

Dear Editor,
We investigated whether screening by whole genome sequencing (WGS) in unselected newborns provides more information of potentially curable or treatable medical conditions than routine newborn screening (NBS). We demonstrated that compared with routine NBS, WGS produced fewer false positive results and identified more actionable pathogenic or likely pathogenic variants in the selective 246 genes.
Previously, WGS has been used to identify mutated genes in newborn children with a suspected disease. 1 However, sequencing of apparently healthy newborns has remained controversial due to technical concerns and ethical issues. 2 In this study, 321 non‐pre‐selected newborns from a cohort of pregnant women in Qingdao, China were recruited (Table 1). DNA from 303 umbilical cord blood samples and 18 umbilical cords was extracted for 40X WGS. For data interpretation, we selected 251 genes associated with 59 Mendelian disorders, 164 primary immunodeficiency diseases (PIDs) and five pharmacogenetic (PGx) genes, following the guidelines by the Recommended Uniform Screening Panel (RUSP), the International Union of Immunologic Societies (IUIS) Expert Committee for Primary Immunodeficiency, the Dutch Pharmacogenetics Working Group (DPWG), and the Clinical Pharmacogenetics Implementation Consortium (CPIC). 3 , 4 , 5 Sequencing protocol, data analysis pipeline, and criteria for sequence variants interpretation following the ACMG/AMP guidelines are described in the Supporting Information. The WGS results were compared with NBS results, including the mandatory checks of hearing impairment and four metabolic diseases, the metabolic testing of 48 inherited metabolic diseases (IMDs), and the genetic screening for 20 hearing loss loci incorporated into the local NBS program in China. 6 , 7
TABLE 1.
Summary of the demographic data collected from the 321 newborns of Qingdao cohort
| Type | Number | Percentage (%) | |
|---|---|---|---|
| Pregnancy | Natural pregnancy | 306 | 95.4 |
| Assisted reproduction technology | 11 | 3.4 | |
| Unspecified | 4 | 1.20 | |
| Gestational weeks | Pre‐term birth | 7 | 2.2 |
| Term birth | 314 | 97.80 | |
| Average delivery gestation | 39 weeks plus 5 days | – | |
| Gender of newborn | Male | 151 | 47.04 |
| Female | 170 | 52.96 | |
| Parental age at delivery | Father's age (ave. year) | 33 | – |
| Mother's age (ave. year) | 32 | – | |
| SD of father's age (year) | 5 | – | |
| SD of mother's age (year) | 4 | – | |
| Mandatory NBS screening of 4 metabolic diseases (PKU, CAH, CH and G6PD) by TRFIA | Phe + | 1 | 0.31 |
| Negative | 320 | 99.69 | |
| Mandatory hearing impairment screening by OAEs or AABR | Not passed | 0 | 0.00 |
| Passed | 321 | 100.00 | |
| 48 IMDs screening by tandem MS/MS | C5‐OH + | 1 | 0.31 |
| Phe + | 1 | 0.31 | |
| Negative | 310 | 96.57 | |
| Unspecified | 9 | 2.80 | |
| Genetic hearing loss screening by MALDI‐TOF | Carrier | 18 | 5.61 |
| Negative | 294 | 91.59 | |
| Unspecified | 9 | 2.80 |
AABR, automated auditory brainstem response; CAH, congenital adrenal hyperplasia; CH, congenital hypothyroidism; CH G6PD, glucose‐6‐phosphate dehydrogenase; C5‐OH+, isovalerylcarnitine positive; MS/MS, mass spectrometry; MALDI‐TOF, matrix‐assisted laser desorption/ionization time of flight; OAEs, otoacoustic emissions; PKU, phenylketonuria; Phe+, phenylalanine positive; SD, standard deviation; TRFIA, time resolved fluoroimmunoassay.
Among the analysed DNA samples of 321 newborns, the average sequencing depth was 47.42X (28.84X–82.90X) and the average coverage was 99.48% (99.01%–99.89%) (Figure 1A). For the 59 Mendelian disorders, a total of 131 pathogenic or likely pathogenic (P/LP) mutations and 5 pathogenic copy number variations were detected in 107 of the 321 newborns (33.33%), corresponding to 106 carriers of 28 diseases and 1 patient with phenylketonuria (PKU) (Figure 1B and Table 2). The 25.23% of newborns (n = 81) carried one P/LP mutations, and 7.17% and 0.93% of newborns (n = 23 and n = 3) carried two or three P/LP mutations, respectively. Hearing loss, methylmalonic acidemia (MMA), primary congenital hypothyroidism (CH), and PKU were diseases with the most carriers, while GJB2 (28/321, 8.72%), MMACHC (11/321, 3.43%), DUOX2 (10/321, 3.12%), PAH (8/321, 2.49%) and SLC26A4 (8/321, 2.49%) were the top five genes with the highest carrier frequencies of P/LP mutations (Table 2).
FIGURE 1.
Overview of the results of newborn WGS in the Qingdao cohort (n = 321). (A) The sequencing quality of WGS results in the Qingdao cohort (n = 321). (B) Overview of carriers and/or patient identified by WGS in the Qingdao cohort (n = 321). A total of 107 carriers and/ or patients associated with 34 inherited disorders were identified in the 321 newborns. Of the 136 P/LP mutations, 42.65% corresponded to genetic disorders, 6.62% were associated with PIDs and 50.74% were related to metabolic disorders, including organic acid conditions (14.71%), amino acid disorders (14.71%), fatty acid oxidation disorders (7.35%) and endocrine disorders (13.97%). Hearing loss, MMA, CH and PKU were the most common diseases with carriers, while GJB2 (28/321, 8.72%), MMACHC (11/321, 3.43%), DUOX2 (10/321, 3.12%), PAH (8/321, 2.49%) and SLC26A4 (8/321, 2.49%) were the top five genes with the highest carrier frequencies of P/LP mutations. For the 164 PIDs recognized by the IUIS, 9 heterozygous P/LP variants in 6 genes, corresponding to 6 diseases, were identified in 9 of the 321 newborn children (2.80%), all in a heterozygous state. Of these, there are two genes (ADA and JAK3) for which we found two carriers. (C) Distribution of actionable PGx variants identified by WGS in the Qingdao cohort (n = 321). A clinical management strategy can be adopted for every carrier according to the DPWG guidelines. Among 321 newborns, 313 newborns (97.51%) in the Qingdao cohort carried at least one clinically relevant variant, while 193 newborns (60.12%) harboured two and/or three variants. From the perspective of the five crucial pharmacogenes, the CYP2D6 gene had the highest carrier frequency, where 266 out of the 321 infants (82.87%) harboured at least one actionable PGx variant. The gene CYP2C19 showed the second‐highest carrier rate, where 209 infants (65.11%) carried at least one clinically relevant variant. In addition, 133 and 122 infants carried actionable PGx variants at the gene UGT1A1 and NUDT15, respectively. No actionable variant was identified in the DPYD gene in the 321 children. Among the selected gene–drug pairs, irinotecan, azathioprine, mercaptopurine and tioguanine were prescription drugs to paediatric patients, while codeine and clopidogrel are restricted for use of children under 18 years old. Our findings suggest that participants may obtain benefit from PGx profiling already in early childhood
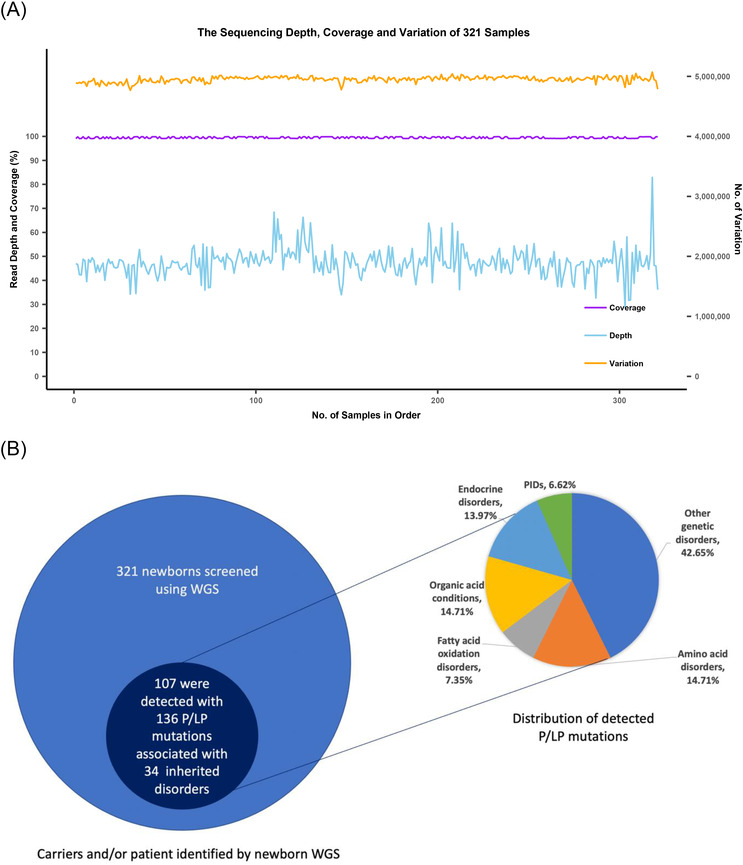
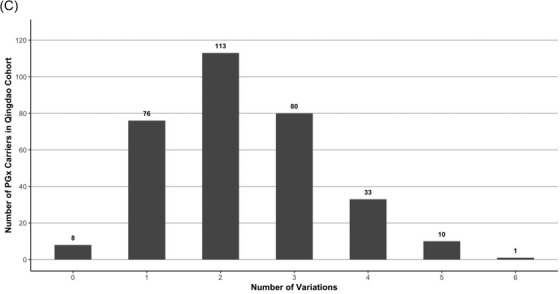
TABLE 2.
Overview of the P/LP mutations identified in 321 newborn children from Qingdao.
| Condition | Inheritance | Gene | Variation | Protein | Classification | Num of newborns | Het/Hom |
|---|---|---|---|---|---|---|---|
| Propionic acidemia | AR | PCCB | c.1364A > G | p.Y455C | P | 1 | Het |
| c.793G > A | p.G265S | LP | 1 | Het | |||
| Methylmalonic acidemia (methylmalonic‐CoA mutase) | AR | MUT | c.2179C > T | p.R727* | P | 1 | Het |
| Methylmalonic acidemia (cobalamin disorders) | AR | MMAA | c.658G > A | p.V220M | LP | 1 | Het |
| AR | MMACHC | c.315C > G | p.Y105* | P | 1 | Het | |
| c.445_446del | p.C149Hfs32 | P | 1 | Het | |||
| c.482G > A | p.R161Q | P | 2 | Het | |||
| c.609G > A | p.W203* | P | 3 | Het | |||
| c.658_660del | p.*220del | P | 3 | Het | |||
| c.80A > G | p.Q27R | P | 1 | Het | |||
| Methylmalonic acidemia (cobalamin disorders)/methylmalonic acidemia with homocystinuria | AR | MMADHC | c.748C > T | p.R250* | P | 1 | Het |
| 3‐Methylcrotonyl‐CoA carboxylase deficiency | AR | MCCC1 | c.639+2T > A | / | P | 1 | Het |
| Holocarboxylase synthase deficiency | AR | HLCS | c.782del | p.G261Vfs20 | P | 2 | Het |
| Glutaric acidemia type I | AR | GCDH | c.1213A > G | p.M405V | P | 1 | Het |
| Carnitine uptake defect/carnitine transport defect | AR | SLC22A5 | c.1472C > G | p.S491C | P | 4 | Het |
| c.468G > A | p.W156* | P | 1 | Het | |||
| Medium‐chain acyl‐CoA Dehydrogenase deficiency | AR | ACADM | c.548_551del | p.T183Rfs4 | P | 2 | Het |
| Trifunctional protein deficiency | AR | HADHB | c.1175C > T | p.A392V | LP | 1 | Het |
| Citrullinemia, type I | AR | ASS1 | c.919C > T | p.R307C | P | 1 | Het |
| c.352G > A | p.A118T | LP | 1 | Het | |||
| Classic phenylketonuria | AR | PAH | c.1301C > A | p.A434D | LP | 1 | 7Het; 1 individual with compound Het |
| c.611A > G | p.Y204C | P | 2 | ||||
| c.728G > A | p.R243Q | P | 4 | ||||
| c.740G > T | p.G247V | P | 1 | ||||
| c.842+2T > A | / | P | 1 | ||||
| Primary congenital hypothyroidism | AR,AD | TSHR | c.1349G > A | p.R450H | P | 4 | Het |
| AR | DUOX2 | c.1588A > T | p.K530* | P | 6 | Het | |
| c.1883del | p.K628Rfs11 | P | 1 | Het | |||
| c.1946C > A | p.A649E | LP | 1 | Het | |||
| c.605_621del | p.Q202Rfs93 | P | 1 | Het | |||
| c.3329G > A | p.R1110Q | P | 1 | Het | |||
| AR | TPO | c.2422del | p.C808Afs24 | P | 1 | Het | |
| Congenital adrenal hyperplasia | AR | CYP21A2 | c.518T > A | p.I173N | P | 2 | Het |
| c.92C > T | p.P31L | P | 2 | Het | |||
| S,S disease (sickle cell anemia)/S, beta‐thalassemia/S,C disease/other haemoglobinopathies | AR | HBB | c.126_129del | p.F42Lfs19 | P | 1 | Het |
| Cystic fibrosis | AR | CFTR | c.2052_2053insA | p.Q685Tfs4 | P | 1 | Het |
| Classic galactosemia | AR | GALT | c.821‐7A > G | / | P | 1 | Het |
| AR | GALT | c.844C > G | p.L282V | LP | 1 | Het | |
| Glycogen storage disease type II (Pompe) | AR | GAA | c.2237G > C | p.W746S | P | 1 | Het |
| c.2662G > T | p.E888* | P | 1 | Het | |||
| c.2647‐7G > A | / | LP | 1 | Het | |||
| Hearing loss | AD, AR, DD (digenic dominant) | GJB2 | c.109G > A | p.V37I | P | 14 | 26Het; 2 individuals with compound Het |
| c.235del | p.L79Cfs3 | P | 10 | ||||
| c.299_300del | p.H100Rfs14 | P | 4 | ||||
| c.605_606insAGAAGACTGTCTTCACAGTGTTCATGATTGCAGTGTCTGGAATTTG | p.C202* | P | 2 | ||||
| AR | SLC26A4 | c.1174A > T | p.N392Y | P | 1 | Het | |
| AR | c.1229C > T | p.T410M | P | 1 | Het | ||
| AR | c.1262A > C | p.Q421P | LP | 1 | Het | ||
| AR | c.2027T > A | p.L676Q | LP | 2 | Het | ||
| AR | c.2168A > G | p.H723R | P | 1 | Het | ||
| AR | c.919‐2A > G | / | P | 2 | Het | ||
| AR | USH2A | c.2802T > G | p.C934W | P | 1 | Het | |
| c.100C > T | p.R34* | P | 1 | Het | |||
| c.8559‐2A > G | / | P | 1 | Het | |||
| Maternal | MT‐RNR1 | m.1095T > C | / | P | 4 | Hom | |
| Spinal muscular atrophy | AR | SMN1 | c.(723+1_724‐1)_(834+1_835‐1)del | p.I242_M278del | P | 4 | Het |
| c.(723+1_724‐1)_(885+1_886‐1)del | p.I242_L294del | P | 1 | Het | |||
| Short‐chain acyl‐CoA dehydrogenase deficiency | AR | ACADS | c.1031A > G | p.E344G | P | 1 | Het |
| Glutaric acidemia type II | AR | ETFDH | c.1211T > C | p.M404T | LP | 1 | Het |
| Citrullinemia, type II | AR | SLC25A13 | c.1180+1G > A | / | P | 1 | Het |
| c.852_855del | p.M285Pfs2 | P | 2 | Het | |||
| Hypermethioninemia | AR | GNMT | c.149T > C | p.L50P | LP | 1 | Het |
| Biopterin defect in cofactor biosynthesis/biopterin defect in cofactor regeneration | AR | PTS | c.166G > A | p.V56M | P | 1 | Het |
| c.259C > T | p.P87S | P | 1 | Het | |||
| c.84‐291A > G | / | P | 3 | Het | |||
| Galactoepimerase deficiency | AR | GALE | c.505C > T | p.R169W | P | 1 | Het |
| Adenosine deaminase (ADA) deficiency | AR | ADA | c.424C > T | p.R142* | P | 1 | Het |
| c.872C > T | p.S291L | P | 1 | Het | |||
| Ataxia‐telangiectasia | AR | ATM | c.67C > T | p.R23* | P | 1 | Het |
| Immunoskeletal dysplasia with neurodevelopmental abnormalities (EXTL3 deficiency) | AR | EXTL3 | c.1970A > G | p.N657S | P | 1 | Het |
| JAK3 deficiency | AR | JAK3 | c.1744C > T | p.R582W | LP | 1 | Het |
| c.307C > T | p.R103C | LP | 1 | Het | |||
| DNA ligase IV deficiency | AR | LIG4 | c.1271_1275del | p.K424Rfs20 | P | 1 | Het |
| TACI deficiency (immunodeficiency, common variable) | AD/AR | TNFRSF13B | c.542C > A | p.A181E | LP | 2 | Het |
Het, heterozygous; Hom, homozgyous. P, pathogenic; LP, likely pathogenic.
For the 164 PIDs, 9 heterozygous P/LP variants in 6 genes were identified in 9 newborns (2.80%), all in a heterozygous state (Table 2). Four newborns were shown to carry heterozygous variant of unknown significance (VUS) in the gene SLC25A13 (c.2T>C, p.M1T), which was predicted as start loss and likely affecting the initiator methionine of the SLC25A13 mRNA. Two newborns carried a VUS in ASS1(c.‐4C>T, p.?). Although these VUSs were not included in the final report to the participants, follow‐up of the children with VUSs will be conducted till 3 years of age.
Sanger sequencing confirmed 143 out of 145 mutations identified by WGS, resulting in an accuracy of 98.62%. Carriers of SMN1 mutations were validated by multiplex ligation‐dependent probe amplification and real‐time quantitative PCR, showing that five out of the six predicted carriers were true.
Of the 321 newborns, 312 (97.20%) had the results of 48 IMDs screening and genetic hearing loss screening on 20 loci, which identified one newborn with PKU and one infant with increased blood level of isovalerylcarnitine (Table 1). In addition, 18 carriers harbouring 20 pathogenic mutations causing hearing impairment were detected by genetic hearting loss screening, albeit all 321 children passed the physical hearing screening at hospital (Supporting Information). The newborn WGS also identified the PKU case and 18 hearing loss carriers (Figure 2A). However, the child with increased level of isovalerylcarnitine was confirmed to be a carrier of 3‐methylcrotonyl‐CoA carboxylase deficiency by WGS. In addition, WGS identified two infants carrying compound heterozygous P/LP variants in GJB2 (Figure 3) and four children carrying pathogenic mutations in MT‐RNR1 (c1095T > C), suggesting an increased risk of late‐onset deafness or drug‐induced hearing loss, respectively. Although currently non‐symptomatic, the two newborns with GJB2 variants were scheduled to undergo hearing tests every 6 months, and the four newborns with the m.1095T mutation in MT‐RNR1 were advised to avoid using aminoglycosides.
FIGURE 2.
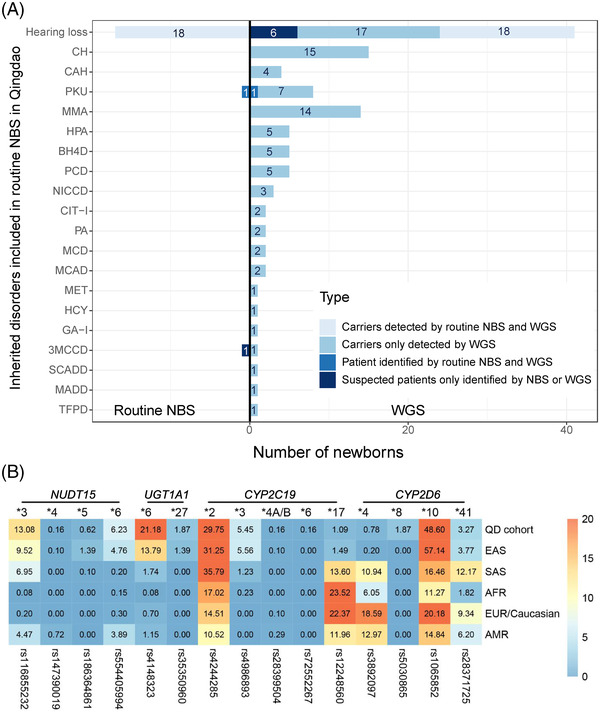
(A) Comparison between the findings of newborn WGS and routine NBS in the Qingdao cohort (n = 321). The findings of routine NBS tests are summarized on the left side of (A). In total, 18 carriers of hearing loss associated genes, one patient with PKU and one false positive result of C5‐OH were detected by existing routine methods. The findings of WGS are shown on the right side of (A). The WGS results confirmed the positive routine NBS findings of 18 carriers of hearing loss and the case with increased level of Phe. However, the infant with a routine NBS showing an increased level of C5‐OH (sample ID 18110806) was found to carry one pathogenic mutation in MCCC1, corresponding to being a carrier of 3‐methylcrotonyl‐CoA carboxylase deficiency (3MCCD). The newborn WGS also identified more infants carrying extra hearing loss mutations that were not identified by the routine NBS method, including 2 newborns carrying compound heterozygous P/LP variants in GJB2, 4 newborns harbouring a pathogenic mutation in MT‐RNR and 17 additional carriers harboured altogether 19 variants. Moreover, newborn WGS identified 59 extra carriers carrying 66 P/LP variants corresponding to 18 inherited metabolic diseases that could not be identified by the routine NBS tests. The abbreviations of diseases and the summary of are listed in Table S1. (B) Comparison of allele frequency of actionable PGx variants between the Qingdao cohort and five subpopulations of the 1000 Genome Project dataset, including East Asians (EAS), South Asians (SAS), Africans (AFR), Europeans (EUR) and Americans (AMR). In most cases, the allele frequency of the Qingdao cohort is consistent with the EAS, but differed significantly with the SAS, the AFR, the EUR and the AMR, such as CYP2C19*2, CYP2C19*3 and NUDT15*6. It should be noted, however, that three common PGx variants in the Qingdao cohort, CYP2D6*10 (48.60%), NUDT15*3 (13.08%) and UGT1A1*6 (21.18%), showed significant frequency differences with the EAS (p < 0.05), indicating population diversity within the East Asians. Notably, two rare variants, CYP2D6*8 (1.87%, n = 11) and CYP2C19*6 (0.16%, n = 1) which have not been reported in any subpopulation in the 1000 Genome phase 3 dataset and were first detected in our Qingdao 321 newborns
FIGURE 3.
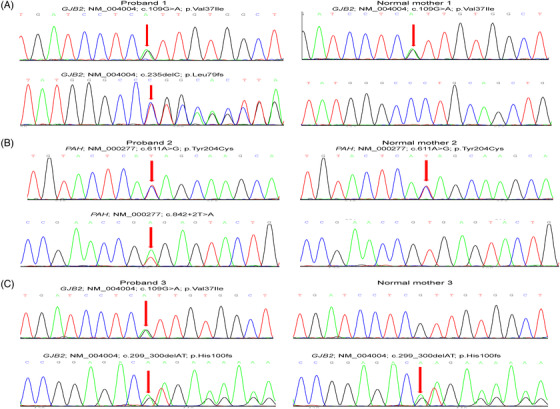
The results of Sanger sequencing and pedigree analysis of three children with compound heterozygous mutations. Three children with compound heterozygous variants were detected by newborn WGS. Two of them had variants at the GJB2 gene (A) (NM_004004.5, c.109G > A, p.V37I; NM_004004.5, c.235del, p.L79Cfs3); (C) (NM_004004.5, c.109G > A, p.V37I; NM_004004.5, c.299_300del, p.H100Rfs14), and one carried two variants at the PAH gene (B) (NM_000277.1, c.611A > G, p.Y204C and NM_000277.1, c.842 + 2T > A). Sanger sequencing confirmed that one variant was inherited from her/his mother. However, as infant father's sample was not available, we could not determine if the small deletion and insertion was inherited from the father or whether it was a de novo variant
Interestingly, we observed that 313 newborns (97.51%) carried at least one actionable PGx variant (Figure 1C). This result is in line with a European 44 000 biobank participants study, where 99.8% of the participants had a genotype associated with increased risks to at least one medication. 8 Furthermore, we found three common PGx variants in the Qingdao cohort, CYP2D6*10 (48.60%), NUDT15*3 (13.08%) and UGT1A1*6 (21.18%) (Figure 2B and Table 3), that showed significant frequency differences as compared to East Asian populations (p < 0.05). An important aspect when screening for disorders in a given population is the use of a matched control database as variants can be highly specific for a given ethnic group. 9 Most databases published to date are based on individuals of European descent and many populations have limited or poor representation.
TABLE 3.
Overview of the actionable PGx variants detected in 321 newborns from Qingdao
| Gene–drug pairs | Number of carriers in the Qingdao cohort | MAF$ | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drug | Gene | Star allele | dbSNP RS ID | Het | Hom | Variant No. | Carrier No. | Qingdao cohort (%) | EAS (%) | SAS (%) | AFR (%) | EUR/Caucasian (%) | AMR (%) |
| Azathioprine, mercaptopurine, tioguanine | NUDT15 | *3 | rs116855232 | 74 | 5 | 79 | 122 | 13.08 | 9.52 | 6.95 | 0.08 | 0.20 | 4.47 |
| *4 | rs147390019 | 1 | 0 | 1 | 0.16 | 0.10 | 0.00 | 0.00 | 0.00 | 0.72 | |||
| *5 | rs186364861 | 4 | 0 | 4 | 0.62 | 1.39 | 0.10 | 0.00 | 0.00 | 0.00 | |||
| *6 | rs554405994 | 36 | 2 | 38 | 6.23 | 4.76 | 0.20 | 0.15 | 0.30 | 3.89 | |||
| Irinotecan | UGT1A1 | *6 | rs4148323 | 106 | 15 | 121 | 133 | 21.18 | 13.79 | 1.74 | 0.08 | 0.70 | 1.15 |
| *27 | rs35350960 | 12 | 0 | 12 | 1.87 | 1.39 | 0.00 | 0.00 | 0.00 | 0.00 | |||
| Clopidogrel | CYP2C19 | *2 | rs4244285 | 141 | 25 | 166 | 209 | 29.75 | 31.25 | 35.79 | 17.02 | 14.51 | 10.52 |
| *3 | rs4986893 | 33 | 1 | 34 | 5.45 | 5.56 | 1.23 | 0.23 | 0.00 | 0.00 | |||
| *4A/B | rs28399504 | 1 | 0 | 1 | 0.16 | 0.10 | 0.00 | 0.00 | 0.10 | 0.29 | |||
| *6 | rs72552267 | 1 | 0 | 1 | 0.16 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |||
| *17 | rs12248560 | 7 | 0 | 7 | 1.09 | 1.49 | 13.60 | 23.52 | 22.37 | 11.96 | |||
| Codeine | CYP2D6 | *4 | rs3892097 | 5 | 0 | 5 | 266 | 0.78 | 0.20 | 10.94 | 6.05 | 18.59 | 12.97 |
| *8 | rs5030865 | 10 | 1 | 11 | 1.87 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |||
| *10 | rs1065852 | 150 | 81 | 231 | 48.60 | 57.14 | 16.46 | 11.27 | 20.18 | 14.84 | |||
| *41 | rs28371725 | 17 | 2 | 19 | 3.27 | 3.77 | 12.17 | 1.82 | 9.34 | 6.20 | |||
1 denotes the default reference (wild type or fully functional) allele or haplotype, while other designations (e.g. *2 or *3) define haplotypes carrying one or more variants.
AFR, africans; AMR, Americans; EAS, East Asians; EUR, Europeans; Het, heterozygous; Hom, homozygous; MAF, minor allelic frequency; SAS, South Asians; $, The MAF of EAS, SAS, AFR, EUR, and AMR refers to the 1000 Genome phase 3 dataset.
Limitations of the current study are the small sample size and restricted metabolic tests. A large‐scale NBS effort is needed to validate our findings and fully investigate the treatable or curable medical conditions in newborns. The technical challenge of newborn WGS is to screen genes with high homology due to the misalignment of short‐read sequencing. Therefore, the customized pipeline is needed to improve the accuracy and sensitivity of SNVs at genes with high‐level homology. Albeit the present cost and turnaround time of WGS is several times more than the present NBS methods, in the forseeable future the pitfalls of WGS cost and turnaround time will likely facilitate the application of newborn WGS in NBS programs.
In our study, selective identification of genomic data, where therapeutic options are available, did not violate the Wilson–Jungner criteria 10 . Our work provides a basis for future research on expanding screening genes and diseases in newborn screening program. Given adequate cost‐effectiveness, WGS should be considered in future newborn screening programs. Further discussion of the interpretation accuracy and ethical use of genomic information needs to take place on a global scale.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGEMENTS
We appreciate the participation of the volunteers and their families. Without their support, this work would not have been possible. This work was also supported by the China National GeneBank (CNGB). This study was funded by the National Natural Science Foundation of China (No.31800765), the Shenzhen Municipal Government of China (JCY20170817145047361) and the Guangdong Provincial Key Laboratory of Genome Read and Write (No. 2017B030301011).
Min Jian, Xiaohong Wang, Yuanyuan Sui, Mingyan Fang, Lennart Hammarström, Xiaojing Jiang, Junnian Liu, Ya Gao contributed equally to this work.
Contributor Information
Lennart Hammarström, Email: lennart.hammarstrom.sr@gmail.com.
Xiaojing Jiang, Email: fuyou015@qd.shandong.cn.
Junnian Liu, Email: chris.liu@genomics.cn.
Ya Gao, Email: gaoya@genomics.cn.
REFERENCES
- 1. Ceyhan‐Birsoy O, Murry JB, Machini K, et al. Interpretation of genomic sequencing results in healthy and ill newborns: results from the BabySeq Project. Am J Hum Genet. 2019;104(1):76‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goldenberg AJ, Sharp RR. The ethical hazards and programmatic challenges of genomic newborn screening. JAMA. 2012;307(5):461‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Picard C, Al‐Herz W, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee report on inborn errors of immunity. J Clin Immunol. 2018;38(1):96‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Newborn screening: toward a uniform screening panel and system. Genet Med. 2006;8(Suppl 1):1s‐252s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Swen JJ, Nijenhuis M, van Rhenen M, et al. Pharmacogenetic information in clinical guidelines: the European perspective. Clin Pharmacol Ther. 2018;103(5):795‐801. [DOI] [PubMed] [Google Scholar]
- 6. Cao Y, Yuan P, Wang YP, Mao M, Zhu J. The profile of newborn screening coverage in China. J Med Screen. 2009;16(4):163‐166. [DOI] [PubMed] [Google Scholar]
- 7. Wang Q, Xiang J, Sun J, et al. Nationwide population genetic screening improves outcomes of newborn screening for hearing loss in China. Genet Med. 2019;21(10):2231‐2238. [DOI] [PubMed] [Google Scholar]
- 8. Reisberg S, Krebs K, Lepamets M, et al. Translating genotype data of 44,000 biobank participants into clinical pharmacogenetic recommendations: challenges and solutions. Genet Med. 2019;21(6):1345‐1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Whiffin N, Ware JS, O'Donnell‐Luria A. Improving the understanding of genetic variants in rare disease with large‐scale reference populations. JAMA. 2019;322(13):1305‐1306. [DOI] [PubMed] [Google Scholar]
- 10. Wilson JM, Jungner YG. Principles and practice of mass screening for disease. Bol Oficina Sanit Panam. 1968;65(4):281‐393. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information