

Abstract
Increasingly, highly multiplexed tissue imaging methods are used to profile protein expression at the single‐cell level. However, a critical limitation is the lack of robust cell segmentation tools for tissue sections. We present Multiplexed Image Resegmentation of Internal Aberrant Membranes (MIRIAM) that combines (a) a pipeline for cell segmentation and quantification that incorporates machine learning‐based pixel classification to define cellular compartments, (b) a novel method for extending incomplete cell membranes, and (c) a deep learning‐based cell shape descriptor. Using human colonic adenomas as an example, we show that MIRIAM is superior to widely utilized segmentation methods and provides a pipeline that is broadly applicable to different imaging platforms and tissue types.
Keywords: cell segmentation, image processing, multiplexed imaging, single cell analysis
1. INTRODUCTION
Single‐cell analytical methods capable of interrogating cellular heterogeneity are now widely available. These methods allow multiple markers to be measured over a large number of cells and can be achieved by disaggregation of tissue into suspension or in situ analysis. Suspension methods, such as single‐cell RNA‐seq [1], multi‐channel flow cytometry [2], and mass cytometry [3], require single‐cell dissociation that results in inherent loss of spatial context. In situ methods such as isotope‐based imaging [4, 5] or the various multiplexed fluorescence imaging methods [6, 7, 8] retain spatial information but require cell identification after data collection.
Robust image segmentation methods are available in vitro [9, 10, 11], but are not generally applicable in tissue due to irregular cell shapes, high cellular density, membrane polarity, and uneven membrane marker coverage. Typical cell segmentation in tissue includes dilation from nuclei [12] or Voronoi tessellation [7]. While adequate for segmenting stereotypically shaped immune cell populations, these methods are often insufficient for segmenting columnar epithelial cells with elongated and irregular morphologies. Segmentation of intestinal epithelial cells proves to be more challenging than their neoplastic counterparts. Loss of apico‐basolateral polarity and cell rounding in tumors frequently result in symmetrically shaped cells that alleviate many segmentation problems encountered in the normal gut. Multiple membrane markers have been used to better define cell borders for seed‐based watershed segmentation [13] as well as other methods [14, 15, 16, 17] including deep learning [18, 19, 20]. However, these methods often fail due to improper or incomplete membrane identification and cell segmentation in tissue remains a significant challenge.
Here, we present multiplexed image resegmentation of internal aberrant membranes (MIRIAM), a pipeline for cell segmentation and quantification on tissue. We apply MIRIAM on multiplexed immunofluorescence‐derived images [6] of precancerous colorectal adenomas [21] and a multiplexed ion beam imaging (MIBI) breast carcinoma data set. Initial pixel classification by random forest‐based machine learning provides an input to a novel method to identify and separate cells with internal membranes. Following cell identification, each marker is then quantified by image intensity over the entire cell as well as in the nucleus, membrane, and cytoplasm. Finally, cell shapes are characterized using an autoencoder neural network.
2. MATERIALS AND METHODS
2.1. Human subjects
The Tennessee Colon Polyp Study (TCPS) [21] was approved by the Vanderbilt University Medical Center (VUMC) and Veterans Affairs Tennessee Valley Health System (VA) institutional review boards and the VA Research and Development Committee. All participants provided written informed consent.
2.2. Tissue processing, antibody staining, and multiplexed immunofluorescence imaging
A de‐identified human colonic adenoma tissue microarray (TMA) derived from the TCPS patient cohort was obtained. The TMA was sectioned (5 μm) prior to deparaffinization, rehydration and antigen retrieval using pH 6.0 citrate buffer (DAKO) at 105°C for 20 min followed by 10 min at room temperature. The slide was incubated in 3% hydrogen peroxide for 10 min to reduce endogenous background signal and subsequently blocked in 3% BSA/10% donkey serum in PBS for 30 min. Multiplexed Immunofluorescence imaging was completed by sequential antibody staining and dye inactivation as described [6]. Briefly, imaging was performed on 124 TMA cores (1 mm diameter) using a Cytell Slide Imaging System (GE Healthcare) at 20× magnification. Images of each core were 5435 × 4473 pixels with a pixel resolution of 0.325 μm. Exposure times were optimized for each antibody stain. Antibody reagents are described in Table S1. Dye inactivation was accomplished with an alkaline peroxide solution, and background images were collected after each round of staining to ensure fluorophore inactivation. Following acquisition, images were processed as described [6, 13]. Briefly, DAPI images for each round were registered to a common baseline, and autofluorescence in staining rounds was removed by subtracting the previous background image for each position. Images were then tiled for each TMA core.
2.3. MIBI data set
MIBI imaging from a breast carcinoma TMA was collected by Keren et al. [22] from 41 patients using 36 protein markers. Images were 2048 × 2048 pixels at 0.5 μM resolution.
2.4. Cell segmentation
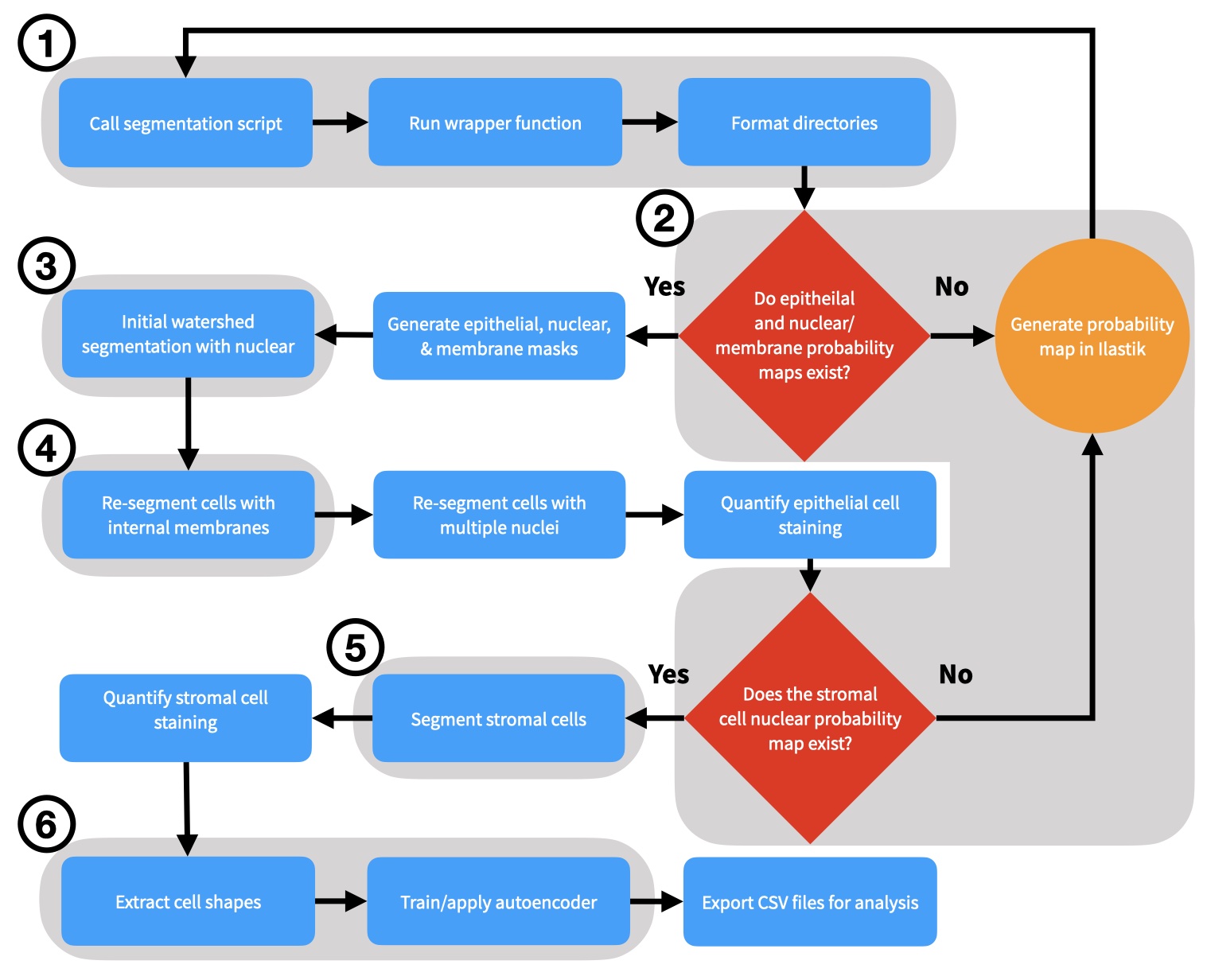
The input to MIRIAM is any type of multiplexed imaging data. All scripts to complete the segmentation pipeline are available at https://github.com/Coffey-Lab/MIRIAM including a step‐by‐step overview of the process and example data. Cell segmentation on each TMA core was conducted using a pipeline utilizing either Matlab (R2018b) or Python (3.7) and Ilastik (1.3.2 or greater). Initialization of the wrapper function generated a number of container folders for each step in the segmentation process. A schematic of the process is shown in Figure 1 and Figure S1.
FIGURE 1.
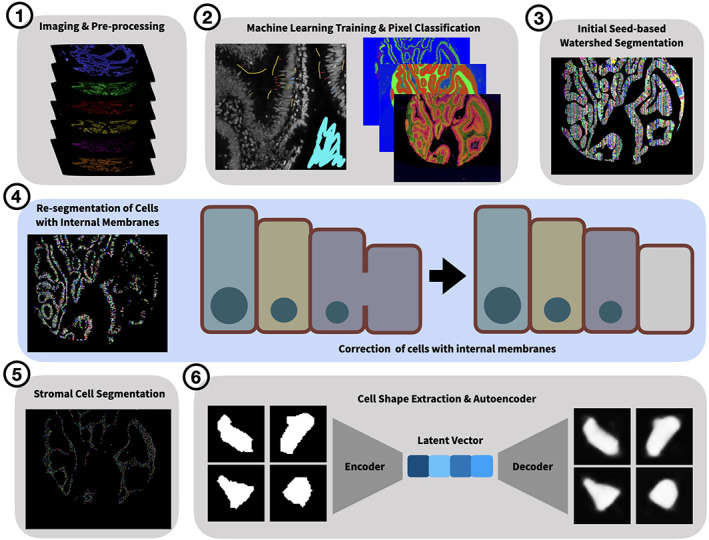
Graphical overview of segmentation pipeline. Key steps in the MIRIAM pipeline are shown pictorially. A cartoon of cell re‐segmentation by connecting internal membranes is highlighted in blue in the middle panel
To facilitate pixel classification machine learning, tiff image stacks containing DAPI and autofluorescence removed images for all markers were generated for each image position after which the script was terminated. The tiff stacks were then manually annotated in Ilastik [23] to generate epithelial/stroma, membrane/cytoplasm/nucleus, and stromal nuclei probability masks using a random forest pixel classification algorithm.
The Matlab or Python script was re‐initialized and binary masks for epithelium, nuclei, and membranes were generated. A watershed was used with nuclei as seed points and learned membranes as boundaries to generate an initial segmentation, which was masked by the epithelial regions to only include epithelial cells.
Subsequently, cells were resegmented using a novel algorithm if they contained greater than 10% internal membranes by area. This algorithm “connects the dots” by finding the endpoints of internal membranes and extends them until they intersect with either the cell border or another extension from a separate internal membrane (Figure 1). The same method is also applied to cells expressing Mucin 2 in order to facilitate segmentation of goblet cells. A flowchart with pseudocode of this algorithm is shown in Figure S2. Following re‐segmentation, cells were assigned unique IDs.
Cell compartments were then defined within each cell. The nucleus was defined as the pixels contained within the cell object and the nuclear mask. Membranes were defined as the pixels contained within the cell object and within 5 pixels of the edge of the cell object that were not already defined as nuclear. The cytoplasm was defined as all other pixels in the cell object that were not already defined as nuclear or membranous. Each marker was then quantified over the entire cell and in each compartment by taking the median image intensity of the autofluorescence removed image. Additionally, the cell centroid coordinates and areas of the entire cell object and subcompartmens were generated. These data were stored in comma‐separated value (CSV) files.
For stromal cell segmentation, a watershed was used with stromal nuclei, defined from the machine learning pixel classification as seed points, was applied. This was then multiplied by a mask created from a 3 pixel dilation of the stromal nuclei mask to define the individual stromal cells. Marker quantification was then performed by calculating the median image intensity for each marker over the entire cell object. Cell location and area were also determined. These data were stored in CSV files.
2.5. Cell shape analysis
An autoencoder neural network was then used to classify cell shapes from the segmented data. Similar methods have been developed to cluster biological [24] and non‐biological data [25]. For each imaging position, all segmented cells were binarized and re‐sized to a 128 × 128 pixel matrix and aligned such that the major axes were all in the same orientation. For autoencoder training, a random subset (20% default) of all cells were chosen and a neural network was run in Matlab using the Deep Learning Toolbox or the Keras library in Python using 256 hidden layers. Following training, all cells were encoded and the latent vectors for all cells were saved for subsequent processing.
2.6. Comparison of segmentation results
Voronoi segmentation [26] was completed using the nuclei derived from pixel‐wise machine learning. Mesmer [20], a deep learning segmentation method, was applied to the colon polyp TMA using DAPI as the nuclear channel and NaKATPase as the membrane channel. Cell membranes were manually annotated on the entirety of three TMA cores. Cell borders were extracted from the final segmentations derived from MIRIAM, Mesmer, Voronoi, and the manual membranes, and dilated using a 5 pixel square kernel. The Dice similarity coefficient (DSC) [27], and Jaccard similarity coefficient (JSC) [28] were calculated to assess segmentation agreement with manually annotated membranes in two ways. First, the cell border images of the three TMA cores with manual annotation were tiled into 512 × 512 pixel sub‐images and the similarity indices were computed within each sub‐image. Sub‐images with no tissue were excluded from analysis. Second, individual cells were extracted and cropped to the bounding box of each cell. The membranes of each cell were then compared to the manually annotated membranes in the same area. To test how varying marker combinations affect MIRIAM segmentation, single markers were excluded or subsets of markers chosen from the entire set of markers. Pixel classification was re‐trained for each permutation using the same annotations for each marker combination and MIRIAM segmentation performed. DSC and JSC were calculated between the final segmentations from each marker subset and the manually annotated cell borders in 512 × 512 pixel sub‐images for each core.
2.7. Data analysis
Data analysis was performed in R. t‐Stochastic Neighbor Embedding was conducted using the Rtsne package [29] and all plots were generated using the ggplot2 package [30].
3. RESULTS
Example raw images, probability masks, and final segmentation are shown in Figure 2. All staining rounds, including NaKATPase (Figure 2A) and DAPI (Figure 2B), were used to derive the epithelial (Figure 2C) and membrane/nucleus (Figure 2D) probability maps. A final cell segmentation mask (Figure 2E) and sub‐cellular segmentation (Figure 2F) shows the spatial extent of each cell border (white), nucleus (blue), cell membrane (red), and cytoplasm (green). Segmentation results (Figure 2G) show generally high correspondence between MIRIAM and Mesmer segmentation in epithelial cells. Voronoi agrees with MIRIAM segmentation in areas where cells are densely packed. However, Voronoi undersegments cells towards the lumen as it does not take into account cell membranes.
FIGURE 2.
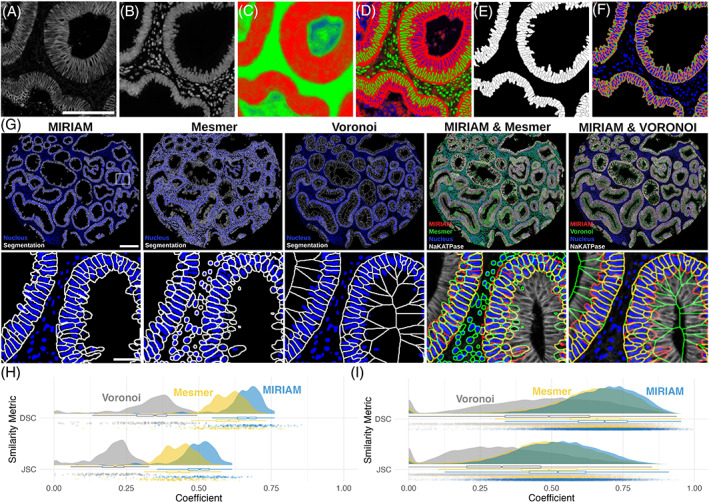
MIRIAM results. Representative (A) NaKATPase and (B) DAPI staining are shown as part of the 16 channel image stack that was used to create probability maps for (C) epithelial (red) and stromal (green) regions, and (D) cellular membrane (red), nucleus (green), and cytoplasm (blue). Final MIRIAM‐derived (E) whole cell and (F) subcellular segmentation are shown. Segmentation results (G) are shown with cell borders in white and blue nuclear masks over an entire tissue microarray core (scale bar: 200 μm) and a zoomed region (scale bar: 50 μm) for MIRIAM, Mesmer, and Voronoi. Comparison of MIRIAM to Mesmer and Voronoi are shown with the nuclear mask and membrane marker NaKATPase. Rain cloud plots show dice similarity coefficient (DSC) and Jaccard similarity coefficient (JSC) comparing segmentation methods to manually annotated cell border images at an image level (H) and in individual cells (I)
On an image level, 240 sub‐images were analyzed across the three manually annotated TMA cores (Figure 2H). For MIRIAM, DSC was 0.65 ± 0.08 (mean ± SD) and JSC was 0.48 ± 0.08. In comparison, Mesmer had a DSC of 0.59 ± 0.10 and JSC of 0.42 ± 0.08, while Voronoi had a DSC of 0.32 ± 0.11 and JSC of 0.20 ± 0.07. Similarly, when comparing individual cells, MIRIAM exhibited higher similarity than Vornonoi (Figure 2I). MIRIAM had a DSC of DSC of 0.67 ± 0.14 and JSC of 0.52 ± 0.15, Mesmer a DSC of 0.61 ± 0.19 and JSC of 0.46 ± 0.18, and Voronoi a DSC of 0.47 ± 0.21 and JSC of 0.33 ± 0.18.
MIRIAM was robust to the composition of markers used for training the pixel classification algorithms. Compared to the full 15 marker data set and DAPI, leaving out a single marker led to minimal change in DSC or DSC when the resulting segmentation was compared to the manually annotated cell borders (Figure S3). Using a subset of only membrane markers (β‐catenin, E‐cadherin, NaKATPase, Pan‐cadherin, and Pan‐cytokeratin) and DAPI similarly showed a small increase in similarity. However, leaving out the membrane markers and DAPI or only using NaKATPase and DAPI resulted in decreased similarity to the manually annotated cell borders.
The quantified segmentation results can be used for downstream data analysis such as defining cellular populations [31] or interrogating cell‐state transition trajectories [32]. Figure 3A shows the results of t‐stochastic neighbor embedding (t‐SNE) using only the intensities for each marker in the data set. Cells with similar protein expression patterns group together in two‐dimensional data space derived from the high dimensional data (Figure S4). For example, cells with p‐ERK staining intensity form a discrete cluster that overlaps with p‐EGFR, PCNA, and Ki67, all of which are features of proliferative cells. When only the encoded latent vectors for cell shape, excluding those used to train the autoencoder, are used as an input to t‐SNE analysis (Figure 3B), cells with similar geometries are near each other in t‐SNE space (Figure 3C). However, our t‐SNE shape analysis did not reveal any discernible cellular populations or clear patterns in marker expression (Figure S5). While cells of different shapes (e.g., columnar, circular, triangular) can be identified, and nearest neighbors confirm the shape groupings, cell shape does not appear to correspond to marker signal intensity, at least in this setting.
FIGURE 3.
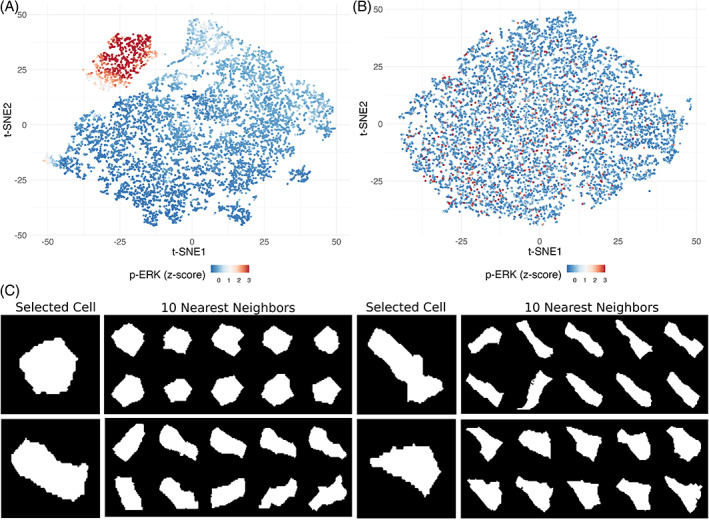
Cell shape similarity. t‐SNE plots using (A) only marker intensity and (B) cell shape latent vectors are shown with p‐ERK staining intensities. (C) The shapes of four selected cells are compared to their 10 nearest neighbors in t‐SNE space
Results of MIRIAM using Matlab or Python implementations show largely similar results (Figure S6). Image correlation between both methods is high for cell membranes over 120 TMA cores with a DSC of 0.84 ± 0.06 and JSC of 0.74 ± 0.09.
MIRIAM performed well on a breast carcinoma MIBI dataset, representative results are demonstrated in Figure S7.
4. DISCUSSION
Cell segmentation represents a difficult task in tissue sections and robust and accurate methods are needed to leverage the rapidly expanding number of highly multiplexed imaging platforms. Significant advances here include a novel algorithm to close missing cell membranes and a method to determine cell shape in tissue.
While deep learning methods have recently been applied to multiplexed image segmentation tasks [20], they may not be suitable for a specific use case without additional training. Machine learning pixel classification, as reported here and elsewhere [33, 34], provides a middle ground between deep learning methods and simpler, yet less time consuming, methods like additive membrane intensities [13]. Using the entire set of markers to define intracellular structures with machine learning resulted in more complete membranes compared to relying on defined membrane or nuclear markers, While not implemented here, the addition of deep learning methods for nuclei detection [11, 18] may help improve initial seeds compared to pixel‐based machine learning.
Despite improved membrane definition, gaps remain in the learned membranes leading to under‐segmentation of cell objects. Oftentimes, the cross section may exclude the nucleus, which can result in under‐segmentation, or a combined cell with no nucleus to one in which a nucleus is detected. To solve this issue, we developed an algorithm that detects objects in the initial watershed segmentation that have internal membranes. For those cells, the algorithm computes the angle of membrane segments within the cell and extends them until another membrane is encountered. This procedure improves segmentation quality in cases where no nucleus is detected in a neighboring cell and is superior to Voronoi in this context both qualitatively (Figure 2G) and quantitatively (Figure 2H, I).
MIRIAM segmentation also outperformed Mesmer deep learning segmentation [20], demonstrating higher similarity to the manually annotated data in this use case (Figure 2H,I). However, while Mesmer was trained on imaging data we provided of normal mouse small intestine, as well normal human colon and colorectal cancer; it was not trained on colorectal adenomas. We expect that segmentation performance gains for Mesmer could potentially be realized if additional training data is included.
The quality of MIRIAM‐derived segmentation is minimally affected by changes in marker composition (Figure S3). Dropping individual membrane markers from the training data, even DAPI, results in small changes in similarity scores compared to the use of all markers. Similar results were shown using a combination of membrane markers and DAPI. Large decreases in performance were observed when all membrane markers were excluded or only NaKATPase was used in conjunction with DAPI. Additionally, MIRIAM performed well in segmenting a breast cancer MIBI data set (Figure S7) composed of a different marker set and lower image quality than the colorectal adenoma data set. Overall, MIRIAM is robust to changes in marker composition and modality of image collection.
Finally, we introduce a deep learning method for classifying cell shape. We chose an autoencoder as other methods that use defined shape descriptors [35], such as circularity and concavity, may not capture the relevant characteristics that a neural network can. In principle, cell shape should provide added information about cell characteristics, for example, crypt base columnar cells have an elongated wedge shape and mucus‐producing goblet cells have their characteristic cup shape. Indeed, cells of similar shape are clustered together (Figure 3C). However, as cells in a tissue section can be bisected in any direction, this is not always the case. As our results in human colonic adenomas show, 2‐dimensional cell shape may not correlate with cell type or protein expression as might be expected if the full 3‐dimensional cell morphology was characterized. While this is the case in our system, cell shape may be more informative in other tissue contexts or when limited to already defined cell types.
Together, these improvements to cell segmentation and quantification present a step forward for single‐cell analysis of highly multiplexed imaging data. Importantly, MIRIAM is tissue‐type and acquisition method agnostic, highly modifiable, and has been previously applied to a large multiplexed immunofluorescence dataset [36]. With implementations in Matlab and Python, we envision this pipeline can be broadly adopted for analysis of multiplexed image data.
AUTHOR CONTRIBUTIONS
Eliot McKinley: Conceptualization (equal); data curation (equal); formal analysis (equal); investigation (equal); methodology (equal); software (equal); validation (equal); visualization (equal); writing – original draft (equal); writing – review and editing (equal). Justin Shao: Software (equal). Samuel T Ellis: Investigation (equal); validation (equal). Cody N Heiser: Software (equal); validation (equal). Joseph T Roland: Investigation (equal); software (equal); validation (equal); writing – review and editing (equal). Mary Catherine Macedonia: Software (equal); validation (equal). Paige Vega: Methodology (equal); validation (equal). Susie Shin: Investigation (equal); validation (equal). Robert J Coffey: Conceptualization (equal); funding acquisition (equal); project administration (equal); writing – original draft (equal); writing – review and editing (equal). Ken Lau: Conceptualization (equal); funding acquisition (equal); methodology (equal); software (equal); supervision (equal); writing – original draft (equal); writing – review and editing (equal).
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
5.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/cyto.a.24541.
Supporting information
Figure S1 Flowchart of the MIRIAM Pipeline. The major steps in the MIRIAM segmentation pipeline are shown. Key steps are shaded and numbered and shown pictorially in Figure 1.
{kind=link}
Figure S2 Cell re‐segmentation algorithm. Major steps in the cell re‐segmentation algorithm are shown with pseudocode.
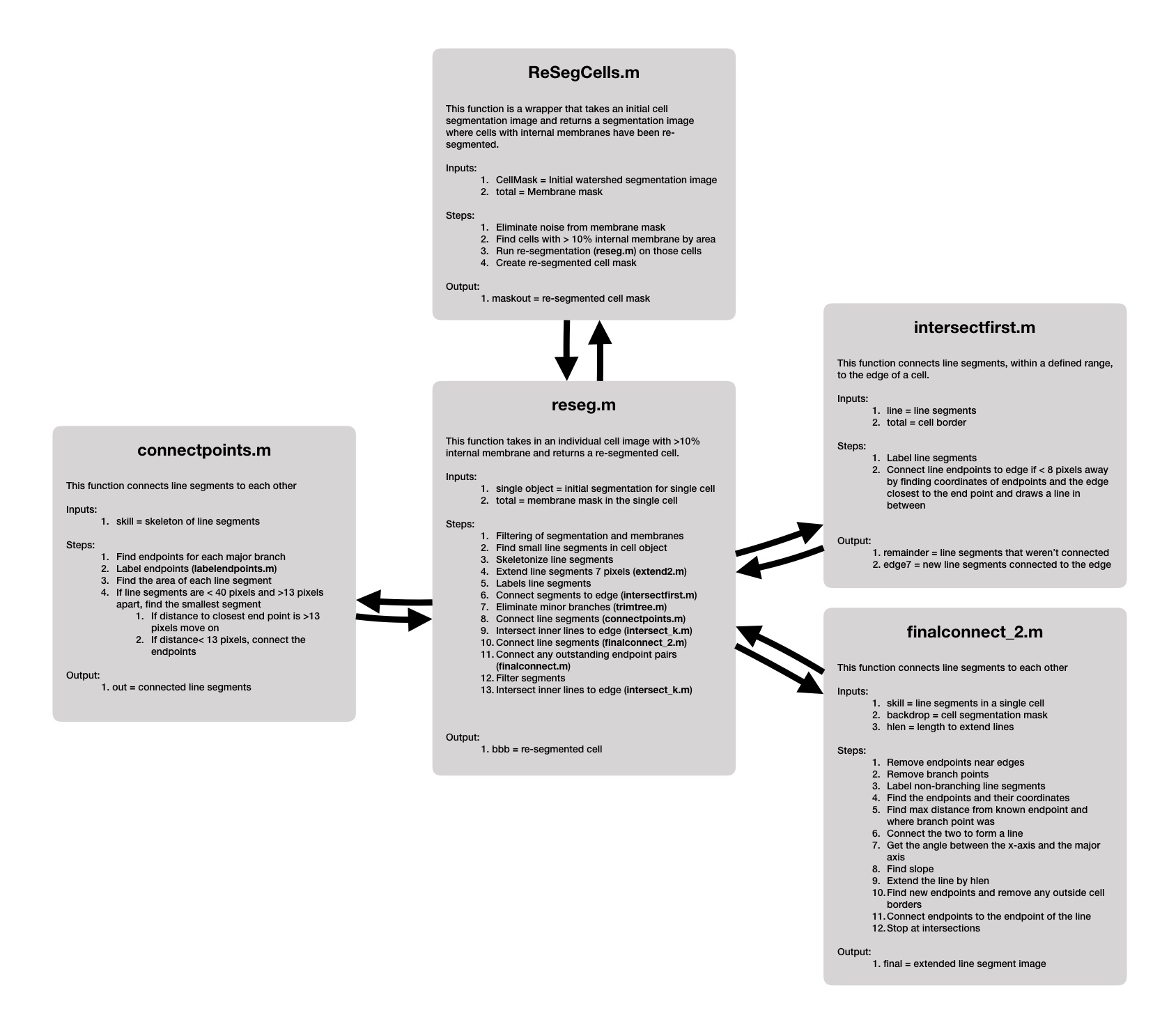{kind=link}
Figure S3 MIRIAM segmentation similarity scores for marker subsets. Rain cloud plots of DSC and JSC coefficients are shown for various subsets of markers used as input to pixel classification (red) compared to those generated using all markers (blue).
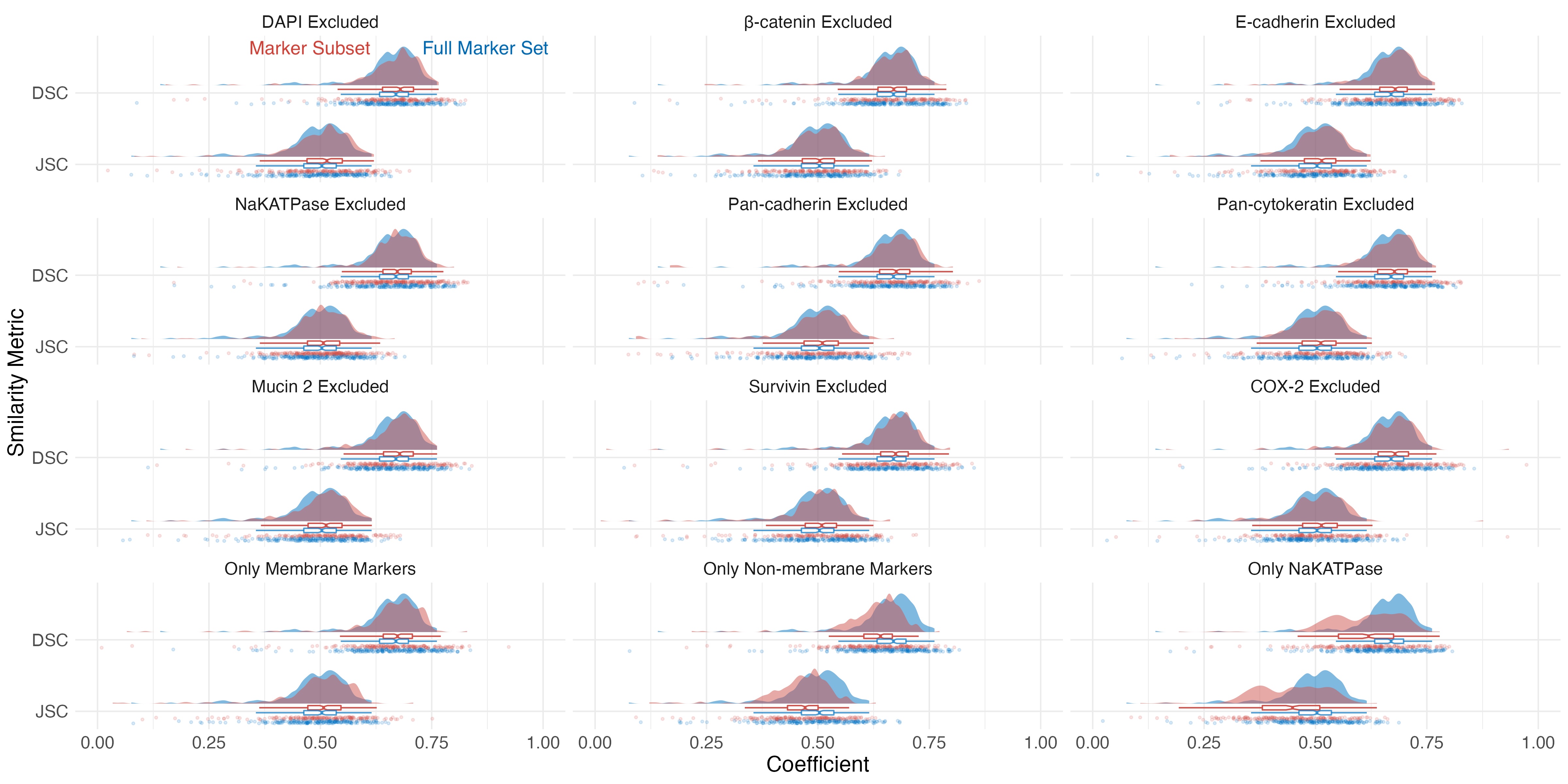{kind=link}
Figure S4 Marker intensity t‐SNE. Plots are shown for each marker used for cell segmentation and t‐SNE analysis using marker intensity only. A strong p‐ERK‐positive population can be identified in the upper left of tSNE space, while other populations are more diffuse.
{kind=link}
Figure S5 Shape t‐SNE. Plots are shown for each marker used for cell segmentation and t‐SNE analysis using only the cell shape latent vector as input. Unlike staining intensity, no identifiable populations were discerned; instead, there was a random distribution for each marker, demonstrating that shape does not reflect marker expression in this instance.
{kind=link}
Figure S6 Comparison between Matlab and Python MIRIAM Implementations. (A) Raincloud plots of Dice similarity coefficient (DSC) and Jaccard similarity coefficient (JSC) between Matlab and Python implementations are shown. (B) In a representative TMA core (scale bar: 100 μm), Matlab segmentation is shown in green, Python segmentation in magenta, and areas of overlap in white. (C) Zoom shows detail (scale bar: 50 μm).
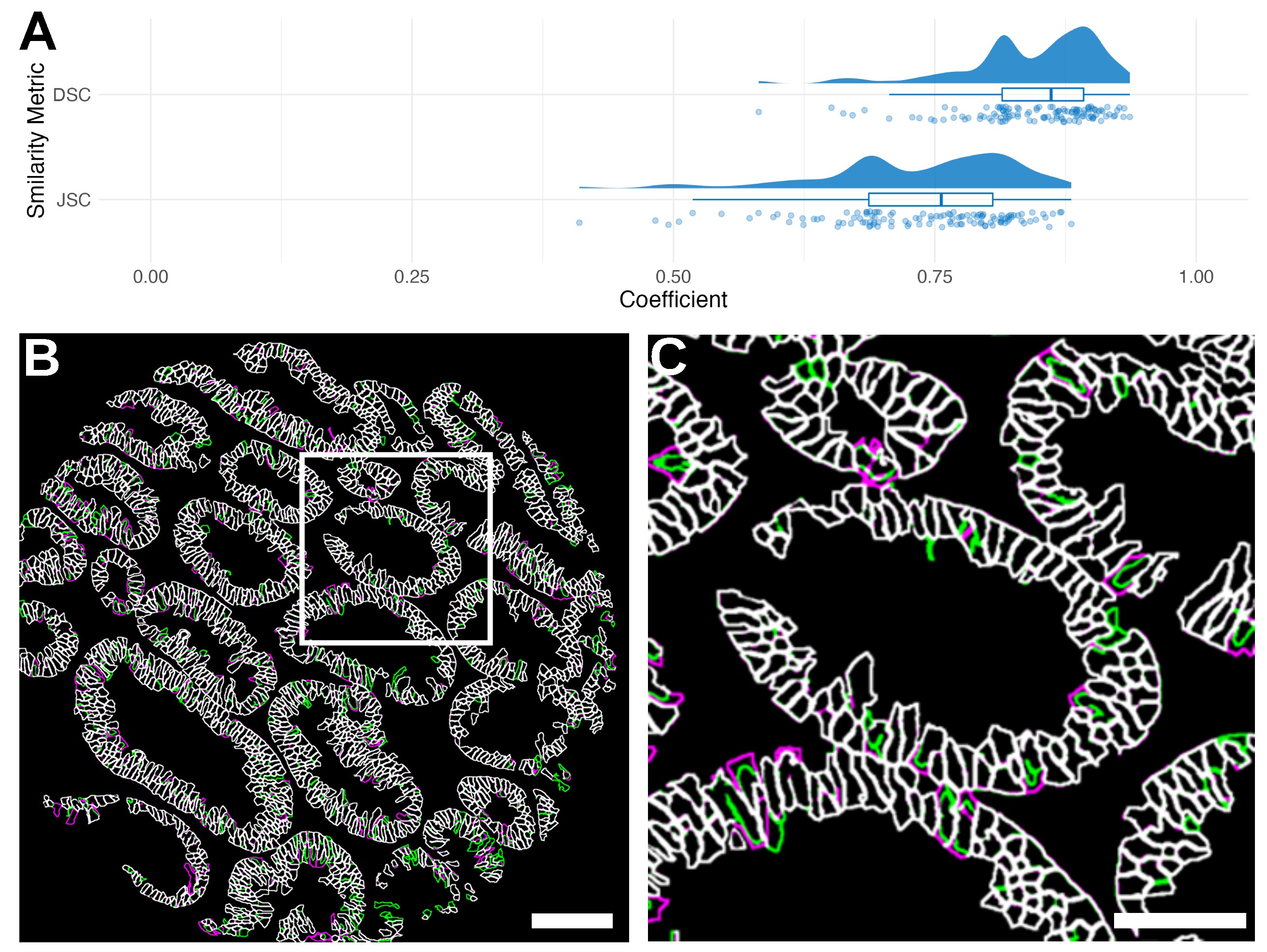{kind=link}
Figure S7 MIRIAM segmentation of MIBI breast carcinoma TMA. Representative β‐catenin (membrane) and dsDNA (nucleus) images are shown from the 36 channel image stack that over the entire TMA core (scale bar: 200 μm) and in a selected region (scale bar: 100 μm). Probability maps for epithelial (red) and stromal (green) regions, and cellular membrane (red), nucleus (green), and cytoplasm (blue) as shown along with final MIRIAM‐derived whole cell and subcellular segmentation.
{kind=link}
Table S1 Antibody reagents.
ACKNOWLEDGMENTS
Special thanks to Benoit Pimpaud for inspiring the shape characterization algorithms and Martha Shrubshole and Tim Su for the TMA used in this study. Research reported in this publication was supported by the National Institutes of Health (NIH) under awards R35CA197570, P50CA236733, and U2CCA233291 to Robert J. Coffey, R01DK103831 to Ken S. Lau, and F31DK127687 to Paige N. Vega. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
McKinley ET, Shao J, Ellis ST, Heiser CN, Roland JT, Macedonia MC, et al. MIRIAM: A machine and deep learning single‐cell segmentation and quantification pipeline for multi‐dimensional tissue images. Cytometry. 2022;101:521–528. 10.1002/cyto.a.24541
Funding information National Institutes of Health, Grant/Award Numbers: F31DK127687, P50CA236733, R01DK103831, R35CA197570, U2CCA233291
REFERENCES
- 1. Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, et al. mRNA‐Seq whole‐transcriptome analysis of a single cell. Nat Methods. 2009;6:377–82. [DOI] [PubMed] [Google Scholar]
- 2. Bradford JA, Buller G, Suter M, Ignatius M, Beechem JM. Fluorescence‐intensity multiplexing: simultaneous seven‐marker, two‐color immunophenotyping using flow cytometry. Cytometry A. 2004;61:142–52. [DOI] [PubMed] [Google Scholar]
- 3. Bendall SC, Simonds EF, Qiu P, Amir E‐AD, Krutzik PO, Finck R, et al. Single‐cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Giesen C, Wang HAO, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods. 2014;11:417–22. [DOI] [PubMed] [Google Scholar]
- 5. Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, et al. Multiplexed ion beam imaging of human breast tumors. Nat Med. 2014;20:436–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, et al. Highly multiplexed single‐cell analysis of formalin‐fixed, paraffin‐embedded cancer tissue. Proc Natl Acad Sci. 2013;110:11982–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lin J‐R, Izar B, Wang S, Yapp C, Mei S, Shah PM, et al. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t‐CyCIF and conventional optical microscopes. Elife. 2018;7:e31657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goltsev Y, Samusik N, Kennedy‐Darling J, Bhate S, Hale M, Vazquez G, et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell. 2018;174:968–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yin Z, Bise R, Chen M, Kanade T. Cell segmentation in microscopy imagery using a bag of local Bayesian classifiers. 2010 IEEE international symposium on biomedical imaging: from nano to macro. Brooklyn, NY: IEEE; 2010. p. 125–8. [Google Scholar]
- 10. Zimmer C, Labruyère E, Meas‐Yedid V, Guillén N, Olivo‐Marin J‐C. Segmentation and tracking of migrating cells in videomicroscopy with parametric active contours: a tool for cell‐based drug testing. IEEE Trans Med Imaging. 2002;21:1212–21. [DOI] [PubMed] [Google Scholar]
- 11. Al‐Kofahi Y, Zaltsman A, Graves R, Marshall W, Rusu M. A deep learning‐based algorithm for 2‐D cell segmentation in microscopy images. BMC Bioinform. 2018;19:365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schmitt O, Hasse M. Morphological multiscale decomposition of connected regions with emphasis on cell clusters. Comput Vis Image Underst. 2009;113:188–201. [Google Scholar]
- 13. McKinley ET, Sui Y, Al‐Kofahi Y, Millis BA, Tyska MJ, Roland JT, et al. Optimized multiplex immunofluorescence single‐cell analysis reveals tuft cell heterogeneity. JCI Insight. 2017;2:e93487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schüffler PJ, Schapiro D, Giesen C, Wang HAO, Bodenmiller B, Buhmann JM. Automatic single cell segmentation on highly multiplexed tissue images. Cytometry A. 2015;87:936–42. [DOI] [PubMed] [Google Scholar]
- 15. Baggett D, Nakaya M‐A, McAuliffe M, Yamaguchi TP, Lockett S. Whole cell segmentation in solid tissue sections. Cytometry A. 2005;67:137–43. [DOI] [PubMed] [Google Scholar]
- 16. Santamaria‐Pang A, Rittscher J, Gerdes M, Padfield D. Cell segmentation and classification by hierarchical supervised shape ranking. 2015 IEEE 12th international symposium on biomedical imaging (ISBI). Brooklyn, NY: IEEE; 2015. p. 1296–9. [Google Scholar]
- 17. Baars MJD, Sinha N, Amini M, Pieterman‐Bos A, van Dam S, Ganpat MMP, et al. MATISSE: a method for improved single cell segmentation in imaging mass cytometry. BMC Biol. 2021;19:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Caicedo JC, Goodman A, Karhohs KW, Cimini BA, Ackerman J, Haghighi M, et al. Nucleus segmentation across imaging experiments: the 2018 data science bowl. Nat Methods. 2019;16:1247–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moen E, Bannon D, Kudo T, Graf W, Covert M, Van Valen D. Deep learning for cellular image analysis. Nat Methods. 2019;16:1233–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Greenwald NF, Miller G, Moen E, Kong A, Kagel A, Dougherty T, et al. Whole‐cell segmentation of tissue images with human‐level performance using large‐scale data annotation and deep learning. Nat Biotechnol. 2021. 10.1038/s41587-021-01094-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shrubsole MJ, Wu H, Ness RM, Shyr Y, Smalley WE, Zheng W. Alcohol drinking, cigarette smoking, and risk of colorectal adenomatous and hyperplastic polyps. Am J Epidemiol. 2008;167:1050–8. [DOI] [PubMed] [Google Scholar]
- 22. Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, et al. A structured tumor‐immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell. 2018;174:1373–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sommer C, Straehle C, Köthe U, Hamprecht FA. Ilastik: Interactive learning and segmentation toolkit. 2011 IEEE international symposium on biomedical imaging: from nano to macro. Chicago, IL: IEEE; 2011. p. 230–3. [Google Scholar]
- 24. Ruan X, Murphy RF. Evaluation of methods for generative modeling of cell and nuclear shape. Bioinformatics. 2019;35:2475–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pimpaud B. After raw stats: exploring possession styles with data embeddings. Medium 2019. Available at: https://towardsdatascience.com/after-raw-stats-exploring-possession-styles-with-data-embeddings-d3ebef718abf. Accessed October 1, 2019.
- 26. Kaliman S, Jayachandran C, Rehfeldt F, Smith A‐S. Limits of applicability of the Voronoi tessellation determined by centers of cell nuclei to epithelium morphology. Front Physiol. 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zou KH, Warfield SK, Bharatha A, Tempany CMC, Kaus MR, Haker SJ, et al. Statistical validation of image segmentation quality based on a spatial overlap index: scientific reports. Acad Radiol. 2004;11:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kobayakawa M, Kinjo S, Hoshi M, Ohmori T, Yamamoto A. Fast computation of similarity based on Jaccard coefficient for composition‐based image retrieval. Advances in multimedia information processing. PCM. 2009;2009:949–55. [Google Scholar]
- 29. Krijthe JH. Rtsne: T‐distributed stochastic neighbor embedding using Barnes‐Hut implementation. R package version 0. 13, https://github.com/jkrijthe/Rtsne. 2015.
- 30. Wickham H. ggplot2: Elegant Graphics for Data Analysis. Springer. 2016. https://ggplot2.tidyverse.org [Google Scholar]
- 31. Maaten L v d, Hinton G. Visualizing Data using t‐SNE. J Mach Learn Res. 2008;9:2579–605. [Google Scholar]
- 32. Herring CA, Banerjee A, McKinley ET, Simmons AJ, Ping J, Roland JT, et al. Unsupervised trajectory analysis of single‐cell RNA‐Seq and imaging data reveals alternative tuft cell origins in the gut. Cell Syst. 2018;6:37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van Ineveld RL, Kleinnijenhuis M, Alieva M, de Blank S, Barrera Roman M, van Vliet EJ, et al. Revealing the spatio‐phenotypic patterning of cells in healthy and tumor tissues with mLSR‐3D and STAPL‐3D. Nat Biotechnol. 2021;39(10):1239–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schapiro D, Sokolov A, Yapp C, Muhlich JL, Hess J, Lin J‐R, et al. MCMICRO: a scalable, modular image‐processing pipeline for multiplexed tissue imaging. Nat Methods. 2021. 10.1038/s41592-021-01308-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Möller B, Poeschl Y, Plötner R, Bürstenbinder K. PaCeQuant: a tool for high‐throughput quantification of pavement cell shape characteristics. Plant Physiol. 2017;175:998–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen B, Scurrah CR, ET MK, Simmons AJ, Ramirez‐Solano MA, Zhu X, et al. Differential pre‐malignant programs and microenvironment chart distinct paths to malignancy in human colorectal polyps. Cell. 2021;184(26):6262–6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Flowchart of the MIRIAM Pipeline. The major steps in the MIRIAM segmentation pipeline are shown. Key steps are shaded and numbered and shown pictorially in Figure 1.
Figure S2 Cell re‐segmentation algorithm. Major steps in the cell re‐segmentation algorithm are shown with pseudocode.
Figure S3 MIRIAM segmentation similarity scores for marker subsets. Rain cloud plots of DSC and JSC coefficients are shown for various subsets of markers used as input to pixel classification (red) compared to those generated using all markers (blue).
Figure S4 Marker intensity t‐SNE. Plots are shown for each marker used for cell segmentation and t‐SNE analysis using marker intensity only. A strong p‐ERK‐positive population can be identified in the upper left of tSNE space, while other populations are more diffuse.
Figure S5 Shape t‐SNE. Plots are shown for each marker used for cell segmentation and t‐SNE analysis using only the cell shape latent vector as input. Unlike staining intensity, no identifiable populations were discerned; instead, there was a random distribution for each marker, demonstrating that shape does not reflect marker expression in this instance.
Figure S6 Comparison between Matlab and Python MIRIAM Implementations. (A) Raincloud plots of Dice similarity coefficient (DSC) and Jaccard similarity coefficient (JSC) between Matlab and Python implementations are shown. (B) In a representative TMA core (scale bar: 100 μm), Matlab segmentation is shown in green, Python segmentation in magenta, and areas of overlap in white. (C) Zoom shows detail (scale bar: 50 μm).
Figure S7 MIRIAM segmentation of MIBI breast carcinoma TMA. Representative β‐catenin (membrane) and dsDNA (nucleus) images are shown from the 36 channel image stack that over the entire TMA core (scale bar: 200 μm) and in a selected region (scale bar: 100 μm). Probability maps for epithelial (red) and stromal (green) regions, and cellular membrane (red), nucleus (green), and cytoplasm (blue) as shown along with final MIRIAM‐derived whole cell and subcellular segmentation.
Table S1 Antibody reagents.