

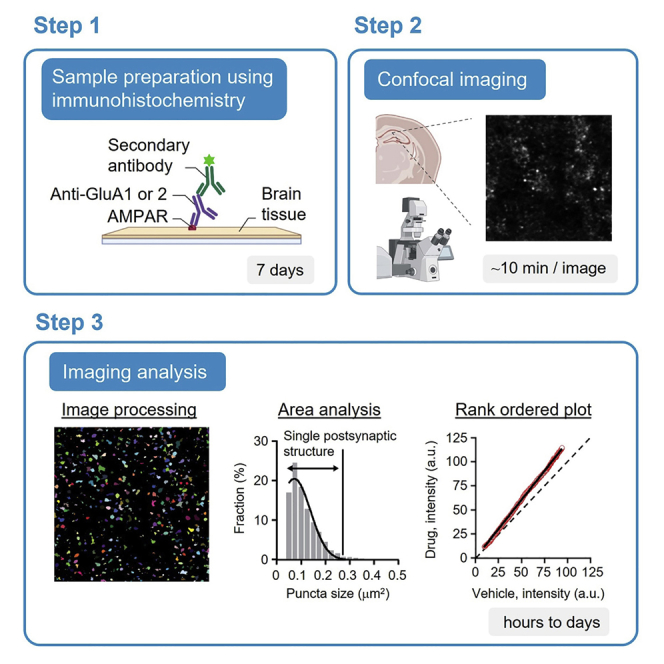
Summary
Immunolabeling of surface AMPA receptors (AMPARs) can be used for in vivo or ex vivo examination of synaptic scaling, a type of homeostatic plasticity. Here, we present a protocol to analyze changes in synaptic weights using immunohistochemistry for surface AMPARs coupled with optical imaging analysis. We detail immunostaining of AMPARs in mouse brain sections, followed by confocal imaging of surface AMPARs in dendritic region of hippocampal CA1. We then describe using Fiji/ImageJ and rank order plots for analyzing synaptic weight.
For complete details on the use and execution of this protocol, please refer to Suzuki et al. (2021).
Subject areas: Cell Biology, Microscopy, Neuroscience
Graphical abstract
Highlights
-
•
Protocol for staining of surface AMPARs in mouse hippocampus using immunohistochemistry
-
•
Strategy for imaging of surface AMPARs in dendritic region of hippocampal CA1
-
•
Isolation of AMPAR puncta using Fiji/ImageJ
-
•
Assessment of synaptic weight with rank order plot
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Immunolabeling of surface AMPA receptors (AMPARs) can be used for in vivo or ex vivo examination of synaptic scaling, a type of homeostatic plasticity. Here, we present a protocol to analyze changes in synaptic weights using immunohistochemistry for surface AMPARs coupled with optical imaging analysis. We detail immunostaining of AMPARs in mouse brain sections, followed by confocal imaging of surface AMPARs in dendritic region of hippocampal CA1. We then describe using Fiji/ImageJ and rank order plots for analyzing synaptic weight.
Before you begin
Synaptic scaling is a form of homeostatic synaptic plasticity. In contrast to Hebbian plasticity, synaptic scaling normalizes synaptic weights in a multiplicative manner such that connections are increased or decreased by the same factor (Kavalali and Monteggia, 2020; Turrigiano, 2008). Electrophysiological recordings, specifically the measurement of mEPSC amplitudes, are commonly used to study synaptic scaling. For example, suppression of neuronal activity in cultured neurons with tetrodotoxin (TTX) for hours to days multiplicatively increases mEPSC amplitudes (Turrigiano et al., 1998). A potential caveat of solely relying on mEPSC recordings to assess synaptic strength is the complexity of dendritic structures and the electrotonic distance of synapses on dendrites to the cell soma. As synaptic upscaling is mediated by an accumulation of AMPA receptors postsynaptically, one alternative approach is the immunolabeling of surface AMPARs as a proxy for synaptic weights irrespective of their distance from the soma (Ibata et al., 2008; Venkatesan et al., 2020; Wierenga et al., 2005). Using an immunolabeling strategy, we have shown the retinoic acid receptor α (RARα) agonist AM580 and the noncompetitive glutamatergic N-methyl-D-aspartate receptor (NMDAR) antagonist ketamine mediate multiplicative synaptic up-scaling in stratum radiatum of the hippocampus CA1 region (Suzuki et al., 2021). Previous studies suggest most AMPARs are localized postsynaptically (Biederer et al., 2017; Graves et al., 2021; Nair et al., 2013), which can be observed as puncta by immunohistochemistry. The current protocol provides detailed experimental steps to quantify surface AMPARs, and analyze the immunolabeling signals using the open source software Fiji/Image J to determine synaptic weight in the CA1 region of the hippocampus in mice.
Installation of software
-
1.
Download and install Fiji/ImageJ from https://imagej.net/software/fiji/downloads.
-
2.
Download and install Interactive Watershed plugin from https://imagej.net/plugins/interactive-watershed.
Note: Watershed is a common tool to separate objects in an image. In the watershed algorithm, an image is interpreted as landscapes of hills and valleys similar to a topographic map. This algorithm calculates the distance from the center to the edges of an object to determine the threshold and then generates the segmentation to separate the objects. While classical watershed is generally used to segment objects, it may not always be sufficient in separating objects. For example, the shape of a synapse and its imaging contrast may be in close proximity to an adjacent synapse or have varying brightness. While synaptic structure in microscopy images is usually detected as a punctum (i.e., a circular object), this may be an oversimplification and insufficient to separate each structure using the classical watershed algorithm. For this reason, we have been exploring ways to determine segmentation to separate synaptic structures (Figures 1, 2, and 3). The Interactive Watershed (Interactive H-Watershed) plugin provides an interactive way to explore local minima (or maxima) and threshold with segmentation parameters. This plugin is utilized to separate structures in detail compared to classical watershed. This Interactive Watershed plugin needs to be installed prior to analyzing the imaging puncta in Fiji/ImageJ.
Figure 1.

Representative images of GluA1 in the stratum radiatum of the hippocampus CA1 region during processing images
(A) Z-stack image subtracted background.
(B) Image applied fast Fourier transform (FFT) bandpass filter (BF).
(C) Image applied interactive H-Watershed.
(D) Binary image was developed from interactive H-Watershed. Regions of interest (ROIs) were determined using particle analysis.
(E) Area and average intensity in the puncta was obtained from Z-stack image subtracted background. Scale bar, 5 μm. Figure adapted with permission Suzuki et al. (2021).
Figure 2.

Parameter optimization during processing of immunolabeled samples
(A) Bandpass filter was applied in the criteria using 50 and 7 pixels for large and small structures. Seed dynamics was set to 3. Intensity threshold was determined by Li’s minimum cross entropy threshold. Peak flooding (in %) was set to 100.
(B) Peak flooding (in %) can be decreased slightly to select punctuate area preciously. Peak flooding (in %) was set to 80.
(C) When bandpass filter was applied in the criteria using 30 for large structures (smaller than 50), the size of H-Watershed segmentation was smaller than (A) image.
(D) When bandpass filter was applied in the criteria using 3 for small structure (smaller than 7), tiny H-Watershed segmentations were detected. Scale bar for the upper and middle images, 5 μm. Scale bar for the bottom images, 2 μm.
Figure 3.

Representative images during processing of Interactive Watershed
(A) Representative image and parameters which were processed during Interactive Watershed.
(B) Determination of threshold using Li’s minimum cross entropy threshold.
Institutional permissions
All animal procedures were performed in accordance with the guide for the care and use of laboratory animals and approved by the Institutional Animal Care and Use Committee at Vanderbilt University.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-GluA1 | Millipore | Cat# ABN241; RRID: AB_2721164 |
| Mouse monoclonal anti-GluA2 | Millipore | Cat# MAB397; RRID: AB_2113875 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A11034 RRID: AB_2576217 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A11029 RRID: AB_2534088 |
| Chemicals, peptides, and recombinant proteins | ||
| 10× PBS | Fisher | Cat# BP399 |
| Sodium phosphate dibasic (Na2HPO4) | Sigma | Cat# S9763 |
| Potassium Phosphate monobasic (KH2PO4) | Fisher | Cat# P285 |
| Sucrose | Sigma | Cat# S5016 |
| Saline | Baxter | Cat# 2F7123 |
| Ketamine | Zoetis | Cat# 10004027 |
| Xylazine | Hanna Pharmaceutical | Cat# 5098923309 |
| AM580 | Cayman | Cat# 15261 |
| DMSO | Sigma | Cat# D2650 |
| Cremophor EL | Sigma | Cat# 238470 |
| Paraformaldehyde | Sigma | Cat# P6148 |
| NaOH | Sigma | Cat# S8045 |
| Tissue-Tek O.C.T Compound | Sakura | Cat# 4583 |
| Normal goat serum | Vector Laboratories | Cat# S-1000 |
| DAPI | Thermo Fisher Scientific | Cat# 01306 |
| Aqua-Poly/Mount | Polysciences, Inc | Cat# 18606 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J, 9 weeks, male | The Jackson Laboratory | Cat# JAX:000664; RRID: IMSR_JAX:000664 |
| Software and algorithms | ||
| Fiji / ImageJ | Schneider et al. (2012) | https://imagej.nih.gov/ij/ |
| Microsoft Excel | Microsoft | https://www.microsoft.com |
| GraphPad Prism | GraphPad | https://www.graphpad.com/ |
| Other | ||
| High Precision Tubing pumps | ISMATEC | Cat# ISM941 |
| LSM 510 | ZEISS | https://www.Zeiss.com/ |
| LSM 510 Laser module | ZEISS | https://www.Zeiss.com/ |
| Objective Plan-Apochromat 63×/1.4 Oil DIC M27 | ZEISS | Cat# 420782-9900 |
| Leica CM1950 Cryostat | Leica | https://www.leicabiosystems.com/ |
Alternatives: Chemicals and reagents obtained from other suppliers could be used for this protocol, although they would need to be tested to ensure the immunoreactive signal is detected. Any confocal microscopies could be used for the proposed experiments.
Materials and equipment
The list of solutions and reagents needs is as follows:
1× PBS
| Reagent | Final concentration | Amount |
|---|---|---|
| 10× PBS | N/A | 100 mL |
| Milli Q H2O | N/A | Up to 1 L |
Note: Store at RT (20°C–22°C) for up to 3 months.
0.5 M phosphate buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Na2HPO4 | 0.8 M | 56.8 g |
| KH2PO4 | 0.2 M | 13.6 g |
| Milli Q H2O | N/A | Up to 1 L |
Note: Adjust the pH to 7.4 using NaOH. Store at RT (20°C–22°C) for up to 3 months.
10% sucrose in PBS
| Regent | Final concentration | Amount |
|---|---|---|
| 10× PBS | N/A | 10 mL |
| Sucrose | 10% | 10 g |
| Milli Q H2O | N/A | Up to 100 mL |
Note: Store at 4°C for up to a week.
20% sucrose in PBS
| Regent | Final concentration | Amount |
|---|---|---|
| 10× PBS | N/A | 10 mL |
| Sucrose | 20% | 20 g |
| Milli Q H2O | N/A | Up to 100 mL |
Note: Store at 4°C for up to a week.
30% sucrose in PBS
| Regent | Final concentration | Amount |
|---|---|---|
| 10× PBS | N/A | 10 mL |
| Sucrose | 30% | 30 g |
| Milli Q H2O | N/A | Up to 100 mL |
Note: Store at 4°C for up to a week.
10 mg/mL ketamine and 1 mg/mL xylazine
| Regent | Final concentration | Amount |
|---|---|---|
| 100 mg/mL ketamine | 10 mg/mL | 200 μL |
| 20 mg/mL xylazine | 1 mg/mL | 100 μL |
| Saline | N/A | 1700 μL |
Note: Freshly prepare before use.
4 mg/mL AM580
| Regent | Final concentration | Amount |
|---|---|---|
| 100 mg/mL AM580 | 4 mg/mL | 40 μL |
| EtOH | 5% | 50 μL |
| Cremophor EL | 10% | 100 μL |
| Saline | N/A | 810 μL |
Preparation
-
•
Dissolve AM580 in DMSO to 100 mg/mL. Make aliquots and store at −20°C.
-
•
To make 4 mg/mL AM580 in saline, mix 40 μL of 100 mg/mL of AM580 with 50 μL of EtOH, and vortex to mix thoroughly.
-
•
Add 100 μL of Cremophor and mix thoroughly.
-
•
Add 810 μL Saline. Vortex to mix thoroughly.
Note: Freshly prepare before use. As a vehicle control, prepare solution containing DMSO instead of AM580.
1 mg/mL ketamine
| Regent | Final concentration | Amount |
|---|---|---|
| 100 mg/mL ketamine | 1 mg/mL | 10 μL |
| Saline | N/A | 990 μL |
Note: As a vehicle control, use saline. Freshly prepare before use.
Blocking solution
| Regent | Final concentration | Amount |
|---|---|---|
| Normal goat serum | 5% | 2.5 mL |
| BSA | 1% | 0.5 g |
| 1× PBS | N/A | Up to 50 mL |
Note: Filter the solution using vacuum filter or filter syringe. Freshly prepare before use.
4% Paraformaldehyde (PFA) in phosphate buffer
| Regent | Final concentration | Amount |
|---|---|---|
| PFA | 4% | 20 g |
| NaOH | N/A | 2 tablets |
| 6 N HCl | N/A | Few drops |
| 0.5 M Phosphate buffer (pH 7.4) | 0.05 M | 50 mL |
| MilliQ H2O | N/A | 450 mL |
Preparation
-
•
Add 450 mL of Milli-Q H2O and 2 tablets of NaOH into a beaker.
-
•
Add 20 g of PFA.
-
•
Stir gently on a heating block at ∼60°C until the PFA is dissolved.
-
•
Adjust pH to pH 7 using pH test indicator papers.
-
•
Add 50 mL of 0.5 M Phosphate buffer (pH 7.4).
-
•
Confirm pH of 7.4 using pH test indicator.
-
•
Filter the solution using vacuum filter.
-
•
Store it at 4°C.
Note: Final pH should be pH 7.4. If pH is changed, adjust the pH to 7.4 with 6 N HCl or 5 M NaOH. Since PFA is toxic, avoid using an electronic pH meter. Freshly prepare and store the 4% PFA at 4°C for no more than one day before use.
CRITICAL: Paraformaldehyde is toxic. The solution should be made inside a fume hood wearing gloves and safety glasses.
Step-by-step method details
Sample preparation using immunohistochemistry
Timing: 7 days
This section describes the protocol to stain surface AMPARs using an antibody recognizing the extracellular domain of AMPARs.
Day 1.
-
1.
Inject mice (9 weeks) intraperitoneally with AM580 (20 mg/kg) or ketamine (5 mg/kg).
Note: Inject 0.05 mL of 4 mg/mL AM580 or 1 mg/mL ketamine per 10 g of body weight.
-
2.
At 2 h after injection of drug, anesthetize mice with ketamine (100 mg/kg)/xylazine (10 mg/kg) via intraperitoneal injection.
Note: Inject 0.1 mL of 10 mg/mL ketamine and 1 mg/mL xylazine per 10 g of body weight.
-
3.Transcardially perfuse mice with ice-cold PBS followed by ice-cold 4% PFA in phosphate buffer using tube pump.
-
a.perfuse mice with ice-cold PBS for 6 min at 3 mL/min.
-
b.perfuse mice with ice-cold 4% PFA for 16 min at 3 mL/min.
-
a.
-
4.
Decapitate mouse, open the skull, and extract the brain.
-
5.
Place the brain in a 15 mL conical tube containing 13 mL of 4% PFA and store for 16–24 h at 4°C.
Day 2–5.
-
6.Cryo-protect and freeze brain.
-
a.Transfer brain into a 15 mL conical tube containing 13 mL of 10% sucrose in PBS and store for 16–24 h at 4°C.
-
b.Transfer brain into a 15 mL conical tube containing 13 mL of 20% sucrose in PBS and store for 16–24 h at 4°C.
-
c.Transfer brain into in a 15 mL conical tube containing 13 mL of 30% sucrose in PBS and store for 16–24 h at 4°C.
-
d.Freeze and embed brain using OCT compound on dry ice.
-
e.Store brain sample at −20°C.
-
a.
Day 6.
-
7.Cut the brain into 25 μm-thick sections using a cryostat.
-
a.Transfer sections using a brush into ice cold PBS and place on ice.
-
a.
Alternatives: A vibratome could be used to section the brain. Brain slices can be cut using a vibratome after post-fixation. Brain tissue may need to be embedded in low-melting agarose gels.
-
8.Wash sections three times in 1× PBS.
-
a.Use a 12 well plate for free-floating sections.
-
b.Use 1 mL of 1× PBS for washing.
-
c.Wash sections with 1× PBS while briefly shaking gently.
-
d.Repeat this step three times.
-
e.When aspirating solution, use clean Pasteur glass pipette with bent tip to carefully hold sections so it is not inadvertently removed.
-
a.
-
9.Block with 5% normal goat serum and 1% BSA without permeabilization.
-
a.Use 1 mL of blocking buffer for 12 well plate.
-
b.Incubate at RT (20°C–22°C) on the shaker at low speed for 1 h.
-
a.
-
10.Prepare primarily antibody solutions. To stain surface AMPARs, an antibody specific to an N-terminal extracellular epitope of GluA1 (1:400) or GluA2 (1:100) is used.
-
a.Primary antibody is diluted with blocking solution.
-
b.Use 1 mL of primary antibody solution per well for a 12 well plate.
-
a.
Note: We use the anti-GluA1 and -GluA2 antibodies at 1:400 and 1:100, respectively. One would need to confirm optimal primary antibody concentrations for staining typically starting at a 1:50–1:1000 dilution.
-
11.Incubate sections with primary antibody solution for overnight (15–20 h) at 4°C.
-
a.Incubate on the shaker at low speed.
-
b.Make sure brain sections are gently and slowly moving on the shaker to avoid tissue damage.
-
a.
Day 7.
-
12.Wash sections with 1× PBS three times.
-
a.Incubate at RT (20°C–22°C) on the shaker at low speed (around 50 rpm) for 10 min.
-
b.Repeat this step three times.
-
a.
-
13.Prepare AlexaFluor 488-conjugated secondary antibody solutions at 1:500 ratio.
-
a.Secondary antibody is diluted with blocking solution.
-
b.Use 1 mL of secondary antibody solution per well for 12 well plate.
-
a.
-
14.Incubate sections with AlexaFluor 488-conjugated secondary antibody for 2 h at RT (20°C–22°C).
-
a.Incubate on the shaker at low speed.
-
a.
-
15.Wash sections with 1× PBS two times.
-
a.Incubate at RT (20°C–22°C) on the shaker at low speed for 10 min.
-
b.Repeat this step once.
-
a.
-
16.Incubate sections with DAPI in 1× PBS.
-
a.Dilute DAPI in 1× PBS at 1:10,000 ratio.
-
b.Incubate at RT (20°C–22°C) on the shaker at low speed for 10 min.
-
a.
-
17.Wash sections with 1× PBS once.
-
a.Incubate at RT (20°C–22°C) on the shaker at low speed for 10 min.
-
a.
-
18.Mount sections using mounting solution.
-
a.Mount free-floating sections in 1× PBS on the glass slide.
-
b.Place the slide with the mounted section in the dark until dry.
-
c.Embed the mounted section using Aqua-Poly/Mount mounting solution and a coverslip.
-
d.Place the samples on a slide tray at RT (20°C–22°C) for overnight in the dark.
-
e.Store the sample at 4°C in the dark.
-
a.
Confocal imaging
This section describes the strategy to provide images of surface AMPARs in dendritic regions of hippocampal CA1.
-
19.Acquire images of the dendritic region of hippocampus neurons using confocal microscopy.
-
a.Take confocal fluorescence z-stack images (0.5 μm intervals, 10 images at scan zoom of 8, 17.86 μm × 17.86 μm image size) of the stratum radiatum of the dorsal hippocampal CA1 in the brain section using a ZEISS LSM 510 with a 63× objective (NA 1.4, oil) at 1.504 × 1.504 pixels resolution.
-
b.Multiple images should be taken from multiple sections of similar stereotaxic coordinate.
-
c.Take images under the same conditions of laser power and gain without saturation.
-
a.
Note: To take confocal images from the stratum radiatum of the dorsal hippocampal CA1 in the brain section, initially locate the pyramidal layer of CA1 with a 10× or 20× objective. Then, focus this region with a 63× objective and move focus to the stratum radiatum. We usually take two z-stack images from 1 section which are localized in proximal dendrites. Expected images are shown in Figure 2 of this paper and Figure 5 of Suzuki et al. (2021).
Imaging analysis
This section explains details to isolate AMPAR puncta using Fiji/ImageJ and build rank order plots for analyzing synaptic weight.
-
20.Open images in Fiji. Project Z-stack images to single image by summation. Save images in TIF file.
-
a.Projection of Z-stack images: Image – Stacks – Z project – Sum slice.
-
b.Save image: Save as – Tiff.
-
a.
-
21.To remove background, threshold images using Li’s minimum cross entropy algorithm which is one of threshold program in Fiji/ImageJ.
-
a.Open image: File | Open.
-
b.Decide threshold: Image | Adjust | Threshold – select Li – apply – set to NaN (Not a Number).
-
c.Subtract background: Process | Math | Subtract minimum intensity.
-
d.Save image: Save as – Tiff.
-
a.
-
22.To enhance contrast, apply a fast Fourier transform (FFT) band pass filter to the image which background is removed (Figures 1A and 1B).
-
a.Convert image to 8 bit: Image | Type – 8 bit.
-
b.Apply FFT band pass filter: Process | FFT – Bandpass Filter – select minimum and maximum feature sizes.
-
a.
-
23.Segment adjacent puncta using the Interactive Watershed program (Figures 1C, 1D, 3A, and 3B).
-
a.Open Interactive Watershed: SCF | Labeling | Interactive H_Watershed.
-
b.Apply Interactive Watershed: Set seed dynamics. Set intensity threshold, which was determined by Li’s minimum cross entropy threshold. Peak flooding (in %) is set to 100. Click allow splitting. Click export regions mask. Then, click export. A new file will be shown as binary image with H-Watershed segmentation.
-
a.
Note: The setting of parameters is crucial to determine the segmentation of each punctum accurately. We have set seed dynamics to 3 (Figure 3A). The intensity threshold is determined by Li’s minimum cross entropy threshold (Figure 3B). While we have used 100% in peak flooding, this can be decreased slightly to select the precise punctuate area (Figures 2A, 2B, and 3A). These criteria need to be optimized depending on the image quality. Always refer to the original image before determining these criteria. The same criteria for seed dynamics and peak flowing should be used in the analysis of all images. See the website which provides a detailed explanation of Interactive Watershed plugin (also, see before you begin), which includes more information about the settings and the criteria.
-
24.Using binary image, determine the regions of interest (ROIs) for individual punctum by particle analysis. Area criteria are set to 0.05–1 μm2. Theoretical lateral resolution in the confocal image is calculated to 178 nm (0.51× λexc/NA=0.51∗488 nm/1.4). 0.05 μm2 is larger than 0.0313 μm2 (Square meter of 177 nm × 177 nm). Then, measure area and intensity of puncta from the image which is subtracted background (Figure 1E).
-
a.Perform particle analysis: Analysis | Analysis particles – set size to 0.05–1.
-
b.Measure area and mean intensities of puncta in image obtained from z-stack image with background subtraction using ROI manager.
-
c.Save these values in an Excel worksheet.
-
a.
Note: We calculate theoretical lateral resolution in confocal image using the following equation (0.51× λexc/NA). If a dye such as AlexaFluor 555 or 647-conjugated secondary antibody is used, area criteria should be changed. Since these secondary antibodies could reduce imaging resolution due to the lateral resolution changes, we recommend to use dye for the green channel. Since background is set to NaN, small puncta (<0.05 μm2) may be detected. If so, do not select these puncta in further analysis.
-
25.Make a list including area and total intensity in Excel.
-
a.Calculate total intensity of puncta by multiplying the area by average intensity.
-
a.
-
26.Generate histogram for area of puncta and perform nonlinear fitting with Gaussian using GraphPad prism. This analysis gives the number of Mean and SD values from Gaussian distribution (Figures 4A and 4B).
-
a.Copy the number of areas in Excel and paste it in GraphPad Prism.
-
b.Make histogram at 0.025 bin width.
-
c.Apply Gaussian using nonlinear fitting.
-
d.Obtain the number of Mean and SD from Gaussian distribution.
-
e.Calculate the number of Mean+3SD.
-
f.Select puncta that exist within that area range in Excel.
-
a.
Note: The value of Mean+3SD from Gaussian distribution is determined when considering the range of area for a single postsynaptic structure. In contrast, large puncta outside these fits may be attributed to fluorescence values originating from multiple adjacent postsynaptic sites.
-
27.Plot rank ordered plot (Figures 4C and 4D).
-
a.To compare total intensity of all selected puncta with vehicle and drug treatment in rank ordered plot, match the number of puncta between vehicle and drug treatment condition using RAND function in Excel. The Excel RAND function gives a random number between 0 and 1.
-
b.Place total intensity of all selected puncta and a random number in parallel in columns in the Excel worksheet. Then, the numbers are sorted from smallest to largest according to the random number column. In this way, the total intensities of puncta can be randomized.
-
c.After matching a number to each punctum total intensity, rank these puncta total intensities from smallest to largest to build the rank order plot.
-
d.Apply linear regression in Prism. Force the line to go through 0 at X and Y and obtain the slope from this linear regression.
-
e.0%–90% fractions of total intensities are shown in figure (Figure 4C) since higher intensities show supralinear or sublinear distribution.
-
f.Repeat this process at least three times since the slope in linear regression may vary slightly depending on the degree of randomization. Take intermediate data and consider it as better randomization.
-
a.
Alternatives: Several other spreadsheet based plotting software options (Sigma Plot etc.) or custom scripts can be used to plot the rank ordered plot.
Figure 4.

Strategy for plotting rank ordered plot
(A) Distribution of individual GluA1 puncta area size in vehicle treatment (n=3824 from 4 mice). The histogram was fitted by Gaussian distribution (Mean + 3SD=0.272 μm2). 3743 puncta were selected as single postsynaptic structure.
(B) Distribution of individual GluA1 puncta area size in AM580 treatment (n=3998 from 4 mice). The histogram was fitted by Gaussian distribution (Mean + 3SD=0.262 μm2). 3902 puncta were selected as single postsynaptic structure.
(C) Rank ordered plot for GluA1 intensity compared between vehicle and AM580 treatment. (Linear regression, slope= 1.21, n=3743). Blue area includes 90% fraction of GluA1 puncta.
(D) Rank order plot is built with 90% of GluA1 puncta. (Linear regression, slope= 1.21, n=3368). Figure adapted with permission Suzuki et al. (2021).
Expected outcomes
It has been proposed that multiplicative up- or down-synaptic scaling is a form of homeostatic synaptic plasticity. When up- or down-synaptic scaling occurs by pharmacological treatment and genetic manipulation, the slope in rank ordered plots can be increased or decreased. We have shown that the RARα agonist AM580 and the noncompetitive glutamatergic NMDAR antagonist ketamine produce multiplicative synaptic up-scaling in the stratum radiatum of the hippocampus CA1 region (Suzuki et al., 2021).
Quantification and statistical analysis
The difference of puncta size between the groups (treatment or genotype) can be compared from the histogram (Figures 4A and 4B). The slope can be obtained from rank ordered plots to determine the change in synaptic weights (Figure 4D).
Limitations
Optical analysis of synaptic scaling provides a widely applicable methodology to determine distribution of synaptic weights across neuronal dendrites. Of note, several processes and analysis should be considered when using this protocol. First, the preparation of the sample needs to be optimized to ensure surface staining. After fixing the brain, a vibratome to prepare brain slices improves the quality of surface staining. Second, the criteria for detection of a single punctum should be carefully evaluated to ensure isolation of a single postsynaptic structure in synapses. In this protocol, we assume the puncta — as identified by Gaussian fitting of the fluorescence amplitude histograms for puncta area — are comprised of individual postsynaptic structures. However, this analysis does not account for whether these postsynaptic structures are functional. Therefore, simultaneous pre- and post-synaptic staining may help increase the confidence that these identified postsynaptic puncta correspond to functional synapses. This approach can also be helpful to validate and isolate single postsynaptic structures. Although we have not studied other pre- and postsynaptic molecules using this protocol, expression of pre- and postsynaptic proteins could also be altered in synaptic homeostasis. Further analytical studies are needed to establish this premise. Third, while we use the slope of the rank ordered plots to evaluate the degree of increase or reduction in synaptic weights, statistical analysis will be needed to ascertain the changes in synaptic weights. Using this method, more than one thousand postsynaptic structures can be detected from several images. While non-parametric test could be used to determine whether intensity distributions are significantly different, large sample size can increase the ability to detect differences. Therefore, statistical tools applicable to large samples are needed for validation.
Troubleshooting
Problem 1
Weak staining (step 10).
Potential solution
GluA1 and GluA2 immunoreactivity could be low. While we use the anti-GluA1 antibody at a ratio of 1:400 or the anti-GluA2 antibody at a ratio of 1:100, these concentrations can be increased to enhance immunoreactivity.
Problem 2
High background (step 10).
Potential solution
Primary antibody concentration may be high. If so, it can be decreased. Also, the number of times the sections are washed can be increased.
Problem 3
Imaging signal is saturated (step 19).
Potential solution
Carefully take confocal images without saturation. One needs to determine the parameter (such as laser power and detection gain) for imaging to avoid signal saturation.
Problem 4
Puncta segmentation is incorrectly generated (steps 22 and 23).
Potential solution
The imaging quality can be influenced by the confocal microscope. To ensure whether puncta segmentation is correctly performed, one may need to test several parameters in FFT bandpass filter and interactive H-Watershed program settings (Figure 2).
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, lisa.monteggia@vanderbilt.edu.
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This work was supported by National Institute of Health, United States grant MH070727 and MH081060 (to L.M.M.) and MH066198 (to E.T.K.) as well as an award from the Brain and Behavior Research Foundation (to K.S.).
Author contributions
K.S., E.T.K., and L.M.M. were responsible for the experimental design. K.S. analyzed data. K.S. wrote and E.T.K. and L.M.M. edited the paper. E.T.K. and L.M.M. supervised this project.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Kanzo Suzuki, Email: kanzo.suzuki@vanderbilt.edu.
Lisa M. Monteggia, Email: lisa.monteggia@vanderbilt.edu.
Data and code availability
This paper does not report original code.
References
- Biederer T., Kaeser P.S., Blanpied T.A. Transcellular nanoalignment of synaptic function. Neuron. 2017;96:680–696. doi: 10.1016/j.neuron.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves A.R., Roth R.H., Tan H.L., Zhu Q., Bygrave A.M., Lopez-Ortega E., Hong I., Spiegel A.C., Johnson R.C., Vogelstein J.T., et al. Visualizing synaptic plasticity in vivo by large-scale imaging of endogenous AMPA receptors. Elife. 2021;10:e66809. doi: 10.7554/elife.66809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair D., Hosy E., Petersen J.D., Constals A., Giannone G., Choquet D., Sibarita J.B. Super-resolution imaging reveals that AMPA receptors inside synapses are dynamically organized in nanodomains regulated by PSD95. J. Neurosci. 2013;33:13204–13224. doi: 10.1523/jneurosci.2381-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K., Sun Q., Turrigiano G.G. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Kavalali E.T., Monteggia L.M. Targeting homeostatic synaptic plasticity for treatment of mood disorders. Neuron. 2020;106:715–726. doi: 10.1016/j.neuron.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Wayne S Rasband W.S., Kevin W Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K., Kim J.W., Nosyreva E., Kavalali E.T., Monteggia L.M. Convergence of distinct signaling pathways on synaptic scaling to trigger rapid antidepressant action. Cell Rep. 2021;37:109918. doi: 10.1016/j.celrep.2021.109918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G.G. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G.G., Leslie K.R., Desai N.S., Rutherford L.C., Nelson S.B. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Venkatesan S., Subramaniam S., Rajeev P., Chopra Y., Jose M., Nair D. Differential scaling of synaptic molecules within functional zones of an excitatory synapse during homeostatic plasticity. eNeuro. 2020;7 doi: 10.1523/eneuro.0407-19.2020. ENEURO.0407-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga C.J., Ibata K., Turrigiano G.G. Postsynaptic expression of homeostatic plasticity at neocortical synapses. J. Neurosci. 2005;25:2895–2905. doi: 10.1523/jneurosci.5217-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This paper does not report original code.