, Noori HR & Heilig M (2014). Stress and alcohol interactions: animal studies and clinical significance. Trends in neurosciences 37(4):219–227. [DOI] [PubMed] [Google Scholar]
- 15.Anderson RI, Lopez MF & Becker HC (2016). Forced swim stress increases ethanol consumption in C57BL/6J mice with a history of chronic intermittent ethanol exposure. Psychopharmacology 233(11):2035–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez MF, Anderson RI & Becker HC (2016). Effect of different stressors on voluntary ethanol intake in ethanol-dependent and nondependent C57BL/6J mice. Alcohol 51:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sommer WH, Rimondini R, Hansson AC, et al. (2008). Upregulation of voluntary alcohol intake, behavioral sensitivity to stress, and amygdala crhr1 expression following a history of dependence. Biological psychiatry 63(2):139–145. [DOI] [PubMed] [Google Scholar]
- 18.Farris SP, Tiwari GR, Ponomareva O, et al. (2020). Transcriptome Analysis of Alcohol Drinking in Non-Dependent and Dependent Mice Following Repeated Cycles of Forced Swim Stress Exposure. Brain Sci 10(5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodberg EM, den Hartog CR, Anderson RI, et al. (2017). Stress Facilitates the Development of Cognitive Dysfunction After Chronic Ethanol Exposure. Alcohol Clin Exp Res 41(9):1574–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rose JH, Karkhanis AN, Chen R, et al. (2016). Supersensitive Kappa Opioid Receptors Promotes Ethanol Withdrawal-Related Behaviors and Reduce Dopamine Signaling in the Nucleus Accumbens. Int J Neuropsychopharmacol 19(5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chavkin C & Koob GF (2016). Dynorphin, Dysphoria, and Dependence: the Stress of Addiction. Neuropsychopharmacology 41(1):373–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karkhanis AN & Al-Hasani R (2020). Dynorphin and its role in alcohol use disorder. Brain Res 1735:146742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tejeda HA & Bonci A (2019). Dynorphin/kappa-opioid receptor control of dopamine dynamics: Implications for negative affective states and psychiatric disorders. Brain Res 1713:91–101. [DOI] [PubMed] [Google Scholar]
- 24.Bruchas MR & Chavkin C (2010). Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology 210(2):137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bruchas MR, Land BB & Chavkin C (2010). The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res 1314:44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crowley NA & Kash TL (2015). Kappa opioid receptor signaling in the brain: Circuitry and implications for treatment. Progress in neuro-psychopharmacology & biological psychiatry 62:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wee S & Koob GF (2010). The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology 210(2):121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knoll AT & Carlezon WA Jr. (2010). Dynorphin, stress, and depression. Brain Res 1314:56–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van’t Veer A & Carlezon WA Jr. (2013). Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology 229(3):435–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Land BB, Bruchas MR, Lemos JC, et al. (2008). The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. The Journal of neuroscience : the official journal of the Society for Neuroscience 28(2):407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shirayama Y, Ishida H, Iwata M, et al. (2004). Stress increases dynorphin immunoreactivity in limbic brain regions and dynorphin antagonism produces antidepressant-like effects. Journal of neurochemistry 90(5):1258–1268. [DOI] [PubMed] [Google Scholar]
- 32.Anderson RI & Becker HC (2017). Role of the Dynorphin/Kappa Opioid Receptor System in the Motivational Effects of Ethanol. Alcohol Clin Exp Res 41(8):1402–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walker BM & Koob GF (2008). Pharmacological evidence for a motivational role of kappa-opioid systems in ethanol dependence. Neuropsychopharmacology 33(3):643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walker BM, Zorrilla EP & Koob GF (2011). Systemic kappa-opioid receptor antagonism by nor-binaltorphimine reduces dependence-induced excessive alcohol self-administration in rats. Addiction biology 16(1):116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson RI, Lopez MF, Griffin WC, et al. (2019). Dynorphin-kappa opioid receptor activity in the central amygdala modulates binge-like alcohol drinking in mice. Neuropsychopharmacology 44(6):1084–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson RI, Lopez MF & Becker HC (2016). Stress-Induced Enhancement of Ethanol Intake in C57BL/6J Mice with a History of Chronic Ethanol Exposure: Involvement of Kappa Opioid Receptors. Front Cell Neurosci 10:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Al-Hasani R, McCall JG, Shin G, et al. (2015). Distinct Subpopulations of Nucleus Accumbens Dynorphin Neurons Drive Aversion and Reward. Neuron 87(5):1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bloodgood DW, Hardaway JA, Stanhope CM, et al. (2020). Kappa opioid receptor and dynorphin signaling in the central amygdala regulates alcohol intake. Mol Psychiatry [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mansour A, Fox CA, Meng F, et al. (1994). Kappa 1 receptor mRNA distribution in the rat CNS: comparison to kappa receptor binding and prodynorphin mRNA. Molecular and cellular neurosciences 5(2):124–144. [DOI] [PubMed] [Google Scholar]
- 40.Marchant NJ, Densmore VS & Osborne PB (2007). Coexpression of prodynorphin and corticotrophin-releasing hormone in the rat central amygdala: evidence of two distinct endogenous opioid systems in the lateral division. The Journal of comparative neurology 504(6):702–715. [DOI] [PubMed] [Google Scholar]
- 41.Poulin JF, Arbour D, Laforest S, et al. (2009). Neuroanatomical characterization of endogenous opioids in the bed nucleus of the stria terminalis. Progress in neuro-psychopharmacology & biological psychiatry 33(8):1356–1365. [DOI] [PubMed] [Google Scholar]
- 42.Erikson CM, Wei G & Walker BM (2018). Maladaptive behavioral regulation in alcohol dependence: Role of kappa-opioid receptors in the bed nucleus of the stria terminalis. Neuropharmacology 140:162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kissler JL, Sirohi S, Reis DJ, et al. (2014). The one-two punch of alcoholism: role of central amygdala dynorphins/kappa-opioid receptors. Biological psychiatry 75(10):774–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nealey KA, Smith AW, Davis SM, et al. (2011). kappa-opioid receptors are implicated in the increased potency of intra-accumbens nalmefene in ethanol-dependent rats. Neuropharmacology 61(1-2):35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haun HL, Griffin WC, Lopez MF, et al. (2020). Kappa opioid receptors in the bed nucleus of the stria terminalis regulate binge-like alcohol consumption in male and female mice. Neuropharmacology 167:107984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reed B, Butelman ER & Kreek MJ (2022). Kappa Opioid Receptor Antagonists as Potential Therapeutics for Mood and Substance Use Disorders. Handb Exp Pharmacol 271:473–491. [DOI] [PubMed] [Google Scholar]
- 47.Krashes MJ, Shah BP, Madara JC, et al. (2014). An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature 507(7491):238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Melendez RI, McGinty JF, Kalivas PW, et al. (2012). Brain region-specific gene expression changes after chronic intermittent ethanol exposure and early withdrawal in C57BL/6J mice. Addiction biology 17(2):351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Solomon MG, Griffin WC, Lopez MF, et al. (2019). Brain Regional and Temporal Changes in BDNF mRNA and microRNA-206 Expression in Mice Exposed to Repeated Cycles of Chronic Intermittent Ethanol and Forced Swim Stress. Neuroscience 406:617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anderson RI, Lopez MF, Griffin WC, et al. (2018). Dynorphin-kappa opioid receptor activity in the central amygdala modulates binge-like alcohol drinking in mice. Neuropsychopharmacology [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stamatakis AM, Sparta DR, Jennings JH, et al. (2014). Amygdala and bed nucleus of the stria terminalis circuitry: Implications for addiction-related behaviors. Neuropharmacology 76 Pt B:320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Girgenti MJ, Pothula S & Newton SS (2021). Stress and Its Impact on the Transcriptome. Biological psychiatry 90(2):102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palmisano M & Pandey SC (2017). Epigenetic mechanisms of alcoholism and stress-related disorders. Alcohol 60:7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pucci M, Micioni Di Bonaventura MV, Wille-Bille A, et al. (2019). Environmental stressors and alcoholism development: Focus on molecular targets and their epigenetic regulation. Neurosci Biobehav Rev 106:165–181. [DOI] [PubMed] [Google Scholar]
- 55.Ferguson LB, Patil S, Moskowitz BA, et al. (2019). A Pathway-Based Genomic Approach to Identify Medications: Application to Alcohol Use Disorder. Brain Sci 9(12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith ML, Lopez MF, Wolen AR, et al. (2020). Brain regional gene expression network analysis identifies unique interactions between chronic ethanol exposure and consumption. PLoS One 15(5):e0233319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou Y, Colombo G, Gessa GL, et al. (2013). Effects of voluntary alcohol drinking on corticotropin-releasing factor and preprodynorphin mRNA levels in the central amygdala of Sardinian alcohol-preferring rats. Neurosci Lett 554:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chung S, Kim HJ, Kim HJ, et al. (2014). Desipramine and citalopram attenuate pretest swim-induced increases in prodynorphin immunoreactivity in the dorsal bed nucleus of the stria terminalis and the lateral division of the central nucleus of the amygdala in the forced swimming test. Neuropeptides 48(5):273–280. [DOI] [PubMed] [Google Scholar]
- 59.Reed B, Fang N, Mayer-Blackwell B, et al. (2012). Chromatin alterations in response to forced swimming underlie increased prodynorphin transcription. Neuroscience 220:109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Torruella-Suarez ML, Vandenberg JR, Cogan ES, et al. (2020). Manipulations of Central Amygdala Neurotensin Neurons Alter the Consumption of Ethanol and Sweet Fluids in Mice. The Journal of neuroscience : the official journal of the Society for Neuroscience 40(3):632–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Guglielmo G, Kallupi M, Pomrenze MB, et al. (2019). Inactivation of a CRF-dependent amygdalofugal pathway reverses addiction-like behaviors in alcohol-dependent rats. Nat Commun 10(1):1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lopez MF, Reasons SE, Carper BA, et al. (2020). Evaluation of the effect of doxasozin and zonisamide on voluntary ethanol intake in mice that experienced chronic intermittent ethanol exposure and stress. Alcohol 89:37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.den Hartog CR, Blandino KL, Nash ML, et al. (2020). Noradrenergic tone mediates marble burying behavior after chronic stress and ethanol. Psychopharmacology 237(10):3021–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Becker JB & Chartoff E (2019). Sex differences in neural mechanisms mediating reward and addiction. Neuropsychopharmacology 44(1):166–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chartoff EH & Mavrikaki M (2015). Sex Differences in Kappa Opioid Receptor Function and Their Potential Impact on Addiction. Front Neurosci 9:466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vijay A, Wang S, Worhunsky P, et al. (2016). PET imaging reveals sex differences in kappa opioid receptor availability in humans, in vivo. Am J Nucl Med Mol Imaging 6(4):205–214. [PMC free article] [PubMed] [Google Scholar]
- 67.Domi E, Barbier E, Augier E, et al. (2018). Preclinical evaluation of the kappa-opioid receptor antagonist CERC-501 as a candidate therapeutic for alcohol use disorders. Neuropsychopharmacology 43(9):1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rorick-Kehn LM, Witkin JM, Statnick MA, et al. (2014). LY2456302 is a novel, potent, orally-bioavailable small molecule kappa-selective antagonist with activity in animal models predictive of efficacy in mood and addictive disorders. Neuropharmacology 77:131–144. [DOI] [PubMed] [Google Scholar]
- 69.Lowe SL, Wong CJ, Witcher J, et al. (2014). Safety, tolerability, and pharmacokinetic evaluation of single- and multiple-ascending doses of a novel kappa opioid receptor antagonist LY2456302 and drug interaction with ethanol in healthy subjects. J Clin Pharmacol 54(9):968–978. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.