

Abstract
Protein kinases regulate fundamental aspects of eukaryotic cell biology, making them attractive chemotherapeutic targets in parasites like Plasmodium spp. and Toxoplasma gondii. To systematically examine the parasite kinome, we developed a high-throughput tagging (HiT) strategy to endogenously label protein kinases with an auxin-inducible degron and fluorophore. Hundreds of tagging vectors were assembled from synthetic sequences in a single reaction and used to generate pools of mutants to determine localization and function. Examining 1,160 arrayed clones, we assigned 40 protein localizations and associated 15 kinases with distinct defects. The fitness of tagged alleles was also measured by pooled screening, distinguishing delayed from acute phenotypes. A previously unstudied kinase, associated with delayed loss, was shown to be a regulator of invasion and egress. We named the kinase Store Potentiating/Activating Regulatory Kinase (SPARK), based on its impact on intracellular Ca2+ stores. Despite homology to mammalian PDK1, SPARK lacks a lipid-binding domain, suggesting a rewiring of the pathway in parasites. HiT screening extends genome-wide approaches into complex cellular phenotypes, providing a scalable and versatile platform to dissect parasite biology.
INTRODUCTION
Apicomplexans are widespread parasites that include the causative agents of toxoplasmosis (Toxoplasma gondii), cryptosporidiosis (Cryptosporidium spp.), and malaria (Plasmodium spp.). Kinases provide important insight into parasite biology as key regulators of cellular processes and attractive drug targets1–3. However, sequence-based functional inference is challenging because evolutionary divergence and parasite-specific adaptations drive the rewiring of signaling pathways. Even broadly conserved kinase families have acquired dramatic lineage-specific adaptations4. The apicomplexan kinome also includes phylum-specific families of secreted kinases that evolved to interfere with host cell signaling5–11. While many studies have explored the function of kinases in isolation, systematic approaches can achieve a global view of the contributions of individual enzymes to parasite biology.
Recent high-throughput knockout screens in T. gondii and Plasmodium spp. have assessed the impact of gene loss on parasite growth12–14. Molecular barcodes have enabled profiling of thousands of genes in a single experiment. Efforts directed at the Plasmodium berghei kinome identified genes required for sexual cycle progression15. However, detailed functional characterization and temporal control remain critical challenges for high-throughput screens.
Precise regulation of T. gondii gene expression has been achieved through several methods. Inducible transcriptional control of target genes can be mediated by heterologous promoters16–18. Alternatively, conditional recombination is possible using a rapamycin-dimerizable Cre recombinase (DiCre). However, the effects of transcriptional regulation or recombination may be delayed for several replication cycles19–21. By contrast, the auxin-inducible degron (AID) system confers post-translational regulation, often achieving knockdown of a target protein within one hour22–24. The AID system is also reversible, retains the target gene’s native promoter, and can be paired with a fluorescent-protein tag. Deploying the AID system at scale could provide both subcellular localization and temporal resolution to the functional dissection of target genes.
High rates of non-homologous end joining (NHEJ) typically preclude efficient homologous recombination (HR) in T. gondii. Deletion of KU80, required for NHEJ, increased the efficiency of HR25,26. High rates of HR in yeast have enabled the generation of arrayed strain collections for the analysis of protein localization and interacting partners27–29. Similar efforts have been undertaken in mammalian cells30,31 and the parasite Trypansoma brucei; however, no such approach has been implemented in apicomplexans32. Stable NHEJ-deficient strains, high rates of transfection, and the adaptation of CRISPR-based genome engineering33 make T. gondii ideal for the development of high-throughput tagging methods.
We present a high-throughput tagging (HiT) strategy amenable to both arrayed and pooled screening in T. gondii. We profiled the kinome of T. gondii assessing subcellular localization and defects in cell division and the lytic cycle. Our system provides spatiotemporal resolution that led to the discovery of regulators of various steps in the lytic cycle, including a previously unstudied kinase critical for invasion and egress from host cells. Our system will be a powerful tool to systematically dissect apicomplexan biology.
RESULTS
Development of high-throughput tagging vectors for T. gondii
Pooled knockout screening fails to capture protein localization, expression levels, and the timing of phenotype development. To that end, we developed a high-throughput tagging (HiT) strategy that uses CRISPR-directed homologous-recombination to site-specifically integrate exogenous sequences (payloads), such as epitope tags or regulatable elements (Fig. 1a). This strategy is scalable based on cloning from entirely synthetic sequences, a modular design, and the flexibility to construct multiple vectors in a pooled format. HiT vectors encode a gRNA and 40-bp homology regions specific to a target site. HR-mediated repair integrates the synthetic construct into the genome, eliminating the gRNA target site. The integrated gRNA sequence provides a molecular barcode to identify each mutant, making it compatible with both pooled and arrayed screening.
Figure 1. Development of high-efficiency tagging (HiT) constructs for protein-centered screening approaches.
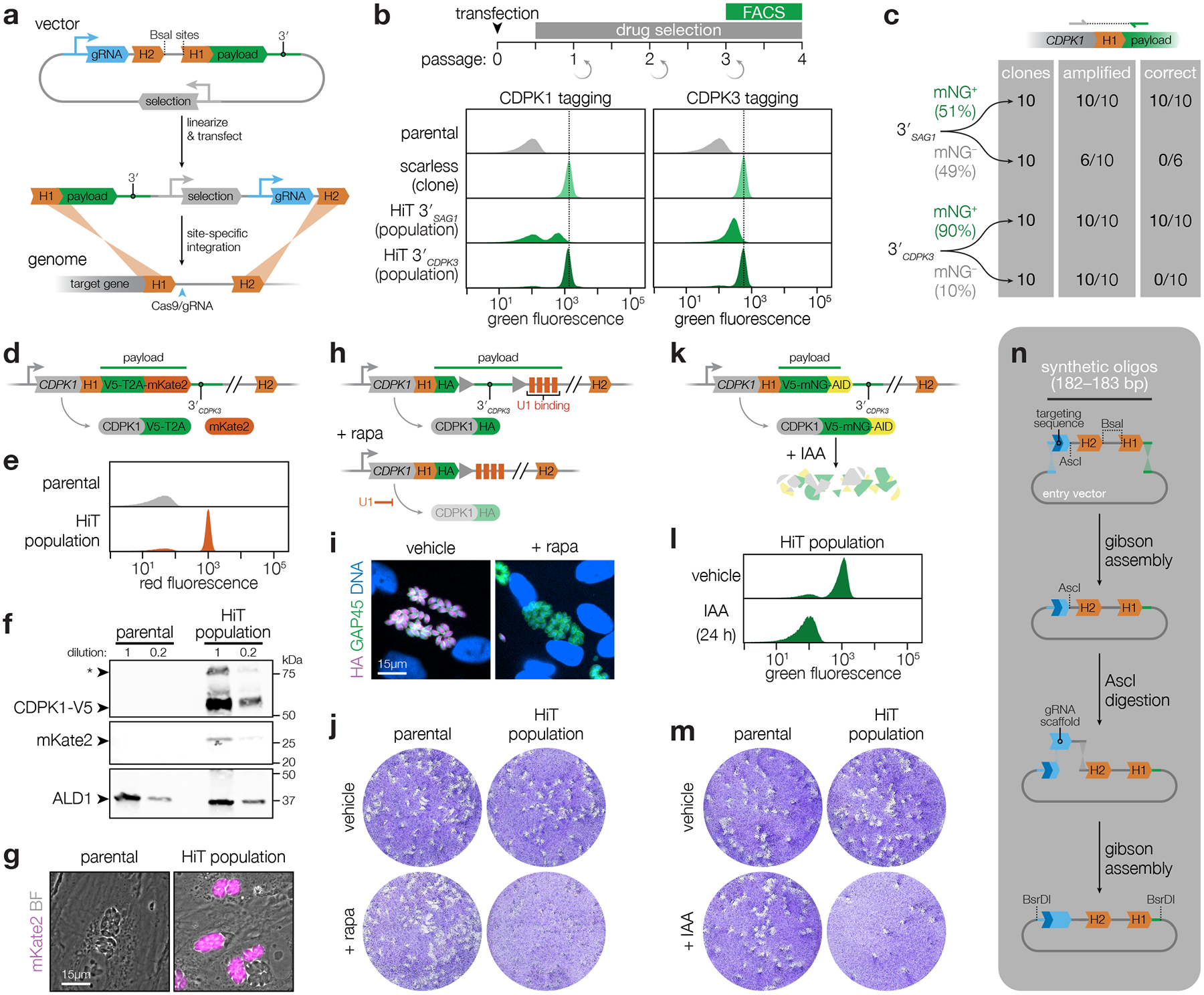
a, Schematic of the high-throughput tagging (HiT) vector. The BsaI-linearized vector is cotransfected with a Cas9-expression plasmid into parasites to mediate homologous recombination of the construct into the target locus. H1 and H2 indicate 40-bp regions of homology to the target locus. Pyrimethamine selection was used to isolate integration events. A heterologous 3′ UTR is included in the vector following the tagging payload. b, Efficiency of HiT vector tagging. Parental TIR1 parasites were transfected with HiT 3′SAG1 or HiT 3′CDPK3 vectors targeting CDPK1 or CDPK3 with a tagging payload encoding mNeonGreen fused to the minimal auxin-induced degron (mAID). Following selection, the populations were analyzed by flow cytometry and compared to clonal strains carrying the same tag with no exogenous sequences (scarless insertion). Dotted line centered on the mode for the fluorescence of the scarless insertion. c, The 5′ integration junctions of 10 mNG positive and 10 mNG negative clones from the CDPK1 HiT 3′SAG1 and HiT 3′CDPK3 populations were amplified and sequenced. Junctions were categorized according to whether they could be amplified and exhibited the correct sequence at the recombination site. d, Schematic of CDPK1 tagged with the V5-T2A-mKate2 HiT vector. e, Flow cytometry for mKate2 fluorescence following CDPK1 tagging with the V5-T2A-mKate2 HiT vector and selection. f, Immunoblot of the CDPK1-V5-T2A-mKate2 population. Expected MW for CDPK1-V5-T2A and mKate2 are 62 kDa and 26 kDa, respectively. Asterisk denotes full-length protein due to incomplete skipping. g, Live-cell microscopy of the CDPK1-V5-T2A-mKate2 HiT population. h, Schematic of CDPK1 tagged with the HA-U1 HiT vector. i, Immunofluorescence assay demonstrating CDPK1 depletion following rapamycin (rapa) treatment. j, Plaque formation following treatment with vehicle or rapamycin for the parental strain or HA-U1 HiT population. k, Schematic of CDPK1 tagged with the V5-mNG-mAID HiT vector. l, Flow cytometry of transfected populations treated with IAA or vehicle for 24 hours. m, Plaque formation following treatment with vehicle or IAA for the parental strain or V5-mNG-mAID HiT population. n, Pooled construction of libraries with linked gRNAs and homology regions.
To optimize the HiT system for screening, we designed vectors to tag genes with the mNeonGreen (mNG) fluorophore fused to a minimal auxin-inducible degron (mAID). In T. gondii strains containing the heterologous F-box protein TIR1, addition of the auxin indole-3-acetic acid (IAA) leads to proteasomal degradation of the mAID-tagged protein22,23,34,35. As test cases, we targeted the kinases CDPK1 and CDPK3, which tolerate C-terminal tags36–38. We also constructed scarless clones in which the tag was integrated in the absence of additional exogenous sequences, to calibrate the expression of genes tagged with our HiT vectors. Tagging vectors were co-transfected with a Cas9-expression plasmid. After selection, populations were predominantly mNG positive (Fig. 1b; Supplementary Fig. 1a). However, HiT-tagged populations were half as fluorescent as their scarless counterparts. Replacing the SAG1 3′ UTR (3′SAG1) with the CDPK3 3′ UTR (3′CDPK3) recovered the expression of the HiT-tagged alleles to near wild-type levels (Fig. 1b; bottom row). The correct localizations and UTR-dependent changes in expression were also apparent by live-cell microscopy (Extended Data Fig. 1a)36,38. Sequencing the gene-tag junctions of isolated clones confirmed correct integration in all the mNG-positive clones (Fig. 1c). Nearly all of the selected parasites integrated the HiT vectors in the correct locus, with most harboring the tag in frame—51% of those generated with 3′SAG1 and 90% of those generated with 3′CDPK3.
To demonstrate the versatility of the HiT vector platform, we generated additional vectors targeting CDPK1. We tagged CDPK1 with a V5 epitope linked to the mKate2 fluorophore via a T2A skip peptide (Fig. 1d). This HiT vector exhibited highly efficient tagging (Fig. 1e–g; Extended Data Fig. 1b; Supplementary Fig. 1b–c), and may be used to select for mutants that incorporate the construct in-frame while avoiding the effect of bulky tags. We also generated a HiT vector for transcriptional regulation, using the U1 system (Fig. 1h)21, which conferred inducible knockdown of CDPK1 (Fig. 1i–j; Extended Data Fig. 1c). Finally, we confirmed the IAA-induced depletion of our previously used mNG-mAID construct (Fig. 1k) and observed complete knockdown (Fig. 1l–m; Extended Data Fig. 1d). The AID system is restricted to downregulation of proteins with cytosolic termini; however, the speed of degradation and compatibility with native regulatory sequences makes it the ideal method to target the parasite kinome22–24,39.
Generation of an array of conditional mutants
Protein kinases localize to distinct subcellular compartments to control diverse cellular functions6. We designed HiT constructs against 147 protein kinases and 8 control genes, which included genes associated with key lytic cycle transitions, genes previously tagged with the AID system, and a dispensable gene (Supplementary Table 1)12,39–43. We excluded kinases with predicted signal peptides, based on the expectation that they are inaccessible to the cytosolic TIR1 and largely dispensable in cell culture12. We designated 40-bp homology regions upstream and downstream from the cut site, which were paired with three different gRNAs per gene, resulting in 465 unique constructs. Homology region 1 (H1) leads into the tag and includes the 40 bp upstream of the stop codon, while homology region 2 (H2) starts 6 bp downstream of the cut site to prevent futile cycles of cutting.
Since the screens were performed in parallel to the analysis of UTR function, we generated libraries with both SAG1 and CDPK3 3′ UTRs. Short oligos encoding the gRNA and matched homology regions were synthesized and cloned into an entry vector (Fig. 1n). The library was linearized to introduce the gRNA scaffold, and then cloned as a unit into the HiT vector to generate the final libraries (Fig. 2a). The strategy maintains library diversity while generating thousands of tagging vectors in a single reaction.
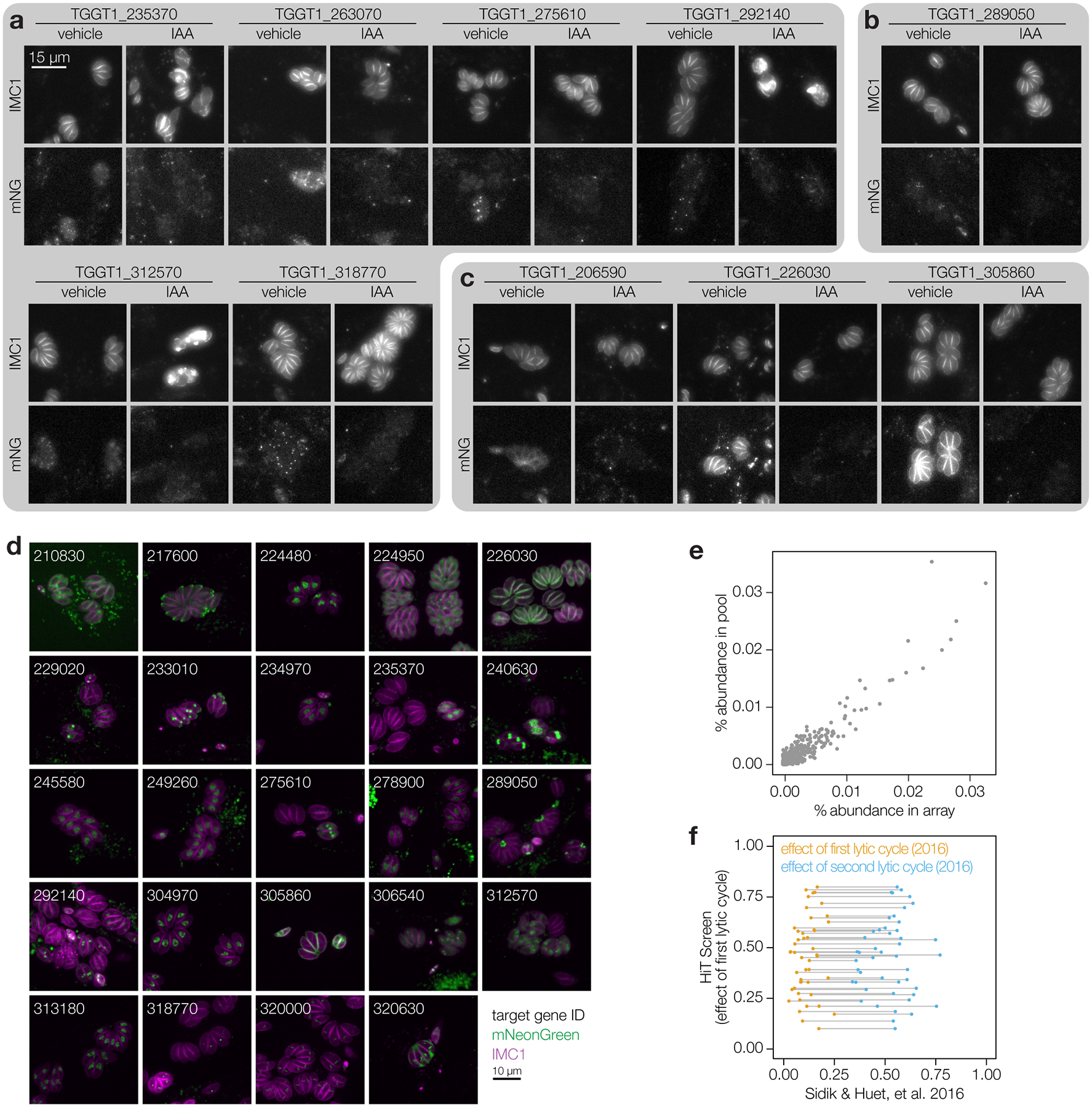
Figure 2. Deconvolution of protein phenotypes and localizations through high-content imaging of arrayed HiT clones.
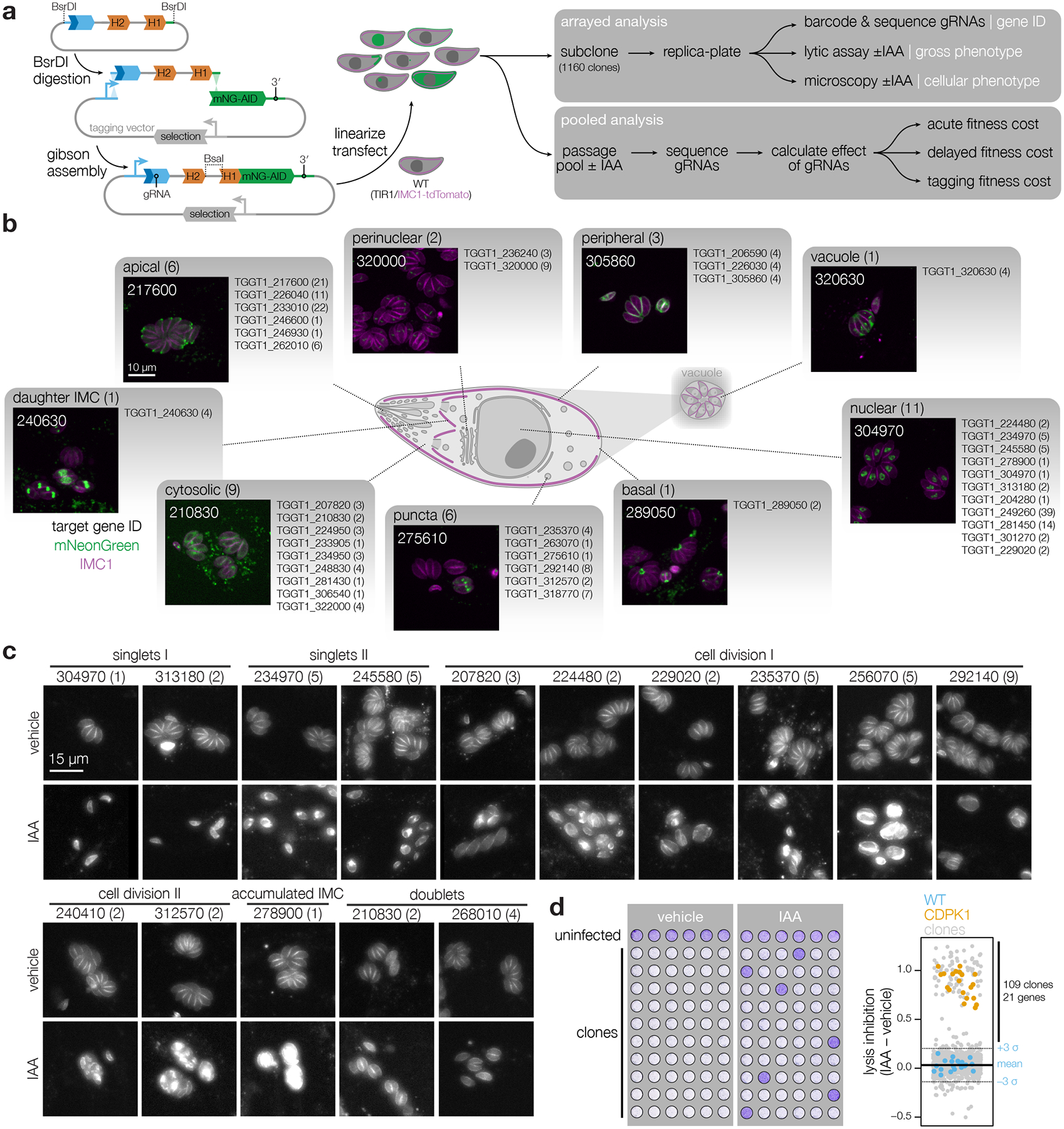
a, Construction of the V5-mNG-mAID HiT vector library and subsequent screening strategy. Following construction and linearization of the library, the vector was co-transfected with a Cas9 expression plasmid into parasites expressing the TIR1 ligase and a fluorescent peripheral marker (TIR1/IMC1-tdTomato). Following selection, the population was analyzed by both pooled and arrayed screening. b, Distribution of subcellular localizations for tagged proteins in the array; number of proteins found in each compartment and clones analyzed for each gene indicated in parentheses. Localizations were assigned to a gene if at least half of the uniquely tagged wells for that gene displayed consistent localizations. Representative confocal images of sample clones are displayed with genes numbered based on their unique identifier (e.g., TGGT1_210830, labeled 210830). Images are maximum intensity z-projections for mNeonGreen (green) and IMC1-tdTomato (magenta). c, Widefield microscopy of representative clones with identified phenotypes. Images are maximum intensity z-projections. The IMC1-tdTomato marker is displayed for cultures treated with either vehicle or IAA for 24 h. Phenotypes were binned into six categories based on their similarity. Number of clones analyzed for each gene indicated in parenthesis. d, The ability of clones to lyse fibroblasts was assayed by infecting monolayers for 72 h in the presence or absence of IAA. Intact monolayers were visualized by crystal violet staining. Normalized absorbance measurements comparing vehicle- and IAA-treated wells are graphed for each clone. Each plate contained the parental strain (WT) and an AID-tagged CDPK1 clone (CDPK1) as controls. Mean ± S.D. for WT controls are shown.
We carried out arrayed and pooled screens using the constructed libraries (Fig. 2a). Pooled screening is highly scalable, whereas arrayed screening can be used to determine phenotypes through replica-plating and microscopy. To generate the clonal array, the 3′CDPK3 library was transfected into parasites expressing TIR122–24,39 and a red-fluorescent inner membrane complex marker (IMC1-tdTomato) to visualize the parasite ultrastructure across the replicative cycle48. Following transfection and selection, the population was subcloned by limiting dilution. 1,160 clonal strains were arrayed and passaged in 96-well plates (Extended Data Fig. 2a).
We employed dual-indexing PCR to sequence the gRNAs from all 1,160 clonal lines in a single next-generation sequencing experiment. We were able to amplify and assign gRNA identities to 917 wells, with 87% (796) of the amplified wells containing a single gRNA representing 49% (228) of gRNAs in the original library and 82% (127) of the targeted genes (Extended Data Fig. 2b). 121 of the amplified wells contained two or more gRNAs. Only singly tagged clones were used for phenotype and localization studies.
Localizations and phenotypes of tagged proteins in the arrays
We employed high-content imaging to visualize the localization and IAA-induced depletion of tagged proteins, and monitor the consequences of their knockdown. Arrayed clones were replica-plated, treated with IAA or vehicle 3 h post-infection, and imaged the following day. 232 (29%) clones displayed a clear mNG signal, enabling unambiguous localization of 40 proteins to diverse subcellular compartments (Fig. 2b; Extended Data Fig. 3–4; Supplementary Table 2). Some proteins, such as TGGT1_275610, consistently displayed heterogeneous expression, likely indicating cell cycle regulation. 36 of the 40 proteins localized showed complete depletion after 24 h of IAA treatment. TGGT1_226040, TGGT1_322000, TGGT1_320630, and TGGT1_234950 showed minimal signal reduction under IAA. Localization to the parasitophorous vacuole (TGGT1_320630) or dense granules (TGGT1_234950)49, suggests inaccessibility to TIR1 may account for the lack of degradation. Nevertheless, HiT screening can generally localize parasite proteins with high-throughput.
Knockdown-induced phenotypes were classified based on parasite structure. Mutants in TGGT1_304970, TGGT1_313180, TGGT1_234970, or TGGT1_245580 arrested early in the lytic cycle (Fig. 2c). Degradation of the latter two caused a discontinuity in the IMC marker (singlets II). Accordingly, TGGT1_304970 is a cyclin–related kinase (TgCrk1) required for daughter cell assembly50 and TGGT1_234970 is a nuclear tyrosine kinase-like protein (TgTKL2)51 related to the human Tousled-like kinase that promotes DNA replication52–54. The two other singlet kinases are also nuclear: TGGT1_313180 is related to a yeast splicing factor55; and TGGT1_245580 is large, exclusively found in coccidians, and lacks any other distinguishing features.
The remaining cell division phenotypes could be broadly categorized as exhibiting a continuous (cell division I) or fragmented (cell division II) IMC distribution (Fig. 2c). Within these categories, we found two previously described cyclin-related kinases, TGGT1_256070 (TgCrk4) and TGGT1_229020 (TgCrk5). Knockdown of TgCrk4 causes major morphological abnormalities and decreased plaque size50. The function of TgCrk5 has not been previously examined, although it was shown to interact with the cell cycle regulator ECR1 at the centrocone56—consistent with our observations (Extended Data Fig. 3a). Indeed, our knockdown of TgCrk5 mirrored the phenotype of a temperature-sensitive allele of ECR156. Knockdown of the centrosome-associated kinases NIMA-related kinase 1 (TgNek1; TGGT1_292140), MAPK-like protein 1 (TgMAPK-L1; TGGT1_312570), and TgMAPK2 (TGGT1_207820) phenocopied previously characterized conditional mutants57–60. TgMAPK-L1 displayed cell cycle–dependent localization to diffuse puncta, consistent with its localization to the pericentrosomal matrix. In addition to the reported cytosolic localization60, TgMAPK2 was observed at paired puncta in a subset of vacuoles, shedding additional light into the function of this kinase.
Cell-cycle defects were also observed for several kinases that have not been previously studied. Knockdown of TGGT1_240410 phenocopied TgMAPK-L1 disruption, despite being conserved only among related coccidians. How other kinases regulate the cell cycle is unknown, but they can be associated with cellular functions based on their localization and similarity to well-studied orthologs: TGGT1_235370 and TgMAPK2, TGGT1_224480 and Cdc-like kinases that regulate the mammalian spliceosome61, and TGGT1_210830 and RIO kinase 1 that participates in ribosomal maturation62. As with TGGT1_210830, knockdown of TGGT1_268010 caused an arrest after a single cell cycle, although its conservation is restricted to coccidians. Accumulation of the IMC marker with grossly normal organization resulted from knockdown of TGGT1_278900, for which homology to yeast BUD32 and association of the Plasmodium falciparum orthologue with the EKC/KEOPS complex components suggest conservation of the complex across eukaryotes63.
We also screened for aggregate defects in the lytic cycle by examining monolayer clearance after knockdown (Fig. 2d). Changes in monolayer clearance were compared to positive and negative controls. All positive controls and 109 clones (11%) scored three standard deviations above the average lysis inhibition score for the negative controls. The 109 clones represented 21 genes (17% of genes tested), including every mutant identified by high-content imaging except TgMAPK2, which displayed an incompletely penetrant phenotype. By contrast, clones in which ERK7 or Doc2.1 were tagged displayed defective monolayer clearance but no morphological changes by microscopy, consistent with reported roles in invasion and egress but not replication42,64,65. TGGT1_306540 shared these phenotypes with ERK7 and Doc2.1, and appears to be a cytosolic ethanolamine kinase (TgEK)66 necessary for the completion of the lytic cycle. Our results highlight that arrayed HiT screening can capture both localization and detailed cellular phenotypes.
Temporal resolution of phenotypes by pooled screening
Pooled screening offers greater scalability and a more sensitive comparison of mutant fitness than arrayed screening. To perform pooled screens, 3′SAG1 or 3′CDPK3 HiT vector libraries were transfected into TIR1 or TIR1/IMC1-tdTomato parasites, respectively. After three passages under pyrimethamine selection, the populations were split and treated with vehicle or IAA, collecting samples to quantify gRNA abundances at each passage (Fig. 3a). The abundance of gRNAs in the array was strongly correlated to their abundance in the 3′CDPK3 pool before IAA treatment (Extended Data Fig. 4e). The relative abundance of a given gRNA could be affected by (i) the efficiency of integration, (ii) locus tolerance to tagging, and (iii) the consequence of protein degradation by IAA. As expected, gRNAs targeting sequences with NAG PAMs were less efficiently integrated than those targeting NGG PAMs67 (Fig. 3b). To regress the effect of the PAM, we normalized the abundance for NAG gRNAs at each time point by the difference in mean abundance between NAG and NGG guides for genes considered dispensable12 within each sample. We calculated the fold change relative to the second passage, when relative gRNA abundances best reflected the composition of subsequent populations, allowing us to merge the results of both screens.
Figure 3. Pooled screening distinguishes between acute and delayed-loss phenotypes.
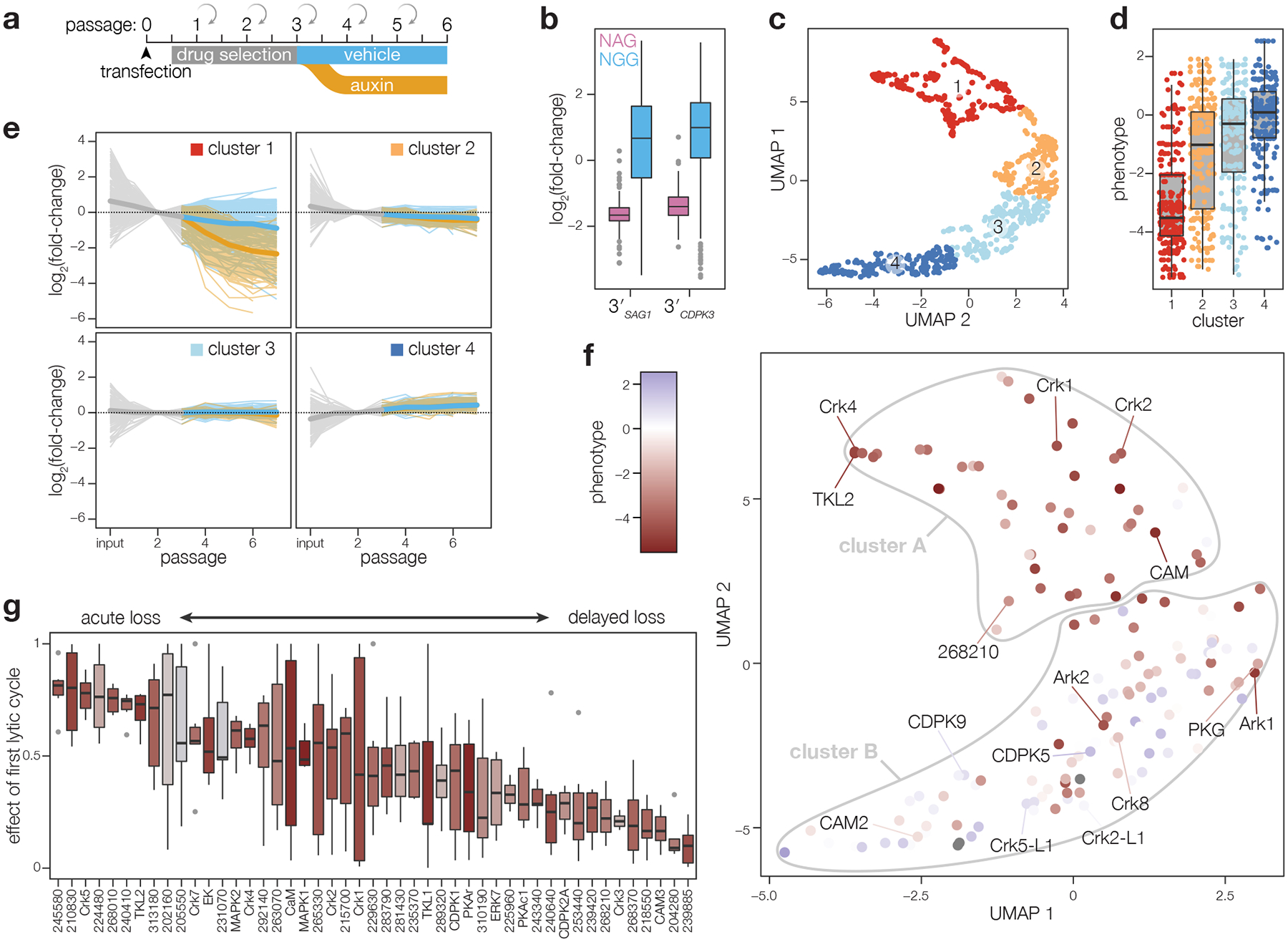
a, Schematic of pooled screening workflow. Transfected populations were selected with pyrimethamine for three passages, after which they were split and cultured in either vehicle- or IAA-containing media. Following each lysis, parasites were split to collect samples for next-generation sequencing and continue propagating in fresh host monolayers. b, Fold-change in gRNA abundance between the vehicle-treated passage 6 and the initial library, plotted by PAM type. Boxplot displays the distribution of each sample by quartiles; outliers highlighted in gray; n = 107 NAG and 358 NGG gRNAs. c, Relative abundances for each guide were corrected for the effect of the PAM used and fold-changes were normalized to passage 2. UMAP was used to compare guides in each screen based on their pattern of fold changes for vehicle- and IAA-treated samples. Clusters were calculated by k-means. d, Comparison of phenotype scores from prior gene-disruption screen for each of the clusters in c. Boxplot displays the distribution of phenotypes in each cluster by quartiles; n = 253, 205, 209, 215 gRNAs per cluster. e, Pattern of fold changes for each guide plotted by cluster. Lines colored by treatment, as in a. Bold lines are the mean for all guides in a given cluster. f–g, Gene centroids in UMAP space based on the effects of targeting guides against each gene (f). Genes were assigned to fitness-conferring (cluster A) or dispensable (cluster B) categories based on k-means clustering. Fraction of the maximum fold-change that is explained by the first lytic cycle following IAA addition for all fitness-conferring genes in cluster A, ordered based on the magnitude of the effect from acute loss to delayed loss (g); n = 6 gRNAs per gene. Individual genes colored based on the phenotype scores from the prior gene-disruption screen12.
We compared the trajectories for gRNAs across time and IAA treatment. Using UMAP for dimensionality reduction followed by k-means clustering68,69, the groupings of gRNAs largely agree with previously determined phenotype scores of the target genes (Fig. 3c–d)12. Notably, cluster 1 was enriched in gRNAs that differentially dropped out of the population following IAA treatment (Fig. 3e), suggesting the targeted genes are fitness-conferring and their gene products are successfully depleted upon IAA treatment.
We next calculated the centroid in UMAP space for gRNAs from both screens to measure the performance of individual genes. Unsupervised clustering on the centroids assigned genes to two classifications, one of which, cluster A, included 47 genes and was highly enriched for contributing to parasite fitness (Fig. 3f). The arrayed screen captured mainly acute phenotypes and 18 of the 22 genes that had deficits by microscopy or lytic assay were found in cluster A. 13 of the cluster A genes were not represented in the array, and the remaining 16 likely display phenotypes difficult to appreciate in isolation or during brief periods of kinase depletion. Discrepancies between the two screens may also result from the analysis of incorrectly integrated vectors in the array; such clones could be theoretically excluded based on lack of tag expression, but that strategy would sacrifice lowly abundant protein kinases. Out-of-frame integration may also be frequent for proteins rendered hypomorphic by the tag and may drive known essential genes like PKG23,70 into cluster B.
Two of the four genes identified by the arrayed screen but not the pooled screen (TGGT1_218720 and TGGT1_250680) likely represent false positives in the arrays, since they were only represented by single clones with modest defects in the lytic assay and were dispensable in previous knockout screens12. By contrast, the two other genes missed by the pooled analysis (TGGT1_278900 and TGGT1_240910) are expected to be fitness-conferring12, and TGGT1_278900 degradation was associated with accumulation of the IMC marker (Fig. 2c). Three clones of TGGT1_240910 (Doc2.1; included as a control based on prior studies42) displayed the expected lytic assay phenotype upon knockdown. For some genes (e.g., Doc2.1), the discrepancy between screens originated from contradictory results between different gRNAs when the population was treated with IAA; however, we noticed that gRNAs with optimal designs—based on proximity to the coding sequence or use of an NGG PAM—were more likely to conform to expectations.
We next investigated the timing of gRNA loss for genes in cluster A. We sorted genes based on the fraction of the maximum effect from IAA treatment that was observed after a single lytic cycle (Fig. 3g). 14 of the 15 genes associated with defects by microscopy (TGGT1_278900 was missed by the pooled screens) dropped out substantially during the first passage in IAA. TGGT1_215700 and TGGT1_270330 (TgCrk7) dropped out acutely but were absent from the array, and the latter is considered essential50. TGGT1_215700 is broadly conserved across eukaryotes and homologous to phosphatidylinositol kinases critical for proliferation in other organisms71,72. This temporal resolution was not observed in conventional CRISPR-based screens (Extended Data Fig. 4f). Our results suggest that this analysis can identify genes whose disruption leads to immediate and catastrophic defects in the parasite.
In contrast to the acute defects associated with some genes, gRNAs against other genes were mostly retained during the first lytic cycle (Fig. 3g). Delayed-loss phenotypes were assigned to several genes linked to invasion or egress: CDPK1, ERK7, PKAc1, TKL1, and CAM324,36,41,43,51,64,65. These genes lacked defects during the brief window selected for microscopy. Another delayed-loss gene, CDPK2A, belongs to a kinase family that has been linked to invasion and egress36–38,73–76. TGGT1_249260 (TgCrk3) similarly displayed no defects by microscopy or lytic assay, consistent with the observation that knockdown reduced plaque size but did not impact morphology50. Pooled screens more easily capture subtle defects that accrue over several lytic cycles and may involve processes that accompany egress and invasion.
Two delayed-loss kinases regulate invasion
We characterized four delayed-loss candidates with patterns of gRNA loss that contrasted with the rapid depletion of representative acute-loss genes (Fig. 4a). All four kinases were fitness-conferring in genome-wide knockout screens12 and remain largely uncharacterized. The four kinases exhibit different phyletic patterns77: TGGT1_204280 is conserved among several single-celled parasitic phyla, including kinetoplastids and amoeba; TGGT1_268210 and TGGT1_239420 are restricted to Apicomplexa; and TGGT1_239885 is restricted to coccidians. TGGT1_239420 was previously linked to the development of artemisinin resistance in vitro78. Intriguingly, the Plasmodium falciparum ortholog of TGGT1_268210 (PfPDK1; Pf3D7_1121900), was classified as fitness-conferring in previous screens and has been linked to the regulation of protein kinase A (PKA)14,79.
Figure 4. Analysis of delayed-loss genes identifies two kinases that impact invasion.
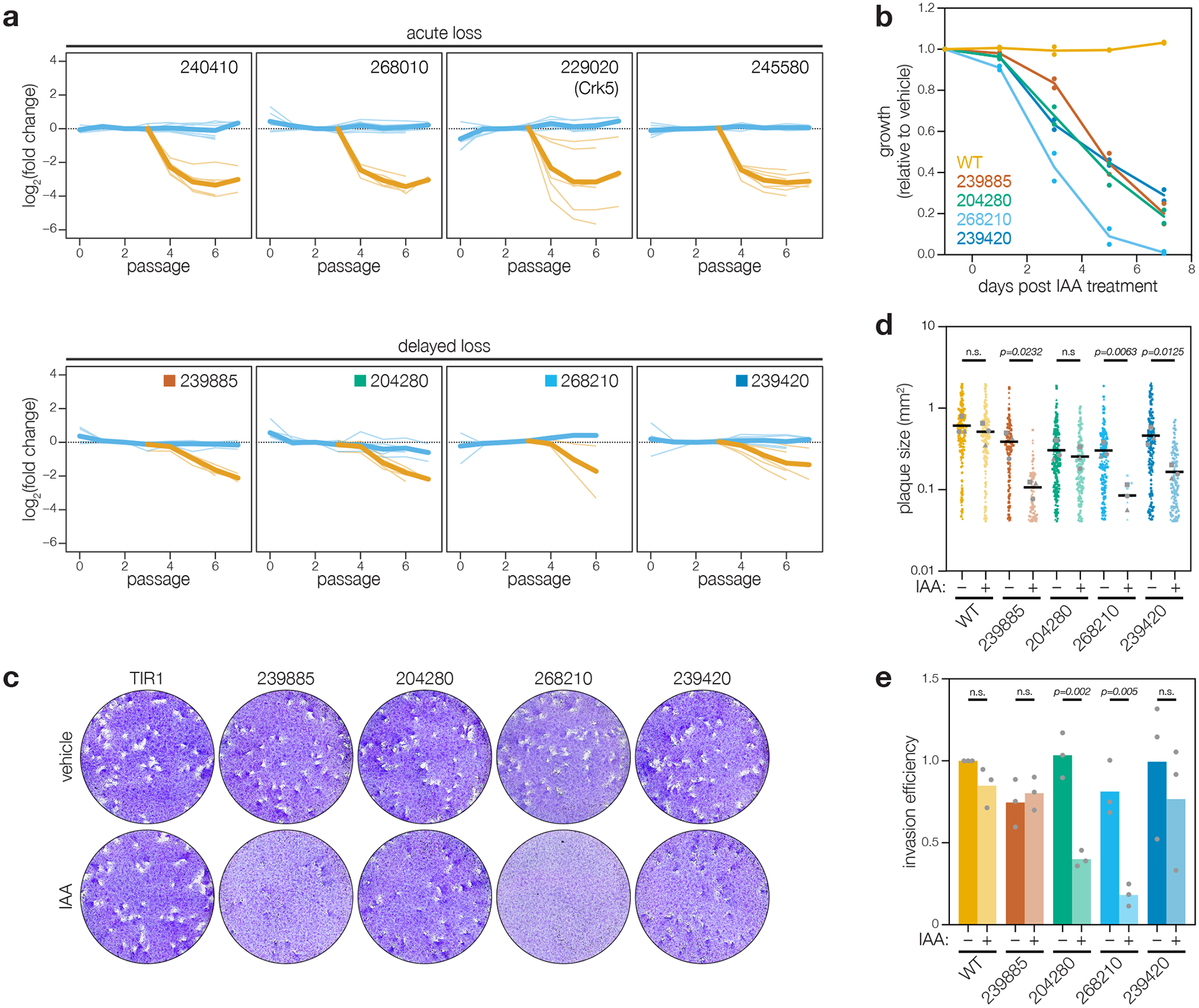
a, Pooled screening traces of selected acute- and delayed-loss genes. The fold change of individual guides in the vehicle- (blue) and IAA- (orange) treated samples are displayed. Bold lines are the mean for all guides in a given condition. b, Competition assays of delayed-loss candidates, compared the relative growth of AID-tagged strains against a wild-type strain in either vehicle- or IAA-containing media. Following each lysis, the proportion of fluorescent parasites within each competing population was measured by flow cytometry and normalized to the vehicle control; n = 2 biological replicates. c–d, Plaque assays of delayed-loss candidates grown in the presence of IAA or vehicle control (c). The parental strain (WT) is included for comparison. Quantification of plaque areas from three separate wells are plotted (d). Means are displayed; n.s. p > 0.05, two-tailed t-test. (e) Invasion assays of delayed loss candidates. AID-tagged strains grown in vehicle- or IAA-containing media for 24 h were incubated on host cells for 10 minutes prior to differential staining of intracellular and extracellular parasites. Parasite numbers were normalized to host cell nuclei for each field. Means graphed for n = 3 biological replicates; n.s. p > 0.05, Welch’s one-tailed t-test.
Although three of the candidates were in the array, we rederived conditional mutants to independently validate the screening results. We placed each mutant in competition with TIR1/IMC1-tdTomato parasites under knockdown conditions. A second wildtype strain was used as a control for the assay. All four mutants were outcompeted by wildtype, demonstrating the kinases contribute to parasite fitness (Fig. 4b; Supplementary Fig. 2). Three of the mutants also showed clear defects in plaque formation when grown in the presence of IAA (Fig. 4c); knockdown of TGGT1_239420 or TGGT1_239885 reduced plaque size, while knockdown of TGGT1_268210 blocked plaque formation (Fig. 4d). The lack of a plaquing defect for TGGT1_204280 suggests a subtle or merely competitive defect in fitness.
We assayed the invasion efficiency of all four mutants, as a potential cause of the delayed-loss phenotype. Both TGGT1_268210 and TGGT1_204280 displayed significant invasion defects upon knockdown (Fig. 4e). Since parasites formed normal plaques upon TGGT1_204280 knockdown, the invasion defect may represent a delay rather than a complete block. By contrast, the severe invasion defect of TGGT1_268210, coupled with effects in plaque formation and competition assays, suggests the gene plays a critical role in the lytic cycle.
SPARK regulates intracellular Ca2+ store release
We examined TGGT1_268210 (hereafter referred to as SPARK) in greater detail. SPARK belongs to the AGC kinase family. Despite homology to mammalian PDK1 (Fig. 5a), SPARK lacks the canonical C-terminal phosphoinositide-binding domain, similarly to related kinases in yeast and nonvascular plants80. Instead, SPARK and its apicomplexan orthologues possess an N-terminal MKXGFL motif absent from canonical PDK1s (Fig. 5b). Free-living alveolates Vitrella brassicaformis and Chromera velia harbor two PDK1-like kinases, but only one paralog has the N-terminal motif. This suggests the SPARK clade may have arisen from gene duplication prior to the split of the Apicomplexa from other Alveolata, followed by loss of the more closely-related PDK1 homologues in the parasitic clade.
Figure 5. SPARK regulates egress and invasion through modulation of intracellular Ca2+ stores.
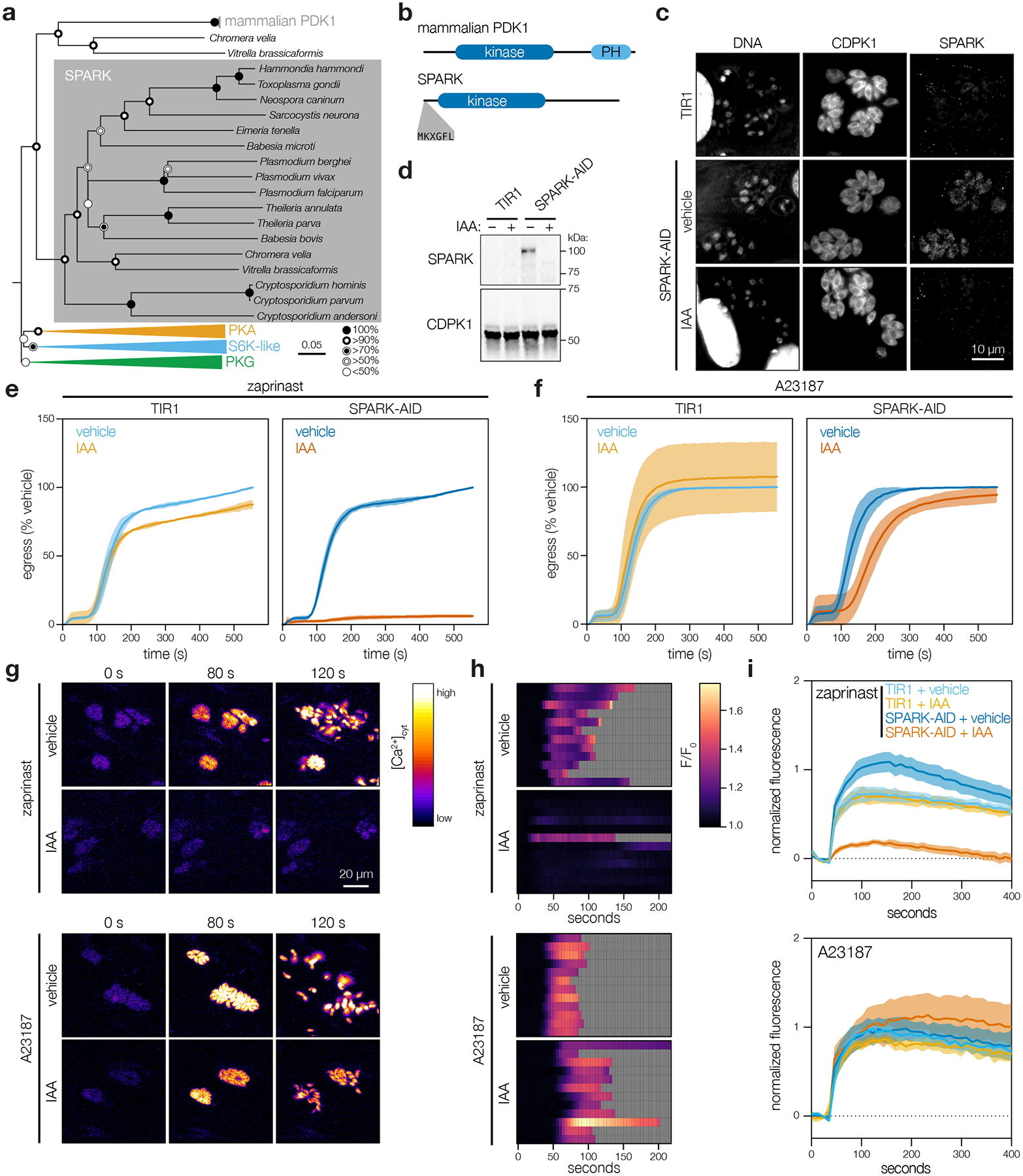
a, Neighbor-joining phylogenetic tree of kinase domains from representative apicomplexan species, along with mammalian PDK1 orthologues and related AGC kinases. Bootstraps determined from 1000 simulations. Scale indicates substitutions per site. b, Models of the canonical mammalian PDK1 and the apicomplexan SPARK proteins. The kinase domains, mammalian pleckstrin homology (PH) domain, and conserved apicomplexan MKXGFL motif are shown. c, SPARK-AID was visualized by immunofluorescence microscopy and immunoblotting using the V5 epitope. SPARK-AID was undetectable after 24 h of IAA treatment. Staining for CDPK1 was used to identify parasites, and nuclei were stained with DAPI. Channels adjusted equivalently across all samples. d, SPARK-AID depletion, as in c, monitored by immunoblot. SPARK-AID is expected to run at 98 kDa. e–f, Parasite egress stimulated with zaprinast (e) or the Ca2+ ionophore A23187 (f) following 24 h of treatment with vehicle or IAA. Egress was monitored by the number of host cell nuclei stained with DAPI over time. Mean graphed for n = 3 biological replicates. Shaded regions represent ± S.D. g, Selected frames from live video microscopy of zaprinast- or A23187-stimulated SPARK-AID parasites expressing the genetically encoded Ca2+ sensor GCaMP6f. Parasites were grown for 24 h with vehicle or IAA prior to the stimulation of egress. h, Kymographs showing normalized fluorescence per vacuole relative to the initial intensity, for 12 vacuoles per strain from the experiments in g. Gray areas represent the period following egress of the vacuole under observation. i, Extracellular parasites in basal Ca2+ buffer stimulated with zaprinast or the Ca2+ ionophore A23187, following 24 h of treatment with vehicle or IAA. Cytosolic Ca2+ flux was measured in bulk as GCaMP6f fluorescence normalized to the initial and maximum fluorescence following aerolysin permeabilization in 2 mM Ca2+. Mean ± S.E. graphed for n = 3–6 biological replicates.
SPARK displayed a diffuse cytosolic localization by immunofluorescence and was depleted without affecting replication after 24 h of IAA treatment in the conditional mutant (Fig. 5c–5d; Extended Data Fig. 5a; Supplementary Fig. 3). Since egress shares several signaling pathways with invasion, we examined the role of SPARK in this process. We induced parasite egress from host cells with the phosphodiesterase inhibitor zaprinast, which increases cGMP levels, activates PKG, and triggers release of intracellular Ca2+ stores81–86. The Ca2+ ionophore A23187 provides an alternative trigger for egress, which also increases cytosolic Ca2+ but circumvents guanylate cyclase activity87–89. Although SPARK knockdown completely blocked zaprinast-induced egress (Fig. 5e), it appeared almost entirely dispensable for A23187-induced egress (Fig. 5f). These results suggest that SPARK loss interferes with the ability of PKG to trigger intracellular Ca2+ release.
To examine SPARK’s role in Ca2+ release, we tagged the endogenous gene with a HiT vector carrying an mCherry-mAID epitope in parasites expressing TIR1 and the genetically-encoded fluorescent Ca2+ sensor GCaMP6f91. Treatment of GCaMP6f/SPARK-AID parasites with IAA attenuated cytosolic Ca2+ fluxes in intracellular parasites following zaprinast-stimulation (Fig. 5g–h). This phenotype was also rescued by A23187. Since cytosolic Ca2+ can originate from multiple sources92,93, we stimulated extracellular parasites in a buffer containing basal Ca2+ concentrations (~100 nM free Ca2+). SPARK knockdown blocked zaprinast-induced release of intracellular Ca2+ stores (Fig. 5i). By contrast, the response to the Ca2+ ionophores A23187 or ionomycin was unchanged (Extended Data Fig. 5b), indicating that intracellular Ca2+ stores are intact but unresponsive to zaprinast in the absence of SPARK. These results establish SPARK as a regulator of the parasite intracellular Ca2+ store discharge that precedes both invasion and egress. We therefore named the kinase “Store Potentiating/Activating Regulatory Kinase” (SPARK), to describe its proposed role in apicomplexan biology.
DISCUSSION
HiT screens benefit from both arrayed and pooled formats. Arrayed screens examine individual clones to characterize subcellular localizations and cellular defects, even in cases when they are heterogeneous or incompletely penetrant. Pooled screening, by contrast, compares all targeted genes simultaneously, detecting subtle defects that are only apparent in competitive settings. Arrayed screens for protein localization in Trypanosoma brucei successfully localized most gene products32. Pooled screens in T. brucei, Plasmodium spp., and T. gondii have examined genetic contributions to fitness, but lack temporal or subcellular resolution12–14,94,95. Arrayed and pooled HiT screens work in tandem to achieve protein localization and phenotypic resolution inaccessible to existing platforms. Further, clonality ensures unambiguous localization, uniform knockdown, and recovery of individual clones from the arrays for follow-up studies.
While false positives in the HiT screens were minimal, 29 genes previously reported to be fitness-conferring12 were missed. False negatives likely originate from poor-quality gRNAs, inaccessibility of the tagged protein to TIR1, and tagging-induced hypomorphism. Reduced abundance of low-quality gRNAs also caused skewed sampling in the array. Future screens should use more gRNAs per gene, avoid targeting NAG PAMs, and array clones soon after transfection. Alternative HiT payloads can constitute regulatory elements instead of a protein tag—as demonstrated by regulation of CDPK1 with the U1 system (Fig. 1h)—achieving regulation of inaccessible proteins or of those rendered hypomorphic by tags.
We identified several previously unstudied regulators of the parasite lytic cycle. Knockdown of each of four nuclear kinases caused early arrests, reminiscent of blocks in G1 or S/M—such kinases likely regulate critical checkpoints. By contrast, later arrests may be associated with a restriction of resources. This is supported by the phenotype of TGGT1_210830, the ortholog of which is critical for ribosomal maturation62. Other phenotypes included abnormalities in parasite morphology. Multiple genes displayed defects similar to those previously observed for TgMAPK-L1 and TgMAPK257–60. Aberrant morphologies allude to failures in daughter cell assembly or cytokinesis. The visualization of cellular consequences following gene knockdown expands upon general fitness screening leading to specific hypotheses regarding gene function. The cellular defects observed also reflect acute and lethal consequences for the parasites, establishing these kinases as promising therapeutic targets.
The temporal resolution of the HiT screens revealed genes displaying a delayed-loss phenotype, including SPARK (TGGT1_268210). The proposed name for the kinase reflects its role in the regulation of Ca2+ stores during the parasite lytic cycle. SPARK is critical for egress and release of intracellular Ca2+ stores following stimulation of the cGMP pathway—phenotypes rescued by Ca2+ ionophores. This pattern mirrors the regulation of filamentous actin translocation during parasite motility (F-actin flux)96, required for invasion and egress. Regulation of Ca2+ stores by proteins such as SPARK likely precedes microneme discharge, and subsequent egress and invasion. However, SPARK’s phenotype is distinct from PKG, loss of which cannot be compensated by Ca2+ ionophores23. This is consistent with models in which PKG regulates the production of diacylglycerol (DAG) and inositol trisphosphate (IP3)37,40,88,97–99. IP3-stimulated release of intracellular Ca2+ stores and DAG conversion to phosphatidic acid (PA) are both thought to mediate secretion of microneme contents100. Altering the activity of the cGMP-producing guanylate cyclase (GC) blocks zaprinast- and BIPPO-induced egress. Reports differ on the effect of A23187 following GC knockdown—ranging from a nearly normal egress to a complete block87–89—making it difficult to definitively place SPARK in the pathway.
The observed phenotypes appear inconsistent with the canonical role of PDK1 activating other AGC kinases101,102. Failure to activate PKG would block both zaprinast- and ionophore-stimulated egress81. Analogously, failure to activate PKA, a negative regulator of egress, would result in premature egress41,43. Whether global downregulation of AGC kinase function would phenocopy the loss of SPARK therefore remains an open question. Our study places SPARK as a positive regulator of invasion and egress via potentiation or activation of intracellular Ca2+ stores, which could be achieved via direct stimulation of Ca2+ channels or modulation of upstream regulators such as PKA. Further work will be needed to distinguish between these models. The characterization of SPARK provides a crucial molecular handle to study the activation of intracellular Ca2+ stores—an event that mediates key transitions in the apicomplexan life cycle.
HiT screens expand current platforms and enable the identification of complex cellular phenotypes. This technology can already handle much larger gene sets (~2000 genes) than the kinome. The AID system is reversible22, which could help distinguish temporary arrests in replication from lethal disruptions. Alternative tags or conditional expression systems, can also extend HiT screening to additional questions and make it compatible with secreted or compartmentalized proteins inaccessible to the AID approach. Concurrently with our work, Jimenez-Ruiz and colleagues developed an alternative strategy for high-throughput phenotypic analysis of T. gondii, which implemented a rapamycin-inducible split Cas9 to precisely time gene disruption103. Together with the HiT screens, these technologies offer unprecedented spatiotemporal resolution to screening in T. gondii and are powerful tools for dissecting the biology of these ubiquitous apicomplexan parasites.
METHODS
Data analysis
All data analysis was performed in either Microsoft Excel (version 16.58), GraphPad Prism (version 9.1.2), RStudio (version 1.2.5033), Perl (version 5.18), FlowJo (version 10.7), ImageJ (version 2.0.0), or SnapGene (version 5.1.5).
Parasite and host cell culture
T. gondii parasites were grown in human foreskin fibroblasts (HFFs) maintained in DMEM (GIBCO) supplemented with 3% inactivated fetal calf serum (IFS) and 10 μg/mL gentamicin (Thermo Fisher Scientific), referred to as D3. Where noted, DMEM supplemented with 10% IFS and 10 μg/mL gentamicin was used, referred to as D10.
Parasite transfection
Parasites were passed through 3 μm filters, pelleted at 1000 × g for 10 min, washed, resuspended in Cytomix (10 mM KPO4, 120 mM KCl, 150 mM CaCl2, 5 mM MgCl2, 25 mM HEPES, 2 mM EDTA, 2 mM ATP, and 5 mM glutathione), and combined with transfected DNA to a final volume of 400 μL. Electroporation used an ECM 830 Square Wave electroporator (BTX) in 4 mm cuvettes with the following settings: 1.7 kV, 2 pulses, 176 μs pulse length, and 100 ms interval.
Strain generation
Oligos were ordered from IDT. All cloning was performed with Q5 2X master mix (NEB) and NEBuilder HiFi assembly (NEB) unless otherwise noted. Primers and plasmids used or generated in this study can be found in (Supplementary Table 1). Oligos, plasmids, and strains generated within this study are available from the corresponding author by request.
Scarless CDPK1-mNG-AID.
The V5-TEV-mNeonGreen-AID-Ty cassette was PCR amplified from plasmid pBM050 with repair homology arms using primers P108 and P109. Oligos P110 and P111 were duplexed and cloned into plasmid pSS013 to create the gRNA/Cas9-expression plasmid. The gRNA/Cas9-expression plasmid was co-transfected with the repair template into TIR1 parasites23,24. Following the first lysis, mNeonGreen positive clones were isolated via fluorescence-activated cell sorting. Single clones were obtained by limiting dilution and verified by PCR amplification using primers P114 and P115 and sequencing with primers P112 and P113.
Scarless CDPK3-mNG-AID.
The CDPK3-mNG-AID scarless strain was generated as CDPK1-mNG-AID above, using primers P116 and P117 for repair template amplification, oligos P118 and P119 for assembly of the gRNA/Cas9-expression plasmid, primers P120 and P121 for validation of clonal isolates, and primers P112 and P113 for the sequencing of tag junctions.
TIR1/IMC1-tdTomato.
The sequence pTUB1_IMC1-tdTomato_DHFR was PCR-amplified with primers P96 and P97 to yield a repair template with homology to a defined, neutral genomic locus104. Approximately 2 × 107 extracellular TIR1 parasites were transfected with 50 μg gRNA/Cas9 plasmid targeting the neutral locus and 6 μg of repair template. Single clones were isolated by fluorescence-activated cell sorting into 96-well plates containing HFFs. IMC1-tdTomato positive clones were subsequently identified by microscopy and verified by PCR amplification of the locus using primers P98 and P99.
TGGT1_268210, TGGT1_204280, TGGT1_239885, and TGGT1_239420 AID-tagged lines.
HiT vector cutting unit gBlocks (IDTDNA) (P122–125) were cloned into the pGL015 mNeonGreen HiT vector backbone. HiT vectors were linearized with BsaI and co-transfected with the pSS014 Cas9-expression plasmid into TIR1 parasites. Clones were selected with pyrimethamine and isolated via limiting dilution. Clones were verified by PCR amplification and sequencing of the junction between the 3′ end of the gene (P100-P103) and 5′ end of the protein tag (P104 for TGGT1_268210 and P105 for TGGT1_204280, TGGT1_239420, and TGGT1_239885).
TIR1/GCaMP6f.
The primers P106 and P107 were used to PCR-amplify the sequence pTUB1-GCaMP6f-DHFR3′UTR from plasmid Genbank MT345687 to yield a repair template with homology to the 5′ and 3′ ends of a defined, neutral genomic locus104. Approximately 1 × 107 extracellular parasites were transfected with 25 μg gRNA/Cas9-expression plasmid pBM041 and 5 μg GCaMP6f repair template. Following two rounds of fluorescence-activated cell sorting, GFP-positive clones were isolated by limiting dilution.
TIR1/GCaMP6f/268210-AID.
The TGGT1_268210 HiT vector cutting unit gBlock (IDTDNA) was cloned into the pALH052 V5-TEV-mCherry-AID HiT vector backbone. The HiT vector was linearized with BsaI and co-transfected with the pSS014 Cas9-expression plasmid into TIR1/GCaMP6f parasites. Parasite populations were selected with 25 μg/mL mycophenolic acid and 50 μg/mL xanthine. Single clones were isolated by limiting dilution. Clones were verified by sequencing of the junction between the 3′ end of the gene and 5′ end of the protein tag.
Analysis of CDPK1- and CDPK3-tagged HiT vector populations
Parasites were co-transfected with 40–50 μg of BsaI-linearized HiT 3′SAG1 or HiT 3′CDPK3 vectors and the Cas9-expression plasmid pSS014. 24 h post-transfection parasite populations were selected with 3 μM pyrimethamine. Following selection populations were analyzed by flow cytometry with a Miltenyi MACSQuant VYB. Populations were imaged by microscopy using a 60X objective and an Eclipse Ti microscope (Nikon) with an enclosure maintained at 37°C and 5% CO2. For IAA-induced depletion experiments, intracellular parasites were treated with either 50 μM IAA or an equivalent dilution of PBS for 24 h. Following treatment, parasites were passed through a 27-gauge needle, isolated by filtration, and analyzed by flow cytometry.
Design and cloning of HiT vector libraries
Three gRNA constructs were designed against each gene in the 155 gene library. Since the efficiency of recombination decreases with the distance of the homology regions from the double-stranded break, we selected gRNAs that cut within a 50 bp window downstream of the stop codon44–47. The 3′ end of the genomic sequences (release 36, ToxoDB.org) were scanned for gRNAs containing either NGG or NAG PAMs. gRNAs were ranked based on predicted on-target activity and off-target activity, as determined by the Rule Set 2 and Cutting Frequency Determination calculators67, respectively, and by the distance of the cutsite from the stop codon. As the Rule Set 2 calculator does not take into account efficiencies of different PAMs, the on-target scores of NAG gRNAs were penalized as predicted by the Cutting Frequency Determination calculator. These ranks were used to create an aggregate rank for gRNA selection. Initially, only the highest-ranking gRNAs were selected with cut sites within 30 bp of the stop codon and with a Rule Set 2 score above 0.2. The criteria were progressively relaxed to allow any gRNA within 30 bp, any gRNA within 50 bp and with a Rule Set 2 score above 0.2, and finally any gRNA within 50 bp, until each gene had 3 gRNAs assigned. A ‘G’ was preprended to gRNAs that did not start with one to ensure proper RNA polymerase III initiation. Synonymous point mutations were introduced to H1 homology regions containing BsaI or AscI restriction sites, in order to prevent restriction enzyme cutting during the cloning process. The guide library was synthesized by Agilent and each oligo includes a gRNA, 40 bp homology regions, an AscI restriction site for insertion of the gRNA scaffold, and tandem BsaI sites for linearization of the final constructs, all flanked by sequences for cloning into empty HiT vectors (Supplementary Table 1). The HiT 3′SAG1 and HiT 3′CDPK3 libraries were cloned as described below. The HiT 3′CDPK3 library protocol resulted in both a slight increase of correctly assembled products and greater library diversity. All PCR steps were performed with iProof High-Fidelity DNA polymerase (Bio-Rad) and cloning products were electroporated into MegaXDH10B T1R electrocompetent cells.
HiT 3′SAG1 library.
The synthesized oligo library was PCR amplified with primers P1 and P2. PCR products were cloned into the pTS018 entry vector via Gibson assembly (VWR). The gRNA scaffold was amplified from pSL001 with primers P3 and P4. The amplified scaffold was inserted into AscI-digested entry vector library via NEBuilder HiFi assembly. Finally, the assembled cutting units were PCR amplified using primers P1 and P2 and cloned into pTS020, the mNeonGreen-AID HiT 3′SAG1 vector, via NEBuilder HiFi assembly. DNA products were isolated from liquid cultures using either a ZymoPURE II Plasmid Maxiprep Kit (Zymo Research) or a Macherey-Nagel Nucleobond Xtra Maxi Kit.
HiT 3′CDPK3 library.
In order to decrease polymerase-induced errors we replaced PCR amplification steps with digestion with the type IIS restriction enzyme BsrDI. The oligo library was PCR amplified with primers P1 and P2, as in the HiT 3′SAG1 library. PCR products were cloned into the pTS031 entry vector via NEBuilder HiFi assembly. The gRNA scaffold was isolated from pTS028 via BsrDI digestion and inserted into AscI-digested entry vector library. Finally, the assembled “cutting units” were BsrDI-digested out of the entry vector library and cloned into pGL015, the mNeonGreen-AID HiT 3′CDPK3 vector. DNA products were isolated from solid agar plate cultures using a ZymoPURE II Plasmid Maxiprep Kit.
Pooled HiT vector screening
For each screen, 500 μg of the HiT vector library was linearized with BsaI-HFv2, cleaned-up using Agencourt RNAClean XP SPRI paramagnetic beads, and co-transfected with the Cas9-expression plasmid pSS014 into 5 × 108 TIR1 parasites in the HiT 3′SAG1 screen and 5 × 108 TIR1/IMC1-tdTomato parasites in the HiT 3′CDPK3 screen. Transfected parasites were used to infect 12 and 10 15-cm2 dishes with HFF monolayers in the HiT 3′SAG1 and 3′CDPK3 screens, respectively. 3 μM pyrimethamine and 10 μg/mL DNaseI was added 24 h later. The parasites were allowed to egress naturally from host cells, isolated by filtration, and passaged onto 8 15-cm2 dishes with fresh monolayers. This process was repeated for two more passages, infecting each dish with approximately 2–3 × 107 parasites. Following the third passage, the population was split into three 15 cm2 dishes containing fresh monolayers in D10 supplemented with 50 μM IAA and 3 15-cm2 dishes supplemented with an equal dilution of vehicle (PBS). The populations were maintained and passaged in their respective conditions for three passages in the HiT 3′SAG1 screen and for four passages in the HiT 3′CDPK3 screen. At select passages approximately 108 parasites were pelleted and stored at −80°C for analysis. Parasite DNA was extracted using the DNeasy Blood and Tissue kit (QIAGEN) and integrated gRNA constructs were amplified with primer P5 and barcoding primers P6–22. The resulting libraries were sequenced using a MiSeq v2 kit (Illumina) with single-reads using custom sequencing primer P23 and custom indexing primer P24.
Sequencing reads were aligned to the gRNA library. Read counts were median normalized and gRNAs in the bottom 5th percentile of the input library were removed. To account for differences in NGG and NAG PAM efficiencies, relative abundances for each gRNA were corrected for the PAM used. The PAM efficiencies were calculated by comparing the abundances of only gRNAs in the selected population that target genes identified as dispensable in a previous genome-wide knockout screen12. Fold-changes were normalized to passage 2. UMAP was used to compare gRNAs in each screen based on their pattern of fold changes for vehicle- and IAA-treated samples. Clusters were calculated by k-means. Gene centroids were calculated in UMAP space and assigned to the dispensable or fitness-conferring class using k-means clustering.
Arrayed HiT vector screening
Generation and passaging of array.
In parallel to the pooled HiT 3′CDPK3 screen, single clones were isolated via limiting dilution after 4 passages of drug selection. Parasites from passage 4 were collected and sequenced as in the pooled screening experiments, using primer P5 and P25 for PCR amplification.. 1160 clonal isolates were arrayed into twenty 96-well plates containing HFFs. Included in each plate was a well containing the TIR1/IMC1-tdTomato parental line and a well containing the CDPK1-AID scarless strain. Arrays were passaged every 3 days with a multichannel pipette by transferring 5% of the total lysed well volume to 96-well plates containing HFF monolayers. Individual wells with incomplete lysis were scraped and passed with 10% of the total well volume.
Arrayed widefield microscopy.
Freshly lysed arrays were replica plated with 12 μL into 96-well plates of HFFs maintained in FluoroBrite DMEM (GIBCO) supplemented with 3% IFS, 4 mM glutamine, and 10 mg/mL gentamicin. Replica plates were centrifuged at 150 × g and 18°C for 5 min and subsequently incubated at 37°C and 5% CO2. At 3 h post-infection, replica plates were supplemented with either PBS or IAA to a final concentration of 50 μM. At 24 h post-IAA or PBS addition, each well was imaged at 4 adjacent fields-of-view using a 40X objective and an Eclipse Ti microscope (Nikon) with an enclosure maintained at 37°C and 5% CO2. Images were acquired using the NIS elements imaging software, W-View Gemini image splitting optics, and a Zyla 4.2 sCMOS camera. FIJI software was used for image analysis and processing.
Arrayed lytic assays.
Freshly lysed arrays were replica plated with 10 μL into 96-well plates of HFFs maintained in D3 supplemented with either 50 μM IAA or PBS. Each replica plate contained 6 uninfected control wells. Replica plates were centrifuged at 150 × g and 18°C for 5 min and incubated for 72 h at 37°C and 5% CO2. Plates were washed 1X with PBS and fixed for 10 minutes with 100% ethanol. Intact monolayers were visualized by staining the plates for 5 min with crystal violet solution (12.5 g crystal violet, 125 mL 100% ethanol, 500 mL 1% ammonium oxalate) followed by two PBS washes, one water wash, and overnight drying. Absorbance at 590 nm was read as a measure of host cell lysis and normalized to the average of a plate’s uninfected control wells.
Dual-indexed sequencing of arrays.
Parasites were harvested from 100 μL of lysed wells by centrifugation at 1000 × g and 18°C for 10 min. Pellets were each resuspended in 25 μL of lysis buffer (1X Q5 buffer supplemented with 0.2 mg/mL Proteinase K) and lysed using the following conditions: 37°C for 1 h, 50°C for 2 h, and 95°C for 15 min. Guides were PCR-amplified from gDNA, with each well utilizing a unique combination of i7 index primers (P26–65) and i5 index primers (P66–95). PCR products from an individual plate were pooled and gel extracted using a Zymoclean Gel DNA Recovery Kit and subsequently pooled at equimolar ratios for sequencing. The final PCR product pool was sequenced with a MiSeq v2 kit (Illumina) using dual-indexed single-reads with primers P23 and P24. Sequencing reads were aligned to the gRNA library. A well was designated as gRNA-containing if a single gRNA had more than 100 reads. Wells containing multiple integrations or mixed populations were defined as those in which a secondary gRNA contained more than 10% the number of reads assigned to the most abundant gRNA.
Confocal microscopy of select clones
Parasites were inoculated into 96-well glass-bottom plates (Cellvis) of HFFs maintained in FluoroBrite DMEM supplemented with 10% IFS, 4 mM glutamine, and 10 μg/mL gentamicin. At 24 h post-infection wells were imaged with an RPI spinning disk confocal microscope and a 63x objective maintained at 37°C and 5% CO2. Images were acquired using the MetaMorph acquisition software and a Hammamatsu ORCA-ER CCD camera. The parental TIR1/IMC1-tdTomato strain was also imaged to control for background fluorescence. ImageJ was used for image analysis and processing.
Plaque assays
Parasites were inoculated into 6-well plates of HFFs maintained in D10 and allowed to grow undisturbed for 9 days. For AID strains the cells were maintained in either 50 μM IAA or PBS. For the CDPK1-HA-U1 population the cells were treated with a 2-hour pulse of either 50 nM rapamycin or DMSO at 24 hours post-infection. The rapamycin and DMSO were subsequently washed out and the cells were grown in D10 for the remainder of the assay. Plates were washed with PBS and fixed for 10 min at room temperature with 100% ethanol. Plates were stained with crystal violet solution for 5 min at room temperature, followed by two washes with PBS, one wash with water, and drying. Plaque area calculations were performed in ImageJ.
Competition assays
Freshly lysed strains were filtered through 5 μm filters and pelleted at 1000 × g and 18°C for 10 min. Pellets were resuspended in D10 and counted. T12.5s of HFFs were infected with 1.5 × 106 parasites of the TIR1/IMC1-tdTomato strain and 1.5 × 106 parasites of the competitor strain. At 24 h post-infection the media was changed with fresh D10. Populations were assayed by flow cytometry following host cell lysis, and each population was passed to two wells of a 6-well plate. At 24 h post-infection the media of one well per strain was changed to D10 and vehicle (PBS) and the media of the second well changed to D10 and 50 μM IAA. Populations were maintained in these conditions for four passages. Following each lysis, the populations were assayed by flow cytometry with a Miltenyi MACSQuant VYB. The fraction of the population that was tdTomato negative was represented as a ratio of the [% tdTomato negative in the IAA sample]/[% tdTomato negative in the vehicle sample] and was normalized to the initial fraction pre-splitting into ± IAA media.
Invasion assays
Strains were each passed to two flasks of HFFs containing D10 media. At 3 h post-infection one flask was supplemented with vehicle (PBS) and the second flask was supplemented with IAA to a final concentration of 50 μM. At 27 h post-infection each flask was syringe-lysed and filtered through 5 μm filters. Parasites were pelleted at 1000 × g and 18°C for 10 min. Pellets were resuspended in invasion media (HEPES-buffered DMEM without phenol red) supplemented with 1% IFS to a concentration of 1 × 106 parasites/mL. 200 μL of each parasite solution was added to 3 wells of a clear-bottom 96-well plate containing HFFs. The plate was centrifuged at 290 × g and room temperature for 5 min. Plates were incubated for 10 min at 37°C to stimulate invasion. Following incubation wells were fixed with 4% formaldehyde and extracellular parasites were stained with mouse anti-SAG1 antibody diluted 1:500105. All parasites were stained by permeabilizing with 0.25% Triton X-100 and staining with guinea pig anti-CDPK1 diluted 1:10,000 (Covance106). Cells were subsequently stained with anti-guinea pig Alexa-594 antibody diluted 1:1000 (Invitrogen), anti-mouse Alexa-488 antibody diluted 1:1000 (Invitrogen), and Hoechst 33258 (Santa Cruz Biotechnology) nuclear dye. Samples were imaged using a Biotek Cytation3 imaging multimode reader. The number of invaded parasites per field of view was counted and normalized to the number of host cells in the same area. The invasion efficiency for each replicate was normalized to the invaded parasites per host cell nuclei of the parental TIR1 vehicle sample.
Phylogenetic analysis of SPARK
SPARK homologs were identified by BLAST search against representative apicomplexan genomes. Protein kinase domains were obtained from EupathDB based on their annotation with Interpro domain IPR011009. Sequences were curated for Theileria spp., Cryptosporidium parvum, Cryptosporidium hominis, Sarcocystis neurona, Vitrella brassicaformis, and Chromera velia to correct errors in the gene model. Domains from the nearest human, mouse, and macaque orthologues (as determined by BLAST) were used as outgroups. Individual domains were aligned using ClustalX2, and the phylogenetic tree was generated by neighbor-joining. Visualizations were generated using FigTree (v1.4.4).
Immunoblotting
For detecting SPARK depletion, TIR1 and SPARK-AID parasites were grown in D10 for 3 h before being treated with either 50 μM IAA or vehicle (PBS). After 24 h of treatment, parasites were passed through 27-gauge needles and isolated via filtration. Parasites were resuspended in lysis buffer (0.8% IGEPAL-CA630, 0.25 U/μL benzonase, and 2X Halt Protease Inhibitor Cocktail in PBS) and incubated on ice for 15 min. Lysates were combined with 1X Laemmli buffer (diluted from 5X buffer containing 10% SDS, 50% glycerol, 300 mM Tris HCl pH 6.8, 0.05% bromophenol blue) with 1% final volume ꞵ-mercaptoethanol and boiled for 10 min. Samples were run on a 7.5% SDS-PAGE gel (BioRad) and transferred onto a nitrocellulose membrane in transfer buffer (25 mM Tris-HCl, 192 mM glycine, 20% methanol). Blocking and all subsequent antibody incubations were performed at room temperature in 5% milk in TBS-T (20 mM Tris, 138 mM NaCl, 1 L PBS, 0.1% Tween-20) unless otherwise noted. The blot was incubated in blocking buffer for 1 h followed by incubation with the mouse anti-V5 primary antibody diluted 1:2000 (R960–25, Invitrogen) for 1 h. The blot was washed three times with TBS-T and incubated for 1 h with the anti-mouse secondary antibody diluted 1:10,000 (LI-COR). Following imaging the blot was incubated overnight at 4°C with the guinea pig anti-CDPK1 primary antibody diluted 1:100,000 (Covance)106. The blot was washed three times with TBS-T, incubated for 1 h with anti-guinea pig secondary antibody diluted 1:10,000 (LI-COR), washed 3X with TBS-T, and imaged using a LI-COR Odyssey CLx.
For detecting CDPK1-V5-T2A-mKate2, Δku80 and the transfected population were harvested from fully lysed host cells. Parasites were lysed, run on a 4–15% gradient SDS-PAGE gel (BioRad), and transferred onto a nitrocellulose membrane as above. The blot was incubated for 1h with mouse anti-V5 primary antibody diluted 1:2000 (R960–25, Invitrogen) and rabbit anti-RFP primary antibody diluted 1:4000 (R10367, Life Technologies). The blot was washed three times with TBS-T and incubated for 1 h with anti-mouse and anti-rabbit secondary antibodies diluted 1:10,000 (LI-COR). Following imaging the blot was incubated with rabbit anti-ALD1 primary antibody diluted 1:10,000 (Zymed Laboratories Inc.)107, for 1 h. Following three washes with TBS-T, the blot was incubated for 1 h with anti-rabbit secondary antibodies diluted 1:10,000 (LI-COR), washed 3X with TBS-T, and imaged.
Immunofluorescence assays
Parasites were inoculated onto coverslips containing HFFs. For AID strains, after 3 h cells were treated with either 50 μM IAA or PBS. At 24 h post-treatment, intracellular parasites were fixed with 4% formaldehyde and permeabilized with 0.25% Triton X-100 in PBS. For the CDPK1-HA-U1 population, after 3 h cells were treated with either 50 nM rapamycin or DMSO for 2h. At 24 h post-rapamycin pulse, intracellular parasites were fixed and permeabilized as above. Nuclei were stained with Hoechst 33342 or Hoescht 33258 (Santa Cruz) and coverslips were mounted in Prolong Diamond (Thermo Fisher). V5 was detected using a mouse monoclonal antibody (R960–25, Invitrogen). CDPK1 was detected using a guinea pig-derived polyclonal antibody diluted 1:10,000 (Covance)106. HA was detected using a mouse monoclonal antibody diluted 1:1000 (901501, BioLegend). GAP45 was detected using a rabbit polyclonal antibody diluted 1:1000 (Eurogentec)108. Primary antibodies were stained with anti-guinea pig, anti-mouse, or anti-rabbit Alexa-Fluor-labeled secondary antibodies diluted 1:1000 (Invitrogen). Images were acquired with an Eclipse Ti microscope (Nikon) using the NIS elements imaging software and a Zyla 4.2 sCMOS camera. ImageJ was used for image analysis and processing.
Replication assays
Parasites were inoculated onto coverslips containing HFFs and after 3 h were treated with either 50 μM IAA or PBS. At 24 h post-IAA addition, intracellular parasites were fixed, permeabilized, and stained and imaged as described under Immunofluorescence assays. For each sample, multiple fields of view were acquired. The number of parasites per vacuole were calculated from 100 vacuoles. Results are the mean of three independent experiments.
Egress assays
Egress was quantified in a plate-based manner90. HFF monolayers in a clear-bottomed 96-well plate infected with 7.5 ×104 parasites per well of parental or SPARK-AID for 3 h were treated with 50 μM IAA or PBS for an additional 24 h. Before imaging, the media was exchanged for FluoroBrite supplemented with 10% IFS. Three images were taken before zaprinast (final concentration 500 μM) or A23187 (final concentration 8 μM) and DAPI (final concentration 5 ng/mL) were added, and imaging of DAPI-stained host cell nuclei continued for 9 additional minutes before 1% Triton X-100 was added to all wells to determine the total number of host cell nuclei. Imaging was performed at 37°C and 5% CO2 using a Biotek Cytation 3 imaging multimode reader. Results are the mean of three wells per condition and are representative of three independent experiments.
Live cell microscopy of GCaMP6f-expressing parasites
To capture egress, SPARK-AID parasites were grown in HFFs in glass-bottom 35 mm dishes (Ibidi) for 3 h at which point they were treated with either 50 μM IAA or PBS for an additional 24 h. Parasites were stimulated to egress with 500 μM zaprinast or 8 μM A23187 in Ringer’s buffer prepared without Ca2+ (155 mM NaCl, 3 mM KCl, 1mM MgCl2, 3 mM NaH2PO4, 10 mM HEPES, 10 mM glucose) and supplemented with 1% BSA (w/v) and recorded every 4 s for 220 s with an Eclipse Ti microscope (Nikon) in an enclosure maintained at 37 °C and 5% CO2. Images were acquired using the NIS elements imaging software and a Zyla 4.2 sCMOS camera. Image analysis and quantification was done using ImageJ.
Extracellular zaprinast and Ca2+ ionophore treatment of GCaMP6f strains
Parasites were passed to 15-cm2 dishes containing HFFs and after 6 h treated with either 50 μM IAA or vehicle (PBS). Following 24 h of treatment the cells were washed once with PBS and harvested in cold Ringer’s Basal Ca2+ (155 mM NaCl, 3 mM KCl, 1 mM MgCl2, 3 mM NaH2PO4, 10 mM HEPES, 10 mM glucose, 250 μM EGTA, 112 μM CaCl2). Parasites were isolated via syringe lysis and filtration, pelleted and washed once with cold Ringer’s Basal Ca2+, and resuspended to 1 × 107 parasites/mL. 100 μL of parasite suspension was added to a clear-bottom 96-well plate and incubated on ice for 5 min. Fluorescence was read with an excitation wavelength of 485 nm and an emission wavelength of 528 nm every 10 s in a BioTek Cytation 3. At 30 s 50 μL of 3X zaprinast (100 μM final concentration), A23187 (2 μM final concentration), ionomycin (1 μM final concentration), or vehicle (DMSO) was added and fluorescence readings were taken for an additional 6 min. 50 μL of 4X aerolysin (3 μg/mL final concentration) and CaCl2 (2 mM final concentration) was added, and the assay plate was incubated at 37 °C for 10 min. Fluorescence was read every 1 min for 40 min. The assay plate was shaken for 1 s before each read. The non-fluorescent TIR1 strain was used to perform a baseline subtraction of background fluorescence. Baseline-subtracted values were normalized to initial fluorescence pre-stimulation and maximum fluorescence post-aerolysin treatment. Results are the mean of three independent experiments in the case of A23187 and ionomycin and six independent experiments in the case of zaprinast and DMSO.
Extended Data
Extended Data Fig. 1. Transfected populations efficiently incorporate a variety of HiT vectors.
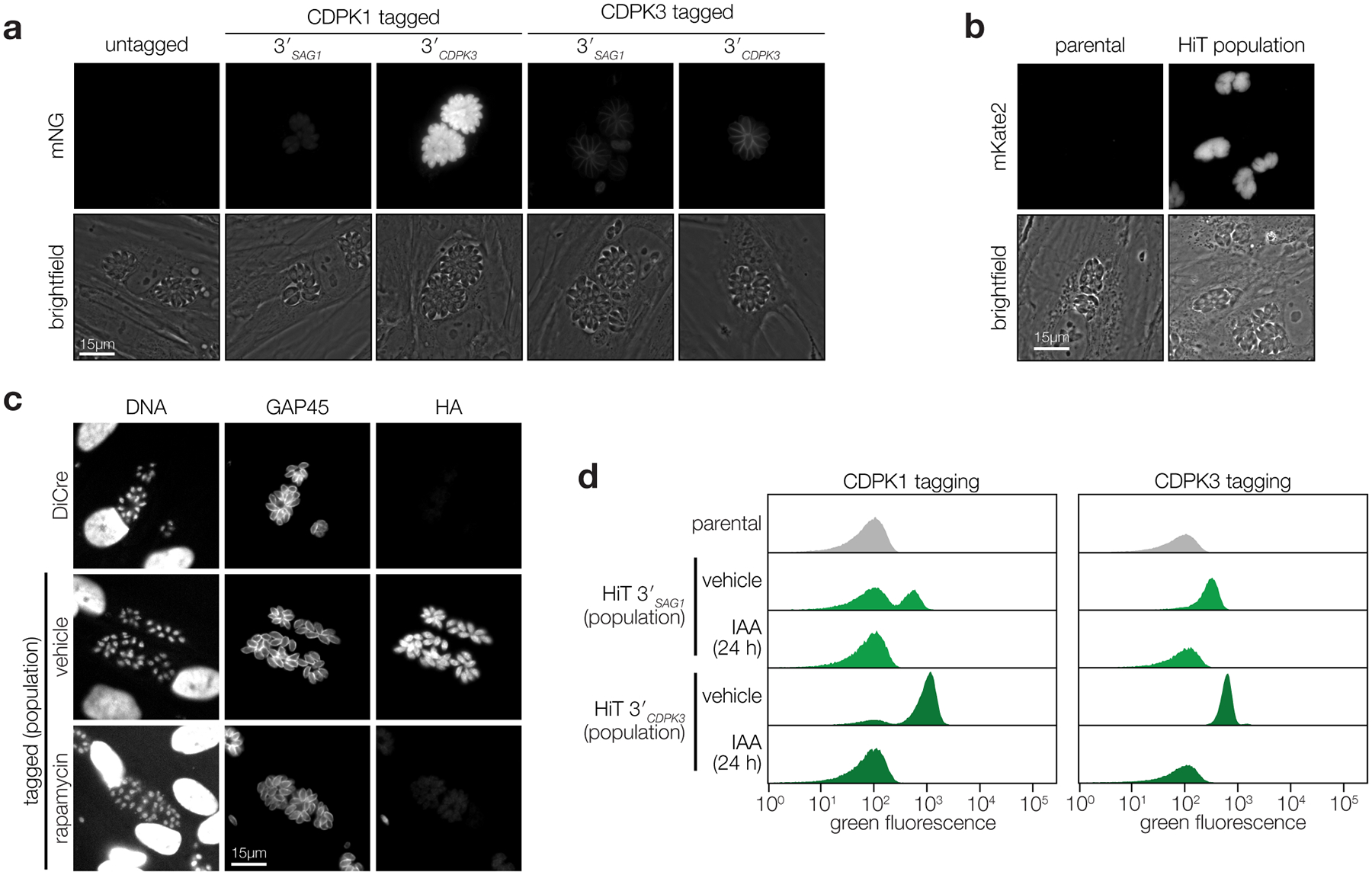
a, Fluorescence microscopy of the tagged populations displaying the correct localization of each kinase and expression levels consistent with flow cytometry (Fig. 1b). b, Live microscopy of V5-T2A-mKate2 HiT-tagged population (merged image in Fig. 1g). c, Immunofluorescence microscopy of population tagged with the HA-U1 HiT vector following treatment with rapamycin or vehicle control (merged image in Fig. 1i). d, Flow cytometry of parasite populations tagged with the V5-mNG-mAID HiT vector targeting CDPK1 or CDPK3 and treated with either IAA or vehicle control for 24 h (excerpt shown in Fig. 1l).
Extended Data Fig. 2. Arrayed screening results.
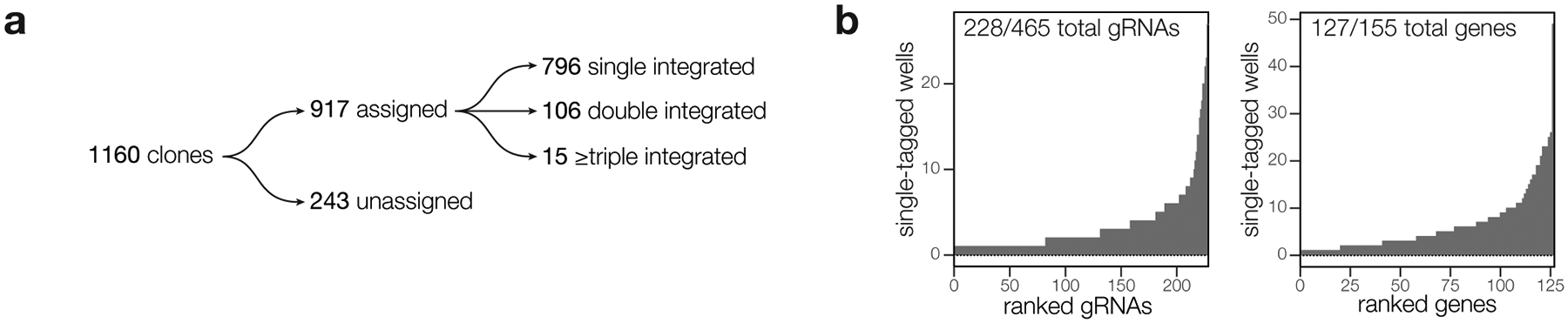
a, Results from dual-indexed sequencing of the arrayed clones. A minimum of 100 reads were required to assign a given gRNA to a particular clone. Cases where a second gRNA reached >10% the abundance of the first gRNA were classified as containing multiple integrations. b, Histogram showing the distribution of gRNAs and genes contained among single-integrated wells within the array. Genes and gRNAs with no representation are omitted from the plot.
Extended Data Fig. 3. Representative images from the arrayed screen.
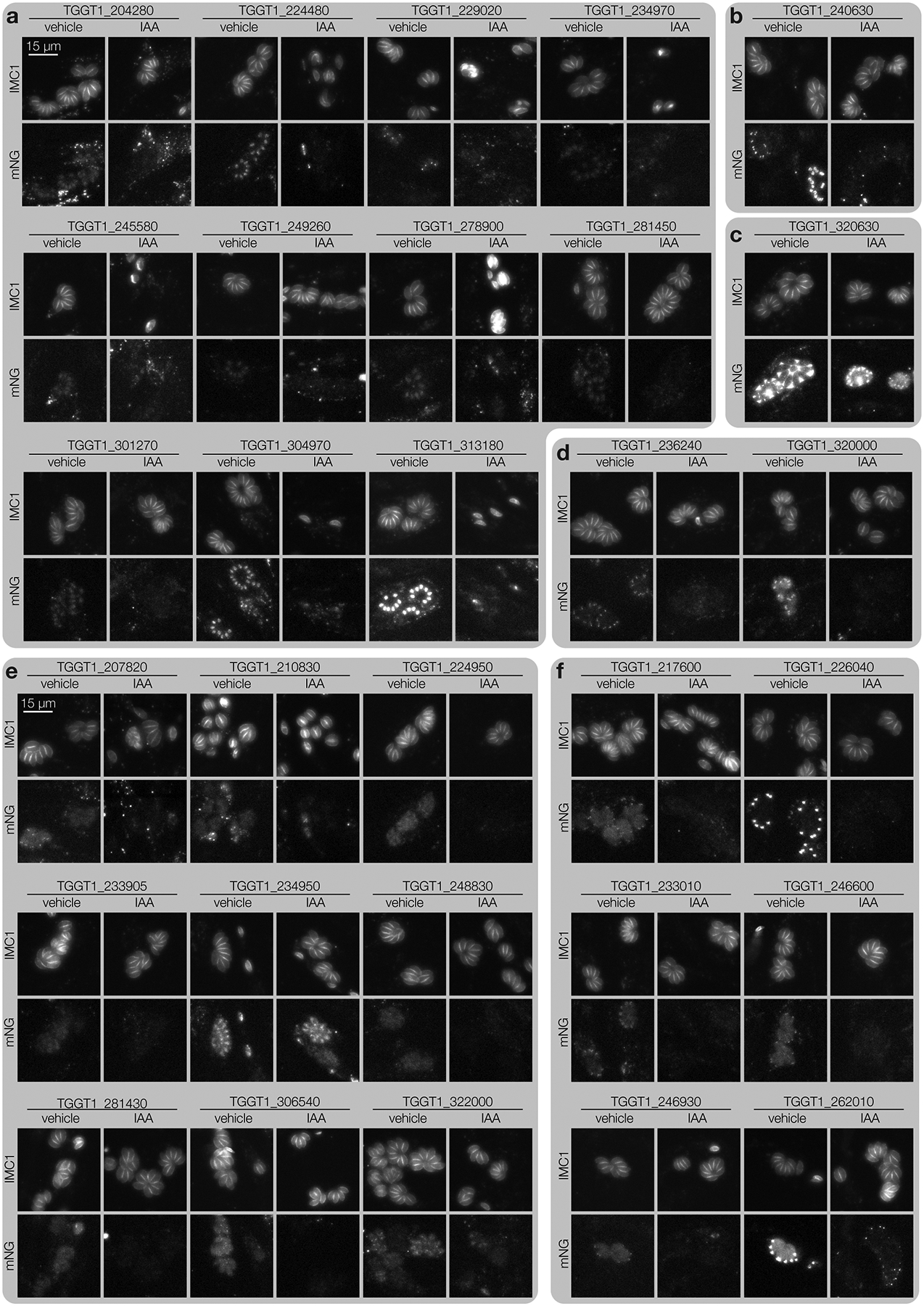
a–f, Widefield microscopy of representative clones. Maximum intensity projections for IMC1-tdTomato and mNeonGreen-tagged targets are displayed for cultures treated with either IAA or vehicle for 24 hours. All images are displayed at the same scale. Localizations to the nucleus (a), daughter cell IMC (b), parasitophorous vacuole (c), perinuclear space (d), cytosol (e) or apical end (f) were assigned to a gene if half or more of single-integrated wells for that gene displayed consistent localizations.
Extended Data Fig. 4. Additional representative images from the arrayed screen and comparisons to the pooled results.
a–c, Widefield microscopy of representative clones. Maximum intensity projections for IMC1-tdTomato and mNeonGreen-tagged targets are displayed for cultures treated with either IAA or vehicle for 24 hours. All images are displayed at the same scale. Localizations to puncta (a), the basal end (b), or peripheral structures (c) were assigned to a gene if half or more of single-integrated wells for that gene displayed consistent localizations. d, Representative confocal images of a sample of clones. mNeonGreen (green); IMC1-tdTomato (magenta). Images are maximum intensity projections. Genes are numbered based on the unique identifier from ToxoDB (e.g., TGGT1_210830, labeled 210830). e, Comparison of relative gRNA abundances in the array compared to the pooled population that was subcloned. Spearman correlation coefficient = 0.77. f, Impact of the initial lytic cycles on gRNA abundance for genes with delayed or acute loss phenotypes in the HiT screen. The effect of the first lytic cycle from the HiT screen is plotted against the effect of the first or second lytic cycles for the genome-wide knockout screen (Sidik & Huet, et al. 2016). Genes are paired across their first and second lytic cycles within the genome-wide knockout screen.
Extended Data Fig. 5. Extended analysis of SPARK depletion.
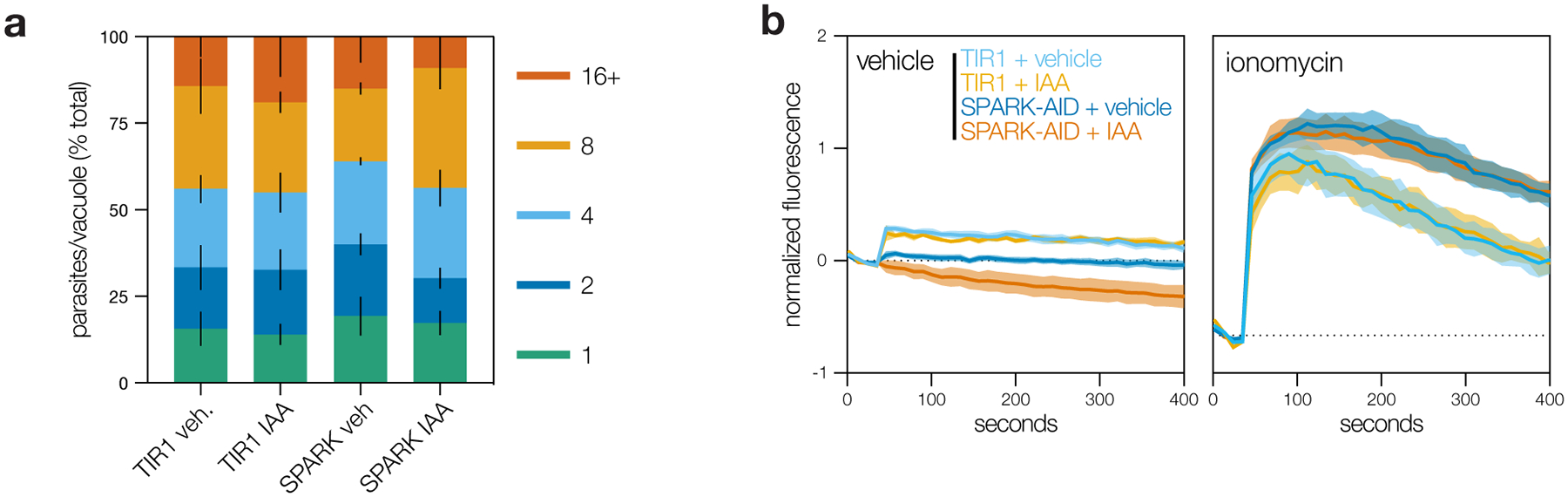
a, Replication assay of SPARK-AID parasites. Parasites were treated with either IAA or vehicle at 3 hours post-invasion and imaged 24 hours later. The number of parasites per vacuole were counted for 100 vacuoles per sample. Mean ± S.E. graphed for n = 3 biological replicates. b, Extracellular parasites in basal Ca2+ buffer stimulated with vehicle or the Ca2+ ionophore ionomycin, following 24 h of treatment with vehicle or IAA. Cytosolic Ca2+ flux was measured in bulk as GCaMP6f fluorescence normalized to the initial and maximum fluorescence following aerolysin permeabilization in 2 mM Ca2+. Mean ± S.E. graphed for n = 3–6 biological replicates.
Supplementary Material
Supplementary Table 1 Oligos and plasmids used in this study.
Supplementary Table 2 Combined results from the HiT screens summarizing data from arrayed and pooled analyses.
Source Data Fig. 4 b, Competition assays of delayed loss candidates. Provided are relative abundances of each strain relative to the WT competitor strain. Values are normalized to the starting ratio. d, Area sizes of individual plaques. Provided values are in mm2. e, Invasion efficiencies of delayed loss candidates. Invaded parasites per nuclei for each replicate are provided, in addition to the values post-normalization to the WT vehicle-treated sample.
Source Data Fig. 5 a, Kinase domain sequences used to generate alignments and the subsequent phylogenetic tree. e–f, Egress efficiencies following either (e) zaprinast or (f) A23187 stimulation. Provided is percent of egress relative to the final percentage of egress of the vehicle-treated sample. h, Quantification of GCaMP6f fluorescence signal following either zaprinast or A23187 treatment. Average fluorescence of each vacuole was quantified relative to initial fluorescence until egress of the vacuole or until the end of the time-course. i, Quantification of GCaMP6f fluorescence from extracellular parasites treated with either zaprinast or A23187. Provided are background-subtracted values normalized to initial fluorescence and the final maximum fluorescence following aerolysin and Ca2+ treatment.
Source Data Extended Data Fig. 5 a, Replication assays of SPARK grown in the presence or absence of IAA. 100 vacuoles were quantified for each sample and condition. Provided are the number of occurrences of each vacuole size for each replicate. b, Quantification of GCaMP6f fluorescence from extracellular parasites treated with either vehicle (DMSO) or ionomycin. Provided are background-subtracted values normalized to initial fluorescence and the final maximum fluorescence following aerolysin and Ca2+ treatment.
ACKNOWLEDGEMENTS
We thank the Whitehead Institute Bioinformatics and Research Computing Core, especially Bingbing Yuan, for assistance implementing gRNA design pipelines; L. David Sibley for the TIR1 strain; Moritz Treeck for the DiCre strain; Wendy Salmon and the W.M. Keck Biological Imaging Facility for confocal microscopy support; Peter W. Reddien for use of the Illumina MiSeq; Benjamin S. Waldman, Elizabeth A. Boydston, Christopher J. Giuliano, Alex W. Chan, Saima Sidik, and Benedikt M. Markus for technical support in generation of the array; VEuPathDB and all contributors to this resource. This work was supported by funds from a National Institutes of Health grant (R01AI144369) to S.L. and National Science Foundation Graduate Research Fellowships to T.A.S. (2018259980) and A.L.H. (174530).
Footnotes
CODE AVAILABILITY
All code is described in the methods section and available from the corresponding author upon request.
COMPETING INTERESTS STATEMENT
The authors declare no competing interests.
DATA AVAILABILITY
All oligos used in this study are available in Supplementary Table 1. All plasmids used or generated in this study are listed with their appropriate GenBank or PMID accession numbers in Supplementary Table 1. Minimally processed pooled and arrayed CRISPR screen sequencing results are available in Supplementary Table 2. Localization assignments, microscopy phenotypes, lytic assay results, and UMAP coordinates and clusters are likewise available in Supplementary Table 2. Data from experimental results is available in the source data files. Additional unprocessed data is available from the corresponding author upon request.
REFERENCES
- 1.Cabrera DG et al. Plasmodial Kinase Inhibitors: License to Cure? J. Med. Chem 61, 8061–8077 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sauvey C, Ehrenkaufer G, Shi D, Debnath A & Abagyan R Antineoplastic kinase inhibitors: A new class of potent anti-amoebic compounds. PLoS Negl. Trop. Dis 15, e0008425 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Merritt C, Silva LE, Tanner AL, Stuart K & Pollastri MP Kinases as druggable targets in trypanosomatid protozoan parasites. Chem. Rev 114, 11280–11304 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Talevich E, Mirza A & Kannan N Structural and evolutionary divergence of eukaryotic protein kinases in Apicomplexa. BMC Evol. Biol 11, 321 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peixoto L et al. Integrative genomic approaches highlight a family of parasite-specific kinases that regulate host responses. Cell Host Microbe 8, 208–218 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gaji RY, Sharp AK & Brown AM Protein kinases in Toxoplasma gondii. Int. J. Parasitol 51, 415–429 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beraki T et al. Divergent kinase regulates membrane ultrastructure of the Toxoplasma parasitophorous vacuole. Proc. Natl. Acad. Sci. U. S. A 116, 6361–6370 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor S et al. A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science 314, 1776–1780 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Fox BA et al. The Toxoplasma gondii Rhoptry Kinome Is Essential for Chronic Infection. MBio 7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fleckenstein MC et al. A Toxoplasma gondii pseudokinase inhibits host IRG resistance proteins. PLoS Biol. 10, e1001358 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niedelman W et al. The rhoptry proteins ROP18 and ROP5 mediate Toxoplasma gondii evasion of the murine, but not the human, interferon-gamma response. PLoS Pathog. 8, e1002784 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sidik SM et al. A Genome-wide CRISPR Screen in Toxoplasma Identifies Essential Apicomplexan Genes. Cell 166, 1423–1435.e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bushell E et al. Functional Profiling of a Plasmodium Genome Reveals an Abundance of Essential Genes. Cell 170, 260–272.e8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang M et al. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 360, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tewari R et al. The systematic functional analysis of Plasmodium protein kinases identifies essential regulators of mosquito transmission. Cell Host Microbe 8, 377–387 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meissner M, Brecht S, Bujard H & Soldati D Modulation of myosin A expression by a newly established tetracycline repressor-based inducible system in Toxoplasma gondii. Nucleic Acids Res. 29, E115 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meissner M, Schlüter D & Soldati D Role of Toxoplasma gondii myosin A in powering parasite gliding and host cell invasion. Science 298, 837–840 (2002). [DOI] [PubMed] [Google Scholar]
- 18.van Poppel NFJ, Welagen J, Duisters RFJJ, Vermeulen AN & Schaap D Tight control of transcription in Toxoplasma gondii using an alternative tet repressor. Int. J. Parasitol 36, 443–452 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Pieperhoff MS et al. Conditional U1 Gene Silencing in Toxoplasma gondii. PLoS One 10, e0130356 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andenmatten N et al. Conditional genome engineering in Toxoplasma gondii uncovers alternative invasion mechanisms. Nat. Methods 10, 125–127 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hunt A et al. Differential requirements for cyclase-associated protein (CAP) in actin-dependent processes of Toxoplasma gondii. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishimura K, Fukagawa T, Takisawa H, Kakimoto T & Kanemaki M An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 6, 917–922 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Brown KM, Long S & David Sibley L Plasma Membrane Association by N-Acylation Governs PKG Function in Toxoplasma gondii. MBio 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Long S et al. Calmodulin-like proteins localized to the conoid regulate motility and cell invasion by Toxoplasma gondii. PLoS Pathog. 13, e1006379 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huynh M-H & Carruthers VB Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot. Cell 8, 530–539 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fox BA, Ristuccia JG, Gigley JP & Bzik DJ Efficient gene replacements in Toxoplasma gondii strains deficient for nonhomologous end joining. Eukaryot. Cell 8, 520–529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krogan NJ et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637–643 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Huh W-K et al. Global analysis of protein localization in budding yeast. Nature 425, 686–691 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Weill U et al. Genome-wide SWAp-Tag yeast libraries for proteome exploration. Nat. Methods 15, 617–622 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leonetti MD, Sekine S, Kamiyama D, Weissman JS & Huang B A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc. Natl. Acad. Sci. U. S. A 113, E3501–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cho NH et al. OpenCell: proteome-scale endogenous tagging enables the cartography of human cellular organization. bioRxiv 2021.03.29.437450 (2021) doi: 10.1101/2021.03.29.437450. [DOI] [Google Scholar]
- 32.Dean S, Sunter JD & Wheeler RJ TrypTag.org: A Trypanosome Genome-wide Protein Localisation Resource. Trends Parasitol. 33, 80–82 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sidik SM, Hackett CG, Tran F, Westwood NJ & Lourido S Efficient genome engineering of Toxoplasma gondii using CRISPR/Cas9. PLoS One 9, e100450 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sathyan KM et al. An improved auxin-inducible degron system preserves native protein levels and enables rapid and specific protein depletion. Genes Dev. 33, 1441–1455 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Long S, Anthony B, Drewry LL & Sibley LD A conserved ankyrin repeat-containing protein regulates conoid stability, motility and cell invasion in Toxoplasma gondii. Nat. Commun 8, 2236 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lourido S et al. Calcium-dependent protein kinase 1 is an essential regulator of exocytosis in Toxoplasma. Nature 465, 359–362 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lourido S, Tang K & Sibley LD Distinct signalling pathways control Toxoplasma egress and host-cell invasion. EMBO J. 31, 4524–4534 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCoy JM, Whitehead L, van Dooren GG & Tonkin CJ TgCDPK3 Regulates Calcium-Dependent Egress of Toxoplasma gondii from Host Cells. PLoS Pathog. 8, e1003066 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown KM, Long S & Sibley LD Conditional Knockdown of Proteins Using Auxin-inducible Degron (AID) Fusions in Toxoplasma gondii. Bio Protoc 8, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bullen HE et al. Phosphatidic Acid-Mediated Signaling Regulates Microneme Secretion in Toxoplasma. Cell Host Microbe 19, 349–360 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Uboldi AD et al. Protein kinase A negatively regulates Ca2+ signalling in Toxoplasma gondii. PLoS Biol. 16, e2005642 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farrell A et al. A DOC2 protein identified by mutational profiling is essential for apicomplexan parasite exocytosis. Science 335, 218–221 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jia Y et al. Crosstalk between PKA and PKG controls pH-dependent host cell egress of Toxoplasma gondii. EMBO J. 36, 3250–3267 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paquet D et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533, 125–129 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Dewari PS et al. An efficient and scalable pipeline for epitope tagging in mammalian stem cells using Cas9 ribonucleoprotein. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bialk P, Rivera-Torres N, Strouse B & Kmiec EB Regulation of Gene Editing Activity Directed by Single-Stranded Oligonucleotides and CRISPR/Cas9 Systems. PLoS One 10, e0129308 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liang X, Potter J, Kumar S, Ravinder N & Chesnut JD Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J. Biotechnol 241, 136–146 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Harding CR et al. Gliding Associated Proteins Play Essential Roles during the Formation of the Inner Membrane Complex of Toxoplasma gondii. PLoS Pathog. 12, e1005403 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barylyuk K et al. A Comprehensive Subcellular Atlas of the Toxoplasma Proteome via hyperLOPIT Provides Spatial Context for Protein Functions. Cell Host Microbe 28, 752–766.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alvarez CA & Suvorova ES Checkpoints of apicomplexan cell division identified in Toxoplasma gondii. PLoS Pathog. 13, e1006483 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Varberg JM, Coppens I, Arrizabalaga G & Gaji RY TgTKL1 Is a Unique Plant-Like Nuclear Kinase That Plays an Essential Role in Acute Toxoplasmosis. MBio 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Silljé HH, Takahashi K, Tanaka K, Van Houwe G & Nigg EA Mammalian homologues of the plant Tousled gene code for cell-cycle-regulated kinases with maximal activities linked to ongoing DNA replication. EMBO J. 18, 5691–5702 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pilyugin M, Demmers J, Verrijzer CP, Karch F & Moshkin YM Phosphorylation-mediated control of histone chaperone ASF1 levels by Tousled-like kinases. PLoS One 4, e8328 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Silljé HH & Nigg EA Identification of human Asf1 chromatin assembly factors as substrates of Tousled-like kinases. Curr. Biol 11, 1068–1073 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Eckert D et al. Prp4 Kinase Grants the License to Splice: Control of Weak Splice Sites during Spliceosome Activation. PLoS Genet. 12, e1005768 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Naumov A et al. The Toxoplasma Centrocone Houses Cell Cycle Regulatory Factors. MBio 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gubbels M-J et al. Forward genetic analysis of the apicomplexan cell division cycle in Toxoplasma gondii. PLoS Pathog. 4, e36 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen C-T & Gubbels M-J The Toxoplasma gondii centrosome is the platform for internal daughter budding as revealed by a Nek1 kinase mutant. J. Cell Sci 126, 3344–3355 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suvorova ES, Francia M, Striepen B & White MW A novel bipartite centrosome coordinates the apicomplexan cell cycle. PLoS Biol. 13, e1002093 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hu X, O’Shaughnessy WJ, Beraki TG & Reese ML Loss of the Conserved Alveolate Kinase MAPK2 Decouples Toxoplasma Cell Growth from Cell Division. MBio 11, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martín Moyano P, Němec V & Paruch K Cdc-Like Kinases (CLKs): Biology, Chemical Probes, and Therapeutic Potential. Int. J. Mol. Sci 21, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berto G, Ferreira-Cerca S & De Wulf P The Rio1 protein kinases/ATPases: conserved regulators of growth, division, and genomic stability. Curr. Genet 65, 457–466 (2019). [DOI] [PubMed] [Google Scholar]
- 63.Mallari JP, Oksman A, Vaupel B & Goldberg DE Kinase-associated endopeptidase 1 (Kae1) participates in an atypical ribosome-associated complex in the apicoplast of Plasmodium falciparum. J. Biol. Chem 289, 30025–30039 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Back PS et al. Ancient MAPK ERK7 is regulated by an unusual inhibitory scaffold required for Toxoplasma apical complex biogenesis. Proc. Natl. Acad. Sci. U. S. A 117, 12164–12173 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.O’Shaughnessy WJ, Hu X, Beraki T, McDougal M & Reese ML Loss of a conserved MAPK causes catastrophic failure in assembly of a specialized cilium-like structure in Toxoplasma gondii. MBoC 31, 881–888 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sampels V et al. Conditional mutagenesis of a novel choline kinase demonstrates plasticity of phosphatidylcholine biogenesis and gene expression in Toxoplasma gondii. J. Biol. Chem 287, 16289–16299 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McInnes L, Healy J & Melville J UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 1–51 (2018). [Google Scholar]
- 69.Hartigan JA & Wong MA Algorithm AS 136: A K-means clustering algorithm. J. R. Stat. Soc. Ser. C Appl. Stat 28, 100 (1979). [Google Scholar]
- 70.Donald RGK et al. Toxoplasma gondii cyclic GMP-dependent kinase: chemotherapeutic targeting of an essential parasite protein kinase. Eukaryot. Cell 1, 317–328 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Katso R et al. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol 17, 615–675 (2001). [DOI] [PubMed] [Google Scholar]
- 72.Balla A & Balla T Phosphatidylinositol 4-kinases: old enzymes with emerging functions. Trends Cell Biol. 16, 351–361 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Dvorin JD et al. A plant-like kinase in Plasmodium falciparum regulates parasite egress from erythrocytes. Science 328, 910–912 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kato N et al. Gene expression signatures and small-molecule compounds link a protein kinase to Plasmodium falciparum motility. Nat. Chem. Biol 4, 347–356 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bansal A et al. Characterization of Plasmodium falciparum calcium-dependent protein kinase 1 (PfCDPK1) and its role in microneme secretion during erythrocyte invasion. J. Biol. Chem 288, 1590–1602 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Green JL et al. The motor complex of Plasmodium falciparum: phosphorylation by a calcium-dependent protein kinase. J. Biol. Chem 283, 30980–30989 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen F, Mackey AJ, Stoeckert CJ, Jr & Roos, D. S. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 34, D363–8 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rosenberg A, Luth MR, Winzeler EA, Behnke M & Sibley LD Evolution of resistance in vitro reveals mechanisms of artemisinin activity in Toxoplasma gondii. Proc. Natl. Acad. Sci. U. S. A (2019) doi: 10.1073/pnas.1914732116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hitz E et al. The 3-phosphoinositide-dependent protein kinase 1 is an essential upstream activator of protein kinase A in malaria parasites. PLoS Biol. 19, e3001483 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dittrich ACN & Devarenne TP Perspectives in PDK1 evolution: insights from photosynthetic and non-photosynthetic organisms. Plant Signal. Behav 7, 642–649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sidik SM et al. Using a Genetically Encoded Sensor to Identify Inhibitors of Toxoplasma gondii Ca2+ Signaling. J. Biol. Chem 291, 9566–9580 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Howard BL et al. Identification of potent phosphodiesterase inhibitors that demonstrate cyclic nucleotide-dependent functions in apicomplexan parasites. ACS Chem. Biol 10, 1145–1154 (2015). [DOI] [PubMed] [Google Scholar]
- 83.Wiersma HI et al. A role for coccidian cGMP-dependent protein kinase in motility and invasion. Int. J. Parasitol 34, 369–380 (2004). [DOI] [PubMed] [Google Scholar]
- 84.Brown KM, Lourido S & Sibley LD Serum Albumin Stimulates Protein Kinase G-dependent Microneme Secretion in Toxoplasma gondii. J. Biol. Chem 291, 9554–9565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McRobert L et al. Gametogenesis in malaria parasites is mediated by the cGMP-dependent protein kinase. PLoS Biol. 6, e139 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Collins CR et al. Malaria parasite cGMP-dependent protein kinase regulates blood stage merozoite secretory organelle discharge and egress. PLoS Pathog. 9, e1003344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang L et al. An apically located hybrid guanylate cyclase-ATPase is critical for the initiation of Ca2+ signaling and motility in Toxoplasma gondii. J. Biol. Chem 294, 8959–8972 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bisio H, Lunghi M, Brochet M & Soldati-Favre D Phosphatidic acid governs natural egress in Toxoplasma gondii via a guanylate cyclase receptor platform. Nat Microbiol 4, 420–428 (2019). [DOI] [PubMed] [Google Scholar]
- 89.Brown KM & Sibley LD Essential cGMP Signaling in Toxoplasma Is Initiated by a Hybrid P-Type ATPase-Guanylate Cyclase. Cell Host Microbe 24, 804–816.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shortt E & Lourido S Plate-Based Quantification of Stimulated Toxoplasma Egress. Methods Mol. Biol 2071, 171–186 (2020). [DOI] [PubMed] [Google Scholar]
- 91.Chen T-W et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pace DA, McKnight CA, Liu J, Jimenez V & Moreno SNJ Calcium entry in Toxoplasma gondii and its enhancing effect of invasion-linked traits. J. Biol. Chem 289, 19637–19647 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Arrizabalaga G & Boothroyd JC Role of calcium during Toxoplasma gondii invasion and egress. Int. J. Parasitol 34, 361–368 (2004). [DOI] [PubMed] [Google Scholar]
- 94.Alsford S et al. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 21, 915–924 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Young J et al. A CRISPR platform for targeted in vivo screens identifies Toxoplasma gondii virulence factors in mice. Nat. Commun 10, 3963 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tosetti N, Dos Santos Pacheco N, Soldati-Favre D & Jacot D Three F-actin assembly centers regulate organelle inheritance, cell-cell communication and motility in Toxoplasma gondii. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brochet M et al. Phosphoinositide metabolism links cGMP-dependent protein kinase G to essential Ca2+ signals at key decision points in the life cycle of malaria parasites. PLoS Biol. 12, e1001806 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Carruthers VB, Moreno SN & Sibley LD Ethanol and acetaldehyde elevate intracellular [Ca2+] and stimulate microneme discharge in Toxoplasma gondii. Biochem. J 342 (Pt 2), 379–386 (1999). [PMC free article] [PubMed] [Google Scholar]
- 99.Lovett JL, Marchesini N, Moreno SNJ & Sibley LD Toxoplasma gondii microneme secretion involves intracellular Ca(2+) release from inositol 1,4,5-triphosphate (IP(3))/ryanodine-sensitive stores. J. Biol. Chem 277, 25870–25876 (2002). [DOI] [PubMed] [Google Scholar]
- 100.Bullen HE, Bisio H & Soldati-Favre D The triumvirate of signaling molecules controlling Toxoplasma microneme exocytosis: Cyclic GMP, calcium, and phosphatidic acid. PLoS Pathog. 15, e1007670 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Leroux AE, Schulze JO & Biondi RM AGC kinases, mechanisms of regulation and innovative drug development. Semin. Cancer Biol 48, 1–17 (2018). [DOI] [PubMed] [Google Scholar]
- 102.Mora A, Komander D, van Aalten DMF & Alessi DR PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell Dev. Biol 15, 161–170 (2004). [DOI] [PubMed] [Google Scholar]
- 103.Li W et al. A phenotypic screen using splitCas9 identifies essential genes required for actin regulation during host cell egress and invasion by Toxoplasma gondii. bioRxiv 2021.09.24.461619 (2021) doi: 10.1101/2021.09.24.461619. [DOI] [Google Scholar]
- 104.Markus BM, Bell GW, Lorenzi HA & Lourido S Optimizing Systems for Cas9 Expression in Toxoplasma gondii. mSphere 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Burg JL, Perelman D, Kasper LH, Ware PL & Boothroyd JC Molecular analysis of the gene encoding the major surface antigen of Toxoplasma gondii. J. Immunol 141, 3584–3591 (1988). [PubMed] [Google Scholar]
- 106.Waldman BS et al. Identification of a Master Regulator of Differentiation in Toxoplasma. Cell 180, 359–372.e16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Starnes GL, Jewett TJ, Carruthers VB & Sibley LD Two separate, conserved acidic amino acid domains within the Toxoplasma gondii MIC2 cytoplasmic tail are required for parasite survival. J. Biol. Chem 281, 30745–30754 (2006). [DOI] [PubMed] [Google Scholar]
- 108.Plattner F et al. Toxoplasma profilin is essential for host cell invasion and TLR11-dependent induction of an interleukin-12 response. Cell Host Microbe 3, 77–87 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 Oligos and plasmids used in this study.
Supplementary Table 2 Combined results from the HiT screens summarizing data from arrayed and pooled analyses.
Source Data Fig. 4 b, Competition assays of delayed loss candidates. Provided are relative abundances of each strain relative to the WT competitor strain. Values are normalized to the starting ratio. d, Area sizes of individual plaques. Provided values are in mm2. e, Invasion efficiencies of delayed loss candidates. Invaded parasites per nuclei for each replicate are provided, in addition to the values post-normalization to the WT vehicle-treated sample.
Source Data Fig. 5 a, Kinase domain sequences used to generate alignments and the subsequent phylogenetic tree. e–f, Egress efficiencies following either (e) zaprinast or (f) A23187 stimulation. Provided is percent of egress relative to the final percentage of egress of the vehicle-treated sample. h, Quantification of GCaMP6f fluorescence signal following either zaprinast or A23187 treatment. Average fluorescence of each vacuole was quantified relative to initial fluorescence until egress of the vacuole or until the end of the time-course. i, Quantification of GCaMP6f fluorescence from extracellular parasites treated with either zaprinast or A23187. Provided are background-subtracted values normalized to initial fluorescence and the final maximum fluorescence following aerolysin and Ca2+ treatment.
Source Data Extended Data Fig. 5 a, Replication assays of SPARK grown in the presence or absence of IAA. 100 vacuoles were quantified for each sample and condition. Provided are the number of occurrences of each vacuole size for each replicate. b, Quantification of GCaMP6f fluorescence from extracellular parasites treated with either vehicle (DMSO) or ionomycin. Provided are background-subtracted values normalized to initial fluorescence and the final maximum fluorescence following aerolysin and Ca2+ treatment.
Data Availability Statement
All oligos used in this study are available in Supplementary Table 1. All plasmids used or generated in this study are listed with their appropriate GenBank or PMID accession numbers in Supplementary Table 1. Minimally processed pooled and arrayed CRISPR screen sequencing results are available in Supplementary Table 2. Localization assignments, microscopy phenotypes, lytic assay results, and UMAP coordinates and clusters are likewise available in Supplementary Table 2. Data from experimental results is available in the source data files. Additional unprocessed data is available from the corresponding author upon request.