

Abstract
The human skin can be affected by a multitude of diseases including inflammatory conditions such as atopic dermatitis and psoriasis. Here, we describe how skin barrier integrity and immunity become dysregulated during these two most common inflammatory skin conditions. We summarise recent advances made in the field of the skin innate immune system and its interaction with adaptive immunity. We review gene variants associated with atopic dermatitis and psoriasis that affect innate immune mechanisms and skin barrier integrity. Finally, we discuss how current and future therapies may affect innate immune responses and skin barrier integrity in a generalized or more targeted approach in order to ameliorate disease in patients.

1. INTRODUCTION
1.1. The skin barrier in healthy human skin
The human skin is the largest and one of the most important immunologically active organs. 1 , 2 Due to its location as the outer surface of the body, the skin must be able to protect the body against all types of environmental threats.
Structure— The skin can roughly be divided into three layers: the epidermis—the outermost layer—consisting mainly of keratinocytes in various differentiation states, the dermis, where blood and lymphatic vessels are found and the majority of immune cells reside, and the inner layer containing the subcutaneous fat. 3 , 4
Physical Barrier function— Keratinocytes form the epidermal skin layer and initially protect from threats, such as UV irradiation, pathogen/allergen entry or water loss. Starting from the inner epidermal layer, one finds rapidly proliferating, undifferentiated keratinocytes. These basal keratinocytes play an important role in the production of proteins and lipids, and later differentiate to form the stratum spinosum, change their shape and proliferate further. 3 Reaching the stratum granulosum, the keratinocytes are at their maximum production of lipids and proteins. Keratinocytes progress through a terminal differentiation programme to form the stratum corneum. The outermost layers of the stratum corneum consist of keratinocytes that are reduced to anucleate cells without organelles (corneocytes) and then shed through enzymatically controlled desquamation. 3 , 4 , 5 Here, corneocytes play a central role in the skin barrier as they prevent external substances such as pathogens and allergens from entering, and water from leaving the skin, thus preventing water loss (xerosis). 3 A key molecule providing structure and integrity here, is the filament‐aggregating protein (filaggrin), a loss of which enhances inflammatory skin conditions, such as atopic dermatitis (AD). 6 , 7 The importance of skin integrity in the pathogenesis of AD was emphasised by the association of loss‐of‐function mutations in the filaggrin gene (FLG) with AD. 8 This work indicated that epidermal barrier dysfunction is a primary aetiological phenomenon in AD rather than a consequence of disrupted immunology. 8 A meta‐analysis of studies on FLG mutations and atopic dermatitis risk showed evidence that FLG mutations have the strongest association with risk of atopic dermatitis due to skin barrier deficiency in genetic variants that have been investigated thus far. 9
The skin‐intrinsic defence against pathogenic microbes is enhanced by the production of antimicrobial peptides (AMPs) by keratinocytes in the deeper layer of the epidermis. 10 AMPs such as LL‐37, human beta‐defensins (hBD) or S100 proteins show a broad antimicrobial activity against bacteria and fungi. Interestingly, interleukin (IL)‐26 produced by subsets of T helper (TH) cells also shows antimicrobial features—while being upregulated in psoriasis—and is thus bridging innate and adaptive immunity. 11 , 12 Interestingly, a recent study indicates that self‐DNA is released in AD skin lesions, which then binds to AMPs and reduces anti‐microbial activity. 13 Thus, AMPs are functionally altered during AD and psoriasis.
1.2. Innate immune mechanisms in the skin
The skin is populated with a complex array of blood‐derived and tissue‐specific immune cells. Several immune cell populations reside in the epidermis: primarily Langerhans cells (LC; specialised epidermal dendritic cells [DC]), monocyte‐derived LC‐like cells and inflammatory dendritic epidermal cells (IDECs), 14 but also tissue‐resident CD8+ T cells. LCs are among the first line of defence, acting as immune sentinels, which migrate to skin‐draining lymph nodes upon pathogen encounter. 15 While LCs and LC‐like cells are found in the skin at healthy steady state, their numbers strongly increase in inflammatory conditions whereas IDECs only populate the skin under inflammatory circumstances. The majority of immune cells are found in the dermis, where—similar to the epidermis—the cell numbers are very low at steady state but show a strong surge in inflammation. The specialised immune cells here are plasmacytoid DCs, dermal DCs, tissue macrophages, different subtypes of CD4+ T cells, such as T helper type 1 (TH1) cells, TH2 cells, and regulatory T cells (Treg), but also natural killer (NK) T cells and different types of innate lymphoid cells (ILC). 4 With regard to their transcription factors and cytokine production, type 1, 2 and 3 ILCs closely resemble the three major TH1, TH2 and TH17 cell subpopulations, respectively. Importantly, ILCs do not require traditional adaptive immune receptor rearrangement and can thus react to innate signals without antigen‐specificity. 16 Similar to classical TH cells, an imbalance of cutaneous ILCs enhances inflammatory skin disease manifestations. 17 , 18 ILC1 are the least well characterised ILC subset and their role in skin inflammation remains elusive. Based on their cytokine profile it is hypothesised that they are involved in allergic contact dermatitis. 19 ILC2 produce the cytokines IL‐5 and IL‐13 and have been implicated in AD, where they were found to be highly enriched in lesional skin. 20 In line with the TH17‐resembling cytokine profile, ILC3 are increased in blood and skin of psoriasis patients. 21 More evidence for their implication in the pathogenesis of psoriasis stems from the observation that mice that lack adaptive lymphocytes still develop psoriasiform inflammation similar to wild‐type controls that possess TH17 cells. 22
Although mostly considered of structural importance to the skin, keratinocytes play their part in the resident skin (innate) immune system. They contribute to immune surveillance by expressing a range of toll‐like receptors (TLRs; TLR1‐6 and 9) and producing AMPs like LL37. 23 Most importantly, keratinocytes are capable of producing a wide range of chemokines and cytokines like CXCL8, 24 chemokine (C‐C motif) ligand 20 (CCL20) 25 and IL‐23. 26 These chemokines and cytokines can in turn attract immune cell and their regulation is key to maintain a homoeostasis in healthy skin.
1.3. Innate immune mechanisms in Cutaneous diseases
1.3.1. Psoriasis
Psoriasis is a common chronic inflammatory skin disease spanning a variety of skin phenotypes and is linked to complex comorbidities including seronegative arthritis, ischaemic heart disease, and metabolic syndrome. 27 , 28 , 29 Histologically, a strikingly thickened epidermis is found, together with deep epidermal ridges reaching into the dermis. 27 In psoriatic lesions, TH1 and TH17 cells are predominantly found together with an increased expression of IL‐17 and IL‐22. 30 A key player accountable for the thickened epidermis is IL‐22, which promotes proliferation of keratinocytes. 31 , 32 , 33 Additionally, an increased amount of IL‐23 and IL‐1β produced by DCs drives TH17 cell differentiation and thereby accelerates the progression of the psoriatic phenotype. 34 While T cells and especially TH17 cells have been seen the crucial culprits in the pathogenesis of psoriasis, an increasing body of evidence shows the role of the innate immune system. 23 , 35 Reports hint towards more influence of the adaptive immune system in mild psoriasis, whereas severe psoriasis is more influenced by actions of the innate immune system. 36 An increasing role in the pathogenesis of the disease has been attributed to ILC3 cells which are found increased in psoriasis. 21 , 37 Due to their ability to produce TH17‐cytokines such as IL‐17 and IL‐22, they bridge adaptive and innate immunity. A recent study has shown that quiescent‐like ILC2 in the skin can transition into pathogenic ILC3‐like cells upon disease initiation. 38 ILC3 numbers in psoriatic skin are reduced after therapeutic treatment with anti‐tumour necrosis factor (TNF) antibody, indicating their contribution to pathogensis. 21 It has further been described that in psoriasis, epidermal LC have impaired migratory capacity to skin‐draining lymph nodes and thus delayed onset of cutaneous immune responses. 39 Additionally, the capacity of the structural keratinocytes to produce a variety of chemokines and cytokines together with the hyperproliferation seen in psoriasis leads to a vicious cycle in the pathogenesis of the disease. 23 The strong upregulation of chemokine ligand CCL20 expression in keratinocytes in presence of TH17‐derived IL‐17A leads to further recruitment of T cells into psoriatic lesions. 25 Additionally, IL‐17 is stimulating the proliferation of keratinocytes and their secretion of AMPs thereby contributing to the hyperproliferation phenotype. 40 While the general action of IL‐17 in the pathogenesis of psoriasis is known, further research is necessary to elucidate cell‐specific contributions such as the role of ILC3‐derived IL‐17.
1.3.2. Atopic dermatitis
AD, also known as atopic eczema, is a chronic inflammatory skin disorder characterised by severe pruritus, dry and scaly skin, as well as raised, red lesions in the bends of arms and legs. 41 , 42 , 43 , 44 The prevalence is approx. 10%–20% in developed countries and in approximately 60% of the cases, onset of disease is in the first year of life. 45 In contrast to psoriasis, AD is a disease with a type 2‐biased phenotype with increased expression of IL‐13 and IL‐5. 46 Chronic AD, however, also displays IFN‐γ, the signature cytokine for TH1 cells and type 1 responses. 47 , 48 In line with TH2 cells, ILC2s have also been reported, to be highly enriched in lesional AD skin. 49 , 50 , 51 Skin ILC2 do not rely on IL‐33 signalling but instead on thymic stromal lymphopoietin (TSLP). 20 TSLP is strongly increased in AD, leads to the production of the TH2 cell attracting chemokine CCL17, 52 and considered to be a trigger factor in the initial stages of the disease. 53 Furthermore, TSLP has been reported to stimulate cutaneous neurons to promote itch and provoke itching. 54 The scratching in response to itching sensation will break down skin barrier functions leaving AD patients largely unarmed against skin infections. It is therefore not suprising that up to 90% of AD patients are colonised with Staphylococcus aureus and are also prone to viral infections caused by herpes simplex virus. 43 , 55 , 56 , 57 Although AD patients with higher S. aureus abundance show significantly higher excoriations and sleep loss, a correlation between (patient reported) itch intensity and S. aureus concentration is not evident. 58 A reason for this might be the lower levels of AMPs (e.g. LL37 and hBD2) in AD compared to psoriasis. 59 , 60 This reduced AMP expression by keratinocytes is partly caused by the inhibitory effects of the TH2 cyokines IL‐4 and IL‐13, 61 as well as IL‐10 62 and TSLP. 63
2. MODULATION OF SKIN BARRIER FUNCTION
2.1. Single nucleotide polymorphisms (SNPs) affecting skin barrier integrity and innate immunity in AD and psoriasis
Both AD and psoriasis are multifactorial diseases with a complex origin. Comparison of SNPs or genetic variants, between healthy controls and persons suffering from AD or psoriasis revealed distinct sets of mutations associated with either disease 64 (Figure 1). These genome‐wide association studies lay the groundwork for the design of novel therapeutic options as they help to dissect the molecules and mechanisms involved in pathogenesis. Relevant genetic variants associated with AD span genes—and their products—involved in the initiation of immune responses, effector cytokines and chemokines, signalling molecules, and, importantly, proteins involved in the maintenance of skin barrier integrity. Important genes and the disease‐associated genetic variants are listed in Table 1. Detailed reviews on the genetics of AD and psoriasis were also recently published by Martin et al. 65 and Ogawa and Okada, 66 respectively.
FIGURE 1.
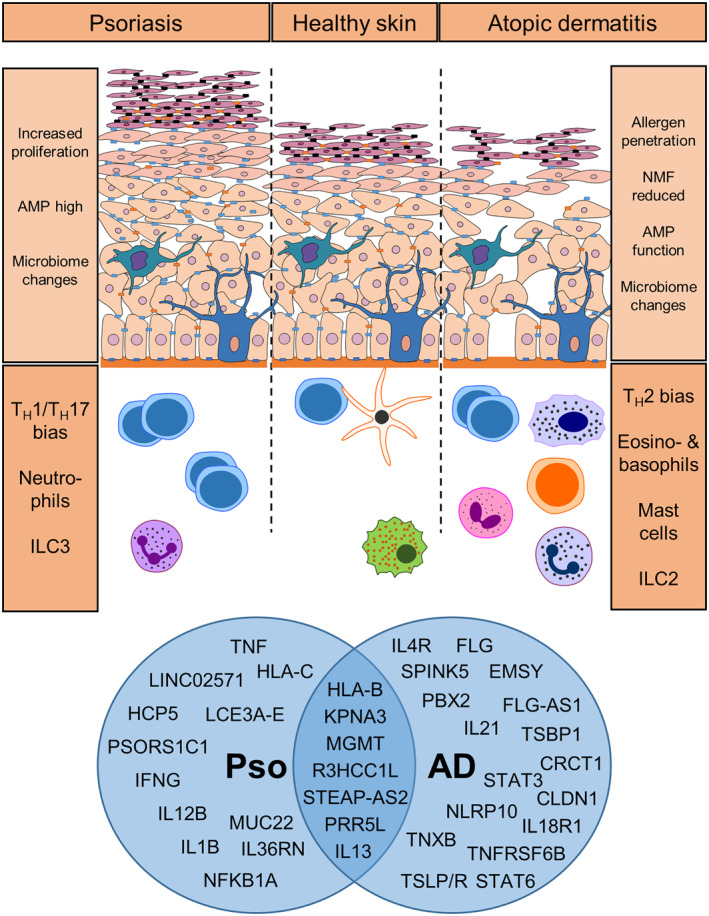
Schematic representation of skin and underlying processes during steady state, psoriasis and atopic dermatitis. In psoriatic lesions increased cell proliferation occurs in the epidermis and elevated production of anti‐microbial peptides (AMPs), while during AD the skin barrier is impaired, which leads to increased allergen penetration, reduced natural moisturising factor (NMF) and may affect AMP abundance or activity. Both diseases are characterised by changes in microbiome composition. During psoriasis pro‐inflammatory TH1 and TH17 cells dominate the affected skin. In contrast, type 2‐associated cells, including TH2 cells, eosinophils, basophils and ILC2 can increase during AD. GWAS analysis (publicly available databases: GWAS database, https://www.ebi.ac.uk/gwas/; GWAS Central, https://www.gwascentral.org/; accessed 15.11.2021; filtered for psoriasis and atopic eczema traits) of the most common SNPs associated with psoriasis (Pso) and atopic dermatitis (AD) are summarised in the Venn diagram
TABLE 1.
Selected SNPs associated with AD and/or psoriasis
| Gene | Reference SNP cluster ID | Reference |
|---|---|---|
| Atopic dermatitis | ||
| IL1RL1 | rs3917265, rs1861246, rs13015714, ‐26999A | [67, 172, 173, 174] |
| TSLP/R | rs111267073, rs10043985, rs2289276, rs1898671, rs11466749, rs2416259, rs1837253, rs3806932, rs2289278, rs36139698, rs36177645, rs36133495 | [68, 175, 176, 177, 178, 179] |
| IL18 R | rs13015714, rs6419573, rs13015714, rs1861246 | [172, 173] |
| IL21 | rs17389644 | [64, 172, 180] |
| IL6R | rs2228145, rs12126142, rs4576655, rs12730935 | [172, 173, 181] |
| IL4/R | rs2107357, rs143021546, ‐590 C/T | [88, 176, 182] |
| STAT6 | rs1059513, rs3024971 | [175, 183] |
| STAT3 | rs4796793, rs17881320, rs12951971 | [172, 184, 185] |
| FLG | rs558269137, rs6661961, rs3126085, 2282del4, rs61816761, rs150597413, rs138726443 | [65, 89, 173, 175, 181] |
| TMEM79 | rs6694514 | [98] |
| EMSY | rs7130588, rs2212434, rs7110818, rs7927894, rs2155219, rs34455012 | [64, 89, 172, 173, 180, 181, 182, 186] |
| CLDN1 | rs893051 | [187] |
| LCE3E | rs10888499, rs61813875 | [64, 172] |
| LCE5A | rs6661961, rs11205006, rs471144, rs12144049, rs12081541 | [64, 89, 184, 188] |
| SPINK5 | rs2303063, rs2303067 | [189, 190] |
| Psoriasis | ||
| PSORS1 region (including HLA‐C) | rs6913137, rs2853950, rs3130573, rs7756521, rs9263717, rs2233959, rs2524096, rs12199223, rs1265078, rs2249742, rs2853961, and many more | [191, 192] |
| TNFAIP3 | rs582757, rs610604, rs6933987, rs643177 | [181, 193, 194, 195, 196] |
| NFKB1A | rs8016947 | [193, 196] |
| IL36RN | rs387906914, rs397514629 | [75, 197, 198] |
| LCE3B | rs4845454, rs11205044, rs1581803 | [64, 199, 200] |
| LCE3D | rs4085613, rs4112788, rs6677595 | [193, 194, 201] |
| AD and psoriasis | ||
| HLA‐A/B | AD: rs148203517, rs4713555, rs28752924, rs28383323, rs200291258, rs9405068, rs4713555, rs2251396; Pso: rs4406273, rs2523619, rs17728338, rs75851973, rs76956521, rs1960278, rs12212594, rs4959062, rs4349859, rs10484554 | [64, 80, 172, 173, 181, 191, 193, 196, 202] |
| CARD14 | AD: rs535171797; Pso: rs11652075 | [193, 203] |
| IL12B | AD: rs3212227, rs393548, rs436857; Pso: rs2082412, rs3212227, rs3213094, rs2546890, rs12188300, rs12188300, rs6887695 | [78, 193, 194, 195, 204, 205, 206] |
| IL13 | AD: rs848, rs1295686, rs847, rs1295685, rs20541, rs12188917; Pso: rs20541, rs1295685, rs847 | [64, 89, 172, 173, 175, 180, 181, 193, 196, 199, 207] |
| MGMT | rs80312298 | [64] |
| PRR5L | rs2592555, rs12295535, rs11033603, rs2218565 | [64, 172, 173, 180] |
| KPNA3 | rs3736830 | [64] |
| R3HCC1L | rs11189494 | [64] |
| STEAP‐AS2 | rs7798970 | [64] |
| PELI2 | rs17761563 | [64] |
The keratinocyte‐derived so‐called ‘alarmin cytokines’ are important inducers of skin immune responses. The −26999 G/A mutation in the distal promoter of IL1RL1 (the receptor for IL‐33; ST2) leads to the increased expression of soluble ST2 (IL‐33 decoy receptor), preferential activation of TH2 cells, high total IgE, and an increased odds ratio (1.87) to develop AD. 67 SNPs in TSLP and its receptor (TSLPR), have also been correlated to the development of AD. 68 Studies in animal models have confirmed a pathogenic role for TSLP, as its overexpression in keratinocytes leads to the development of AD‐like inflammation. SNPs in IL18 are also associated with AD. 69 , 70 In psoriasis, IL‐18 is produced by activated keratinocytes and can act on DC and promote type‐1 and type‐3 responses. In AD, IL‐18 can act in concert with IL‐12 to drive type‐1 responses or promote type‐2 responses together with TSLP. Recently, it was found that IL‐18 can activate ILC2, which may contribute to skin inflammation through their production of IL‐13. 71 These SNPs highlight the importance of molecules that signal the presence of potentially harmful events as they are generally released upon tissue damage. The functional characterisation of IL‐25, IL‐33 and TSLP in the outset of the cutaneous inflammation has led to the development of biologics targeting each cytokine or their cognate receptor for AD and indeed other allergic disorders.
In psoriasis, alarmins do not seem to play such a prominent role in the initiation phase of the immune response. Instead, human leucocyte antigen (HLA)‐related gene variants correlate with an increased odds ratio to develop psoriasis. 72 The PSORS1 locus maps to the major histocompatibility complex (MHC) region and spans nine genes. For example, a variant of HLA‐C (HLA‐Cw6) 73 can—in addition to conventional antigens—present autoantigens to CD8 T cells. Activation of pro‐inflammatory pathways is also involved in the pathogenesis of psoriasis, including TNFAIP3, NFKBIA and CARD14. 74 Here, the immunoregulatory or inhibitory functions of TNFAIP3 and IkB are decreased and promote pro‐inflammatory NFkB activation. Similarly, gain‐of‐function mutations in CARD14 increase NFkB activation. Mutations in the gene encoding for IL‐36 receptor antagonist (IL36RN) are associated with the development of generalised pustular psoriasis, raising potential for targeting IL‐36—a member of the IL‐1 family—in psoriasis. 75 Indeed, targeted deletion of IL‐36R on keratinocytes led to decreased expression of pro‐inflammatory cytokines. 76
Downstream of the initiation of an immune response, SNPs in the signalling cascade of cytokines—mainly the respective JAK‐STAT pathway—are associated with AD. Here, the divergent nature of AD (type‐2‐biased) and psoriasis (type‐1/3‐biased) becomes apparent. IL‐12B is a subunit of the cytokine IL‐12 that promotes the differentiation of TH1 cells. Genetic variations in IL12B are associated with psoriasis in a cohort of Danish patients, 77 while SNPs in IL12B decrease the risk of developing AD. 78 Similarly, IL‐21 can drive TH17 differentiation in psoriasis, 79 with higher levels of IL‐21 reported in lesional skin of psoriatic patients. The genomic region (4q27) including the IL21 gene is associated with psoriasis and SNPs in this locus are associated with higher IL‐21 levels in other inflammatory disorders. 80 In addition to TH17 and TH1 activation, IL‐21 can cause epidermal hyperplasia. While increased levels of IL‐21 are found in acute lesions of AD patients, 81 the implication for AD pathogenesis requires further study. In mouse models of AD‐like inflammation, lower levels of the IL‐17‐induced TH2‐recruiting chemokine CCL17 have been observed, 82 and a protective role for the IL‐17RA in AD has been described. 83 Certain SNPs in IL6—required for balancing TH17/TREG cells 84 —are associated with a decreased risk to develop psoriasis, 85 while it may increase the risk for AD 86 and disruption of IL‐6‐receptor signalling improved AD. 87
Opposed to the pro‐inflammatory type‐1 and type‐3 responses, IL‐4 is the main cytokine driving TH2 polarization and is associated with allergic diseases including AD. 88 IL‐13, another hallmark cytokine of type 2 immune responses, is also associated with AD. 89 In contrast, IL‐4 is not directly associated with psoriasis but repeated IL‐4 application in psoriatic patients can divert the pro‐inflammatory type‐1/‐3 response towards type‐2. 90 Both IL‐4 and IL‐13 signal through STAT6, which is also linked to AD pathogenesis, 91 and mice that express a constitutively active form of STAT6 were shown to develop AD‐like lesions. 92
Genes involved in the skin barrier are linked to AD and psoriasis. In the PSORS4 region, more than 60 genes that control keratinocyte differentiation are encoded. 93 Deletion of LCE3B and LCE3C (late cornified envelope proteins) is associated with psoriasis. 94 Importantly, variants of genes involved in the maintenance of the skin barrier are strongly associated with AD. Among the most important affected genes are FLG (encoding for filaggrin), TMEM79 (mattrin), and EMSY. First described in 2006, filaggrin is one of the most prominent proteins, in which a SNP can cause ichthyosis vulgaris 95 and predispose for AD. 8 To date, more than 50 mutations in FLG in all human populations studied around the globe (>1% allele frequency) are reported, suggesting an evolutionary benefit of a mildly leaky barrier. Loss‐of‐function of filaggrin leads to increased abnormal structure and physiology of the skin, increased trans‐epidermal water loss (TEWL) and dry skin, as natural moisturising factor (NMF) is a catabolic product of filaggrin. 96 Similarly, Tmem79 mutations were found to increase barrier leakage and AD‐like inflammation in mice. 97 , 98 Recently, a possible function of EMSY in maintaining the skin barrier was described. Using skin organoids, it was found that EMSY acts as a transcriptional regulator in keratinocytes. 99 In skin biopsies of AD patients, EMSY located in the nucleus, actively repressing gene expression. 99 Furthermore, De Benedetto et al. showed that genetic variants in the tight junction gene CLDN1 are associated with AD. 100 The strong associations of the described SNPs with AD and psoriasis highlights the necessity of an intact barrier to maintain tissue homoeostasis. Despite the recent advances in the field of 3D‐organ cultures, skin‐on‐a‐chip models and in silico‐prediction models, functional studies on the identified loci are required to work towards novel therapeutics. Thus, animal models of skin disease and clinical studies are still essential for scientific progress.
2.2. Keratinocytes recruit immune cells
Keratinocytes are important central modulators of immune responses in the skin. They are not only capable of producing AMPs that act as a first line of defence but can also release chemotactic factors. In psoriasis, keratinocytes release chemokines CXCL9, ‐10, ‐11 and ‐20, as well as CXCL‐1 and ‐8, attracting LC and neutrophils to the skin, respectively. Keratinocytes are also able to sense microbial patterns via TLRs. As a consequence of TLR signalling, keratinocytes can release pro‐inflammatory cytokines, such as IL‐1β and IL‐18, both of which are cleaved via the NLRP‐inflammasome into the active form. IL‐1β can upregulate ICAM‐1 on dermal endothelial cells and may facilitate entry of leukocytes into the skin. 101 In AD, keratinocytes synthesise increased amounts of chemokines, such as CSF‐2, RANTES/CCL5 and MCP‐1/CCL2, promoting the infiltration of eosinophils, dendritic cells and monocytes, as well as T cells into the skin. 102 In order to counteract the loss of the skin's barrier function, the stratum corneum increases in thickness. Hyperkeratosis is a hallmark of chronic AD and palmoplantar hyperkeratotic psoriasis and develops from an imbalance of protease–protease inhibitors interactions. 103 While kallikrein‐(KLK)7 protein levels were increased during AD, its activity was not elevated. Instead the increased levels of lymphoepithelial Kazal‐type‐related inhibitor (LEKTI, encoded by SPINK5) were thought to prevent corneodesosome degradation by KLK7. Similarly, unregulated KLK5 activity in the absence of LEKTI led to the development of AD‐like inflammation in mice. 104 Thus, regulation of proteolytic pathways contribute to skin barrier function and pathogenesis of inflammatory skin disease.
2.3. Innate cells can regulate skin barrier function
Innate immune cells in the skin are the first responders to tissue damage and invading pathogens. Innate cells are potent producers of an array of cytokines. The predominant cytokines linked to skin diseases are IL‐17, IL‐4, and IL‐13. IL‐17A can be produced by γδ T cells, iNKT and ILC3, and IL‐17 can activate the release of AMP. 105 Keratinocytes are a major source of AMPs. 106 Defective function of these AMPs, such as cathelicidin or β‐defensins may contribute to AD. 107 Interestingly, decreased AMP production is associated with predisposition to AD, while high AMP expression is observed in psoriatic lesions. 61 , 108 LL‐37, a member of the cathelicidin family, which was increased during atopic eczema, 109 has an essential role in angiogenesis and wound healing 110 and is released in response to injury. 111
IL‐4 and IL‐13 are produced by both innate and adaptive immune cells. For example, basophils can release large amounts of IL‐4 upon engagement of the Fcε receptor by IgE crosslinking. Eosinophils are also a significant source of IL‐4 and can be activated by IL‐33 or CSF‐2 as well as TLR and interferons. ILC2 can produce large amounts of IL‐13 and IL‐5, but also—depending on the context—IL‐4. IL‐4 has been shown to massively alter barrier function. 92 In IL‐4‐deficient animals, expression of skin barrier proteins, such as loricrin, involucrin and transglutaminase‐3, was two‐ to three‐fold increased. Importantly, IL‐4‐deficiency also increased filaggrin expression in the skin. Indeed, keratinocytes decrease expression of filaggrin, loricrin, involucrin, and hornerin in response to IL‐4 and IL‐13, while the peptidase KLK7 was induced. Thus, IL‐4 leads to skin desquamation through degradation of corneodesmosomal proteins. In a recent study, basophil‐derived IL‐4 was also shown to reduce IL‐1 and IL‐23 production from keratinocytes that impaired γδ T cell activation and thus promoted S. aureus colonisation. 112 IL‐13 signals also through the IL‐4Rα chain (heterodimeric receptor with IL‐13Rα1) and probably exerts similar functions, although they are still under debate. 113 In addition, the second receptor, IL‐13Rα2, functions as a decoy receptor scavenging IL‐13 ameliorating skin barrier defects and cutaneous inflammation. 114 However, it was also recently shown that homoeostatic IL‐13 from ILC2 in healthy skin—while fostering a noninflammatory skin environment – may predispose for allergic sensitisation through the activation of TH2‐priming dermal DC2 subsets. 115 Interestingly, IL‐33 was shown to disturb skin barrier integrity independently of mast cell and TH2‐cell‐derived cytokines. 116 Whether IL‐33 directly modulates keratinocyte‐function, which increase expression of ST2 in AD lesions 117 or via the activation of ILC2 118 remains to be determined.
Pruritus is a major cause for the breakdown of the skin barrier and the crosstalk of innate immune cells with the nervous system is an emerging field. The alarmins TSLP and IL‐33 can induce itch by acting directly on sensory neurons, 54 activation of ILC2 and basophils, 119 IL‐31 release from T cells, reducing skin barrier protein expression, 116 , 120 or histamine release from mast cells. 121 Histamine and serotonin release from mast cells activated by IgE‐crosslinking of the FcE‐receptor causes histaminergic itch. Recently, it was discovered that mast cells are also activated by PAMP9‐20, a peptide released by various cell populations, including keratinocytes, acting on Mrgprb2 in mice or MRGPRX2 in humans. Mast cells then only secrete small amounts of histamine and serotonin but instead release tryptase and thereby trigger non‐histaminergic itch. 122 The release of IL‐4 and IL‐13 by mast cells, eosinophils and basophils may also directly contribute to scratching behaviour as it has been demonstrated that IL‐4 injection induces scratching via signalling through IL‐4Ra and JAK1. 123 Confirmation of IL‐4Ra‐JAK pathways mediating itch was provided with Dupilumab 124 and JAK inhibitors 125 both improving pruritus in treatment of AD for patients. 126
Taken together, the release of cytokines by immune cells during psoriasis and AD alters both the composition and function of the skin barrier and further may aggravate disease through neuronal circuits. Targeting cytokines and the signalling pathways of the skin neuro‐inflammatory network with monoclonal antibodies and inhibitors have become promising areas of research to develop novel therapies.
3. EFFECTS OF THERAPEUTIC APPROACHES ON THE INNATE IMMUNE SYSTEM
In this section, we will highlight novel developments in the treatment of AD and psoriasis that affect mechanisms related to innate immunity and barrier function of the skin.
3.1. Topical and systemic therapies
Emollients and topical barrier treatments are used in AD and psoriasis to maintain the skin barrier function, combined with avoidance of detergents. Improvement of the barrier in skin barrier deficiency seen in AD, via disease‐specific, barrier corrective topical treatments such as ceramide‐dominant mixtures with barrier lipids, down‐regulates pro‐inflammatory signalling mechanisms involved in barrier repair. The ingress of further haptens, which drive TH2‐type responses are increasingly blocked by skin barrier improvement. A lipid mixture gives an acidic pH on the skin surface adding to the barrier function and blocking the activation of proinflammatory serine proteases. 10 Itch is a feature of some dermatoses that are associated with skin barrier deficiency. The inclusion of anti‐pruritic ingredients in emollients can supplement the barrier restorative factors of the emollient by decreasing itch, which is a factor in skin barrier deficiency. 103 , 127
Topical corticosteroids (TCS) accompanied by emollients have been the mainstay of the treatment of AD since their introduction in the 1950s. 128 The anti‐inflammatory effect of TCS is mediated through a cytoplasmic glucocorticoid receptor (GCR) in target cells. 42 One feature of how innate immunity influences anti‐inflammatory effects with TCS is after ligand binding, when the corticosteroid/GCR complex translocates to the nucleus. Various transcription factors including nuclear factor κB (NF–κB), inhibit the transcriptional activity of genes encoding cytokines such as IL‐1, IL‐4, IL‐5, IL‐13, TNF as well as chemotactic proteins and adhesion molecules. 42 While this immunosuppression limits skin inflammatory processes, TCS therapy is associated with skin atrophy and leads to an impaired skin barrier. Thus, emollients are used in combination to promote barrier restoration.
A similar mode of action is observed when treating psoriasis with TCS. Immune cells and pro‐inflammatory cytokines are repressed by signalling via the GCR. Corticosteroids commonly repress maturation and differentiation of DC and macrophages, thereby further reducing pro‐inflammatory type‐1 and type‐3 responses. In addition, TCS has antimitotic properties reducing the hyperproliferation but also lead to skin atrophy. The impaired skin barrier facilitates TCS penetration and promotes systemic adverse events. Additional treatment with Vitamin D analogues, which corrects epidermal hyperproliferation and induces apoptosis in inflammatory cells, can prevent some of the adverse effects of TCS. 129 Vitamin D affects the production of AMPs, which are involved in maintenance of the skin barrier, decreased levels of which can results in exacerbations of AD, particulary infective flares of inflammation. 105 , 130
Topical calcineurin inhibitors (TCIs), such as tacrolimus or pimecrolimus, act as steroid‐sparing agents. 131 They inhibit inflammatory cytokine transcription in activated T cells and other inflammatory cells through inhibition of calcineurin. 131 With this anti‐inflammatory activity, topical calcineurin inhibitors help to allow the skin barrier to be restored. 106 , 132 Topical calcineurin inhibitors do have the potential for local immunosuppression, however, clinical trials have shown no increase in systemic or local skin infections. Yet, TCI treatment impairs skin barrier function through decreasing epidermal lipid synthesis, suppression of IL‐1α and reducing AMP synthesis. 136 While TCIs are approved in the treatment of mild‐to‐moderate AD, they show limited efficacy in psoriasis. 137 Psoriasis that affects facial skin or genital skin can show improvement with the use of TCIs.
Traditional systemic therapies: Despite the usual success of topical therapies at gaining control of a patient's AD, there is a minority of patients for whom systemic therapy is a necessary next step in AD management. 128 Systemic medications include azathioprine, methotrexate, cyclosporine as discussed in Table 2, which act as steroid‐sparing immunosuppressants, 138 , 139 , 140 which are also indicated in moderate‐to‐severe psoriasis.
TABLE 2.
Advanced therapeutics for atopic dermatitis and psoriasis
| Compound proprietary name | Target | Details | References | |
|---|---|---|---|---|
| Atopic dermatitis | Dupilumab | IL‐4Rα | IL‐13/IL‐4 inhibitor, monoclonal antibody | [124, 148, 208] |
| Topical crisaborale | Phosphodiesterase 4 (PDE4) enzymes | Phosphodiesterase 4 inhibitor, small molecules | [144, 145, 146, 209] | |
| Oral apremilast | ||||
| Tofacitinib | JAK/STAT pathway | JAK inhibitors, small molecules | [161, 162] | |
| Baricitinib | ||||
| Upadacitinib | ||||
| Ruxolitinib | ||||
| Nemolizumab | IL‐31Rα | IL‐31 inhibitor, monoclonal antibody | [210] | |
| Tralokinumab | IL‐13 | IL‐13 inhibitor, monoclonal antibody | [150] | |
| Lebrikizumab | ||||
| Tezepelumab | TSLP | TSLP inhibitor, monoclonal antibody | [143] | |
| Etokimab | IL‐33 | IL‐33 inhibitor, monoclonal antibody | [159] | |
| Fezakinumab | IL‐22 | IL‐22 inhibitor, monoclonal antibody | [211, 212] | |
| Psoriasis | Adalimumab | TNFα | TNF inhibitor, monoclonal antibody | [213, 214, 215, 216] |
| Certolizumab pegol | ||||
| Golimumab | ||||
| Infliximab | ||||
| Etanercept | TNFα | TNF inhibitor, fusion protein decoy receptor | [217] | |
| Brodalumab | IL‐17A | IL‐17A inhibitor, monoclonal antibody | [218, 219, 220, 221, 222, 223] | |
| Ixekinumab | ||||
| Secukinumab | ||||
| Guselkumab | IL‐23 | IL‐23 inhibitor, monoclonal antibody | [224, 225, 226] | |
| Risankizumab | ||||
| Tildrakizumab | ||||
| Ustekinumab | IL‐12/IL‐23 | IL‐12/IL‐23 inhibitor | [227] | |
| Spesolimab | IL‐36R | IL‐36 receptor inhibitor, monoclonal antibody | [167] |
Advanced therapeutics in AD including biologic agents and small molecule inhibitors.
3.2. Advanced therapeutics in AD including biologic agents and small molecule inhibitors
Biologic agents allow specific targeting of molecules, which can be further upstream in inflammatory pathways. As discussed above, targeting key proteins in the initiation (TSLP, IL‐18), effector phase (IL‐4, IL‐13) or the crosstalk to neurons (IL‐31) may interfere with the innate‐adaptive interaction, relieve symptoms and restore barrier integrity. Some have been used in Dermatology more than two decades for other inflammatory cutaneous disorders 141 and especially to great effect for treating psoriasis. However the development of this line of therapeutics for AD has been slower. 142 Reasons for this slower development for AD include the complex heterogeneity of acute and chronic inflammation seen in AD, the multiple as yet poorly characterised endotypes and the multifactorial causes and exacerbators of AD, including bacterial, viral and fungal dysbiosis.
TSLP: Proinflammatory stimuli generate TSLP. TH2 cytokine production by DCs is induced by TSLP, which is upstream from IL‐4, ‐5 and ‐13. Tezepelumab is a human IgG monoclonal antibody that binds TSLP and stops further interactions with the receptor complex. A phase 2b, clinical study showed that there was a trend of improvements in clinical scorings for patients treated with tezepelumab and topical corticosteroid versus placebo; however, significance was not reached. 143 Considering that TSLP is a driver of one of the pathways that innate immunity influences the downstream inflammation that occurs in AD and other parts of the atopic march such as asthma and allergic rhinitis, further investigations may demonstrate how influencing the inflammatory pathway at an earlier stage can reduce the range of severity of acute and chronic atopic inflammation.
PDE4: Studies have shown that phosphodiesterase (PDE) inhibitors altered inflammatory pathways stimulated in AD and could be considered as a therapeutic target for a non‐steroid based topical treatment for AD. 144 , 145 , 146 Crisaborale is a topical PDE4 inhibitor which has been authorised for the treatment of atopic dermatitis in the US and the European Union. Proinflammatory cytokine responses arise from the conversion by PDE4 of intracellular messenger cyclic adenosine monophosphate (cAMP), into adenosine monophosphate. In AD there is up‐regulation of PDE4 with over‐expression of cytokines such as IL‐4, ‐13, ‐31, released by both innate and adaptive immune cells. 145
IL‐4/IL‐13: Dupilumab, now licenced for AD treatment in the United States, Europe, China and Japan is a fully human anti‐IL‐4 receptor alpha monoclonal antibody, which inhibits IL‐4 and IL‐13. 147 Both cytokines are potently produced by cells of the innate immune system and act on innate and adaptive immune cells. Thus the cell‐specific contribution of IL‐4RA‐blockade remains to be determined. Considerable decreases in clinical scores and pruritus of AD were seen, and no systemic side effects were noted, but conjunctivitis incidence was increased. 148 Importantly, dupilumab therapy reduced type 2 inflammation, reversed AD‐induced epidermal abnormalities and increased gene expression of barrier‐associated proteins. 126 , 149
IL‐13: A clinical trial by Wollenberg et al. showed that inhibiting IL‐13 in adults with moderate to severe AD, led to significant clinical improvements in Eczema Area and Severity Index (EASI) and Dermatology Life Quality Index scoring. 150 Tralokinumab is a fully human IgG4 monoclonal antibody, which targets IL‐13 and was administered in a phase 2b randomised study with concomitant topical glucocorticoids. The safety profile in this trial was in line with those of previous trials of tralokinumab in patients with asthma. None of the adverse events reported were associated with the study drug. 150 Similarly, lebrikizumab, developed for the treatment of asthma, selectively targets IL‐13 and appears to be effective in moderate‐to‐severe AD. 151 , 152 Because ILC2 are implicated in AD pathogenesis and can produce large amounts of IL‐13, future research should include the identification of strategies to interfere with ILC2 function.
IL‐18: IL‐18 contributes to the change from an acute TH2‐driven AD endotype towards TH1 polarization in chronic disease. 153 The receptor for IL‐18 signals via the innate inflammatory MyD88‐pathway and can activate TH1 cells, basophils, NK cells, mast cells. 154 Hu et al. reported that serological IL‐18 and IL‐18 binding protein was found in increased amounts—specially during worsening pathology—n patients with AD. In a murine model of eczema increased mast cells in lesional skin and elevated levels of IL‐18BP+ mast cells in lesional skin were found. 154 Allergen challenge resulted in amplified expression of IL‐18, IL‐18BP and IL18 R mRNA. This study suggested that IL‐18 inhibitor agents may be a therapeutic option for AD. 154
IL‐31: IL‐31 is associated with pruritus that occurs with AD and plays a pathogenic role in the progress of inflammation. Nemolizumab is an IL‐31RA humanised monoclonal antibody which blocks the effects of IL‐31. 155 Ruzicka et al. highlighted the potential for looking at the IL‐31 pathway as treatment option in AD when significant improvements were seen in pruritus scoring in a phase 2, randomised, double‐blind, placebo‐controlled clinical trial. No specific safety signals were noted however the authors discuss that limited size of a trial precludes clear conclusions about potential side effects of nemolizumab. 155 Thus, interference with the immune cell‐neuron‐crosstalk may be an important step to break the itch‐scratch‐inflammation cycle and restore the physical skin barrier.
IL‐33: IL‐33 is an inflammatory cytokine associated with innate immunity and can activate ILC2s. 156 IL‐33 is overexpressed in keratinocytes of AD patients. 157 , 158 The activation of ILC2s may contribute to IL‐33‐driven AD‐like inflammation in mice with increases in IL‐5 and IL‐13. 156 Initial studies on the efficacy of etokimab targeting IL‐33 in human moderate‐to‐severe AD showed moderate but sustained improvement in disease severity. 159 While this improvement is associated with reducing innate inflammatory pathways by inhibiting IL‐33, basophils can also activate ILC2s induced by IL‐33, which work via IL‐4 in AD‐like inflammation in mouse models. 119 IL‐33 has been shown to decrease the expression of filaggrin in the stratum corneum, which decreases the barrier function of the epidermis. 116 Markers that are only expressed on human ILC2s have not been established. By inhibiting IL‐33 the impact on AD of innate and acquired immunity can be modified.
JAK‐STAT: The JAK‐STAT pathway mediates translation of cytokine stimulation into cellular effector function. Recent studies have delivered evidence for the use of JAK inhibitors in treating alopecia areata, psoriasis, vitiligo and AD. 160 , 161
Both topical and oral formulations of JAK inhibitors have been assessed in clinical trials. Topical tofacitinib—targeting JAK1 and JAK3, which are downstream of the receptors for IL‐2, IL‐7, IL‐4, IL‐13, IL‐6, IL‐21, type I and II interferons, and others—was shown to bring about a reduction in EASI versus placebo at week 4 (p < 0.001) as well as significant results in physician global assessment (PGA), body surface area (BSA) and pruritus scores. 162 A pilot study of oral tofacitinib showed a decrease in the scoring of AD during treatment for AD. 163
The nature of broad inhibition of cytokine signalling increases the risk of infections as innate and adaptive immune responses are effectively inhibited. Indeed, infections of the upper respiratory tract are among the most common adverse events. Whether inhibition of JAKs will improve barrier function remains to be determined.
3.3. Advanced therapeutics in psoriasis
TNF: Etanercept (TNF inhibitor), infliximab (chimaeric TNF‐neutralising antibody), adalimumab (anti‐TNFα), certolizumab pegol (pegylated anti‐TNF‐Fab fragment) interfere with the actions of TNF during moderate‐to‐severe psoriasis. As TNF downregulates filaggrin and loricrin, inhibition of TNF can help to restore skin barrier integrity. 164
PDE4: As discussed above, inhibiting PDE4 works in an anti‐inflammatory action rather than immunosuppressant activity. Proinflammatory cytokines are decreased allowing greater expression of anti‐inflammatory mediators by intracellular inhibition of cAMP degradation and increased levels of cAMP at the intracellular level. 165
IL‐12/IL‐23: Ustekinumab binds the p40‐subunit shared by IL‐12 and IL‐23 and inhibits binding to their receptors, thus interfering with the polarization of T cells by innate immunity but increasing the risk of infections as innate immune cells cannot polarise T cells towards TH1. Guselkumab, tildrakizumab and risankizumab bind the p19‐subunit of IL‐23 and prevents the TH17‐polarization of T cells, and subsequently epidermal hyperproliferation, keratinocyte activation and inflammation.
IL‐17: Interference with the IL‐17‐mediated inflammation is achieved by treatment with secukinumab (anti‐IL‐17A), ixekizumab (anti‐IL‐17A), and brodalumab (anti‐IL‐17RA). As IL‐17 can downregulate filaggrin expression in keratinocytes, 166 inhibition of the IL‐17‐pathway alleviates psoriatic symptoms and facilitates skin barrier restoration.
IL‐36R: Targeting the IL‐36 pathway may pose a novel treatment option for generalized pustular psoriasis. 167 A single dose of BI655130/Spesolimab improved skin symptoms in study participants within two weeks. 167 Mechanistically, IL‐36R expressed by IL‐17A‐activated keratinocytes may be blocked by Spesolimab, which may break the pro‐inflammatory cycle during chronic psoriasis and GPP. 168
3.4. Gene polymorphisms and prediction of response to biologicals and other therapies
Similar to the role of gene polymorphisms with regard to disease susceptibility, these polymorphisms also play an important role in the response to therapy. Two extensive reviews of how and which gene polymorphisms influence the efficiency of different therapies for psoriasis have been published by Linares‐Pineda et al. 169 and more recently by Membrive Jiménez et al. 170 Additionally, Prieto‐Perez and co‐workers reviewed anti‐TNF‐treatment efficacy in psoriasis with a focus on related autoimmune disorders, such as rheumatoid arthritis. 171
The majority of the described gene polymorphisms are in relation to TNF and its intracellular signalling, which is probably attributable to the fact that anti‐TNF treatments were first approved and the most prescribed biologic treatments for psoriasis. Even though there are some studies on the association of gene polymorphisms with treatment response to other biologicals (e.g. IL‐12/23 inhibitor ustekinumab), this field requires more in‐depth research to clearly elucidate the roles of different SNPs on the efficacy of advanced treatment options. Currently, there is a lack of research on the influence of SNPs in treatments for diseases other than psoriasis and thus limiting the potential of gene polymorphisms as useful biomarkers in personalised medicine.
4. CONCLUSION
The global research effort to advance our understanding of inflammatory skin diseases, such as psoriasis and AD, led to the recognition of the importance of immunological processes underlying pathogenesis (Figure 1). The realisation that skin barrier integrity can be modified by immunological factors unlocked a new perspective on the genesis of AD. Thus, restoration of an intact functional skin barrier needs to be one of the main objectives in successfully ameliorating AD. The advent of novel treatments targeting single components or shared pathways involved in genesis of skin inflammation will allow us to tailor therapies to the patient across the AD to psoriasis disease spectrum.
CONFLICT OF INTEREST
None to declare.
AUTHOR CONTRIBUTIONS
Heike C. Hawerkamp: Investigation‐Equal, Writing – original draft‐Equal, Writing – review & editing‐Equal; Caoimhe M. R. Fahy: Investigation‐Supporting, Writing – original draft‐Equal, Writing – review & editing‐Equal; Padraic G. Fallon: Conceptualization‐Equal, Investigation‐Equal, Supervision‐Supporting, Writing – original draft‐Equal, Writing – review & editing‐Supporting; Christian Schwartz: Conceptualization‐Equal, Investigation‐Equal, Supervision‐Lead, Visualization‐Lead, Writing – original draft‐Equal, Writing – review & editing‐Equal.
ACKNOWLEDGEMENT
We thank Prof. Alan D. Irvine for critical reading of the manuscript and helpful comments. HCH is an Irish Research Council Government of Ireland Postdoctoral Fellow (GOIPD/2019/193). PGF is supported by the National Children's Research Centre and Science Foundation Ireland (10/IN.1/B3004). CS is supported by the Interdisciplinary Centre for Clinical Research (IZKF) at the University Hospital of the University of Erlangen‐Nuremberg (Junior project J79) and the Else Kröner‐Fresenius‐Stiftung (2019_A181).
Hawerkamp HC, Fahy CMR, Fallon PG, Schwartz C. Break on through: the role of innate immunity and barrier defence in atopic dermatitis and psoriasis. Skin Health Dis. 2022;2(2):e99. 10.1002/ski2.99
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- 1. Salmon JK, Armstrong CA, Ansel JC. The skin as an immune organ. West J Med. 1994;160(2):146–52. [PMC free article] [PubMed] [Google Scholar]
- 2. Eyerich S, Eyerich K, Traidl‐Hoffmann C, Biedermann T. Cutaneous barriers and skin immunity: differentiating A connected network. Trends Immunol. 2018;39(4):315–27. [DOI] [PubMed] [Google Scholar]
- 3. Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ. Skin immune sentinels in health and disease. Nat Rev Immunol. 2009;9(10):679–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heath WR, Carbone FR. The skin‐resident and migratory immune system in steady state and memory: innate lymphocytes, dendritic cells and T cells. Nat Immunol. 2013;14(10):978–85. [DOI] [PubMed] [Google Scholar]
- 5. Hirobe T. Keratinocytes regulate the function of melanocytes. Dermatol Sin. 2014;32(4):200–4. [Google Scholar]
- 6. Sandilands A, Sutherland C, Irvine AD, McLean WH. Filaggrin in the frontline: role in skin barrier function and disease. J Cell Sci. 2009;122(Pt 9):1285–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Elias PM, Steinhoff M. “Outside‐to‐inside” (and now back to “outside”) pathogenic mechanisms in atopic dermatitis. J Invest Dermatol. 2008;128(5):1067–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Palmer CN, Irvine AD, Terron‐Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss‐of‐function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38(4):441–6. [DOI] [PubMed] [Google Scholar]
- 9. Rodriguez E, Baurecht H, Herberich E, Wagenpfeil S, Brown SJ, Cordell HJ, et al. Meta‐analysis of filaggrin polymorphisms in eczema and asthma: robust risk factors in atopic disease. J Allergy Clin Immunol. 2009;123(6):1361–70. [DOI] [PubMed] [Google Scholar]
- 10. Coates M, Blanchard S, MacLeod AS. Innate antimicrobial immunity in the skin: a protective barrier against bacteria, viruses, and fungi. PLoS Pathog. 2018;14(12):e1007353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meller S, Di Domizio J, Voo KS, Friedrich HC, Chamilos G, Ganguly D, et al. T(H)17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat Immunol. 2015;16(9):970–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hawerkamp HC, van Geelen L, Korte J, Di Domizio J, Swidergall M, Momin AA, et al. Interleukin‐26 activates macrophages and facilitates killing of Mycobacterium tuberculosis. Sci Rep. 2020;10(1):17178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kopfnagel V, Dreyer S, Zeitvogel J, Pieper DH, Buch A, Sodeik B, et al. Free human DNA attenuates the activity of antimicrobial peptides in atopic dermatitis. Allergy. 2021;76(10):3145–54. [DOI] [PubMed] [Google Scholar]
- 14. Otsuka M, Egawa G, Kabashima K. Uncovering the mysteries of Langerhans cells, inflammatory dendritic epidermal cells, and monocyte‐derived Langerhans cell‐like cells in the epidermis. Front Immunol. 2018;9:1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Deckers J, Hammad H, Hoste E. Langerhans cells: sensing the environment in health and disease. Front Immunol. 2018;9(93). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim BS. Innate lymphoid cells in the skin. J Invest Dermatol. 2015;135(3):673–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kobayashi T, Ricardo‐Gonzalez RR, Moro K. Skin‐resident innate lymphoid cells ‐ cutaneous innate guardians and regulators. Trends Immunol. 2020;41(2):100–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou S, Li Q, Wu H, Lu Q. The pathogenic role of innate lymphoid cells in autoimmune‐related and inflammatory skin diseases. Cell Mol Immunol. 2020;17(4):335–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim BS. Innate lymphoid cells in the skin. J Invest Dermatol. 2015;135(3):673–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, et al. TSLP elicits IL‐33‐independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med. 2013;5(170):170ra16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, et al. Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J Invest Dermatol. 2014;134(4):984–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pantelyushin S, Haak S, Ingold B, Kulig P, Heppner FL, Navarini AA, et al. Rorgammat+ innate lymphocytes and gammadelta T cells initiate psoriasiform plaque formation in mice. J Clin Invest. 2012;122(6):2252–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sweeney CM, Tobin AM, Kirby B. Innate immunity in the pathogenesis of psoriasis. Arch Dermatol Res. 2011;303(10):691–705. [DOI] [PubMed] [Google Scholar]
- 24. Kondo S, Kono T, Sauder DN, McKenzie RC. IL‐8 gene expression and production in human keratinocytes and their modulation by UVB. J Invest Dermatol. 1993;101(5):690–4. [DOI] [PubMed] [Google Scholar]
- 25. Harper EG, Guo C, Rizzo H, Lillis JV, Kurtz SE, Skorcheva I, et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol. 2009;129(9):2175–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Piskin G, Sylva‐Steenland RM, Bos JD, Teunissen MB. In vitro and in situ expression of IL‐23 by keratinocytes in healthy skin and psoriasis lesions: enhanced expression in psoriatic skin. J Immunol. 2006;176(3):1908–15. [DOI] [PubMed] [Google Scholar]
- 27. Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007;445:866. [DOI] [PubMed] [Google Scholar]
- 28. Henseler T, Christophers E. Disease concomitance in psoriasis. J Am Acad Dermatol. 1995;32(6):982–6. [DOI] [PubMed] [Google Scholar]
- 29. Griffiths CEM, Barker JNWN. Pathogenesis and clinical features of psoriasis. Lancet. 2007;370(9583):263–71. [DOI] [PubMed] [Google Scholar]
- 30. Lowes MA, Kikuchi T, Fuentes‐Duculan J, Cardinale I, Zaba LC, Haider AS, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol. 2008;128(5):1207–11. [DOI] [PubMed] [Google Scholar]
- 31. Boniface K, Bernard F‐X, Garcia M, Gurney AL, Lecron J‐C, Morel F. IL‐22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005;174(6):3695–702. [DOI] [PubMed] [Google Scholar]
- 32. Van Belle AB, de Heusch M, Lemaire MM, Hendrickx E, Warnier G, Dunussi‐Joannopoulos K, et al. IL‐22 is required for Imiquimod‐induced psoriasiform skin inflammation in mice. J Immunol. 2012;188(1):462–9. [DOI] [PubMed] [Google Scholar]
- 33. Wolk K, Haugen HS, Xu W, Witte E, Waggie K, Anderson M, et al. IL‐22 and IL‐20 are key mediators of the epidermal alterations in psoriasis while IL‐17 and IFN‐γ are not. J Mol Med. 2009;87(5):523–36. [DOI] [PubMed] [Google Scholar]
- 34. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, cytokine profile and function of human interleukin 17‐producing helper T cells. Nat Immunol. 2007;8(9):950–7. [DOI] [PubMed] [Google Scholar]
- 35. Schon MP. Adaptive and innate immunity in psoriasis and other inflammatory disorders. Front Immunol. 2019;10:1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Christophers E, van de Kerkhof PCM. Severity, heterogeneity and systemic inflammation in psoriasis. J Eur Acad Dermatol Venereol. 2019;33(4):643–7. [DOI] [PubMed] [Google Scholar]
- 37. Pantelyushin S, Haak S, Ingold B, Kulig P, Heppner FL, Navarini AA, et al. Rorγt+ innate lymphocytes and γδ T cells initiate psoriasiform plaque formation in mice. J Clin Investig. 2012;122(6):2252–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bielecki P, Riesenfeld SJ, Hutter JC, Torlai Triglia E, Kowalczyk MS, Ricardo‐Gonzalez RR, et al. Skin‐resident innate lymphoid cells converge on a pathogenic effector state. Nature. 2021;592(7852):128–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cumberbatch M, Singh M, Dearman RJ, Young HS, Kimber I, Griffiths CEM. Impaired Langerhans cell migration in psoriasis. J Exp Med. 2006;203(4):953–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Furue M, Furue K, Tsuji G, Nakahara T. Interleukin‐17A and keratinocytes in psoriasis. Int J Mol Sci. 2020;21(4):1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leung DYM, Bieber T. Atopic dermatitis. Lancet. 2003;361(9352):151–60. [DOI] [PubMed] [Google Scholar]
- 42. Leung DY, Boguniewicz M, Howell MD, Nomura I, Hamid QA. New insights into atopic dermatitis. J Clin Invest. 2003;113(5):651–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Leung DY. New insights into atopic dermatitis: role of skin barrier and immune dysregulation. Allergol Int. 2013;62(2):151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Novak N, Bieber T, Leung DYM. Immune mechanisms leading to atopic dermatitis. J Allergy Clin Immunol. 2003;112(6):S128–S39. [DOI] [PubMed] [Google Scholar]
- 45. Weidinger S, Novak N. Atopic dermatitis. Lancet. 2016;387(10023):1109–22. [DOI] [PubMed] [Google Scholar]
- 46. Hamid Q, Boguniewicz M, Leung DY. Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J Clin Invest. 1994;94(2):870–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Grewe M, Gyufko K, Schopf E, Krutmann J. Lesional expression of interferon‐gamma in atopic eczema. Lancet. 1994;343(8888):25–6. [DOI] [PubMed] [Google Scholar]
- 48. Grewe M, Bruijnzeel‐Koomen CAFM, Schöpf E, Thepen T, Langeveld‐Wildschut AG, Ruzicka T, et al. A role for Th1 and Th2 cells in the immunopathogenesis of atopic dermatitis. Immunol Today. 1998;19(8):359–61. [DOI] [PubMed] [Google Scholar]
- 49. Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, et al. TSLP elicits IL‐33‐independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med. 2013;5(170):170ra16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Orimo K, Tamari M, Saito H, Matsumoto K, Nakae S, Morita H. Characteristics of tissue‐resident ILCs and their potential as therapeutic targets in mucosal and skin inflammatory diseases. Allergy. 2021;76(11):3332–48. [DOI] [PubMed] [Google Scholar]
- 51. Bartemes KR, Kita H. Roles of innate lymphoid cells (ILCs) in allergic diseases: the 10‐year anniversary for ILC2s. J Allergy Clin Immunol. 2021;147(5):1531–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3(7):673–80. [DOI] [PubMed] [Google Scholar]
- 53. Indra AK. Epidermal TSLP: a trigger factor for pathogenesis of atopic dermatitis. Expert Rev Proteomics. 2013;10(4):309–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wilson SR, The L, Batia LM, Beattie K, Katibah GE, McClain SP, et al. The epithelial cell‐derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell. 2013;155(2):285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Leyden JJ, Marples RR, Kligman AM. Staphylococcus aureus in the lesions of atopic dermatitis. Br J Dermatol. 1974;90(5):525–30. [DOI] [PubMed] [Google Scholar]
- 56. Leung DY. Infection in atopic dermatitis. Curr Opin Pediatr. 2003;15(4):399–404. [DOI] [PubMed] [Google Scholar]
- 57. McGirt LY, Beck LA. Innate immune defects in atopic dermatitis. J Allergy Clin Immunol. 2006;118(1):202–8. [DOI] [PubMed] [Google Scholar]
- 58. Blicharz L, Usarek P, Mlynarczyk G, Skowronski K, Rudnicka L, Samochocki Z. Is itch intensity in atopic dermatitis associated with skin colonization by Staphylococcus aureus? Indian J Dermatol. 2020;65(1):17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. de Jongh GJ, Zeeuwen PLJM, Kucharekova M, Pfundt R, van der Valk PG, Blokx W, et al. High expression levels of keratinocyte antimicrobial proteins in psoriasis compared with atopic dermatitis. J Invest Dermatol. 2005;125(6):1163–73. [DOI] [PubMed] [Google Scholar]
- 60. Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347(15):1151–60. [DOI] [PubMed] [Google Scholar]
- 61. Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347(15):1151–60. [DOI] [PubMed] [Google Scholar]
- 62. Howell MD, Novak N, Bieber T, Pastore S, Girolomoni G, Boguniewicz M, et al. Interleukin‐10 downregulates anti‐microbial peptide expression in atopic dermatitis. J Invest Dermatol. 2005;125(4):738–45. [DOI] [PubMed] [Google Scholar]
- 63. Lee H, Ryu WI, Kim HJ, Bae HC, Ryu HJ, Shin JJ, et al. TSLP down‐regulates S100A7 and ss‐defensin 2 via the JAK2/STAT3‐dependent mechanism. J Invest Dermatol. 2016;136(12):2427–35. [DOI] [PubMed] [Google Scholar]
- 64. Baurecht H, Hotze M, Brand S, Buning C, Cormican P, Corvin A, et al. Genome‐wide comparative analysis of atopic dermatitis and psoriasis gives insight into opposing genetic mechanisms. Am J Hum Genet. 2015;96(1):104–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Martin MJ, Estravis M, Garcia‐Sanchez A, Davila I, Isidoro‐Garcia M, Sanz C. Genetics and epigenetics of atopic dermatitis: an updated systematic review. Genes. 2020;11(4):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ogawa K, Okada Y. The current landscape of psoriasis genetics in 2020. J Dermatol Sci. 2020;99(1):2–8. [DOI] [PubMed] [Google Scholar]
- 67. Shimizu M, Matsuda A, Yanagisawa K, Hirota T, Akahoshi M, Inomata N, et al. Functional SNPs in the distal promoter of the ST2 gene are associated with atopic dermatitis. Hum Mol Genet. 2005;14(19):2919–27. [DOI] [PubMed] [Google Scholar]
- 68. Gao PS, Rafaels NM, Mu D, Hand T, Murray T, Boguniewicz M, et al. Genetic variants in thymic stromal lymphopoietin are associated with atopic dermatitis and eczema herpeticum. J Allergy Clin Immunol. 2010;125(6):1403–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Novak N, Kruse S, Potreck J, Maintz L, Jenneck C, Weidinger S, et al. Single nucleotide polymorphisms of the IL18 gene are associated with atopic eczema. J Allergy Clin Immunol. 2005;115(4):828–33. [DOI] [PubMed] [Google Scholar]
- 70. Novota P, Kolostova K, Pinterova D, Novak J, Treslova L, Andel M, et al. Interleukin IL‐18 gene promoter polymorphisms in adult patients with type 1 diabetes mellitus and latent autoimmune diabetes in adults. Immunol Lett. 2005;96(2):247–51. [DOI] [PubMed] [Google Scholar]
- 71. Ricardo‐Gonzalez RR, Van Dyken SJ, Schneider C, Lee J, Nussbaum JC, Liang HE, et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat Immunol. 2018;19(10):1093–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nair RP, Stuart PE, Nistor I, Hiremagalore R, Chia NVC, Jenisch S, et al. Sequence and haplotype analysis supports HLA‐C as the psoriasis susceptibility 1 gene. Am J Hum Genet. 2006;78(5):827–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Henseler T, Christophers E. Psoriasis of early and late onset: characterization of two types of psoriasis vulgaris. J Am Acad Dermatol. 1985;13(3):450–6. [DOI] [PubMed] [Google Scholar]
- 74. Nograles KE, Davidovici B, Krueger JG. New insights in the immunologic basis of psoriasis. Semin Cutan Med Surg. 2010;29(1):3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Marrakchi S, Guigue P, Renshaw BR, Puel A, Pei XY, Fraitag S, et al. Interleukin‐36‐receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med. 2011;365(7):620–8. [DOI] [PubMed] [Google Scholar]
- 76. Hernandez‐Santana YE, Leon G, St Leger D, Fallon PG, Walsh PT. Keratinocyte interleukin‐36 receptor expression orchestrates psoriasiform inflammation in mice. Life Sci Alliance. 2020;3(4):e201900586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Loft ND, Skov L, Rasmussen MK, Gniadecki R, Dam TN, Brandslund I, et al. Genetic polymorphisms associated with psoriasis and development of psoriatic arthritis in patients with psoriasis. PLoS One. 2018;13(2):e0192010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tsunemi Y, Saeki H, Nakamura K, Sekiya T, Hirai K, Fujita H, et al. Interleukin‐12 p40 gene (IL12B) 3'‐untranslated region polymorphism is associated with susceptibility to atopic dermatitis and psoriasis vulgaris. J Dermatol Sci. 2002;30(2):161–6. [DOI] [PubMed] [Google Scholar]
- 79. Wang Y, Wang LL, Yang HY, Wang FF, Zhang XX, Bai YP. Interleukin‐21 is associated with the severity of psoriasis vulgaris through promoting CD4+ T cells to differentiate into Th17 cells. Am J Transl Res. 2016;8(7):3188–96. [PMC free article] [PubMed] [Google Scholar]
- 80. Liu Y, Helms C, Liao W, Zaba LC, Duan S, Gardner J, et al. A genome‐wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet. 2008;4(3):e1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Caruso R, Botti E, Sarra M, Esposito M, Stolfi C, Diluvio L, et al. Involvement of interleukin‐21 in the epidermal hyperplasia of psoriasis. Nat Med. 2009;15(9):1013–15. [DOI] [PubMed] [Google Scholar]
- 82. Vandeghinste N, Klattig J, Jagerschmidt C, Lavazais S, Marsais F, Haas JD, et al. Neutralization of IL‐17C reduces skin inflammation in mouse models of psoriasis and atopic dermatitis. J Invest Dermatol. 2018;138(7):1555–63. [DOI] [PubMed] [Google Scholar]
- 83. Floudas A, Saunders SP, Moran T, Schwartz C, Hams E, Fitzgerald DC, et al. IL‐17 receptor A maintains and protects the skin barrier to prevent allergic skin inflammation. J Immunol. 2017;199(2):707–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kimura A, Kishimoto T. IL‐6: regulator of Treg/Th17 balance. Eur J Immunol. 2010;40(7):1830–5. [DOI] [PubMed] [Google Scholar]
- 85. Boca AN, Talamonti M, Galluzzo M, Botti E, Vesa SC, Chimenti S, et al. Genetic variations in IL6 and IL12B decreasing the risk for psoriasis. Immunol Lett. 2013;156(1‐2):127–31. [DOI] [PubMed] [Google Scholar]
- 86. Gharagozlou M, Farhadi E, Khaledi M, Behniafard N, Sotoudeh S, Salari R, et al. Association between the interleukin 6 genotype at position ‐174 and atopic dermatitis. J Investig Allergol Clin Immunol. 2013;23(2):89–93. [PubMed] [Google Scholar]
- 87. Navarini AA, French LE, Hofbauer GF. Interrupting IL‐6‐receptor signaling improves atopic dermatitis but associates with bacterial superinfection. J Allergy Clin Immunol. 2011;128(5):1128–30. [DOI] [PubMed] [Google Scholar]
- 88. de Guia RM, Ramos JD. The ‐590C/TIL4 single‐nucleotide polymorphism as a genetic factor of atopic allergy. Int J Mol Epidemiol Genet. 2010;1(1):67–73. [PMC free article] [PubMed] [Google Scholar]
- 89. Weidinger S, Willis‐Owen SA, Kamatani Y, Baurecht H, Morar N, Liang L, et al. A genome‐wide association study of atopic dermatitis identifies loci with overlapping effects on asthma and psoriasis. Hum Mol Genet. 2013;22(23):4841–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ghoreschi K, Thomas P, Breit S, Dugas M, Mailhammer R, van Eden W, et al. Interleukin‐4 therapy of psoriasis induces Th2 responses and improves human autoimmune disease. Nat Med. 2003;9(1):40–6. [DOI] [PubMed] [Google Scholar]
- 91. Tamura K, Suzuki M, Arakawa H, Tokuyama K, Morikawa A. Linkage and association studies of STAT6 gene polymorphisms and allergic diseases. Int Arch Allergy Immunol. 2003;131(1):33–8. [DOI] [PubMed] [Google Scholar]
- 92. Sehra S, Yao Y, Howell MD, Nguyen ET, Kansas GS, Leung DY, et al. IL‐4 regulates skin homeostasis and the predisposition toward allergic skin inflammation. J Immunol. 2010;184(6):3186–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Oh IY, de Guzman Strong C. The molecular revolution in cutaneous biology: EDC and locus control. J Invest Dermatol. 2017;137(5):e101–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pajic P, Lin YL, Xu D, Gokcumen O. The psoriasis‐associated deletion of late cornified envelope genes LCE3B and LCE3C has been maintained under balancing selection since human denisovan divergence. BMC Evol Biol. 2016;16(1):265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Smith FJ, Irvine AD, Terron‐Kwiatkowski A, Sandilands A, Campbell LE, Zhao Y, et al. Loss‐of‐function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet. 2006;38(3):337–42. [DOI] [PubMed] [Google Scholar]
- 96. Riethmuller C, McAleer MA, Koppes SA, Abdayem R, Franz J, Haftek M, et al. Filaggrin breakdown products determine corneocyte conformation in patients with atopic dermatitis. J Allergy Clin Immunol. 2015;136(6):1573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Saunders SP, Floudas A, Moran T, Byrne CM, Rooney MD, Fahy CMR, et al. Dysregulated skin barrier function in Tmem79 mutant mice promotes IL‐17A‐dependent spontaneous skin and lung inflammation. Allergy. 2020. [DOI] [PubMed] [Google Scholar]
- 98. Saunders SP, Goh CS, Brown SJ, Palmer CN, Porter RM, Cole C, et al. Tmem79/Matt is the matted mouse gene and is a predisposing gene for atopic dermatitis in human subjects. J Allergy Clin Immunol. 2013;132(5):1121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Elias MS, Wright SC, Remenyi J, Abbott JC, Bray SE, Cole C, et al. EMSY expression affects multiple components of the skin barrier with relevance to atopic dermatitis. J Allergy Clin Immunol. 2019;144(2):470–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. De Benedetto A, Rafaels NM, McGirt LY, Ivanov AI, Georas SN, Cheadle C, et al. Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol. 2011;127(3):773–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Patel NA, Patel JA, Stins MF, Kim KS, Chang SL. Dexamethasone affects cytokine‐mediated adhesion of HL‐60 human promyelocytic leukemia cells to cultured dermal microvascular endothelial cells. Clin Immunol. 2001;99(3):387–94. 10.1006/clim.2001.5029 [DOI] [PubMed] [Google Scholar]
- 102. Giustizieri ML, Mascia F, Frezzolini A, De Pita O, Chinni LM, Giannetti A, et al. Keratinocytes from patients with atopic dermatitis and psoriasis show a distinct chemokine production profile in response to T cell‐derived cytokines. J Allergy Clin Immunol. 2001;107(5):871–7. [DOI] [PubMed] [Google Scholar]
- 103. Igawa S, Kishibe M, Minami‐Hori M, Honma M, Tsujimura H, Ishikawa J, et al. Incomplete KLK7 secretion and upregulated LEKTI expression underlie hyperkeratotic stratum corneum in atopic dermatitis. J Invest Dermatol. 2017;137(2):449–56. [DOI] [PubMed] [Google Scholar]
- 104. Briot A, Deraison C, Lacroix M, Bonnart C, Robin A, Besson C, et al. Kallikrein 5 induces atopic dermatitis‐like lesions through PAR2‐mediated thymic stromal lymphopoietin expression in Netherton syndrome. J Exp Med. 2009;206(5):1135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi‐Joannopoulos K, Collins M, et al. Interleukin (IL)‐22 and IL‐17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203(10):2271–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Harder J, Bartels J, Christophers E, Schroder JM. A peptide antibiotic from human skin. Nature. 1997;387(6636):861. [DOI] [PubMed] [Google Scholar]
- 107. Wollenberg A, Rawer HC, Schauber J. Innate immunity in atopic dermatitis. Clin Rev Allergy Immunol. 2011;41(3):272–81. [DOI] [PubMed] [Google Scholar]
- 108. de Jongh GJ, Zeeuwen PL, Kucharekova M, Pfundt R, van der Valk PG, Blokx W, et al. High expression levels of keratinocyte antimicrobial proteins in psoriasis compared with atopic dermatitis. J Invest Dermatol. 2005;125(6):1163–73. [DOI] [PubMed] [Google Scholar]
- 109. Ballardini N, Johansson C, Lilja G, Lindh M, Linde Y, Scheynius A, et al. Enhanced expression of the antimicrobial peptide LL‐37 in lesional skin of adults with atopic eczema. Br J Dermatol. 2009;161(1):40–7. [DOI] [PubMed] [Google Scholar]
- 110. Zanetti M. Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol. 2004;75(1):39–48. [DOI] [PubMed] [Google Scholar]
- 111. Schauber J, Dorschner RA, Coda AB, Buchau AS, Liu PT, Kiken D, et al. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D‐dependent mechanism. J Clin Invest. 2007;117(3):803–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Leyva‐Castillo JM, Das M, Kane J, Strakosha M, Singh S, Wong DSH, et al. Basophil‐derived IL‐4 promotes cutaneous Staphylococcus aureus infection. JCI Insight. 2021;6(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Strid J, McLean WHI, Irvine AD. Too much, too little or just enough: a goldilocks effect for IL‐13 and skin barrier regulation? J Invest Dermatol. 2016;136(3):561–4. [DOI] [PubMed] [Google Scholar]
- 114. Sivaprasad U, Warrier MR, Gibson AM, Chen W, Tabata Y, Bass SA, et al. IL‐13Ralpha2 has a protective role in a mouse model of cutaneous inflammation. J Immunol. 2010;185(11):6802–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Mayer JU, Hilligan KL, Chandler JS, Eccles DA, Old SI, Domingues RG, et al. Homeostatic IL‐13 in healthy skin directs dendritic cell differentiation to promote TH2 and inhibit TH17 cell polarization. Nat Immunol. 2021;22(12):1538–50. [DOI] [PubMed] [Google Scholar]
- 116. Seltmann J, Roesner LM, von Hesler FW, Wittmann M, Werfel T. IL‐33 impacts on the skin barrier by downregulating the expression of filaggrin. J Allergy Clin Immunol. 2015;135(6):1659–61. [DOI] [PubMed] [Google Scholar]
- 117. Seltmann J, Werfel T, Wittmann M. Evidence for a regulatory loop between IFN‐gamma and IL‐33 in skin inflammation. Exp Dermatol. 2013;22(2):102–7. [DOI] [PubMed] [Google Scholar]
- 118. Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska‐Owsiak D, Wang X, et al. A role for IL‐25 and IL‐33‐driven type‐2 innate lymphoid cells in atopic dermatitis. J Exp Med. 2013;210(13):2939–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Imai Y, Yasuda K, Nagai M, Kusakabe M, Kubo M, Nakanishi K, et al. IL‐33‐Induced atopic dermatitis‐like inflammation in mice is mediated by group 2 innate lymphoid cells in concert with basophils. J Invest Dermatol. 2019;139(10):2185–94. [DOI] [PubMed] [Google Scholar]
- 120. Ryu WI, Lee H, Bae HC, Jeon J, Ryu HJ, Kim J, et al. IL‐33 down‐regulates CLDN1 expression through the ERK/STAT3 pathway in keratinocytes. J Dermatol Sci. 2018;90(3):313–22. [DOI] [PubMed] [Google Scholar]
- 121. Imai Y, Yasuda K, Sakaguchi Y, Haneda T, Mizutani H, Yoshimoto T, et al. Skin‐specific expression of IL‐33 activates group 2 innate lymphoid cells and elicits atopic dermatitis‐like inflammation in mice. Proc Natl Acad Sci U S A. 2013;110(34):13921–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Meixiong J, Anderson M, Limjunyawong N, Sabbagh MF, Hu E, Mack MR, et al. Activation of mast‐cell‐expressed mas‐related G‐protein‐coupled receptors drives non‐histaminergic itch. Immunity. 2019;50(5):1163–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Oetjen LK, Mack MR, Feng J, Whelan TM, Niu H, Guo CJ, et al. Sensory neurons Co‐opt classical immune signaling pathways to mediate chronic itch. Cell. 2017;171(1):217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Simpson EL, Bieber T, Guttman‐Yassky E, Beck LA, Blauvelt A, Cork MJ, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375(24):2335–48. [DOI] [PubMed] [Google Scholar]
- 125. Nakagawa H, Nemoto O, Yamada H, Nagata T, Ninomiya N. Phase 1 studies to assess the safety, tolerability and pharmacokinetics of JTE‐052 (a novel Janus kinase inhibitor) ointment in Japanese healthy volunteers and patients with atopic dermatitis. J Dermatol. 2018;45(6):701–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Langan SM, Irvine AD, Weidinger S. Atopic dermatitis. Lancet. 2020;396(10247):345–60. [DOI] [PubMed] [Google Scholar]
- 127. Yosipovitch G, Misery L, Proksch E, Metz M, Stander S, Schmelz M. Skin barrier damage and itch: review of mechanisms, topical management and future directions. Acta Derm Venereol. 2019;99(13):1201–9. [DOI] [PubMed] [Google Scholar]
- 128. Bingham. Guidelines to management of atopic dermatitis. Wiley; 2005. [Google Scholar]
- 129. O’Neill JL, Feldman SR. Vitamine D analogue‐based therapies for psoriasis. Drugs Today. 2010;46(5):351–60. [DOI] [PubMed] [Google Scholar]
- 130. Reinholz M, Schauber J. [Vitamin D and innate immunity of the skin]. Dtsch Med Wochenschr. 2012;137(46):2385–9. [DOI] [PubMed] [Google Scholar]
- 131. Grassberger M, Baumruker T, Enz A, Hiestand P, Hultsch T, Kalthoff F, et al. A novel anti‐inflammatory drug, SDZ ASM 981, for the treatment of skin diseases: in vitro pharmacology. Br J Dermatol. 1999;141(2):264–73. [DOI] [PubMed] [Google Scholar]
- 132. Jensen JM, Pfeiffer S, Witt M, Brautigam M, Neumann C, Weichenthal M, et al. Different effects of pimecrolimus and betamethasone on the skin barrier in patients with atopic dermatitis. J Allergy Clin Immunol. 2009;123(5):1124–33. [DOI] [PubMed] [Google Scholar]
- 133. Kawashima M, Nakagawa H, Ohtsuki M, Tamaki K, Ishibashi Y. Tacrolimus concentrations in blood during topical treatment of atopic dermatitis. Lancet. 1996;348(9036):1240–1. [DOI] [PubMed] [Google Scholar]
- 134. Alaiti S, Kang S, Fiedler VC, Ellis CN, Spurlin DV, Fader D, et al. Tacrolimus (FK506) ointment for atopic dermatitis: a phase I study in adults and children. J Am Acad Dermatol. 1998;38(1):69–76. [DOI] [PubMed] [Google Scholar]
- 135. Fleischer AB, Jr. , Ling M, Eichenfield L, Satoi Y, Jaracz E, Rico MJ, et al. Tacrolimus ointment for the treatment of atopic dermatitis is not associated with an increase in cutaneous infections. J Am Acad Dermatol. 2002;47(4):562–70. [DOI] [PubMed] [Google Scholar]
- 136. Kim M, Jung M, Hong SP, Jeon H, Kim MJ, Cho MY, et al. Topical calcineurin inhibitors compromise stratum corneum integrity, epidermal permeability and antimicrobial barrier function. Exp Dermatol. 2010;19(6):501–10. [DOI] [PubMed] [Google Scholar]
- 137. Torsekar R, Gautam MM. Topical therapies in psoriasis. Indian Dermatol Online J. 2017;8(4):235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Berth‐Jones J, Takwale A, Tan E, Barclay G, Agarwal S, Ahmed I, et al. Azathioprine in severe adult atopic dermatitis: a double‐blind, placebo‐controlled, crossover trial. Br J Dermatol. 2002;147(2):324–30. [DOI] [PubMed] [Google Scholar]
- 139. Schram ME, Roekevisch E, Leeflang MM, Bos JD, Schmitt J, Spuls PI. A randomized trial of methotrexate versus azathioprine for severe atopic eczema. J Allergy Clin Immunol. 2011;128(2):353–9. [DOI] [PubMed] [Google Scholar]
- 140. Khattri S, Shemer A, Rozenblit M, Dhingra N, Czarnowicki T, Finney R, et al. Cyclosporine in patients with atopic dermatitis modulates activated inflammatory pathways and reverses epidermal pathology. J Allergy Clin Immunol. 2014;133(6):1626–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Oh CJ, Das KM, Gottlieb AB. Treatment with anti‐tumor necrosis factor alpha (TNF‐alpha) monoclonal antibody dramatically decreases the clinical activity of psoriasis lesions. J Am Acad Dermatol. 2000;42(5 Pt 1):829–30. [DOI] [PubMed] [Google Scholar]
- 142. Wollenberg A, Barbarot S, Bieber T, Christen‐Zaech S, Deleuran M, Fink‐Wagner A, et al. Consensus‐based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part II. J Eur Acad Dermatol Venereol. 2018;32(6):850–78. [DOI] [PubMed] [Google Scholar]
- 143. Simpson EL, Parnes JR, She D, Crouch S, Rees W, Mo M, et al. Tezepelumab, an anti‐thymic stromal lymphopoietin monoclonal antibody, in the treatment of moderate to severe atopic dermatitis: a randomized phase 2a clinical trial. J Am Acad Dermatol. 2019;80(4):1013–21. [DOI] [PubMed] [Google Scholar]
- 144. Hanifin JM, Chan SC, Cheng JB, Tofte SJ, Henderson WR Jr., Kirby DS, et al. Type 4 phosphodiesterase inhibitors have clinical and in vitro anti‐inflammatory effects in atopic dermatitis. J Invest Dermatol. 1996;107(1):51–6. [DOI] [PubMed] [Google Scholar]
- 145. Yang H, Wang J, Zhang X, Zhang Y, Qin ZL, Wang H, et al. Application of topical phosphodiesterase 4 inhibitors in mild to moderate atopic dermatitis: a systematic review and meta‐analysis. JAMA Dermatol. 2019;155(5):585–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Samrao A, Berry TM, Goreshi R, Simpson EL. A pilot study of an oral phosphodiesterase inhibitor (apremilast) for atopic dermatitis in adults. Arch Dermatol. 2012;148(8):890–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Beck LA, Thaci D, Hamilton JD, Graham NM, Bieber T, Rocklin R, et al. Dupilumab treatment in adults with moderate‐to‐severe atopic dermatitis. N Engl J Med. 2014;371(2):130–9. [DOI] [PubMed] [Google Scholar]
- 148. de Bruin‐Weller M, Thaci D, Smith CH, Reich K, Cork MJ, Radin A, et al. Dupilumab with concomitant topical corticosteroid treatment in adults with atopic dermatitis with an inadequate response or intolerance to ciclosporin A or when this treatment is medically inadvisable: a placebo‐controlled, randomized phase III clinical trial (LIBERTY AD CAFE). Br J Dermatol. 2018;178(5):1083–101. [DOI] [PubMed] [Google Scholar]
- 149. Guttman‐Yassky E, Bissonnette R, Ungar B, Suarez‐Farinas M, Ardeleanu M, Esaki H, et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J Allergy Clin Immunol. 2019;143(1):155–72. [DOI] [PubMed] [Google Scholar]
- 150. Wollenberg A, Howell MD, Guttman‐Yassky E, Silverberg JI, Kell C, Ranade K, et al. Treatment of atopic dermatitis with tralokinumab, an anti‐IL‐13 mAb. J Allergy Clin Immunol. 2019;143(1):135–41. [DOI] [PubMed] [Google Scholar]
- 151. Simpson EL, Flohr C, Eichenfield LF, Bieber T, Sofen H, Taieb A, et al. Efficacy and safety of lebrikizumab (an anti‐IL‐13 monoclonal antibody) in adults with moderate‐to‐severe atopic dermatitis inadequately controlled by topical corticosteroids: a randomized, placebo‐controlled phase II trial (TREBLE). J Am Acad Dermatol. 2018;78(5):863–71. [DOI] [PubMed] [Google Scholar]
- 152. Guttman‐Yassky E, Blauvelt A, Eichenfield LF, Paller AS, Armstrong AW, Drew J, et al. Efficacy and safety of lebrikizumab, a high‐affinity interleukin 13 inhibitor, in adults with moderate to severe atopic dermatitis: a phase 2b randomized clinical trial. JAMA Dermatol. 2020;156(4):411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Oyoshi MK, He R, Kumar L, Yoon J, Geha RS. Cellular and molecular mechanisms in atopic dermatitis. Adv Immunol. 2009;102:135–226. [DOI] [PubMed] [Google Scholar]
- 154. Hu Y, Wang J, Zhang H, Xie H, Song W, Jiang Q, et al. Enhanced expression of IL‐18 and IL‐18BP in plasma of patients with eczema: altered expression of IL‐18BP and IL‐18 receptor on mast cells. Mediat Inflamm. 2017;2017:3090782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Ruzicka T, Mihara R. Anti‐Interleukin‐31 receptor A antibody for atopic dermatitis. N Engl J Med. 2017;376(21):2093. [DOI] [PubMed] [Google Scholar]
- 156. Imai Y. Interleukin‐33 in atopic dermatitis. J Dermatol Sci. 2019;96(1):2–7. [DOI] [PubMed] [Google Scholar]
- 157. Savinko T, Matikainen S, Saarialho‐Kere U, Lehto M, Wang G, Lehtimaki S, et al. IL‐33 and ST2 in atopic dermatitis: expression profiles and modulation by triggering factors. J Invest Dermatol. 2012;132(5):1392–400. [DOI] [PubMed] [Google Scholar]
- 158. Nakamura N, Tamagawa‐Mineoka R, Yasuike R, Masuda K, Matsunaka H, Murakami Y, et al. Stratum corneum interleukin‐33 expressions correlate with the degree of lichenification and pruritus in atopic dermatitis lesions. Clin Immunol. 2019;201:1–3. [DOI] [PubMed] [Google Scholar]
- 159. Chen YL, Gutowska‐Owsiak D, Hardman CS, Westmoreland M, MacKenzie T, Cifuentes L, et al. Proof‐of‐concept clinical trial of etokimab shows a key role for IL‐33 in atopic dermatitis pathogenesis. Sci Transl Med. 2019;11(515). [DOI] [PubMed] [Google Scholar]
- 160. Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76(4):736–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Cotter DG, Schairer D, Eichenfield L. Emerging therapies for atopic dermatitis: JAK inhibitors. J Am Acad Dermatol. 2018;78(3 Suppl 1):S53–62. [DOI] [PubMed] [Google Scholar]
- 162. Bissonnette R. JAK inhibitors appear to have a bright future in the treatment of atopic dermatitis. Br J Dermatol. 2018;178(2): 321. [DOI] [PubMed] [Google Scholar]
- 163. Levy LL, Urban J, King BA. Treatment of recalcitrant atopic dermatitis with the oral Janus kinase inhibitor tofacitinib citrate. J Am Acad Dermatol. 2015;73(3):395–9. [DOI] [PubMed] [Google Scholar]
- 164. Kim BE, Howell MD, Guttman‐Yassky E, Gilleaudeau PM, Cardinale IR, Boguniewicz M, et al. TNF‐alpha downregulates filaggrin and loricrin through c‐Jun N‐terminal kinase: role for TNF‐alpha antagonists to improve skin barrier. J Invest Dermatol. 2011;131(6):1272–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Torres T, Puig L. Apremilast: a novel oral treatment for psoriasis and psoriatic arthritis. Am J Clin Dermatol. 2018;19(1):23–32. [DOI] [PubMed] [Google Scholar]
- 166. Gutowska‐Owsiak D, Schaupp AL, Salimi M, Selvakumar TA, McPherson T, Taylor S, et al. IL‐17 downregulates filaggrin and affects keratinocyte expression of genes associated with cellular adhesion. Exp Dermatol. 2012;21(2):104–10. [DOI] [PubMed] [Google Scholar]
- 167. Bachelez H, Choon SE, Marrakchi S, Burden AD, Tsai TF, Morita A, et al. Inhibition of the interleukin‐36 pathway for the treatment of generalized pustular psoriasis. N Engl J Med. 2019;380(10):981–3. [DOI] [PubMed] [Google Scholar]
- 168. Madonna S, Girolomoni G, Dinarello CA, Albanesi C. The significance of IL‐36 hyperactivation and IL‐36R targeting in psoriasis. Int J Mol Sci. 2019;20(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Linares‐Pineda TM, Canadas‐Garre M, Sanchez‐Pozo A, Calleja‐Hernandez MA. Gene polymorphisms as predictors of response to biological therapies in psoriasis patients. Pharmacol Res. 2016;113(Pt A):71–80. [DOI] [PubMed] [Google Scholar]
- 170. Membrive Jimenez C, Perez Ramirez C, Sanchez Martin A, Vieira Maroun S, Arias Santiago SA, Ramirez Tortosa MDC, et al. Influence of genetic polymorphisms on response to biologics in moderate‐to‐severe psoriasis. J Pers Med. 2021;11(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Prieto‐Perez R, Cabaleiro T, Dauden E, Abad‐Santos F. Gene polymorphisms that can predict response to anti‐TNF therapy in patients with psoriasis and related autoimmune diseases. Pharmacogenomics J. 2013;13(4):297–305. [DOI] [PubMed] [Google Scholar]
- 172. Paternoster L, Standl M, Waage J, Baurecht H, Hotze M, Strachan DP, et al. Multi‐ancestry genome‐wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet. 2015;47(12):1449–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Sliz E, Huilaja L, Pasanen A, Laisk T, Reimann E, Magi R, et al. Uniting biobank resources reveals novel genetic pathways modulating susceptibility for atopic dermatitis. J Allergy Clin Immunol. 2021. [DOI] [PubMed] [Google Scholar]
- 174. Hirota T, Takahashi A, Kubo M, Tsunoda T, Tomita K, Sakashita M, et al. Genome‐wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nat Genet. 2012;44(11):1222–6. [DOI] [PubMed] [Google Scholar]
- 175. Kichaev G, Bhatia G, Loh PR, Gazal S, Burch K, Freund MK, et al. Leveraging polygenic functional enrichment to improve GWAS power. Am J Hum Genet. 2019;104(1):65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Johansson A, Rask‐Andersen M, Karlsson T, Ek WE. Genome‐wide association analysis of 350 000 Caucasians from the UK Biobank identifies novel loci for asthma, hay fever and eczema. Hum Mol Genet. 2019;28(23):4022–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Ko EJ, Heo WI, Park KY, Lee MK, Seo SJ. Genetic polymorphism of thymic stromal lymphopoietin in Korean patients with atopic dermatitis and allergic march. J Eur Acad Dermatol Venereol. 2018;32(12):e468–70. [DOI] [PubMed] [Google Scholar]
- 178. Heo WI, Park KY, Lee MK, Moon NJ, Seo SJ. TSLP polymorphisms in atopic dermatitis and atopic march in Koreans. Ann Dermatol. 2018;30(5):529–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Wang IJ, Wu LS, Lockett GA, Karmaus WJ. TSLP polymorphisms, allergen exposures, and the risk of atopic disorders in children. Ann Allergy Asthma Immunol. 2016;116(2):139–45. [DOI] [PubMed] [Google Scholar]
- 180. Ellinghaus D, Baurecht H, Esparza‐Gordillo J, Rodriguez E, Matanovic A, Marenholz I, et al. High‐density genotyping study identifies four new susceptibility loci for atopic dermatitis. Nat Genet. 2013;45(7):808–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Sakaue S, Kanai M, Tanigawa Y, Karjalainen J, Kurki M, Koshiba S, et al. A cross‐population atlas of genetic associations for 220 human phenotypes. Nat Genet. 2021;53(10):1415–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Hinds DA, McMahon G, Kiefer AK, Do CB, Eriksson N, Evans DM, et al. A genome‐wide association meta‐analysis of self‐reported allergy identifies shared and allergy‐specific susceptibility loci. Nat Genet. 2013;45(8):907–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Ziyab AH, Davies GA, Ewart S, Hopkin JM, Schauberger EM, Wills‐Karp M, et al. Interactive effect of STAT6 and IL13 gene polymorphisms on eczema status: results from a longitudinal and a cross‐sectional study. BMC Med Genet. 2013;14:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Schaarschmidt H, Ellinghaus D, Rodriguez E, Kretschmer A, Baurecht H, Lipinski S, et al. A genome‐wide association study reveals 2 new susceptibility loci for atopic dermatitis. J Allergy Clin Immunol. 2015;136(3):802–6. [DOI] [PubMed] [Google Scholar]
- 185. Sobczyk MK, Richardson TG, Zuber V, Min JL, Gaunt TR, Paternoster L, et al. Triangulating molecular evidence to prioritize candidate causal genes at established atopic dermatitis loci. J Invest Dermatol. 2021;141(11):2620–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Esparza‐Gordillo J, Weidinger S, Folster‐Holst R, Bauerfeind A, Ruschendorf F, Patone G, et al. A common variant on chromosome 11q13 is associated with atopic dermatitis. Nat Genet. 2009;41(5):596–601. [DOI] [PubMed] [Google Scholar]
- 187. Asad S, Winge MC, Wahlgren CF, Bilcha KD, Nordenskjold M, Taylan F, et al. The tight junction gene Claudin‐1 is associated with atopic dermatitis among Ethiopians. J Eur Acad Dermatol Venereol. 2016;30(11):1939–41. [DOI] [PubMed] [Google Scholar]
- 188. Marenholz I, Esparza‐Gordillo J, Ruschendorf F, Bauerfeind A, Strachan DP, Spycher BD, et al. Meta‐analysis identifies seven susceptibility loci involved in the atopic march. Nat Commun. 2015;6:8804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Kato A, Fukai K, Oiso N, Hosomi N, Murakami T, Ishii M. Association of SPINK5 gene polymorphisms with atopic dermatitis in the Japanese population. Br J Dermatol. 2003;148(4):665–9. [DOI] [PubMed] [Google Scholar]
- 190. Nishio Y, Noguchi E, Shibasaki M, Kamioka M, Ichikawa E, Ichikawa K, et al. Association between polymorphisms in the SPINK5 gene and atopic dermatitis in the Japanese. Genes Immun. 2003;4(7):515–17. [DOI] [PubMed] [Google Scholar]
- 191. Lee KY, Leung KS, Tang NLS, Wong MH. Discovering Genetic Factors for psoriasis through exhaustively searching for significant second order SNP‐SNP interactions. Sci Rep. 2018;8(1) 15186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Bejaoui Y, Witte M, Abdelhady M, Eldarouti M, Abdallah NMA, Elghzaly AA, et al. Genome‐wide association study of psoriasis in an Egyptian population. Exp Dermatol. 2019;28(5):623–7. [DOI] [PubMed] [Google Scholar]
- 193. Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet. 2012;44(12):1341–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Genetic Analysis of Psoriasis C, the Wellcome Trust Case Control C, Strange A, Capon F, Spencer CC, Knight J, et al. A genome‐wide association study identifies new psoriasis susceptibility loci and an interaction between HLA‐C and ERAP1. Nat Genet. 2010;42(11):985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, et al. Genome‐wide scan reveals association of psoriasis with IL‐23 and NF‐kappaB pathways. Nat Genet. 2009;41(2):199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Yin X, Low HQ, Wang L, Li Y, Ellinghaus E, Han J, et al. Genome‐wide meta‐analysis identifies multiple novel associations and ethnic heterogeneity of psoriasis susceptibility. Nat Commun. 2015;6:6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Farooq M, Nakai H, Fujimoto A, Fujikawa H, Matsuyama A, Kariya N, et al. Mutation analysis of the IL36RN gene in 14 Japanese patients with generalized pustular psoriasis. Hum Mutat. 2013;34(1):176–83. [DOI] [PubMed] [Google Scholar]
- 198. Kanazawa N, Nakamura T, Mikita N, Furukawa F. Novel IL36RN mutation in a Japanese case of early onset generalized pustular psoriasis. J Dermatol. 2013;40(9):749–51. [DOI] [PubMed] [Google Scholar]
- 199. Stuart PE, Nair RP, Tsoi LC, Tejasvi T, Das S, Kang HM, et al. Genome‐wide association analysis of psoriatic arthritis and cutaneous psoriasis reveals differences in their genetic architecture. Am J Hum Genet. 2015;97(6):816–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Patrick MT, Stuart PE, Zhang H, Zhao Q, Yin X, He K, et al. Causal relationship and shared genetic loci between psoriasis and type 2 diabetes through trans‐disease meta‐analysis. J Invest Dermatol. 2021;141(6):1493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Zhang XJ, Huang W, Yang S, Sun LD, Zhang FY, Zhu QX, et al. Psoriasis genome‐wide association study identifies susceptibility variants within LCE gene cluster at 1q21. Nat Genet. 2009;41(2):205–10. [DOI] [PubMed] [Google Scholar]
- 202. Tomer Y, Dolan LM, Kahaly G, Divers J, D’Agostino RB Jr., Imperatore G, et al. Genome wide identification of new genes and pathways in patients with both autoimmune thyroiditis and type 1 diabetes. J Autoimmun. 2015;60:32–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Peled A, Sarig O, Sun G, Samuelov L, Ma CA, Zhang Y, et al. Loss‐of‐function mutations in caspase recruitment domain‐containing protein 14 (CARD14) are associated with a severe variant of atopic dermatitis. J Allergy Clin Immunol. 2019;143(1):173–81. [DOI] [PubMed] [Google Scholar]
- 204. Wu Y, Lu Z, Chen Y, Xue F, Chen X, Pan M, et al. Association of IL‐12B gene rs6887695 polymorphism with hereditary susceptibility and clinical characterization of psoriasis vulgaris in the Chinese Han population. Arch Dermatol Res. 2013;305(6):477–82. [DOI] [PubMed] [Google Scholar]
- 205. Ellinghaus E, Ellinghaus D, Stuart PE, Nair RP, Debrus S, Raelson JV, et al. Genome‐wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat Genet. 2010;42(11):991–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Yoon NY, Wang HY, Jun M, Jung M, Kim DH, Lee NR, et al. Simultaneous detection of barrier‐ and immune‐related gene variations in patients with atopic dermatitis by reverse blot hybridization assay. Clin Exp Dermatol. 2018;43(4):430–6. [DOI] [PubMed] [Google Scholar]
- 207. Ishigaki K, Akiyama M, Kanai M, Takahashi A, Kawakami E, Sugishita H, et al. Large‐scale genome‐wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat Genet. 2020;52(7):669–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Simpson EL, Gadkari A, Worm M, Soong W, Blauvelt A, Eckert L, et al. Dupilumab therapy provides clinically meaningful improvement in patient‐reported outcomes (PROs): a phase IIb, randomized, placebo‐controlled, clinical trial in adult patients with moderate to severe atopic dermatitis (AD). J Am Acad Dermatol. 2016;75(3):506–15. [DOI] [PubMed] [Google Scholar]
- 209. Guttman‐Yassky E, Hanifin JM, Boguniewicz M, Wollenberg A, Bissonnette R, Purohit V, et al. The role of phosphodiesterase 4 in the pathophysiology of atopic dermatitis and the perspective for its inhibition. Exp Dermatol. 2019;28(1):3–10. [DOI] [PubMed] [Google Scholar]
- 210. Ruzicka T, Hanifin JM, Furue M, Pulka G, Mlynarczyk I, Wollenberg A, et al. Anti‐Interleukin‐31 receptor A antibody for atopic dermatitis. N Engl J Med. 2017;376(9):826–35. [DOI] [PubMed] [Google Scholar]
- 211. Lou H, Lu J, Choi EB, Oh MH, Jeong M, Barmettler S, et al. Expression of IL‐22 in the skin causes Th2‐biased immunity, epidermal barrier dysfunction, and pruritus via stimulating epithelial Th2 cytokines and the GRP pathway. J Immunol. 2017;198(7):2543–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL‐22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005;174(6):3695–702. [DOI] [PubMed] [Google Scholar]
- 213. Goedkoop AY, Kraan MC, Teunissen MB, Picavet DI, de Rie MA, Bos JD, et al. Early effects of tumour necrosis factor alpha blockade on skin and synovial tissue in patients with active psoriasis and psoriatic arthritis. Ann Rheum Dis. 2004;63(7):769–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Mease PJ, Gladman DD, Ritchlin CT, Ruderman EM, Steinfeld SD, Choy EH, et al. Adalimumab for the treatment of patients with moderately to severely active psoriatic arthritis: results of a double‐blind, randomized, placebo‐controlled trial. Arthritis Rheum. 2005;52(10):3279–89. [DOI] [PubMed] [Google Scholar]
- 215. Mazumdar S, Greenwald D. Golimumab. mAbs. 2009;1(5):422–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Mease PJ, Fleischmann R, Deodhar AA, Wollenhaupt J, Khraishi M, Kielar D, et al. Effect of certolizumab pegol on signs and symptoms in patients with psoriatic arthritis: 24‐week results of a Phase 3 double‐blind randomised placebo‐controlled study (RAPID‐PsA). Ann Rheum Dis. 2014;73(1):48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Mease PJ, Goffe BS, Metz J, VanderStoep A, Finck B, Burge DJ. Etanercept in the treatment of psoriatic arthritis and psoriasis: a randomised trial. Lancet. 2000;356(9227):385–90. [DOI] [PubMed] [Google Scholar]
- 218. Blauvelt A, Prinz JC, Gottlieb AB, Kingo K, Sofen H, Ruer‐Mulard M, et al. Secukinumab administration by pre‐filled syringe: efficacy, safety and usability results from a randomized controlled trial in psoriasis (FEATURE). Br J Dermatol. 2015;172(2):484–93. [DOI] [PubMed] [Google Scholar]
- 219. Paul C, Lacour JP, Tedremets L, Kreutzer K, Jazayeri S, Adams S, et al. Efficacy, safety and usability of secukinumab administration by autoinjector/pen in psoriasis: a randomized, controlled trial (JUNCTURE). J Eur Acad Dermatol Venereol. 2015;29(6):1082–90. [DOI] [PubMed] [Google Scholar]
- 220. Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, et al. Secukinumab in plaque psoriasis‐‐results of two phase 3 trials. N Engl J Med. 2014;371(4):326–38. [DOI] [PubMed] [Google Scholar]
- 221. Griffiths CE, Reich K, Lebwohl M, van de Kerkhof P, Paul C, Menter A, et al. Comparison of ixekizumab with etanercept or placebo in moderate‐to‐severe psoriasis (UNCOVER‐2 and UNCOVER‐3): results from two phase 3 randomised trials. Lancet. 2015;386(9993):541–51. [DOI] [PubMed] [Google Scholar]
- 222. Papp KA, Reich K, Paul C, Blauvelt A, Baran W, Bolduc C, et al. A prospective phase III, randomized, double‐blind, placebo‐controlled study of brodalumab in patients with moderate‐to‐severe plaque psoriasis. Br J Dermatol. 2016;175(2):273–86. [DOI] [PubMed] [Google Scholar]
- 223. Lebwohl M, Strober B, Menter A, Gordon K, Weglowska J, Puig L, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373(14):1318–28. [DOI] [PubMed] [Google Scholar]
- 224. Blauvelt A, Papp KA, Griffiths CE, Randazzo B, Wasfi Y, Shen YK, et al. Efficacy and safety of guselkumab, an anti‐interleukin‐23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double‐blinded, placebo‐ and active comparator‐controlled VOYAGE 1 trial. J Am Acad Dermatol. 2017;76(3):405–17. [DOI] [PubMed] [Google Scholar]
- 225. Gordon KB, Strober B, Lebwohl M, Augustin M, Blauvelt A, Poulin Y, et al. Efficacy and safety of risankizumab in moderate‐to‐severe plaque psoriasis (UltIMMa‐1 and UltIMMa‐2): results from two double‐blind, randomised, placebo‐controlled and ustekinumab‐controlled phase 3 trials. Lancet. 2018;392(10148):650–61. [DOI] [PubMed] [Google Scholar]
- 226. Reich K, Papp KA, Blauvelt A, Tyring SK, Sinclair R, Thaci D, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. 2017;390(10091):276–88. [DOI] [PubMed] [Google Scholar]
- 227. Leonardi CL, Kimball AB, Papp KA, Yeilding N, Guzzo C, Wang Y, et al. Efficacy and safety of ustekinumab, a human interleukin‐12/23 monoclonal antibody, in patients with psoriasis: 76‐week results from a randomised, double‐blind, placebo‐controlled trial (PHOENIX 1). Lancet. 2008;371(9625):1665–74. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.