

Abstract
Late-onset Alzheimer’s disease (LOAD) is significantly more frequent in Hispanics than in non-Hispanic Whites. Ancestry may explain these differences across ethnic groups. To this end, we studied a large cohort of Caribbean Hispanics (CH, N=8,813) and tested the association between Local Ancestry (LA) and LOAD (“admixture mapping”) to identify LOAD-associated ancestral blocks, separately for ancestral components (European [EUR], African [AFR], Native American[NA]) and jointly (AFR+NA). Ancestral blocks significant after permutation were fine-mapped employing multi-ethnic whole-exome sequencing (WES) to identify rare variants associated with LOAD (SKAT-O) and replicated in the UK Biobank WES dataset. Candidate genes were validated studying A) protein expression in human LOAD and control brains; B) two animal AD models, Drosophila and Zebrafish. In the joint AFR+NA model, we identified four significant ancestral blocks located on chromosomes 1 (p-value=8.94E-05), 6 (p-value=8.63E-05), 21 (p-value=4.64E-05) and 22 (p-value=1.77E-05). Fine-mapping prioritized the GCAT gene on chromosome 22 (SKAT-O p-value=3.45E-05) and replicated in the UK Biobank (SKAT-O p-value=0.05). In LOAD brains, a decrease of 28% in GCAT protein expression was observed (p-value=0.038), and GCAT knockdown in Amyloid-β42 Drosophila exacerbated rough eye phenotype (68% increase, p-value=4.84E-09). In zebrafish, gcat expression increased after acute amyloidosis (34%, p-value=0.0049), and decreased upon anti-inflammatory Interleukin-4 (39%, p-value=2.3E-05). Admixture mapping uncovered genomic regions harboring new LOAD-associated loci that might explain the observed different frequency of LOAD across ethnic groups. Our results suggest that the inflammation-related activity of GCAT is a response to amyloid toxicity, and reduced GCAT expression exacerbates AD pathology.
INTRODUCTION.
Late-onset Alzheimer’s disease (LOAD) is the most common cause of neurodegenerative disease worldwide, but it is not equally distributed among racial and ethnic groups. The frequency of the disease in African Americans (AA) and Caribbean Hispanics (CH) is significantly higher than in non-Hispanic Whites (NHW) [1], and such increased risk is observed also for family members of affected individuals [2]. An established genetic risk locus, the APOE-ɛ4 allele, alone does not explain the different frequency of the disease. In fact, AA and CH non-carriers still show a two-to-four-fold higher incidence of LOAD, compared with NHW [3]. Studies of genetic association between loci other than APOE and LOAD further confirmed the heterogeneity across racial and ethnic groups [4].
What factors underlie these disparities is of critical importance. Ancestry is an understudied yet a critical aspect of health disparities, as it may explain observed differences in frequency of LOAD across ethnic groups. To this end, we aimed to investigate the role of ancestry on LOAD risk in a cohort of CH from New York, Dominican Republic, and Puerto Rico. To do so, we inferred global and local ancestry and tested their association with LOAD (admixture mapping). Admixture mapping compares differences in ancestry frequencies at a specified genetic locus when performing association with a disease, in contrast to genome-wide association studies (GWAS), which measure differences in allele frequencies between cases and controls. Admixture mapping may have more power than standard GWAS for several reasons; 1) the multiple-testing burden is greatly reduced as in practice only genomic regions with same parental ancestors (“ancestral blocks”) are being tested, compared to GWAS where millions of single-nucleotide polymorphisms (SNPs) are being studied independently; 2) while GWAS pinpoints a single genetic marker, admixture mapping identifies large genomic regions (up to several megabases) that might harbor multiple risk and/or protective loci.
To our knowledge, this is the largest investigation in CH aiming to tackle the role of ancestry in LOAD.
METHODS.
Subjects.
The data analyzed were derived from three studies recruiting individuals of Caribbean Hispanic ancestry. 1) the Washington Heights and Inwood Columbia Aging Project (WHICAP study); 2) Estudio Familiar de Influencia Genetica en Alzheimer (EFIGA); 3) Puerto Rico 10/66 study (10/66 PR). Detailed description of the cohorts, diagnosis and demographics are already described elsewhere [5] and briefly summarized in the Supplementary file. In brief, we excluded individuals because of a) missing or non-LOAD diagnosis or missing covariates; b) PSEN1-G206A carriers (a relatively frequent mutation in the Caribbean population); c) individuals that failed to meet the criterion for three-way admixed ancestry; d) samples that were determined to be duplicates. No outliers (±6 standard deviations) were observed after principal components estimation.
For the WHICAP and EFIGA studies, informed consent and Recruitment was approved by the Institutional Review Board of Columbia University Medical Center and the national research ethics committee of the Dominican Republic. The study protocol for the 10/66 population-based study and the consent procedures were approved by the King’s College London research ethics committee and University of Puerto Rico, Medical Sciences Campus Institutional Review Board (IRB). Informed consent was documented in writing in all cases. All studies were conducted according to the principles expressed in the Declaration of Helsinki.
Outcomes.
For EFIGA and WHICAP, LOAD diagnosis was carried out according to the National Institute of Neurological and Communication Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) [6]. For 10/66 PR, diagnosis of dementia was assigned according to 10/66 protocol [7]. Sex and age (age at onset for incident cases, age at baseline for prevalent cases, age at last evaluation for cognitive healthy controls) were used as main covariates in all statistical models.
Genetic data.
Detailed description of genotyping, quality control and imputation procedures have already been reported in a recent publication from our group [5]. All analyses were conducted on autosomal chromosomes only. Common single-nucleotide polymorphism (SNPs, MAF>1%) were quality controlled by applying standard filters for missingness and Hardy-Weinberg equilibrium using plink software [8]; data were then phased and imputed using Michigan imputation server [9] employing Topmed reference panel [10]. Data were phased with Eagle (v2) [11], imputed with Minimac4 [12]. We only retained high-quality SNPs (i.e. “Rsq” metric ≥0.80). Details for genotype QC and imputation can be found elsewhere [13] and summarized in Supplementary Table 1.
Global ancestry analysis.
Global ancestry (GA) for each individual was estimated using the ADMIXTURE (v1.3.0) software [14]; methods and detailed code can be found in the Supplementary File.
Local ancestry Estimation
Overlapping SNPs in HGDP reference panel and CH cohort were extracted and employed within the efficient-local ancestry inference (ELAI) software (v0.99) Guan (15) to infer locus-specific haplotypes derived for EUR, AFR and NAT parental population. Detailed code could be found in the Supplementary File. ELAI exploits a two-layer HMM model by estimating cluster-switch rates which enhances estimation of recombination hotspots. ELAI generated output with values ranging from 0–2 per genomic position per individual per ancestry (“ancestral allele dosage”). These were used as the main predictors in the admixture mapping models (see below).
Admixture Mapping.
The association between local ancestral allele dosage (i.e. 0, 1, or 2 copies of EUR, AFR or NAT ancestry at a particular genomic position) and LOAD case-control status was tested in two association models:
“Single-ancestry” association analyses. We tested the association between local ancestry dosage (independently for EUR, AFR, NAT) with LOAD. To do so, we employed a linear-mixed regression model (Wald test) as implemented in GEMMA [16] with two sets of covariates: Model 1) age, sex, principal components (PCs) and genotyping batch (fixed effects). Model 2) age, sex, PCs, genotyping batch, APOE-ɛ4 allele. For both models, the genetic relationship matrix (kinship) generated by GEMMA (v0.98) [16] was used as a random effect to adjust for relatedness.
“Joint-ancestry” association analyses. We employed a fisher test approach to test the association between jointly NAT + AFR local ancestries and LOAD. We used the admixMap function implemented in the GENESIS (v 1.24.1) [17] R (v3.5) package with default values. Detailed code is available in Supplementary File.
Calculation of p-value threshold for genome-wide admixture association
To identify the significant p-value threshold after multiple correction adjustment, the number of recombination events were estimated, i.e. the effective number of transitions as proposed by Shriner and colleagues [18]. We also employed an alternative permutation method, employing the MultiTrans software [19], designed for studies with related samples. Detailed methods and coding can be found in Supplementary File.
Ancestral blocks
An ancestral block is a genomic region where haplotypes are derived from common ancestors that resulted from recombination events during meiosis. The age of the population (number of generations) defines ancestral blocks and its length. The higher the age of the population, the smaller the size of the ancestral haplotypes that can be observed due to more recombination events. To define the borders of an admixture block we scanned regions to identify a decay of at least an order of magnitude in p-value observed [20] in the association analyses for the index local ancestry. The index local ancestry is defined as the genomic position with the lowest p-value within the ancestral block. For instance, if the index local ancestry’s p-value= 1E-05, we scanned the region until a p-value of 1E-04 is observed. An in-house script was used to perform scanning to identify start and end coordinates of the admixture genomic regions.
Fine mapping.
Ancestral blocks prioritized by admixture mapping were fine-mapped (i.e. the region is analyzed to identify a specific gene or variants that are likely to causally influence the examined trait) using a multiethnic whole-exome sequencing (WES) experiment performed on the WHICAP cohort (N=845 non-Hispanic Whites; N=1,051 African Americans; N=1,699 Caribbean Hispanics). All CH individuals with WES overlap with the GWAS cohort employed in the current project, whereas the non-Hispanic Whites and AA are completely independent of the CH individuals. Details about recruitment, QC, methods and results are already reported elsewhere [21]. Briefly, we restricted our analyses to rare and uncommon variants (i.e. MAF≤5%). Quality-controlled variants were annotated by Variant Effect Predictor (VEP) and filtered in according to in silico functional prediction with combined annotation dependent depletion (CADD>20). For those genes lying within the ancestral blocks prioritized by admixture mapping, we tested for single-marker (Firth’s logistic regression method) and gene-based association (SKAT-O) using seqMETA and SKAT R packages, respectively. Results are presented by ethnic group and for the combined transethnic meta-analysis.
Replication in UK Biobank WES data.
Candidate genes prioritized in WHICAP were then replicated using publicly available UK biobank WES dataset using AD (ICD-10, code F00) as the selected outcome (N= 1,959; N cases=335; Version Mar 2021). Gene-based SKAT-O tests for loss-of-function variants are available through the web platform https://genebass.org/ [22].
Human brain tissue protein expression.
To determine whether the genetic association was correlated with differential expression in LOAD vs. non-LOAD brains, we performed quantitative Western blot analysis on autopsy brain tissue, obtained from the New York Brain Bank at Columbia University Medical Center. Methods regarding brain tissue preparation and Western blotting are reported in the Supplementary File.
Drosophila rough eye phenotype assessment.
Drosophila were used to functionally validate candidate genes prioritized by admixture mapping analyses. We used a Drosophila model of Amyloid beta (Aβ42)-induced toxicity and a conditional expression system; we expressed a short interfering RNA (siRNA) against GCAT. Drosophila GCAT-RNAi line and GFP-RNAi control line were obtained from Bloomington Drosophila Stock Center. For Amyloid beta overexpression, we used the Drosophila line previously generated by Dr. Fernandez-Funez [23] and kindly shared with us. The GMR-GAL4 driver was used for expression of transgenes in the eye. For light microscopy imaging of the Drosophila eyes, 7-day old adult flies were collected and eye images recorded, in a blinded fashion, by three independent observers as described before [24] [25]. Full description of experimental methods can be found in the Supplementary File.
Transcriptomics in adult zebrafish brain models of neurodegeneration and regeneration.
We investigated adult zebrafish brain models generated by cerebroventricular microinjection of human Aβ42 monomers or IL-4 protein [26–29]. These are well-established models for experimental neurodegeneration and regeneration in adult zebrafish brain [26, 27, 29]. Aβ42 peptide injection is a model for cellular pathology and opens subsequent protective/regenerative mechanisms while IL-4 injection is a model for immune suppression as well as enhanced regenerative neurogenesis. Full description of experimental methods can be found in the Supplementary File.
RESULTS
Sample characteristics.
The total number of samples included in the current study was 8,813 (EFIGA N=5,375; WHICAP N=1,896; PR N=1,542). Sample characteristics are detailed in Supplementary Table 2.
Global Ancestry.
Supervised admixture analyses showed that the EUR ancestry accounted for 57%, followed by the AFR (33%) and the NAT ancestry (8%) (Supplementary Figure 1).
We observed an association between the Global African proportion and LOAD (Beta=0.36, SE=0.023, p-value<0.01), whereas neither European nor Native proportion showed evidence of association.
Local Ancestry Inference (LAI)
Supplementary Figure 2 shows EUR, AFR and NAT ancestry profiles across autosomal chromosomes (averaged over individuals). Our findings for local ancestry are consistent with global admixture, i.e. averaging local ancestry across chromosomes as estimated by ELAI (which employed imputed SNPs) is highly correlated with global ancestry estimated by ADMIXTURE using a small set of genotyped SNPs. In the HLA region we observed an excess of AFR more and consequently a decrease of EUR ancestry (~40% AFR and ~40% EUR ancestry compared to genome-wide 33% and 57%, respectively; detailed figure of chromosome 6 in Supplementary Figure 3). In order to investigate if this observation was due to a methodological artifact, we LD pruned the SNPs located in the HLA region before re-estimating the LA (data not shown). Our findings indicate that AFR proportion remains high in the HLA region before and after LD pruning. Our findings mirror those already reported by Meyer and colleagues [30] where an excess of AFR ancestral proportion was reported in the HLA region for Latino populations (Mexicans, Puerto Ricans, Colombians etc.). It is difficult to reach to a conclusion if the LA inference is biased by the highly polymorphic regions or whether AFR ancestral component truly follows this trajectory; therefore, this aspect remains to be investigated in the future.
Admixture mapping p-value threshold
We estimated 415 upper switches transitions. Using Bonferroni correction, genome-wide significant p-value threshold was set at 1.20E-04 (0.05/415). The MultiTrans approach retrieved a p-value threshold of 1.49E-04. Because these two p-values were almost identical, we employed the former as the genome-wide significant threshold for the study.
Admixture Mapping
Univariate ancestry analysis
Type-1 errors were well controlled genome-wide for all models (lambdas and Q-Q plots can be found in the Supplementary figure 4). For EUR univariate ancestry analysis, we identified a significant ancestral block on chromosome 22 that spanned from 37Mb to 39Mb (Model 1: OR=0.87 95%CI=0.80–0.93); consistently, we observed opposite signal in the NAT univariate ancestry analysis (OR=1.21 95%CI=1.11–1.30) along with an additional ancestral block on chromosome 17 in Model 2. No LA survived multiple testing correction for the AFR univariate ancestry analyses (Manhattan plots in Supplementary figure 5). The Supplementary table 3 reports the top index LA and associated statistics.
Joint ancestry analysis
In model 1, we identified three ancestral blocks significant after multiple testing correction, on chromosome 22 (37.46Mb to 39.35Mb; p-value=2.63E-06), chromosome 21 (23.09Mb to 25.34Mb; p-value=4.64E-05) and chromosome 19 (45.21 Mb to 47.33 Mb; p-value=7.03E-05). In model 2, we confirmed the first two ancestral blocks from model 1 and identified two additional: chromosome 6 (164.18Mb to 166.90Mb; p-value=3.78E-05) and chromosome 1 (111.58Mb to 113.75Mb; p-value=1.11E-04). Details about admixture block statistics and length could be found in Table 1. Figure 1 shows Manhattan plot for joint analysis.
Table 1:
Joint analysis of NAT+AFR ancestry (EUR as reference). We also report the OR (CI) for the single-ancestry association analyses within blocks prioritized by the joint analyses. Index LA represents SNP with the top association statistics within those ancestral blocks.
| Ancestral block (Mb start – Mb end) |
Index LA | CHR | Joint Analysis p-value |
AFR | NAT | |||
|---|---|---|---|---|---|---|---|---|
| p-value | OR (95%CI) | p-value | OR (95%CI) | |||||
| Model 1 | 37.46 – 39.35 | 22:38647406 | 22 | 2.63E-06 | 3.94E-02 | 1.08 (1.00–1.15) | 2.78E-04 | 1.19 (1.10–1.29) |
| 23.10 – 25.35 | 21:23374870 | 21 | 4.64E-05 | 2.56E-01 | 0.95 (0.88–1.03) | 1.52E-03 | 0.84 (0.73–0.94) | |
| 45.21 – 47.34 | 19:47037620 | 19 | 7.03E-05 | 3.63E-02 | 1.07 (1.00–1.14) | 3.63E-02 | 1.12 (1.02 – 1.21) | |
| Model 2 | 37.42 – 39.47 | 22:38647406 | 22 | 3.30E-06 | 5.02E-02 | 1.07 (1.00–1.14) | 3.39E-04 | 1.19 (1.09–1.29) |
| 23.05 – 25.42 | 21:23374870 | 21 | 2.95E-05 | 2.16E-01 | 0.95 (0.88–1.02) | 1.09E-03 | 0.84 (0.73–0.94) | |
| 164.18 – 166.90 | 6:166240666 | 6 | 3.78E-05 | 2.05E-04 | 0.87 (0.80 – 0.94) | 9.32E-02 | 1.08 (0.98– 1.18) | |
| 111.58 – 113.75 | 1:112789288 | 1 | 1.11E-04 | 7.27E-03 | 1.10 (1.03–1.17) | 2.68E-02 | 1.11 (1.01– 1.20) | |
EUR =European ancestry, AFR = African ancestry, and NAT=Native American ancestry. OR= odds ratio. We report index LA in CHR:BP format, where CHR represents Chromosome and BP represents base-pair position (human genome hg19 build), respectively.
Figure 1. Joint Admixture Mapping analyses.
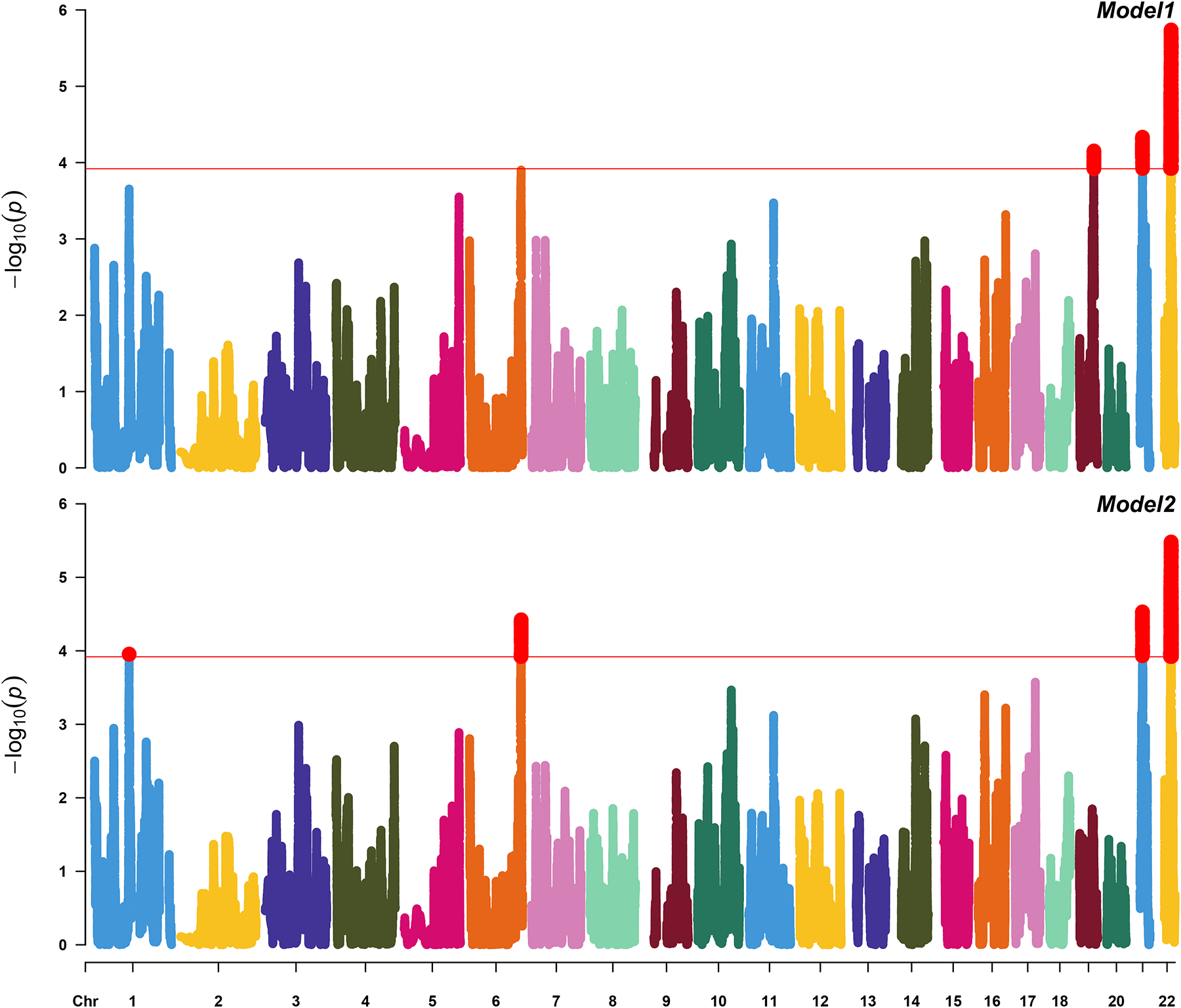
Manhattan plots of analyses conducted in admixMap (Model 1 (upper panel): LOAD~sex+age+genotype batch+PCs+kin; Model 2 (lower panel): LOAD~sex+age+genotype batch+APOE-e4+PCs+kin)
Fine mapping
Results from the transethnic meta-analysis (metaSKAT) in WHICAP within the ancestral blocks prioritized by the admixture mapping identified GCAT on chromosome 22 (CADD20 annotation SKAT-Op-value= 3.45E-05 in Model 1; SKAT-Op-value= 3.55E-05 in Model 2). The association observed in the meta-analysis was driven by association in both CH (SKAT-Op-value= 4.0E-04) and AA (SKAT-Op-value= 7.0E-04). No association was observed in NHW (SKAT-Op-value= 0.55). Effect sizes (betas) for the ancestral block on chromosome 22 are reported in Figure 2, separately for AFR and NAT univariate ancestry analyses.
Figure 2. Chromosome 22 ancestral block.
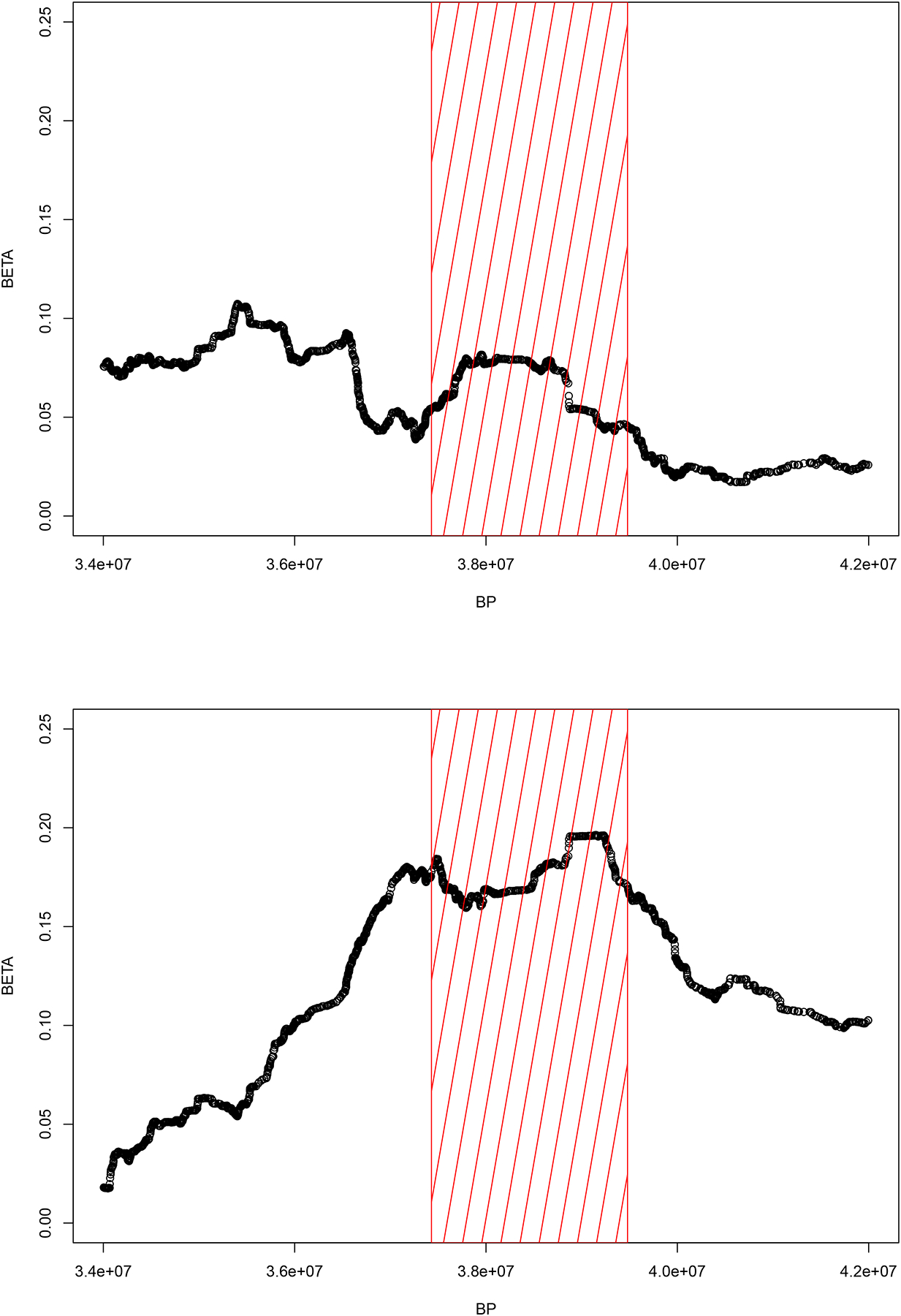
Beta coefficient (Y axis) against base-pair position (X axis) for AFR (upper panel) and NAT (lower panel) ancestry within the chromosome 22 ancestral block prioritized by the joint admixture mapping analyses (red rectangular panel).
Within GCAT, twenty-seven SNVs were annotated as highly deleterious (CADD score >20). Single-marker analyses identified one variant significant after multiple testing correction in CH (rs17856459, MAF=0.01, BP=38,212,624, OR=2.61, 95%CI= 2.24–4.16, padj=0.037). This is a non-synonymous SNV located in exon 9 (p.Arg387Gly), annotated as deleterious by several independent algorithms (CADD=35; SIFT=D; Polyphen=D; MutationTaster=D).
None of the genes lying within other ancestral blocks survived multiple testing correction.
Replication using UKBB
GCAT was replicated in the UKBB WES dataset (pLOF annotation SKAT-Op-value= 0.051, SKATp-value= 0.029, NSNPs=18).
GCAT differential protein expression.
Characteristics of human brain samples are reported in Supplementary Table 4. Western Blot showed a significant decrease in the GCAT protein levels in the prefrontal cortex in LOAD brains with respect to control brains (28% reduction, p=0.038, Figure 3).
Figure 3. GCAT protein is dysregulated in Late Onset Alzheimer’s Disease.
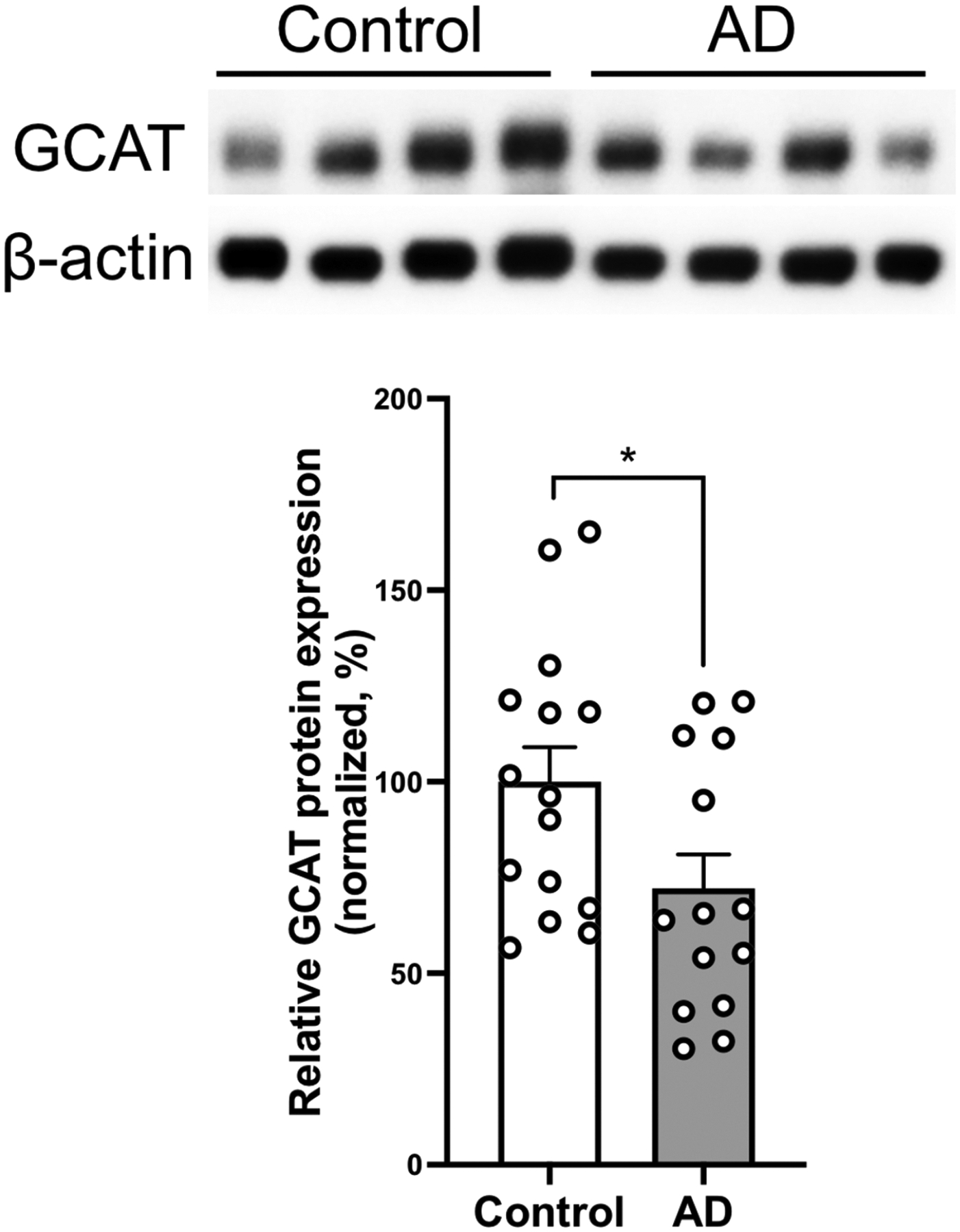
Western blot quantification of GCAT protein levels in human prefrontal cortex of controls (n = 15) and AD (n = 14). Top portion of the panel shows representative Western blots. Histograms show densitometric quantification of GCAT protein abundance with respect to control at the bottom of the panel. GCAT is normalized by β-actin in all samples. Mann Whitney U Test; * p-value=0.038. Data represent mean ± SEM.
Gcat deficiency exacerbates amyloid beta toxicity in a Drosophila AD model.
Aβ42 was first co-expressed with a siRNA control in the Drosophila eye, producing a rough eye phenotype and confirming the model system was functioning (Figure 4A, left panels). Next, Aβ42 and Gcat siRNA were co-expressed: quantitative assessment (Figure 4A, right panel) showed a significant exacerbation of the of the eye phenotypes (68% increase, p-value=4.84E-09, Figure 4B).
Figure 4. GCAT exacerbates Amyloid-β mediated toxicity in vivo.
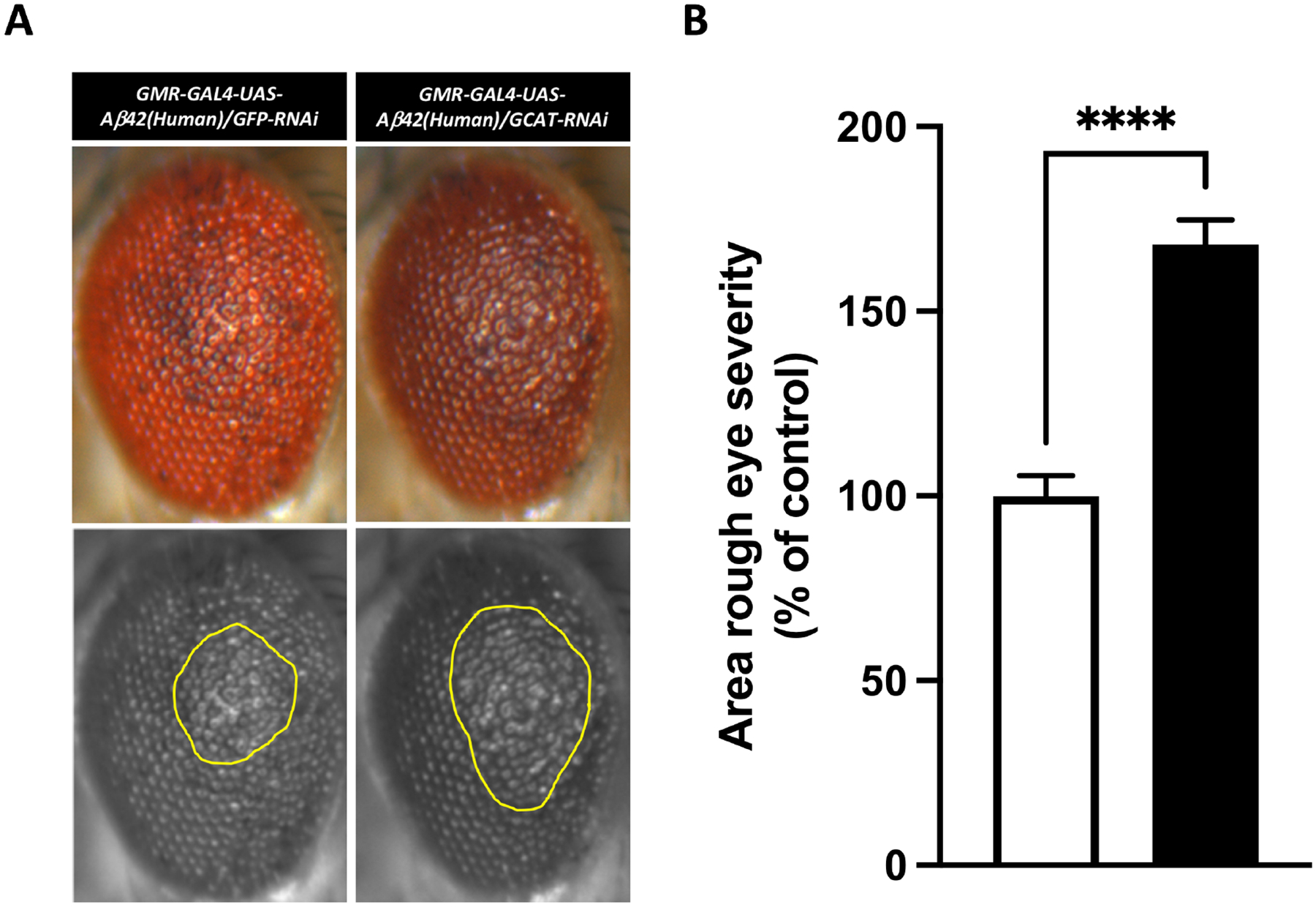
(A) Co-expression of NSun2 RNAi (n = 17) with human Amyloid-β1–42 exacerbated the rough eye phenotype compared with that observed in the GFP RNAi control (n = 19). The yellow marked area shows degenerated part of eyes. (B) Histograms show quantitative assessment of eye phenotypes (****p-value=4.84E-09 by Mann-Whitney U test). Data represent mean ± standard error.
Expression of gcat in adult zebrafish brain model of neurodegeneration and regeneration
Transcriptomics analysis in the adult zebrafish brain showed significant reduction of gcat expression levels in glial (35% decrease, p-value=3.16E-05) and neuronal (39% decrease, p-value=2.3E-05) populations after treatment with IL-4, whereas Aβ42 increased gcat expression (34% increase, p-value=0.0048).
DISCUSSION.
Admixed populations present a complex socio-demographical, clinical, and genetic profile because of social, geographical, and historical events. The native population of the Caribbean islands experienced the invasion by European colonists and subsequently the trading of Africans slaves to the islands. Consequently, studies of Hispanics have shown wide ranges of admixture estimates, even within the same country [31]. Results in our cohort are consistent with those previously reported for Caribbean populations.
Previous investigations showed that ancestry is associated with several traits in African Americans and Hispanics (e.g. glaucoma) [32]. The role of African ancestry, and even less, Native ancestry in LOAD has been understudied. We observed that higher African ancestry is associated with risk of LOAD. This observation matches previous reports finding a similar association in African Americans[33] although results are conflictive.
Our admixture mapping analyses identified four genomic regions associated with LOAD. Ancestral blocks span several Mb that can potentially harbor multiple pathogenic or protective genes/variants. Thus, fine mapping is critical to prioritize disease-associated genetic loci. We employed WES data, providing compelling results for GCAT as a strong gene candidate. This gene is also the top associated locus at genome-wide level for the WES transethnic metanalysis[34]. Importantly, we replicated GCAT in the UKBB WES dataset, ultimately showing that ancestry-specific findings in one population are associated with the disease in other populations, albeit with attenuated effect sizes.
The role of GCAT in aging has been established in vivo and in vitro. Ravichandran et al [35] conducted a comprehensive transcriptomic screening for genes downregulated during aging across three different species, C. elegans, zebrafish, and mice. GCAT was identified as the top physiologically-downregulated gene during aging. GCAT effect on lifespan seems to be mediated through methylglyoxal-mediated proteohormesis, a toxic pyruvic acid derivative which in turn seems to affect lifespan in a nonlinear dose-response manner. Interestingly, two main mechanisms have been proposed to explain methylglyoxal and methylglyoxal-derived advanced glycation end products (AGEs) neurotoxicity in LOAD: a) AGEs induced cross-linking of cytoskeletal proteins promoting neuronal dysfunction and cell death and b) AGEs inducing accumulation of Aβ42, one of the main hallmarks of LOAD [36] [37] and chronic activation of astroglial and microglial cells [38].
We have found a significant gcat protein reduction in LOAD brains when compared to healthy controls, suggesting accumulation of toxic derivatives might play a role in the pathological worsening and neurodegeneration of brain vulnerable areas [39]. To further explore the involvement of GCAT in LOAD pathogenesis, we asked whether the gene’s deficiency influences Aβ42 associated toxicity in vivo. We conducted two experiments in non-mammalian invertebrates (Drosophila) and vertebrates (Zebrafish), which are optimal animal models to study AD. Indeed, many molecular mechanisms are conserved between Drosophila and humans [40] and Aβ42 toxicity Drosophila models have proven to be useful for modeling essential mechanisms of AD [41]. Zebrafish instead offers a novel perspective on how new neurons can be regenerated after neurodegeneration[42, 43]. Our result in Drosophila supports the observation in human brain transcriptome, i.e. Gcat deficiency promotes AD-related neurotoxicity, suggesting that Gcat activity is required to counteract AD pathology (Supplementary Figure 6). In zebrafish, Amyloid toxicity enhanced gcat activity, suggesting that regenerative vertebrate brains could respond to AD by activating gcat. This supports the clinical and functional observations in humans and Drosophila that GCAT might have beneficial roles for counteracting AD pathology, and vertebrate brains might response to amyloid toxicity by enhancing gcat activity. Zebrafish findings, where IL-4 reduced GCAT expression points towards an inflammation-related aspect of gcat activity. Innate immune response positively affects neural regeneration and neuroprotective mechanisms in zebrafish [28, 29, 44–47], and gcat expression might also be related to active immune response [26, 48] as it is downregulated when a hypoinflammatory state is induced by IL-4. Considering that IL-4 signaling is a second-stage mechanisms that links initial Amyloid-pathology to the resolution of inflammation and regenerative neurogenesis in zebrafish brain, reciprocal activity of amyloid and IL4 in zebrafish AD model suggests that GCAT expression is required for early response to the pathology in zebrafish and might be required to initiate the early mechanisms for subsequent neuro-regeneration. Overall, GCAT’s downregulation in human AD brains may impair a neuroprotective outcome and promote neurodegeneration. Therefore, expression levels of GCAT could be a critical factor for the cellular outcome of the neuropathology in AD and the brain resilience against the disease. Further studies are needed to advance our understanding of the regulation of GCAT and the mechanistic role that GCAT plays in the disease process, which may guide us toward the development of novel therapeutic strategies.
Supplementary Material
ACKNOWLEDGMENTS.
The National Institutes of Health (NIH), National Institute on Aging (NIH-NIA) and National Institute of Neurological Disorders and Stroke supported this work through the following grants: R56AG069118, R56AG066889, R56AG059756, R01AG056531, and R01NS095922.
For WHICAP: Data collection and sharing for this project was supported by the Washington Heights-Inwood Columbia Aging Project (WHICAP, R01AG037212, RF1AG054023, RF1AG066107) funded by the NIH-NIA and by the National Center for Advancing Translational Sciences, NIH, through Grant Number UL1TR001873. This manuscript has been reviewed by WHICAP investigators for scientific content and consistency of data interpretation with previous WHICAP Study publications. We acknowledge the WHICAP study participants and the WHICAP research and support staff for their contributions to this study.
For EFIGA: Data collection for this project was supported by the Genetic Studies of Alzheimer’s disease in Caribbean Hispanics (EFIGA) funded by the NIH-NIA and by the NIH (5R37AG015473, RF1AG015473, R56AG051876, R01AG067501, R56AG063908, RF1AG015473. We acknowledge the EFIGA study participants and the EFIGA research and support staff for their contributions to this study.
For ADI 10/66 PR Alzheimer’s Disease International Epidemiological Study: Data collection for this project was supported by a recurrent PR Legislature grant, Pfizer Co. Grant # GA9001NE, and for PR Apo-E labs: Human Genetics Core Award from Columbia University Irving Institute for Clinical and Translational Research.
Zebrafish work was supported by grants from Helmholtz Association (VH-NG-1021) and Schaefer Research Scholars Award. We thank Drs. Mehmet Ilyas Cosacak and Prabesh Bhattarai for gene expression analyses in zebrafish.
Footnotes
Conflict of interests.
C.K. has executive role in Neuron-D GmbH, which had no academic or financial relationship to the design and execution of this project.
Supplementary information is available at MP’s website
REFERENCES.
- 1.Tang MX, Cross P, Andrews H, Jacobs DM, Small S, Bell K, et al. Incidence of AD in African-Americans, Caribbean Hispanics, and Caucasians in northern Manhattan. Neurology. 2001;56(1):49–56. doi: 10.1212/wnl.56.1.49. [DOI] [PubMed] [Google Scholar]
- 2.Vardarajan BN, Faber KM, Bird TD, Bennett DA, Rosenberg R, Boeve BF, et al. Age-specific incidence rates for dementia and Alzheimer disease in NIA-LOAD/NCRAD and EFIGA families: National Institute on Aging Genetics Initiative for Late-Onset Alzheimer Disease/National Cell Repository for Alzheimer Disease (NIA-LOAD/NCRAD) and Estudio Familiar de Influencia Genetica en Alzheimer (EFIGA). JAMA Neurol. 2014;71(3):315–23. doi: 10.1001/jamaneurol.2013.5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang MX, Stern Y, Marder K, Bell K, Gurland B, Lantigua R, et al. The APOE-epsilon4 allele and the risk of Alzheimer disease among African Americans, whites, and Hispanics. JAMA. 1998;279(10):751–5. doi: 10.1001/jama.279.10.751. [DOI] [PubMed] [Google Scholar]
- 4.Tosto G, Reitz C. Genome-wide association studies in Alzheimer’s disease: a review. Curr Neurol Neurosci Rep. 2013;13(10):381. Epub 2013/08/21. doi: 10.1007/s11910-013-0381-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sariya S, Felsky D, Reyes-Dumeyer D, Lali R, Lantigua RA, Vardarajan B, et al. Polygenic risk score for Alzheimer’s Disease in Caribbean Hispanics. Ann Neurol. 2021. Epub 2021/05/27. doi: 10.1002/ana.26131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr., Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prince M, Ferri CP, Acosta D, Albanese E, Arizaga R, Dewey M, et al. The protocols for the 10/66 dementia research group population-based research programme. BMC Public Health. 2007;7:165. doi: 10.1186/1471-2458-7-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48(10):1284–7. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590(7845):290–9. doi: 10.1038/s41586-021-03205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loh PR, Palamara PF, Price AL. Fast and accurate long-range phasing in a UK Biobank cohort. Nat Genet. 2016;48(7):811–6. doi: 10.1038/ng.3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuchsberger C, Abecasis GR, Hinds DA. minimac2: faster genotype imputation. Bioinformatics. 2015;31(5):782–4. doi: 10.1093/bioinformatics/btu704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sariya S, Lee JH, Mayeux R, Vardarajan BN, Reyes-Dumeyer D, Manly JJ, et al. Rare Variants Imputation in Admixed Populations: Comparison Across Reference Panels and Bioinformatics Tools. Front Genet. 2019;10:239. Epub 2019/04/20. doi: 10.3389/fgene.2019.00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexander DH, Lange K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinformatics. 2011;12:246. doi: 10.1186/1471-2105-12-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guan Y Detecting structure of haplotypes and local ancestry. Genetics. 2014;196(3):625–42. Epub 2014/01/07. doi: 10.1534/genetics.113.160697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44(7):821–4. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gogarten SM, Sofer T, Chen H, Yu C, Brody JA, Thornton TA, et al. Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics. 2019;35(24):5346–8. doi: 10.1093/bioinformatics/btz567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shriner D, Adeyemo A, Rotimi CN. Joint ancestry and association testing in admixed individuals. PLoS Comput Biol. 2011;7(12):e1002325. doi: 10.1371/journal.pcbi.1002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joo JW, Hormozdiari F, Han B, Eskin E. Multiple testing correction in linear mixed models. Genome Biol. 2016;17:62. doi: 10.1186/s13059-016-0903-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Z, Shriner D, Hansen NF, Rotimi CN, Mullikin JC, Program NCS. Admixture mapping identifies genetic regions associated with blood pressure phenotypes in African Americans. PLoS One. 2020;15(4):e0232048. doi: 10.1371/journal.pone.0232048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tosto G, Vardarajan B, Sariya S, Brickman AM, Andrews H, Manly JJ, et al. Association of Variants in PINX1 and TREM2 With Late-Onset Alzheimer Disease. JAMA Neurol. 2019. doi: 10.1001/jamaneurol.2019.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karczewski K, Solomonson M, Chao KR, Goodrich JK, Tiao G, Lu W, et al. Systematic single-variant and gene-based association testing of 3,700 phenotypes in 281,850 UK Biobank exomes. medRxiv. 2021:2021.06.19.21259117. doi: 10.1101/2021.06.19.21259117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casas-Tinto S, Zhang Y, Sanchez-Garcia J, Gomez-Velazquez M, Rincon-Limas DE, Fernandez-Funez P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum Mol Genet. 2011;20(11):2144–60. Epub 2011/03/11. doi: 10.1093/hmg/ddr100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reid DW, Muyskens JB, Neal JT, Gaddini GW, Cho LY, Wandler AM, et al. Identification of genetic modifiers of CagA-induced epithelial disruption in Drosophila. Front Cell Infect Microbiol. 2012;2:24. doi: 10.3389/fcimb.2012.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santa-Maria I, Alaniz ME, Renwick N, Cela C, Fulga TA, Van Vactor D, et al. Dysregulation of microRNA-219 promotes neurodegeneration through post-transcriptional regulation of tau. J Clin Invest. 2015;125(2):681–6. doi: 10.1172/JCI78421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhattarai P, Cosacak MI, Mashkaryan V, Demir S, Popova S, Govindarajan N, et al. Neuron-glia interaction through Serotonin-BDNF-NGFR axis enables regenerative neurogenesis in Alzheimer’s model of adult zebrafish brain.. PLoS Biol. 2020;18(1):e3000585. doi: 10.1371/journal.pbio.3000585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cosacak MI, Bhattarai P, Reinhardt S, Petzold A, Dahl A, Zhang Y, et al. Single-Cell Transcriptomics Analyses of Neural Stem Cell Heterogeneity and Contextual Plasticity in a Zebrafish Brain Model of Amyloid Toxicity. Cell Rep. 2019;27(4):1307–18 e3. doi: 10.1016/j.celrep.2019.03.090. [DOI] [PubMed] [Google Scholar]
- 28.Bhattarai P, Thomas AK, Zhang Y, Kizil C. The effects of aging on Amyloid-β42-induced neurodegeneration and regeneration in adult zebrafish brain. Neurogenesis. 2017;4(1):e1322666. Epub 2 May 2017. doi: 10.1080/23262133.2017.1322666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhattarai P, Thomas AK, Papadimitriou C, Cosacak MI, Mashkaryan V, Froc C, et al. IL4/STAT6 signaling activates neural stem cell proliferation and neurogenesis upon Amyloid-β42 aggregation in adult zebrafish brain. Cell Reports. 2016;17(4):941–8. doi: 10.1016/j.celrep.2016.09.075. [DOI] [PubMed] [Google Scholar]
- 30.Meyer D, A VRC, BD Bitarello, B DYC, Nunes K. A genomic perspective on HLA evolution. Immunogenetics. 2018;70(1):5–27. doi: 10.1007/s00251-017-1017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moreno-Estrada A, Gravel S, Zakharia F, McCauley JL, Byrnes JK, Gignoux CR, et al. Reconstructing the population genetic history of the Caribbean. PLoS Genet. 2013;9(11):e1003925. doi: 10.1371/journal.pgen.1003925. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ancestry.com, 23andMe’s “Roots into the Future” project, and Personalis, Inc. He is on the medical advisory board of Invitae and Med-tek. None of these entities played any role in the project or research results reported here.
- 32.Bonnemaijer PWM, Cook C, Nag A, Hammond CJ, van Duijn CM, Lemij HG, et al. Genetic African Ancestry Is Associated With Central Corneal Thickness and Intraocular Pressure in Primary Open-Angle Glaucoma. Invest Ophthalmol Vis Sci. 2017;58(7):3172–80. Epub 2017/06/28. doi: 10.1167/iovs.17-21716. [DOI] [PubMed] [Google Scholar]
- 33.Hohman TJ, Cooke-Bailey JN, Reitz C, Jun G, Naj A, Beecham GW, et al. Global and local ancestry in African-Americans: Implications for Alzheimer’s disease risk. Alzheimers Dement. 2016;12(3):233–43. Epub 2015/06/21. doi: 10.1016/j.jalz.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tosto G, Vardarajan B, Sariya S, Brickman AM, Andrews H, Manly JJ, et al. Association of Variants in PINX1 and TREM2 With Late-Onset Alzheimer Disease. JAMA Neurol. 2019;76(8):942–8. Epub 2019/05/07. doi: 10.1001/jamaneurol.2019.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ravichandran M, Priebe S, Grigolon G, Rozanov L, Groth M, Laube B, et al. Impairing L-Threonine Catabolism Promotes Healthspan through Methylglyoxal-Mediated Proteohormesis. Cell Metab. 2018;27(4):914–25 e5. doi: 10.1016/j.cmet.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 36.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123(1):1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–12. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 38.Krautwald M, Munch G. Advanced glycation end products as biomarkers and gerontotoxins - A basis to explore methylglyoxal-lowering agents for Alzheimer’s disease? Exp Gerontol. 2010;45(10):744–51. doi: 10.1016/j.exger.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 39.Angeloni C, Zambonin L, Hrelia S. Role of methylglyoxal in Alzheimer’s disease. Biomed Res Int. 2014;2014:238485. doi: 10.1155/2014/238485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pandey UB, Nichols CD. Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol Rev. 2011;63(2):411–36. doi: 10.1124/pr.110.003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prussing K, Voigt A, Schulz JB. Drosophila melanogaster as a model organism for Alzheimer’s disease. Mol Neurodegener. 2013;8:35. doi: 10.1186/1750-1326-8-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kizil C, Bhattarai P. Is Alzheimer’s Also a Stem Cell Disease? - The Zebrafish Perspective. Front Cell Dev Biol. 2018;6:159. doi: 10.3389/fcell.2018.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kizil C Mechanisms of Pathology-Induced Neural Stem Cell Plasticity and Neural Regeneration in Adult Zebrafish Brain. Curr Pathobiol Rep. 2018;6(1):71–7. doi: 10.1007/s40139-018-0158-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kizil C, Kyritsis N, Brand M. Effects of inflammation on stem cells: together they strive? EMBO reports. 2015;16(4):416–26. doi: 10.15252/embr.201439702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kizil C, Dudczig S, Kyritsis N, Machate A, Blaesche J, Kroehne V, et al. The chemokine receptor cxcr5 regulates the regenerative neurogenesis response in the adult zebrafish brain. Neural Dev. 2012;7:27. Epub 2012/07/25. doi: 1749-8104-7-27 [pii] 10.1186/1749-8104-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kizil C, Kyritsis N, Dudczig S, Kroehne V, Freudenreich D, Kaslin J, et al. Regenerative neurogenesis from neural progenitor cells requires injury-induced expression of Gata3. Dev Cell. 2012;23(6):1230–7. Epub 2012/11/22. doi: S1534–5807(12)00477–7 [pii] 10.1016/j.devcel.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 47.Kyritsis N, Kizil C, Zocher S, Kroehne V, Kaslin J, Freudenreich D, et al. Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science. 2012;338(6112):1353–6. Epub 2012/11/10. doi: science.1228773 [pii] 10.1126/science.1228773. [DOI] [PubMed] [Google Scholar]
- 48.Jurisch-Yaksi N, Yaksi E, Kizil C. Radial glia in the zebrafish brain: Functional, structural, and physiological comparison with the mammalian glia. Glia. 2020. doi: 10.1002/glia.23849. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.