

Abstract
L-ergothioneine (ERGO) is a potent antioxidant with cytoprotective effects. To study ERGO biodistribution and detect oxidative stress in vivo, we report an efficient and reproducible preparation of [11C]-labeled ERGO PET radioligand based on protecting the histidine carboxylic group with a methyl ester. Overall, this new protection approach using methyl ester improved the chemical yield of a 4-step reaction from 14% to 24% compared to the previous report using t-butyl ester. The [11C]CH3 methylation of the precursor provided the desired product with 55 ± 10% radiochemical purity and a molar activity of 450 ± 200 TBq/mmol. The [11C]ERGO radioligand was able to detect threshold levels of oxidative stress in a preclinical animal model of Alzheimer’s disease.
Keywords: Ergothioneine, PET imaging, [11C]CH3 labeling, oxidative stress, GFAP, IBA1, Alzheimer, ROS
Graphical Abstract

L-Ergothioneine (ERGO) is a natural antioxidant capable of scavenging reactive oxygen species and transition metals. This work reports the robust synthesis of a novel [11C]ERGO PET radioligand to visualize ERGO in action. Here, using the preclinical animal models, we show that the probe could report the threshold levels of oxidative stress in Alzheimer’s disease.
Introduction
L-ergothioneine (ERGO) is a potent antioxidant synthesized by actinomycetes, cyanobacteria, methylbacteria, and some fungi, like mushrooms [1, 2]. It has been reported that ERGO can prevent cell and tissue damage, a key contributor to aging, by protecting against free radicals and oxidative stress [3–5]. The chemical structure of ERGO is similar to that of histidine but with the presence of a thione group on the imidazole ring [6]. It is reported that ERGO protects against cytotoxicity by scavenging singlet oxygen, hydroxyl radicals [7–9], hypochlorous acid (HOCl), peroxyl radicals [9, 10], and acting as an inhibitor of iron or copper ion-dependent generation of hydroxyl radicals from hydrogen peroxide [7]. This protection is derived from its conspicuous affinity for metal cations, such as Fe and Cu, permitting capture and neutralization of associated radicals [11]. Thus, ERGO is considered a potent antioxidant with potential therapeutic implications. In particular, this antioxidant might play essential roles in the central nervous system [12–14], given the substantial research data indicating that ERGO can be distributed to the brain via the OCTN1 transporter [11].
Our group recently reported the synthesis of an [11C]ERGO PET radioligand to facilitate in vivo imaging of the biodistribution and pharmacokinetics using preclinical mouse models [6]. The work also demonstrated the specificity of the probe for the detection of LPS-induced inflammation and oxidative stress. In the previous synthesis, the carboxylic group of histidine was protected as a t-butyl ester for the synthesis of the precursor. After labeling with a [11C]CH3 positron emitter, all of the acidic-labile protecting groups, including the t-butyl ester, were cleaved by treating with hydrochloric acid at 80°C. The advantage of using t-butyl ester is its sensitivity to an acidic condition, albeit with sufficient stability to survive very mild acidic conditions at low temperature. This feature serves best when robust deprotection conditions are required. The t-butyl ester’s strength, however, is also its shortcoming. Its sensitivity in an acidic condition made reaction workup harder than usual, and thus, low reaction yield is inevitable.
The issue mentioned above is further compounded by the reproducibility problem pertaining to the presence of the t-butyl ester. To overcome these drawbacks, we switched the t-butyl ester group to a more stable methyl ester counterpart, although this new chemistry needs one additional step during radioisotope labeling. The reproducibility of this design is the real impetus behind this work. This article describes an improved synthesis with an overall yield of 24% starting from the methyl ester to the precursor, compared to 14% as reported in the past [6].
Given the apparent implications of reactive oxygen species (ROS) in mitochondrial dysfunction related to the pathogenesis of AD [15], we used the resultant [11C]ERGO PET radioligand to test the incipient hypothesis that oxidative stress could potentially serve as a biomarker for AD [16–19]. Here, we present the data demonstrating that the probe can detect oxidative stress in the brains of a common mouse model for Alzheimer’s disease (AD) neuropathology 5XFAD mice. The in vivo PET imaging showed the probe was retained in the brain of 5XFAD mice with a higher concentration than wild-type (WT) counterparts. As expected, this is due to the trapping of the probe, given its high affinity for ROS products and oxidized metals associated with AD [20–24]. Overall, the [11C]ERGO PET radioligand enables us to observe, for the first time, the dynamics of this antioxidant in action noninvasively in a live subject.
Results
Development of a precursor.
The carboxylic group of protected histidine 1 was capped as a methyl ester by a routine procedure using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) with a trace 4-dimethylaminopyridine (DMAP) as a catalyst in methanolic dichloromethane (Fig.1). Since three active groups were orthogonally protected with a different group, the tert-butyloxycarbonyl (Boc) protecting group could be cleaved independently from the others using trifluoroacetic acid (TFA) to provide a high reaction yield. Next, reductive methylation of a primary amine with aldehyde and sodium cyanoborohydride to produce intermediate 4 with good yield. The incorporation of the thioketone into the imidazole ring and protection of the active functional groups in imidazole ring with Boc groups were performed as described in the past to provide precursor 6 [6].
Figure 1.
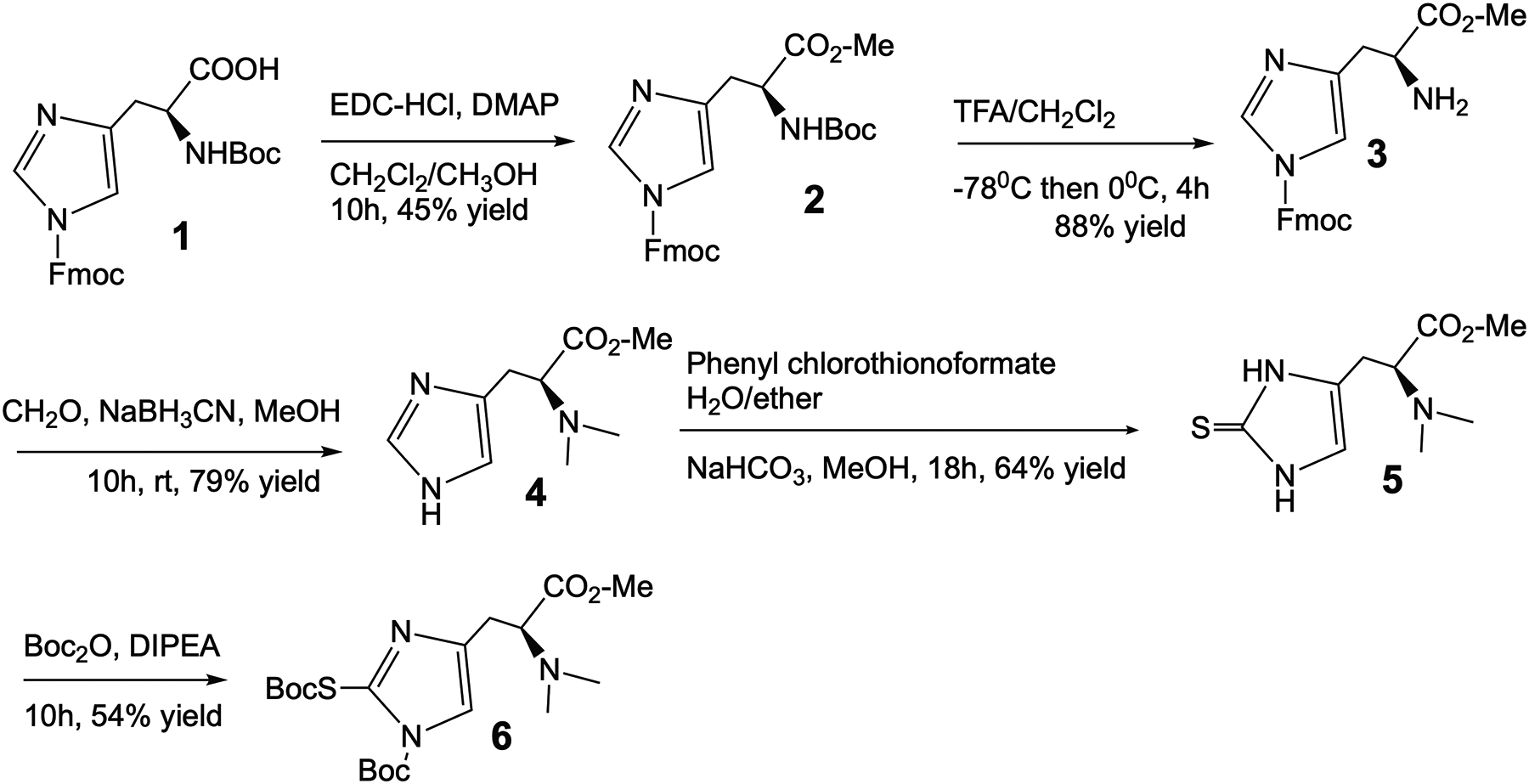
Design and synthesis of an ERGO precursor for [11C]CH3 labeling.
Radiosynthesis of [11C]ERGO PET radioligand.
Direct methylation of precursor 6 was achieved using [11C]MeOTf in acetonitrile (ACN) (Fig.2). The Boc protecting groups were removed by treatment with TFA, followed by ester hydrolysis in the presence of 5.0 M aqueous sodium hydroxide at 70°C for 4 min. The product was produced with 55 ± 10% radiochemical purity (decay corrected, n=5) confirmed by TLC and HPLC, and a molar activity of 450 ± 200 TBq/mmol. The identity of [11C]ERGO was confirmed by comparison with non-radioactive reference compounds. The retention time of [11C]ERGO was ~ 7.6 min, while the reference compound was 7.3 min.
Figure 2.
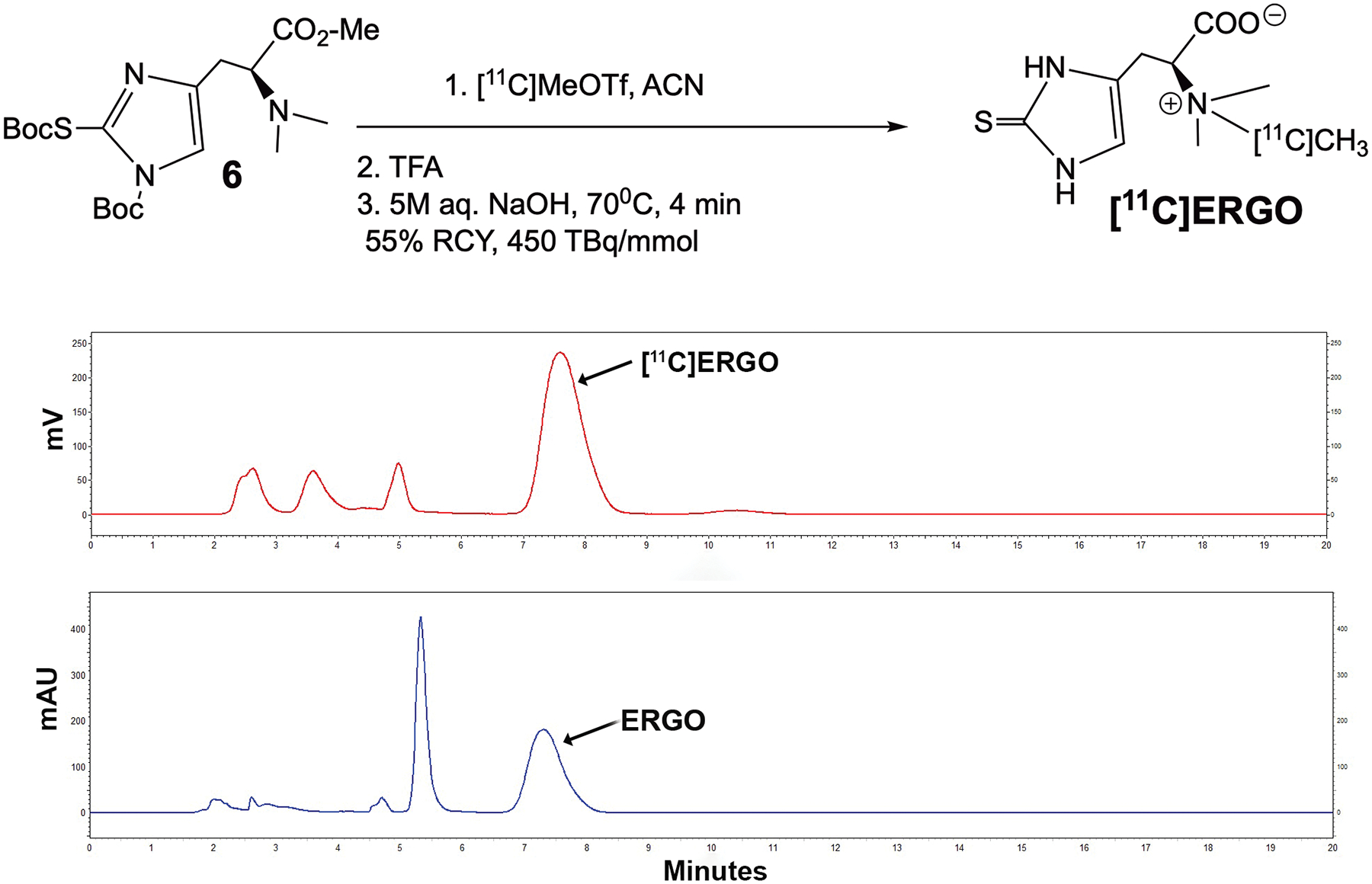
Radiolabeling to generate [11C]ERGO radioligand. After labeling, the Boc groups were deprotected in TFA, while the methyl ester was cleaved in 5M NaOH at an elevated temperature. The RP-HPLC chromatogram of the co-injection data of the ERGO compound, which was detected in the UV channel (254 nm) along with [11C]ERGO radioligand, detected in the radiometric channel. The radioactive compound [11C]ERGO elutes at 7.6 min, while the “cold” compound elutes at 7.3 min.
Dynamic uptake and retention of [11C]ERGO radioligand.
Two cohorts of age-matched mice (10-month-old, n=4, each), including 5XFAD and WT animals, were injected with a consistent amount of 14.8 MBq of [11C]ERGO in 0.1mL via the tail vein prior to acquiring dynamic imaging. Our previous PET imaging data indicated that ERGO started to present in the brain 5 min post-injection, and it continued to accumulate until 30 min before saturation [6]. In this work, we decided to run 20-min dynamic scans to test, as a proof-of-concept, whether the affinity of ERGO for radicals and/or metals, which are integrated in or very closely associated with Abeta aggregates [25–30], would trap the probe. As shown in a representative set of dynamic scans along with the corresponding time-activity curves (TAC) (Fig.3), [11C]ERGO radioligand accumulated significantly higher in every subregions of 5XFAD brain (Fig.3A,B,C,D) compared to WT mice (Fig.3E,F,G,H). The mean percent of ID/g in the brains of 5XFAD mice (n=4) were 0.81 ± 0.09, 0.72 ± 0.09, 0.72 ± 0.10, 0.71 ± 0.12, 0.72 ± 0.08, 0.83 ± 0.10 for cortex, hippocampus, striatum, thalamus, cerebellum and whole brain, respectively, whereas for WT mice (n=4), the values were 0.52 ± 0.13, 0.47 ± 0.18, 0.47 ± 0.15, 0.42 ± 0.18, 0.51 ± 0.13, 0.55 ± 0.14. Overall, the uptake in 5XFAD brains was significantly (p < 0.05) higher than that of WT control.
Figure 3.

A set of representative data of a 30-min dynamic microPET imaging of the distribution and retention of [11C]ERGO radioligand with a focus on the brains of 5XFAD mice (A, B, C,D) versus WT control (E, F,G,H) (n = 4, each). Each animal received 14.8 MBq of [11C]ERGO in 0.1 mL via the lateral tail vein. The uptake was quantified and compared using ImageJ (G). Data are shown as the mean ± SEM for all brain subregions, p<0.05.
5XFAD brains are associated with high levels of activated astrocytes and microglia.
After PET/CT imaging, animals underwent through the cardiac perfusion process, and the brains were embedded in OCT solution for micro-sectioning. Fig.4 shows the representative photomicrographs of brain slices (10μm thickness) stained for astrocyte (green) (Fig.4A,B) and microglia (red) (Fig.4D,E) protein markers in DAPI (blue) positive cells. The hippocampal regions of 5XFAD mice, including the dentate gyrus (DG), cornu ammonis (CA) and the surrounding regions are populated with astrocytes, which were detected and quantified by immunostaining using anti-GFAP antibody. In contrast, very low amounts of reactive astrocytes were observed in the brains of WT mice. Quantitatively, about 2-fold higher levels of this inflammatory marker is found in 5XFAD versus control mice (*p<0.05) (Fig.4C). In a similar observation, 5XFAD mouse brains have greater numbers of IBA1-positive activated microglia (arrows) compared to WT counterparts. The level of activated microglia in WT mice is nearly negligible. Detailed pixel count and quantitative analysis show that the number of activated microglia cells in the hippocampus of 5XFAD mice over 20-fold (**p<0.01) (Fig.4F).
Figure 4.

Representative fluorescence images of the mouse hippocampus stained with activated astrocytes (A,B), microglia (D,E), and OCTN1 (G-J) using anti-GFAP, anti-IBA1 protein markers, and anti-OCTN1/2 antibodies, respectively in WT (A,D,G,H) versus 5XFAD (B,E,I,J). The coronal sections were stained with DAPI (nuclear staining, blue, 465 nm channel) to provide anatomical laminar landmarks. The GFAP expression (green, 517nm channel, scale bar 100μm) is prominent across the hippocampal regions of 5XFAD compared to those from WT counterparts. The GFAP signal was quantified, and the signal distribution was scored on an ordinal scale after thresholding using the Otsu method and presented in the bar graph (C). An asterisk indicates significant differences between WT vs. 5XFAD (*p<0.05). Meanwhile, IBA1 immunohistochemistry (red, 696 nm channel, scale bar 200μm) reveals a significant increase in microglial in 5XFAD vs. WT mice (F) (**p<0.01). The OCTN1 immunohistochemistry (green, 517 nm channel) was observed with a magnification of 20x (G,I; scale bar 50μm) and 63x oil immersion (H,J; scale bar 10μm).
OCTN1 transporters are strongly expressed in the hippocampus of WT and 5XFAD mice.
Consecutive coronal brain slides (10μm thickness) of those shown in Fig.4A–E were processed for OCTN1 staining using anti-OCTN1/2 antibodies (Santa Cruz Biotechnology Inc., Dallas, Texas, USA). Data shown in Fig.4G–J the abundant overexpression of OCTN1 transporters in the hippocampal regions of both WT and 5XFAD mice. This observation reiterates our hypothesis that [11C]ERGO PET probe is distributed to the brain, but only ROS and metals in the oxidative stress milieu trap the probe and resulted in enhanced PET signals.
Experimental methods
General
All solvents and chemicals were purchased from common vendors at reagent grade and were used without further purifications. 1H- and 13C-NMR spectra were obtained using a Bruker 600-MHz NMR spectrometer equipped with cryogenic radio frequency probe.
Compound 2.
To a stirring solution of 1 (535 mg, 1.12 mmol) in CH2Cl2 (10 mL) and methanol (5 mL) at 0°C was added with EDC-HCl (257 mg, 1.3 mmol), and DMAP (25 mg, 0.22 mmol. This solution was allowed to warm up to room temperature (r.t.) and stirred overnight. The reaction was diluted with H2O and CH2Cl2. The product was extracted 3x with CH2Cl2. The organic layers were combined, washed with 1M HCl, saturated sodium bicarbonate, brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. Purified by flash chromatography (0–50% CH2Cl2/(20%MeOH/CH2Cl2)), (241 mg, 45% yield). 1H CDCl3 (600.13 mHz): 7.97 (d, J=0.6 Hz, 1H); 7.80 (d, J=7.2 Hz, 2H); 7.57 (d, J=7.8 Hz, 2H); 7.44, (t, J=7.2 Hz, 2H); 7.35 (t, J=7.8 Hz, 2H); 7.16 (s, 1H); 5.66 (d, J=7.8 Hz, 1H); 4.72 (d, J1=6.6 Hz, 2H); 4.60 (m, 1H); 4.35 (t, J=6.6 Hz, 1H); 3.72 (s, 3H); 3.09 (dd, J1=15.0 Hz, J2=5.4 Hz, 1H); 3.04 (dd, J1=15.0 Hz, J2=4.8 Hz, 1H); 1.44 (s, 9H). 13C CDCl3 (150.9 mHz): 172.3, 155.6, 148.5, 142.8, 141.5, 139.5, 137.0, 128.4, 127.6, 124.9, 120.5, 114.6, 80.0, 69.9, 53.2, 52.5, 46.7, 30.4, 28.5.
Compound 3.
To a stirring solution of 2 (241 mg, 0.50 mmol) in CH2Cl2 (20 mL) at −78°C was added TFA (2 mL) dropwise. This solution was allowed to warm up to 0°C and stirred until completion. The solution was then neutralized with sodium bicarbonate and diluted with H2O and CH2Cl2. The product was extracted 3x with CH2Cl2. The organic layers were combined, washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The product was purified by flash chromatography (0–50% CH2Cl2/(20%MeOH/CH2Cl2)), (172 mg, 88% yield). 1H CDCl3 (600.13 mHz): 8.24 (s, 1H); 7.79 (d, J=7.2 Hz, 2H); 7.54 (d, J=7.8 Hz, 2H); 7.43, (t, J=7.2 Hz, 2H); 7.40, (s, 1H); 7.33 (t, J=7.2 Hz, 2H); 4.78 (dd, J1=6.6 Hz, J2=1.8 Hz, 2H); 4.43 (br.s, 1H); 4.35 (t, J=6.6 Hz, 1H); 3.79 (s, 3H); 3.38 (d, J=13.8 Hz, 1H); 3.30 (dd, J1=15.6 Hz, J2=6.6 Hz, 1H). 13C CDCl3 (150.9 mHz): 168.4, 147.3, 142.4, 141.5, 136.9, 134.8, 128.5, 127.6, 124.8, 120.5, 116.6, 71.1, 53.8, 52.9, 46.6, 26.4.
Compound 4.
To a stirring solution of 3 (172 mg, 0.44 mmol) in MeOH (10mL) was added NaBH3CN (63 mg, 1.0 mmol) and CH2O (37% in H20, 90 mg, 3.0 mmol). The reaction was capped and stirred overnight. The resulting solution was then concentrated via rotovap. Purified by reverse phase flash chromatography (0–50% H2O/ACN), (68 mg, 79% yield). The FMOC group was removed in this step through interaction with the NaBH3CN. 1H CDCl3 (400.13 mHz): 7.47 (s, 1H); 6.75 (s, 1H); 3.61 (s, 3H); 3.48 (t, J=7.6 Hz, 1H); 3.01 (dd, J1=14.8 Hz, J2=8.0 Hz, 1H); 2.88 (dd, J1=14.8 Hz, J2=6.8 Hz, 1H); 2.33 (s, 6H). 13C CDCl3 (100.6 mHz): 172.0, 134.8, 133.1, 118.5, 67.6, 51.3, 41.7, 26.5.
Compound 5.
To a stirring solution of 4 (68 mg, 0.34 mmol) in H2O (2 mL) and diethyl ether (2 mL) was added NaHCO3 (211 mg, 2.5 mmol), and phenyl chlorothionoformate (158 mg, 0.42 mmol) and stirred overnight. Reaction was diluted with H2O and diethyl ether. The product was extracted 3x with diethyl ether. The organic layers were combined, washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting oil was then redissolved in MeOH (5mL) and triethylamine (174 μL) was added. This solution was stirred overnight. The resulting solution was then concentrated under reduced pressure. The final product was purified by flash chromatography (0–50% CH2Cl2/(20%MeOH/CH2Cl2)), (50 mg, 64% yield).
Compound 6.
To a stirring solution of 5 (50 mg, 0.218 mmol) in CH2Cl2 (6 mL) was added: Boc anhydride (190 mg, 0.873 mmol), and DIPEA (113 mg, 0.873 mmol) was heated to 37°C overnight. The reaction was cooled to r.t. and diluted with CH2Cl2 and H2O. The product was extracted 3x with CH2Cl2. The organic layers were combined, washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. Purified by flash chromatography (0–50% CH2Cl2/(20%MeOH/CH2Cl2)), (50 mg, 54% yield). 1H CDCl3 (400.13 mHz): 7.36 (s, 1H); 3.65 (s, 3H); 3.63 (m, 1H); 2.88 (dd, J1=14.4 Hz, J2=8.4 Hz, 1H); 2.86 (dd, J1=14.4 Hz, J2=6.0 Hz, 1H); 2.35 (s, 6H); 1.58 (s, 9H), 1.46 (s, 9H). 13C CDCl3 (100.6 mHz): 171.9, 165.3, 146.7, 139.7, 135.2, 119.8, 86.8, 85.9, 66.7, 51.3, 41.8, 29.8, 28.2, 27.9.
Radiosynthesis of [11C]ERGO PET radioligand
The [11C]ERGO radioligand was prepared using the GE Healthcare Tracerlab FXC-Pro, a commercially supplied reaction platform, modified to directly connect V7nc to V11no. The [11C]CO2 is made by irradiating a target filled with nitrogen and 1% oxygen gas with protons. The [11C]CO2 is then trapped on nickel Shimalite with molecular sieves at room temperature. The [11C]CO2 is then converted to [11C]CH4 by heating the trapped [11C]CO2 to 400°C in the presence of hydrogen gas. The [11C]CH4 is then released from the nickel Shimalite at 400°C and isolated on molecular sieves at −75°C. The [11C]CH4 is then converted to [11C]MeI via a recirculation through gaseous iodine at ~720°C, with the [11C]MeI being trapped on Porapak N with each cycle. The [11C]MeI is then released from the Porapak N by heating with a gentle flow of helium that is passed through an AgOTf impregnated column at ~200°C to convert the [11C]MeI to [11C]MeOTf; this [11C]MeOTf is bubbled into a solution of precursor in 250 μl acetonitrile at −10°C. After the activity transfer is complete the reaction mixture was heated to 80°C for 2 minutes. At this time trifluoroacetic acid (250 μl) was added, the reaction mixture heated at 70°C for 3 minutes, 5M of aqueous NaOH (1 mL) added and heated at 70°C for 4 minutes. The reaction mixture was cooled to room temperature, passed through an ion-retardation resin (Ag11-A8, 3g) into the product vial and the resin was rinsed with water (5 mL) into the final product vial.
We labeled the precursor 6 with [11C]CH3OTf using a commercial automated radiosynthesis module (GE TRACERlab FXc Pro). [11C]CO2 was converted to [11C]MeOTf using the standard reaction conditions, reacted with precursor 6 at 80°C to give the protected intermediate. The Boc-groups were removed under acidic conditions followed by the use of 5M aqueous sodium hydroxide both to neutralize the TFA, and for the saponification of the methyl ester to provide an [11C]ERGO radioligand. The [11C]ERGO was passed through an ion-retardation resin, the pH was adjusted with aqueous HCl before use.
Animals
All animal experiments performed complied with institutional guidelines and were conducted according to the protocol approved by the Vanderbilt Institutional Animal Care and Use Committee. The 5XFAD mice were maintained at Vanderbilt University under standard conditions, in a 12-h light/dark cycle and with free access to food and water. The 5XFAD mice over express both mutant human APP and PS1 genes and it correlates with high APP levels correlating with high burden and accelerated accumulation of β-amyloid (Abeta). A colony of 5XFAD transgenic mice obtained from Jackson Laboratories was maintained by crossing 5XFAD mice with a wild-type (WT) C57BL/6J strain. The mice were genotyped by a standard polymerase chain reaction (PCR) using DNA isolated from tail tips with the following primers: PSEN1 forward, 5’–TCATGACTATCCTCCTGGTGG-3’ and reverse, 5’-CGTTATAGGTTTTAAACACTTCCCC-3’. For APP, forward, 5’-AGGACTGACCACTCGACCAG-3’ and reverse, 5’-CGGGGGTCTAGTTCTGCAT-3’. The Pde6brd1 mutation using forward, 5’-AAGCTAGCTGCAGTAACGCCATTT-3’ and reverse, 5’-ACCTGCATGTGAACCCAGTATTCTATC-3’. Amplified PCR products were then analyzed by size fractionation using agarose (1%) gel electrophoresis; with band sizes for Pde6b mutant = 560 bp, APP transgene = 377 bp and PSEN1 transgene = 608 bp. The 5XFAD mice were maintained as heterozygous.
Dynamic PET imaging.
The dynamic acquisition was divided into twelve 5 sec frames, four 60 sec frames, five 120 sec frames, three 5 min frames, and six 10 min scans. The data from all possible lines of response (LOR) were saved in the list mode raw data format. The raw data were then binned into 3D sinograms with a span of 3 and ring difference of 47. The images were reconstructed into transaxial slices (128 × 128 × 159) with voxel sizes of 0.0815 × 0.0815 × 0.0796 cm3, using the MAP algorithm with 16 subsets, 4 iterations, and a beta of 0.0468. For anatomical co-registration, immediately following the PET scans, the mice received a CT scan in a NanoSPECT/CT (Mediso, Washington DC) at an X-ray beam intensity of 90 mAs and x-ray peak voltage of 45 kVp. The CT images were reconstructed into 170 × 170 × 186 voxels at a voxel size of 0.4 × 0.4 × 0.4 mm3. The PET/CT images were uploaded into Amide software (www.sourceforge.com), co-registered to an MRI template made in-house, and volumetric regions-of-interest were drawn around the cortex, hippocampus, striatum, thalamus, and cerebellum in addition to the whole brain. The PET images were normalized to the injected dose, and the time-activity-curves (TACs) of the mean activity within the ROIs were estimated for the entire duration of the scans.
Cardiac perfusion procedure and tissue collection.
A sharp incision into the abdomen of the anesthetized mouse was made, followed by a longitudinal cut with a scalpel to open the thoracic cavity, which then was stabilized with a retractor. Perfusion began with a 20-gauge syringe containing ice cold PBS (30 ml, pH 7.4) in the left ventricle while the atrium was snipped off. This was followed by injection of paraformaldehyde (PFA) solution (4%). Once the perfusion was completed, the animals were decapitated, and the brains were quickly removed and fixed in PFA overnight at 4°C followed by sucrose precipitation (30%) overnight at 4°C. The brains were then embedded in Cryo-OCT compound (Fisher Scientific) before sectioning.
Ex vivo brain cross-sections processed for GFAP, IBA1, and OCTN1 Immunohistochemistry.
The embedded brains in OCT were cut into coronal sections (10 μm) using a Tissue-Tek cryostat and mounted onto charged glass slides. Prior to staining, slides were washed with PBS (3x, 5,5, and 10 min); then, they were treated with blocking buffer (5% normal goat serum, 0.2% Triton X-100, 0.5% bovine albumin in PBS) for 1h at room temperature. The treated sections were then incubated overnight at 4°C with primary anti-GFAP antibody (1:100 dilution, Biolegend San Diego, CA, USA) or anti-IBA1 antibody (1:250 dilution, FUJIFILM Wako Chemicals U.S.A Corporation Richmond, VA, USA). For OCTN1 staining, the slides were treated with OCTN1/2 (H-9) antibodies (1:100 dilution, Santa Cruz Biotechnology, Dallas, TX, USA). Then, the slides were washed with PBS (3x) for 10 min each, the sections stained for GFAP and OCTN1 were subsequently incubated with secondary antibody goat anti-mouse Alexa Fluor 488 (1:200 dilution, Thermo Fisher Scientific, Carlsbad, CA, USA). Sections stained for IBA1 were incubated with secondary antibody goat anti-rabbit Alexa Fluor 680 (1:200 dilution, Thermo Fisher Scientific, Carlsbad, CA, USA) for 30 min at room temperature. The sections were then washed with PBS twice for 10 minutes and once for 30 minutes, and cover slipped with an antifade mounting medium with DAPI (Vector Laboratories, Burlingame, CA) before observation under a fluorescence microscope.
Statistical analysis.
Data were converted to 8-bit grayscale and thresholding using Otsu’s method; the pixels were quantified using ImageJ software (version 1.53). The data were then imported to GraphPad software (GraphPad Prism version 9.2.0 for Mac) for statistical analysis. The results are presented as mean ± SEM. Differences were analyzed with student’s t-test, and the results were considered significant at p values <0.05. For the IBA1 data, since the pixels are small due low expression, the RGB channels were separated, and the red channel, which indicates IBA1-positive signal were thresholded and analyzed as described.
Discussion
ERGO possesses a unique chemical structure, which contains multiple functional groups. Thus, extensive protection and deprotection maneuvers are necessary to generate a precursor for [11C]CH3 labeling. In that case, many potential combinations of protecting groups have been explored to identify the most compatible deployment of these groups and their order. After several attempts, we reported a successful sequential order to reach to the dimethyl amine precursor 6. Basically, to set the stage for the crucial incorporation of the thiol group onto imidazole, the amino and carboxyl groups of histidine were blocked with acidic labile moieties, while the imidazole amine was protected with an Fmoc group. We previously reported that the amine-protecting Boc groups could be removed using TFA in the presence the t-butyl ester at −78°C [6]. This reaction confers good yield, albeit only with stringent operations and attention to detail. And thus, the reproducibility is moderate in this case, which should come as no surprise since both the Boc and t-butyl ester share a similar acid-catalyzed hydrolysis mechanism (Fig.5). In essence, both processes yield t-Bu+ adduct after C-O cleavage, followed by the formation of a more stable Me2C=CH2 moiety. In the case of the Boc group, the formation of t-Bu+ occurs with the presence of the carbamic acid, which is accompanied by the release of CO2 and the desired free amine. Thus, changing to other esters would alleviate this problem. While methyl ester could be simply achieved through Fischer esterification by treating carboxylic 1 with methanol in the presence of acid. However, this will affect the Boc group, and thus we opted to use Steglich esterification using EDC and DMAP as a catalyst.
Figure 5.
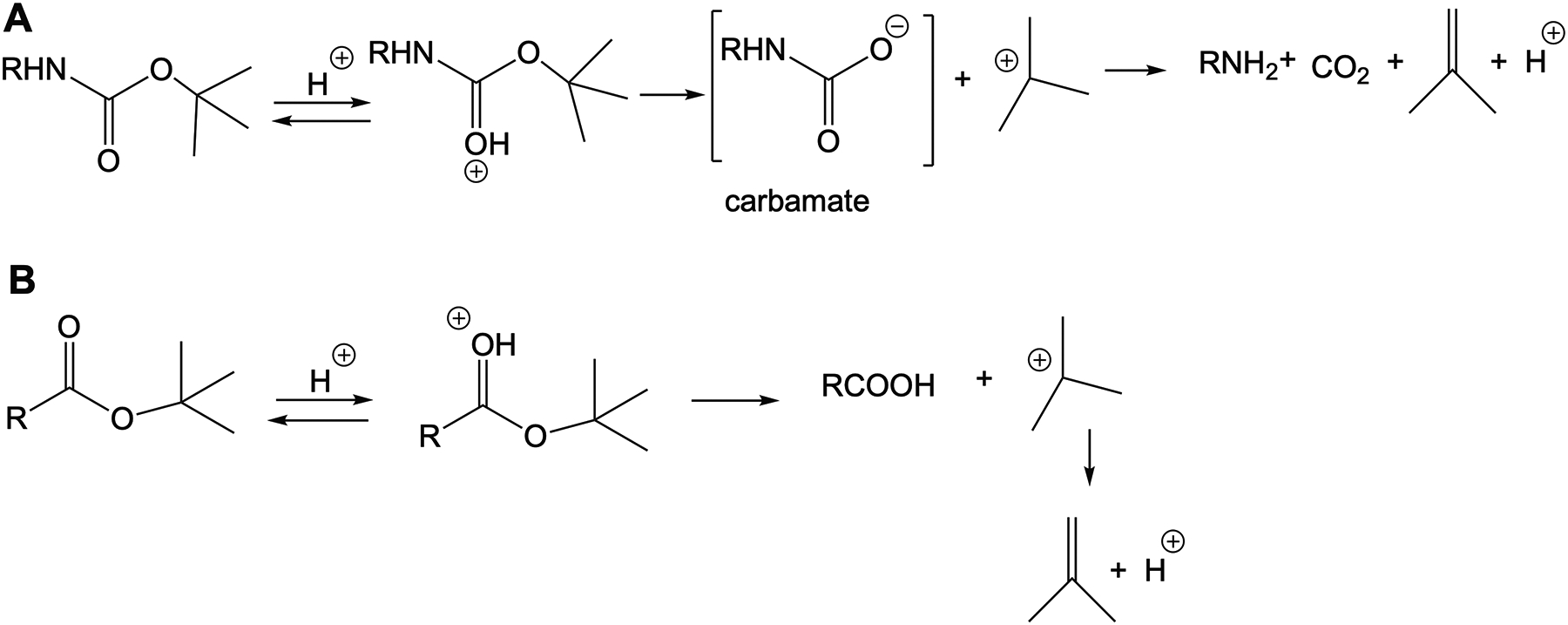
Mechanism of acidic catalyzed hydrolysis of (A) a Boc group and (B) t-butyl ester.
When the carboxylic group was protected as a methyl ester, the stability of this ester contributes to better overall reaction yield, 24% versus 14% in the case of tert-butyl ester starting from compounds 2 to 6. But that is only a part of the motivation for this development. We found that this new chemistry is more reliable and reproducible, a main feature necessary for any radiopharmaceutical development. It is certain that this improved and robust chemistry will foster more translational value of the probe for in vivo imaging applications.
We have also demonstrated the specificity of the probe for in vivo detection of oxidative stress using an acute inflammatory LPS mouse model [6]. To further understand the implications of oxidative stress in the context of Abeta cascade mechanism, we want to assess whether this probe could report specific activity relevant to a disease that manifests chronic presentation of an array of ROS-induced inflammation like AD [31–35]. As shown in Fig.3, the probe was distributed to the brain and its retention in 5XFAD is more significant than that of control animals, in all brain subregions. The high uptake in AD brains conflates a number of reasons. First, the data suggest that Abeta-associated free radical oxidative stress might trap the probe, and thus its accumulation in the brain was reported in the PET images with enhanced spatial and temporal resolution compared to WT counterparts. Second, the existence of metals, such as Zn and Cu, which potentiate AD by participating in the Abeta aggregation by the generation of ROS [36], might offer an additional reason why the probe was trapped in AD mice’s brains. Third, our data is consistent with the current hypothesis in this area that ERGO could serve as an adaptive antioxidant, and thus it can be found at high concentration in pathological tissues [2]. In essence, the detection of the probe retention via noninvasive PET imaging alludes to this being visualization of the probe in action.
Oxidative stress and inflammation are upregulated in the brains of AD patients [37]. And this relationship has been verified in animal models [38], but the 5XFAD mouse model. We are interested to know whether the brain of 5XFAD mice also express biomarkers related to inflammation, such as reactive astrocytes and microglia. In corroboration with the PET imaging data, the immunohistochemistry staining of the brain sections showed that the hippocampus of 5XFAD mice exhibit higher expression levels of activated astrocytes and microglia compared to WT mice. The background level of GFAP-positive astrocytes in WT mice was low, but it could be detected; while IBA1-positive microglia (resting state) in WT mice were barely detectable at high resolution (data not shown). The upregulation of activated microglia and astrocytes support the hypothesis that neuroinflammation could be one of the mechanisms by which AD pathology, including Abeta, leads to neuronal death and dysfunction [39]. Particularly, the pro-inflammatory GFAP marker of the reactive astrocytes, those expression has been linked to Abeta plaque load [40–42]. Furthermore, accumulated data in the past showed that GFAP could serve as an early marker associated with brain Abeta pathology [43]. The detection of oxidative stress using the [11C]ERGO radioligand confirms a past report, which showed that the formation of Abeta plaques trigger a proinflammatory response from microglia and astrocytes [44]. Taken together the data obtained from this work suggest that neuroinflammation reflected in activated microglia and astrocytes in this Abeta accumulating model may trap the [11C]ERGO radioligand contributing to the enhanced PET signal in the 5XFAD mice. Based on the data obtained herein, it is beneficial to combine oxidative stress and neuroinflammation in one cohesive strategy for preventing and treating AD. Further, aside from the [11C]ERGO PET probe, other inflammatory PET agents should be developed and used in tandem for the early detection of AD. These imaging capabilities using PET tracers with sub-pharmacological doses are safe for clinical translation. At the same time, noninvasive PET assessments of AD progress during therapy can serve as a critical tool at the juncture between precision and personalized medicine.
In summary, we report an improved synthesis for the robust and reliable production of [11C]ERGO PET radioligand. Further, the in vivo imaging of an AD phenotype using this probe provides a proof-of-principle to demonstrate for the first time that the neuroinflammation associated with AD could be detected non-invasively, and thereby establishes a novel platform for additional work in the future. As said, a few caveats worth mentioning that AD is a complex disease, it is intuitive to predict that no single probe could report the whole picture of what is going with the early onset of AD. This is compounded by the fact that neuroinflammation and oxidative stress are also manifested in other neurodegenerative diseases. Thus, for the early detection and assessment of the response of AD, a combination of probes targeting relevant pathways along the mechanism of AD should be used, and [11C]ERGO PET radioligand could serve as one of them. Another remark worth mentioning is that we used the anti-OCTN1/2 antibodies for the immunohistochemical staining of the brain specimens. Thus, we cannot conclude OCTN1 is the sole transporter for this probe. The availability of specific OCTN1 antibodies in the future will help to resolve this issue.
Acknowledgements.
This work was partially supported by the R01 AG061138 (W.P.) and The NIH 1S10 OD016245 for the procurement of the Inveon microPET scanner.
Abbreviations:
- 5XFAD
5-gene familial AD
- Abeta
amyloid-β
- AD
Alzheimer’s disease
- Boc
tert-butyloxycarbonyl
- CA
cornu ammonis
- CT
computed tomography
- Cu
copper
- DG
dentate gyrus
- DMAP
dimethylaminopyridine
- ERGO
ergothioneine
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- Fe
iron
- GFAP
glial fibrillary acidic protein
- HPLC
high performance liquid chromatography
- IBA1
ionized calcium binding adaptor molecule 1
- LPS
lipopolysaccharide
- MeOTf
methyl trifluoromethanesulfonate
- MRI
magnetic resonance imaging
- NMR
nuclear magnetic resonance
- OCTN1
novel organic cation transporter 1
- PFA
paraformaldehyde
- PET
positron emission tomography
- t-butyl
tertiary butyl
- TBq
terabecquerel
- TLC
thin layer chromatography
- TFA
trifluoroacetic acid
- ROS
reactive oxygen species
- WT
wild type
Footnotes
Competing interests. The authors declare no competing interests.
Data Availability
The data that support the findings of this study are available from the corresponding author (wellington.pham@vumc.org) upon reasonable request.
References
- [1].Cumming BM, Chinta KC, Reddy VP, Steyn AJC (2018) Role of Ergothioneine in Microbial Physiology and Pathogenesis. Antioxid Redox Signal 28, 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Halliwell B, Cheah IK, Drum CL (2016) Ergothioneine, an adaptive antioxidant for the protection of injured tissues? A hypothesis. Biochem Biophys Res Commun 470, 245–250. [DOI] [PubMed] [Google Scholar]
- [3].Borodina I, Kenny LC, McCarthy CM, Paramasivan K, Pretorius E, Roberts TJ, van der Hoek SA, Kell DB (2020) The biology of ergothioneine, an antioxidant nutraceutical. Nutr Res Rev 33, 190–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kerley RN, McCarthy C, Kell DB, Kenny LC (2018) The potential therapeutic effects of ergothioneine in pre-eclampsia. Free Radic Biol Med 117, 145–157. [DOI] [PubMed] [Google Scholar]
- [5].Samuel P, Tsapekos M, de Pedro N, Liu AG, Casey Lippmeier J, Chen S (2020) Ergothioneine Mitigates Telomere Shortening under Oxidative Stress Conditions. J Diet Suppl, 1–14. [DOI] [PubMed] [Google Scholar]
- [6].Behof WJ, Whitmore CA, Haynes JR, Rosenberg AJ, Tantawy MN, Peterson TE, Harrison FE, Beelman RB, Pham W (2021) A novel antioxidant ergothioneine PET radioligand for in vivo imaging applications. Scientific Reports 11, 18450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Akanmu D, Cecchini R, Aruoma OI, Halliwell B (1991) The antioxidant action of L-Ergo. Arch Biochem Biophys 288, 10–16. [DOI] [PubMed] [Google Scholar]
- [8].Motohashi N, Mori I (1986) Thiol-induced hydroxyl radical formation and scavenger effect of thiocarbamides on hydroxyl radicals. J Inorg Biochem 26, 205–212. [DOI] [PubMed] [Google Scholar]
- [9].Asmus KD, Bensassion RV, Bernier JL, Houssin R, Land EJ (1996) One electron oxidation of L-Ergo and analogues investigated by pulsed radiolysis: redox reactions involving L-Ergo and vitamin C. Biochem J 315, 625–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Aruoma OI, Whiteman M, England TG, Halliwell B (1997) Antioxidant action of ergothioneine: assessment of its ability to scavenge peroxynitrite. Biochem Biophys Res Commun 231, 389–391. [DOI] [PubMed] [Google Scholar]
- [11].Grundemann D, Harlfinger S, Golz S, Geerts A, Lazar A, Berkels R, Jung N, Rubbert A, Schomig E (2005) Discovery of the ergothioneine transporter. Proc Natl Acad Sci U S A 102, 5256–5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Crossland J, Mitchell J, Woodruff GN (1966) The presence of ergothioneine in the central nervous system and its probable identity with the cerebellar factor. J Physiol 182, 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nakamichi N, Nakayama K, Ishimoto T, Masuo Y, Wakayama T, Sekiguchi H, Sutoh K, Usumi K, Iseki S, Kato Y (2016) Food-derived hydrophilic antioxidant ergothioneine is distributed to the brain and exerts antidepressant effect in mice. Brain Behav 6, e00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Teruya T, Chen YJ, Kondoh H, Fukuji Y, Yanagida M (2021) Whole-blood metabolomics of dementia patients reveal classes of disease-linked metabolites. Proc Natl Acad Sci U S A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Benzi G, Moretti A (1995) Are reactive oxygen species involved in Alzheimer’s disease? Neurobiol Aging 16, 661–674. [DOI] [PubMed] [Google Scholar]
- [16].Abramov AY, Canevari L, Duchen MR (2004) Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci 24, 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bermejo P, Martin-Aragon S, Benedi J, Susin C, Felici E, Gil P, Ribera JM, Villar AM (2008) Peripheral levels of glutathione and protein oxidation as markers in the development of Alzheimer’s disease from Mild Cognitive Impairment. Free Radic Res 42, 162–170. [DOI] [PubMed] [Google Scholar]
- [18].Casado A, Encarnacion Lopez-Fernandez M, Concepcion Casado M, de La Torre R (2008) Lipid peroxidation and antioxidant enzyme activities in vascular and Alzheimer dementias. Neurochem Res 33, 450–458. [DOI] [PubMed] [Google Scholar]
- [19].Govoni S, Lanni C, Racchi M (2001) Advances in understanding the pathogenetic mechanisms of Alzheimer’s disease. Funct Neurol 16, 17–30. [PubMed] [Google Scholar]
- [20].Atwood CS, Huang X, Moir RD, Tanzi RE, Bush AI (1999) Role of free radicals and metal ions in the pathogenesis of Alzheimer’s disease. Met Ions Biol Syst 36, 309–364. [PubMed] [Google Scholar]
- [21].Atwood CS, Scarpa RC, Huang X, Moir RD, Jones WD, Fairlie DP, Tanzi RE, Bush AI (2000) Characterization of copper interactions with alzheimer amyloid beta peptides: identification of an attomolar-affinity copper binding site on amyloid beta1-42. J Neurochem 75, 1219–1233. [DOI] [PubMed] [Google Scholar]
- [22].Bush AI (2000) Metals and neuroscience. Curr Opin Chem Biol 4, 184–191. [DOI] [PubMed] [Google Scholar]
- [23].Bush AI (2003) The metallobiology of Alzheimer’s disease. Trends Neurosci 26, 207–214. [DOI] [PubMed] [Google Scholar]
- [24].Huang X, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JD, Hanson GR, Stokes KC, Leopold M, Multhaup G, Goldstein LE, Scarpa RC, Saunders AJ, Lim J, Moir RD, Glabe C, Bowden EF, Masters CL, Fairlie DP, Tanzi RE, Bush AI (1999) Cu(II) potentiation of alzheimer abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem 274, 37111–37116. [DOI] [PubMed] [Google Scholar]
- [25].Butterfield DA (1997) Beta-Amyloid-associated free radical oxidative stress and neurotoxicity: implications for Alzheimer’s disease. Chem Res Toxicol 10, 495–506. [DOI] [PubMed] [Google Scholar]
- [26].Butterfield DA, Boyd-Kimball D (2020) Mitochondrial Oxidative and Nitrosative Stress and Alzheimer Disease. Antioxidants (Basel) 9, 818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Butterfield DA, Halliwell B (2019) Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci 20, 148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F (2018) Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol 14, 450–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Christen Y (2000) Oxidative stress and Alzheimer disease. Am J Clin Nutr 71, 621S–629S. [DOI] [PubMed] [Google Scholar]
- [30].Smith DG, Cappai R, Barnham KJ (2007) The redox chemistry of the Alzheimer’s disease amyloid beta peptide. Biochim Biophys Acta 1768, 1976–1990. [DOI] [PubMed] [Google Scholar]
- [31].Ahmad W, Ijaz B, Shabbiri K, Ahmed F, Rehman S (2017) Oxidative toxicity in diabetes and Alzheimer’s disease: mechanisms behind ROS/ RNS generation. J Biomed Sci 24, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ganguly U, Kaur U, Chakrabarti SS, Sharma P, Agrawal BK, Saso L, Chakrabarti S (2021) Oxidative Stress, Neuroinflammation, and NADPH Oxidase: Implications in the Pathogenesis and Treatment of Alzheimer’s Disease. Oxid Med Cell Longev 2021, 7086512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Huang WJ, Zhang X, Chen WW (2016) Role of oxidative stress in Alzheimer’s disease. Biomed Rep 4, 519–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kaur U, Banerjee P, Bir A, Sinha M, Biswas A, Chakrabarti S (2015) Reactive oxygen species, redox signaling and neuroinflammation in Alzheimer’s disease: the NF-kappaB connection. Curr Top Med Chem 15, 446–457. [DOI] [PubMed] [Google Scholar]
- [35].Verri M, Pastoris O, Dossena M, Aquilani R, Guerriero F, Cuzzoni G, Venturini L, Ricevuti G, Bongiorno AI (2012) Mitochondrial alterations, oxidative stress and neuroinflammation in Alzheimer's disease. Int J Immunopathol Pharmacol 25, 345–353. [DOI] [PubMed] [Google Scholar]
- [36].Adlard PA, Bush AI (2006) Metals and Alzheimer’s disease. J Alzheimers Dis 10, 145–163. [DOI] [PubMed] [Google Scholar]
- [37].Uruno A, Matsumaru D, Ryoke R, Saito R, Kadoguchi S, Saigusa D, Saito T, Saido TC, Kawashima R, Yamamoto M (2020) Nrf2 Suppresses Oxidative Stress and Inflammation in App Knock-In Alzheimer’s Disease Model Mice. Mol Cell Biol 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Saito T, Saido TC (2018) Neuroinflammation in mouse models of Alzheimer’s disease. Clin Exp Neuroimmunol 9, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hopperton KE, Mohammad D, Trepanier MO, Giuliano V, Bazinet RP (2018) Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: a systematic review. Mol Psychiatry 23, 177–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hanzel DK, Trojanowski JQ, Johnston RF, Loring JF (1999) High-throughput quantitative histological analysis of Alzheimer’s disease pathology using a confocal digital microscanner. Nat Biotechnol 17, 53–57. [DOI] [PubMed] [Google Scholar]
- [41].Muramori F, Kobayashi K, Nakamura I (1998) A quantitative study of neurofibrillary tangles, senile plaques and astrocytes in the hippocampal subdivisions and entorhinal cortex in Alzheimer’s disease, normal controls and non-Alzheimer neuropsychiatric diseases. Psychiatry Clin Neurosci 52, 593–599. [DOI] [PubMed] [Google Scholar]
- [42].Vehmas AK, Kawas CH, Stewart WF, Troncoso JC (2003) Immune reactive cells in senile plaques and cognitive decline in Alzheimer’s disease. Neurobiol Aging 24, 321–331. [DOI] [PubMed] [Google Scholar]
- [43].Pereira JB, Janelidze S, Smith R, Mattsson-Carlgren N, Palmqvist S, Teunissen CE, Zetterberg H, Stomrud E, Ashton NJ, Blennow K, Hansson O (2021) Plasma GFAP is an early marker of amyloid-beta but not tau pathology in Alzheimer’s disease. Brain 144, 3505–3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Huat TJ, Camats-Perna J, Newcombe EA, Valmas N, Kitazawa M, Medeiros R (2019) Metal Toxicity Links to Alzheimer’s Disease and Neuroinflammation. J Mol Biol 431, 1843–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author (wellington.pham@vumc.org) upon reasonable request.