

Abstract
TP53 mutations in acute myeloid leukemia (AML) are associated with resistance to standard treatments and dismal outcomes. The incidence and prognostic impact of the emergence of newly detectable TP53 mutations over the course of AML therapy has not been well described. We retrospectively analyzed 200 patients with newly diagnosed TP53 wild type AML who relapsed after or were refractory to frontline therapy. Twenty-nine patients (15%) developed a newly detectable TP53 mutation in the context of relapsed/refractory disease. The median variant allelic frequency (VAF) was 15% (range, 1.1% - 95.6%). TP53 mutations were more common after intensive therapy versus lower-intensity therapy (23% versus 10%, respectively; P=0.02) and in patients who had undergone hematopoietic stem cell transplant versus those who had not (36% versus 12%, respectively; P=0.005). Lower TP53 VAF was associated with an increased likelihood of complete remission (CR) or CR with incomplete hematologic recovery (CRi) compared to higher TP53 VAF (CR/CRi rate of 41% for VAF <20% versus 13% for VAF ≥ 20%, respectively). The median overall survival (OS) after acquisition of TP53 mutation was 4.6 months, with a 1-year OS rate of 19%. TP53 VAF at relapse was significantly associated with OS; the median OS of patients with TP53 VAF ≥20% was 3.5 months versus 6.1 months for those with TP53 VAF <20% (P<0.05). In summary, new TP53 mutations may be acquired throughout the course of AML therapy. Sequential monitoring for TP53 mutations is likely to be increasingly relevant in the era of emerging TP53-targeting therapies for AML.
Keywords: TP53 mutated AML, emergence, treatment, relapsed/refractory
Introduction
Although most patients with newly diagnosed acute myeloid leukemia (AML) achieve complete remission after induction chemotherapy, approximately 50% relapse, after which outcomes are generally poor1. These relapses are often driven by expansion of a previously existing mutations or emergence of new ones2. Several of these mutations, including FLT3, IDH1 and IDH2, are targetable with commercially available small molecular inhibitors, and others may be targetable with new, emerging therapeutic approaches3. Accurate detection of these mutations therefore plays an integral role in the current management of AML, and as the treatment landscape of AML expands, the number of potentially actionable mutations is likely to increase.
Mutations in the tumor protein 53 (TP53) gene are detectable in approximately half of human cancers and in approximately 20% of newly diagnosed AML4–7. These mutations often result in p53 protein accumulation are frequently associated with therapy-related AML and complex karyotype8–10. TP53 mutations confer resistance to currently available therapies, and particularly to intensive chemotherapy11. Consequently, the outcomes of TP53-mutated AML are poor, with high rates of relapse and dismal outcome; it is therefore important to identify these mutations from a prognostic standpoint, as their presence warrants strong consideration of hematopoietic stem cell transplant (HSCT) in first remission11–14. New novel therapies currently in clinical trials are poised to become important strategies for TP53-mutated AML. These agents include eprenetapopt (APR-246) and magrolimab, both of which have shown promising data in the treatment of TP53-mutated myeloid malignancies15,16. Alternatively, other emerging agents such as MDM2 inhibitors work through TP53-dependent mechanisms and require intact p53 function in order to be effective4. In this context, detection of TP53 mutations is important in order to select alternative strategies.
Addition of these agents to our therapeutic armamentarium now offers new avenues to treat TP53-mutated AML. However, there are limited data regarding the emergence of newly detectable TP53 mutations over the course of AML therapy and whether repeat testing for this mutation might have clinical implications in the emerging therapeutic landscape of AML. Hence, we sought to evaluate the frequency of TP53 mutations emerging over the course of therapy in patients with TP53 wild type (WT) AML who relapsed or were refractory to initial therapy.
Methods
Patients and methods
We retrospectively reviewed the electronic medical records of 1293 patients with newly diagnosed TP53 WT AML between December 2012 and March 2020 who were treated with frontline therapy at our institution. AML was diagnosed per the World Health Organization criteria17. Patients with TP53 mutation at baseline, core binding factor (CBF) AML, and those who did not have mutation data at baseline were excluded (Fig. 1). Among 924 patients with TP53 WT AML, we limited our analysis to 200 patients who were relapsed or refractory to frontline therapy and had additional mutation profiling performed at relapse or treatment failure. Refractoriness to frontline therapy was defined as lack of response to at least 1 cycle of intensive induction chemotherapy or at least 2 cycles of lower intensity therapy (unless clear evidence of disease progression after 1 cycle). Clinical, cytogenetic, and molecular data were collected through chart review. The study was approved by the institutional review board of The University of Texas MD Anderson Cancer Center.
Fig. 1 -. Patient selection flow chart.
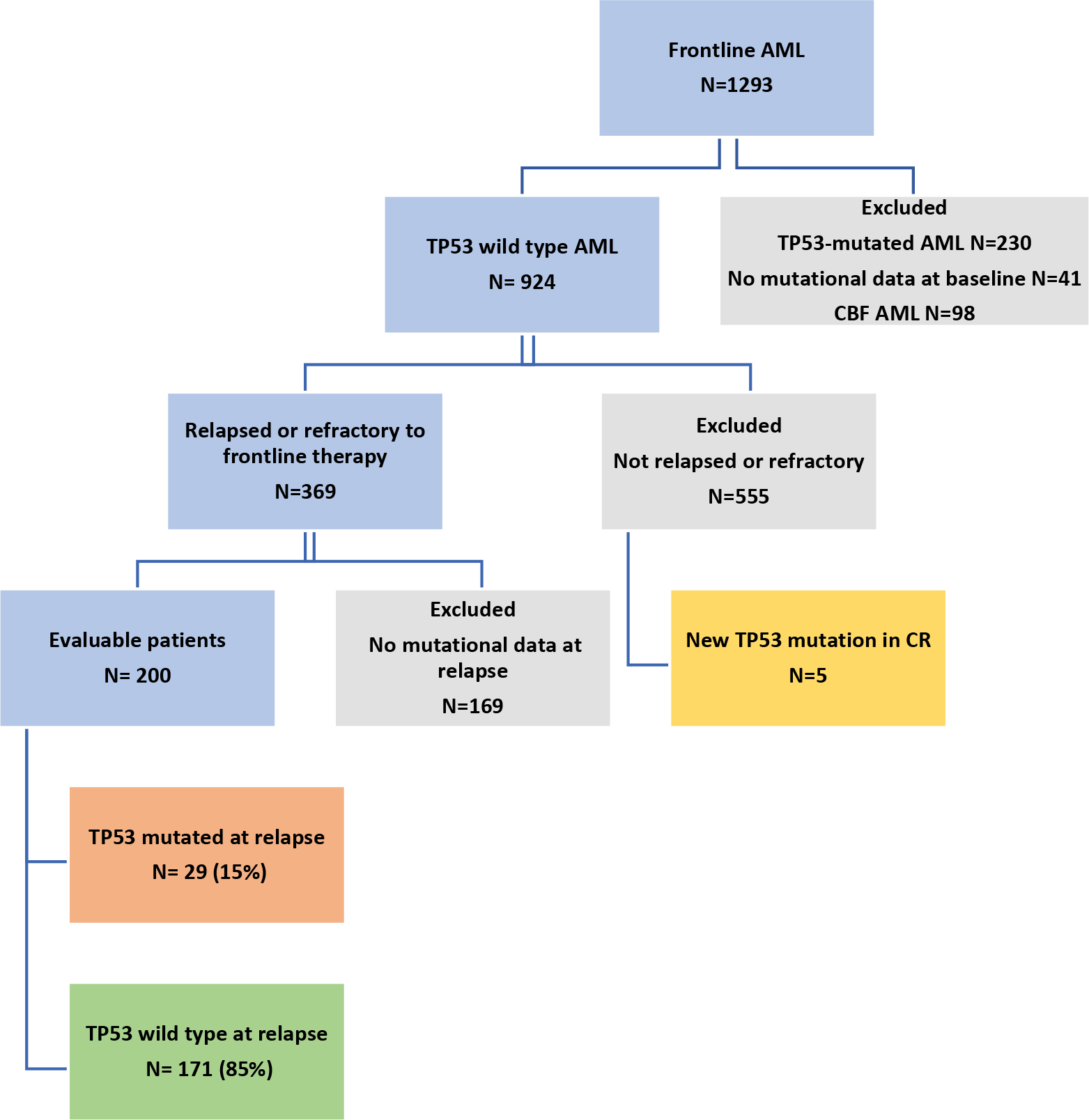
Abbreviations: AML, acute myeloid leukemia; CBF, core-binding factor
Karyotyping, target gene sequencing, and immunohistochemistry
Conventional cytogenetic analysis was conducted in the Clinical Cytogenetic Laboratory at MDACC following standard protocols. Cytogenetic results were interpreted and reported according to the International System for Human Cytogenetic nomenclature18. To identify TP53 mutations at baseline or relapse, a targeted next-generation sequencing (NGS) platform covering 28, 53, or 81 genes was performed. The analytical sensitivity of this NGS panel was established at 1–2% mutant reads in a background of WT reads. Mutations were manually reviewed to exclude any artefacts. The European Leukemia Net (ELN) 2017 risk classification was used to categorize patients into their risk group depending on their mutational status and cytogenetic abnormalities12. Immunohistochemistry (IHC) was performed on baseline and relapse samples in order to assess p53 protein expression patterns. IHC was performed on automated Leica Bond stainers (Leica Biosystems, Buffalo Gove, Illinois) using 3–4 μm sections from formalin-fixed paraffin-embedded bone marrow tissue samples using the monoclonal anti-p53 antibody clone DO-7 (Dako, Carpinteria, CA, USA) as described previously 19,20. The extent (percentage) and intensity (0, 1+, 2+, 3+) of staining was evaluated independently by two hematopathologists (J.D.K. and M.T.). Mutant p53 expression pattern (accumulation) was defined as uniform 3+ staining in at least 5% of cells8,21,22. Patients with mutant p53 expression patterns were further subdivided into low-burden mutation pattern (≤ 10% of cellularity) or high-burden mutation pattern (>10% of cellularity). The other cases were considered to have WT p53 expression pattern.
Response definitions and statistical methods
Complete remission (CR), CR with incomplete hematological recovery (CRi), and morphologic leukemia-free state (MLFS) were defined according to the ELN 2017 criteria12. Relapse was defined as recurrence of >5% blasts in the bone marrow. Patient characteristics were summarized using descriptive statistics including median (range) for continuous variables and frequency (%) for categorical variables. The fisher’s exact test and Wilcoxon rank-sum test were used to compare categorical and continuous variables, respectively. Overall survival (OS) was defined as the time from the date of first detected TP53 mutation until death due to any cause or censored at last follow-up. The Kaplan Meier method was performed to estimate OS. All statistical analysis was performed using the IBM SPSS Statistics 23.0 software.
Results
Patient characteristics
We identified 200 adult patients with non-CBF TP53 WT AML at diagnosis who relapsed after or were refractory to frontline therapy and are the subject of this analysis (Fig. 1). The median age at diagnosis was 69 years (range, 17–94 years) (Table 1). At diagnosis, 87 patients (44%) had normal karyotype, 41 (21%) had complex karyotype, 15 (8%) had −5/del5q, and 31 (16%) had −7. Eighty-five patients (43%) had ELN adverse risk AML, 94 (47%) were intermediate risk, and 21 (11%) were favorable risk. The most common mutations at baseline were SRSF2 (24%), DNMT3A (23%), IDH2 mutation (20%), and NRAS (18%). Sixty-nine patients (35%) received frontline intensive chemotherapy (defined as cytarabine and anthracycline-based induction) and 131 (66%) received lower-intensity therapy. Among those patients who received lower-intensity therapy, 108 patients (82%) received a hypomethylating agent-based regimen. Twenty-two patients (11%) had undergone prior HSCT in first remission. Eighty-five patients (43%) were refractory to frontline therapy, and 115 (58%) relapsed after initial response to frontline therapy. Among the relapsed patients, the median time to first relapse was 6 months (range 1–53 months).
Table 1.
Baseline patient characteristics of patients with TP53-wild type AML who were refractory to or relapsed after frontline therapy
| Characteristic (N=200) | Median [range] / N (%) |
|---|---|
| Age, years | 69 [17–94] |
| Therapy-related AML | 26 (13) |
| Frontline therapy | |
| Intensive chemotherapy | 69 (35) |
| Low-intensity therapy | 131 (66) |
| Response to frontline therapy | |
| Relapsed | 115 (58) |
| Refractory | 85 (43) |
| European LeukemiaNet 2017 risk | |
| Favorable | 21 (11) |
| Intermediate | 94 (47) |
| Adverse | 85 (43) |
| Cytogenetics | |
| Diploid | 87 (44) |
| 11q23 rearrangement | 7 (4) |
| −7 | 31 (16) |
| −5/−5q | 15 (8) |
| Complex | 41 (21) |
| Other abnormalities | 19 (10) |
| Mutations * | |
| SRSF2 | 1¼6 (24) |
| DNMT3A | 46 (23) |
| IDH2 | 39 (20) |
| NRAS | 36 (18) |
| ASXL1 | 35 (18) |
| RUNX1 | 33 (17) |
| FLT3-ITD | 32 (16) |
| TET2 | 30 (15) |
| NPM1 | 29 (15) |
| DDX41 | 3/23 (13) |
| IDH1 | 22 (11) |
| CEBPA | 22 (11) |
| SF3B1 | 3/31 (10) |
| PTPN11 | 14 (7) |
| U2AF1 | ¾6 (7) |
| ETV6 | 3/51 (6) |
| FLT3-D835 | 10 (5) |
| KRAS | 10 (5) |
Only mutations present in ≥5% of cases are included
TP53 mutation acquisition and association with baseline features
Overall, 29 patients (15%) developed a newly detectable TP53 mutation by NGS at any point over the course of therapy in the context of relapsed/refractory disease. Nineteen of these pts (66%) acquired a detectable mutation after the first line of therapy, 6 patients (21%) after two lines of therapy, and 4 patients (14%) after three lines of therapy. Sixteen patients (55%) with newly detectable TP53 mutation had received frontline intensive chemotherapy, and 13 patients (45%) had received a frontline lower-intensity regimen; only 2 of these patients (13%) received venetoclax plus a hypomethylating agent. Twenty-four patients (83%) acquired one TP53 mutation, among these patients; 21 patients acquired a missense mutation, and 3 patients acquired a frameshift mutation. Five patients (17%) acquired 2 TP53 mutations; 3 of these patients acquired 2 missense mutations, 1 patient acquired 2 frameshift mutations, and 1 patient acquired 1 missense and 1 nonsense mutation. The median variant allelic frequency (VAF) of the TP53 mutation at the time of first detection was 15% (range 1.1–95.6%). Five patients (17%) had a VAF >40%, 5 (17%) had a VAF of 20%–40%, 6 (21%) had a VAF of 10%–20%, and 13 (45%) had a VAF of <10%. Overall, 11 patients (38%) had a VAF <5%. The median time from diagnosis to first detection of TP53 mutation was 10 months (range 1–23 months).
We identified factors at baseline associated with increased likelihood of developing a newly detectable TP53 mutations. New TP53 mutations were more common in patients with a baseline chromosome 5 abnormality versus those without a chromosome 5 abnormality (23% versus 10%, respectively; P=0.02) and in those with a baseline IDH2 mutations versus no IDH2 mutation (28% versus 12%, respectively; P=0.02). Newly detected TP53 mutations were also more common after intensive therapy versus lower-intensity therapy (23% versus 10%, respectively; P=0.02) and in patients who had undergone HSCT in first remission versus those who had not (36% versus 12%, respectively; P=0.005). No other baseline features were associated with the development of a newly detectable TP53 mutation, including age or other cytogenetic/molecular features. Interestingly, there was no association with the development of TP53 mutation in patients with complex karyotype versus non-complex karyotype at baseline (15% versus 17%, respectively; P=0.95). Additionally, even though therapy-related AML is commonly associated with TP53 mutations, among the 26 patients with therapy-related TP53 WT AML with relapsed/refractory disease, only 1 patient (4%) developed a detectable TP53 mutation over the course of therapy.
At the time of emergence of TP53 mutation, 13 of the newly detectable TP53 mutations (45%) occurred in the context of complex cytogenetics. In 7 of the 13 cases, both the cytogenetic complexity and TP53 mutation emerged concomitantly and were not present at baseline. Among the 13 total cases where TP53 mutations occurred with complex cytogenetics, the median VAF was 20.9% (range, 1.4% - 95.6%). Eight patients (28%) had diploid cytogenetics at time of detection of TP53 mutation, with a median VAF of 3.2% (range, 1.2% - 36.5%). Among patients who developed a newly detectable TP53 mutation, the most common co-mutations at the time of TP53 detection were DDX41, DNMT3A, IDH2 and NRAS, each of which was mutated in 30%, 22%, 22%, and 18% of TP53-mutated relapses, respectively. In most cases, the co-mutations had also been present at diagnosis. However, at time of detection of TP53 mutation, 3 patients acquired a newly detectable NRAS mutation.
p53 protein accumulation by IHC and relationship with NGS
We performed IHC for p53 protein accumulation in baseline and relapse samples as an orthogonal method to assess TP53 mutation status. The relationship between TP53 VAF by NGS and p53 protein expression by IHC at baseline and relapse is shown in (Table 2). Among the 29 patients who relapsed with a newly detectable TP53 mutation by NGS, 17 patients (59%) had available bone marrow samples at baseline or at relapse for assessment of p53 protein expression by IHC, and 16 patients (55%) had paired baseline and relapse samples. Evaluation of p53 accumulation by IHC at baseline demonstrated a WT p53 expression pattern at baseline in 12 patients (71%) and a mutant p53 staining pattern in 5 patients (29%); 4 of these patients had IHC findings consistent with a low-frequency mutant TP53 clones (≤10% of marrow cellularity) and 1 patient had a mutant high-frequency clone estimated to comprise 20% of marrow cellularity. Conversely, at time of TP53 development by NGS, mutant p53 expression by IHC was identified in 14 patients (82%), 6 patients with a low-frequency clone and 8 patients with a high-frequency clone. Of note, 3 patients (18%) out of the 17 patients with a detectable TP53 mutation by NGS had WT p53 expression pattern by IHC; the TP53 VAFs in these patients were 2.7%, 5.5% and 36.5%. At the time of TP53 mutation detection by NGS, the VAF of TP53 mutations in patients with WT p53 protein expression or mutant-low p53 protein expression by IHC was numerically lower than in patients with mutant-high p53 protein expression (5.5% versus 20.4%, respectively; P=0.3). At this same time point, patients with mutant-high p53 protein expression by IHC were more likely to have complex cytogenetics than patients with WT or mutant-low p53 protein expression by IHC (75% versus 22%, respectively; P=0.05). These data suggest that p53 expression could support conclusions regarding p53 pathway altering the pathogenic impact of TP53 mutations when detected.
Table 2.
Paired assessment of patients with newly detectable TP53 mutation at time of relapse or refractory disease and of patients in remission at time of new TP53 mutation detection
| Paired assessment of patients with newly detectable TP53 mutation at baseline and at time of relapsed or refractory disease | |||||
| Patient a | p53 protein expression by IHC at baseline | Cytogenetics at baseline | p53 protein expression by IHC at relapse | Cytogenetics at relapse | TP53 VAF (%) |
| 1 | Wild-type pattern | Complex | Wild-type pattern | Complex | 5.5 |
| 2 | Mutant-high pattern | Complex | Mutant-high pattern | Complex | 95.6 |
| 3 | Mutant-low pattern | Other | Mutant-low pattern | Other | 16.7 |
| 4 | Mutant-low pattern | Other | Wild-type pattern | Diploid | 36.5 |
| 5 | Mutant-low pattern | Diploid | Mutant-high pattern | Complex | 43.3 |
| 6 | Wild-type pattern | Other | Mutant-low pattern | Other | 15.0 |
| 7 | Wild-type pattern | Diploid | Wild-type pattern | Diploid | 2.7 |
| 8 | n/a | Diploid | Mutant-high pattern | Complex | 19.1 |
| 9 | Wild-type pattern | Diploid | Mutant-high pattern | Complex | 19.9 |
| 10 | Wild-type pattern | Other | Mutant-low pattern | Diploid | 3.2 |
| 11 | Wild-type pattern | Other | Mutant-high pattern | Other | 1.1 |
| 12 | Wild-type pattern | Other | Mutant-high pattern | Other | 2.7 |
| 13 | Wild-type pattern | Other | Mutant-low pattern | Other | 9.3 |
| 14 | Wild-type pattern | Other | Mutant-low pattern | Diploid | 1.5 |
| 15 | Wild-type pattern | Diploid | Mutant-high pattern | Complex | 20.9 |
| 16 | Wild-type pattern | Other | n/a | Other | 1.1 |
| 17 | Wild-type pattern | Diploid | Mutant-low pattern | Complex | 1.4 |
| 18 | Mutant-low pattern | Complex | Mutant-high pattern | Complex | 95.2 |
| Paired assessment of patients at baseline and at time of new TP53 mutation detection while in remission | |||||
| Patient b | p53 protein expression by IHC at baseline | Cytogenetics at baseline | p53 protein expression by IHC at time of new TP53 mutation | Cytogenetics at time of new TP53 mutation | TP53 VAF (%) |
| 1 | Wild-type pattern | Diploid | Wild-type pattern | Diploid | 1.6 |
| 2 | Wild-type pattern | Diploid | Mutant-low pattern | Diploid | 5.7 |
| 3 | Wild-type pattern | Other | Wild-type pattern | Diploid | 1.0 |
| 4 | Wild-type pattern | Diploid | Mutant-low pattern | Diploid | 1.6 |
Abbreviations: IHC, immunhistochemistry; VAF, variant allelic frequency
11 patients did not have samples available for IHC at baseline or relapse/refractory disease
1 patient did not have samples available for IHC at baseline or at time of new TP53 mutation detection in remission
Treatment response and overall survival of TP53-mutated patients
After TP53 mutation acquisition, 23 patients (79%) received low intensity chemotherapy, 2 patients (7%) received high intensity chemotherapy and 4 patients (14%) did not receive any treatment. Among the 25 patients who received salvage therapy, 6 patients (24%) received a venetoclax-based salvage regimen. Seventeen patients (68%) responded to salvage therapy. The CR/CRi/MLFS rate to salvage therapy was similar regardless of TP53 VAF (71% for VAF <20% and 63% for VAF ≥ 20%). However, there was a trend towards a higher rate of CR/CRi for those with lower TP53 VAF (41% for VAF <20% and 13% for VAF ≥ 20%; P=0.15). Seven of these responders underwent subsequent HSCT. However, all transplanted patients relapsed post-transplant. With a median follow-up of 35 months, the median overall survival (OS) after acquisition of TP53 mutation was 4.6 months, with a 1-year OS rate of 19% (Fig. 2A). TP53 VAF was significantly associated with OS; the median OS of patients with TP53 VAF ≥20% was 3.5 months versus 6.1 months for those with TP53 VAF <20% (P<0.05) (Fig. 2B). At the time of TP53 detection by NGS, evaluation of p53 protein staining by IHC was correlated with OS. Patients with mutant-high p53 protein expression showed strong trend towards worse outcomes than those with mutant-low or WT p53 protein expression (3.4 months versus 7.4 months, respectively; P=0.09) (Fig. 2C).
Fig. 2. Overall survival from the time of TP53 mutation.
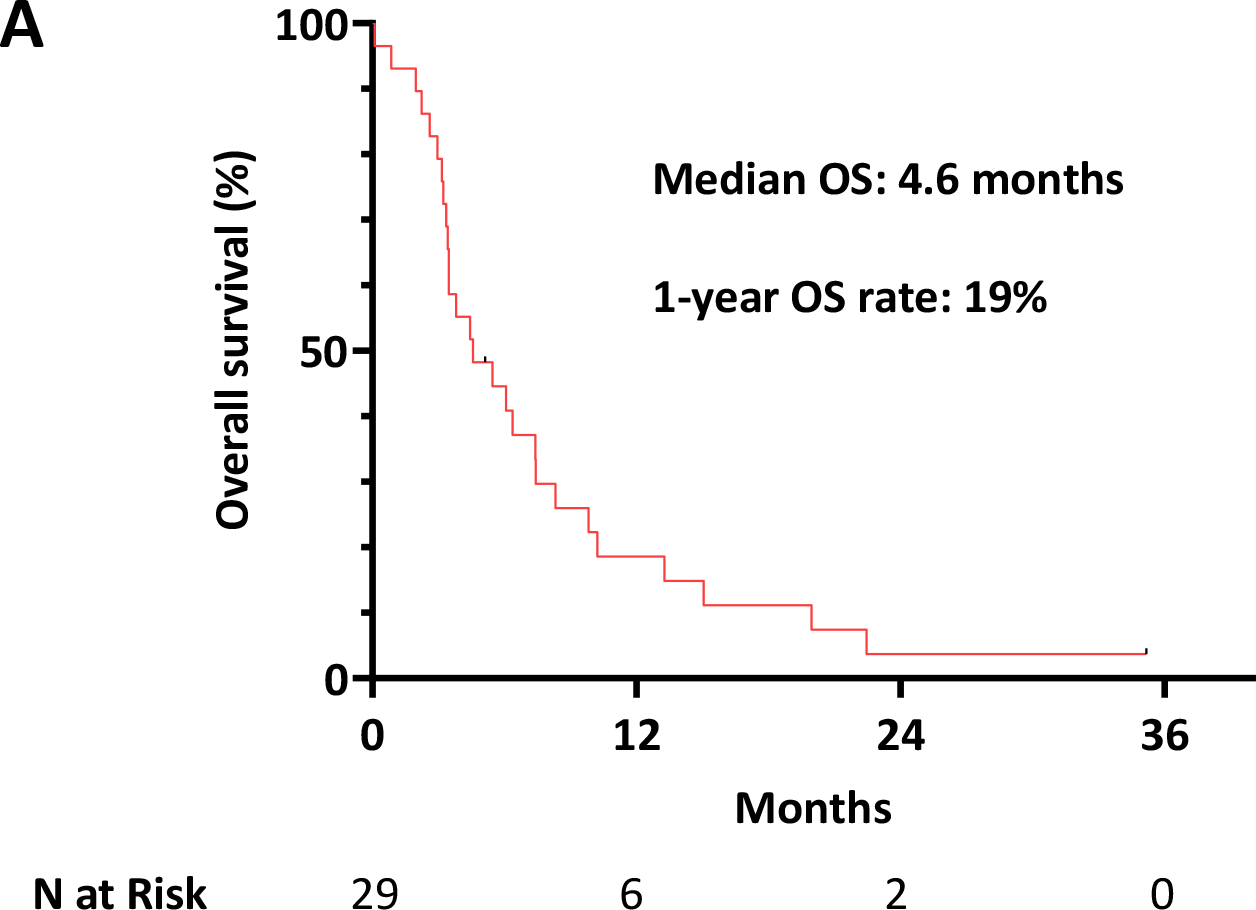
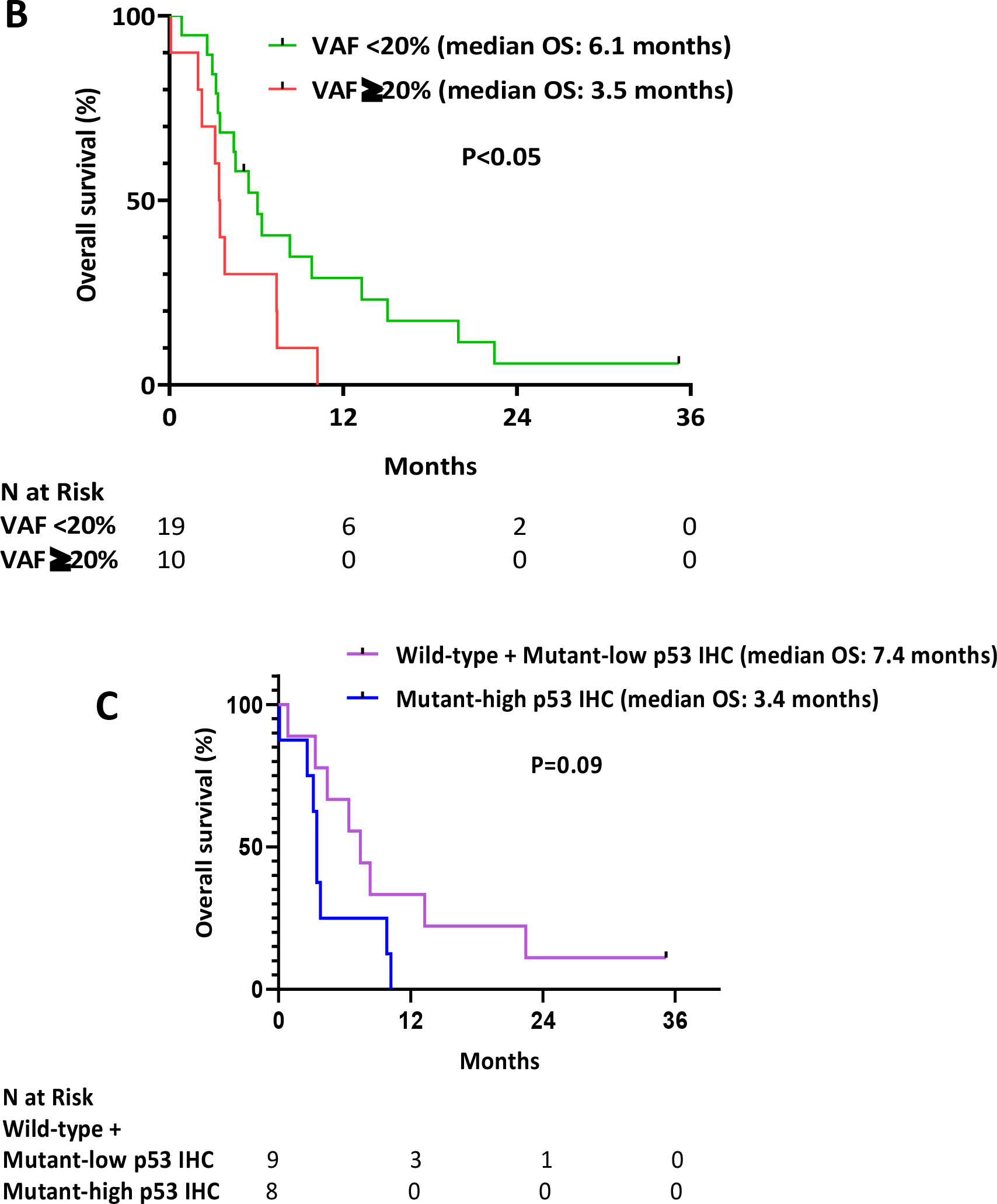
A.) Overall survival for the whole population, B.) Overall survival stratified by TP53 variant allelic frequency <20% and ≥ 20%, C.) Overall survival stratified by p53 protein expression by immunohistochemistry
TP53 acquisition during complete remission
We also identified a newly detectable TP53 mutation while in CR in 5 patients (1%) out of the 555 patients who responded to frontline therapy and were not included in the primary analysis (Fig. 1). The median VAF at the time of TP53 mutation detection was 2.5% (range 1.0% - 3.3%). Three of these patients had a detectable TP53 mutation after HSCT. A fourth patient had a TP53 mutation detected while receiving consolidation therapy; this patient subsequently underwent HSCT, after which the mutation was no longer detectable. A fifth patient developed a newly detectable TP53 mutation in the setting of flow cytometric measurable residual disease (MRD)-positive disease. This patient received nivolumab maintenance and subsequently achieved flow MRD negativity, although the TP53 mutation persisted.
Four patients had available bone marrow samples for IHC assessment at baseline and at time of TP53 mutation detection by NGS (Table 2). All of these patients demonstrated a WT p53 protein expression pattern by IHC at baseline. At the time of TP53 detection by NGS, two patients had a mutant-low p53 protein expression pattern and 2 patients had a WT p53 protein expression pattern. All 4 patients had diploid karyotype at time of TP53 detection. With a median follow-up of 27 months since detection of a TP53 mutation by NGS, none of these patients have developed hematologic relapse, 4 patients are still alive, and one patient died from GVHD complication 4 months after HSCT.
Discussion
In this single-center, retrospective study, we showed that (15%) of patients with WT TP53 AML acquired a newly detectable TP53 mutation over the course of therapy. These mutations tended to be subclonal with a median VAF of 15%. Acquisition of a newly detectable TP53 mutation was associated with receiving frontline intensive chemotherapy or HSCT, suggesting that these therapies may select for development of TP53 mutations and/or TP53 clonal expansion. Interestingly, despite the strong association of TP53 mutations with complex karyotype,10 the rate of TP53 mutations was the same between patients with complex or non-complex cytogenetics at baseline. Conversely, at relapse, 45% of cases of newly detectable TP53 mutations were associated with complex karyotype, with both the TP53 mutation and cytogenetic complexity arising concurrently in most of these cases. Taken together, these findings support the concept that TP53 mutations generally precede cytogenetic complexity rather than vice versa. We also observed significantly higher TP53 VAF in patients with complex karyotype compared to those with diploid karyotype, further supporting the association between cytogenetic complexity and TP53 mutations, which has been described predominantly in the context of newly diagnosed AML10.
Several studies has shown the inferior outcome of TP53-mutated AML11,13,23–25. Interestingly, in our study, the outcomes of patients who relapsed with TP53 mutations were better than expected, with a CR/CRi/MLFS rate of 68%. However, only 32% of patients achieved CR/CRi, and these responses were very short lived, with a median OS of only 4.6 months. The relatively high marrow response rate in these patients may reflect the subclonal nature of these mutations, as the median VAF was only 15% and patients with lower TP53 VAF had a higher rate of CR/CRi to salvage therapy. It is possible that these very small TP53-mutated clones therefore may not impart the same degree of chemotherapy resistance as larger clones. This relationship between TP53 VAF and clinical outcomes was further supported by our analysis which showed that patients with TP53 VAF ≥20% had significantly worse OS than those with VAF <20% (median OS: 3.5 months versus 6.1 months; P<0.05). This is consistent with other studies which demonstrated that patients with lower mutant TP53 burden are more likely to respond to frontline therapy and have better outcomes, particularly with conventional cytarabine-based regimens.11,26–28 Interestingly, the median OS of 6.1 months for patients with TP53 mutation with VAF <20% is approximately what would be expected in an unselected relapsed/refractory AML population, and therefore the clinical significance of these very low level subclonal mutations at relapse remains unclear. Our analysis is consistent with another study in the frontline setting that suggested that TP53 VAF may impact prognosis, and it therefore raises further questions about whether TP53 VAF should be considered in consensus risk stratification guidelines.11
We detected TP53 mutations in 5 patients who had no morphological disease and who did not subsequently relapse. Overall, this constituted approximately 1% of evaluable patients, suggesting this is a relatively uncommon phenomenon. These mutations were of very low VAFs (median 2.5%). Together, the indolent nature of these mutations and their subclonal nature suggest that these may represent clonal hematopoiesis of indeterminate potential (CHIP).29 With longer follow-up, it remains uncertain whether these persistent low-level TP53 mutations could contribute to later relapse or the development of a secondary therapy-related neoplasm.
Interestingly, when we performed IHC on the baseline samples that were TP53 WT by NGS, we found 5 patients with evidence of mutant p53 expression by IHC (4 with evidence of low-level mutant p53 protein expression and 1 with high-level mutant p53 protein expression). In contrast, at time of relapse and TP53 mutation detection by NGS, we identified 3 patients with WT p53 protein expression by IHC. While these two methods of mutant p53 detection were largely concordant, the few cases of discordant findings suggest these two methods highlight the potentially complementary roles of NGS and IHC in detecting mutant TP53, particularly in the era of new TP53-specific therapies. More comprehensive studies are needed to further define the relative roles of NGS and IHC in the detection of mutated TP53 and p53, respectively.
Novel therapies are needed to improve the duration of response and outcome in this group of patients with TP53-mutated AML3. Two such promising drugs in the treatment of TP53-mutated myeloid malignancies are eprenetapopt (APR-246) and magrolimab. Eprenetapopt has been proposed to work through restoration of transcriptional transactivation function of mutant p53, although its precise mechanism is still not fully established30. Similarly, the anti-CD47 monoclonal antibody magrolimab has been studied in combination with azacitidine in newly diagnosed AML, including a substantial subgroup of patients with TP53 mutations16. In the 29 patients with TP53-mutated AML, the median OS was 12.9 months in patients with TP53-mutated AML, which compares very favorably to the median OS of <8 months achieved with a hypomethylating agent plus venetoclax31,32. Given the 15% rate of newly detectable TP53 mutations that we observed in this study, one could consider evaluating these agents in the maintenance setting for patients with high-risk TP53 WT AML. Such an approach might be especially important for patients treated with chemotherapy and/or HSCT, since these are the main factors that determined TP53-mutated relapse in our study. Conversely, the use MDM2 inhibitors should be avoided in patients with TP53-mutated disease, as these agents require the presence of functional p53 proteins to be effective3. The development of these new agents that may have TP53-specific or TP53-preferential activity highlights the importance of repeat molecular profiling at time of relapse in order to select optional salvage therapies.
As a retrospective study, this analysis has certain limitations. In our study, we excluded patients with CBF AML because these patients have a distinctly superior OS even in the relapsed/refractory setting, which may have skewed the findings of our post-relapse analysis; a future analysis evaluating these patients and their rates of TP53 mutation acquisition may be informative. Our analysis specifically focused on the detection of TP53 mutations by NGS over the course of therapy, and therefore we did not assess the development of new TP53 deletions, which might also have prognostic or therapeutic importance. Repeated molecular testing over the course of the disease also was not performed regularly and was conducted only in a subset of patients. This could have caused an underestimation of the frequency of newly emergent TP53 mutations and may have limited our assessment of the exact time of the development of a new mutation. The sensitivity of our TP53 NGS assay was 1–2%; however most newly detectable TP53 mutations were of very low VAF (for example, 38% had VAF <5%). Therefore, we cannot definitively determine whether these mutations were truly newly developed mutations or expansion of an already existing subclone at relapse. Higher sensitivity panels or single-cell sequencing could provide more accurate and comprehensive information in a subsequent analysis.
In conclusion, this study demonstrated that 15% of patients with TP53 WT AML can acquire one or more newly detectable TP53 mutation(s) over the course of their treatment, particularly after intensive chemotherapy and/or HSCT. These newly detectable TP53 mutations were associated with a complex karyotype in 45% of cases, with both the TP53 mutation and cytogenetic complexity arising concurrently in a majority of these cases. Post-relapse OS was very poor if the TP53 mutation was present at a VAF >20%, although the outcomes of patients with lower TP53 VAF appear similar to previous reports of unselected patients in the relapsed/refractory setting, and thus the impact of these small subclonal mutations remains uncertain. Overall, our findings suggest that sequential monitoring for new, emergent TP53 mutations over the course of AML therapy might have clinical utility. Such monitoring may be particularly relevant in the era of novels therapies with the potential to target TP53-mutated myeloid malignancies.
Funding Statement:
Supported by an MD Anderson Cancer Center Support Grant (CA016672) and SPORE. N.J.S. is supported by the K12 Paul Calabresi Clinical Oncology Scholar Award and the American Society of Hematology Junior Faculty Scholar Award in Clinical Research.
Footnotes
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Institutional review board approval: Obtained
Data Availability Statement:
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- 1.Walter RB, Othus M, Burnett AK, et al. Resistance prediction in AML: analysis of 4601 patients from MRC/NCRI, HOVON/SAKK, SWOG and MD Anderson Cancer Center. Leukemia. 2015;29(2):312–320. doi: 10.1038/leu.2014.242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506–510. doi: 10.1038/nature10738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Short NJ, Konopleva M, Kadia TM, et al. Advances in the treatment of acute myeloid leukemia: New drugs and new challenges. Cancer Discov. 2020;10(4):506–525. doi: 10.1158/2159-8290.CD-19-1011 [DOI] [PubMed] [Google Scholar]
- 4.Klein C, Vassilev LT. Targeting the p53-MDM2 interaction to treat cancer. Br J Cancer. Published online 2004. doi: 10.1038/sj.bjc.6602164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. Published online 2018. doi: 10.1038/nrc.2017.109 [DOI] [PubMed] [Google Scholar]
- 6.Prokocimer M, Molchadsky A, Rotter V. Dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: Projections on diagnostic workup and therapy. Blood. Published online 2017. doi: 10.1182/blood-2017-02-763086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuykendall A, Duployez N, Boissel N, Lancet JE, Welch JS. Acute Myeloid Leukemia: The Good, the Bad, and the Ugly. Am Soc Clin Oncol Educ B. 2018;38(38):555–573. doi: 10.1200/edbk_199519 [DOI] [PubMed] [Google Scholar]
- 8.Fernandez-Pol S, Ma L, Ohgami RS, Arber DA. Immunohistochemistry for p53 is a useful tool to identify cases of acute myeloid leukemia with myelodysplasia-related changes that are TP53 mutated, have complex karyotype, and have poor prognosis. Mod Pathol. 2017;30(3):382–392. doi: 10.1038/modpathol.2016.206 [DOI] [PubMed] [Google Scholar]
- 9.Cole AJ, Dwight T, Gill AJ, et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci Rep. 2016;6. doi: 10.1038/srep26191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kadia TM, Jain P, Ravandi F, et al. TP53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer. 2016;122(22):3484–3491. doi: 10.1002/cncr.30203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Short NJ, Montalban-Bravo G, Hwang H, et al. Prognostic and therapeutic impacts of mutant TP53 variant allelic frequency in newly diagnosed acute myeloid leukemia. Blood Adv. 2020;4(22):5681–5689. doi: 10.1182/bloodadvances.2020003120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447. doi: 10.1182/blood-2016-08-733196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kadia TM, Jain P, Ravandi F, et al. TP53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer. Published online 2016. doi: 10.1002/cncr.30203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mrózek K, Eisfeld AK, Kohlschmidt J, et al. Complex karyotype in de novo acute myeloid leukemia: typical and atypical subtypes differ molecularly and clinically. Leukemia. 2019;33(7):1620–1634. doi: 10.1038/s41375-019-0390-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sallman DA, DeZern AE, Garcia-Manero G, et al. Eprenetapopt (APR-246) and Azacitidine in TP53 -Mutant Myelodysplastic Syndromes. J Clin Oncol. Published online January 15, 2021:JCO.20.02341. doi: 10.1200/JCO.20.02341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sallman D The First-in-Class Anti-CD47 Antibody Magrolimab Combined with Azacitidine Is Well-Tolerated and Effective in AML Patients: Phase 1b Results. Published online December 6, 2020. [Google Scholar]
- 17.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi: 10.1182/blood-2016-03-643544 [DOI] [PubMed] [Google Scholar]
- 18.Willatt L, Morgan SM. Shaffer LG, Slovak ML, Campbell LJ (2009): ISCN 2009 an international system for human cytogenetic nomenclature. Hum Genet. Published online 2009. doi: 10.1007/s00439-009-0726-6 [DOI] [Google Scholar]
- 19.Khoury JD, Wang WL, Prieto VG, et al. Validation of immunohistochemical assays for integral biomarkers in the NCI-MATCH EAY131 clinical trial. Clin Cancer Res. 2018;24(3):521–531. doi: 10.1158/1078-0432.CCR-17-1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sukswai N, Khoury JD. Immunohistochemistry Innovations for Diagnosis and Tissue-Based Biomarker Detection. Curr Hematol Malig Rep. 2019;14(5):368–375. doi: 10.1007/s11899-019-00533-9 [DOI] [PubMed] [Google Scholar]
- 21.Loghavi S, Al-Ibraheemi A, Zuo Z, et al. TP53 overexpression is an independent adverse prognostic factor in de novo myelodysplastic syndromes with fibrosis. Br J Haematol. 2015;171(1):91–99. doi: 10.1111/bjh.13529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Assi R, Gur HD, Loghavi S, et al. P53 protein overexpression in de novo acute myeloid leukemia patients with normal diploid karyotype correlates with FLT3 internal tandem duplication and worse relapse-free survival. Am J Hematol. 2018;93(11):1376–1383. doi: 10.1002/ajh.25255 [DOI] [PubMed] [Google Scholar]
- 23.Haferlach C, Dicker F, Herholz H, Schnittger S, Kern W, Haferlach T. Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia. 2008;22(8):1539–1541. doi: 10.1038/leu.2008.143 [DOI] [PubMed] [Google Scholar]
- 24.Seifert H, Mohr B, Thiede C, et al. The prognostic impact of 17p (p53) deletion in 2272 adults with acute myeloid leukemia. Leukemia. 2009;23(4):656–663. doi: 10.1038/leu.2008.375 [DOI] [PubMed] [Google Scholar]
- 25.Bowen D, Groves MJ, Burnett AK, et al. TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia. 2009;23(1):203–206. doi: 10.1038/leu.2008.173 [DOI] [PubMed] [Google Scholar]
- 26.Sabapathy K, Lane DP. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol. 2018;15(1):13–30. doi: 10.1038/nrclinonc.2017.151 [DOI] [PubMed] [Google Scholar]
- 27.Montalban-Bravo G, Kanagal-Shamanna R, Benton CB, et al. Genomic context and TP53 allele frequency define clinical outcomes in TP53-mutated myelodysplastic syndromes. Blood Adv. 2020;4(3):482–495. doi: 10.1182/bloodadvances.2019001101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Welch JS, Petti AA, Miller CA, et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N Engl J Med. 2016;375(21):2023–2036. doi: 10.1056/nejmoa1605949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen S, Wang Q, Yu H, et al. Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway. Nat Commun. 2019;10(1):1–14. doi: 10.1038/s41467-019-13542-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bykov VJN, Zhang Q, Zhang M, Ceder S, Abrahmsen L, Wiman KG. Targeting of mutant P53 and the cellular redox balance by APR-246 as a strategy for efficient cancer therapy. Front Oncol. 2016;6(FEB). doi: 10.3389/fonc.2016.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7–17. doi: 10.1182/blood-2018-08-868752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shoukier M, Konopleva M, Dinardo CD, et al. Activity of venetoclax-based therapy in TP53 -mutated acute myeloid leukemia. . J Clin Oncol. Published online 2019. doi: 10.1200/jco.2019.37.15_suppl.7034 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.