

Abstract
Glypican-1 (GPC1) is a cell surface proteoglycan that is upregulated in multiple types of human cancers including pancreatic cancer. Here, we investigated whether GPC1 could be a target of antibody-toxin fusion proteins (i.e. immunotoxins) for treating pancreatic cancer. We constructed a panel of GPC1-targeted immunotoxins derived from a functional domain of Pseudomonas exotoxin A. An albumin-binding domain (ABD) was also introduced into the anti-GPC1 immunotoxin to improve serum half-life. Small molecule screening was performed to identify irinotecan that shows synergistic efficacy with the immunotoxin. We demonstrated that GPC1 was internalized upon antibody binding. Anti-GPC1 immunotoxins alone inhibited tumor growth in a pancreatic cancer xenograft model. The immunotoxin treatment reduced active β-catenin expression in tumor cells. Furthermore, immunotoxins containing an ABD in combination with irinotecan caused pancreatic tumor regression. GPC1 expression was reduced by the immunotoxin treatment due to the degradation of the internalized GPC1 and its short cellular turnover rate. Our data indicate that the GPC1-targeted immunotoxin inhibits pancreatic tumor growth via degradation of internalized GPC1, downregulation of Wnt signaling, and inhibition of protein synthesis. The anti-GPC1 immunotoxin in combination with irinotecan thus provides a potential new treatment strategy for patients with pancreatic tumors.
Keywords: Glypican-1, immunotoxin, irinotecan, pancreatic cancer, Wnt signaling
Introduction
Pancreatic cancer is a devastating malignancy with a 5-year survival rate of only 9% (1). Gemcitabine is a first-line treatment for patients with locally advanced or metastatic pancreatic cancers (2). However, a large number of patients do not respond to gemcitabine. A second cytotoxic agent, such as a platinum analog (3), fluoropyrimidine (4), taxoid (5), or a taxane (6) is combined with gemcitabine. Yet, such combinational therapies provide only a modest improvement in survival (7). Therefore, therapeutics with different mechanisms of action are urgently needed for the treatment of pancreatic cancer.
Glypican-1 (GPC1) is a heparan sulfate proteoglycan anchored to the cell surface via a glycosylphosphatidylinositol (GPI) linkage (8,9). It has been reported to be overexpressed in multiple types of cancers, including pancreatic cancer (10). The high expression of GPC1 is also correlated with a poorer prognosis (11). Moreover, GPC1 is specifically enriched on pancreatic cancer cell-derived exosomes and may serve as a potential non-invasive diagnostic and screening tool (12). A recent report has revealed low to no expression of GPC1 in normal tissues by immunohistochemistry. Chimeric antigen receptor (CAR) T cells targeting GPC1 showed anti-tumor activity in pancreatic cancer xenograft and syngeneic mouse models (13). Recombinant immunotoxins are chimeric proteins composed of the variable fragment of an antibody and a portion of a toxin such as Pseudomonas exotoxin A (PE) (14,15). Immunotoxins can induce potent cytotoxicity on cancer cells known to be resistant to standard chemotherapy (16). The anti-CD22 immunotoxin, moxetumomab pasudotox, is the first example of a recombinant immunotoxin that has been approved by the US Food and Drug Administration (FDA) for the treatment of relapsed or refractory hairy cell leukemia patients (17). After internalization of the antigen/immunotoxin complex, the PE fragment is released into the cytosol and irreversibly modifies elongation factor 2 (EF2) by ADP-ribosylation. It will then lead to loss of the anti-apoptotic protein myeloid cell leukemia 1 (Mcl-1) (18), and ultimately apoptotic cell death (19). PE38, which is composed of domain II and domain III of PE, is a widely used PE fragment for immunotoxin construction (15,20). LR, a truncated PE38 fragment, contains domain III, the catalytic domain, and lacks all of domain II except for an 11-residue furin cleavage site (21,22). Immunotoxins derived from LR have shown high activity, low off-target toxicity, and reduced antigenicity when compared with those derived from PE38 (22).
Several characteristics of a target antigen are required for immunotoxins. Firstly, the density of a target antigen may influence immunotoxin efficacy. Our previous study demonstrated that liver cancer cells displayed an average of 104 – 105 GPC3 molecules per cell and the immunotoxin effect correlated with GPC3 expression levels (23). In addition, the internalization rate of a target antigen may be another critical characteristic for antibody-toxin conjugates. Anti-CD22 immunotoxin, moxetumomab pasudotox, is impressively active in treating leukemia patients in the clinic, partly due to the fast and effective internalization of CD22 antigen (19). GPC3 could be internalized into liver cancer cells with a relatively slower rate in comparison to CD22, but additional GPC3 molecules were recruited to the cell surface over time allowing more GPC3 internalization, making it a promising target by immunotoxins for treating hepatocellular carcinoma (HCC) (23). Besides the toxin part, the re-directing role of the antibody fragment is also essential for the therapeutical potential of an immunotoxin. Camelid VHHs (heavy-chain variable domains; also known as single-domain antibodies or nanobodies) have several advantages over conventional antibodies. They are easy to produce, highly soluble, and stable in a wide range of pH and temperatures. They can also be easily engineered due to their small size (12–15 kDa) (24). We previously isolated HN3, a human single-domain antibody specific for GPC3, from a phage-display engineered VH domain antibody library (25). Our data demonstrated that HN3-based immunotoxins were quite potent in treating mice bearing hepatocellular carcinoma (HCC) (23,26).
In the present study, we investigated the potential of GPC1 to be a new target by immunotoxins for treating pancreatic cancer. We constructed a panel of anti-GPC1 immunotoxins and evaluated their anti-tumor effect in the pancreatic cancer cell and xenograft mouse models. We improved the serum half-life of the immunotoxin by introducing an albumin-binding domain (ABD). Furthermore, we identified small-molecule compounds that showed synergistic efficacy with anti-GPC1 immunotoxins. Our mouse testing data suggest that the immunotoxins targeting GPC1 with irinotecan showed the best synergistic efficacy in pancreatic cancer.
Materials and Methods
Cell culture.
A431 is a human epidermoid carcinoma cell line supplied by Ira Pastan (NCI) (CLS Cat# 300112/p753_A-431, RRID: CVCL_0037) (27). GPC1+/A431 (clone H8) was derived from the A431 cell line and was transfected to stably express human GPC1. The T3M4 cell line (RCB Cat# RCB1021, RRID: CVCL_4056) was provided by Dr. Udo Rudloff (NCI) and was engineered to express luciferase and green fluorescence protein (GFP). The KLM1 cell line (TKG Cat# TKG 0490, RRID: CVCL_5146) and the Hep3B cell line were purchased from American Type Culture Collection. The cells were cultured in DMEM (A431 cells) or RPMI 1640 (T3M4 and KLM1 cells) supplemented with 10% fetal bovine serum, 100 U ml−1 penicillin, 0.1 mg ml−1 streptomycin, and 2 mmol l−1 L-glutamine. All cell lines were passaged less than 15 times at the time of usage. All cell lines were confirmed to be mycoplasma- and mouse pathogen-free by the Animal Diagnostic Laboratory Services at NCI in Frederick, Maryland.
Expression and purification of the immunotoxins.
The construction and production of immunotoxins followed our published protocol (15). Briefly, the pRB98 vector was linearized by NdeI and EcoRI restriction enzymes. The synthetic DNA fragments were subcloned into the linearized vector to produce the indicated immunotoxin expression plasmids: pMH334 (D4-LR), pMH335 (D4-AAA-D4-LR), pMH336 (D4-GGS-D4-LR), pMH337 (HM2-LR), pMH386 (D4-PE38), pMH387 (D4-ABD-LR), and pMH388 (D4-ABD-T20). The D4 nucleotide (SEQ ID No: 5) and amino acid sequences (SEQ ID No: 6) are described in the international patent application PCT/US2020/013739.
Octet binding kinetic analysis.
Bio-Layer Interferometry (BLI) technology was used to measure interactions between the immunotoxins and GPC-1 antigen. GPC1-His at 2 μg/ml and 100 nM or 33 nM of immunotoxins were prepared in Octet binding buffer (PBS, 0.1% bovine serum albumin, and 0.05% Tween-20). Specific settings for the assay were baseline 1 (180 sec), GPC1-His loading (300 sec), baseline 2 (300 sec), immunotoxin association (600 sec), and dissociation (1800 sec). The kinetic binding assay was performed on Octet RED96e at the Biophysics Core (National Heart, Lung and Blood Institute or NHLBI) and affinities were calculated by Octet System Data Analysis 8.2 software.
ADP ribosylation assay.
ADP-ribosylation buffer (20 mM Tris at pH 7.5 and 1 mM EDTA) was prepared. Four µl of ADP-ribosylation buffer, 1 µl of 1 M DTT, 20 ng of the immunotoxin, 5 µg of cell lysate, and 1 µl of Biotin-NAD+ (Trevigen, 250 µM) was mixed, and nuclease-free water was used to bring the volume up to 20 µl. The mixture was left at room temperature for 60 minutes, then equal parts of Laemmli buffer (BioRad) were added to stop the reaction. Twenty microliters of protein were separated by SDS-PAGE gel and transferred to a nitrocellulose blot. The blot membrane was washed with TBST (50 mM Tris, 150 mM NaCl, 0.1% Tween 20) followed by incubation with HRP-Streptavidin (Invitrogen) in TBST at 1:100,000 dilution for 30 minutes. After washing, the blot was covered with ECL buffer and imaged on the Chemidoc (BioRad).
Combination screening.
We initially screened a library of 1912 clinical-grade small compounds (NCATS)(28,29) using an anti-GPC3 immunotoxin (T19) on Hep3B cells. We then narrowed down to a group of eleven small compounds for synergy screening with an anti-GPC1 immunotoxin (D4-LR) in T3M4 cells. The small compounds were added to 1536-well white flat-bottom plates using the Echo 525 acoustic dispenser (Beckman Coulter) to create 10 ×10 blocks. Briefly, the compounds were transferred by acoustic dispensing (10 nl/well) into plates containing 1 µL of media / well with each compound plated in a ten-point (including a zero concentration) dilution series at two-fold serial dilutions. An anti-GPC1 immunotoxin diluted in the same way was transferred to the screen plates (30 nl/well) using the Echo 525 acoustic dispenser. T3M4 cells were dispensed at a density of 500 cells in 4 µl of media, (MultiDrop Combi, Thermofisher Scientific), making a total volume of 5 µl media per well. After 72 hours of incubation, 2.5 µl of Cell-TiterGlo reagent (Promega) were added to each well. Following a 10-minute incubation, luminescence was read using the Viewlux microplate reader (PerkinElmer). Data were normalized to in-plate controls (bortezomib as positive control, DMSO as negative control) and the normalized data were deconvoluted to individual dose combination matrices using in-house software. Bliss model of additivity (Bliss, 1956) was employed to characterize the presence or absence of synergy for each combination. Scatter plots were generated using the derived delta bliss values from the bliss model that was then inputted into TIBCO’s Spotfire 6.0. All the compounds are purchased from Selleck Chemicals LLC, dissolved in DMSO at 10 mmol/L stock concentration, and stored frozen at −20°C.
Animal Studies.
All the procedures used in the animal studies were approved by the Institutional Animal Care and Use Committee at the National Institutes of Health (NIH) under the protocol (LMB-059). Five-week-old female athymic nu/nu mice were provided by NCI CCR Animal Resource Program from NCI-Frederick (Frederick, Maryland). To generate a subcutaneous tumor mouse model, GPC1+/A431 cells (5 × 106) were resuspended in 200 μl of DMEM medium and injected in the right dorsal flank of the mice. Tumor volume calculations and experimental endpoints were conducted as previously reported (30). The T3M4 pancreatic model was generated through intraperitoneal injection of T3M4 cells (2 × 106) suspended in 200 μl of RPMI-1640 medium. Tumors were measured by total bioluminescent flux using a Xenogen IVIS Lumina (PerkinElmer). Mice were randomized based on their bioluminescence signal strength and grouped for immunotoxin treatment. Irinotecan (IRT, also known as CPT-11), is the prodrug of SN-38. Irinotecan was purchased from NIH pharmacy (Bethesda, Maryland).
Statistical analysis.
All experiments in the present were repeated at least three times. Statistical analyses were performed using GraphPad Prism 8 (GraphPad Prism, RRID: SCR_002798). Results were analyzed by unpaired Student’s t-test (2-tailed). For comprehensive serum analysis, complete blood counts, and necropsy, a one-way ANOVA analytical method was used. Asterisks were used to indicate significance (*P < 0.05, **P < 0.01, ***P < 0.001).
ELISA.
The detailed procedures are described in previously published reports (30,31). Briefly, 2 μg/ml of human GPC1-His (50 μl/well) were coated on ELISA plates and incubated at 4oC overnight. Serially diluted immunotoxins were added to the plates and incubated at room temperature for 1 hr. Plates were then washed five times with PBST followed by incubation with rabbit anti-Pseudomonas exotoxin A antibody (Sigma) at 1:200 dilution at room temperature for 1 hr. After washing, 50 μl/well of HRP-labeled goat anti-rabbit antibody (Jackson Laboratory) at 1:5000 dilution was added to wells. After 1 hr incubation at room temperature, 50 μl/well of 3,3,5,5’-tetramethybezidine dihydrochloride detection buffer was added to the wells for 10-minute substrate development. The reaction was stopped by the addition of 0.1 N sulfuric acid. Absorbance was read at 450 nm.
Western blot.
For all the western blot assays, the target cells, untreated or treated as designed, were washed with PBS and then lysed with RIPA buffer (Cell Signaling Technology). Novex 4–20% Tris-Glycine mini gels were used to separate extracted proteins. Lysates transferred to nitrocellulose filter membranes were blocked with 5% non-fat milk/PBS for 1 hour at room temperature with shaking and probed with proper primary antibodies at 4oC overnight with shaking. After three times’ washing with 0.05% PBST, 10-min for each time, the primary antibodies were detected with an HRP-labeled goat anti-mouse or anti-rabbit secondary antibody (Jackson Immunoresearch). For the measurement of the expression levels of GPC1, lysates derived from GPC1 expressing or knockout cells were stained with 1 μg/ml of the anti-GPC1 mouse antibody (clone HM2, in-house generated). For the apoptosis analysis, T3M4 cells were plated in 6-well plates and cultured overnight then were treated with different simulators, including 333 ng/mL of D4-LR, 0.2 µM of SN-38, 5 µM of ABT-263, D4-LR+SN38, or D4-LR+ABT-263, at 37oC for 24 hours. The primary antibodies (Cell Signaling Technology) used included rabbit anti-Mcl-1 (#5453), Bcl-xL (#2764), Bak (#12105), Bax (#5023), caspase-7 (#9492), cleaved caspase-7 (#9491), caspase-9 (#9502), cleaved caspase-9 (#9505), poly (ADP-ribose) polymerase (PARP, #9542) and cleaved PARP (#5625). In the assays to investigate the correlation between GPC1 and beta-catenin, T3M4 cells were cultured at 37oC for 24 hours in a media containing different components, for example, D4-VHH (10 nM), D4-rabbit Fc (10 nM), HM2-LR (10 nM), or HM2-mAb (10 nM). Mouse anti-active β-catenin was purchased from MilliporeSigma (#05–665), rabbit anti-β-catenin was purchased from Cell Signaling Technology (#9582).
Flow cytometry.
Target cells were trypsinized into single-cell suspensions and then incubated with 1 μg/ml of the HM2 mouse antibody or 10 μg/ml of the D4 VHH nanobody in FACS buffer (PBS / 5% BSA / 0.02% NaN3) for 1 hr on ice. The bound HM2 was detected by goat anti-mouse IgG-PE secondary antibody, while the bound D4 VHH was detected by mouse anti-flag antibody followed by goat anti-mouse IgG-PE, in FACS buffer for 30 mins on ice. For binding activity measurement of immunotoxins, a single cell suspension of GPC1+/A431 cells was prepared and incubated with serially diluted immunotoxins in FACS buffer for 1 hr on ice. The cells were then washed with PBS and incubated with rabbit anti-Pseudomonas exotoxin A antibody (Sigma-Aldrich) at 1:200 dilution for 1 hr on ice. After washing with PBS, the cells were incubated with PE-conjugated goat anti-rabbit IgG at 1:200 dilution for 30 mins on ice. The average number of GPC1 sites per cell was measured using BD Quantibrite™ PE beads (BD Biosciences) according to the manufacturer’s instructions. The internalization rate measurement was described as previously reported (23). Cells were analyzed by Canto or Sony SA3800.
Cell proliferation assay.
Target cells were seeded into 96-well plates at a concentration of 5000 cells per well. Serially diluted immunotoxins were added after overnight culture. After a 3-day incubation, the cell supernatant was removed, and cell growth inhibition was detected by the addition of 100 μl/well WST-8 reagent (Dojindo Molecular Technologies) prepared in Opti-MEM medium at 1:10 dilution. Absorbance was read at 450 nm using a microplate reader. Cytotoxicity was presented as IC50, which is the toxin concentration that reduced cell proliferation by 50% when compared with the untreated cells.
Construction of GPC1 knockout single-cell clones by CRISPR Cas9 technology.
GPC1 knockout single-cell clones were constructed according to previously established protocols (32). Several sgRNAs targeting the predicted promoter regions of GPC1 were designed using the website http://crispor.tefor.net/. Three pairs of sgRNAs with high on-target but low off-target effects were selected. The sequences of the GPC1 promoters and the sgRNAs were shown in Supplementary Fig. S1. The three sgRNAs were subcloned into the lentiCRISPRv2 backbone according to the Target Guide Sequence Cloning Protocol. The lentiCRISPRv2 plasmid, together with pMD2G and psPAX2, were co-transfected into 293T cells to produce lentivirus particles, which were then used to transfect the target GFP- and luciferase-expressing T3M4 cells. Single-cell clones not expressing GPC1 were isolated. Flow cytometry and western blot analysis were performed to further identify GPC1−/− single-cell clones. Genomic DNA was extracted using Blood & Cell Culture DNA Mini Kit (Qiagen). The amplicons containing target genes were amplified, extracted, and sequenced. The sequences of GPC1−/− single-cell clones were aligned with the wild-type GPC1 promoter sequence to identify the specific deletions.
IncuCyte Real-Time Growth Assay.
T3M4 cells were seeded in 384-well clear-bottom black tissue culture plates (ViewPlate, PerkinElmer) at a density of 1,000 cells per well in 4a 0 μL culture medium using a Multidrop Combi peristaltic dispenser (ThermoFisher). Echo 525 acoustic dispenser (Beckman Coulter) was used to dispense 3-fold serially diluted SN-38 starting from 1000 nM. Anti-GPC1 immunotoxin was serially diluted (2-fold, starting from 1000 ng/ml) and added to the plate manually. Each combination of SN-38 and immunotoxin was performed in triplicate. Cells were grown in an IncuCyte ZOOM live cell analysis system (Essen Bioscience) placed in an incubator at 37oC supplemented with 5% CO2. Wells were continuously monitored for cell confluency every two hours over 5 days. The percentage of cell confluency was plotted, and statistical analysis was conducted using GraphPad Prism 8 (La Jolla). To evaluate the synergistic effect of SN-38 and immunotoxin, the growth rate was measured as log2 (fold change) per two hours (n = 3 for each dose combination). The data points with less than 0.5% confluency due to out-of-focus imaging were excluded from the analysis. Exponential growth was found between 10 hr and 96 hr in DMSO. Therefore, we normalized confluency to the corresponding T = 10 hr for each replicate, and log2 transformed. Then growth rate (fold change per hour) was fitted by these normalized confluency values at different time points (10–96 hr) using a linear model. Positive (or negative growth rate) was indicative of cells proliferating (or dying) during the experiment.
Cycloheximide (CHX) chase assay.
Targeted cells were seeded in 6-well plates and incubated in a CO2 incubator overnight. After 12-hrs incubation, the medium was removed and replenished with a complete medium with 20 μg/ml cycloheximide (Cell Signaling Technology, #2112). Cell lysates were collected at different time points (t=0, 0.25, 0.5, 0.75, 1, 2, 4, 8, 12, and 24 hrs) according to the experimental design. Cell lysates were centrifuged at 12,000 rpm at 4 oC for 30 mins. Protein concentration was determined by the BCA protein assay kit (Pierce). The results were then analyzed by SDS-PAGE assay and Western blotting. The Western bands of GPC1 and β-actin were quantified using Image Lab Software (BioRad).
Results
Internalization of GPC1 from the surface of pancreatic tumor cells
To investigate whether GPC1 could be a suitable target for an immunotoxin, we measured antigen density and internalization rate in various tumor cell lines. A431 cells exhibited a very low surface GPC1 expression. To enhance the expression of GPC1, we engineered a GPC1+/A431 stable cell line with a high expression of GPC1 on the cell surface. This pair of A431 and GPC1+/A431 was used as the reference cell lines in the following in vitro and in vivo experiments. In addition, we examined the surface expression of GPC1 in two human pancreatic tumor cell lines, T3M4 and KLM1. They displayed moderate protein expression of GPC1 on the cell surface compared to that of GPC1+/A431 cells (Fig. 1A). The overexpressing cell line GPC1+/A431 had over 3×105 recombinant GPC1 sites per cell, whereas only ~7,000 copies of GPC1 were detected on the T3M4 and KLM1 cells (Fig. 1B). The endogenous GPC1 expression levels were then evaluated by western blot (Fig. 1C). To confirm the specificity of antibodies, GPC1 knockout cells, GPC1−/− T3M4, were also included in western blot (the construction of GPC1−/− T3M4 cells was described in Supplementary Fig. S1). T3M4 and KLM1 expressed modest levels of GPC1, while GPC1−/− T3M4 cells had no visible GPC1 band. Though GPC1 is absent on the surface of A431 cells (Fig. 1A), its protein expression can be detected by immunoblot (Fig. 1C), suggesting that the surface level of GPC1 on A431 cells is below the detection limit of the flow cytometer.
Figure 1. GPC1 expression and internalization in pancreatic tumor cells.
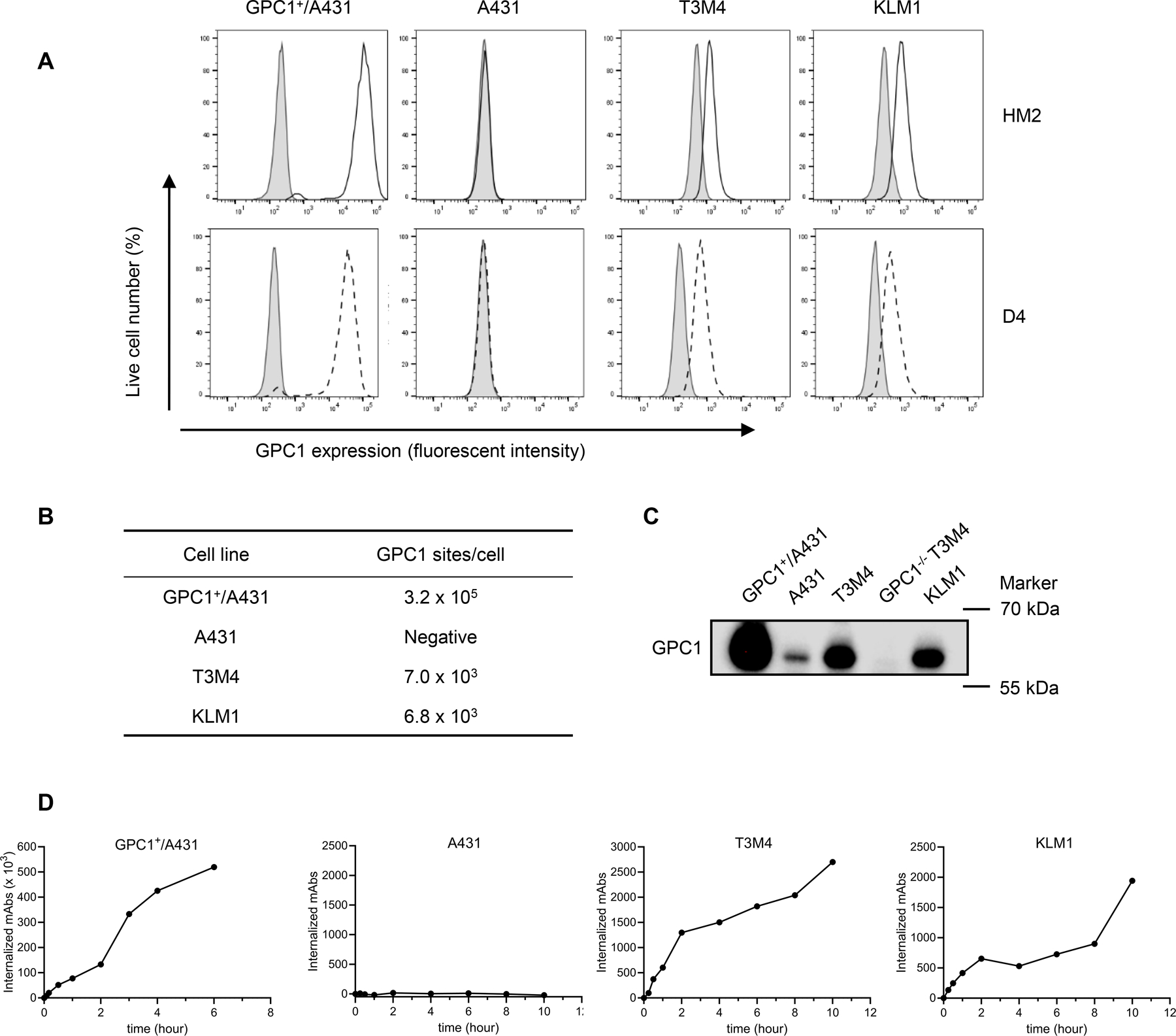
A, Cell surface GPC1 expression in tumor cell lines analyzed by flow cytometry. A431 cells and GPC1+/A431 cells are used as the negative and positive control, respectively. The solid open histograms or dashed open histograms represent cell surface staining of GPC1 with HM2 or D4 antibodies, respectively; shaded gray histograms represent cell surface staining with isotype control. B, Quantification of GPC1 sites per cell using QuantiBrite PE beads. C, Endogenous GPC1 expression level in pancreatic tumor cells and GPC1 knockout cells analyzed by western blot. 1µg of total protein lysate for GPC1+/A431 and 15 µg of total protein lysates for other samples were loaded to the gel. D, Internalization rate of GPC1 detected by flow cytometry (isotype control antibody was not included due to its failure to bind to cell surface GPC1). Western blot and flow cytometry data are representative of three independent experiments.
Next, the internalization rate of GPC1 in pancreatic tumor cells was evaluated by flow cytometry (Fig. 1D). In GPC1+/A431 cells, nearly 25% of surface GPC1 (77,846 molecules) was internalized after 1 hour. Interestingly, more than 100% of surface GPC1 (333,366 molecules) was internalized after 3 hours and additional GPC1 molecules continued to be recruited to the cell surface over time allowing more GPC1 internalization. In contrast, T3M4 and KLM1 showed a much slower GPC1 internalization rate. After 10 hours, approximately 38% and 29% of GPC1 molecules were internalized by T3M4 and KLM1 tumor cells, respectively. No internalization signal was observed on GPC1-negative A431 cells (Fig. 1D). Overall, these data demonstrate that GPC1 is expressed on the cell surface of pancreatic tumor cells and internalized upon the binding of anti-GPC1 antibodies.
Design of anti-GPC1 immunotoxins
D4 (camel VHH) and HM2 (mouse Fv) are two antibodies with specific binding to GPC1 (Supplementary Fig. S2A and B). LR is a PE catalytic domain with an 11-residue linker that contains a furin cleavage site. We fused the variable fragments of D4 or HM2 with LR and generated two immunotoxins against GPC1, D4-LR, or HM2-LR, respectively (Fig. 2A). Then, to obtain immunotoxins with improved binding avidity and efficacy, we generated two bivalent immunotoxins (D4-AAA-D4-LR and D4-GGS-D4-LR) by combining two D4 VHHs with either an AAA or GGS linker (Fig. 2A). All proteins had predicted molecular weights and had high purity with > 95% homogeneity (Fig. 2B). To evaluate interactions between the immunotoxins and GPC1 antigen, the kinetic binding assay was performed to determine the binding affinity or avidity of those four immunotoxins. D4-LR and HM2-LR exhibited high and comparable binding affinity to GPC1 (KD = 6.3–6.9 nM). We observed the enhanced avidity to GPC1 for the bivalent immunotoxins when compared to the monovalent D4-LR, with a KD of 0.09 nM for D4-AAA-D4-LR and 0.08 nM for D4-GGS-D4-LR, respectively (Fig. 2C). Then, ELISA and flow cytometry analysis were performed to further validate the increased binding of D4-AAA-D4-LR and D4-GGS-D4-LR to GPC1 (Fig. 2D and E). In addition, an ADP-ribosylation assay revealed that bivalent immunotoxins had comparable levels of eEF2 modification when compared with the monovalent immunotoxin (Fig. 2F). Taken together, we have generated immunotoxins targeting GPC1 with high purity and high affinity and demonstrated that the engineered bivalent immunotoxins show enhanced avidity and retain enzymatic activity similar to that of the monovalent immunotoxin.
Figure 2. Generation of anti-GPC1 immunotoxins.
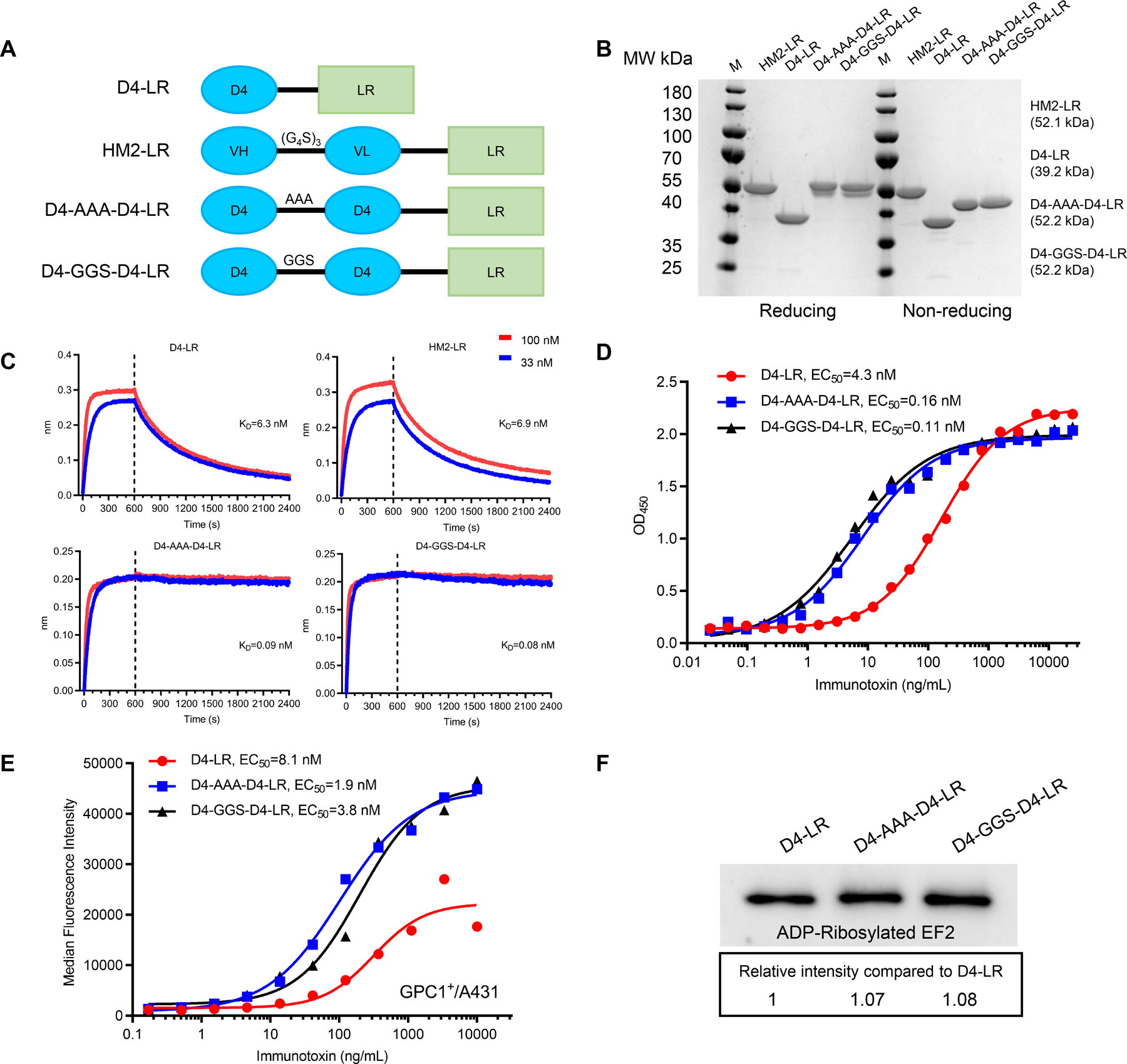
A, Schematic of anti-GPC1 immunotoxins. LR is a truncated PE that lacks domain II but retains catalytic domain III. B, SDS-PAGE gel analysis of purified immunotoxins under reducing and non-reducing conditions. C, Octet binding kinetic analysis, detecting affinity or avidity of anti-GPC1 immunotoxins. GPC1-His at 2 µg/ml was loaded to Ni-NTA sensor, and the binding of each immunotoxin was tested in two different concentrations, 100 nM in red line and 33 nM in the blue line. The association period (0–600 sec) and dissociation period (601–2400 sec) are separated by the vertical dotted line. D, ELISA results show the ability of D4-LR and the bivalent immunotoxins, D4-AAA-D4-LR and D4-GGS-D4-LR, to bind GPC1. E, Cell binding ability of D4-LR and the bivalent immunotoxins to GPC1 positive cells analyzed by flow cytometry. F, Enzymatic activity of D4-LR and the bivalent immunotoxins as assessed by ADP-ribosylated EF2 intensity. All the data are representative of three independent experiments.
Cytotoxicity of immunotoxins on GPC1-positive cancer cells
To evaluate the inhibition of cell proliferation by immunotoxins, we treated GPC1-positive cancer cells with increasing concentrations of immunotoxins. As shown in Fig. 3A, all the anti-GPC1 immunotoxins showed cytotoxicity on GPC1+/A431 cells but exhibited minimal cytotoxic activity on A431 cells. D4-LR and HM2-LR had comparable cytotoxicity, while the bivalent immunotoxins D4-AAA-D4-LR (IC50=0.18 nM) and D4-GGS-D4-LR (IC50=0.15 nM) showed slightly higher inhibitory activity on GPC1+/A431 cells when compared to D4-LR. Nearly 100% of cell death of GPC1+/A431 was observed under microscope at high concentrations of immunotoxins, indicating that the lack of signals reflects cell death due to the activation of apoptosis, not the inhibition of cell proliferation. Moreover, two immunotoxins targeting two closely related glypicans, GPC2 and GPC3 (an anti-GPC2 immunotoxin, CT3-LR, and an anti-GPC3 immunotoxin, HN3-LR) had no cytotoxicity on GPC1+/A431 cells (Fig. 3A), suggesting that the cytotoxicity of our new anti-GPC1 immunotoxins was highly antigen-specific.
Figure 3. Anti-GPC1 immunotoxins showed antigen-dependent cytotoxicity towards pancreatic tumor cells.
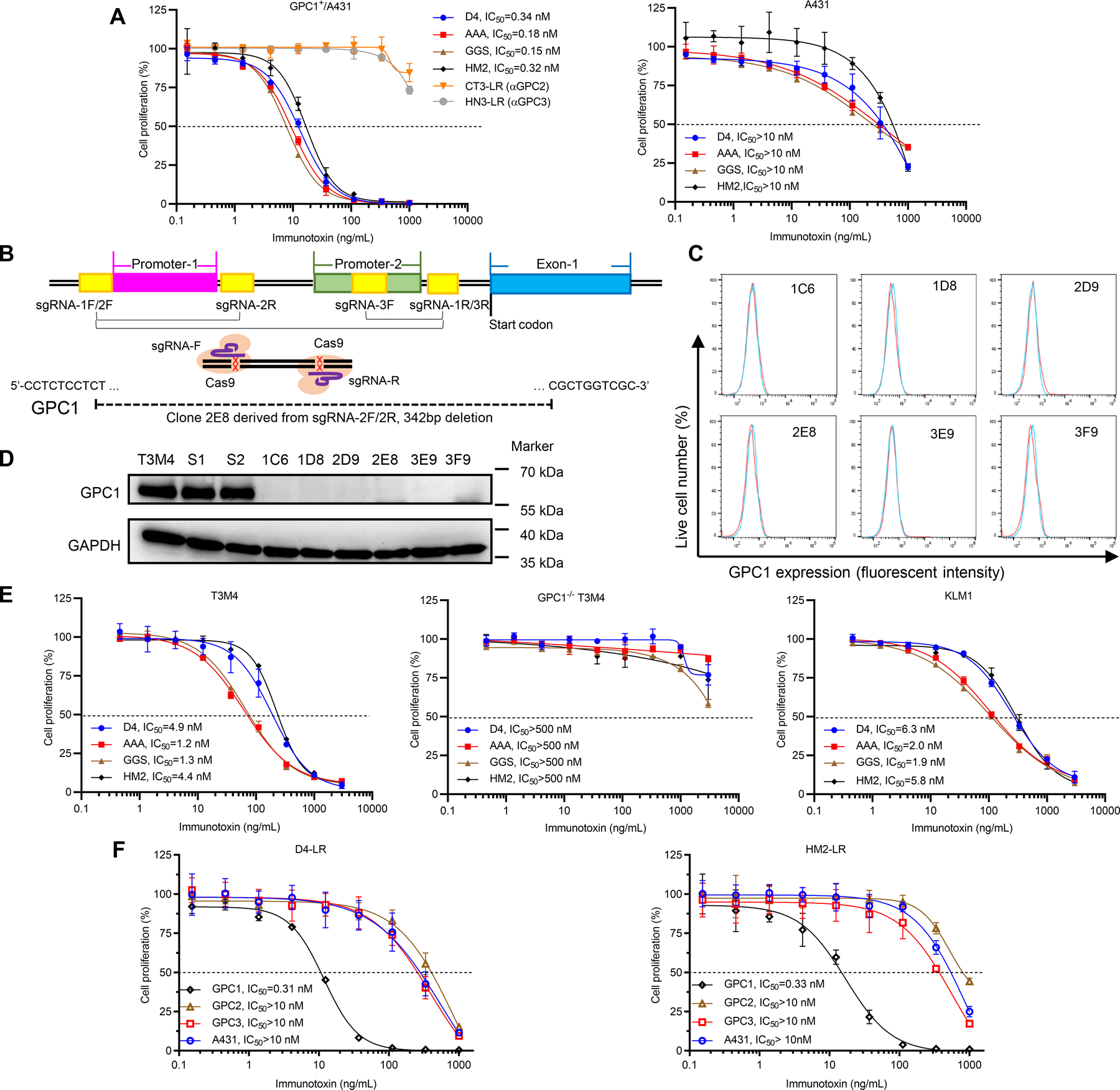
A, Cell proliferation inhibitory activity of immunotoxins on GPC1+/A431 and A431 cells determined by WST-8 reagent after 3-day co-culture. CT3-LR (an anti-GPC2 immunotoxin) and HN3-LR (an anti-GPC3 immunotoxin) were two non-GPC1 targeting controls. Horizontal dashed lines indicate the IC50 value. Data points represent mean ± SD. B, Schematic overview of the construction of GPC1 knockout single-cell clones. Two predicted GPC1 promoter regions are indicated in magenta and green, respectively. GPC1 exon1 is indicated in blue. Three pairs of sgRNAs are indicated in yellow. GPC1 gene deletions with 342 base pairs in clone 2E8 are indicated proportionally. C, GPC1 expression on six single-cell clones detected by flow cytometry. The histograms in red represent cell surface staining of GPC1 with HM2 antibody; the histograms in blue represent cell surface staining of GPC1 with isotype control. D, Levels of GPC1 on six single-cell clones detected by western blot analysis. S1 and S2 represent T3M4 cells transfected with scrambled sgRNA1 and sgRNA2, respectively. E, Cytotoxicity of immunotoxins on T3M4 cells, GPC1 knockout T3M4 cells, and KLM1 cells. Data points represent mean ± SD. F, Cell proliferation inhibition of D4-LR and HM2-LR on A431 cells overexpressing different glypicans (GPC1+/A431, GPC2+/A431, and GPC3+/A431) as well as wild-type A431 cells. Data points represent mean ± SD. Flow cytometry, western blot, and cytotoxicity data are representative of at least three independent experiments.
To further confirm that the cytotoxicity of anti-GPC1 immunotoxins is antigen-dependent, we constructed GPC1 knockout T3M4 single-cell clones using CRISPR Cas9 technology (Fig. 3B). Single-cell clones were GPC1 negative as analyzed by both flow cytometry and western blot (Fig. 3C and D). Genomic DNA sequencing was performed to confirm the deletion of promoter regions of the GPC1 gene as we previously described for GPC3 knockout in liver cancer cells (32). Deletions of the predicted promoters (and part of exon-1) of the GPC1 gene were identified in the 2E8 clone (Fig. 3B). The 2E8 clone is used as the GPC1 knockout T3M4 cell model in the present study. Subsequently, two human pancreatic tumor cell lines, T3M4 and KLM1, as well as GPC1−/− T3M4 cells were used to evaluate the cytotoxic properties of anti-GPC1 immunotoxins in a cell proliferation assay. D4-LR (IC50=4.9 nM) and HM2-LR (IC50=4.4 nM) showed less cytotoxic activity on T3M4 cells compared to GPC1+/A431 cells (Fig. 3E). Of note, D4-AAA-D4-LR (IC50=1.2 nM) and D4-GGS-D4-LR (IC50=1.3 nM) exhibited better efficacy (3-fold) on T3M4 cells when compared with D4-LR. Similar cytotoxic activity results could be found on KLM1 cells (Fig. 3E). None of the immunotoxins affected the growth of GPC1−/− T3M4 cells (Fig. 3E). We also examined the activity of anti-GPC1 immunotoxins on A431 cell lines overexpressing other glypican family members, including GPC2+/A431 (clone name G10) and GPC3+/A431 (clone name G1). The growth of these two cell lines was minimally affected by anti-GPC1 immunotoxins (Fig. 3F). Together, we conclude that the cytotoxic activity of anti-GPC1 immunotoxins is antigen-dependent.
Immunotoxins targeting GPC1 inhibits tumor growth in mice
To evaluate the anti-tumor efficacy of the immunotoxins, two different mouse models were established. In the GPC1+/A431 subcutaneous model, anti-GPC1 immunotoxins including D4-LR (5 mg/kg), HM2-LR (5 mg/kg), D4-AAA-D4-LR (3 mg/kg) were administered to the mice (Supplementary Fig. S3A). Treatment with any of the three GPC1-specific immunotoxins alone inhibited tumor growth (Supplementary Fig. S3B) and extended mice survival (Supplementary Fig. S3C). In another pancreatic tumor model, T3M4-GFP/luciferase tumor cells were inoculated into mice by intraperitoneal injection. Six days after inoculation, mice were treated with 3 mg/kg of D4-LR, 3 mg/kg of HM2-LR, or 1.5 mg/kg of D4-AAA-D4-LR (Supplementary Fig. S3D). Groups that received immunotoxins treatment showed evidence of tumor burden reduction (Supplementary Fig. S3E) and survival increase (Supplementary Fig. S3F). However, D4-AAA-D4-LR failed to show better efficacy than D4-LR even though it showed enhanced cytotoxic activity on T3M4 cells in vitro. Taken together, our findings demonstrate that anti-GPC1 immunotoxins alone inhibit GPC1-positive tumor growth, yet could not induce tumor regression in mice.
Anti-GPC1 immunotoxin in combination with irinotecan causes pancreatic tumor regression in mice
To improve the efficacy of immunotoxin therapy, we performed an in vitro screening to identify small-molecule compounds showing a synergistic effect with the immunotoxin against tumor cells. We screened an anti-GPC3 immunotoxin (HN3-PE)(26) on Hep3B cells using a library of 1912 clinical-grade small molecule compounds. We then narrowed down to a group of 11 compounds (Supplementary Table 1) for further screening with anti-GPC1 (D4-LR) on T3M4 tumor cells based on two criteria: (a) top synergistic cytotoxicity with an anti-GPC3 immunotoxin on Hep3B tumor cells and (b) standard care medication for pancreatic cancer. Bcl-2 family inhibitor, ABT-263, and the topoisomerase inhibitor, SN-38, were the top two candidates that showed good synergism with the anti-GPC1 immunotoxin on the T3M4 cell model (Fig. 4A and B). The combination index (CI) theorem offers a quantitative definition for additive effect (CI = 1), synergism (CI < 1), and antagonism (CI > 1) in drug combinations (33). Both the combinations provided a synergistic effect on T3M4 cell proliferation inhibition (Fig. 4C). To further validate the synergism provided by SN-38 with immunotoxin, the IncuCyte assay was performed and T3M4 cell confluency was continuously monitored for five days (Supplementary Fig. S4). The corresponding dose-response curve (DRC) of D4-LR plotted by the area under the curve (AUC) is shown in Fig. 4D. Consistent with the single timepoint combination data (Fig. 4A), we observed synergy of growth rate reduction from cells treated with SN-38 and D4-LR. The growth rate was significantly reduced as compared to SN-38 or D4-LR alone (Supplementary Fig. S5).
Figure 4. D4-LR in combination with irinotecan regressed pancreatic tumor in mice.
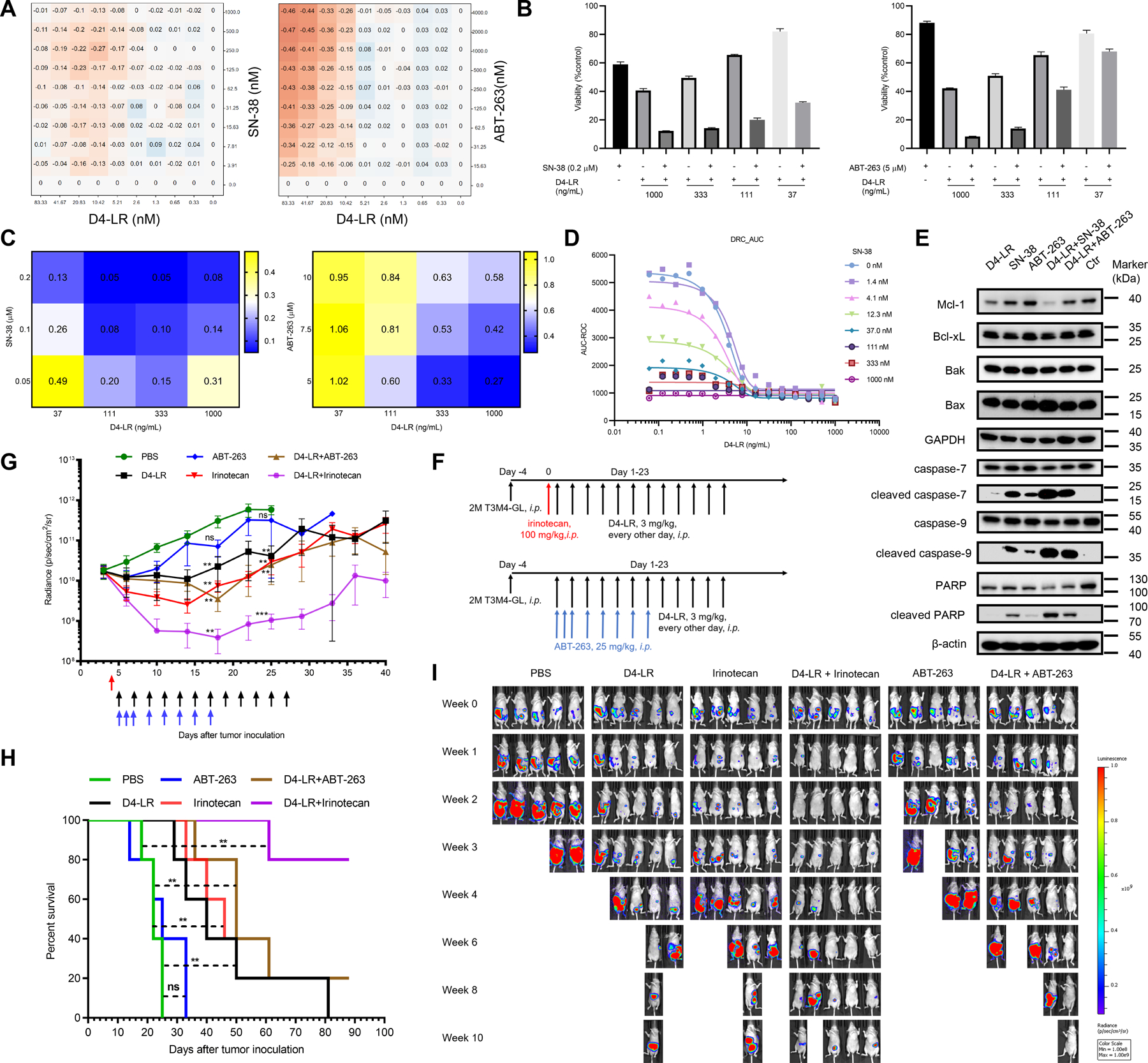
A, In vitro screening results of SN-38 and ABT-263 in combination with D4-LR towards T3M4 cells. Shaded orange represents synergism. B, Viability of T3M4 cells after co-culture with D4-LR for 48 hours at 37℃ in the presence or absence of SN-38 (0.2 μM, left panel) or ABT-263 (5 μM, right panel). Values represent mean ± SD. Cell viability data are representative of at least three independent experiments. C, The calculated combination index for drug combinations, D4-LR + SN-38 or D4-LR + ABT-263, based on the viability of T3M4 cells in B. CI = 1 (additive effect), CI < 1 (synergism), and CI > 1 (antagonism). D, Dose-response curve (DRC) of D4-LR plotted by Area under the curve (AUC). E, Western blot analysis showing the expression levels of key components in cell apoptotic signaling pathways in T3M4 cells after co-culture with D4-LR, SN-38, ABT-263, or combinations for 24 hours. Ctr represents T3M4 cells only. 15µg of total cell lysates were loaded for each sample. Data are representative of three independent experiments. F, Six-week-old female athymic nude mice were intraperitoneally injected with 2 × 106 T3M4-GL cells (T3M4 cells tagged with GFP and luciferase). One single injection of irinotecan at 100 mg/kg (indicated with red arrow) was administered to the mice 24-hours before immunotoxins treatment. ABT-263 at 25 mg/kg was intraperitoneally injected on the days indicated by blue arrows. D4-LR at 3 mg/kg was intraperitoneally injected on the days indicated by black arrows. N=5 for each group. G, Average tumor volume ± SEM for each group. H, Kaplan-Meier survival curve of mice upon treatment with indicated immunotoxins. I, Bioluminescent imaging showing tumor burden of an individual mouse. D4-LR in combination with irinotecan demonstrated potent anti-tumor activity in T3M4 xenograft tumors, D4-LR, irinotecan, or D4-LR combined with ABT-263 modestly inhibited the tumor growth, while ABT-263 alone failed to suppress tumor growth.
Then, to study the underlying mechanism of the combinational therapy that showed better cytotoxicity, we performed western blot analysis and determined the expression levels of key components of the apoptotic cell death signaling pathway (Fig. 4E). No significant difference in the expression of Bcl-xL, Bax, or Bak was observed. Rapid degradation of Mcl-1 occurred after anti-GPC1 immunotoxin treatment. The loss of Mcl-1 was even more significant in the group treated with immunotoxin in combination with SN-38 (Fig. 4E). Levels of cleaved caspase-7, −9, and -PARP were upregulated in T3M4 cells treated with SN-38 or ABT-263, and to a greater extent in both combinational groups (Fig. 4E). These data reveal that the higher activation of the apoptotic signaling pathway may be one of the mechanisms underlying the enhanced efficacy of combinational therapy.
To evaluate the anti-tumor efficacy of the combinational therapy in mice, we established the T3M4 pancreatic tumor model and treated the mice as designed (Fig. 4F). Irinotecan is the prodrug of SN-38 and is the second-line treatment for patients with metastatic pancreatic cancer after gemcitabine-based chemotherapy. Our previous study demonstrated that the combination of irinotecan with an immunotoxin against GPC3 significantly reduced tumor burden in the HCC xenograft mice model (23). Therefore, irinotecan was used in mice. Mice were treated with single agents (ABT-263, irinotecan, or D4-LR) or combinations (D4-LR + ABT-263, or D4-LR + irinotecan). As shown in Fig. 4G, the administration of D4-LR in combination with irinotecan potently reduced tumor burden in mice while D4-LR, irinotecan, or D4-LR combined with ABT-263 modestly inhibited T3M4 xenograft tumor growth. ABT-263 alone showed minimal anti-tumor activity. Parallelly, D4-LR + irinotecan significantly extended mice survival; 80% of the mice were still alive at the experimental endpoint (Fig. 4H). All the treatment groups showed evidence of decreased tumor burden (Fig. 4I). Four weeks post-treatment, all the mice untreated or treated with ABT-263 were lost due to a large tumor burden. The tumors in mice received D4-LR, irinotecan, or D4-LR + ABT-263 grew; 100% of mice from those groups died after 10 weeks except one mouse in D4-LR + ABT-263 group that experienced tumor regression. Interestingly, D4-LR in combination with irinotecan caused tumor regression in 80% of mice two weeks post-treatment, and at the end of the study, there were 80% of mice alive with relatively small tumor burden (Fig. 4I). Taken together, we may conclude that immunotoxin targeting GPC1 in combination with irinotecan shows the synergistic killing capacity and induces T3M4 pancreatic tumor regression in mice, possibly due to the significant loss of Mcl-1 and the activation of apoptosis signaling.
Downregulation of active β-catenin levels in pancreatic tumor cells by targeting GPC1
In addition to the apoptosis signaling pathway, we also measured GPC1 expression levels in pancreatic cancer cells treated with anti-GPC1 immunotoxin, SN-38, ABT-263 alone or in combination (Fig. 5A). The levels of GPC1 were reduced after immunotoxin or SN-38 treatment, and the levels were undetectable after treatment with immunotoxin combined with SN-38. We hypothesized that one of the mechanisms might be related to a short turnover rate of GPC1 in cells. T3M4 cells were then treated with cycloheximide (CHX) at different time points to determine the half-life of GPC1. The CHX chase assay revealed that GPC1 indeed had a short half-life of around 15 minutes (Fig. 5B). In contrast, the half-life of other glypicans, e.g., GPC2 and GPC3, was determined to be approximately one hour and two hours, respectively, longer than that of GPC1 (Supplementary Fig. S6 A-D). In addition to the short half-life of GPC1 in cells, we hypothesized that reduction of GPC1 might be due to the internalization and lysosomal degradation of the immunotoxin/GPC1 complex. To test this hypothesis, T3M4 cells were treated with immunotoxin targeting GPC1 at different time points, and the levels of GPC1 expression were monitored. We found that GPC1 levels continued to decrease as the incubation time increased (Fig. 5B). We compared the reduction of GPC1 after immunotoxin treatment with the GPC1 internalization rate and found that these two parameters were highly correlated, suggesting that the decrease of GPC1 levels may be due to the internalization of GPC1 upon immunotoxin binding and subsequent complex degradation in the lysosome (Fig. 5B).
Figure 5. Anti-GPC1 immunotoxin downregulated active β-catenin levels in pancreatic tumor cells by targeting GPC1.
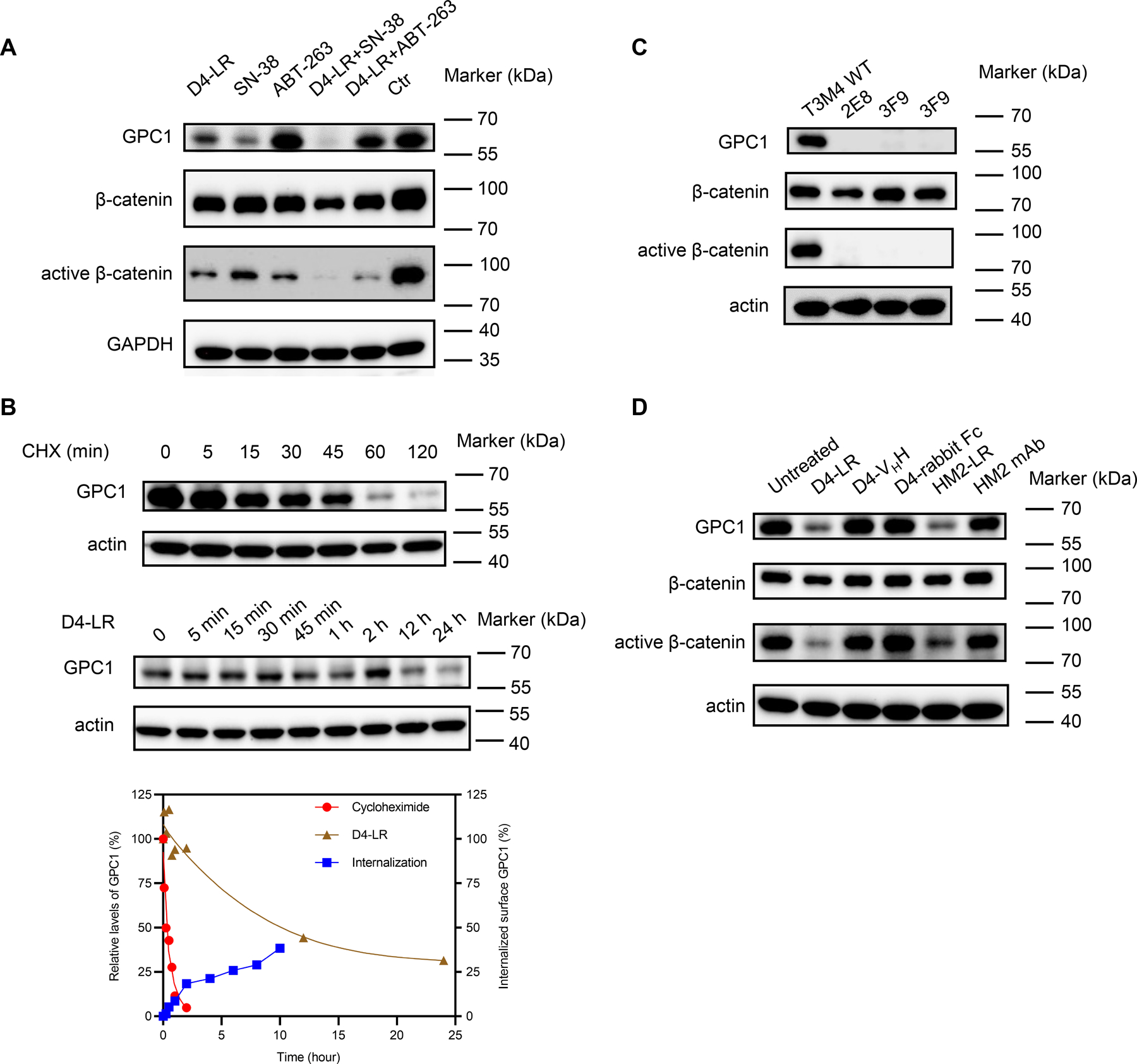
A, The expression levels of GPC1, β-catenin, and active β-catenin in T3M4 cells treated with D4-LR, SN-38, ABT-263 alone or in combination. B, Measurement of GPC1 expression levels in T3M4 after treatment with cycloheximide (upper panel) or D4-LR (middle panel) at different time points. β-actin was stained as a positive control for sample loading. Signals of the target bands were quantified with Image Lab software (Bio-Rad). GPC1 bands were normalized to β-actin, and then normalized to the t=0 controls and plotted in the bottom panel. C, The expression levels of GPC1, β-catenin, and active β-catenin in wild-type T3M4, and GPC1 knockout T3M4 single cells clones. D, The expression levels of GPC1, β-catenin, and active β-catenin in T3M4 cells treated with anti-GPC1 antibodies (D4-VHH, D4-rabbit Fc or HM2 mAb) or immunotoxins (D4-LR or HM2-LR).
Glypicans (e.g., GPC2 and GPC3) have been reported to be extracellular co-receptors for Wnt proteins, playing an essential role in the Wnt/β-catenin signaling pathway (23,34,35). As shown in Fig. 5A, the expression of active β-catenin was substantially reduced following anti-GPC1 immunotoxin treatment, while total β-catenin expression was minimally affected. To further investigate whether GPC1 could affect Wnt signaling in pancreatic tumor cells, we assessed the levels of total β-catenin and active β-catenin in GPC1 knockout T3M4 single-cell clones. No significant differences in total β-catenin levels were observed between knockout and wild-type cells. Interestingly, active β-catenin levels were greatly decreased in GPC1 knockout cells when compared with wild-type T3M4 cells, suggesting that the Wnt/β-catenin signaling pathway may be inhibited by GPC1 removal (Fig. 5C).
Next, to investigate whether the antibody component of the immunotoxin was enough to downregulate active β-catenin, we treated T3M4 cells with anti-GPC1 antibodies in different formats or anti-GPC1 immunotoxins. Active β-catenin levels remained unchanged when treated with anti-GPC1 antibodies only, but decreased after immunotoxin treatment. These data suggest that the binding of GPC1 antigen by antibodies was not sufficient for active β-catenin reduction. However, GPC1 degradation upon the treatment of immunotoxins reduces active β-catenin. (Fig. 5D). These findings suggest that similar to GPC3 in liver cancer (35), GPC1 may be associated with the Wnt/β-catenin signaling pathway in pancreatic cancer. Taken together, our data indicate that anti-GPC1 immunotoxin could downregulate active β-catenin levels in pancreatic tumor cells following GPC1 reduction by immunotoxins.
Anti-GPC1 immunotoxin containing ABD exhibits greater activity in mice
To further optimize the efficacy of anti-GPC1 immunotoxins, we engineered three more constructs based on D4-LR (Supplementary Fig. S7A). The addition of ABD to immunotoxins has been reported to improve the anti-tumor efficacy in mice via serum half-life extension (26,36). In the present study, we incorporated a 54-amino acid-long ABD isolated from the streptococcal protein G into D4-LR to generate D4-ABD-LR. Furthermore, we replaced the LR fragment of D4-ABD-LR with the T20 fragment (26,37,38) containing 6-point mutations targeting T cell activation to create D4-ABD-T20 for reduced immunogenicity. We also produced D4-PE38 containing both domain II and domain III of PE. All the immunotoxins showed great purity (Supplementary Fig. S7B) and comparable binding affinities to GPC1 (Supplementary Fig. S7C). In contrast to D4-LR or D4-PE38, the ABD-containing immunotoxins, D4-ABD-LR or D4-ABD-T20 showed strong binding to both human and mouse serum albumin (Supplementary Fig. S7D). In addition, the cell proliferation inhibition assay was performed to compare the cytotoxicity of D4-based immunotoxins (Supplementary Fig. S7E). In summary, D4-ABD-LR with strong binding to serum albumin is a promising candidate with potentially enhanced efficacy.
To evaluate the efficacy of D4-ABD-LR as a single treatment or in combination with irinotecan, mice bearing T3M4 pancreatic tumors were established and treated as planned (Fig. 6A). In combinational groups, twenty-four hours post-irinotecan administration, mice were administered either D4-LR at 3 mg/kg or D4-ABD-LR at 1 mg/kg intraperitoneally every other day. While the tumors from the control group continued to grow and the single-agent treatment groups could only stabilize tumor growth, the combinational groups, D4-LR + irinotecan, and D4-ABD-LR + irinotecan, significantly reduced tumor burden (Fig. 6B) and increased mice survival (Fig. 6C). As shown in Fig. 6D, all mice in the PBS control group were removed by week-4 since the tumor burden exceeded the cut-off value of 5×1011 p/sec/cm2/sr. At the same time point, only one mouse in the D4-LR group and two mice in the irinotecan group bear large tumors with radiance signals over 1×1011 p/sec/cm2/sr. Mice from the two combinational groups (D4-LR + irinotecan, D4-ABD-LR + irinotecan) showed little to no tumor burden. After nine weeks, all mice treated with a single agent (D4-LR, D4-ABD-LR, or irinotecan) died due to large tumor burden except one mouse bearing small tumor (4.8×107 p/sec/cm2/sr) in the D4-ABD-LR group. Fifteen weeks post-treatment, there was still one mouse alive in D4-LR + irinotecan group, and two mice in D4-ABD-LR + irinotecan group (Fig. 6D). No difference in the tumor volume or survival rate for the D4-LR and the D4-ABD-LR groups were observed, likely due to the lower dose we used for D4-ABD-LR (1 mg/kg) compared to D4-LR (3 mg/kg). Similarly, for combinational therapies, 1 mg/kg of D4-ABD-LR in combination with irinotecan showed enhanced, though not significant, survival benefit when compared to D4-LR at 3 mg/kg combined with irinotecan. These data suggest that D4-ABD-LR with a one-third dose of D4-LR retains similar anti-tumor activity in mice either as a single agent or in combination, probably due to the improved half-life of D4-ABD-LR in mice.
Figure 6. D4-ABD-LR in combination with irinotecan showed enhanced anti-tumor efficacy.
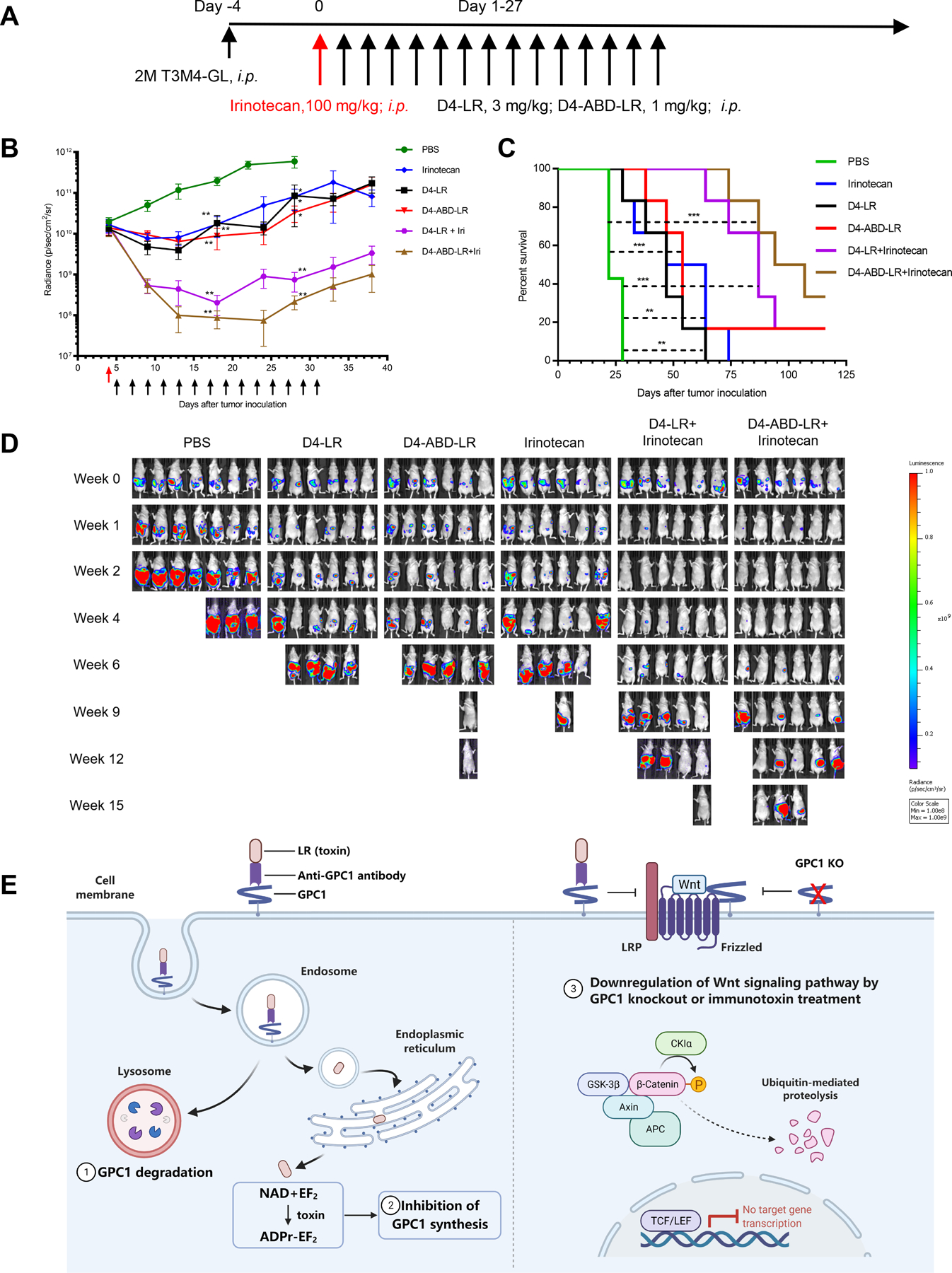
A Timeline of the T3M4 mouse model treated with D4-LR or D4-ABD-LR as a single agent or combined with irinotecan. Six-week-old female athymic nude mice were intraperitoneally injected with 2 × 106 T3M4 cells. The red arrow represents the administration of irinotecan at 100 mg/kg. D4-LR at 3 mg/kg or D4-ABD-LR at 1 mg/kg was intraperitoneally injected into mice on the days indicated by black arrows. N=7 for PBS control group, n=6 for other treating groups. B, Average tumor volume ± SEM for each group. C, Kaplan-Meier survival curve. D, Bioluminescent imaging showing tumor burden of an individual mouse. E, A schematic describing three potential mechanisms employed by anti-GPC1 immunotoxins to inhibit pancreatic tumor cells growth. Upon binding to GPC1 on the tumor cell surface, the GPC1/immunotoxin complex is internalized by endocytosis. In the endosome, the immunotoxin is processed by the protease furin to separate the antibody fragment from the toxin. The antibody fragment together with GPC1 goes to the lysosome where it is degraded. In contrast, the toxin is transferred to the endoplasmic reticulum and then enters the cytosol. After reaching the cytosol, the toxin resulted in the inhibition of GPC1 synthesis via mediating ADP-ribosylation of EF2. In addition, GPC1 knockout or immunotoxin treatment reduces Wnt binding to Frizzed/LRP, leading to the downregulation of Wnt/β-catenin signaling. Abbreviations: ADPr, ADP-ribose; APC, adenomatous polyposis coli; CKIα, casein kinase Iα; EF2, elongation factor 2; GSK-3β, glycogen synthase kinase-3β; KO, knockout; LRP, low-density lipoprotein receptor-related protein; NAD, nicotinamide adenine dinucleotide; TCF/LEF, T cell factor/lymphoid enhancer factor. Figure 6E was created with BioRender (https://app.biorender.com).
The representative images of tumor mass tagged with GFP fluorescence in a mouse from the control group were shown in Supplementary Fig. S8A. Significantly large tumors were found from the mice in the untreated group (Supplementary Fig. S8B); whereas in the D4-LR or irinotecan treated groups, only small nodules were found from the peritoneal wall (Supplementary Fig. S8C). The harvested tumors (from two individual mice per group) were lysed and investigated for the expression levels of β-catenin, active β-catenin, and GPC1, by western blot analysis (Supplementary Fig. S9). The levels of GPC1 in tumors from treatment groups were reduced than those of the PBS control group. We can also see a slight reduction of active β-catenin in tumor cells derived from mice treated with immunotoxin and/or irinotecan in comparison with that from the PBS group. The limitation of the WB analysis using tumor samples harvested from mice is that the commercial antibodies bind both mouse and human total β-catenin and active β-catenin. Therefore, the amount of total and active β-catenin detected in tumor samples might be less accurate due to the influence of other cells including mouse cells in the xenograft tumors.
To further assess the toxicity of single-agent or combinational therapy, after 11 injections of immunotoxins, three mice from each group were selected for comprehensive mouse blood analysis and necropsy (Supplementary Table 2). No obvious adverse effects were found in mice though D4 could bind to mouse GPC1. No significant weight change was observed for the other internal organs. Collectively, these results demonstrate that D4-ABD-LR in combination with irinotecan shows high potency and low toxicity, making it a promising therapeutic for pancreatic tumor treatment.
Discussion
In the present study, we generated and tested novel immunotoxins targeting the cell surface proteoglycan GPC1 and demonstrated that GPC1 may be a promising target of antibody toxin conjugates for pancreatic cancer. We also tested several different strategies to improve the efficacy of anti-GPC1 immunotoxins.
Since single domain antibodies can be easily engineered into multivalent structures with improved avidity, we firstly constructed bivalent immunotoxins based on D4-LR by fusing two tandem D4 domains to the LR toxin domain. As expected, the re-engineered bivalent immunotoxins showed greatly improved binding avidity and had 3–4-fold higher cytotoxicity in T3M4 cells in culture. However, D4-AAA-D4-LR failed to exhibit increased anti-tumor efficacy in mice as compared with monomeric D4-LR. The size and valency of immunotoxins may influence the uptake and intracellular transport of an antigen. It has been reported that a ligand-nanoparticle conjugate induced different endocytic mechanisms when compared with the free ligand (39). In the present study, the bivalent anti-GPC1 immunotoxins with enlarged size and valency may somehow employ a different antigen uptake and intracellular transport compared to the monovalent immunotoxin, which may attenuate the efficacy despite achieving enhanced binding.
Different antigen densities and cell types may contribute to different internalization rates or levels. In the present study, GPC1+/A431 shows the highest expression levels of GPC1 (3.2E5), followed by T3M4 (7.0E3) and KLM1(6.8E3). More GPC1 molecules on the cell surface may induce faster internalization. In addition, pancreatic tumor cells lines may naturally bear resistance to the receptor-mediated endocytosis. A previous study has revealed that Erbitux could mediate EGFR internalization in MIA PaCa-2 cells after 2 hours of incubation, whereas it did not promote EGFR internalization in BxPC-3 cells, though both pancreatic cancer cell lines expressed EGFR (40). Similarly, T3M4 and KLM1 may have different mechanisms for internalization, leading to a reduced internalization rate when compared to GPC1+/A431 cells.
To obtain better anti-tumor efficacy, a combination of immunotoxins with other agents is a viable strategy (41). The mechanism of action for immunotoxins is protein synthesis inhibition, which is unique from other cancer treatment regimens. Thus, immunotoxins can be useful to combine with other therapeutics for anti-tumor synergism. We initially screened a library of 1912 small compounds with a PE-based immunotoxin targeting GPC3 on the HCC tumor cell line (Hep3B). The standard of care drugs used in pancreatic cancer such as Abraxane (taxol/paclitaxel), Cisplatin, Oxaliplatin, and 5-FU were tested but showed no synergistic effect with the GPC3-specific immunotoxin on Hep3B tumor cells. We speculated that these drugs, among many others, would unlikely have a synergistic effect with immunotoxins for killing cancer cells in general. Therefore, we decided to focus on the compounds based on the two criteria: (1) top synergistic with the GPC3-specific immunotoxin on Hep3B cells and (2) standard care for pancreatic cancer. However, we understand the limitation of our screening during the ongoing pandemic. Future more comprehensive screening of a large library with our immunotoxin targeting GPC1 in multiple pancreatic cell lines would be useful to identify potentially more compounds for treating pancreatic cancer. In the present study, ABT-263 and SN-38 were identified as two top candidates showing synergism with anti-GPC1 immunotoxin. SN38 is the active metabolism of irinotecan and acts as a topoisomerase I inhibitor by inhibiting DNA synthesis. Irinotecan is a key component of a chemotherapy combination (FOLFIRINOX), which is commonly used for pancreatic cancer treatment. In the present study, the combination of immunotoxin with irinotecan resulted in pancreatic tumor regression in mice and significantly extended survival rate. Significant reduction of Mcl-1 expression and the activation of caspases may be one of the factors leading to tumor cell apoptosis when treated with immunotoxin in combination with irinotecan. Future studies using immunocompetent pancreatic tumor mouse models would be useful to further evaluate the therapeutic effect of the anti-GPC1 immunotoxin.
We also investigated the underlying mechanisms by which immunotoxins against GPC1 inhibit pancreatic tumor growth (Fig. 6E). The levels of active β-catenin were substantially reduced after GPC1 was knocked out in pancreatic tumor cells. Importantly, in the present study, we found that GPC1 has a uniquely much shorter intracellular half-life (15 minutes) (Fig. 5B) than those of GPC2 (1 hour) and GPC3 (2 hours) (Supplementary Fig. S6), other members of the glypican family. Immunotoxin treatment could reduce the expression levels of short-lived GPC1 through lysosomal degradation following internalization. The generation of new GPC1 molecules is blocked by immunotoxin-induced protein synthesis inhibition. The downregulated GPC1 can inhibit the downstream Wnt signaling cascade by reducing the levels of active-β-catenin. The precise mechanism of GPC1 in Wnt/ β-catenin signaling regulation remains to be further studied.
In conclusion, our results indicate that GPC1 is a target for immunotoxin therapy in pancreatic cancer. Anti-GPC1 immunotoxins inhibit tumor cell growth via degradation of internalized GPC1, downregulation of Wnt signaling, and blockade of protein synthesis. The anti-GPC1 immunotoxin in combination with irinotecan is a promising therapeutic strategy to treat pancreatic cancer.
Supplementary Material
Acknowledgments
We thank the NCI CCR Animal Resource Program/NCI Biological Testing Branch and NCI CCR/Leidos Animal Facility for assisting in animal support and the NIH Fellows Editorial Board for editorial assistance. We also thank Daniel Urban (NCATS) for conducting an initial screening of our anti-GPC3 immunotoxin on the Hep3B cell line using a library of 1912 small compounds. The anti-GPC1 antibodies, including HM2 and D4, are the subject of pending patent applications assigned to the NIH and are available for license in certain fields of use to qualified candidates. Please contact Dr. Mitchell Ho (NCI) at homi@nih.gov if you are interested in pursuing a license.
Financial support
This work was supported by the Intramural Research Program of the National Institutes of Health (NIH), National Cancer Institute (NCI), Center for Cancer Research (CCR) (Z01 BC010891, ZIA BC010891 and ZIC BC011891 to M.H.). JP was a recipient of a predoctoral fellowship from the China Scholarship Council and supported by the CCR at the NCI and the NIH Graduate Partnerships Program in Bethesda, Maryland.
Footnotes
Conflict of Interest
Mitchell Ho, Nan Li, and Jiajia Pan are inventors on the international patent application no. PCT/US2020/013739, “High affinity monoclonal antibodies targeting glypican-1 and methods of use thereof”. Any patent awarded will be the property of the NIH. All authors declare no potential conflict of interest.
Ethics approval
All the procedures used in the animal studies were approved by the Institutional Animal Care and Use Committee at the National Institutes of Health (NIH) under the protocol (LMB-059).
Data availability statement
Data sharing does not apply to this article as no datasets were generated or analyzed during the current study. Materials from the present study are available from the corresponding author Dr. Mitchell Ho at homi@mail.nih.gov under material transfer agreements.
Patient consent for publication
Not applicable.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70(1):7–30 doi 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15(6):2403–13 doi 10.1200/jco.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 3.Colucci G, Labianca R, Di Costanzo F, Gebbia V, Cartenì G, Massidda B, et al. Randomized phase III trial of gemcitabine plus cisplatin compared with single-agent gemcitabine as first-line treatment of patients with advanced pancreatic cancer: the GIP-1 study. J Clin Oncol 2010;28(10):1645–51. [DOI] [PubMed] [Google Scholar]
- 4.Ueno H, Ioka T, Ikeda M, Ohkawa S, Yanagimoto H, Boku N, et al. Randomized phase III study of gemcitabine plus S-1, S-1 alone, or gemcitabine alone in patients with locally advanced and metastatic pancreatic cancer in Japan and Taiwan: GEST study. J Clin Oncol 2013;31(13):1640–8. [DOI] [PubMed] [Google Scholar]
- 5.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. The New England journal of medicine 2013;369(18):1691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Philip PA, Lacy J, Portales F, Sobrero A, Pazo-Cid R, Manzano Mozo JL, et al. Nab-paclitaxel plus gemcitabine in patients with locally advanced pancreatic cancer (LAPACT): a multicentre, open-label phase 2 study. Lancet Gastroenterol Hepatol 2020;5(3):285–94 doi 10.1016/s2468-1253(19)30327–9. [DOI] [PubMed] [Google Scholar]
- 7.Sun C, Ansari D, Andersson R, Wu D-Q. Does gemcitabine-based combination therapy improve the prognosis of unresectable pancreatic cancer? World journal of gastroenterology 2012;18(35):4944–58 doi 10.3748/wjg.v18.i35.4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li N, Gao W, Zhang YF, Ho M. Glypicans as Cancer Therapeutic Targets. Trends Cancer 2018;4(2405–8025 (Electronic)):741–54 doi 10.1016/j.trecan.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan J, Ho M. Role of glypican-1 in regulating multiple cellular signaling pathways. American Journal of Physiology-Cell Physiology 2021;321(5):C846–C58 doi 10.1152/ajpcell.00290.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleeff J, Ishiwata T, Kumbasar A, Friess H, Buchler MW, Lander AD, et al. The cell-surface heparan sulfate proteoglycan glypican-1 regulates growth factor action in pancreatic carcinoma cells and is overexpressed in human pancreatic cancer. J Clin Invest 1998;102(9):1662–73 doi 10.1172/JCI4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu H, Niu F, Liu F, Gao J, Sun Y, Zhao X. Elevated glypican-1 expression is associated with an unfavorable prognosis in pancreatic ductal adenocarcinoma. Cancer Med 2017;6(6):1181–91 doi 10.1002/cam4.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015;523(7559):177–82 doi 10.1038/nature14581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kato D, Yaguchi T, Iwata T, Katoh Y, Morii K, Tsubota K, et al. GPC1 specific CAR-T cells eradicate established solid tumor without adverse effects and synergize with anti-PD-1 Ab. eLife 2020;9 doi 10.7554/eLife.49392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pastan I, Willingham MC, FitzGerald DJ. Immunotoxins. Cell 1986;47(5):641–8 doi 10.1016/0092-8674(86)90506-4. [DOI] [PubMed] [Google Scholar]
- 15.Pastan I, Ho M. Recombinant Immunotoxins for Treating Cancer. In: Kontermann R, Dübel S, editors. Antibody Engineering Berlin, Heidelberg: Springer Berlin Heidelberg; 2010. p 127–46. [Google Scholar]
- 16.Kreitman RJ, Wilson WH, Bergeron K, Raggio M, Stetler-Stevenson M, FitzGerald DJ, et al. Efficacy of the Anti-CD22 Recombinant Immunotoxin BL22 in Chemotherapy-Resistant Hairy-Cell Leukemia. The New England journal of medicine 2001;345(4):241–7 doi 10.1056/nejm200107263450402. [DOI] [PubMed] [Google Scholar]
- 17.Lin AY, Dinner SN. Moxetumomab pasudotox for hairy cell leukemia: preclinical development to FDA approval. Blood advances 2019;3(19):2905–10 doi 10.1182/bloodadvances.2019000507 %J Blood Advances. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andersson Y, Juell S, Fodstad Ø. Downregulation of the antiapoptotic MCL-1 protein and apoptosis in MA-11 breast cancer cells induced by an anti-epidermal growth factor receptor–Pseudomonas exotoxin a immunotoxin. Int J Cancer 2004;112(3):475–83 doi 10.1002/ijc.20371. [DOI] [PubMed] [Google Scholar]
- 19.Kreitman RJ, Pastan I. Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin Cancer Res 2011;17(20):6398–405 doi 10.1158/1078-0432.CCR-11-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang J, Fitzgerald DJ, Adhya S, Pastan I. Functional domains of Pseudomonas exotoxin identified by deletion analysis of the gene expressed in E. coli. Cell 1987;48(1):129–36 doi 10.1016/0092-8674(87)90363-1. [DOI] [PubMed] [Google Scholar]
- 21.Weldon JE, Xiang L, Chertov O, Margulies I, Kreitman RJ, FitzGerald DJ, et al. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood 2009;113(16):3792–800 doi 10.1182/blood-2008-08-173195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weldon JE, Xiang L, Zhang J, Beers R, Walker DA, Onda M, et al. A recombinant immunotoxin against the tumor-associated antigen mesothelin reengineered for high activity, low off-target toxicity, and reduced antigenicity. Mol Cancer Ther 2013;12(1):48–57 doi 10.1158/1535-7163.MCT-12-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao W, Tang Z, Zhang YF, Feng M, Qian M, Dimitrov DS, et al. Immunotoxin targeting glypican-3 regresses liver cancer via dual inhibition of Wnt signalling and protein synthesis. Nature communications 2015;6:6536 doi 10.1038/ncomms7536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bannas P, Hambach J, Koch-Nolte F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front Immunol 2017;8:1603 doi 10.3389/fimmu.2017.01603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng M, Gao W, Wang R, Chen W, Man YG, Figg WD, et al. Therapeutically targeting glypican-3 via a conformation-specific single-domain antibody in hepatocellular carcinoma. Proc Natl Acad Sci U S A 2013;110(12):E1083–91 doi 10.1073/pnas.1217868110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fleming BD, Urban DJ, Hall MD, Longerich T, Greten TF, Pastan I, et al. Engineered Anti-GPC3 Immunotoxin, HN3-ABD-T20, Produces Regression in Mouse Liver Cancer Xenografts Through Prolonged Serum Retention. Hepatology 2020;71(5):1696–711 doi 10.1002/hep.30949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merlino GT, Xu YH, Ishii S, Clark AJ, Semba K, Toyoshima K, et al. Amplification and enhanced expression of the epidermal growth factor receptor gene in A431 human carcinoma cells. Science 1984;224(4647):417–9 doi 10.1126/science.6200934. [DOI] [PubMed] [Google Scholar]
- 28.Mott BT, Eastman RT, Guha R, Sherlach KS, Siriwardana A, Shinn P, et al. High-throughput matrix screening identifies synergistic and antagonistic antimalarial drug combinations. Sci Rep 2015;5:13891 doi 10.1038/srep13891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang R, Zhu H, Shinn P, Ngan D, Ye L, Thakur A, et al. The NCATS Pharmaceutical Collection: a 10-year update. Drug Discov Today 2019;24(12):2341–9 doi 10.1016/j.drudis.2019.09.019. [DOI] [PubMed] [Google Scholar]
- 30.Wang C, Gao W, Feng M, Pastan I, Ho M. Construction of an immunotoxin, HN3-mPE24, targeting glypican-3 for liver cancer therapy. Oncotarget 2017;8(20):32450–60 doi 10.18632/oncotarget.10592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ho M, Kreitman RJ, Onda M, Pastan I. In vitro antibody evolution targeting germline hot spots to increase activity of an anti-CD22 immunotoxin. J Biol Chem 2005;280(1):607–17 doi 10.1074/jbc.M409783200. [DOI] [PubMed] [Google Scholar]
- 32.Li N, Wei L, Liu X, Bai H, Ye Y, Li D, et al. A frizzled-like cysteine rich domain in glypican-3 mediates Wnt binding and regulates hepatocellular carcinoma tumor growth in mice. Hepatology 2019. doi 10.1002/hep.30646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010;70(2):440–6 doi 10.1158/0008-5472.Can-09-1947. [DOI] [PubMed] [Google Scholar]
- 34.Li N, Fu H, Hewitt SM, Dimitrov DS, Ho M. Therapeutically targeting glypican-2 via single-domain antibody-based chimeric antigen receptors and immunotoxins in neuroblastoma. Proc Natl Acad Sci U S A 2017;114(32):E6623–E31 doi 10.1073/pnas.1706055114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolluri A, Ho M. The Role of Glypican-3 in Regulating Wnt, YAP, and Hedgehog in Liver Cancer. Frontiers in oncology 2019;9:708 doi 10.3389/fonc.2019.00708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei J, Bera TK, Liu XF, Zhou Q, Onda M, Ho M, et al. Recombinant immunotoxins with albumin-binding domains have long half-lives and high antitumor activity. Proc Natl Acad Sci U S A 2018;115(15):E3501–E8 doi 10.1073/pnas.1721780115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mazor R, Eberle JA, Hu X, Vassall AN, Onda M, Beers R, et al. Recombinant immunotoxin for cancer treatment with low immunogenicity by identification and silencing of human T-cell epitopes. Proc Natl Acad Sci U S A 2014;111(23):8571–6 doi 10.1073/pnas.1405153111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mazor R, Zhang J, Xiang L, Addissie S, Awuah P, Beers R, et al. Recombinant Immunotoxin with T-cell Epitope Mutations That Greatly Reduce Immunogenicity for Treatment of Mesothelin-Expressing Tumors. Mol Cancer Ther 2015;14(12):2789–96 doi 10.1158/1535-7163.MCT-15-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iversen TG, Frerker N, Sandvig K. Uptake of ricinB-quantum dot nanoparticles by a macropinocytosis-like mechanism. Journal of nanobiotechnology 2012;10:33 doi 10.1186/1477-3155-10-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arnoletti JP, Buchsbaum DJ, Huang ZQ, Hawkins AE, Khazaeli MB, Kraus MH, et al. Mechanisms of resistance to Erbitux (anti-epidermal growth factor receptor) combination therapy in pancreatic adenocarcinoma cells. J Gastrointest Surg 2004;8(8):960–9; discussion 9–70 doi 10.1016/j.gassur.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 41.Fitzgerald DJ, Moskatel E, Ben-josef G, Traini R, Tendler T, Sharma A, et al. Enhancing immunotoxin cell-killing activity via combination therapy with ABT-737. Leukemia & Lymphoma 2011;52(sup2):79–81 doi 10.3109/10428194.2011.569961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.