

Abstract
Systemic lupus erythematosus (SLE) is a chronic and often progressive autoimmune disorder marked clinically by a variable constellation of symptoms including fatigue, rash, joint pains, and kidney damage. The lungs, heart, gastrointestinal system, and brain can also be impacted, and individuals with lupus are at higher risk for atherosclerosis, thrombosis, thyroid disease, and other disorders associated with chronic inflammation [1, 2]. Autoimmune diseases are marked by erroneous immune responses in which the target of the immune response is a “self”-antigen, or autoantigen, driven by the development of antigen-specific B or T cells that have overcome the normal systems of self-tolerance built into the development of B and T cells. SLE is specifically characterized by the production of autoantibodies against nucleic acids and their binding proteins, including anti-double stranded DNA, anti-Smith (an RNA binding protein), and many others [3]. These antibodies bind their nuclear-derived antigens to form immune complexes that cause injury and scarring through direct deposition in tissues and activation of innate immune cells [4]. In over 50% of SLE patients, immune complex aggregation in the kidneys drives intrarenal inflammation and injury and leads to lupus nephritis, a progressive destruction of the glomeruli that is one of the most common causes of lupus-related death [5]. To counter this pathology increasing attention has turned to developing approaches to reduce the development and continued generation of such autoantibodies. In particular, the molecular and cellular events that lead to long term, continuous activation of such autoimmune responses have become the focus of new therapeutic strategies to limit renal and other pathologies in lupus patients. The focus of this review is to consider how the innate immune system is involved in the development and progression of lupus nephritis [6] and how a novel approach to inhibit innate immune activation by neutralizing the activators of this response, called Damage Associated Molecular Patterns (DAMPs), may represent a promising approach to treat this and other autoimmune disorders.
Keywords: Damage Associated Molecular Pattern (DAMP), Toll Like Receptors (TLRs), Innate Immune Activation, Nucleic Acid Scavenger, Systemic Lupus Erythematosus
Overview
Systemic lupus erythematosus (SLE) is a chronic and often progressive autoimmune disorder marked clinically by a variable constellation of symptoms including fatigue, rash, joint pains, and kidney damage. The lungs, heart, gastrointestinal system, and brain can also be impacted, and individuals with lupus are at higher risk for atherosclerosis, thrombosis, thyroid disease, and other disorders associated with chronic inflammation [1, 2]. Autoimmune diseases are marked by erroneous immune responses in which the target of the immune response is a “self”-antigen, or autoantigen, driven by the development of antigen-specific B or T cells that have overcome the normal systems of self-tolerance built into the development of B and T cells. SLE is specifically characterized by the production of autoantibodies against nucleic acids and their binding proteins, including anti-double stranded DNA, anti-Smith (an RNA binding protein), and many others [3]. These antibodies bind their nuclear-derived antigens to form immune complexes that cause injury and scarring through direct deposition in tissues and activation of innate immune cells [4]. In over 50% of SLE patients, immune complex aggregation in the kidneys drives intrarenal inflammation and injury and leads to lupus nephritis, a progressive destruction of the glomeruli that is one of the most common causes of lupus-related death [5]. To counter this pathology increasing attention has turned to developing approaches to reduce the development and continued generation of such autoantibodies. In particular, the molecular and cellular events that lead to long term, continuous activation of such autoimmune responses have become the focus of new therapeutic strategies to limit renal and other pathologies in lupus patients. The focus of this review is to consider how the innate immune system is involved in the development and progression of lupus nephritis [6] and how a novel approach to inhibit innate immune activation by neutralizing the activators of this response, called Damage Associated Molecular Patterns (DAMPs), may represent a promising approach to treat this and other autoimmune disorders.
Innate immunity and the pathogenesis of lupus nephritis
While seemingly an adaptive immunity-driven process, SLE progression and pathogenesis also relies on innate immune activity. Innate immunity both triggers the defective adaptive response and is a direct actor with the simultaneous involvement of innate cells, associated signaling, and inflammation all being consequential in SLE and associated lupus nephritis [6, 7]. For instance, clonal expansion of autoreactive B cells and T cells is directed by dendritic cell presentation of autoantigens coupled with appropriate inflammatory co-stimulatory signals [7]. The particularly pro-inflammatory neutrophils that are elevated in SLE patients produce more cytokines such as type I interferons, that lead to further immune dysregulation, and are prone to the release of neutrophil extracellular traps or NETs [8]. The associated NETosis products have been implicated in the etiology of SLE because the nuclear antigens released by this mechanism are protected from nuclease degradation and are accompanied by inflammatory cytokines that provide costimulatory signals that can assist the break in immune tolerance [9, 10]. Of significance, many clinical studies have found that bacterial and especially viral infections trigger SLE presentation or exacerbate symptoms, pointing to connection between pathogen driven activation of innate immunity, systemic inflammation and the break in immune tolerance [11]. Finally, innate immune cells can directly exacerbate disease. For example, neutrophils, activated by immune complex deposition, release proteases and reactive oxygen species that directly damage renal tissue [12, 13].
PAMPs and DAMPs as triggers of inflammation
Small molecular motifs from microbes like bacterial lipopolysaccharides or viral double stranded RNAs bind to Pattern Recognition Receptors (PRRs) of innate immune cells including neutrophils and monocytes and many non-hematopoietic cells, triggering a pro-inflammatory signaling cascade in response to this “non-self” signal. The pathogen-associated molecular patterns are termed PAMPs. Also recognized by the PRRs, when present in atypical locations or concentrations indicative of cellular damage, are endogenous nucleic acids, proteins, and metabolites from damaged or dying cells, collectively termed damage-associated molecular patterns or DAMPs [14]. The best characterized family of PRRs are toll-like receptors (TLRs). TLR1, 2, 4, 5, 6, and 10 sense proteins, lipids, or glycans and are primarily found on the cellular membrane and while TLRs 3, 7, 8, and 9 are nucleic acid sensing and are usually located on endosomal membranes. Their varied specificities are demonstrated by the recognition of viral double-stranded RNA and RNAs released from necrotic cells by TLR3 [15] and the recognition of bacterial lipopolysaccharides as well as host cell products like fibrinogen, nuclear protein HMGB1, and heparan sulfate fragments by TLR4 [16]. Upon binding these PAMPs and DAMPs, PRRs initiate signaling cascades of cellular adaptors and kinase activity that results in the activation and translocation of various transcription factors, like NFκB, AP-1, and IRFs [17]. These in turn induce the transcription, translation, and/or activation of effectors like cytokines, chemokines, interferons, bioactive amines (like histamine), arachidonic acid metabolites (leukotrienes and prostaglandins) and inflammatory mediators like bradykinins [18, 19]. The cardinal signs of inflammation, namely redness, heat, pain and swelling, are the direct consequence of this signaling cascade in innate immune cells as well as non-hematologic cells including endothelial cells lining the vasculature and epithelial cells within the tissue. The functional outcome of this inflammatory signaling cascade is the recruitment of additional immune effector cells to the site of infection and injury. With infection, innate immune cells reduce the spread of pathogens through direct elimination, consumption of infected cells, and release of systemic inflammatory signals to make the environment less hospitable to the invader (i.e. through elevation of body temperature or sequestration of nutrients like iron). Furthermore, activated innate immune cells present the pathogenic material and appropriate co-stimulatory signals to engage the slower but more specific adaptive immune response. Inflammation is generally self-limited and its resolution is a tightly controlled process that begins shortly after the initiation of the inflammatory response [20, 21]. Inflammation thus does not simply fade out but instead is balanced by anti-inflammatory signals that accompany the removal of noxious stimuli.
DAMPs in SLE and lupus nephritis
While important for tissue repair, excess production of DAMPs and activation of PRR-DAMP pathways triggers chronic inflammation and resulting diseases [14]. A central basis of lupus is the loss of tolerance for nuclear DAMPs that have emerged from dying and dead cells not effectively cleared after infection or sterile inflammation [22, 23]. Progression ensues as endocytosed immune complexes containing nucleic acids and pre-formed autoantibodies engage TLR7 (sensor of single stranded RNA) and TLR9 (sensor of CpG DNA), causing NFkB translocation and pro-inflammatory transcription initiation in dendritic cells [24] and proliferation of autoantibody producing B cells [25, 26]. The amplified production of autoantibodies against DNA or DNA associated proteins leads to the deposition of DNA containing immune complexes in the kidney that then further stimulate both innate and adaptive immune cells. The consequent DAMP-stimulated inflammation is implicated in the acceleration of renal scarring and fibrosis leading to renal failure [7].
cfDNA
A representative DAMP that can serve as an autoantigen is cell-free DNA (cfDNA), or circulating extracellular DNA. DNA is “the central autoantigen in SLE” [27] with antibodies to double stranded DNA considered diagnostic for SLE [28]. While hypomethylated CpG DNA from bacteria is the appropriate ligand, endogenous DNA can stimulate the TLR9 axis in error [29, 30]. The cfDNA in the blood nucleome that triggers and modulates immune response in SLE patients is associated with three categories of cell death: apoptosis, necrosis, and NETosis. It is the impaired clearance of apoptotic cells, secondary necrosis, and increased NETs that leads to the increased DAMPs that then amplifies autoimmunity and inflammation [31]. cfDNA arising from apoptosis is digested by nucleases, producing low molecular weight DNA strands while necrosis and NETosis, which are comparatively much faster mechanisms, produce higher weight, more intact cfDNA [27]. While the most studied cfDNA in the blood nucleome is linear, other topologies such as circular DNAs are present and the extrachromosomal circular DNA found in circulation tends to be longer than linear DNA [32]. Mitochondria contribute to the circular DNA as well as nuclease-digested linear DNA species. Notably, mitochondrial DNA is particularly inflammatory because it has a high content of unmethylated CpG motifs, similar to bacterial DNA [29] and CpG motif containing DNA is enriched in immune complexes from SLE patients [33]. Characterization of circulating cfDNA in SLE patients found that those with higher cfDNA concentration, more fragmentation, and certain characteristic fragment lengths, had worse glomerular filtration rates and more severe lupus nephritis in comparison to SLE patients with cfDNA profiles comparable to healthy individuals [34].
Other DAMPs
Beyond DNA DAMPs, other molecules such as RNA and intracellular proteins are associated with cell damage and contribute to SLE inflammation. Upon release from nuclear compartments RNAs and histones interact with endosomal and cell surface TLRs respectively [35]. HMGB1, a nuclear protein affecting chromatin structure and transcription, is released through NETosis and necroptosis and interacts with TLRs 2, 4, and 9 to activate the NF-κB pathway and RAGE to activate pro-inflammatory genes [31, 35]. Originating in the cytosol, S100 proteins released during phagocytosis and heat shock proteins s from necrosing cells are released as DAMPs and interact with cell surface TLRs 2 and 4 [35]. In addition, while cfDNA and other nucleic acid DAMPs can exist freely and are often modeled as such in various in vitro assays, nucleic acids in vivo closely associate with other molecules. Nucleic acids are intrinsically protein binding and can associate with extracellular vesicles such as microparticles [36] and apoptotic bodies. Thus, nucleic acid DAMPs exist to a large extent in complexes in vivo which further facilitates multiple interactions with TLRs as well as immune complex formation.
Nucleic acid degradation enzymes
Nucleic acid degradation enzymes have pivotal roles in ligand availability for TLR interaction as documented in a recent comprehensive review by Santa et al. [37]. Cell-free nucleic acids are degraded for clearance by various DNases and RNases. In general, deactivation or mutation of nucleases promotes autoimmune onset as DAMPs then evade degradation and interact with TLRs. Deficiencies in DNASEIL3, an extracellular DNAse capable of digesting cfDNA as well as DNA in NETs and microparticles, are particularly associated with SLE. Mutations of this nuclease in humans are associated with pediatric onset of SLE, and knockouts in murine models display strong SLE-like serologic features [37].
TLR signaling in SLE and lupus nephritis
Crucial to the response to viral pathogens is signaling through TLRs leading to production of interferons and inflammatory cytokines. Such signaling, particularly the induction of type I interferon alpha, is also implicated in chronic inflammatory diseases such as SLE and associated lupus nephritis [38, 39]. Given the central role of TLR signaling in SLE and lupus nephritis pathogenesis, TLR perturbations have been examined to elucidate specific pathways and identify possible interventional targets. Included in these efforts are TLR knockout studies in murine models and polymorphism analysis of TLR pathway genes in SLE patients.
Mouse studies to dissect the significance of the different TLRs in lupus nephritis
Mouse studies, particularly those utilizing MRL/MPlpr/lpr mice, have highlighted the involvement of several TLRs in lupus. In these mice exposure to TLR3 agonists exacerbates renal pathology and Tlr3 mRNA expression increases with worsening glomerulonephritis [40-42]. Knockout of TLR7, a sensor for single stranded RNA, partially ameliorates SLE-like pathology in MRL/MPlpr/lpr mice [43], and TLR7 deficiency in pristane-induced lupus mice is associated with a decrease in anti-snRNP antibody production, IgG immune complex glomerular deposition, and attenuated glomerulonephritis [44]. Signaling through TLR8, also activated by single stranded RNA, is sufficient in the absence of TLR7 for the loss of B-cell tolerance in an autoantibody knockin mouse strain [45]. The consequences of manipulating TLR9, the sensor of DNA featuring unmethylated CpG motifs, have also been assessed in the MRL/Mplpr/lpr mice. Exposure to the TLR9 agonists CpG and bacterial DNA leads to lupus nephritis progression [46] and ablation of TLR9 inhibits anti-dsDNA and antichromatin antibody production [47]. Loss of TLR9, however, exacerbates lupus nephritis [43], possibly because its absence leads to enhanced TLR7 signaling and a net promotion of disease [48].
TLR pathway polymorphisms linked to SLE
Many genes associated with susceptibility to lupus encode innate immune functions involved in the clearance of cellular debris and immune complexes and the activation of TLR/interferon/NFκB signaling pathways [49]. Aberrantly high DAMP levels can be caused by nuclease deficiency, as mentioned above, and by defects in the clearance of apoptotic cells and antibody complexes. Such defects lead to increased availability of autoantigens and are linked to the development of lupus [50]. For instance, a missense integrin subunit encoded by a mutant ITGAM gene leads to impaired phagocytosis of complement-opsonized targets by macrophages, monocytes and neutrophils resulting in reduced clearance [51] and this particular ITGAM gene variant is associated with susceptibility to lupus in women of European descent [52]. Lupus risk is also associated with mutations causing elevated expression or, more rarely, altered function of various TLRs. A 3’ untranslated region mutation in the TLR7 gene leading to increased TLR7 transcripts and TLR7 protein is associated with development of lupus in humans [53, 54]. Based on dosage studies in mice, the X-linked expression of TLR8 has been theorized to underly the female predominance of SLE [45]. Higher TLR9 mRNA expression in leukocytes is associated with lupus nephritis [55]. Common polymorphisms of the TLR9 encoding gene have been found to be associated with increased risk for asthma and Crohn’s disease in humans. While studies assessing the association of TLR9 gene polymorphisms with an increased risk of SLE are conflicting [56, 57], intense TLR9 staining in the kidney is associated with lupus nephritis [57]. A polymorphism causing a premature stop codon in the TLR5 gene that truncates TLR5 is associated with reduced lupus nephritis [56-58]. Given its primary role in interacting with bacterial flagella, the mechanism by which TLR5 mutations affect SLE pathogenesis requires additional characterization. Factors further downstream of the TLRs in the type I interferon and pro-inflammatory cytokine pathways also feature variants associated with lupus. The gene variants include those encoding the IRF transcription factors and factors regulating NFκB activity [49].
Current therapeutics and unmet clinical need
Current clinical treatments for SLE utilize immunomodulators and immunosuppressants to regulate immune activity [59] as well as lifestyle modifications to reduce inflammation. Lifestyle modifications can be made to avoid triggers, minimize damage and inflammation, and manage symptoms. For example, minimizing exposure to sunlight prevents cellular injury and subsequent DAMP release following damage by ionizing UV light. Fibromyalgia and fibromyalgia-like symptoms are common in SLE patients, and the associated fatigue, pain, and cognitive dysfunction can be alleviated through exercise and cognitive rehabilitation [59, 60]. Beyond lifestyle modifications, the antimalarial drug hydroxychloroquine is the current pharmacologic standard of care for SLE [61]. Given the widespread role of TLR signaling in SLE pathogenesis, hydroxychloroquine functions in part by inhibiting acid-dependent receptor processing in endosomes [62] and/or shielding the nucleic acid ligands [63] and thereby muting the activation of TLRs 7 and 9 [64]. Hydroxychloroquine also blocks MHCII-mediated autoantigen presentation and binds internalized nucleic acids, preventing their binding to nucleic acid sensor cyclic GMP-AMP synthase, which if bound mediates type 1 IFN transcription [64]. These mechanisms work in tandem to prevent excessive production of pro-inflammatory cytokines, ameliorating SLE symptoms and slowing lupus nephritis onset [61]. Hydroxychloroquine is generally safe and effective in lupus patients, although side effects include GI distress [65] and dose and duration dependent risk of retinopathy [66, 67]. Furthermore, corticosteroids are still required acutely for management of flares and chronically given at lower doses for many patients. Though potent as an anti-inflammatory and immune suppressant, corticosteroid treatment is accompanied by a wide range of side effects, including metabolic dysregulation [68], bone loss [69], increased risk of infection and other conditions [70]. Thus, additional therapies that can effectively and specifically target innate immune activation with fewer side effects would be welcomed for management of this challenging autoimmune disease process.
Targeting of DAMPs and TLRs
As described above, DAMPs and PRR signaling is centrally involved in inflammation and has pathologic roles associated with many infectious and inflammatory diseases. Many approaches have been made to pharmacologically intervene in this axis. Given that many of the lupus susceptibility genes are associated with increased activity of TLR/interferon/NFκB pathways [49], targeting TLR signaling in its role in initiating and perpetuating chronic inflammation is a sound focus of therapeutic development. TLR axis inhibition using therapeutics generally focuses on one of two mechanisms: preventing the interaction of DAMPs with TLR receptors, and blocking intracellular signal transduction following pathway activation [71]. Oligonucleotides are one class of molecules that have been studied as inhibitors of DAMP-TLR interactions. Treatment of lupus-prone NZB/W mice with synthetic, guanine-rich nucleic acid sequences that inhibit activation of TLRs 3, 7, 8, and 9 delayed progression of glomerulonephritis and proteinuria in conjunction with decreased production of Th1 cytokines and anti-dsDNA antibodies [72]. In addition, as reviewed by Gao et al. [71], small molecule inhibitors (SMIs) targeting TLRs 7, 8, and 9 and the MyD88 protein that activates IFN transcription factors after TLR-ligand binding have been tested for their abilities to interfere in TLR signaling. As discussed above, hydroxychloroquine is an SMI that is in wide clinical use for SLE treatment. Antibodies targeting specific DAMPs like anti-HMGB1 have been trialed for neutralization of DAMPs in the extracellular space [73, 74]. These approaches have largely remained in the preclinical phase or have not been approved for clinical use [71].
Nucleic acid scavengers
Despite the sound biological premise of intervening on the TLR axis to interrupt autoimmune inflammation, most preclinical work inhibiting TLR signaling has not yielded translatable results. The redundancy of TLRs and other PRRs likely limits efficacy when trying to inhibit a single TLR receptor to mitigate the hyperinflammatory response to multifactorial cellular damage and debris [75]. A therapeutic agent that neutralizes a wide range of DAMPs may be more effective as it could theoretically prevent interactions of this complex mixture of proinflammatory ligands with a wide range of TLRs and PRRs, thereby interrupting the feedforward cycle of hyperinflammation that underpins lupus nephritis and other autoimmune conditions.
Molecular scavengers that bind nucleic acid-containing DAMPs have emerged as a potential therapeutic solution to the challenge of targeting multiple TLRs for inhibition. A subset of nucleic acid binding polymers, these nanoparticles are able to bind and neutralize a wide range of DAMPs from activating their respective PRRs. The binding properties of these polymers is enabled by cationic surface groups, most commonly amines, that are positively charged via protonation at physiologic pH. The negatively charged phosphate backbone of DNAs and RNAs bind to these cationic polymers, an ionic interaction that has a wide range of applications. The best studied polymer in this class of nanoparticles is polyamidoamine, or PAMAM. Originally synthesized by Tomalia et al. in 1985 [76] this “starburst dendrimer” was recognized for its biocompatability and ability to bind and deliver nucleic acids and other agents to cells as described in a recent review [77]. Herein, unless otherwise specified, PAMAM will refer to PAMAM species with all terminal amines as end groups. Nucleic acid binding polymers like PAMAM are used in many commercially available transfection reagents because of their ability to load nucleic acid cargo and cross cellular membranes [78]. Larger generations of PAMAM have container properties, and have been studied extensively as agents for targeted drug delivery [78].
Pharmacologic utility of nucleic acid scavengers
This property of high affinity binding to nucleic acids attracted our group to test the pharmacologic utility of PAMAM as a universal-reversal agent to RNA aptamers. While aptamers can often be readily inactivated by their antisense “antidote,” which binds and unfolds the functional three-dimensional confirmation of the original aptamer [79-81], Oney et al. demonstrated that PAMAM G3 and certain other cationic polymers could also reverse function for multiple high affinity protein-binding aptamers [82]. Exploiting these nucleic acid binding polymers would preclude the cost-prohibitive clinical development of multiple antisense oligos for each of the many aptamers that eventually may be used clinically. These studies were intriguing because the administration of such nucleic acid binding polymers rapidly neutralized aptamers in vivo despite the fact that the aptamers bound their target proteins with low nanomolar affinities. Thus such binding agents were able to reversibly inhibit high affinity protein-nucleic acid interactions in the blood even when the nucleic acid and protein were preassembled into a complex in circulation in large animals [82].
Pre-clinical anti-inflammatory functionality in vitro and in vivo
Our group then wondered if the ability to disrupt extracellular nucleic acid-protein interactions could be utilized as an intervention in inflammatory disorders and thromboinflammation. As discussed earlier, DAMPs and PAMPs, exemplified by host and pathogen derived DNAs and RNAs, induce inflammatory responses through binding to their target proteins, pattern-recognition receptors (PRRs) including TLRs, just as aptamers bind to their target proteins with high affinity and specificity [83, 84]. Often in injury and disease, excessive inflammation is induced, leading the immune system to cause collateral damage in its attempt to heal or sequester infection. First, our group observed that certain nucleic acid scavengers, like PAMAM G3, can bind and sequester mediators of cellular damage, preventing the immune stimulating interactions of these DAMPs and PAMPs with TLRs, and reducing mortality in a DAMP-mediated murine model of toxic shock-induced liver injury [85]. In vitro imaging revealed that PAMAM G3 both reduces uptake of the pro-inflammatory synthetic oligonucleotide CpG 1668 and alters its intracellular distribution away from co-localization with TLR9 [85]. PAMAM G3 was also found to have antithrombotic properties, as the pro-thrombotic DAMPs released by vascular wall injury, and neutrophil and platelet activation can be bound and neutralized by PAMAM’s cationic surface groups [86, 87]. PAMAM G3 treatment also reduced wound scarring [88], and cancer metastasis in murine models of pancreatic cancer and breast cancer [89-91]. As well, PAMAM or other scavenger variants have been shown to limit inflammation in models of rheumatoid arthritis [92, 93] and psoriasis [94]. Thus PAMAM-mediated nucleic acid-DAMP scavenging has proven effective at inhibiting a variety of pathological thromboinflammatory insults.
Nucleic acid scavengers for potential treatment of SLE, lupus nephritis and cutaneous lupus
The observation that nucleic acid DAMP scavengers could limit activation of TLRs and other PRRs by prototypical nucleic acid-containing DAMPs led us to evaluate the potential utility of this approach to limit innate immune activation and associated pathologies in chronic autoimmune and inflammatory disorders, such as SLE. As discussed above, autoimmune diseases like SLE are disorders of aberrant immune response to one’s own tissues that are also mediated by excessive DAMP-driven inflammation. As well, a DAMP targeting strategy would likely be helpful to a lupus patient with a defect in the clearance of cellular debris or with abnormally elevated TLR activity driven by genetic polymorphisms. In vitro, PAMAM G3 and other cationic polymers are able to inhibit the binding of anti-DNA antibodies to DNA [95] and inhibit dendritic cell and B cell responsiveness to nucleic acid TLR agonists without blocking T-cell or anti-viral responses [96]. These in vitro results suggested that the nucleic acid DAMP-scavenger approach should be evaluated in animal models of lupus. To determine if PAMAM G3 limits pathological inflammation in lupus-prone mice, we determined the maximal lethal dose of the polymer in NZBW F1 mice, 100 to 200 mg/kg, and then performed all studies at 5-10 fold below this level at 20 mg/kg [97]. First the DAMP scavenger approach was evaluated in a cutaneous lupus erythematosus (CLE) model using NZBW F1 mice (Figure 1). Following dermal injury using tape stripping, it was observed that PAMAM G3 (20 mg/kg twice per week) when delivered by subcutaneous administration around the site of injury over 14-21 days reduces skin inflammation [97]. Blinded pathological analyses of PAMAM G3 treated animals reveals significantly reduced disease grades compared to PBS-treated animals. Although these results were encouraging, we noticed in subsequent experiments that delivering PAMAM G3 at this dose occasionally induces skin irritation at sites of injection in mice with the most advanced lupus. Therefore, we sought to evaluate alternative nucleic acid scavenging agents in the CLE model. A linear β-cyclodextrin-containing polymer, termed CDP, had been reported by Mark Davis’s laboratory to have a good safety profile including in clinical studies [98, 99]. As we had previously observed that the CDP could limit TLR activation by nucleic acid DAMPs [85], it was evaluated in the same CLE murine tape stripping injury model (Figure 1). Results from these studies indicated that CDP significantly improves skin healing following dermal damage of lupus prone mice and moreover CDP is associated with limited toxicity compared to PAMAM G3 [100].
Figure 1. Overview of Nucleic Acid DAMP Scavengers Reducing Inflammation and Improving Skin Healing in a Mouse Model of Cutaneous Lupus Erythematosus.
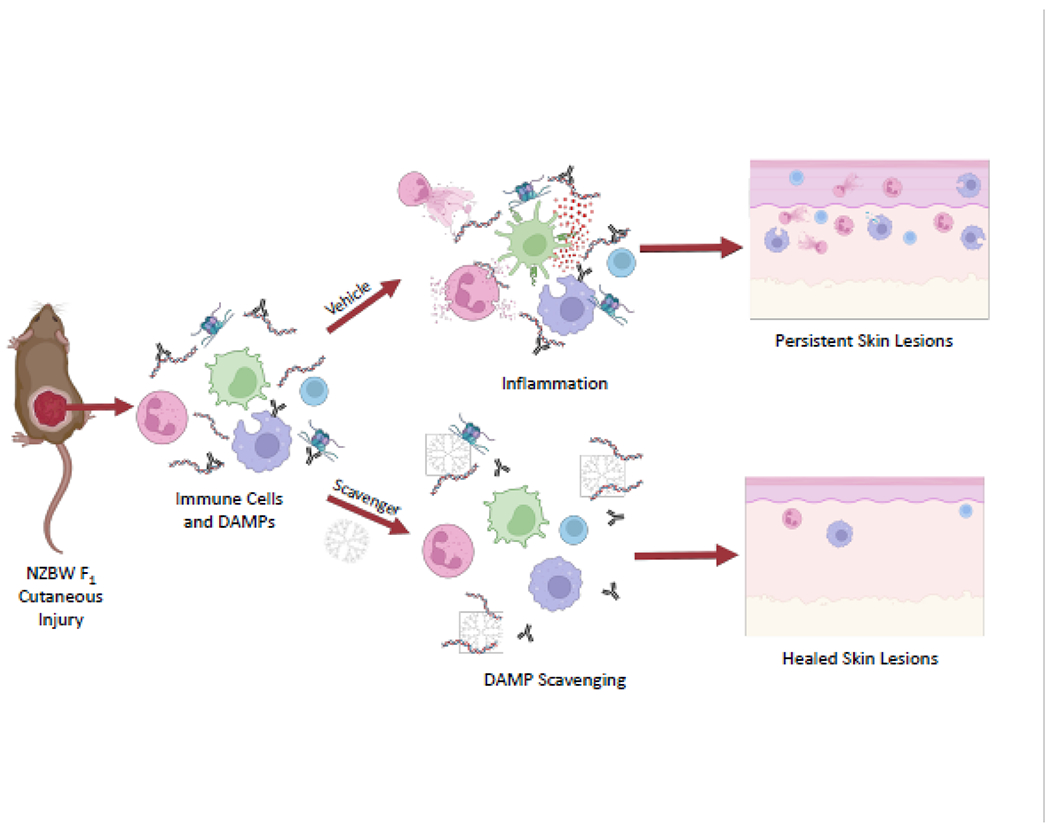
When lupus-prone NZBW F1 mice undergo tape stripping, they develop hyper-inflamed cutaneous lesions. Nucleic acid DAMPs are released and anti-nuclear antibodies and immune cells such as neutrophils, dendritic cells, macrophages and T cells, flood the wound zone. Mice treated with subcutaneous injections of vehicle (saline) exhibit continued inflammation that leads to persistent leukocyte infiltration of the dermis and epidermis, epidermal hyperplasia, and slowed wound healing (top right). In contrast, the skin of scavenger-treated mice shows reduced inflammation and improved wound healing (bottom right). Figure created with BioRender.com.
Abbreviations in figure:
The New Zealand Black White hybrid mouse strain abbreviated as NZBW F1.
Damage associated molecular patterns abbreviated as DAMPs.
In vivo, long term systemic PAMAM G3 treatment (20 mg/kg i.p., twice per week) slowed the progression of glomerulonephritis in the kidneys of lupus-prone male MRLlpr mice [97]. These studies also demonstrated that a 10-week treatment regimen with PAMAM G3 reduces immune complex and complement C3c deposits in the kidneys in the nucleic acid scavenger treated animals at 5 months of age. When this treatment period was extended however, we started to observe some toxicity revealed by weight loss associated with PAMAM G3 administration at this dose. Thus, for prolonged exposure, lower doses of PAMAM G3 or alternative nucleic acid scavengers such as CDP should be evaluated.
To determine if PAMAM G3 or CDP treatment causes immune suppression, mice being systemically treated with either of the DAMP scavengers were challenged with PR8 influenza infection. Such treatment did not make the mice more susceptible to influenza but rather reduced systemic symptoms of infection, ameliorating weight loss and hypothermia, and improving survival [97, 100]. This observation was initially somewhat surprising. However, given that much of the morbidity and mortality associated with influenza infections is thought to result from a cytokine storm elicited from DAMPs and PAMPs released from virus infected cells, the nucleic acid scavengers utilized to reduce DAMP induced inflammation in the setting of lupus could well have potential as antiviral agents.
Barriers to translation
Despite the promising preclinical studies using PAMAM G3, concerns about its potential toxicity have precluded immediate translation of it as a systemic anti-inflammatory agent. As a rule, the higher the generation of PAMAM, the greater the risk. Studies of cationic PAMAM interaction with lipid bilayers show that cationic PAMAM, especially of larger generations, can form pores in cellular membranes [101, 102]. Cationic PAMAM G4 and larger generations can cause immediate toxicity when administered at high doses intravenously, notably causing thrombosis and hemorrhage. While we have used PAMAM G3 as an antithrombotic in an injury-induced thrombosis model, intravenous delivery of this polymer can also induce acute toxicity when delivered at high doses (>20 mg/kg) to mice [86]. When dosed i.p., PAMAM G3 at 20 mg/kg induces, on average, a transient 5% dip in weight over the next 24 hours [100]. Within the literature, side effects of treatments (when reported) appear to be strain, age, and insult dependent. In our hands, some strains appear more sensitive to PAMAM treatment and have more weight loss following treatment, while other strains seem to adapt faster to any harmful impact of treatment. Toxicity is also more common in older mice or mice with severe illness. These findings demonstrate the narrow therapeutic window of cationic PAMAM G3. This rather limits the translational potential of this polymer, especially for use in chronic inflammatory illnesses like autoimmune disease such as SLE which require dosing over long spans of time. For this reason, we believe that evaluation of alternative nucleic acid DAMP scavengers may reveal more viable candidates for clinical translation. As mentioned above, Kelly et al. found that the linear β-cyclodextrin-containing polymer, termed CDP, appears to have an improved therapeutic window compared to PAMAM-G3 [100].
Summary
Scavenging of nucleic acid-containing DAMPs has been shown to induce a significant therapeutic effect in multiple animal models of thromboinflammation including lupus nephritis and CLE models of SLE. Much of this work was performed with first generation scavengers such as PAMAM G3. More recent studies using optimized nucleic acid scavengers suggests DAMP scavengers with wider therapeutic windows are now being identified. Their discovery will almost assuredly lead to the translation of this novel and innovative approach to limiting the induction and pathological progression of inflammation in several disease settings including the treatment of patients with lupus.
Acknowledgements
This work was supported by National Institutes of Health grant RO1AR073935 (to B.A.S.) We thank David Pisetsky and Diane Spencer for useful discussions about Lupus and DAMPs.
Abbreviations:
- SLE
systemic lupus erythematosus
- PRRs
Pattern Recognition Receptors
- PAMPs
pathogen-associated molecular patterns
- DAMPs
damage-associated molecular patterns
- TLRs
Toll-like receptors
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
All authors have read Translational Research’s authorship agreement and policy on disclosure of potential conflicts of interest.
Duke University has applied for patents on the strategy to reduce inflammation via nucleic acid scavengers. All authors are listed as inventors on such patents.
References
- [1].Maidhof W, Hilas O. Lupus: an overview of the disease and management options. P T. 2012;37:240–9. [PMC free article] [PubMed] [Google Scholar]
- [2].Cojocaru M, Cojocaru IM, Silosi I, Vrabie CD. Manifestations of systemic lupus erythematosus. Maedica (Bucur). 2011;6:330–6. [PMC free article] [PubMed] [Google Scholar]
- [3].Dema B, Charles N. Autoantibodies in SLE: Specificities, Isotypes and Receptors. Antibodies (Basel). 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Herrada AA, Escobedo N, Iruretagoyena M, Valenzuela RA, Burgos PI, Cuitino L, et al. Innate Immune Cells’ Contribution to Systemic Lupus Erythematosus. Front Immunol. 2019;10:772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Parikh SV, Almaani S, Brodsky S, Rovin BH. Update on Lupus Nephritis: Core Curriculum 2020. Am J Kidney Dis. 2020;76:265–81. [DOI] [PubMed] [Google Scholar]
- [6].Frangou E, Georgakis S, Bertsias G. Update on the cellular and molecular aspects of lupus nephritis. Clin Immunol. 2020;216:108445. [DOI] [PubMed] [Google Scholar]
- [7].Lorenz G, Anders HJ. Neutrophils, Dendritic Cells, Toll-Like Receptors, and Interferon-alpha in Lupus Nephritis. Semin Nephrol. 2015;35:410–26. [DOI] [PubMed] [Google Scholar]
- [8].Yu Y, Su K. Neutrophil Extracellular Traps and Systemic Lupus Erythematosus. J Clin Cell Immunol. 2013;4:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA–peptide complexes in systemic lupus erythematosus. Science translational medicine. 2011;3:73ra19–73ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-α in systemic lupus erythematosus. Science. 2001;294:1540–3. [DOI] [PubMed] [Google Scholar]
- [11].Rigante D, Esposito S. Infections and Systemic Lupus Erythematosus: Binding or Sparring Partners? Int J Mol Sci. 2015;16:17331–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Johnson RJ, Couser WG, Alpers CE, Vissers M, Schulze M, Klebanoff SJ. The human neutrophil serine proteinases, elastase and cathepsin G, can mediate glomerular injury in vivo. J Exp Med. 1988;168:1169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Suzuki Y, Gomez-Guerrero C, Shirato I, Lopez-Franco O, Gallego-Delgado J, Sanjuan G, et al. Pre-existing glomerular immune complexes induce polymorphonuclear cell recruitment through an Fc receptor-dependent respiratory burst: potential role in the perpetuation of immune nephritis. J Immunol. 2003;170:3243–53. [DOI] [PubMed] [Google Scholar]
- [14].Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020;20:95–112. [DOI] [PubMed] [Google Scholar]
- [15].Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. 2004;279:12542–50. [DOI] [PubMed] [Google Scholar]
- [16].Molteni M, Gemma S, Rossetti C. The Role of Toll-Like Receptor 4 in Infectious and Noninfectious Inflammation. Mediators Inflamm. 2016;2016:6978936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li X, Jiang S, Tapping RI. Toll-like receptor signaling in cell proliferation and survival. Cytokine. 2010;49:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22:240–73, Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vanarsa K, Henderson J, Soomro S, Qin L, Zhang T, Jordan N, et al. Upregulation of Proinflammatory Bradykinin Peptides in Systemic Lupus Erythematosus and Rheumatoid Arthritis. J Immunol. 2020;205:369–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of Inflammation: What Controls Its Onset? Frontiers in immunology. 2016;7:160-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Neurath MF. Resolution of inflammation: from basic concepts to clinical application. Seminars in Immunopathology. 2019;41:627–31. [DOI] [PubMed] [Google Scholar]
- [22].Kruse K, Janko C, Urbonaviciute V, Mierke CT, Winkler TH, Voll RE, et al. Inefficient clearance of dying cells in patients with SLE: anti-dsDNA autoantibodies, MFG-E8, HMGB-1 and other players. Apoptosis. 2010;15:1098–113. [DOI] [PubMed] [Google Scholar]
- [23].Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. 2016;12:716–30. [DOI] [PubMed] [Google Scholar]
- [24].Kim WU, Sreih A, Bucala R. Toll-like receptors in systemic lupus erythematosus; prospects for therapeutic intervention. Autoimmun Rev. 2009;8:204–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wu Y-w, Tang W, Zuo J-p. Toll-like receptors: potential targets for lupus treatment. Acta Pharmacol Sin. 2015;36:1395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Tolllike receptors. Nature. 2002;416:603–7. [DOI] [PubMed] [Google Scholar]
- [27].Pisetsky DS. The origin and properties of extracellular DNA: from PAMP to DAMP. Clin Immunol. 2012;144:32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].de Leeuw K, Bungener L, Roozendaal C, Bootsma H, Stegeman CA. Auto-antibodies to double-stranded DNA as biomarker in systemic lupus erythematosus: comparison of different assays during quiescent and active disease. Rheumatology (Oxford). 2017;56:698–703. [DOI] [PubMed] [Google Scholar]
- [29].Duvvuri B, Lood C. Cell-Free DNA as a Biomarker in Autoimmune Rheumatic Diseases. Front Immunol. 2019;10:502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lamphier MS, Sirois CM, Verma A, Golenbock DT, Latz E. TLR9 and the recognition of self and non-self nucleic acids. Ann N Y Acad Sci. 2006;1082:31–43. [DOI] [PubMed] [Google Scholar]
- [31].Yang F, He Y, Zhai Z, Sun E. Programmed Cell Death Pathways in the Pathogenesis of Systemic Lupus Erythematosus. J Immunol Res. 2019;2019:3638562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lo YMD, Han DSC, Jiang P, Chiu RWK. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science. 2021;372. [DOI] [PubMed] [Google Scholar]
- [33].Sano H, Takai O, Harata N, Yoshinaga K, Kodama-Kamada I, Sasaki T. Binding properties of human anti-DNA antibodies to cloned human DNA fragments. Scand J Immunol. 1989;30:51–63. [DOI] [PubMed] [Google Scholar]
- [34].Truszewska A, Wirkowska A, Gala K, Truszewski P, Krzemien-Ojak L, Perkowska-Ptasinska A, et al. Cell-free DNA profiling in patients with lupus nephritis. Lupus. 2020;29:1759–72. [DOI] [PubMed] [Google Scholar]
- [35].Roh JS, Sohn DH. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018;18:e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mobarrez F, Svenungsson E, Pisetsky DS. Microparticles as autoantigens in systemic lupus erythematosus. Eur J Clin Invest. 2018;48:e13010. [DOI] [PubMed] [Google Scholar]
- [37].Santa P, Garreau A, Serpas L, Ferriere A, Blanco P, Soni C, et al. The Role of Nucleases and Nucleic Acid Editing Enzymes in the Regulation of Self-Nucleic Acid Sensing. Front Immunol. 2021;12:629922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Migliorini A, Anders HJ. A novel pathogenetic concept-antiviral immunity in lupus nephritis. Nat Rev Nephrol. 2012;8:183–9. [DOI] [PubMed] [Google Scholar]
- [39].Ronnblom L, Alm GV, Eloranta ML. Type I interferon and lupus. Curr Opin Rheumatol. 2009;21:471–7. [DOI] [PubMed] [Google Scholar]
- [40].Devarapu SK, Anders H-J. Toll-like receptors in lupus nephritis. Journal of Biomedical Science. 2018;25:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Patole PS, Pawar RD, Lech M, Zecher D, Schmidt H, Segerer S, et al. Expression and regulation of Toll-like receptors in lupus-like immune complex glomerulonephritis of MRL-Fas(lpr) mice. Nephrol Dial Transplant. 2006;21:3062–73. [DOI] [PubMed] [Google Scholar]
- [42].Patole PS, Gröne HJ, Segerer S, Ciubar R, Belemezova E, Henger A, et al. Viral double-stranded RNA aggravates lupus nephritis through Toll-like receptor 3 on glomerular mesangial cells and antigen-presenting cells. J Am Soc Nephrol. 2005;16:1326–38. [DOI] [PubMed] [Google Scholar]
- [43].Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–28. [DOI] [PubMed] [Google Scholar]
- [44].Savarese E, Steinberg C, Pawar RD, Reindl W, Akira S, Anders HJ, et al. Requirement of Toll-like receptor 7 for pristane-induced production of autoantibodies and development of murine lupus nephritis. Arthritis Rheum. 2008;58:1107–15. [DOI] [PubMed] [Google Scholar]
- [45].Umiker BR, Andersson S, Fernandez L, Korgaokar P, Larbi A, Pilichowska M, et al. Dosage of X-linked Toll-like receptor 8 determines gender differences in the development of systemic lupus erythematosus. European Journal of Immunology. 2014;44:1503–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Anders HJ, Vielhauer V, Eis V, Linde Y, Kretzler M, Perez de Lema G, et al. Activation of toll-like receptor-9 induces progression of renal disease in MRL-Fas(lpr) mice. FASEB J. 2004;18:534–6. [DOI] [PubMed] [Google Scholar]
- [47].Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Fukui R, Saitoh S, Matsumoto F, Kozuka-Hata H, Oyama M, Tabeta K, et al. Unc93B1 biases Toll-like receptor responses to nucleic acid in dendritic cells toward DNA- but against RNA-sensing. J Exp Med. 2009;206:1339–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dai C, Deng Y, Quinlan A, Gaskin F, Tsao BP, Fu SM. Genetics of systemic lupus erythematosus: immune responses and end organ resistance to damage. Curr Opin Immunol. 2014;31:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Munoz LE, Gaipl US, Franz S, Sheriff A, Voll RE, Kalden JR, et al. SLE--a disease of clearance deficiency? Rheumatology (Oxford). 2005;44:1101–7. [DOI] [PubMed] [Google Scholar]
- [51].Fossati-Jimack L, Ling GS, Cortini A, Szajna M, Malik TH, McDonald JU, et al. Phagocytosis is the main CR3-mediated function affected by the lupus-associated variant of CD11b in human myeloid cells. Plos One. 2013;8:e57082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].International Consortium for Systemic Lupus Erythematosus G, Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Shen N, Fu Q, Deng Y, Qian X, Zhao J, Kaufman KM, et al. Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2010;107:15838–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Deng Y, Zhao J, Sakurai D, Kaufman KM, Edberg JC, Kimberly RP, et al. MicroRNA-3148 modulates allelic expression of toll-like receptor 7 variant associated with systemic lupus erythematosus. PLoS Genet. 2013;9:e1003336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Chauhan SK, Singh VV, Rai R, Rai M, Rai G. Distinct autoantibody profiles in systemic lupus erythematosus patients are selectively associated with TLR7 and TLR9 upregulation. J Clin Immunol. 2013;33:954–64. [DOI] [PubMed] [Google Scholar]
- [56].Rahman AH, Eisenberg RA. The role of toll-like receptors in systemic lupus erythematosus. Springer Semin Immunopathol. 2006;28:131–43. [DOI] [PubMed] [Google Scholar]
- [57].Elloumi N, Fakhfakh R, Abida O, Ayadi L, Marzouk S, Hachicha H, et al. Relevant genetic polymorphisms and kidney expression of Toll-like receptor (TLR)-5 and TLR-9 in lupus nephritis. Clin Exp Immunol. 2017;190:328–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hawn TR, Wu H, Grossman JM, Hahn BH, Tsao BP, Aderem A. A stop codon polymorphism of Toll-like receptor 5 is associated with resistance to systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2005;102:10593–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Fava A, Petri M. Systemic lupus erythematosus: Diagnosis and clinical management. J Autoimmun. 2019;96:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Seet D, Allameen NA, Tay SH, Cho J, Mak A. Cognitive Dysfunction in Systemic Lupus Erythematosus: Immunopathology, Clinical Manifestations, Neuroimaging and Management. Rheumatol Ther. 2021;8:651–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wilhelmus S, Bajema IM, Bertsias GK, Boumpas DT, Gordon C, Lightstone L, et al. Lupus nephritis management guidelines compared. Nephrol Dial Transplant. 2016;31:904–13. [DOI] [PubMed] [Google Scholar]
- [62].Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, et al. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008;456:658–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kuznik A, Bencina M, Svajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol. 2011;186:4794–804. [DOI] [PubMed] [Google Scholar]
- [64].Schrezenmeier E, Dörner T. Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nature Reviews Rheumatology. 2020;16:155–66. [DOI] [PubMed] [Google Scholar]
- [65].Wallace DJ, Tse K, Hanrahan L, Davies R, Petri MA. Hydroxychloroquine usage in US patients, their experiences of tolerability and adherence, and implications for treatment: survey results from 3127 patients with SLE conducted by the Lupus Foundation of America. Lupus Science & Medicine. 2019;6:e000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Marmor MF, Carr RE, Easterbrook M, Farjo AA, Mieler WF, American Academy of O. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy: a report by the American Academy of Ophthalmology. Ophthalmology. 2002;109:1377–82. [DOI] [PubMed] [Google Scholar]
- [67].Marmor MF, Kellner U, Lai TY, Melles RB, Mieler WF, American Academy of O. Recommendations on Screening for Chloroquine and Hydroxychloroquine Retinopathy (2016 Revision). Ophthalmology. 2016;123:1386–94. [DOI] [PubMed] [Google Scholar]
- [68].Hwang JL, Weiss RE. Steroid-induced diabetes: a clinical and molecular approach to understanding and treatment. Diabetes Metab Res Rev. 2014;30:96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kipen Y, Buchbinder R, Forbes A, Strauss B, Littlejohn G, Morand E. Prevalence of reduced bone mineral density in systemic lupus erythematosus and the role of steroids. J Rheumatol. 1997;24:1922–9. [PubMed] [Google Scholar]
- [70].Grennan D, Wang S. Steroid Side Effects. Jama. 2019;322:282. [DOI] [PubMed] [Google Scholar]
- [71].Gao W, Xiong Y, Li Q, Yang H. Inhibition of Toll-Like Receptor Signaling as a Promising Therapy for Inflammatory Diseases: A Journey from Molecular to Nano Therapeutics. Frontiers in Physiology. 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Graham KL, Lee LY, Higgins JP, Steinman L, Utz PJ, Ho PP. Treatment with a toll-like receptor inhibitory GpG oligonucleotide delays and attenuates lupus nephritis in NZB/W mice. Autoimmunity. 2010;43:140–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Stevens NE, Chapman MJ, Fraser CK, Kuchel TR, Hayball JD, Diener KR. Therapeutic targeting of HMGB1 during experimental sepsis modulates the inflammatory cytokine profile to one associated with improved clinical outcomes. Scientific Reports. 2017;7:5850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Nishibori M, Mori S, Takahashi HK. Anti-HMGB1 monoclonal antibody therapy for a wide range of CNS and PNS diseases. Journal of Pharmacological Sciences. 2019;140:94–101. [DOI] [PubMed] [Google Scholar]
- [75].Anwar MA, Shah M, Kim J, Choi S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med Res Rev. 2019;39:1053–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Tomalia DA, Baker H, Dewald J, Hall M, Kallos G, Martin S, et al. A New Class of Polymers: Starburst-Dendritic Macromolecules. Polymer Journal. 1985;17:117–32. [Google Scholar]
- [77].Chauhan AS. Dendrimers for Drug Delivery. Molecules. 2018;23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Araujo RV, Santos SDS, Igne Ferreira E, Giarolla J. New Advances in General Biomedical Applications of PAMAM Dendrimers. Molecules. 2018;23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Rusconi CP, Roberts JD, Pitoc GA, Nimjee SM, White RR, Quick G Jr., et al. Antidote-mediated control of an anticoagulant aptamer in vivo. Nat Biotechnol. 2004;22:1423–8. [DOI] [PubMed] [Google Scholar]
- [80].Powell Gray B, Kelly L, Ahrens DP, Barry AP, Kratschmer C, Levy M, et al. Tunable cytotoxic aptamer-drug conjugates for the treatment of prostate cancer. Proc Natl Acad Sci U S A. 2018;115:4761–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Gunaratne R, Kumar S, Frederiksen JW, Stayrook S, Lohrmann JL, Perry K, et al. Combination of aptamer and drug for reversible anticoagulation in cardiopulmonary bypass. Nat Biotechnol. 2018;36:606–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Oney S, Lam RTS, Bompiani KM, Blake CM, Quick G, Heidel JD, et al. Development of universal antidotes to control aptamer activity. Nat Med. 2009;15:1224–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Nimjee SM, Dornbos D, 3rd, Pitoc GA, Wheeler DG, Layzer JM, Venetos N, et al. Preclinical Development of a vWF Aptamer to Limit Thrombosis and Engender Arterial Recanalization of Occluded Vessels. Mol Ther. 2019;27:1228–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Nimjee SM, White RR, Becker RC, Sullenger BA. Aptamers as Therapeutics. Annu Rev Pharmacol Toxicol. 2017;57:61–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Lee J, Sohn JW, Zhang Y, Leong KW, Pisetsky D, Sullenger BA. Nucleic acid-binding polymers as anti-inflammatory agents. Proceedings of the National Academy of Sciences. 2011;108:14055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Jain S, Pitoc GA, Holl EK, Zhang Y, Borst L, Leong KW, et al. Nucleic acid scavengers inhibit thrombosis without increasing bleeding. Proceedings of the National Academy of Sciences. 2012;109:12938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Lee J, Jackman JG, Kwun J, Manook M, Moreno A, Elster EA, et al. Nucleic acid scavenging microfiber mesh inhibits trauma-induced inflammation and thrombosis. Biomaterials. 2017;120:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Holl EK, Bond JE, Selim MA, Ehanire T, Sullenger B, Levinson H. The nucleic acid scavenger polyamidoamine third-generation dendrimer inhibits fibroblast activation and granulation tissue contraction. Plast Reconstr Surg. 2014;134:420e–33e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Naqvi I, Gunaratne R, McDade JE, Moreno A, Rempel RE, Rouse DC, et al. Polymer-Mediated Inhibition of Pro-invasive Nucleic Acid DAMPs and Microvesicles Limits Pancreatic Cancer Metastasis. Molecular Therapy. 2018;26:1020–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Holl EK, Frazier V, Landa K, Boczkowski D, Sullenger B, Nair SK. Controlling cancer-induced inflammation with a nucleic acid scavenger prevents lung metastasis in murine models of breast cancer. Mol Ther. 2021;29:1772–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Eteshola EOU, Landa K, Rempel RE, Naqvi IA, Hwang ES, Nair SK, et al. Breast cancer-derived DAMPs enhance cell invasion and metastasis, while nucleic acid scavengers mitigate these effects. Mol Ther Nucleic Acids. 2021;26:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Liang H, Peng B, Dong C, Liu L, Mao J, Wei S, et al. Cationic nanoparticle as an inhibitor of cell-free DNA-induced inflammation. Nat Commun. 2018;9:4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Peng B, Liang H, Li Y, Dong C, Shen J, Mao HQ, et al. Tuned Cationic Dendronized Polymer: Molecular Scavenger for Rheumatoid Arthritis Treatment. Angew Chem Int Ed Engl. 2019;58:4254–8. [DOI] [PubMed] [Google Scholar]
- [94].Liang H, Yan Y, Wu J, Ge X, Wei L, Liu L, et al. Topical nanoparticles interfering with the DNA-LL37 complex to alleviate psoriatic inflammation in mice and monkeys. Sci Adv. 2020;6:eabb5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Stearns NA, Lee J, Leong KW, Sullenger BA, Pisetsky DS. The Inhibition of Anti-DNA Binding to DNA by Nucleic Acid Binding Polymers. Plos One. 2012;7:e40862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Holl EK, Shumansky KL, Pitoc G, Ramsburg E, Sullenger BA. Nucleic acid scavenging polymers inhibit extracellular DNA-mediated innate immune activation without inhibiting antiviral responses. Plos One. 2013;8:e69413–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Holl EK, Shumansky KL, Borst LB, Burnette AD, Sample CJ, Ramsburg EA, et al. Scavenging nucleic acid debris to combat autoimmunity and infectious disease. Proceedings of the National Academy of Sciences. 2016;113:9728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Davis ME, Pun SH, Bellocq NC, Reineke TM, Popielarski SR, Mishra S, et al. Self-assembling nucleic acid delivery vehicles via linear, water-soluble, cyclodextrin-containing polymers. Curr Med Chem. 2004;11:179–97. [DOI] [PubMed] [Google Scholar]
- [99].Zuckerman JE, Gritli I, Tolcher A, Heidel JD, Lim D, Morgan R, et al. Correlating animal and human phase Ia/Ib clinical data with CALAA-01, a targeted, polymer-based nanoparticle containing siRNA. Proc Natl Acad Sci U S A. 2014;111:11449–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Kelly L, Olson LB, Rempel RE, Everitt JI, Levine D, Nair SK, et al. beta-Cyclodextrin-containing polymer treatment of cutaneous lupus and influenza improves outcomes. Mol Ther. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Lee H, Larson RG. Molecular dynamics simulations of PAMAM dendrimer-induced pore formation in DPPC bilayers with a coarse-grained model. The journal of physical chemistry B. 2006;110:18204–11. [DOI] [PubMed] [Google Scholar]
- [102].Majoros IJ, Williams CR, Becker AC, Baker JR. Surface interaction and behavior of poly (amidoamine) dendrimers: deformability and lipid bilayer disruption. Journal of computational and theoretical nanoscience. 2009;6:1430–6. [DOI] [PMC free article] [PubMed] [Google Scholar]