

Abstract
To find a cure for cystic fibrosis, there has been tremendous progress in the development of treatments that target the basic defect in the protein channel, CFTR. However, 10% of cystic fibrosis patients have rare CFTR mutations that are still without an approved CFTR-targeting drug. To identify relevant therapies for these patients, culture models using nasal, bronchial, and rectal tissue from individual patients allow functional, biochemical, and cellular detection of drug-rescued CFTR. Additionally, novel systems such as induced pluripotent stem cell-derived models are utilized to characterize CFTR mutations and identify treatments. State-of-the art translational models were instrumental for CFTR modulator development and may become important for gene-based drug discovery and other novel therapeutic strategies.
Keywords: CFTR, modulator, primary human epithelial cells, cell model, organoid, spheroid
Introduction
Cystic fibrosis (CF) is an autosomal recessive disease resulting from mutations in the CF transmembrane conductance regulator (CFTR) gene [1], which encodes an ion channel that transports chloride and bicarbonate, playing an important role in hydration of many epithelial surfaces. After discovery of the CFTR gene in 1989, preclinical research with in vitro human cell models paved the way for the development of CFTR-targeting therapeutics that permit successful treatment of the basic defect in CF. These CFTR modulators are small-molecular compounds known as correctors that augment transfer of mutant CFTR to the apical membrane, and potentiators that increase CFTR channel activity [2].
90% of people with CF (pwCF) in North America, and 80% worldwide carry the F508del CFTR mutation. Unfortunately, 10% of the CF population or more, depending on ethnicity, have rare mutations for which CFTR modulator therapies are not available. To reach these patients and to improve upon available therapies, numerous CFTR-targeting compounds and reagents are currently in the clinical pipeline including novel mRNA- and DNA-based gene therapy therapeutics, read-through reagents for premature stop codons, and advanced modulator compounds (https://www.cff.org/Trials/Pipeline), and thus, personalized models for CF research remain in high demand for predicting drug efficacy for CF individuals.
CF research has developed and employed specific, physiologically relevant human assay systems to advance discovery of CF drugs. Primary human bronchial epithelial (HBE) and nasal epithelial (HNE) cells are typically grown at air-liquid interface (ALI) as planar cultures to study electrophysiological properties that reflect the function of epithelial ion channels such as CFTR [3–7]. Such models are imperative for efficiently identifying and screening compounds before they enter clinical trials, maximizing the likelihood of achieving clinically meaningful improvements in CFTR function, thus facilitating rapid progression of clinical trials toward more effective CF treatments. This review will highlight new and improved models that have been utilized for identifying effective CFTR-targeting therapies for pwCF.
CF Therapeutics
The first FDA-approved CFTR modulator was the potentiator ivacaftor (IVA), which is the active ingredient of a drug that improves the function of the CFTR gating mutant G551D [8,9]. While IVA or the CFTR corrector lumacaftor (LUM) alone did not significantly improve lung function in the CFTR folding mutant, F508del [10], combining LUM with IVA or combining the newer corrector tezacaftor (TEZ) with IVA resulted in modest lung function improvements in clinical trials in pwCF homozygous for F508del CFTR [11–14]. The recently approved triple therapy drug (next-generation corrector elexacaftor (ELX) with TEZ and IVA; ELX/TEZ/IVA) showed substantial efficacy in phase 3 clinical trials [15,16] and was more robust than IVA and dual combination therapies in some populations. The improvement in lung function measured as forced expiratory volume in 1 second percent predicted compared to sex- and age-matched healthy lungs (FEV1pp) in pwCF receiving ELX/TEZ/IVA is significant, with an overall improvement of FEV1pp of at least 10% [16,17], which is comparable to what was observed in G551D pwCF treated with IVA. In addition, improvement in the gastrointestinal (GI) system was observed in patients taking ELX/TEZ/IVA.
Personalized Models
The FDA approval of CFTR modulators for certain CFTR mutations was based on FRT cell line data. Although cell lines are useful to study mechanistic defects of CFTR mutations, they may not constitute a reliable physiological system for predicting all drug effects that are observed in epithelial tissues [18–21]. Some CFTR therapeutics such as read-through reagents prevented premature termination mutations in cell lines but did not work in primary epithelia where extensive nonsense-mediated mRNA decay is observed [22–25]. Identifying therapeutics that rescue CFTR splicing mutations are important for pwCF with such mutations. Studies on splicing mutations were previously conducted in cell lines but are more complicated in vivo and are therefore being intensively pursued [26]. In differentiated epithelia, CFTR localization and function are determined by cell-type specific expression and spatial interactions [27] and highly modulated by environmental factors such as inflammation [28].
Human in vitro models have been crucial for testing CFTR rescue in a relevant physiological environment. As patients respond differently to drugs, the most effective models for predicting clinical responses to therapeutics are cultures derived from tissue samples of individual patients; a personalized medicine approach. These cultures can be used in multiple assays to identify, optimize, and confirm therapies. In addition, it is important to have models available for examination of pharmacokinetics and pharmacodynamics of drugs [29].
Well-established in vitro models are differentiated cultures of HBE and HNE cells on membranes at ALI [30]. ALI culture protocol details (i.e., passage number of cells, differentiation time, type of media, supplements and insert type) may affect cell type composition, the magnitude of ion channel expression and activity, and the amount of CFTR protein that is rescued [31,32]. A common method to assess mutant CFTR rescue in these cultures is to observe electrophysiological responses of CFTR measured as short-circuit currents in Ussing chambers by treatment with forskolin that leads to CFTR activation by cAMP-dependent protein kinase, PKA. Basal CFTR currents before further activation by forskolin may also be indicative of restored CFTR function, as well as a diminution of the epithelial sodium channel (ENaC) activity. Furthermore, activation of CFTR by nucleotides (i.e., UTP or ATP) in the presence of the calcium-activated chloride channel (CaCC) TMEM16A inhibitor offers the possibility to evaluate an additional physiological activation route. Restoration of mucociliary clearance (MCC) of CF HNE and HBE ALI cultures is less commonly used for primary drug screenings; however, they can be investigated to evaluate the ability of a CFTR-targeting drug to restore airway surface liquid homeostasis, normalize mucin concentrations, and ciliary dynamics [33,34], which are all important components of a well-functioning MCC. A recent publication demonstrates the importance of CFTR modulators on airway hydration and mucus properties [33].
Patient specimens from airway and GI tissue can be used to generate organoids for testing rescue of mutant CFTR function [35–37]. Depending on the culture method, airway organoids can be oriented such that the apical membrane faces inward or outward, and rescue of mutant CFTR can be quantitated by changes in organoid size. These methods vary by tissue type and are described in more detail, below. An overview of the most commonly used models to evaluate CFTR therapeutics is shown in Figure 1.
Figure 1: Translational human in vitro models for CF research and drug discovery.
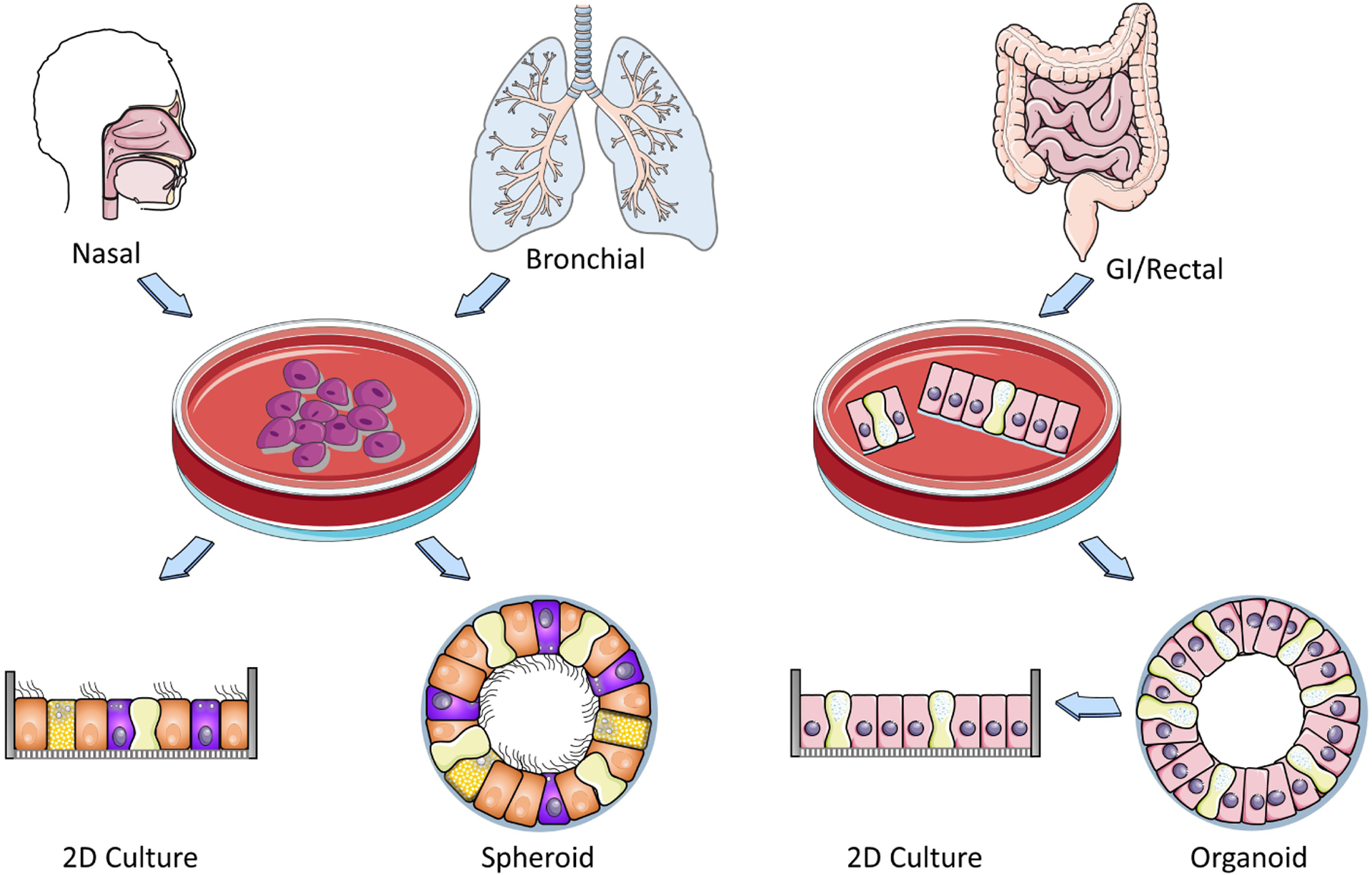
Processing and expansion of nasal bronchial and GI epithelial tissues to form 2D planar and 3D spheroid cultures. Expanded HNE and HBE cells are either seeded on membranes form planar cultures or in matrigel to form spheroids. GI organoids develop directly from partially digested tissues. Planar and spheroid cultures can be utilized to evaluate pharmacological rescue of CFTR rescue by various assays that evaluate CFTR function and maturation. Some image panels were obtained from Servier Medical Art (smart.servier.com).
Bronchial Cultures
Primary HBE cells derived from explant lungs of pwCF and cultured at ALI have been the gold standard for studying the efficacy of CFTR modulators [8,38,39]. In addition, HBE can be obtained from living patients via bronchial brushings, which provide the potential to identify optimal therapeutics for individual patients (personalized medicine). To expand resources available for testing of CFTR modulators, CF HBE cells can be conditionally reprogrammed and maintained to higher passage numbers [32]. Organoids/spheroids from airway epithelial cells are obtained by seeding of CF HBE in matrigel, in which the movement of ions by luminal CFTR channels drives fluid toward the center, resulting in swelling that is not observed in CF patient-derived organoids where CFTR is defective. These models are suitable to quantitate CFTR-targeting therapeutics that induce CFTR-dependent spheroid swelling and are also an appropriate model to study CF pathophysiology [37,40]. In addition, a novel method for generating bronchial organoids with externally oriented apical membranes in mixed matrix components was recently published [41]. Expanded cultures of bronchial epithelial cells can be utilized as an additional source of tissue to create spheroids for more testing of CFTR-targeting therapeutics.
Nasal Cultures
HNE cells have many properties in common with HBE cells and form polarized, pseudostratified epithelia mimicking in vivo airways and the expression of ion channels including CFTR, ENaC, and CaCCs. Similar to HBE cells, HNE cells can be derived by brushing or scraping but the collection is far less invasive than for HBE cells [42]. Thus, patient-derived HNE cultures differentiated at ALI have become a standard model in CFTR modulator testing [42–49]. Epithelial cell types such as ionocytes, ciliated cells, and secretory cells differ between nasal cells and large (bronchi) and small (bronchioles) airways [50–53]; however, HNE cultures appear to recapitulate many of the bioelectric properties of differentiated HBE and respond in a similar fashion to CFTR modulators [42,44,54]. HBE and HNE cultures can be used to evaluate the effects of CFTR modulators on reversing secondary CF phenotypes such as decreased MCC. Similar to HBE, conditionally reprogramming of HNE is available as a method to expand patient culture lifetime without majorly affecting CFTR function [55,56]. CFTR-mediated chloride currents in HNE cells correlated with patients’ sweat chloride concentrations, a common method to detect CF [55]. Thus, HNE cells are recognized as a non-invasive surrogate for HBE cells in many preclinical studies of CFTR modulators [36,42–46,55,57–63]. Patient-derived nasal tissue can be grown in suspension, creating nasospheroids with CFTR channels on the outer surface [36]. Upon rescue of mutant CFTR, ion transport and fluid will move outward toward the media, causing the spheroids to shrink, which can be quantitated [36].
Intestinal Cultures
In 2013, it was shown that organoids derived from patients’ intestines can be utilized to study drug rescue of mutant CFTR function [64]. A recent study showed that intestinal organoids can be used to test for CFTR rescue of nonsense mutations, in which 5 different therapies were combined: the 3 CFTR modulators in ELX/TEZ/IVA plus a compound that induces translational readthrough and a compound that inhibits nonsense mRNA-mediated decay. This is very promising for pwCF with nonsense CFTR mutations that are not eligible for ELX/TEZ/IVA [65]. Another recent study using intestinal organoids showed the importance of testing compounds from different companies to optimize efficacy of F508del CFTR rescue [66]. Although F508del CFTR in patient-derived cultures typically responds well to rescue by triple therapy, cultures from different patients with the same CFTR genotype do not respond in a similar fashion, which may be due to genetic traits other than CFTR that may affect CFTR modulator efficacy and therefore should be further examined [67,68]. Additional models using different intestinal tissues and different culture methods/scaffolds may also have the potential to be used for CF research [69–71].
As clinical improvement of pwCF and in vitro readout by rectal organoids appear to be correlated [65,72,73], large efforts such as HIT-CF Europe aim to expand organoid-based screenings of drugs from multiple companies to pwCF with rare mutations (https://www.hitcf.org/). Furthermore, GI organoids are not only utilized for selection of optimized treatments for rare CFTR mutations but also for evaluation of novel read-through therapies for nonsense mutations and gene therapeutic approaches [74,75]. Additionally, GI organoids can be seeded on membranes to form monolayers that develop to differentiated cultures that are analyzed electrophysiologically in Ussing chambers to study restoration of CFTR-mediated currents in CF planar cultures [76]. Recent publications demonstrate the power of directly comparing clinical data with in vitro data from patient-derived intestinal cultures [77,78]. GI organoids are the most straightforward model to develop from only partially digested tissue specimens. These can be subsequently cultured on inserts as monolayers, offering the following advantages: 1) they can be analyzed in Ussing chambers, and 2) the apical surface is directly accessible for treatments.
Other Gastrointestinal Cultures
CF patients frequently suffer from CF-related diabetes (CFRD), which can lead to glucose imbalance that can in turn, augment the severity of CF disease. It is therefore important to develop a relevant pancreas model for testing potential therapies that improve these glucose imbalances. A pancreas-on-a-chip model was developed using patient-derived pancreatic ductal epithelial cells (PDECs) and pancreatic islets, allowing for examination of the relationship between these cell types [79]. CFTR is expressed in PDECs and inhibition of CFTR channel function leads to a decrease in insulin secretion. To alleviate the effects of CFRD, this pancreas-on-a-chip model can be utilized to test CFTR modulators for their ability to improve glucose imbalances in individual patients. In addition, patient-derived cultures of biliary tissue (cholangiocytes) can also be used to test for rescue of CFTR function [80,81].
Induced Pluripotent Stem Cells
Induced pluripotent stem cells (iPSCs) are adult somatic (e.g., skin or blood) cells that have been reprogrammed, bringing the cells back to an embryonic-like pluripotent state [82]. This allows the creation of an unlimited source of any type of human cell for therapeutic testing. Although creating lung tissue from stem cells is very complicated, in 2015, iPSCs were created from F508del pwCF, corrected with wild-type CFTR gene sequences, and then differentiated into airway epithelial cells [83]. iPSC-derived lung epithelium can be used to generate lung organoids that mimic lung tissue [84–86]. Furthermore, iPSCs can be used to model defects in ciliary function, which can be beneficial for measuring defective MCC in CF cultures [87]. iPSC-derived lung progenitor cells can be set up in a high-throughput platform, allowing studies that measure rescue of mutant CFTR function [88]. In addition, for GI studies, iPSCs were used to create pancreatic duct-like organoids that expressed CFTR [89]. Overall, iPSC technology may be particularly beneficial for pwCF with rare CFTR mutations such as nonsense mutations that do not yet have approved CFTR-targeting therapies.
Preclinical Models in Development
Exciting developments are ongoing using cells not only from various GI segments, but also other tissues such as sweat ducts, submucosal glands, and liver [90–92] for translation modeling of CF. As CF disease is thought to be initiated in small airways, HBE cells from small and large airways are utilized as separate models to display typical characteristics of cell populations found in these specific regions [50]. To study engraftment with HBE cultures expressing wild-type CFTR, a cell therapy approach was developed involving repopulation CF HBE cultures, thereby creating a population of cells that express CFTR with normal function [93]. Advanced models aim to incorporate environmental conditions found in the CF lungs such as infection, inflammation, mucus burden, and hypoxia [28,94]. In testing various drugs for their ability to repair the defects of CFTR mutations, it is important to consider how the efficacy of drugs may be affected by nearby tissues, cells, and secreted reagents such as the endothelium, immune system cells and cytokines, and bacteria. To address this, a CF Airway Chip was created that includes CF bronchial epithelial cells grown at air-liquid interface, lung endothelium, and dynamic fluid flow that can deliver immune cells [95]. These CF Airway Chips accurately represent the human airway in vivo, allowing direct testing of various drugs to rescue rare CFTR mutations. Other organs-on-a-chip are also in development for CF research, including multi-tissue organs-on-a-chip to address the effects of different organs on each other [96,97].
Conclusions
In vitro models derived from tissues of pwCF are vital for the development of effective CFTR therapeutics. Personalized models are important as patients respond differently to treatments. Using tissue that is relevant to CF (bronchial, nasal, and GI epithelial cells) collected from pwCF for drug testing is an effective method for identifying and optimizing therapeutics for each patient. Cultures grown in planar (2D) and spheroid or organoid (3D) formats are used as models of CF disease in multiple drug testing assays. These in vitro translational models have been crucial for understanding CF pathophysiology and CFTR regulation and were instrumental in identifying effective CFTR modulators for pwCF. Newer models such as iPSC-derived cultures and organs-on-a-chip allow the use of additional material and more advanced methods, respectively. In the future, researchers will continue to tackle remaining complicated cellular issues with models that simulate inflammation, infection, mucus, and MCC, and enable gene therapy studies that accurately predict clinical outcomes for pwCF in need of effective therapies.
Highlights.
Relevant models of CF are critical for predicting clinical outcomes of therapeutics
Nasal, bronchial, and rectal 2D and 3D cultures have been key for drug discovery
Personalized medicine is important as individuals respond differently to drugs
Novel models such as iPSCs and organs-on-a-chip will improve drug testing
Advanced personalized models will be useful for future gene therapy drug discovery
Acknowledgements
We thank members of the Marsico Lung Institute, UNC Cystic Fibrosis Research and Translation Core Center and Research and Development Program Cores for their contributions to translation research discussed here. Related research was supported by the Cystic Fibrosis Foundation (GENTZS18P0, GENTZS19I0, BOUCHE19R0) and the National Institutes of Health (P30DK065988).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest:
M. Gentzsch and D. M. Cholon declare no potential conflict of interest.
References
- 1.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, et al. : Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989, 245:1066–1073. [DOI] [PubMed] [Google Scholar]
- 2.Gentzsch M, Mall MA: Ion Channel Modulators in Cystic Fibrosis. Chest 2018, 154:383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Randell SH, Fulcher ML, O’Neal W, Olsen JC: Primary epithelial cell models for cystic fibrosis research. Methods Mol Biol 2011, 742:285–310. [DOI] [PubMed] [Google Scholar]
- 4.Muller L, Brighton LE, Carson JL, Fischer WA 2nd, Jaspers I: Culturing of human nasal epithelial cells at the air liquid interface. J Vis Exp 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Courcey F, Zholos AV, Atherton-Watson H, Williams MT, Canning P, Danahay HL, Elborn JS, Ennis M: Development of primary human nasal epithelial cell cultures for the study of cystic fibrosis pathophysiology. Am J Physiol Cell Physiol 2012, 303:C1173–1179. [DOI] [PubMed] [Google Scholar]
- 6.Wiszniewski L, Jornot L, Dudez T, Pagano A, Rochat T, Lacroix JS, Suter S, Chanson M: Long-term cultures of polarized airway epithelial cells from patients with cystic fibrosis. Am J Respir Cell Mol Biol 2006, 34:39–48. [DOI] [PubMed] [Google Scholar]
- 7.Mosler K, Coraux C, Fragaki K, Zahm JM, Bajolet O, Bessaci-Kabouya K, Puchelle E, Abely M, Mauran P: Feasibility of nasal epithelial brushing for the study of airway epithelial functions in CF infants. J Cyst Fibros 2008, 7:44–53. [DOI] [PubMed] [Google Scholar]
- 8.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, et al. : Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A 2009, 106:18825–18830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, Griese M, McKone EF, Wainwright CE, Konstan MW, et al. : A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011, 365:1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, Ballmann M, Boyle MP, Bronsveld I, Campbell PW, et al. : Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012, 67:12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyle MP, Bell SC, Konstan MW, McColley SA, Rowe SM, Rietschel E, Huang X, Waltz D, Patel NR, Rodman D: A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med 2014, 2:527–538. [DOI] [PubMed] [Google Scholar]
- 12.Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, Colombo C, Davies JC, De Boeck K, Flume PA, et al. : Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med 2015, 373:220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donaldson SH, Pilewski JM, Griese M, Cooke J, Viswanathan L, Tullis E, Davies JC, Lekstrom-Himes JA, Wang LT: Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am J Respir Crit Care Med 2018, 197:214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor-Cousar JL, Munck A, McKone EF, van der Ent CK, Moeller A, Simard C, Wang LT, Ingenito EP, McKee C, Lu Y, et al. : Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N Engl J Med 2017, 377:2013–2023. [DOI] [PubMed] [Google Scholar]
- 15.Heijerman HGM, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, Mall MA, Welter JJ, Ramsey BW, McKee CM, et al. : Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet 2019, 394:1940–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Middleton PG, Mall MA, Drevinek P, Lands LC, McKone EF, Polineni D, Ramsey BW, Taylor-Cousar JL, Tullis E, Vermeulen F, et al. : Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med 2019, 381:1809–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zemanick ET, Taylor-Cousar JL, Davies J, Gibson RL, Mall MA, McKone EF, McNally P, Ramsey BW, Rayment JH, Rowe SM, et al. : A Phase 3 Open-Label Study of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 through 11 Years of Age with Cystic Fibrosis and at Least One F508del Allele. Am J Respir Crit Care Med 2021, 203:1522–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pedemonte N, Tomati V, Sondo E, Galietta LJ: Influence of cell background on pharmacological rescue of mutant CFTR. Am J Physiol Cell Physiol 2010, 298:C866–874. [DOI] [PubMed] [Google Scholar]
- 19.Rowe SM, Pyle LC, Jurkevante A, Varga K, Collawn J, Sloane PA, Woodworth B, Mazur M, Fulton J, Fan L, et al. : DeltaF508 CFTR processing correction and activity in polarized airway and non-airway cell monolayers. Pulm Pharmacol Ther 2010, 23:268–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ostedgaard LS, Rogers CS, Dong Q, Randak CO, Vermeer DW, Rokhlina T, Karp PH, Welsh MJ: Processing and function of CFTR-DeltaF508 are species-dependent. Proc Natl Acad Sci U S A 2007, 104:15370–15375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bebok Z, Collawn JF, Wakefield J, Parker W, Li Y, Varga K, Sorscher EJ, Clancy JP: Failure of cAMP agonists to activate rescued deltaF508 CFTR in CFBE41o- airway epithelial monolayers. J Physiol 2005, 569:601–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haggie PM, Phuan PW, Tan JA, Xu H, Avramescu RG, Perdomo D, Zlock L, Nielson DW, Finkbeiner WE, Lukacs GL, et al. : Correctors and Potentiators Rescue Function of the Truncated W1282X-Cystic Fibrosis Transmembrane Regulator (CFTR) Translation Product. J Biol Chem 2017, 292:771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mutyam V, Libby EF, Peng N, Hadjiliadis D, Bonk M, Solomon GM, Rowe SM: Therapeutic benefit observed with the CFTR potentiator, ivacaftor, in a CF patient homozygous for the W1282X CFTR nonsense mutation. J Cyst Fibros 2017, 16:24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamosh A, Rosenstein BJ, Cutting GR: CFTR nonsense mutations G542X and W1282X associated with severe reduction of CFTR mRNA in nasal epithelial cells. Hum Mol Genet 1992, 1:542–544. [DOI] [PubMed] [Google Scholar]
- 25.Aksit MA, Bowling AD, Evans TA, Joynt AT, Osorio D, Patel S, West N, Merlo C, Sosnay PR, Cutting GR, et al. : Decreased mRNA and protein stability of W1282X limits response to modulator therapy. J Cyst Fibros 2019, 18:606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dang Y, van Heusden C, Nickerson V, Chung F, Wang Y, Quinney NL, Gentzsch M, Randell SH, Moulton HM, Kole R, et al. : Enhanced delivery of peptide-morpholino oligonucleotides with a small molecule to correct splicing defects in the lung. Nucleic Acids Res 2021, 49:6100–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gentzsch M, Farinha CM: Revisiting CFTR Interactions: Old Partners and New Players. Int J Mol Sci 2021, 22:13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ribeiro CMP, Gentzsch M: Impact of Airway Inflammation on the Efficacy of CFTR Modulators. Cells 2021, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guhr Lee TN, Cholon DM, Quinney NL, Gentzsch M, Esther CR Jr.: Accumulation and persistence of ivacaftor in airway epithelia with prolonged treatment. J Cyst Fibros 2020, 19:746–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fulcher ML, Randell SH: Human nasal and tracheo-bronchial respiratory epithelial cell culture. Methods Mol Biol 2013, 945:109–121. [DOI] [PubMed] [Google Scholar]
- 31.Rayner RE, Makena P, Prasad GL, Cormet-Boyaka E: Optimization of Normal Human Bronchial Epithelial (NHBE) Cell 3D Cultures for in vitro Lung Model Studies. Sci Rep 2019, 9:500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gentzsch M, Boyles SE, Cheluvaraju C, Chaudhry IG, Quinney NL, Cho C, Dang H, Liu X, Schlegel R, Randell SH: Pharmacological Rescue of Conditionally Reprogrammed Cystic Fibrosis Bronchial Epithelial Cells. Am J Respir Cell Mol Biol 2017, 56:568–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morrison CB, Shaffer KM, Araba KC, Markovetz MR, Wykoff JA, Quinney NL, Hao S, Delion MF, Flen AL, Morton LC, et al. : Treatment of cystic fibrosis airway cells with CFTR modulators reverses aberrant mucus properties via hydration. Eur Respir J 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chioccioli M, Feriani L, Kotar J, Bratcher PE, Cicuta P: Phenotyping ciliary dynamics and coordination in response to CFTR-modulators in Cystic Fibrosis respiratory epithelial cells. Nat Commun 2019, 10:1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boj SF, Vonk AM, Statia M, Su J, Vries RR, Beekman JM, Clevers H: Forskolin-induced Swelling in Intestinal Organoids: An In Vitro Assay for Assessing Drug Response in Cystic Fibrosis Patients. J Vis Exp 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guimbellot JS, Leach JM, Chaudhry IG, Quinney NL, Boyles SE, Chua M, Aban I, Jaspers I, Gentzsch M: Nasospheroids permit measurements of CFTR-dependent fluid transport. JCI Insight 2017, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sachs N, Papaspyropoulos A, Zomer-van Ommen DD, Heo I, Bottinger L, Klay D, Weeber F, Huelsz-Prince G, Iakobachvili N, Amatngalim GD, et al. : Long-term expanding human airway organoids for disease modeling. EMBO J 2019, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, et al. : Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A 2011, 108:18843–18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keating D, Marigowda G, Burr L, Daines C, Mall MA, McKone EF, Ramsey BW, Rowe SM, Sass LA, Tullis E, et al. : VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N Engl J Med 2018, 379:1612–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cholon DM, Gentzsch M: Recent progress in translational cystic fibrosis research using precision medicine strategies. J Cyst Fibros 2018, 17:S52–S60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boecking CA, Walentek P, Zlock LT, Sun DI, Wolters PJ, Ishikawa H, Jin BJ, Haggie PM, Marshall WF, Verkman AS, et al. : A simple method to generate human airway epithelial organoids with externally-oriented apical membranes. Am J Physiol Lung Cell Mol Physiol 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brewington JJ, Filbrandt ET, LaRosa FJ 3rd, Moncivaiz JD, Ostmann AJ, Strecker LM, Clancy JP: Brushed nasal epithelial cells are a surrogate for bronchial epithelial CFTR studies. JCI Insight 2018, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pranke IM, Hatton A, Simonin J, Jais JP, Le Pimpec-Barthes F, Carsin A, Bonnette P, Fayon M, Stremler-Le Bel N, Grenet D, et al. : Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci Rep 2017, 7:7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kmit A, Marson FAL, Pereira SV, Vinagre AM, Leite GS, Servidoni MF, Ribeiro JD, Ribeiro AF, Bertuzzo CS, Amaral MD: Extent of rescue of F508del-CFTR function by VX-809 and VX-770 in human nasal epithelial cells correlates with SNP rs7512462 in SLC26A9 gene in F508del/F508del Cystic Fibrosis patients. Biochim Biophys Acta Mol Basis Dis 2019, 1865:1323–1331. [DOI] [PubMed] [Google Scholar]
- 45.Clarke LA, Awatade NT, Felicio VM, Silva IA, Calucho M, Pereira L, Azevedo P, Cavaco J, Barreto C, Bertuzzo C, et al. : The effect of premature termination codon mutations on CFTR mRNA abundance in human nasal epithelium and intestinal organoids: a basis for read-through therapies in cystic fibrosis. Hum Mutat 2019, 40:326–334. [DOI] [PubMed] [Google Scholar]
- 46.Leir SH, Yin S, Kerschner JL, Xia S, Ahmadi S, Bear C, Harris A: An organoid model to assay the role of CFTR in the human epididymis epithelium. Cell Tissue Res 2020, 381:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laselva O, Moraes TJ, He G, Bartlett C, Szarics I, Ouyang H, Gunawardena TNA, Strug L, Bear CE, Gonska T: The CFTR Mutation c.3453G > C (D1152H) Confers an Anion Selectivity Defect in Primary Airway Tissue that Can Be Rescued by Ivacaftor. J Pers Med 2020, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laselva O, Bartlett C, Popa A, Ouyang H, Gunawardena TNA, Gonska T, Moraes TJ, Bear CE: Emerging preclinical modulators developed for F508del-CFTR have the potential to be effective for ORKAMBI resistant processing mutants. J Cyst Fibros 2021, 20:106–119. [DOI] [PubMed] [Google Scholar]
- 49.Pedemonte N: Nasal epithelial cells as a gold-standard predictive model for personalized medicine in cystic fibrosis. J Physiol 2022. [DOI] [PubMed] [Google Scholar]
- 50.Okuda K, Dang H, Kobayashi Y, Carraro G, Nakano S, Chen G, Kato T, Asakura T, Gilmore RC, Morton LC, et al. : Secretory Cells Dominate Airway CFTR Expression and Function in Human Airway Superficial Epithelia. Am J Respir Crit Care Med 2021, 203:1275–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scudieri P, Musante I, Venturini A, Guidone D, Genovese M, Cresta F, Caci E, Palleschi A, Poeta M, Santamaria F, et al. : Ionocytes and CFTR Chloride Channel Expression in Normal and Cystic Fibrosis Nasal and Bronchial Epithelial Cells. Cells 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Plasschaert LW, Zilionis R, Choo-Wing R, Savova V, Knehr J, Roma G, Klein AM, Jaffe AB: A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560:377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, Yuan F, Chen S, Leung HM, Villoria J, et al. : A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560:319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anderson JD, Liu Z, Odom LV, Kersh L, Guimbellot JS: CFTR function and clinical response to modulators parallel nasal epithelial organoid swelling. Am J Physiol Lung Cell Mol Physiol 2021, 321:L119–L129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Noel S, Servel N, Hatton A, Golec A, Rodrat M, Ng DRS, Li H, Pranke I, Hinzpeter A, Edelman A, et al. : Correlating genotype with phenotype using CFTR-mediated whole-cell Cl(−) currents in human nasal epithelial cells. J Physiol 2021. [DOI] [PubMed] [Google Scholar]
- 56.Sette G, Lo Cicero S, Blacona G, Pierandrei S, Bruno SM, Salvati V, Castelli G, Falchi M, Fabrizzi B, Cimino G, et al. : Theratyping cystic fibrosis in vitro in ALI culture and organoid models generated from patient-derived nasal epithelial conditionally reprogrammed stem cells. Eur Respir J 2021, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McCarthy C, Brewington JJ, Harkness B, Clancy JP, Trapnell BC: Personalised CFTR pharmacotherapeutic response testing and therapy of cystic fibrosis. Eur Respir J 2018, 51. [DOI] [PubMed] [Google Scholar]
- 58.Brewington JJ, Filbrandt ET, LaRosa FJ 3rd, Ostmann AJ, Strecker LM, Szczesniak RD, Clancy JP: Detection of CFTR function and modulation in primary human nasal cell spheroids. J Cyst Fibros 2018, 17:26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brewington JJ, Filbrandt ET, LaRosa FJ 3rd, Moncivaiz JD, Ostmann AJ, Strecker LM, Clancy JP: Generation of Human Nasal Epithelial Cell Spheroids for Individualized Cystic Fibrosis Transmembrane Conductance Regulator Study. J Vis Exp 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McCravy MS, Quinney NL, Cholon DM, Boyles SE, Jensen TJ, Aleksandrov AA, Donaldson SH, Noone PG, Gentzsch M: Personalised medicine for non-classic cystic fibrosis resulting from rare CFTR mutations. Eur Respir J 2020, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gentzsch M, Ren HY, Houck SA, Quinney NL, Cholon DM, Sopha P, Chaudhry IG, Das J, Dokholyan NV, Randell SH, et al. : Restoration of R117H CFTR folding and function in human airway cells through combination treatment with VX-809 and VX-770. Am J Physiol Lung Cell Mol Physiol 2016, 311:L550–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Laselva O, McCormack J, Bartlett C, Ip W, Gunawardena TNA, Ouyang H, Eckford PDW, Gonska T, Moraes TJ, Bear CE: Preclinical Studies of a Rare CF-Causing Mutation in the Second Nucleotide Binding Domain (c.3700A>G) Show Robust Functional Rescue in Primary Nasal Cultures by Novel CFTR Modulators. J Pers Med 2020, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laselva O, Bartlett C, Gunawardena TNA, Ouyang H, Eckford PDW, Moraes TJ, Bear CE, Gonska T: Rescue of multiple class II CFTR mutations by elexacaftor+tezacaftor+ivacaftor mediated in part by the dual activities of elexacaftor as both corrector and potentiator. Eur Respir J 2021, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dekkers JF, Wiegerinck CL, de Jonge HR, Bronsveld I, Janssens HM, de Winter-de Groot KM, Brandsma AM, de Jong NW, Bijvelds MJ, Scholte BJ, et al. : A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med 2013, 19:939–945. [DOI] [PubMed] [Google Scholar]
- 65.de Poel E, Spelier S, Suen SWF, Kruisselbrink E, Graeber SY, Mall MA, Weersink EJM, van der Eerden MM, Koppelman GH, van der Ent CK, et al. : Functional Restoration of CFTR Nonsense Mutations in Intestinal Organoids. J Cyst Fibros 2021. [DOI] [PubMed] [Google Scholar]
- 66.de Poel E, Spelier S, Korporaal R, Lai KW, Boj SF, Conrath K, van der Ent CK, Beekman JM: CFTR Rescue in Intestinal Organoids with GLPG/ABBV-2737, ABBV/GLPG-2222 and ABBV/GLPG-2451 Triple Therapy. Front Mol Biosci 2021, 8:698358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Furstova E, Dousova T, Beranek J, Libik M, Fila L, Modrak M, Cinek O, Macek M Jr., Drevinek P: Response to elexacaftor/tezacaftor/ivacaftor in intestinal organoids derived from people with cystic fibrosis. J Cyst Fibros 2021. [DOI] [PubMed] [Google Scholar]
- 68.Gong J, Wang F, Xiao B, Panjwani N, Lin F, Keenan K, Avolio J, Esmaeili M, Zhang L, He G, et al. : Genetic association and transcriptome integration identify contributing genes and tissues at cystic fibrosis modifier loci. PLoS Genet 2019, 15:e1008007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Y, DiSalvo M, Gunasekara DB, Dutton J, Proctor A, Lebhar MS, Williamson IA, Speer J, Howard RL, Smiddy NM, et al. : Self-renewing Monolayer of Primary Colonic or Rectal Epithelial Cells. Cell Mol Gastroenterol Hepatol 2017, 4:165–182 e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gunasekara DB, Speer J, Wang Y, Nguyen DL, Reed MI, Smiddy NM, Parker JS, Fallon JK, Smith PC, Sims CE, et al. : A Monolayer of Primary Colonic Epithelium Generated on a Scaffold with a Gradient of Stiffness for Drug Transport Studies. Anal Chem 2018, 90:13331–13340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nikolaev M, Mitrofanova O, Broguiere N, Geraldo S, Dutta D, Tabata Y, Elci B, Brandenberg N, Kolotuev I, Gjorevski N, et al. : Homeostatic mini-intestines through scaffold-guided organoid morphogenesis. Nature 2020, 585:574–578. [DOI] [PubMed] [Google Scholar]
- 72.Ramalho AS, Furstova E, Vonk AM, Ferrante M, Verfaillie C, Dupont L, Boon M, Proesmans M, Beekman JM, Sarouk I, et al. : Correction of CFTR function in intestinal organoids to guide treatment of cystic fibrosis. Eur Respir J 2021, 57. [DOI] [PubMed] [Google Scholar]
- 73.Berkers G, van Mourik P, Vonk AM, Kruisselbrink E, Dekkers JF, de Winter-de Groot KM, Arets HGM, Marck-van der Wilt REP, Dijkema JS, Vanderschuren MM, et al. : Rectal Organoids Enable Personalized Treatment of Cystic Fibrosis. Cell Rep 2019, 26:1701–1708 e1703. [DOI] [PubMed] [Google Scholar]
- 74.Crawford DK, Mullenders J, Pott J, Boj SF, Landskroner-Eiger S, Goddeeris MM: Targeting G542X CFTR nonsense alleles with ELX-02 restores CFTR function in human-derived intestinal organoids. J Cyst Fibros 2021, 20:436–442. [DOI] [PubMed] [Google Scholar]
- 75.Geurts MH, de Poel E, Amatngalim GD, Oka R, Meijers FM, Kruisselbrink E, van Mourik P, Berkers G, de Winter-de Groot KM, Michel S, et al. : CRISPR-Based Adenine Editors Correct Nonsense Mutations in a Cystic Fibrosis Organoid Biobank. Cell Stem Cell 2020, 26:503–510 e507. [DOI] [PubMed] [Google Scholar]
- 76.Zomer-van Ommen DD, de Poel E, Kruisselbrink E, Oppelaar H, Vonk AM, Janssens HM, van der Ent CK, Hagemeijer MC, Beekman JM: Comparison of ex vivo and in vitro intestinal cystic fibrosis models to measure CFTR-dependent ion channel activity. J Cyst Fibros 2018, 17:316–324. [DOI] [PubMed] [Google Scholar]
- 77.Arora K, Yang F, Brewington J, McPhail G, Cortez AR, Sundaram N, Ramananda Y, Ogden H, Helmrath M, Clancy JP, et al. : Patient personalized translational tools in cystic fibrosis to transform data from bench to bed-side and back. Am J Physiol Gastrointest Liver Physiol 2021, 320:G1123–G1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Muilwijk D, de Poel E, van Mourik P, Suen SWF, Vonk AM, Brunsveld JE, Kruisselbrink E, Oppelaar H, Hagemeijer MC, Berkers G, et al. : Forskolin-induced Organoid Swelling is Associated with Long-term CF Disease Progression. Eur Respir J 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shik Mun K, Arora K, Huang Y, Yang F, Yarlagadda S, Ramananda Y, Abu-El-Haija M, Palermo JJ, Appakalai BN, Nathan JD, et al. : Patient-derived pancreas-on-a-chip to model cystic fibrosis-related disorders. Nat Commun 2019, 10:3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bijvelds MJC, Roos FJM, Meijsen KF, Roest HP, Verstegen MMA, Janssens HM, van der Laan LJW, de Jonge HR: Rescue of chloride and bicarbonate transport by elexacaftor-ivacaftor-tezacaftor in organoid-derived CF intestinal and cholangiocyte monolayers. J Cyst Fibros 2021. [DOI] [PubMed] [Google Scholar]
- 81.Verstegen MMA, Roos FJM, Burka K, Gehart H, Jager M, de Wolf M, Bijvelds MJC, de Jonge HR, Ardisasmita AI, van Huizen NA, et al. : Human extrahepatic and intrahepatic cholangiocyte organoids show region-specific differentiation potential and model cystic fibrosis-related bile duct disease. Sci Rep 2020, 10:21900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brennan LC, O’Sullivan A, MacLoughlin R: Cellular Therapy for the Treatment of Paediatric Respiratory Disease. Int J Mol Sci 2021, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Firth AL, Menon T, Parker GS, Qualls SJ, Lewis BM, Ke E, Dargitz CT, Wright R, Khanna A, Gage FH, et al. : Functional Gene Correction for Cystic Fibrosis in Lung Epithelial Cells Generated from Patient iPSCs. Cell Rep 2015, 12:1385–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen YW, Huang SX, de Carvalho A, Ho SH, Islam MN, Volpi S, Notarangelo LD, Ciancanelli M, Casanova JL, Bhattacharya J, et al. : A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat Cell Biol 2017, 19:542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hawkins F, Kramer P, Jacob A, Driver I, Thomas DC, McCauley KB, Skvir N, Crane AM, Kurmann AA, Hollenberg AN, et al. : Prospective isolation of NKX2–1-expressing human lung progenitors derived from pluripotent stem cells. J Clin Invest 2017, 127:2277–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Miller AJ, Hill DR, Nagy MS, Aoki Y, Dye BR, Chin AM, Huang S, Zhu F, White ES, Lama V, et al. : In Vitro Induction and In Vivo Engraftment of Lung Bud Tip Progenitor Cells Derived from Human Pluripotent Stem Cells. Stem Cell Reports 2018, 10:101–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sone N, Konishi S, Igura K, Tamai K, Ikeo S, Korogi Y, Kanagaki S, Namba T, Yamamoto Y, Xu Y, et al. : Multicellular modeling of ciliopathy by combining iPS cells and microfluidic airway-on-a-chip technology. Sci Transl Med 2021, 13. [DOI] [PubMed] [Google Scholar]
- 88.Jiang JX, Wellhauser L, Laselva O, Utkina I, Bozoky Z, Gunawardena T, Ngan Z, Xia S, Di Paola M, Eckford PDW, et al. : A new platform for high-throughput therapy testing on iPSC-derived lung progenitor cells from cystic fibrosis patients. Stem Cell Reports 2021, 16:2825–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wiedenmann S, Breunig M, Merkle J, von Toerne C, Georgiev T, Moussus M, Schulte L, Seufferlein T, Sterr M, Lickert H, et al. : Single-cell-resolved differentiation of human induced pluripotent stem cells into pancreatic duct-like organoids on a microwell chip. Nat Biomed Eng 2021, 5:897–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Klaka P, Grudl S, Banowski B, Giesen M, Sattler A, Proksch P, Welss T, Forster T: A novel organotypic 3D sweat gland model with physiological functionality. PLoS One 2017, 12:e0182752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McMahon DB, Carey RM, Kohanski MA, Tong CCL, Papagiannopoulos P, Adappa ND, Palmer JN, Lee RJ: Neuropeptide regulation of secretion and inflammation in human airway gland serous cells. Eur Respir J 2020, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Leite SB, Roosens T, El Taghdouini A, Mannaerts I, Smout AJ, Najimi M, Sokal E, Noor F, Chesne C, van Grunsven LA: Novel human hepatic organoid model enables testing of drug-induced liver fibrosis in vitro. Biomaterials 2016, 78:1–10. [DOI] [PubMed] [Google Scholar]
- 93.Lee RE, Miller SM, Mascenik TM, Lewis CA, Dang H, Boggs ZH, Tarran R, Randell SH: Assessing Human Airway Epithelial Progenitor Cells for Cystic Fibrosis Cell Therapy. Am J Respir Cell Mol Biol 2020, 63:374–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.O’Toole GA, Crabbe A, Kummerli R, LiPuma JJ, Bomberger JM, Davies JC, Limoli D, Phelan VV, Bliska JB, DePas WH, et al. : Model Systems to Study the Chronic, Polymicrobial Infections in Cystic Fibrosis: Current Approaches and Exploring Future Directions. mBio 2021, 12:e0176321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Plebani R, Potla R, Soong M, Bai H, Izadifar Z, Jiang A, Travis RN, Belgur C, Dinis A, Cartwright MJ, et al. : Modeling pulmonary cystic fibrosis in a human lung airway-on-a-chip: Cystic fibrosis airway chip. J Cyst Fibros 2021. [DOI] [PubMed] [Google Scholar]
- 96.Ogden HL, Kim H, Wikenheiser-Brokamp KA, Naren AP, Mun KS: Cystic Fibrosis Human Organs-on-a-Chip. Micromachines (Basel) 2021, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rajan SAP, Aleman J, Wan M, Pourhabibi Zarandi N, Nzou G, Murphy S, Bishop CE, Sadri-Ardekani H, Shupe T, Atala A, et al. : Probing prodrug metabolism and reciprocal toxicity with an integrated and humanized multi-tissue organ-on-a-chip platform. Acta Biomater 2020, 106:124–135. [DOI] [PMC free article] [PubMed] [Google Scholar]