

Abstract
Background:
Ataluren was developed for potential treatment of nonsense-mutation cystic fibrosis (CF). A previous phase 3 ataluren study failed to meet its primary efficacy endpoint, but post-hoc analyses suggested that aminoglycosides may have interfered with ataluren’s action. Thus, this subsequent trial (NCT02139306) was designed to assess the efficacy and safety of ataluren in patients with nonsense-mutation CF not receiving aminoglycosides.
Methods:
Eligible subjects with nonsense-mutation CF (aged ≥6 years; percent predicted (pp) FEV1 ≥40 and ≤90) from 75 sites in 16 countries were randomly assigned in double-blinded fashion to receive oral ataluren or matching placebo thrice daily for 48 weeks. The primary endpoint was absolute change in average ppFEV1 from baseline to the average of Weeks 40 and 48.
Findings:
279 subjects were enrolled; 138 subjects in the ataluren arm and 136 in the placebo arm were evaluable for efficacy. Absolute ppFEV1 change from baseline did not differ significantly between the ataluren and placebo groups at Week 40 (−0.8 vs −1.8) or Week 48 (−1.7 vs −2.4). Average ppFEV1 treatment difference from baseline to Weeks 40 and 48 was 0.6 (95% CI −1.3, 2.5; p = 0.54). Pulmonary exacerbation rate per 48 weeks was not significantly different (ataluren 0.95 vs placebo 1.13; rate ratio p = 0.40). Safety was similar between groups. No life-threatening adverse events or deaths were reported.
Interpretation:
Neither ppFEV1 change nor pulmonary exacerbation rate over 48 weeks were statistically different between ataluren and placebo groups. Development of a nonsense-mutation CF therapy remains elusive.
Keywords: Cystic fibrosis, Ataluren, Clinical trial
1. Introduction
More than 2000 different mutations of the cystic fibrosis transmembrane conductance regulator (CFTR) gene have been identified within the global CF population, of which ~8% are nonsense mutations resulting in premature termination of CFTR protein synthesis [1] occurring in ~10% of CF cases worldwide [2,3]. These CFTR nonsense mutations are categorized as ‘severe’, in that little to no protein product is produced by the ribosome, and patients carrying two of these types of mutations have very poor prognoses [4–8]. Drug developers have made remarkable progress in developing small molecule therapies that increase mutant CFTR protein function in persons with CF, but these advances have largely been limited to individuals with mutations resulting in complete mutant CFTR proteins that are improperly processed and/or catalytically diminished. Pharmacologic approaches to increasing CFTR protein activity in individuals with premature termination of CFTR protein synthesis from mRNA must necessarily target nucleic acids, either by changing the coding sequence of the CFTR gene or its mRNA or by modulating the translation of mRNA by the ribosome to ‘read-through’ the premature termination codon.
Ataluren (PTC124) is an oral agent that has been shown to allow ribosomes to read through premature termination codons [9–12] and produce full-length, functional CFTR protein [12,13]. A randomized, double-blind, placebo-controlled 48-week multinational trial of ataluren in 238 persons with CF and at least one nonsense CFTR mutation (NCT00803205) showed no statistically significant difference in change from baseline in percent predicted forced expiratory volume in 1 s (ppFEV1) or rate of pulmonary exacerbations between treatment arms [14]. However, a post hoc subgroup analysis of study data suggested that subjects not receiving chronic inhaled tobramycin as a concomitant medication experienced ataluren-associated benefits, including a 5.7% difference in ppFEV1 change from baseline and 40% fewer pulmonary exacerbations [14]. Affinity of aminoglycosides such as tobramycin for both the prokaryotic [15] and eukaryotic [16] ribosome supports a hypothesis that the activity of ataluren, which binds to the eukaryotic ribosome to facilitate stop codon readthrough [17], may be inhibited by intracellular tobramycin. This Phase 3 clinical trial (NCT02139306, the Ataluren Confirmatory Trial in Cystic Fibrosis; ACT CF) was designed to evaluate the efficacy and safety of ataluren in persons with CF and nonsense CFTR mutations not receiving concomitant inhaled aminoglycosides.
2. Methods
2.1. Study oversight
The protocol was reviewed and approved by the ethics committee or institutional review board of each participating institution. Written informed consent was obtained from patients or their custodians prior to patient screening. External oversight of the study was provided by a Study Steering Committee and an independent Data Safety Monitoring Committee.
2.2. Study design and participants
This randomized, double-blind, placebo-controlled, phase 3 trial was performed between August 2014 and November 2016 at 75 sites in 16 countries in North America, South America, Europe, and Israel. Included subjects were ≥6 years old with a body weight ≥16 kg, a sweat chloride >60 mEq/L, and documentation of the presence of a nonsense mutation in at least 1 allele of the CFTR gene. They also had the ability to perform a valid, reproducible spirometry tests, a screening ppFEV1 ≥ 40 and ≤ 90, confirmed screening laboratory values within pre-specified central laboratory ranges, and a resting oxygen saturation by pulse oximetry of ≥92% on room air. Sexually active subjects had to be willing to abstain from sexual intercourse or employ a barrier or medical method of contraception during the study and 60-day follow-up period.
Major exclusion criteria were known hypersensitivity to any study drug ingredients or excipients, previous participation in the ataluren Phase 3 trial PTC124-GD-009-CF, changes in a chronic treatment/prophylaxis regimen for CF or for CF-related conditions within 4 weeks prior to screening or any change in acute therapy between screening and randomization, chronic use of inhaled or systemic tobramycin within 4 weeks prior to screening, exposure to another investigational drug within 4 weeks prior to screening, evidence of pulmonary exacerbation or acute upper or lower respiratory tract infection (including viral illnesses) within 3 weeks prior to screening or between screening and randomization, treatment with intravenous (IV) antibiotics within 3 weeks prior to screening, ongoing immunosuppressive therapy (other than corticosteroids), ongoing warfarin, phenytoin, or tolbutamide therapy, history of solid organ or hematological transplantation, major complications of lung disease (including massive hemoptysis, pneumothorax, or pleural effusion) within 8 weeks prior to screening, known portal hypertension, positive hepatitis B surface antigen, hepatitis C antibody test or human immunodeficiency virus (HIV) test, pregnancy or breast-feeding, smoking or a smoking history of ≥10 pack-years, and prior or ongoing medical conditions, medical history, physical findings, ECG findings, or laboratory abnormality that, in the investigator’s opinion, could have adversely affected the safety of the patient, made it unlikely that the course of treatment or follow up would be completed, or could have impaired the assessment of study results.
2.3. Randomization and masking
Eligible subjects were randomized in a 1:1 ratio to ataluren or placebo by means of interactive response technology, using a block size of 4 within the cells created by the interaction of the 3 stratification factors (current use of a chronic regimen of inhaled antibiotic [yes vs no], baseline age [<18 years versus ≥18 years], and baseline ppFEV1 [< 65 vs ≥65]). Subsequently enrolled family members (e.g., siblings) were assigned to the same treatment group as the first family member enrolled to minimize unblinding potential and to reduce the chance of inadvertent dosing errors. The random allocation sequence was generated by the contract research organization. Patients, medical and ancillary staff, the study investigators, and the sponsor were masked to treatment assignment. Only designated personnel at the contract research organization had access to treatment assignments.
2.4. Procedures
The active study medication ataluren, a powder for oral suspension, was dosed thrice daily at 10, 10, 20 mg/kg (total daily dose 40 mg/kg) every day during the treatment period as morning, mid-day, and evening doses, respectively, for 48 weeks. Placebo was identical in appearance. The primary efficacy endpoint of the study was absolute ppFEV1 change from baseline to Week 48, defined as the average of the change at Week 40 and that at Week 48. Spirometry was performed at screening, at randomization, and every eight weeks during the 48-week study duration. All sites used a study-specific spirometer for FEV1, FVC, and FEF25–75 assessments following American Thoracic Society/European Respiratory Society guidelines [18,19]. At baseline and each subsequent study visit, subjects completed the Cystic Fibrosis Questionnaire-Revised (CFQ-R) [20].
2.5. Statistical analysis
A sample size of 252 subjects (126/treatment group) was chosen to detect a 3% predicted greater average change from baseline ppFEV1 (SD = 7) among subjects randomized to receive ataluren with >90% power using a two-sided t-test at a 0.05 significance level. Assuming a 10% premature discontinuation rate, an enrollment maximum of 280 patients was set. Efficacy analyses were performed on the intent-to-treat (ITT) population, defined as those patients who had at least 1 valid post-baseline spirometry measurement.
The primary efficacy endpoint (difference in average ppFEV1 absolute change from baseline at Weeks 40 and 48) tested in the ITT population using mixed-model repeated-measures (MMRM) analysis of covariance (ANCOVA), with an unstructured variance/covariance matrix and ppFEV1 change from baseline (defined as the ppFEV1 average at screening and randomization) as the dependent variable. If the unstructured variance/covariance matrix did not converge, the analysis was to be replaced with an MMRM analysis with multiple imputation. Rate of pulmonary exacerbation (PEx), defined as clinician decision to treat with (oral, inhaled, and/or IV) antibiotics in the presence of ≥4 Fuchs sign and symptom criteria [21] was tested by a negative binomial regression model with independent variables including treatment group, 3 stratification factors, and baseline ppFEV1. Maximum likelihood methods were used to fit the model. The analysis was performed using the generalized linear model as implemented in SAS by the GENMOD procedure [22]. All analyses were 2-sided at the 0.05 significance level, with analyses and data tabulations performed using SAS (Version 9.1 or higher).
2.6. Role of the funding source
The study sponsor oversaw trial management, data collection, statistical analyses, and the writing and review of the clinical study report. The corresponding author had full access to all data in the study and had final responsibility for the decision to submit for publication.
3. Results
In all, 279 subjects were randomly assigned to the ataluren treatment arm (N = 140) or to the placebo arm (N = 139) (Fig. 1). Treatment groups were generally well-balanced at randomization with respect to presence of CFTR nonsense mutation alleles and other demographic characteristics (Table 1). Use of concomitant CF medications, and notably chronic inhaled antibiotics, was somewhat more frequent among subjects randomized to receive ataluren: 86 ataluren subjects (61.4%) were receiving chronic inhaled antibiotics at randomization versus only 66 placebo subjects (47.5%)(Table 1). Five patients did not have a valid post-baseline spirometry measurement, resulting in an ITT population of 274 subjects, 138 subjects in the ataluren arm and 136 in the placebo arm. Thirteen subjects withdrew from the ataluren treatment group and 14 withdrew from the placebo group; 252 subjects completed the study (Fig. 1).
Fig. 1.

Subject disposition. Subjects include in the intent-to-treat population were required to have at least one spirometry measure at Week 8.
Table 1.
Subject demographics at randomization.
| Ataluren (N = 140) | Placebo (N = 139) | Total (N = 279) | |
|---|---|---|---|
|
| |||
| Mean Age, years (SD) | 22.0 (11.00) | 22.0 (10.44) | 22.0 (10.70) |
| Min, Max, years | 6, 52 | 7, 52 | 6, 52 |
| Age Categories, n (%) | |||
| ≥6 – < 12 years | 25 (17.9) | 19 (13.7) | 44 (15.8) |
| ≥12 – <18 years | 34 (24.3) | 37 (26.6) | 71 (25.4) |
| ≥18 years | 81 (57.9) | 83 (59.7) | 164 (58.8) |
| Females, n (%) | 59 (42.1) | 74 (53.2) | 133 (47.7) |
| Race category, n (%) | |||
| Caucasian | 136 (97.1) | 133 (95.7) | 269 (96.4) |
| Asian | 1 (0.7) | 4 (2.9) | 5 (1.8) |
| Hispanic | 3 (2.1) | 2 (1.4) | 5 (1.8) |
| CFTR nonsense mutation category, n (%) | |||
| G542X | 42 (30.4) | 43 (31.6) | 85 (31.0) |
| W1282X | 33 (23.9) | 34 (25.0) | 67 (24.5) |
| R553X | 21 (15.2) | 17 (12.5) | 38 (13.9) |
| R1162X | 9 (6.5) | 13 (9.6) | 22 (8.0) |
| Other nonsense mutations | 37 (26.8) | 31 (22.8) | 68 (24.8) |
| Mean ppFEV1 at Screening (SD) | 63.5 (13.46) | 62.2 (14.34) | 62.9 (13.90) |
| Min, Max | 40.0, 88.0 | 40.0, 90.0 | 40.0, 90.0 |
| Baseline ppFEV1 Categories, n (%) | |||
| 40 to <65 | 78 (55.7) | 78 (56.1) | 156 (55.9) |
| 65 to 90 | 62 (44.3) | 61 (43.9) | 123 (44.1) |
| Mean Sweat Chloride, mEq/L (SD) | 100.7 (2.87) | 100.0 (2.99) | 100.4 (2.94) |
| Min, Max, mEq/L | 95, 120 | 93, 107 | 93, 120 |
| Pancreatic Insufficiency, n (%) | 115 (82.1) | 124 (89.2) | 239 (85.7) |
| P. aeruginosa Infection History, n (%) | 113 (80.7) | 110 (79.1) | 223 (79.9) |
| P aeruginosa in Sputum at Screening, n (%) | 37 (26.4) | 29 (20.9) | 66 (23.7) |
| History of Diabetes, n (%) | 28 (20.0) | 34 (24.5) | 62 (22.2) |
| CF Medications used at Randomization, n (%) | |||
| Bronchodilators | 122 (87.1) | 119 (85.6) | 241 (86.4) |
| Dornase Alfa | 102 (72.9) | 97 (69.8) | 199 (71.3) |
| Chronic Azithromycin | 76 (54.3) | 71 (51.1) | 147 (52.7) |
| Inhaled Hypertonic Saline | 72 (51.4) | 73 (52.5) | 145 (52.0) |
| Inhaled Glucocorticoids | 30 (21.4) | 24 (17.3) | 54 (19.4) |
| Chronic Inhaled Antibiotic Use at randomizationa, n (%) | 86 (61.4) | 66 (47.5) | 152 (54.5) |
| Colistimethateb | 60 (42.9) | 49 (35.3) | 109 (39.1) |
| Aztreonamb | 33 (23.6) | 22 (15.8) | 55 (19.7) |
| Tobramycinb | 0 | 0 | 0 |
| Otherb | 2 (1.4) | 3 (2.2) | 5 (1.8) |
Subjects who received inhaled antibiotics (excluding aminoglycosides) up to 56 days prior to their randomization date.
Subjects who took multiple inhaled antibiotics prior to randomization are counted once for each inhaled antibiotic received.
3.1. Primary endpoint
At baseline, mean ppFEV1 for the ITT population was 63.2 for the ataluren group and 61.6 for the placebo group. Both treatment groups experienced a mean loss in lung function during the 48-week study (Fig. 2). LS means for ppFEV1 change from baseline to Week 40 and Week 48 averages (MMRM modeling of the ITT Population) were −1.396 for the ataluren group and −1.992 for the placebo group. The MMRM-based average treatment difference between ataluren and placebo groups in ppFEV1 change from baseline to Weeks 40 and 48 was 0.597 (95% CI −1.3, 2.5; p = 0.53; Table 2.
Fig. 2.
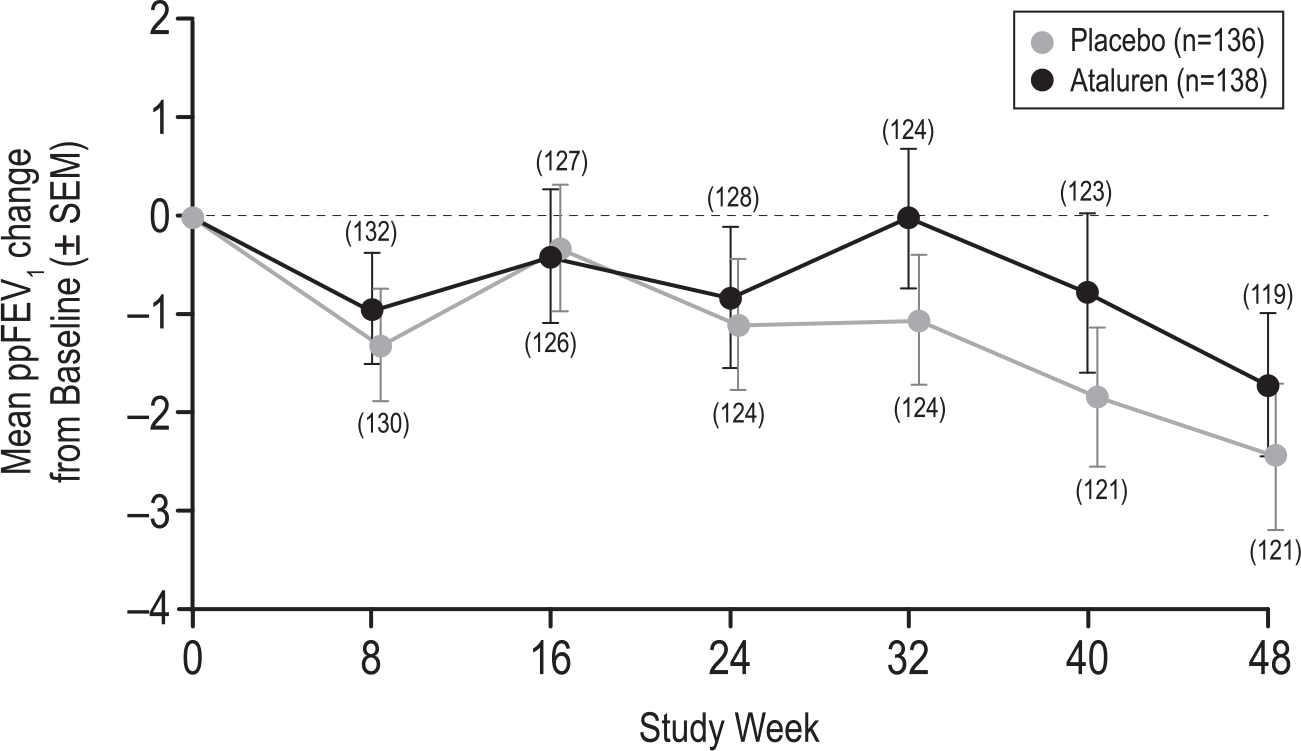
Modeled Mean ppFEV1 Change in from Baseline over 48 Weeks (ITT Population). Plotted values are observed means; bars represent ±SEM. Covariates were baseline ppFEV1, treatment, visit, treatment-by-visit interaction, ppFEV1-by-visit interaction, and the stratification factors of baseline inhaled antibiotics (yes vs no), baseline age (<18 vs ≥18 years), and baseline ppFEV1 (40 to <65% vs ≥65 to 90%). Abbreviations: ppFEV1 = percent predicted forced expiratory volume in 1 s, SEM = standard error of the mean.
Table 2.
Primary and secondary efficacy results.
| Efficacy endpoint | Endpoint hierarchy | LS mean | Difference (ataluren minus placebo) | Std error | 95% CI | p-Value |
|---|---|---|---|---|---|---|
|
| ||||||
| ppFEV1 change from baseline a | Primary | |||||
| Ataluren | −1.396 | 0.6996 | −2.7735, −0.0180 | |||
| Placebo | −1.992 | 0.6777 | −3.3271, −0.6576 | |||
| Ataluren vs Placebo | 0.597 | 0.957 | −1.2881, 2.4813 | 0.5336 | ||
| PEx rate, events/48 weeks | 1st Secondary | |||||
| Ataluren | 0.950 (1.4038)b | 0.1195 | 0.7136, 1.1862 | |||
| Placebo | 1.127 (2.5241)b | 0.2164 | 0.6986, 1.5547 | |||
| Ataluren/Placebo | 0.8567c | 0.4008 | ||||
| BMI change from baseline, kg/m2 | 2nd Secondary | |||||
| Ataluren | 0.296 | 0.093 | 0.1126, 0.4789 | |||
| Placebo | 0.361 | 0.0938 | 0.1759, 0.5455 | |||
| Ataluren vs Placebo | −0.065 | 0.1312 | −0.3233, 0.1934 | 0.6208 | ||
| Respiratory HRQL change from baseline | 3rd Secondary | |||||
| Ataluren | −0.76 | 1.3691 | −3.4566, 1.9364 | |||
| Placebo | −1.032 | 1.3733 | −3.7368, 1.6728 | |||
| Ataluren vs Placebo | 0.272 | 1.93 | −3.5292, 4.0731 | 0.8881 | ||
ppFEV1, forced expiratory volume in 1 s; PEx, pulmonary exacerbation; BMI, body mass index; HQRL, health-related quality of life.
Mean of Week 40 and 48 minus baseline.
Mean (SD).
Ratio of ataluren to placebo rate.
3.2. Secondary endpoints
Mean (SD) protocol-defined pulmonary exacerbation rates per 48 weeks were 0.95 (1.404) and 1.13 (2.524) for ataluren and placebo subjects, respectively (Table 2). The 48-week ataluren/placebo exacerbation rate ratio of 0.86 was not statistically significant (p = 0.40 by negative binomial regression). Both treatment groups experienced modest BMI increases during the study (Table 2). At Week 48, the MMRM-modeled mean BMI change from baseline was 0.30 kg/m2 for the ataluren group and 0.36 kg/m2 for the placebo group, a difference (ataluren minus placebo) of −0.06 kg/m2 (95% CI −0.32, 0.19; p = 0.62). Both treatment groups experienced a net decline in their respiratory quality of life during the study (Table 2). At Week 48, the MMRM-modeled mean change in the respiratory domain of the CFQ-R from baseline was −0.76 for the ataluren group and −1.03 for the placebo group, a difference (ataluren minus placebo) of 0.272 (95% CI −3.53, 4.07; p = 0.89).
3.3. Safety and study drug compliance
The as-treated population comprised 279 patients: 140 subjects treated at least once with ataluren and 139 subjects treated at least once with placebo. The mean [range] duration of study drug treatment was 45.39 weeks [0.3–53.3 weeks] among the 140 ataluren subjects and 44.68 weeks [1.1–54.9 weeks] among the 139 placebo subjects. Median compliance rate, based on study drug accountability, was 95.7% for ataluren and 94.9% for placebo.
Ataluren was generally well-tolerated, with the numbers of subjects experiencing treatment-emergent adverse events (TEAEs) similar between treatment groups (Table 3). However, more TEAEs were reported among subjects receiving ataluren (905) than among those receiving placebo (768). No clinically significant differences in vital sign measurements were observed between treatment groups and no clinically meaningful abnormalities were identified based on physical examinations, ECGs, and renal ultrasound. The most common TEAEs (≥15% in either treatment group) were infective pulmonary exacerbation of CF, cough, viral upper respiratory tract infection, and upper respiratory tract infection (Table 3). There was a somewhat higher incidence of gastrointestinal disorders (diarrhea, abdominal pain, nausea, and vomiting) in the ataluren group (31 subjects, 22.1%) than in the placebo group (22 subjects, 15.8%). Three subjects receiving ataluren (2.1%) and four subjects receiving placebo (2.9%) experienced TEAEs that led to discontinuation.
Table 3.
Summary of treatment-emergent adverse events occurring in ≥5% of subjects; as-treated population.
| System organ class Preferred terma | Ataluren (N = 140) n (%) |
Placebo (N = 139) n (%) |
Total (N = 279) n (%) |
|---|---|---|---|
|
| |||
| Number of Subjects with at Least One Such Treatment Emergent Adverse Eventb | 125 (89.3) | 127 (91.4) | 252 (90.3) |
| INFECTIONS AND INFESTATIONS | 116 (82.9) | 119 (85.6) | 235 (84.2) |
| Infective pulmonary exacerbation of cystic fibrosis | 84 (60.0) | 92 (66.2) | 176 (63.1) |
| Viral upper respiratory tract infection | 27 (19.3) | 21 (15.1) | 48 (17.2) |
| Upper respiratory tract infection | 21 (15.0) | 21 (15.1) | 42 (15.1) |
| Sinusitis | 16 (11.4) | 13 (9.4) | 29 (10.4) |
| Nasopharyngitis | 10 (7.1) | 8 (5.8) | 18 (6.5) |
| Rhinitis | 10 (7.1) | 8 (5.8) | 18 (6.5) |
| Influenza | 7 (5.0) | 8 (5.8) | 15 (5.4) |
| Pharyngitis | 7 (5.0) | 2 (1.4) | 9 (3.2) |
| Pseudomonas infection | 6 (4.3) | 9 (6.5) | 15 (5.4) |
| Staphylococcal infection | 4 (2.9) | 8 (5.8) | 12 (4.3) |
| RESPIRATORY, THORACIC AND MEDIASTINAL DISORDERS | 38 (27.1) | 35 (25.2) | 73 (26.2) |
| Cough | 26 (18.6) | 25 (18.0) | 51 (18.3) |
| Haemoptysis | 14 (10.0) | 11 (7.9) | 25 (9.0) |
| GASTROINTESTINAL DISORDERS | 31 (22.1) | 22 (15.8) | 53 (19.0) |
| Diarrhoea | 13 (9.3) | 12 (8.6) | 25 (9.0) |
| Abdominal pain | 11 (7.9) | 5 (3.6) | 16 (5.7) |
| Nausea | 11 (7.9) | 10 (7.2) | 21 (7.5) |
| Vomiting | 8 (5.7) | 4 (2.9) | 12 (4.3) |
Abbreviations: MedDRA = Medical Dictionary for Regulatory Activities; N = total number of patients; n = number of patients.
Adverse Events were coded using MedDRA, Version 17.0.
A treatment-emergent adverse event is defined as an adverse event that occurs or worsens in the period extending from the day of a subject’s first dose of study drug to 4 weeks after the last dose of study drug in this study. The total number of AEs counts all treatment-emergent AEs for subjects. At each level of subject summarization, a subject is counted once if the subject reported one or more events.
Across both treatment arms, 63.1% of subjects experienced Grade 2 (moderate) TEAEs, 17.2% experienced Grade 3 (severe) TEAEs, and 15.8% experienced Grade 1 (mild) TEAEs. No subjects experienced Grade 4 (life-threatening) TEAEs and there were no subject deaths during the study. Grade 3 infective pulmonary exacerbations of CF occurred in 5 subjects receiving ataluren (3.6%) and 22 subjects receiving placebo (15.8%). Two subjects receiving ataluren (1.4%) experienced Grade 3 nephrolithiasis, which was not observed among subjects receiving placebo. One ataluren subject experienced nephrolithiasis and acute renal failure, the latter associated with dehydration.
4. Discussion
A previous double-blinded, placebo-controlled phase 3 study of ataluren failed to meet its primary efficacy endpoint of change in FEV1 over 48 weeks, but post hoc analyses suggested that study participants not receiving concomitant inhaled aminoglycosides experienced ataluren-associated FEV1 and exacerbation benefits during the study [14]. The goal of the current study was to prospectively assess ataluren’s efficacy in CF subjects with at least one nonsense CFTR mutation not concomitantly receiving inhaled aminoglycosides, again using improvement in pulmonary function and reduction in pulmonary exacerbation rate as primary and secondary efficacy endpoints.
Neither ppFEV1 change from baseline nor pulmonary exacerbation rate over 48 weeks were statistically different between ataluren and placebo groups in this trial, suggesting that results observed from the previous post hoc subgroup analyses occurred by chance. Both treatment groups experienced a net loss in ppFEV1 over the course of the study, reflecting the severity of CF lung disease associated with CFTR nonsense mutations. However, subjects receiving ataluren had an increased TEAE incidence.
Further development of ataluren for treating persons with nonsense-mutation CFTR has been halted [23]. There may be several reasons why ataluren failed to be associated with clinical benefit in this study. In addition to the possibility of poor ataluren bioavailability in the lung, mRNA transcripts carrying nonsense mutations are subjected to nonsense mediated decay (NMD), a surveillance mechanism which detects and degrades transcripts carrying nonsense mutations, preventing the synthesis of truncated proteins [24] and protecting the cell from potentially deleterious truncated proteins [25]. This process may also regulate the response to read-though treatment with ataluren [26]. Inhibition of the NMD pathway has been associated with upregulated the CFTR RNA, protein, and surface-localized protein in cells homozygous for the W1282X mutation [27], and may be a viable approach to augment the efficacy of read-through [28]. Inefficient NMD may also activate the unfolded protein response (UPR), which restores cellular homeostasis by activating chaperons and foldases in the endoplasmic reticulum, inhibiting new substrate translation [29]. Because translation attenuation also inhibits NMD by a feedback mechanism, NMD and UPR may modulate response to ataluren read-through treatment. Observations of UPR differences in patients correlating with readthrough response appear to support this suggestion [30]. Finally, although successful readthrough with ataluren would allow translation elongation to continue in the correct reading frame, elongation is achieved by insertion of near-cognate tRNAs at the nonsense codon, which could result in a non-functional “neo-formed” protein [31] or a protein with reduced CFTR activity requiring a CFTR modulator for restored activity [32].
Although negative in outcome, the results of this study should be of value for the CF community, in that they demonstrated what we all should accept: that post hoc subgroup analyses, no matter how rational and well-justified, are hypothesis forming with results unverified in the absence of a prospective test. It has been suggested that ataluren’s initial discovery in cell-based firefly luciferase assays may have been an artifact resulting from direct interaction of ataluren with the reporter system, as use of a different luciferase reporter did not confirm ataluren’s activity with respect to nonsense codon suppression [33]. In retrospect, these observations in addition to the lack of sweat chloride changes associated with ataluren exposure in the previous Phase 3 study (including in the subgroup not receiving aminoglycosides) should have raised suspicions as to the probability of success of the current study. Similarly, developers of future stop codon read-through candidates might place greater weight on observing sweat chloride changes early in clinical development before proceeding to large pivotal trials.
Supplementary Material
Acknowledgments
The authors thank the patients and families, study investigators and coordinators, the Cystic Fibrosis Foundation Therapeutics Development Network, the Cystic Fibrosis Data Safety Monitoring Board, and the European Cystic Fibrosis Society Clinical Trials Network for their support of this trial.
Funding source
PTC Therapeutics, Cystic Fibrosis Foundation, US Food and Drug Administration’s Office of Orphan Products Development.
Footnotes
Declaration of Competing Interest
JM is an employee of PTC Therapeutics, the funder of this clinical trial, and holds financial interests in the company. MWK, SMR, and DRV received compensation for consultant services from PTC Therapeutics prior to and/or during this study. EK, MW, IS-G, and KDB received compensation for travel expenses for meetings related to the study. All other authors declare no competing interests.
Members listed in the online supplement.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.jcf.2020.01.007.
References
- [1].http://www.genet.sickkids.on.ca/StatisticsPage.html.
- [2].https://www.ecfs.eu/projects/efcs-patient-registry/guidelines.
- [3].Cystic Fibrosis Foundation Patient Registry. 2017 annual data report to the center directors. Bethesda (MD): Cystic Fibrosis Foundation; 2018. [Google Scholar]
- [4].Shoshani T, Augarten A, Gazit E, Bashan N, Yahav Y, Rivlin Y, Tal A, Seret H, Yaar L, Kerem E, et al. Association of a nonsense mutation (W1282X), the most common mutation in the Ashkenazi Jewish cystic fibrosis patients in Israel, with presentation of severe disease. Am J Hum Genet 1992;50(1):222–8. [PMC free article] [PubMed] [Google Scholar]
- [5].Cystic Fibrosis Genotype-Phenotype Consortium Correlation between genotype and phenotype in patients with cystic fibrosis. N Engl J Med 1993;329(18):1308–13. [DOI] [PubMed] [Google Scholar]
- [6].Kerem E, Kerem B. Genotype-phenotype correlations in cystic fibrosis. Pediatr Pulmonol 1996;22(6):387–95. [DOI] [PubMed] [Google Scholar]
- [7].de Gracia J, Mata F, Alvarez A, Casals T, Gatner S, Vendrell M, de la Rosa D, Guarner L, Hermosilla E. Genotype-phenotype correlation for pulmonary function in cystic fibrosis. Thorax 2005;60(7):558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].McKone EF, Goss CH, Aitken ML. CFTR genotype as a predictor of prognosis in cystic fibrosis. Chest 2006;130(5):1441–7. [DOI] [PubMed] [Google Scholar]
- [9].Peltz SW, Morsy M, Welch Jacobson A. Ataluren as an agent for therapeutic nonsense suppression. Annu Rev Med 2013;64:407–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, ConnM M, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007;447(7140):87–91. [DOI] [PubMed] [Google Scholar]
- [11].Gonzalez-Hilarion S, Beghyn T, Jia J, Debreuck N, Berte G, Mamchaoui K, Mouly V, Gruenert DC, Déprez B, Lejeune F. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J Rare Dis 2012;7:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Du M, Liu X, Welch EM, Hirawat S, Peltz SW, Bedwell DM. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc Natl Acad Sci USA 2008;105(6):2064–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sermet-Gaudelus I, Boeck KD, Casimir GJ, Vermeulen F, Leal T, Mogenet A, Roussel D, Fritsch J, Hanssens L, Hirawat S, Miller NL, Constantine S, Reha A, Ajayi T, Elfring GL, Miller LL. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am J Respir Crit Care Med 2010;182(10):1262–72. [DOI] [PubMed] [Google Scholar]
- [14].Kerem E, Konstan MW, De Boeck K, Accurso FJ, Sermet-Gaudelus I, Wilschanski M, Elborn JS, Melotti P, Bronsveld I, Fajac I, Malfroot A, Rosenbluth DB, Walker PA, McColley SA, Knoop C, Quattrucci S, Rietschel E, Zeitlin PL, Barth J, Elfring GL, Welch EM, Branstrom A, Spiegel RJ, Peltz SW, Ajayi T, Rowe SMCystic Fibrosis Ataluren Study Group. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med 2014;2(7):539–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Neu HC. Tobramycin: an overview. J Infect Dis 1976;134(Suppl):S3–19. [DOI] [PubMed] [Google Scholar]
- [16].Prokhorova I, Altman RB, Djumagulov M, Shrestha JP, Urzhumtsev A, Ferguson A, Chang CT, Yusupov M, Blanchard SC, Yusupova G. Aminoglycoside interactions and impacts on the eukaryotic ribosome. Proc Natl Acad Sci USA 2017;114(51):E10899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Roy B, Friesen WJ, Tomizawa Y, Leszyk JD, Zhuo J, Johnson B, Dakka J, Trotta CR, Xue X, Mutyam V, Keeling KM, Mobley JA, Rowe SM, Bedwell DM, Welch EM, Jacobson A. Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc Natl Acad Sci USA 2016;113(44):12508–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med 1999;159(1):179–87. [DOI] [PubMed] [Google Scholar]
- [19].Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, Crapo R, Enright P, van der Grinten CP, Gustafsson P, Jensen R, Johnson DC, MacIntyre N, McKay R, Navajas D, Pedersen OF, Pellegrino R, Viegi G, Wanger JATS/ERS Task Force. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–38. [DOI] [PubMed] [Google Scholar]
- [20].Quittner AL, Buu A, Messer MA, Modi AC, Watrous M. Development and validation of the cystic fibrosis questionnaire in the United States: a health-related quality-of-life measure for cystic fibrosis. Chest 2005;128(4):2347–54. [DOI] [PubMed] [Google Scholar]
- [21].Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, Rosenstein BJ, Smith AL, Wohl ME. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med 1994;331(10):637–42. [DOI] [PubMed] [Google Scholar]
- [22].Der G, Everitt BS. Statistical analysis of medical data using SAS. Boca Raton, FL: Chapman Hall/CRC; 2006. [Google Scholar]
- [23].http://ir.ptcbio.com/news-releases/news-release-details/ptc-therapeutics-announces-results-pivotal-phase-3-clinical.
- [24].Frischmeyer HC, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet 1999;8:1893–900. [DOI] [PubMed] [Google Scholar]
- [25].Maquat LE. When cells stop making sense: effects of nonsense codons on RNA metabolism in vertebrate cells. RNA 1995;1:453–65. [PMC free article] [PubMed] [Google Scholar]
- [26].Linde L, Boelz S, Nissim-Raffinia M, Oren YS, Wilschanski M, Yaacov Y, Virgilis D, Neu-Yilik G, Kulozik AE, Kerem E, Kerem B. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J Clin Invest 2007;117:683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Keenan MM, Huang L, Jordan NJ, Wong E, Cheng Y, Valley HC, Mahiou J, Liang F, Bihler H, Mense M, Guo S, Monia BP. Nonsense-mediated RNA decay pathway inhibition restores expression and function of W1282X CFTR. Am J Respir Cell Mol Biol 2019;61:290–300. [DOI] [PubMed] [Google Scholar]
- [28].Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011;334:1081–6. [DOI] [PubMed] [Google Scholar]
- [29].Keenan MM, Huang L, Jordan NJ, Wong E, Cheng Y, Valley HC, Mahiou J, Liang F, Bihler H, Mense M, Guo S, Monia BP. Nonsense-mediated RNA decay pathway inhibition restores expression and function of W1282X CFTR. Am J Respir Cell Mol Biol 2019;61:290–300. [DOI] [PubMed] [Google Scholar]
- [30].Oren YS, McClure ML, Rowe SM, Sorscher EJ, Bester AC, Manor M, Kerem E, Rivlin J, Zahdeh F, Mann M, Geiger T, Kerem B. The unfolded protein response affects readthrough of premature termination codons. EMBO Mol Med 2014;6:685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yeh JT, Hwang TC. Positional effects of premature termination codons on the biochemical and biophysical properties of CFTR. J Physiol 2019. [Epub ahead of print]. doi: 10.1113/JP278418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xue X, Mutyam V, Thakerar A, Mobley J, Bridges RJ, Rowe SM, Keeling KM, Bedwell DM. Identification of the amino acids inserted during suppression of CFTR nonsense mutations and determination of their functional consequences. Hum Mol Genet 2017;26(16):3116–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Auld DS, Thorne N, Maguire WF, Inglese J. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc Natl Acad Sci USA 2009;106(9):3585–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.