

Abstract
Ischemic stroke is a leading cause of death and disability and remains without effective treatment options. Improved treatment of stroke requires efficient delivery of multimodal therapy to ischemic brain tissue with high specificity. Here, this article reports the development of multifunctional polymeric nanoparticles (NPs) for both stroke treatment and drug delivery. The NPs are synthesized using an reactive oxygen species (ROS)-reactive poly (2,2′-thiodiethylene 3,3′-thiodipropionate) (PTT) polymer and engineered for brain penetration through both thrombin-triggered shrinkability and AMD3100-mediated targeted delivery. It is found that the resulting AMD3100-conjugated, shrinkable PTT NPs, or ASPTT NPs, efficiently accumulate in the ischemic brain tissue after intravenous administration and function as antioxidant agents for effective stroke treatment. This work shows ASPTT NPs are capable of efficient encapsulation and delivery of glyburide to achieve anti-edema and antioxidant combination therapy, resulting in therapeutic benefits significantly greater than those by either the NPs or glyburide alone. Due to their high efficiency in brain penetration and excellent antioxidant bioactivity, ASPTT NPs have the potential to be utilized to deliver various therapeutic agents to the brain for effective stroke treatment.
Keywords: anti-edema, antioxidants, blood–brain barrier, shrinkable nanoparticles, stroke
1. Introduction
Stroke is a major cause of death and disability worldwide.[1] Intravenous administration of tissue-type plasminogen activator (tPA) within 4.5 h of symptom onset is the only currently available therapeutic for clinical management of this disease.[1,2] Unfortunately, due to such a narrow therapeutic window, over 90% of patients are ineligible for this treatment.[2] The major hurdle that complicates the development of effective pharmacological treatment for stroke is the blood-brain barrier (BBB). As a biological barrier that protects the brain from toxins and infections, the BBB limits the penetration of most therapeutic drugs to the brain.[3] Although the BBB is compromised after ischemic insult,[4,5] the degree of leakage cannot meet the requirement for delivery of a pharmacologically significant amount of therapeutics to the brain for effective stroke treatment. Therefore, improved treatment of stroke requires the development of novel approaches to enhance drug delivery to the ischemic brain.
To enhance drug delivery to the ischemic brain, nanotechnology approaches are promising, as nanoparticles (NPs) can be potentially engineered through surface conjugation of ligands that interact with receptors highly expressed in the BBB or enriched in the ischemic microenvironment.[6–8] However, accumulating evidence suggests that limited by the availability and activity of receptor molecules, the ligand-mediated approach alone may not provide sufficient efficiency for stroke treatment.[9–11] To overcome this limitation, we recently developed a stimuli-responsive approach, through which NPs were engineered to respond to the ischemic microenvironment by shrinking in size. We demonstrated that, although the stimuli-responsiveness may not provide greater efficiency than the ligand-mediated approach, it significantly enhances brain penetration and further augment the efficiency of ligand-mediated targeting.[12]
In this study, we developed multifunctional NPs consisting of reactive oxygen species (ROS) consumable poly(2,2′-thiodiethylene 3,3′-thiodipropionate) (PTT) polymer for both stroke treatment and drug delivery. Unlike polymeric NPs consisting of conventional polymers, such as poly(lactic-co-glycolic acid), which function as a carrier without biological activities,[13,14] the multifunctional polymeric NPs have dual functions as both a carrier and as an antioxidant agent. The NPs were further functionalized for brain penetration through both stimuli-responsiveness and ligand-receptor interaction. We found that the resulting NPs efficiently penetrated the brain through the disrupted BBB and effectively promote stroke recovery in mouse models. We showed that the NPs could encapsulate and deliver glyburide, an anti-edema agent which demonstrated promise in human patients but suffers from limited brain penetration,[15,16] and that the encapsulation of glyburide provided additional therapeutic benefits.
2. Results
ROS dramatically increases after ischemic insult and represents a major cause of post-stroke cell death in the neurovascular unit.[17,18] To eliminate locoregional ROS, we synthesized a group of poly(ethylene glycol) (PEG)-terminated PTT (PEG-PTT) polymers, which contain ROS-reactive thioether groups. To enable stimuli-responsiveness, the polymers were conjugated with a second PEG through a thrombin-cleavable peptide, NH2-norleucine-TPRSFL-C-SH.[19] When utilized for NP construction, the resulting NPs respond to thrombin, a protease highly enriched in the ischemic microenvironment, by shrinking in size. To further enhance the brain penetrability, the NPs were surface conjugated with AMD3100, which recognizes CXCR4, a chemokine receptor preferentially expressed in ischemic brain tissue.[12] The resulting AMD3100-conjugated, shrinkable PTT NPs, terminated as ASPTT NPs, were evaluated in stroke-bearing mice induced by middle cerebral artery occlusion (MCAO) as both a therapeutic agent and as a carrier for drug delivery to stroked regions by using glyburide as a model therapeutic (Figure 1).
Figure 1.
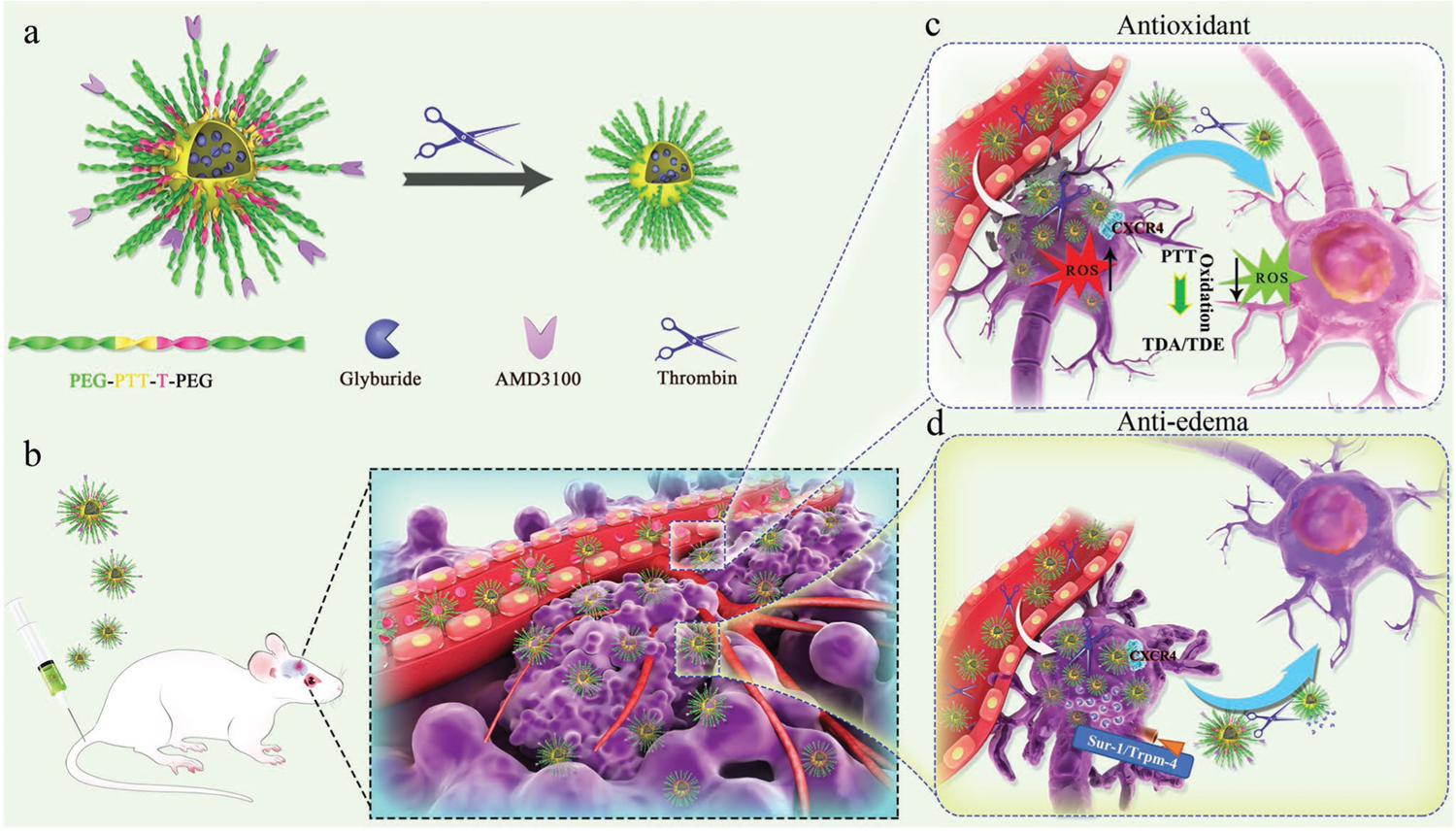
Schematic diagrams of ASPTT NP design and combination therapy for stroke treatment. a) Construction of ASPTT NPs through self-assembly of PEG-PTT-T-PEG block copolymer and size shrink behavior of ASPTT NPs in response to thrombin. b) Brain targeting and BBB penetration of ASPTT NPs in stroke-bearing mice. c) Antioxidant therapy of ASPTT NPs in ischemic brain by reacting with excess ROS. d) Anti-edema of ASPTT NPs through delivering glyburide.
2.1. Synthesis and Characterization of Thrombin-Responsive Polymers
We recently developed enzyme-catalyzed polymerization chemistry for metal-free synthesis of multifunctional biopolymers.[20–22] By taking advantage of the extraordinary tolerance of lipase toward organic functional groups, PEG-PTT block copolymers bearing thioether functionalities in the main chain were synthesized through direct copolymerization of 3,3′-thiodipropionic acid (TDA), 2,2′-thiodiethanol (TDE), with MeOPEG2K-OH (Figure 2a). An excessive amount of TDA versus (TDE + PEG) was used to control polymer molecular weight and to ensure that polymers were terminated with carboxyl groups. Calculated amounts of MeO-PEG2K-OH were added to allow the formation of copolymers with ≈20 wt% PEG content. Through this reaction, three PEG-PTT copolymers, termed PEG-PTT-1, PEG-PTT-2, and PEG-PTT-3, were obtained in a yield of 82–85% with a molecular weight of 18 300, 12 900, and 9200 Da, respectively (Figure 2b).
Figure 2.
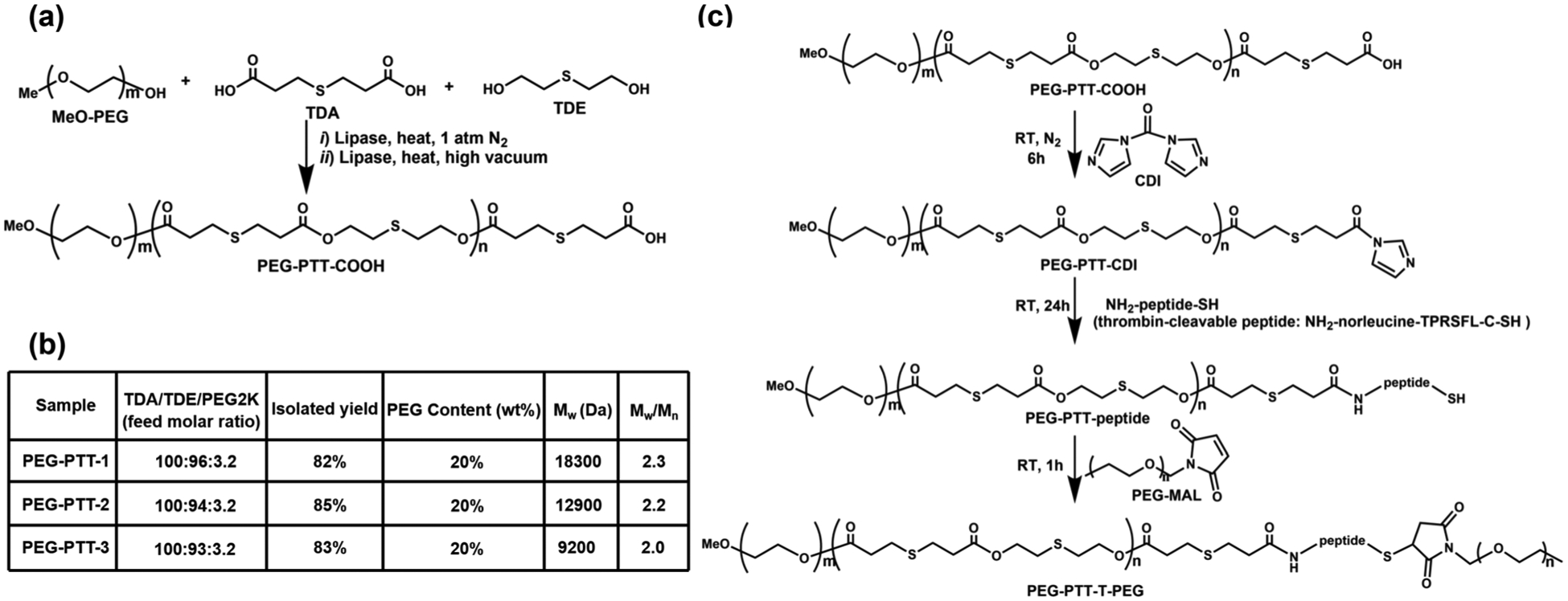
Synthesis and characterization of polymers. a) Enzyme-catalyzed synthesis of carboxyl-terminated PEG-PTT block copolymer. b) Characterization of PEG-PTT block copolymers. c) Chemical synthesis of PEG-PTT-peptide and PEG-PTT-T-PEG.
We confirmed the structures of PEG-PTT copolymers using 1H and 13C NMR spectroscopy. The structural assignments for the proton and carbon-13 resonances of the polymers are illustrated in Figure S1, Supporting Information. To enable thrombin-responsiveness, we further conjugated a second PEG to the end of polymers via a thrombin-cleavable linker peptide in the sequence of NH2-norleucine-TPRSFL-C-SH (Figure 2c). Briefly, a given carboxy-terminated PEG-PTT polymer was activated by N, N′-carbonyl diimidazole (CDI) and reacted with the peptide, resulting in cysteine-terminated PEG-PTT-peptide. The product was then mixed with maleimide-terminated PEG2k. One hour later, the resultant product was purified, and a block copolymer bearing thrombin-cleavable peptide, designated as PEG-PTT-T-PEG, was obtained. Control nonresponsive polymers were synthesized through the reaction of CDI activated PEG-PTT with amine-terminated PEG2k directly without the peptide.
2.2. Construction and Characterization of PEG-PTT-T-PEG NPs for Drug Delivery to Stroke
PEG-PTT-T-PEG polymers were designed to form NPs that respond to thrombin by shrinking in size. This is because NPs consisting of PEG-PTT-T-PEG triblock copolymers include two layers of PEG. After exposure to thrombin, those PEG molecules conjugated via the thrombin-responsive peptide are cleaved and removed. As a result, the balance of hydrophobicity and hydrophilicity is shifted, leading to the formation of a new micellular structure. Compared to the original NPs, the newly formed NPs contain a higher percentage of hydrophobic components, which shrink the NP size. To validate the design, we synthesized PEG-PTT-T-PEG NPs through the standard nanoprecipitation method. The shrinkability of NPs was tested by incubating NPs in PBS containing 100 nm thrombin at 37 °C. Change of size and morphology of NPs with time were monitored by dynamic light scattering (DLS) and transmission electron microscopy (TEM). DLS analysis found that the size of all three PEG-PTT-T-PEG NPs started to change 10 min after incubation with thrombin. After 24 h, the hydrodynamic size shrank from 128 to 87 nm, 112 to 75 nm, and 98 to 66 nm for NPs consisting of PEG-PTT1-T-PEG, PEG-PTT2-T-PEG, and PEG-PTT3-T-PEG, respectively. Analysis by TEM confirmed the changes in size, while the morphology of NPs remained spherical (Figure 3a–c). The observed size reduction is not because the polymers or the NPs are unstable. Analysis by gel permeation chromatography (GPC) found that the molecular weight of PEG-PTT-T-PEG did not significantly change over the 24 h incubation time (Figure S2a,b, Supporting Information). Analysis by DLS showed that PEG-PTT-T-PEG NPs maintained similar sizes over 10 days in PBS without thrombin or with prothrombin (Figure S2c,d, Supporting Information). In contrast, the same treatment did not alter the size of control nonresponsive NPs, which were synthesized using PEG-PTT-PEG or PEG-PTT (Figures S3 and S4, Supporting Information).
Figure 3.
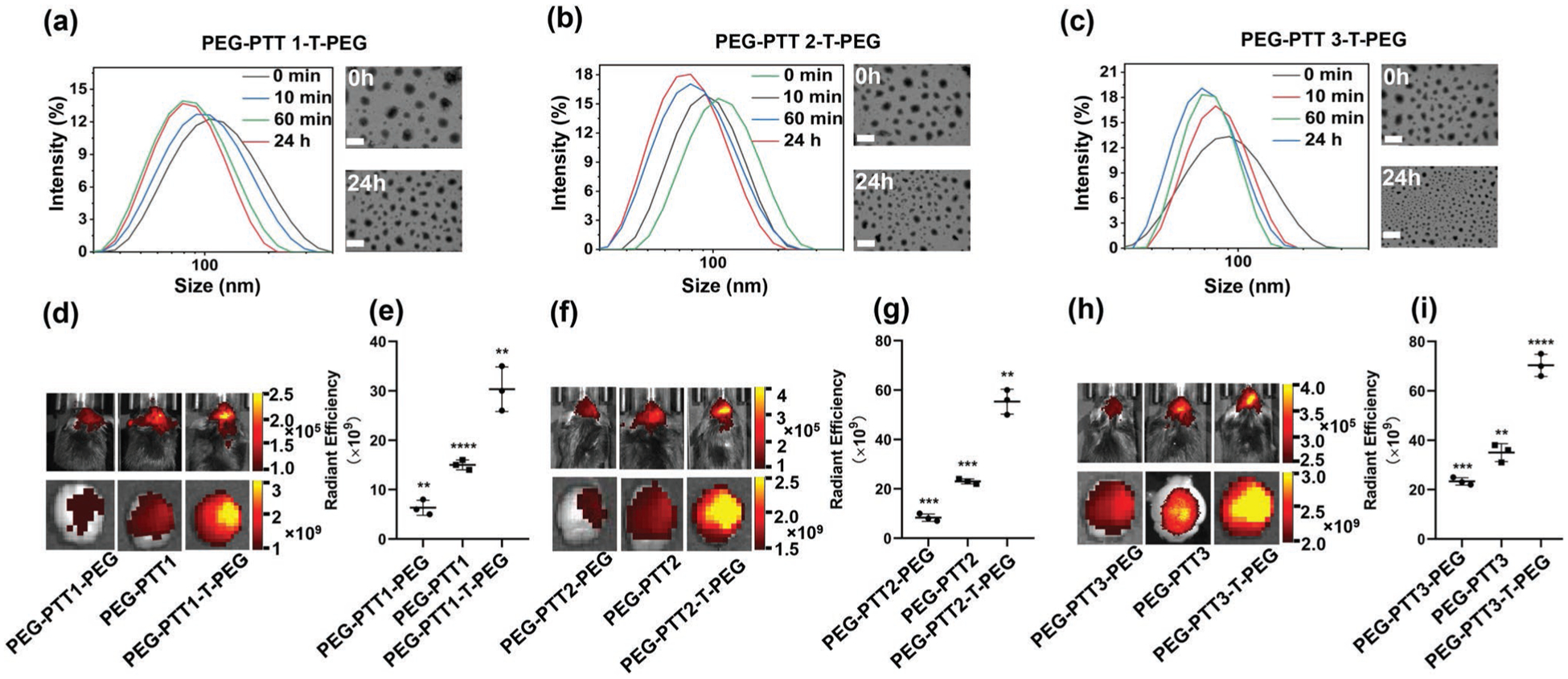
Characterization and evaluation of thrombin responsive PEG-PTT-T-PEG NPs. a–c) DLS (left) and TEM (right) analyses of PEG-PTT-T-PEG NPs in PBS with thrombin (100 nm). Scale bar: 100 nm. d–i) Representative images (d,f,h) and semi-quantification (e,g,i) of NPs in stroke mice receiving the indicated treatments.
We evaluated if the thrombin-responsiveness enhances the delivery of PEG-PTT-T-PEG NPs to the ischemic brain. NPs were synthesized with the encapsulation of IR780, a nearinfrared fluorescence dye allowing for non-invasive imaging, and administered to mice after MCAO surgery through tail vein injection. Twenty-four hours later, the mice were imaged using an in vivo imaging system (IVIS). We found that compared to control nonresponsive PEG-PTT-PEG or PEG-PTT NPs, all three PEG-PTT-T-PEG NPs exhibited significantly greater efficiency in brain penetration. The average fluorescence intensity in the ischemic area treated with PEG-PTT1-T-PEG shrinkable NPs was 2.1-fold and 5-fold greater than those of PEG-PTT1 and PEG-PTT1-PEG NPs, respectively (Figure 3d,e). PEG-PTT2-T-PEG and PEG-PTT3-T-PEG NPs demonstrated a similar trend of enhancement (Figure 3f–i). The observation that PEG-PTT-T-PEG NPs have greater efficiency than PEG-PTT NPs can be potentially explained by two reasons. First, although detailed mechanisms are yet to be determined, shrinking size in response to disease microenvironment has shown to improve NP penetration in various disease models.[12,23,24] To confirm the critical role of shrinkability in this specific study, we compared the penetration of PEG-PTT-T-PEG NPs with and without co-administration of heparin, a thrombin inhibitor, that inhibits thrombin activity and thus prevents the NPs from shrinking. We found that PEG-PTT-T-PEG NPs co-administrating with heparin, a thrombin inhibitor, and PEG-PTT3 NP had comparable brain penetration, which was significantly less than that for PEG-PTT-T-PEG NPs (Figure S5a–c, Supporting Information). Second, we found that, compared to PEG-PTT3 NPs, PEG-PTT3-T-PEG NPs have a longer blood circulation time (Figure S5d, Supporting Information). This is likely because the density of PEG on the surface of PEG-PTT3-T-PEG NPs is two times of that on the surface of PEG-PTT3 NPs. The prolonged blood circulation increases the chance of PEG-PTT3-T-PEG NPs penetrating ischemic brain tissue. Due to their high efficiency in brain penetration, PEG-PTT3-T-PEG NPs were selected for the remainder of the study.
2.3. Characterization of PEG-PTT-T-PEG NPs for Stroke Treatment
Ischemic insults dramatically increase the production of ROS, which overwhelms the endogenous antioxidant systems.[17] Without prompt elimination, the excess ROS induces significant toxicity to cells in the neurovascular unit, resulting in cellular apoptosis, BBB disruption, and an inflammatory response.[18] PEG-PTT-T-PEG polymers contain thioether groups, which react with ROS,[25] and likely function as antioxidants to improve stroke recovery after delivery to the ischemic region. To test the hypothesis, we first determined the antioxidant effect of PEG-PTT-T-PEG NPs by reacting the NPs with H2O2 and quantifying the remaining H2O2 using 2′,7′-dichlorofluorescein diacetate (DCFDA). We found that all three PEG-PTT-T-PEG NPs reduced H2O2 in a dose-dependent manner and consumed up to 90% of H2O2 at the concentration of 1 μg μL−1 (Figure 4a). Next, we evaluated PEG-PTT-T-PEG NPs for stroke treatment in mice. Stroke-bearing mice were established and treated with PEG-PTT-T-PEG NPs through intravenous administration at 0, 24, and 48 h after surgery. On day 4, the mice were euthanized. The brains were isolated and subjected to 2,3,5-triphenyltetrazolium chloride staining. We found that, compared to that in control mice, the average infarct volume in mice receiving treatment with PEG-PTT1-T-PEG NPs, PEG-PTT2-T-PEG NPs, and PEG-PTT3-T-PEG NPs was reduced by 19%, 26%, and 30%, respectively (Figure 4b,d). Consistently, Western Blot analysis showed that the expression levels of Nrf2, a key mediator of the antioxidant signaling, and heme oxygenase-1 (HO-1), a major antioxidant enzyme, were significantly up-regulated by PEG-PTT-T-PEG NP treatments (Figure 4c). These results suggest that systemic treatment with PEG-PTT-T-PEG NPs promoted stroke recovery through regulation of the antioxidant pathway. Among the three NPs, PEG-PTT3-T-PEG NPs demonstrated the greatest infarct reduction effect, which can be attributed to its highest efficiency in brain penetration (Figure 4e,f), and, therefore, was chosen for further characterization. We found that consistent with that for brain penetration, thrombin-mediated shrinkability is important for the efficacy of PEG-PTT-T-PEG NPs, as inhibition of thrombin activity through pre-treatment with heparin significantly reduced the efficacy of the NPs (Figure S6, Supporting Information).
Figure 4.
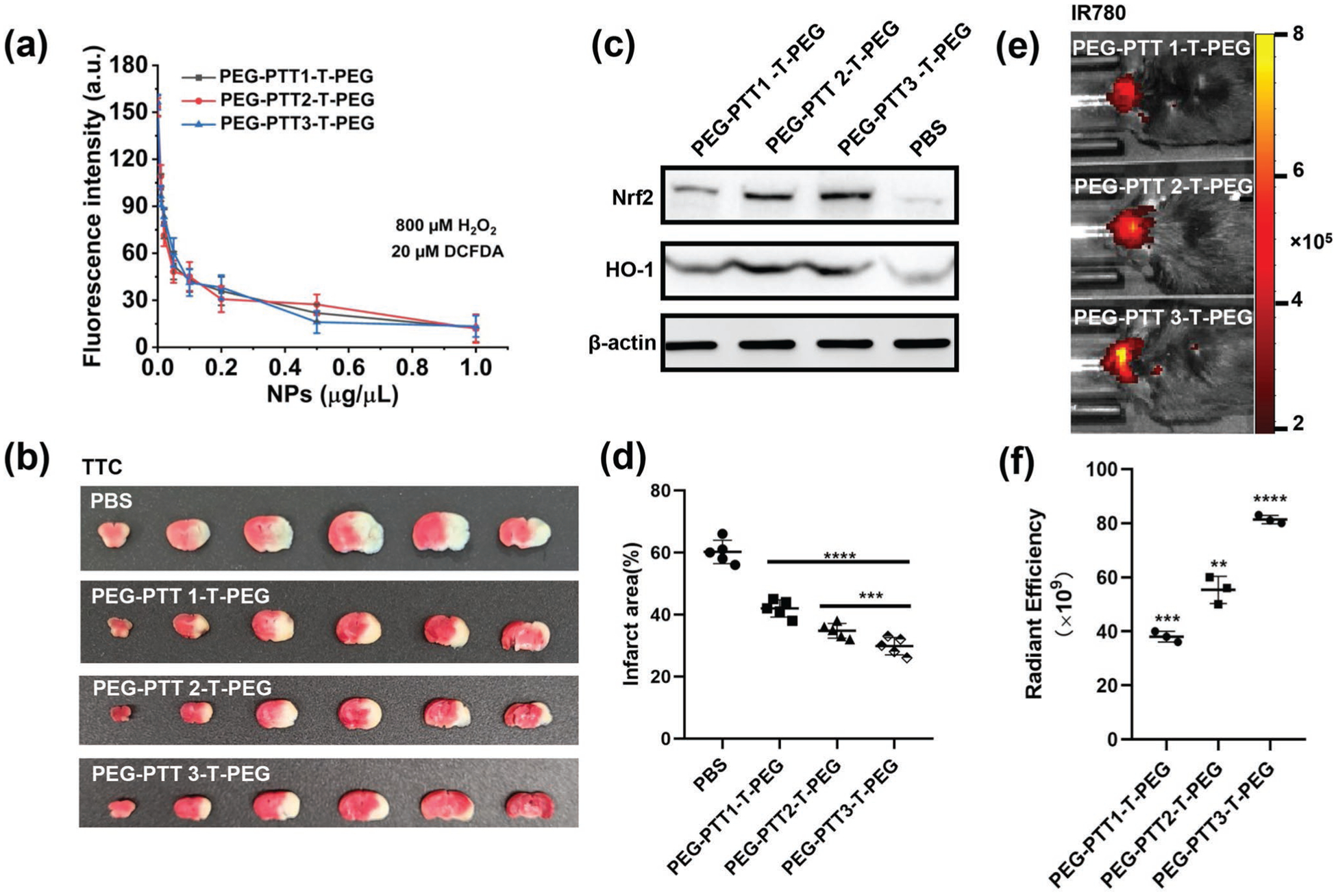
Characterization of PEG-PTT-T-PEG NPs for stroke treatment. a) Characterization of the ROS scavenging effect of PEG-PTT-T-PEG NPs by DCFDA assay. Representative images (b) and quantification (d) of cerebral infarction in MCAO mice receiving treatment of the indicated PEG-PTT-T-PEG NPs. c) Western blot analysis of ischemic brain tissues isolated from mice with and without PEG-PTT-T-PEG NPs treatment. Representative images (e) and semi-quantification (f) of NPs in the brains of MCAO mice received with the indicated treatment.
2.4. Engineering PEG-PTT-T-PEG NPs for Targeted Delivery to the Ischemic Brain
PEG-PTT3-T-PEG NPs penetrated the ischemic brain through local BBB disruption and thrombin-mediated shrinkability (Figures 3 and 4). To further improve its efficiency in brain penetration, we focused on CXCR4, a chemokine receptor that is significantly highly expressed in ischemic brain tissue after stroke (Figure 5a). To enable targeting CXCR4, we synthesized maleimide-terminated PEG-PTT3-T-PEG or PEG-PTT3-T-PEG-Mal. The synthesis was carried out through the same procedures that were developed for PEG-PTT3-T-PEG synthesis, except the use of MAL-PEG-OH instead of MeO-PEG-OH. Next, PEG-PTT3-TPEG-Mal NPs were synthesized and conjugated with AMD3100 on the surface via the maleimide groups through our previously published procedures.[12,26] The resulting AMD3100-conjugated, shrinkable PTT NPs, or ASPTT NPs, were evaluated for brain penetration in stroke-bearing mice. ASPTT NPs were synthesized with encapsulation of IR780 and intravenously administered to mice after MCAO surgery. Control mice were treated with PEG-PTT3-T-PEG NPs without AMD3100. The amounts of NPs were normalized to ensure each mouse received the same amount of IR780 fluorescence. Twenty-four hours later, the mice were imaged by IVIS, after which the mice were euthanized. Major organs, including the brain, heart, liver, spleen, lung, and kidney, were isolated and imaged ex vivo. We found that conjugation of AMD3100 significantly improved the accumulation of NPs in the ischemic region by 2.1-fold (Figure 5b,d). In particular, the amount of ASPTT NPs in the brain is greater than that in the liver (Figure S7, Supporting Information). We confirmed the uptake of NPs in the ischemic brain region after intravenous administration. The experiment was performed by the same procedures except NPs loaded with CY5.5. The brains were isolated and sliced 24 h after NPs administration. Fluorescence imaging identified a significant number of NPs in the ischemic brain region. The intensity of red fluorescence represented NPs in the ASPTT group is much higher than that of the PEG-PTT3-T-PEG group (Figure 5c). Due to their high efficiency in penetrating the ischemic brain, ASPTT NPs were selected for further study.
Figure 5.
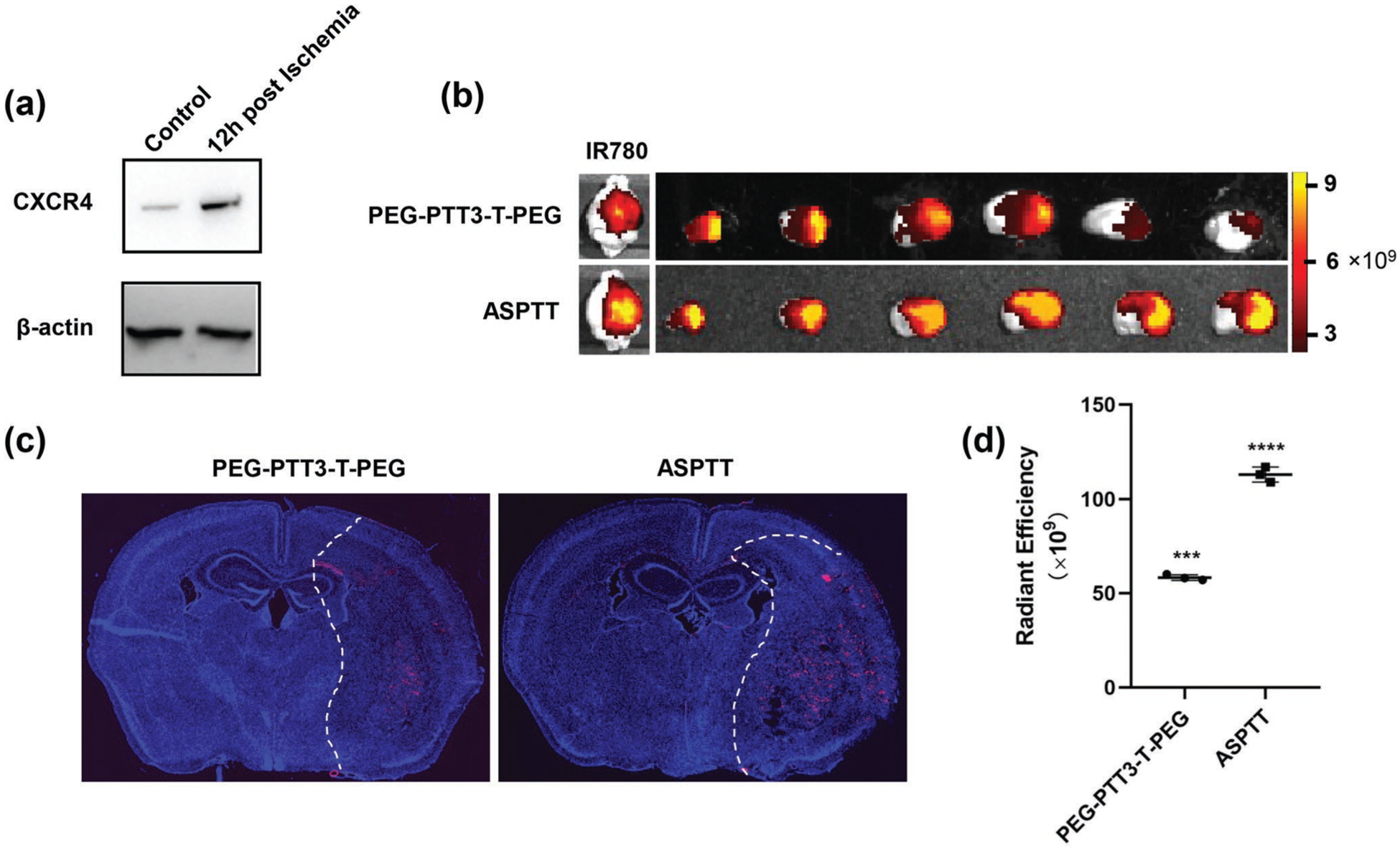
Enhancing delivery of PEG-PTT-T-PEG NPs to the ischemic brain through CXCR4 targeting. a) Western Blot analysis of the expression of CXCR4 in the ischemic and normal brain. b) Representative images of brain and brain slices and d) semi-quantification of NPs in the brains from mice receiving the indicated treatment. c) Representative fluorescence images of CY5.5-loaded NPs (red) in the stroke regions (right side of dotted line).
2.5. Targeted Delivery of Antioxidant and Anti-Edema Combination Therapy via Glyburide-Loaded ASPTT NPs for Stroke Treatment
Edema represents one of the major causes of stroke-associated death and disability.[27] In a recent clinical test,[15] we showed that infusion of glyburide improved patient survival through reduction of cerebral edema. Despite the promise, we found that the efficacy of glyburide is limited by its low efficiency in brain penetration and full capitalization of glyburide as a stroke therapeutic requires enhancing delivery to the brain.[12,16] To test if ASPTT NPs could be employed to deliver glyburide to achieve antioxidant and anti-edema combination therapy, we synthesized glyburide-loaded ASPTT NPs, which encapsulated glyburide at an efficiency of 63.5%. As expected, ASPTT NPs released encapsulated glyburide at an accelerated rate in the presence of both thrombin and ROS. Twenty-four hours after incubation in PBS with thrombin and ROS, 91.6% of glyburide was released; in contrast, only 23.6% was released in PBS without thrombin and ROS at the same condition (Figure 6a). As glyburide functions as an anti-edema therapeutic targeting the acute phase, the release of glyburide at a fast rate is beneficial for stroke treatment. We characterized glyburide-loaded ASPTT NPs for stroke treatment by treating stroke-bearing mice at 0, 24, and 48 h after MCAO surgery. NPs were given at a dose equivalent to 5 μg kg−1 of glyburide per injection, which was found to be safe for mouse treatment without induction of hypoglycemia.[12,16] Control mice were treated with PBS, free glyburide, PEG-PTT3-T-PEG NPs, or ASPTT NPs without glyburide. Mice were monitored for survival and behavior and were euthanized on day 8. We found that, compared to control mice receiving PBS, treatment with glyburide-loaded ASPTT NPs markedly reduced infarct volumes by 41% (Figure 6b,c; Figure S9, Supporting Information), improved mouse neurological scores (Figure 6d) and survival (Figure 6e, p < 0.01). In contrast, treatment with free glyburide, PEG-PTT3-T-PEG NPs or ASPTT NPs without glyburide, while improving stroke recovery, had significantly less efficacy. H&E staining did not identify detectable myocardial toxicity nor tissue damage in major organs isolated from the treated mice (Figure S8, Supporting Information), suggesting that glyburide-loaded ASPTT NPs are safe for intravenous administration.
Figure 6.
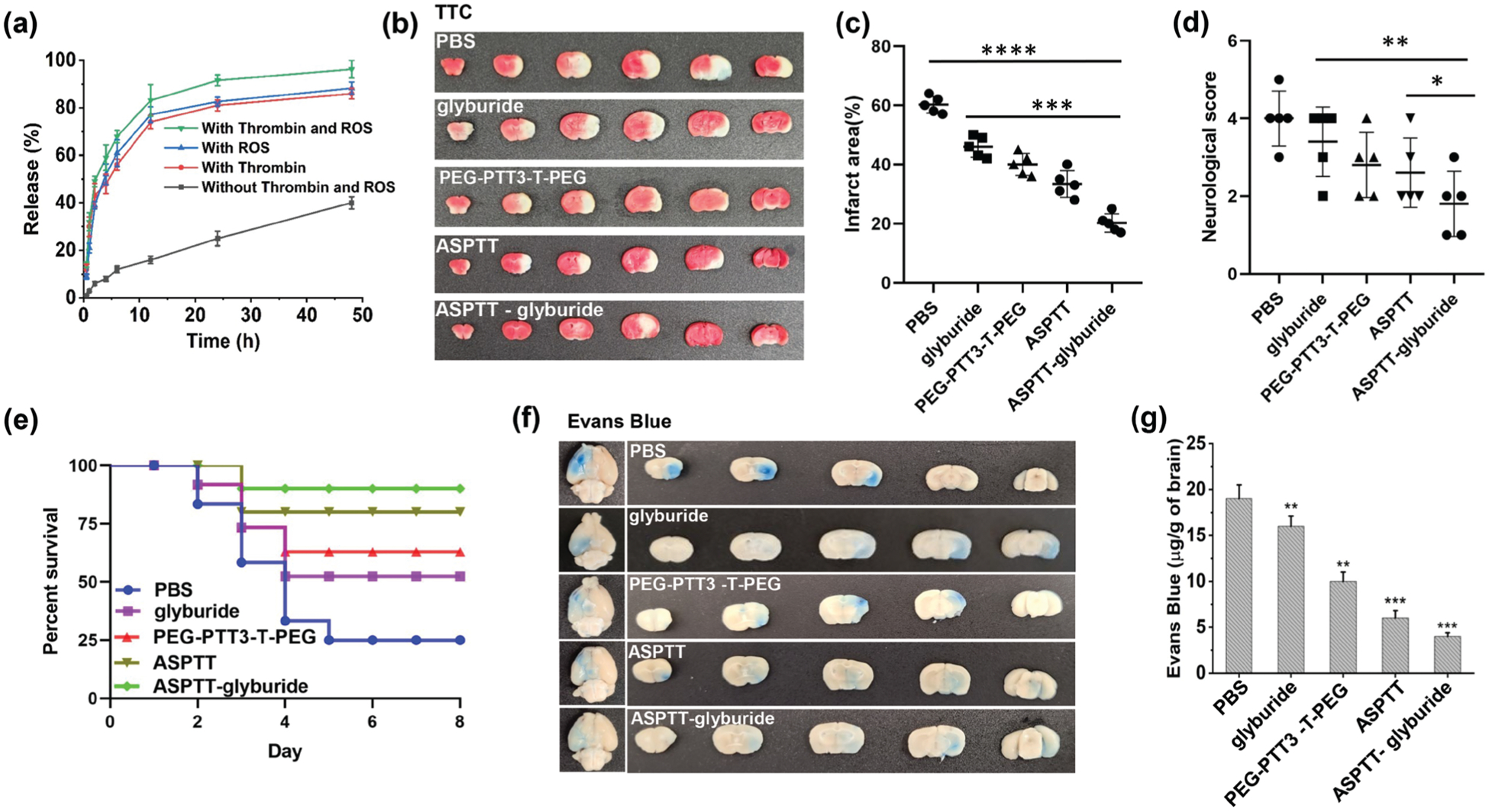
Characterization of glyburide-loaded ASPTT NPs for stroke treatment. a) Release of glyburide in PBS at 37 °C with/without thrombin (100 nm) and ROS (10 μm). b) Representative images and c) quantification of cerebral infarction in MCAO mice receiving the indicated treatments. d) Neurological scores (day 3 after surgery, n = 5) and e) Kaplan–Meier survival analysis (n = 10) of MCAO mice receiving the indicated treatments. Representative images (f) and quantification (g) of Evans blue extravasation in MCAO mice received the indicated treatments.
We studied the mechanism accounting for the significant therapeutic effects of glyburide-loaded ASPTT NP treatment. Results showed that, in addition to the observed antioxidant effect (Figure 4), the treatment effectively reduced brain edema, repaired the compromised BBB, and promoted the growth of new vessels. Measurement of the weight of the brain found that the treatment reduced brain tissue water content by 11% (Figure S10, Supporting Information). Analysis by Western Blot and immunostaining showed that the treatment significantly improved the expression of tight junction protein ZO-1 in the BBB (Figures S11 and S12, Supporting Information). Consistently, Evans-blue leakage assay found that the treatment markedly reduced BBB leakage by 15% (Figure 6f,g). A separate cohort of mice were euthanized 15 days after treatment. The brains were harvested and subjected to CD31 immunohistochemistry (IHC) and immunofluorescence (IF) analysis. IHC staining and immunostaining showed that the amount of new blood vessels in the brain of mice treated with glyburide-loaded ASPTT NPs were significantly more than those in control groups (Figure S13, Supporting Information).
3. Discussion
Stroke is a major public health problem and remains without effective pharmacological therapies. Improved treatment of stroke requires the development of multimodal therapy, which should also be efficiently delivered to the ischemic brain. To achieve this goal, we developed glyburide-loaded ASPTT NPs, which enable efficient delivery of antioxidant and anti-edema combination therapy to the ischemic brain for effective stroke treatment. ASPTT NPs are unique in that, unlike most traditional polymeric or lipid NPs, which do not have biological activities, ASPTT NPs function as both an antioxidant therapeutic and a brain-penetrating drug carrier. ASPTT NPs mainly consisted of PTT polymers, which consume ROS through reaction with thioether groups in the polymer chain. The ROS consumption effect of ASPTT NPs was confirmed by incubating the NPs with H2O2 (Figure S14, Supporting Information). ASPTT NPs were further engineered through two distinct but complementary mechanisms for brain penetration, including shrinkability in response to thrombin and surface conjugation of AMD3100 to target CXCR4. Both thrombin and CXCR4 are preferentially expressed and accumulated in the ischemic but not normal brain (Figure 5a; Figure S15a, Supporting Information). Although the concentration of thrombin in the blood was elevated and peaked at 4.2 nm 12 h after ischemic insult (Figure S15b, Supporting Information), it remains significantly below that needed for cleaving the thrombin-responsive peptide in PEG-PTT-T-PEG NPs (Figure 1a–c). The stability of NPs at 4.2 nm was confirmed by DLS analysis (Figure S15c, Supporting Information). Therefore, it is likely that the PEG-PTTT-PEG NPs maintained intact in structure in blood circulation, reached the ischemic brain through the interaction of AMD3100 with CXCR4, and responded to thrombin by shrinking size, leading to enhanced brain penetration. The interaction between AMD3100 with CXCR4 is critical for brain targeting, as pre-treatment with AMD3100 significantly decreased the accumulation of PEG-PTT-T-PEG NPs in the ischemic brain (Figure S16, Supporting Information). We showed that, as a result of the composition and dual-targeting mechanisms, ASPTT NPs were able to efficiently penetrate the already disrupted BBB, accumulate in the ischemic brain tissue after intravenous administration, and effectively promote stroke recovery through regulation of the antioxidant pathway (Figure 4c).
To further enable combination therapy, we employed ASPTT NPs for targeted delivery of glyburide. In a recently completed GAMES-RP clinical trial, we demonstrated that intravenous infusion of glyburide significantly prolonged the survival of stroke patients.[28] However, we also found that the trial did not meet its primary endpoint, as the treatment failed to significantly prevent decompressive craniectomy or increase favorable clinical outcomes. Further analysis shows that the unmet efficacy could be largely attributed to the fact that glyburide has a limited ability to penetrate the brain.[16,29,30] We showed that delivery of glyburide via ASPTT NPs effectively enhanced mouse survival, reduced infarct area, and improved neurological functions; in contrast, delivery of a dose-equivalent amount of ASPTT NPs without glyburide or free glyburide had significantly less efficacy (Figure 6).
4. Conclusion
In summary, we developed a unique antioxidant ASPTT NP as a carrier for efficient drug delivery to the ischemic brain. We demonstrated that delivery of glyburide via ASPTT NPs allows achieving an anti-edema and antioxidant combination therapy in a simple formulation. Due to their high efficiency in brain penetration and great therapeutic benefits, we anticipate the potential to employ ASPTT NPs as a platform for targeted delivery of anti-stroke therapeutic agents and the potential to translate glyburide-loaded ASPTT NPs as a therapeutic for effective treatment of stroke.
5. Experimental Section
PEG-PTT-COOH Synthesis:
Various reaction mixtures were prepared by adding TDA, TDE, MeO-PEG2K-OH, and Novozym 435 into diphenyl ether solvent. The amounts of the enzyme catalyst and solvent were respectively 10 and 200 wt% versus total substrate. During the polymerization reactions, the reaction mixtures were stirred at 90 °C initially under 1 atmospheric pressure of nitrogen gas for 20 h and subsequently under 1.6 mmHg vacuum for 70 h. The formed polymer products were isolated by adding n-hexane to precipitate the copolymers. After washing twice with n-hexane solvent, the polymers were dissolved in chloroform and were filtered to remove the catalyst particles. The filtrates were concentrated under vacuum and then dropwise added to stirring n-hexane. The re-precipitated copolymers were finally dried at 30 °C overnight under a high vacuum (< 1.0 mmHg). The polymer molecular weights were measured by GPC, calibrated by narrow polydispersity polystyrene standards.
Synthesis of PEG-PTT-T-PEG:
First, PEG-PTT-COOH was activated by CDI. PEG-PTT-COOH (0.01 mmol) and CDI (0.1 mmol) were dissolved in anhydrous DCM under the protection of N2 atmosphere and reacted at room temperature by magnetic stirring overnight. The unreacted CDI was removed by adding excess cold ethanol. PEG-PTT-CDI was dried under vacuum to obtain the product. Next, NH2-norleucine-TPRSFL-C-SH (0.004 mmol) and PEG-PTT-CDI (0.002 mmol) were dissolved in anhydrous DMSO and stirred overnight. Finally, MPEG-Mal/Mal-PEG-Mal (0.005 mmol) was added to the above reaction system for 1 h.
Construction of NPs:
NPs were fabricated via the nanoprecipitation method. To prepare the PEG-PTT-T-PEG NPs, the polymer was added dropwise into deionized water (DMSO/water = 1:10) under magnetic stirring. After 30 min of balance process, the solution was transferred to a dialysis bag (100 000 MWCO) and dialyzed against deionized water. The NPs were formed with the removal of DMSO and unreacted PEG and peptide. AMD3100 was conjugated on the surface of NP via maleimide group according to previously reported method.[31] The IR780/Glyburide-loaded NPs were prepared similar to the empty NP fabrication method, except polymer and IR780/Glyburide were co-dissolved in an organic solvent. IR780 (IR780/polymer = 2%) w/w was loaded as an in vivo imaging dye. Glyburide (glyburide/polymer = 0.2% – 1%) w/w was loaded as an anti-edema agent.
Characterization of Thrombin-Responsiveness:
DLS (Malvern Zetasizer 90) and TEM (JEOL 1230) were used to analyze the thrombin-triggered size change of the NPs. 1 mg mL−1 PEG-PTT-T-PEG NPs and PEGPTT-PEG NPs without thrombin sensitive peptide were incubated with 100 nm thrombin at 37 °C separately. 100 μL of NP solution was taken at different time points (0 min, 10 min, 60 min, 24 h) and diluted to 1 mL with ddH2O water. DLS measures the change in NP size over time. Changes in the size and morphology of NPs were also observed by TEM after 2% phosphotungstic acid negatively staining.
Characterization of ROS Scavenging In Vitro by DCFDA Assay:
DCFDA)-based ROS assay kit was used to test the reaction between H2O2 and PEG-PTT-T-PEG NPs. 20 μm freshly prepared DCFDA and 800 μm H2O2 was added to 300 μL of NPs solution with different contents (0, 5, 10, 20, 30, 60, 150, 300 μg) and incubated for 30 min at 37 °C. The fluorescence signal of oxidation product DCF was detected at 535 nm by the multimode microplate reader (Spectramax M5, Molecular devices, USA) using a 488 nm excitation laser.
Middle Cerebral Artery Occlusion:
Male C57BL/6 mice (Charles River Laboratories, USA), ≈20 g each, were given free access to food and water before the experiment. All animal experiments were approved by the Yale University Institutional Animal Care and Utilization Committee. MCAO models were established according to the methods previously reported.[32,33] First, mice were anesthetized through a tabletop anesthesia system (Harvard Apparatus) with 5% isoflurane (Aerrane, Baxter, Deerfield, IL) in 30% O2/70% N2. Next, Mice were placed in the supine position and maintained body temperature at 37.0 ± 0.5 °C using a heating pad. The isoflurane was then adjusted to 1.5% after induction, and the regional cerebral blood flow (rCBF) was monitored using a laser Doppler flowmeter (AD Instruments Inc.) during the whole surgery. The surgical site was disinfected with povidone-iodine solution and alcohol pads. The right external carotid was exposed by making a middle neck incision under a dissecting microscope (Leica A60). After that, the right common carotid artery, external carotid artery (ECA), and internal carotid artery (ICA) were carefully dissected from the surrounding tissue and ligated with silk thread. The ECA was cut, and a nylon blunted monofilament (Ducal Corporation) was inserted from ECA into the ICA at 8–10 mm beyond the bifurcation. The basic principle of occlusion is the reduction of rCBF by over 80%. After 1.5 h, reperfusion of blood flow was allowed by withdrawing the monofilament.
Evaluation of the Therapeutic Benefits:
Mice with successful MCAO surgeries were randomly divided into four groups (n = 10 for each group), which received treatment of PBS, PEG-PTT-T-PEG NPs, PEG-PTT-T-PEG NPs with AMD3100 targeting, and glyburide-loaded PEG-PTT-T-PEG NPs with AMD3100 targeting, respectively. Treatment injections were administrated intravenously at three time points, 0, 24, and 48 h after surgery. Mice were monitored for survival for 8 days or were euthanized if one of the following criteria was observed: 1) the mouse’s body weight dropped below 15% of its initial weight; 2) the mouse became lethargic or sick of if the mouse was unable to feed. The determination of anti-edema effect was carried out 24 h after surgery. The mice were sacrificed, and the brains were excised. Then, wet weight and lyophilized dry weight of the brain were obtained. Brain water content was calculated by the formula described as: brain tissue water (%) = (wet weight − dry weight)/wet weight × 100. The analysis on infarct area and neurological score (n = 5) was conducted 3 days after surgery. The following formula was used to calculate the infarct area: Corrected infarct area (%) = (contralateral hemisphere area − non-infarcted ipsilateral hemisphere)/contralateral hemisphere area × 100. The neurologic outcome of each mouse was evaluated by a standard behavioral test[34] and was scored: 1) normal motor function, 2) flexion of the torso and contralateral forelimb when the animal was lifted by the tail, 3) hemiparalysis resulting in circling to the contralateral side when held by the tail on a flat surface, but normal posture at rest, 4) leaning to the contralateral side at rest, and 5) no spontaneous motor activity. An unbiased approach was used to evaluate the therapeutic effect. The reviewer was unaware of the group number of mice that need to be scored.
Western Blot:
For characterization of CXCR4, brains were harvested 12 h after MCAO surgery, and the right hemispheres containing the ischemic area were excised. Normal brain without surgery was used as a control. To determine the BBB repair effect in vivo, mice with successful MCAO surgery were received different treatments. After 72 h, the brains were harvested, and the right hemispheres containing the ischemic area were excised. The brains from normal mice without surgery were used as controls. Western Blot was conducted according to the standard procedures as described in the previous report.[35] Antibodies used in this study included anti-CXCR4 (NB100–74396, Novus), Anti-ZO1 (ab216880, Abcam), Anti-Claudin 5 (34–1600, Invitrogen), Anti-Nrf2 (Novus Biologicals), Anti-HO-1 (Novus Biologicals), HRP anti-rabbit IgG (656 120, Invitrogen), HRP anti-mouse IgG (AP307P, Sigma-Aldrich), and beta-actin (c643802, BioLegend).
Statistical Analysis:
All data were collected in triplicate and reported as mean and standard deviation. Comparison of different groups was evaluated by the unpaired t-test. One-way ANOVA was used to analyze multiple comparisons by GraphPad Prism 8.0. p < 0.05 (*), 0.01 (**), 0.001 (***), and 0.0001 (****) were considered significant.
Supplementary Material
Acknowledgements
H.W. and B.P. contributed equally to this work. This work was supported by NIH grant NS095147 and AHA grants 15GRNT25290018 and 18TPA34170180.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Haoan Wu, Department of Neurosurgery, Yale University, New Haven, CT 06510, USA.
Bin Peng, Department of Neurosurgery, Yale University, New Haven, CT 06510, USA.
Farrah S. Mohammed, Department of Biomedical Engineering, Yale University, New Haven, CT 06510, USA
Xingchun Gao, Department of Neurosurgery, Yale University, New Haven, CT 06510, USA.
Zhenpeng Qin, Department of Mechanical Engineering, Department of Bioengineering, Center for Advanced Pain Studies, University of Texas, Dallas–UTD, TX 75080, USA.
Kevin N. Sheth, Department of Neurosurgery, Yale University, New Haven, CT 06510, USA
Jiangbing Zhou, Department of Neurosurgery, Yale University, New Haven, CT 06510, USA; Department of Biomedical Engineering, Yale University, New Haven, CT 06510, USA.
Zhaozhong Jiang, Department of Biomedical Engineering, Yale University, New Haven, CT 06510, USA; Integrated Science and Technology Center, Yale University, 600 West Campus Drive, West Haven, CT 06516, USA.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- [1].Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Circulation 2017, 135, e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Krishnamurthi RV, Feigin VL, Forouzanfar MH, Mensah GA, Connor M, Bennett DA, Moran AE, Sacco RL, Anderson LM, Truelsen T, Lancet Glob. Health 2013, 1, e259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Patel T, Zhou J, Piepmeier JM, Saltzman WM, Adv. Drug Delivery Rev 2012, 64, 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kuntz M, Mysiorek C, Pétrault O, Pétrault M, Uzbekov R, Bordet R, Fenart L, Cecchelli R, Bérézowski V, Cereb J. Blood Flow Metab. 2014, 34, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Luo T, Wang J, Hao S, Guo T, Ren P, Cheng Z, Gao F, Gong Y, Wang B, Curr. Pharm. Des 2017, 23, 2258. [DOI] [PubMed] [Google Scholar]
- [6].Yemisci M, Caban S, Gursoy-Ozdemir Y, Lule S, Novoa-Carballal R, Riguera R, Fernandez-Megia E, Andrieux K, Couvreur P, Capan Y, Cereb J. Blood Flow Metab 2015, 35, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Deshane J, Garner CC, Sontheimer H, J. Biol. Chem 2003, 278, 4135. [DOI] [PubMed] [Google Scholar]
- [8].Marsh JN, Hu G, Scott MJ, Zhang H, Goette MJ, Gaffney PJ, Caruthers SD, Wickline SA, Abendschein D, Lanza GM, Nanomedicine 2011, 6, 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhou J, Atsina K-B, Himes BT, Strohbehn GW, Saltzman WM, Cancer J 2012, 18, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sarmah D, Saraf J, Kaur H, Pravalika K, Tekade RK, Borah A, Kalia K, Dave KR, Bhattacharya P, Micromachines 2017, 8, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Domínguez A, Suárez-Merino B, Goñi-de-Cerio F, J. Nanosci. Nanotechnol 2014, 14, 766. [DOI] [PubMed] [Google Scholar]
- [12].Guo X, Deng G, Liu J, Zou P, Du F, Liu F, Chen AT, Hu R, Li M, Zhang S, Tang Z, Han L, Liu J, Sheth KN, Chen Q, Gou X, Zhou J, ACS Nano 2018, 12, 8723. [DOI] [PubMed] [Google Scholar]
- [13].Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC, Chem. Soc. Rev 2012, 41, 2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zielińska A, Carreiró F, Oliveira AM, Neves A, Pires B, Venkatesh DN, Durazzo A, Lucarini M, Eder P, Silva AM, Molecules 2020, 25, 3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sheth KN, Elm JJ, Molyneaux BJ, Hinson H, Beslow LA, Sze GK, Ostwaldt A-C, Del Zoppo GJ, Simard JM, Jacobson S, Kimberly WT, Lancet Neurol. 2016, 15, 1160. [DOI] [PubMed] [Google Scholar]
- [16].Deng G, Ma C, Zhao H, Zhang S, Liu J, Liu F, Chen Z, Chen AT, Yang X, Avery J, Zou P, Du F, Lim KP, Holden D, Li S, Carson RE, Huang Y, Chen Q, Kimberly WT, Simard JM, Sheth KN, Zhou J, Theranostics 2019, 9, 6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sun MS, Jin H, Sun X, Huang S, Zhang FL, Guo ZN, Yang Y, Oxid. Med. Cell. Longevity 2018, 2018, 3804979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Margaill I, Plotkine M, Lerouet D, Free Radic. Biol. Med 2005, 39, 429. [DOI] [PubMed] [Google Scholar]
- [19].Whitney M, Savariar EN, Friedman B, Levin RA, Crisp JL, Glasgow HL, Lefkowitz R, Adams SR, Steinbach P, Nashi N, Angew. Chem 2013, 125, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhou J, Liu J, Cheng CJ, Patel TR, Weller CE, Piepmeier JM, Jiang Z, Saltzman WM, Nat. Mater 2012, 11, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chen Y, Su M, Li Y, Gao J, Zhang C, Cao Z, Zhou J, Liu J, Jiang Z, ACS Appl. Mater. Interfaces 2017, 9, 30519. [DOI] [PubMed] [Google Scholar]
- [22].Zhou Y, Zhang S, Chen Z, Bao Y, Chen AT, Sheu WC, Liu F, Jiang Z, Zhou J, Adv. Sci. (Weinh) 2020, 7, 1901866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shu M, Tang J, Chen L, Zeng Q, Li C, Xiao S, Jiang Z, Liu J, Biomaterials 2021, 268, 120574. [DOI] [PubMed] [Google Scholar]
- [24].Cheng X, Li H, Ge X, Chen L, Liu Y, Mao W, Zhao B, Yuan WE, Front. Mol. Biosci 2020, 7, 576420. D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li W, Yang S, Brain Circ. 2016, 2, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang S, Deng G, Liu F, Peng B, Bao Y, Du F, Chen AT, Liu J, Chen Z, Ma J, Tang X, Chen Q, Zhou J, Adv. Funct. Mater 2020, 30, 1910651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Thorén M, Azevedo E, Dawson J, Egido JA, Falcou A, Ford GA, Holmin S, Mikulik R, Ollikainen J, Wahlgren N, Stroke 2017, 48, 2464. [DOI] [PubMed] [Google Scholar]
- [28].Sheth KN, Elm JJ, Molyneaux BJ, Hinson H, Beslow LA, Sze GK, Ostwaldt AC, Del Zoppo GJ, Simard JM, Jacobson S, Kimberly WT, Lancet Neurol 2016, 15, 1160. [DOI] [PubMed] [Google Scholar]
- [29].Tournier N, Saba W, Cisternino S, Peyronneau M-A, Damont A, Goutal S, Dubois A, Dolle F, Scherrmann J-M, Valette H, Kuhnast B, Bottlaender M, AAPS J. 2013, 15, 1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lahmann C, Kramer HB, Ashcroft FM, PLoS One 2015, 10, e0134476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li J, Zhu Y, Hazeldine ST, Li C, Oupický D, Angew. Chem 2012, 124, 8870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Deng G, Ma C, Zhao H, Zhang S, Liu J, Liu F, Chen Z, Chen AT, Yang X, Avery J, Theranostics 2019, 9, 6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Guo X, Deng G, Liu J, Zou P, Du F, Liu F, Chen AT, Hu R, Li M, Zhang S, ACS Nano 2018, 12, 8723. [DOI] [PubMed] [Google Scholar]
- [34].Han L, Cai Q, Tian D, Kong DK, Gou X, Chen Z, Strittmatter SM, Wang Z, Sheth KN, Zhou J, Nanomed.: Nanotechnol. Biol. Med 2016, 12, 1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chen Y, Gou X, Kong DK, Wang X, Wang J, Chen Z, Huang C, Zhou J, Oncotarget 2015, 6, 32575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.