

Abstract
Mitochondria‐associated endoplasmic reticulum membranes (MAM) are specialized subcellular compartments that are shaped by endoplasmic reticulum (ER) subdomains placed side by side to the outer membrane of mitochondria (OMM) being connected by tethering proteins in mammalian cells. Studies showed that MAM has multiple physiological functions. These include regulation of lipid synthesis and transport, Ca2+ transport and signaling, mitochondrial dynamics, apoptosis, autophagy, and formation and activation of an inflammasome. However, alterations of MAM integrity lead to deleterious effects due to an increased generation of mitochondrial reactive oxygen species (ROS) via increased Ca2+ transfer from the ER to mitochondria. This, in turn, causes mitochondrial damage and release of mitochondrial components into the cytosol as damage‐associated molecular patterns which rapidly activate MAM‐resident Nod‐like receptor protein‐3 (NLRP3) inflammasome components. This complex induces the release of pro‐inflammatory cytokines that initiate low‐grade chronic inflammation that subsequently causes the development of metabolic diseases. But, the mechanisms of how MAM is involved in the pathogenesis of these diseases are not exhaustively reviewed. Therefore, this review was aimed to highlight the contribution of MAM to a variety of cellular functions and consider its significance pertaining to the pathogenesis of inflammation‐mediated metabolic diseases.
Keywords: ER‐stress, inflammatory mediated metabolic diseases, MAM, NLRP3‐inflammasome
Aberrations of MAM integrity are considered as a cornerstone in the pathogenesis of several inflammation‐mediated metabolic diseases neurodegenerative diseases, CVD, and cancer.
1. INTRODUCTION
Subcellular organelles have been viewed as separate entities with defined compositions and organizations that equip them with specialized functions. 1 However, studies proved that there are interorganellar membrane contact sites in organelles with close tethered proximity. 2 , 3 Among such sites, the interaction between the outer membrane of mitochondria (OMM) and that of endoplasmic reticulum (ER) was one of the best characterized. 3 , 4
Bernhard et al. came up with the first reported evidence for the existence of sites of physical interaction between ER and OMM from electron microscopic studies of rat liver cells in 1952. 5 A similar experimental approach by Bernhard and Rouiller in 1956 reassured this finding. 6 Another study of pseudobranch gland cells of Atlantic killifish in 1959 also reported the existence of this site. 7 However, the unique membrane that corresponded to this site was isolated as fraction X from a crude rat liver mitochondrial preparation in Vance laboratory in 1990 8 and later named as mitochondrial‐associated endoplasmic reticulum membranes (MAM) in the paper published in 1994. 9
Structurally, MAM is composed of ER subdomains placed side by side with OMM but are biochemically distinct from either pure ER or mitochondria membrane. 10 , 11 Electron tomography images revealed that ER and mitochondria are linked by tethers formed from specific protein–protein interactions (Figure 1). 10
Figure 1.
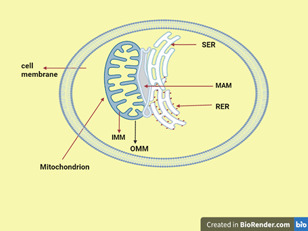
Structure of mitochondrial‐associated endoplasmic membranes. IMM, inner membrane of mitochondrion; MAM, mitochondria associated endoplasmic reticulum membrane; OMM, the outer membrane of mitochondria; RER, rough endoplasmic reticulum; SER, smooth endoplasmic reticulum. The picture is created at https://biorender.com/.
Studies showed that MAM has multiple functions including regulation of lipid synthesis and transport, 8 , 9 cellular apoptosis, 11 initiation of autophagy, 12 Ca2+ transport and signaling, 13 mitochondrial dynamics, 14 and insulin signaling. 15 Most importantly, it serves as a platform for inflammasome formation and activation which play a significant role in initiating inflammatory responses as Thoudam et al. 16 elegantly explained in their review published in 2018.
Aberrations of MAM integrity were considered as a cornerstone in the pathogenesis of several inflammatory‐mediated metabolic diseases like type 2 diabetes mellitus (T2DM), neurodegenerative diseases including Alzheimer's disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis/frontotemporal dementia (ALS/FTD), cardiovascular diseases (CVD), and cancer. 17 , 18 , 19 , 20
2. STRUCTURE AND COMPOSITION OF MAM
2.1. Structure of MAM
When observed under a wide‐field three‐dimensional deconvolution microscope, approximately 5%–20% of the total surface of the mitochondrial network is estimated to be close to the ER membrane. 21 Moreover, the electron micrograph image revealed that overlapping apposition distances between the ER and OMM vary approximately between 10 and 25 nm. 10 This variation emanates from the fact that OMM is attached differently to smooth ER and rough ER (Figure 1). The distance of rough ER from OMM is greater than its distance from smooth ER. This is because ribosomes are attached to rough ER and act as spacers, limiting the minimum distance between them to about 20 nm. 22 The distance can also be varied by the influence of intracellular Ca2+ signaling as studies of live cell imaging revealed. 23 , 24 , 25
2.2. Composition of MAM
Biochemical methods like subcellular fractionation on a percoll gradient or microscopic techniques like fluorescence microscopy were used for the analysis of the composition MAM fraction. 23 When the fraction was subjected to proteolysis with trypsin or proteinase K, it detached from mitochondria. 10 Further analysis using the mass spectroscopy identified more than 1000 MAM resident proteins with variable functions. 26 Of these proteins, some are involved in tethering the two organelles. MAM tethering proteins form paired complexes (Figure 2). These include Mitofusin‐2 (Mfn2)‐Mitofusin‐1/2 (Mfn1/2), inositol‐1,4,5‐trisphosphate receptor (IP3R3)‐glucose‐regulated protein‐75 (Grp75)‐voltage‐dependent anion channel 1 (VDAC1), vesicle‐associated membrane protein‐associated protein B (VAPB)‐protein tyrosine phosphatase‐interacting protein‐51 (PTPIP51), and Fission 1 (Fis1)‐B cell‐associated protein 31 (Bap31).
Figure 2.
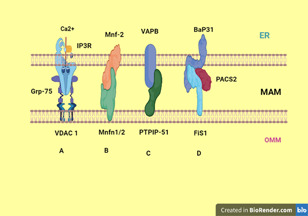
Proposed ER–mitochondria tethering protein complexes. (A) IP3R and VDAC1 interact via GRP75. (B) ER‐located Mfn‐2 interacts with mitochondrial Mfn1/2. (C) VAPB binds to PTPIP51. (D) Bap31 binds to Fis1 and their binding is stabilized by PACS2. The picture is created at https://biorender.com/.
ER‐located Mfn‐2 interacts in trans with mitochondrial mitofusins to form a tethering complex to bridge the ER and mitochondria and allow efficient Ca2+ transfer between them. 27 Silencing of Mfn‐2 in embryonic fibroblasts has been shown to increase the distance between them Furthermore, the absence of Mfn‐2 consistently cause a loosening of their connection. 18
The VDAC1 of the OMM interacts with the ER‐Ca2+ release channel IP3R3 via the molecular chaperone Grp75 and forms the VDAC1–Grp75–IP3R3 complex serving as a conduit of Ca2+ transfer from the ER to mitochondria. It may not have a tethering role, but rather a contact site spacing/filling function. Sigma1R (Sig‐1R), another MAM resident protein, stabilizes MAM by interacting with VDAC1 and IP3R3. 27 , 28
VAPB interacts with OMM protein tyrosine phosphatase‐interacting protein‐51 (PTPIP51) and forms the VAPB–PTPIP51 tethering complex. 28 , 29 Overexpression of either protein increase ER–mitochondria tethering and Ca2+ exchange between them, while their knockout decrease it. 28 , 30
Bap31 interacts with Fis1 and forms the Bap31–Fis1 MAM complex. 31 , 32 , 33 Simmen et al. demonstrated that another protein called phosphofurin acidic cluster sorting protein 2 (PACS‐2) modulates the role of Bap31 in tethering the two organelles. However, depletion of PACS‐2 was reported to cause Bap‐31‐dependent mitochondrial fragmentation and uncoupling from the ER along with inhibition of Ca2+signal transmission. 34 Mammalian target of rapamycin complex 2 (mTORC2) also regulates the integrity of MAM by Akt‐dependent phosphorylation of PACS‐2. 35
2.3. Functions of MAM
The existence of contact sites between mitochondria and ER suggests that the structures that are localized to these two different organelles can come together and synergize to provide additional functions at these specialized domains called MAM. 18 , 27 , 36 , 37
2.3.1. MAM and Ca2+ signaling
Ca2+ is released from the ER and transferred to mitochondria using MAM as a conduit. 21 Moderate loading of mitochondria with Ca2+ stimulates ATP production via Ca2+‐dependent activation of the key metabolic enzymes such as pyruvate dehydrogenase (PDH), isocitrate dehydrogenase, and α‐ketoglutarate dehydrogenase. 27 However, prolonged overflow of Ca2+ into mitochondria activates apoptosis whereas its reduction cellular causes energy crisis by decreasing oxidative phosphorylation. 22 , 27
The mechanism of Ca2+ transfer from ER to mitochondria is mediated by four major proteins which include IP3R, VDAC1, Grp75, and mitochondrial Ca2+ uniporter (MCU) reside in MAM, OMM, cytosol, and inner mitochondrial membrane (IMM), respectively. 38 The VDAC1 of the OMM interacts with IP3R via Grp75 (ref. 27 ) and increases the efficiency of mitochondrial Ca2+ uptake. 39 Although OMM is permeable to Ca2+ through VDAC1, the IMM is not. Thus, Ca2+ needs to go through MCU, a low‐affinity Ca2+ channel that requires high Ca2+ levels, to reach the mitochondrial matrix. 27 , 39 , 40
Sig‐1R and glucose‐regulated protein 78 (GRP78) are also involved in this process. Sig‐1R physically associate with GRP78 at MAM where they regulate Ca2+ flux via IP3R3, stabilizing it and prolonging Ca2+ signaling from the ER to mitochondria. 23 Other proteins also take part in this process. For instance, Akt phosphorylates IP3R and suppresses IP3R‐mediated Ca2+ release, while tumor suppressors phosphatase and tensin homolog (PTEN) directly dephosphorylates IP3R and promyelocytic leukemia protein (PML) indirectly dephosphorylates IPR3 via sequestration of protein phosphatase 2A (PP2A). 20
2.3.2. MAM and lipid synthesis and transfer
Phospholipid transport and synthesis is the first recognized function of MAM. 9 The ER is the main site of phospholipid biosynthesis and plays a significant role in intracellular vesicular trafficking. Because mitochondria are not connected to this trafficking, they require direct lipid transfer from the ER 18 or they might utilize MAM as a conduit. 24 On top of this, MAM are also enriched in major enzymes that are involved in the biosynthesis of the two most abundant phospholipids namely phosphatidylcholine and phosphatidylethanolamine. These enzymes include phosphatidylserine synthase‐1 or ‐2 and phosphatidylethanolamine N‐methyltransferase 2. 9 , 41 , 42
Studies reported that MAM is also the site of triacylglycerol synthesis and steroidogenesis. 43 Long‐chain‐fatty‐acid‐CoA ligase 4 that mediates the ligation of fatty acids to coenzyme A also enriched at MAM. 42 An enzyme catalyzing the formation of cholesterol esters and diacylglycerol, Acyl‐coenzyme A: cholesterol acyltransferase‐1, is also found in MAM. 42 , 44 A MAM resident steroidogenic acute regulatory protein interacts with VDAC2, another MAM protein, and facilitates its translocation to the MAM before it is targeted to mitochondria for its role in steroidgenesis. 45
2.3.3. MAM and insulin signaling
MAM is also involved in the insulin signaling pathway. 10 However, several proteins involved in the insulin signaling pathway are enriched in MAM. For example, Akt which phosphorylates IP3R and reduces Ca2+ release and prevents apoptosis, 10 , 35 , 46 mTORC2 which maintains MAM integrity, 35 , 47 PTEN which sensitizes cells to apoptosis by dephosphorylating IP3R and restoring Ca2+ release 48 are localized in MAM. PML which modulates its sensitivity to apoptosis by sequestering PP2A and blocking Akt phosphorylation and Ca2+ release by IP3R is also found in this site. 49 Likewise, mitochondrial Ca2+ uptake was found crucial for effective insulin signaling in skeletal muscle cells 50 and cardiac myocytes. 51
2.3.4. MAM and mitochondrial dynamics
Under normal conditions, mitochondrion changes morphology to create a fragmented or tubular network and to move along the cytoskeleton with coordinated mitochondrial fission and fusion processes. 15 Mitochondrial fusion assists cells to recover from stressful conditions whereas fission promotes mitophagy to remove mitochondria that are damaged or unable to regain their function to undergo apoptosis. 17
Mfn‐1, Mfn‐2, and optic atrophy 1 (Opa1) are the most studied of several proteins known to involve in mitochondrial fusion. Mfn‐1 exclusively localizes to mitochondria, whereas Mfn‐2 resides in MAM and mitochondria. While Mfn‐1 and Mfn2 are responsible for the fusion of the OMM, Opa1 is responsible for the fusion of the IMM. 15 Likewise, the fission process also involves several proteins of which dynamin‐related protein 1 (Drp1) is well studied and recruited from the cytosol to the OMM by various adaptor proteins including mitochondrial fission protein 1 (Fis‐1) which are present on the OMM. DRP1 is translocated to the MAM site, where it can cleave mitochondria efficiently and target damaged mitochondria for mitophagy. 15 , 20
2.3.5. MAM and autophagy
“Autophagy is a mechanism for the degradation of cellular material either as a way to provide nutrients during times of starvation or as a quality control system that eliminates unneeded proteins and/or organelles during normal growth and development” 13 These wastes are isolated by double‐membrane vesicles called autophagosomes which fuse with lysosomes to form autolysosomes and eventually degraded by lysosomal enzymes. 52 The formation and development of autophagosomes involve autophagy‐related genes, which encode proteins that regulate autophagy as discussed in various articles. 53 , 54 , 55 , 56
Hamasaki et al. 54 reviewed that the origin and formation of autophagosomes remain obscure for scientists though independent studies have pointed to several different organelles as potential membrane sources. However, a recent study showed that autophagosomes form at MAM. 13
Gomez‐Suaga et al. reported that VAPB‐PTPIP51 tethers are also in regulating autophagy. However, overexpression of VAPB or PTPIP51 tightens ER–mitochondria contacts and impairs autophagosome formation. However, small interfering RNA (siRNA)‐mediated loss of VAPB or PTPIP51 loosens contacts and stimulates autophagosome formation. 57
2.3.6. MAM and cellular apoptosis
The transfer of Ca2+ from ER to mitochondria is accomplished by MAM and excessive mitochondrial Ca2+ uptake can trigger Ca2+‐mediated apoptosis. 58 Higher matrix Ca2+ levels sensitize mitochondria to undergo mitochondrial outer membrane permeabilization (MOMP), a process preceding apoptosis. 59 Increased uptake of Ca2+ by mitochondria may result in changes in the permeability of the IMM. This is caused by the prolonged opening of the mitochondrial permeability transition pore which induces mitochondrial swelling and OMM rupture. This is followed by the release of apoptosis‐inducing caspase‐activating factors such as cytosolic cytochrome C. 58 The released cytochrome C amplifies caspase activation by binding to the IP3R and exacerbating its Ca2+ leaking properties. 60
Bap31–Fis1 complex also play role in apoptosis by recruiting caspase‐8 which enables the cleavage of Bap31 into its pro‐death fragment, p20Bap31. This fragment favors the emptying of ER–Ca2+ stores and induces cell death. 33 Moreover, PTEN has been known to interact with IP3R/Akt complex and reduce their phosphorylation. This, in turn, results in increased Ca2+ release and apoptosis. 48
2.3.7. MAM and ER stress
“The ER plays an indispensable role in protein folding. This role is facilitated by the presence of chaperone proteins capable of binding to newly synthesized, but as yet unfolded, proteins to facilitate optimal protein folding and prevent protein‐protein aggregation under normal physiological conditions.” 61 However, in pathological conditions, the accumulation of misfolded or unfolded proteins may occur and cause cellular dyshomeostasis. This triggers ER to elicit an adaptive or protective response called unfolded protein response (UPR) which restores cellular homeostasis. 60 , 61 If the homeostasis is not restored, the UPR switches to promote apoptosis. Nevertheless, in some pathophysiological situations, the homeostatic capacity of the ER and the UPR may not meet cellular demands and may even become a detrimental condition called ER stress in which structural uncoupling of ER from mitochondria may also induce it. 61 , 62 , 63
2.3.8. MAM in the formation and activation of the NLRP3 inflammasome
Cells require the capacity to sense and respond to the danger presented by extrinsic threats. Pattern recognition receptors (PRRs) recognize conserved molecular patterns expressed by invading pathogens (pathogen‐associated molecular patterns, PAMPs) or endogenous ligands derived from cellular damage resulting from infection or tissue injury (danger‐associated molecular patterns, DAMPs). Activation of PRRs by PAMPs or DAMPs triggers downstream signaling cascades and causes the production of Type I interferon (interferon‐α and interferon‐β) and pro‐inflammatory cytokines resulting in inflammation. DAMP‐triggered inflammation was reported to play a crucial role in the pathogenesis of inflammation‐mediated metabolic diseases. 61
One of the innate immunity sensors that mediate this inflammatory response are cytosolic multiprotein complexes termed inflammasomes 23 and the formation of this inflammasome involves MAM as a platform. 60 , 65 The most studied inflammasome was the NOD‐like receptor family protein 3 (NLRP3) inflammasome. 65
In an inactive state, NLRP3 localizes to the ER membrane and cytosol. However, in its active state, both NLRP3 and its adaptor apoptosis‐associated speck‐like protein containing a CARD (ASC) relocate to the MAM fraction where they are strategically assembled and located to sense signals emanating from mitochondria like increased ROS and mitochondrial‐derived DAMPs like mitochondrial DNA (mtDNA), ATP, cardiolipin, cytochrome C, and succinate. 19 NLRP3 oligomerizes and exposes its effector domain to interact with ASC. ASC in turn recruits pro‐caspase‐1 which is cleaved and becomes matured caspase‐1. Finally, activated caspase‐1 cleaves pro‐interleukin‐1β (pro‐IL‐1β) and pro‐IL‐18 to generate mature IL‐1β and IL‐18 (Figure 3). 20
Figure 3.
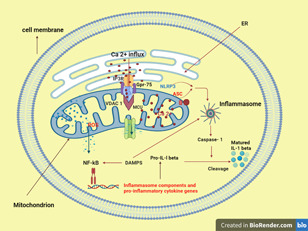
MAM are important sites for NLRP3‐inflammasome formation and activation (described in Section 2.3.8). The picture is created at https://biorender.com/.
Recently, Zang et al. reported that the NLRP3 inflammasome has been shown to be activated by a variety of distinct stimuli, including K+ efflux, mitochondrial dysfunction, lysosomal disruption, and trans‐Golgi disassembly. However, the most widely accepted stimuli was K+ efflux‐induced NLRP3 inflammasome activation. This mechanism has been thought to involve mitochondria. This is supported by the fact that PAMPs such as bacterial lipopolysaccharide (LPS) induced the expression of genes involved in mitochondrial biogenesis and mitophagy, resulting in an increase in mitochondrial mass and mitochondrial membrane potential. To back up their claim, the researchers silenced the mitochondrial transcription factor A (Tfam), and genetic ablation of Tfam abolished the NLRP3 inflammasome activation induced by K+ efflux via release of mtDNA as deprivation of cellular mtDNA by ethidium bromide treatment could reverse inflammasome activation induced by K+ efflux. They also revealed that mtDNA release induced by K+ efflux in macrophages activates NLRP3 inflammasome. 66
It has also been shown that ER stress activates the NLRP3 inflammasome in both peripheral and central immune cells. ER stress‐induced NLRP3 inflammasome activation occurs via a Ca2+‐dependent and ROS‐independent mechanism in monocytes, which is associated with upregulation of MAMs‐resident chaperones, closer ER–mitochondrial contacts, mitochondrial depolarization, and impaired dynamics. MAM thus plays an important role in the innate immune cells' response to ER stress. 67
3. MAM AND PATHOGENESIS OF INFLAMMATION‐MEDIATED DISEASES
3.1. Neurodegenerative diseases
Neurodegenerative diseases, such as AD, PD, and ALS/FTD, occur when nerve cells in the brain or peripheral nervous system lose function over time and ultimately die. 68 While they involve distinct protein pathologies, they share similar features that involve MAM disruption including mitochondrial damage, Ca2+ homeostasis, lipid metabolism, axonal transport, UPR activation, autophagy, and inflammatory responses. 36
Inflammatory response proteins have been most commonly implicated in neurodegenerative diseases. For example, a continuous release of IL‐1β negatively modulates the integrity of the brain–blood barrier, which results in the infiltration of immune cells into the central nervous system.19 The same cytokine amplifies the generation of other pro‐inflammatory factors by stimulating the activation of microglia and astrocytes. 69 Moreover, Fogal et al. 70 have demonstrated that overexpression of IL‐1β mediates neuronal injury and cell death throughout glutamate excitotoxicity.
Misfolded protein aggregates and excessive accumulation of metabolites are also critical determinants for the activation of ER‐stress and NLRP3 inflammasome which in turn initiates neurodegeneration including AD and PD. 64
3.1.1. Alzheimer's disease
The pathogenesis of AD and the series of events underlying it are unknown. The most widely accepted hypothesis is called the amyloid cascade, based on the observation that the brain of AD patients contains high levels of extracellular plaques called β‐amyloid (Aβ) composed of 40–42 amino acids and neurofibrillary tangles (NFTs) which are composed of hyperphosphorylated forms of the microtubule‐associated protein tau in the cerebrum. Aβ is produced by cleavage of the amyloid precursor protein (APP) by presenilin (presenilin‐1 and/or presenilin‐2), both of which are active components of the γ‐secretase complex. 69 Notably, dominant mutations both in the presenilins and in APP are currently the only known causes of the familial form of AD (FAD). 71 , 72 As summarized in references 17 , 73 these two isoenzymes of presenilins were found to be enriched in MAM fractions from neuronal and non‐neuronal cells. Yu et al. 74 briefly explained that significant mutations in APP or/and PSEN1/2 might lead to the excessive generation of Aβ42 and the increased ratio of Aβ42/40 which result in AD in their recent review.
AD that is linked with presenilin mutation is also characterized by increased levels of monocyte chemoattractant protein 1 (MCP‐1), IL‐6, and IL‐8 while a Presenilin1 mutation in microglial cells amplified tumor necrosis factor α, IL‐1α, IL‐1β, and IL‐6 gene expression. 72 It was also reported that APP and its catabolites are also found in MAM, where they interact with other MAM‐resident proteins and modulate ER functions. 75
The relationship between MAM and NLRP3 inflammasome is already described in Section 2.3.8. Moreover, researchers reported the intimate relationship between amyloid‐β and NLRP3 inflammasome as oligomerized Aβ originating from nontoxic Aβ monomers directly interacted with NLRP3, leading to the activation of the NLRP3 inflammasome. 76 , 77 Heneka et al. demonstrated that the deposition of Aβ drives cerebral neuro‐inflammation by activating microglia. Indeed, Aβ activation of the NLRP3 inflammasome in microglia is fundamental for IL‐1β maturation. The researchers explained their claim with a piece of evidence from NLRP3−/− or caspase‐1−/− mice carrying mutations associated with familial AD. These mice, which were largely protected from loss of spatial memory, demonstrated reduced brain caspase‐1 and IL‐1β activation, enhanced Aβ clearance, and NLRP3 inflammasome deficiency skewed microglial cells to an M2 phenotype and resulted in the decreased deposition of Aβ. 78
3.1.2. Parkinson's disease
PD is the most common movement disorder and the second most common neurodegenerative disease after AD. 23 , 36 It is characterized by an excessive death of dopaminergic neurons in the substantia nigra pars compacta together with intraneuronal inclusions termed “Lewy bodies” which are mainly formed from aggregates of a protein called α‐synuclein. Most recently, it has been shown that α‐synuclein localizes at the MAM. 36 , 75 The overexpression of both wild type and mutant α‐synuclein isoforms disrupt the VAPB‐PTPIP51 tethers, thus decreasing MAM formation. This causes decreases in Ca2+ exchanges between the two organelles that, in turn, lowers mitochondrial ATP production. 28
Additionally, pathogenic mutations of α‐synuclein causes downregulation of MAM functions while activating inflammasome. Indeed, α‐synuclein aggregates were found to be sufficient to provoke IL‐1β production by activating microglia and astrocytes. The fibrillary and monomeric forms of this protein showed differences in their capacity to induce inflammation. The monomeric form only induces the expression of pro‐IL‐1β whereas the fibrillary form can provoke caspase‐1 activation and maturation of IL‐1β and fully activates the inflammasome. 79 , 80
In fact, similar to AD, stimulating caspase‐1 activation and the release of IL‐1β is necessary to induce the production of ROS and activity of cathepsin‐B. 81 Accordingly, through specific inhibition of cathepsin‐B; it is possible to interfere with the inflammasome assembly though this finding was not validated.
More notably, Yan et al. showed that dopamine‐producing neurons and NLRP3 inflammasome are tightly interconnected and are able to regulate each other. They further showed that the neurotransmitter dopamine has the potential to inhibit NLRP3 inflammasome activation and subsequent IL‐1β production. This inhibitory activity of dopamine occurs via the dopamine D1 receptor signaling through an autophagic dependent process. 82
3.1.3. ALS with associated front temporal dementia
ALS/FTD is a neurodegenerative disease caused by the loss of motor neurons, resulting in the gradual deterioration of muscles. The exact cause of ALS is still not clear. However, a mutation in SigR1 is discovered in a juvenile form of ALS. In the SigR1 knockout mouse, ALS phenotypes such as muscle weakness and motor neuron loss were exhibited. 27
Another MAM protein, VAPB is also mutated in familial ALS. A mutant VAPB increases its affinity to PTPIP51 and strengthens VAPB‐PTPIP51 tethering, which alters Ca2+ shuttling between ER and mitochondria as elegantly summarized in a recently published review by Lee and Min. 83 Dominantly inherited forms of the disease were caused by deposits of Tar DNA‐binding protein 43 (TDP‐43) gene mutation. Notably, TDP‐43‐induce alteration of MAM involves breaking of the VAPB–PTPIP51, causing aberrant cellular Ca2+ homeostasis and decreased rates of ATP production. 36
Expression, activation, and co‐localization of the NLRP3 inflammasome were observed in the spinal cord of male SOD1 (G93A) mice carrying a mutant human superoxide dismutase 1 (SOD1). 84 It was also demonstrated that both aggregated and soluble SOD1G93A activates the inflammasome in primary mouse microglia. 85 However, SOD1G93A was unable to induce IL‐1β secretion from microglia pretreated with NLPR3 or deficient for NLRP3,r, confirming NLRP3 as the key inflammasome complex mediating SOD1‐induced microglial IL‐1β secretion. 86 Microglial NLRP3 upregulation was also observed in the TDP‐43 mutant mice model. TDP‐43 could also activate microglial inflammasomes in an NLRP3‐dependent manner. Mechanistically, they could identify the generation of ROS and ATP as key events required for SOD1G93A‐mediated NLRP3 activation. 84 , 87
3.2. Diabetic mellitus
Insulin resistance and pancreatic β‐cell dysfunction in T2DM are widely associated with derangement of MAM compartments as there is a strong linkage between MAM integrity and insulin action in hepatic cells. 88 It was also demonstrated in vitro and in vivo that defective MAM is closely associated with impaired hepatic insulin sensitivity and restoration of MAM integrity by cyclophilin D overexpression improved insulin signaling in primary hepatocytes of diabetic mice. 89
Notably, an in vivo experimental study reported that in the skeletal muscle of obese and diabetic humans, the expression levels of the ER–mitochondria tethering protein Mfn‐2 are reduced. Indeed, the livers of transgenic mice deleted for Mfn‐2 possessed a low insulin response and a reduction in mitochondrial respiration resulting in increased production of ROS which cause subsequent accumulation of mutation at the level of mtDNA. 86 An increase in the level of ROS was found to be a primary contributor to inflammation in T2DM. In fact, pro‐inflammatory cytokines also exacerbate ER and oxidative stress events, leading to β‐cell loss, recruitment of NLRP3 inflammasome, and finally the pathogenesis of T2DM. Moreover, increased ROS also stimulates conformational changes in thioredoxin‐interacting protein (TXNIP) and subsequent loss of the complex thyroidotoxin (TRX)–TRXNIP that binds and activates NLRP3 resulting in the generation of IL‐1β. 23
3.3. Cardiovascular diseases
3.3.1. Mitochondrial dynamics and cardiovascular disease
MAM and mitochondrial dynamics are also recognized as key factors in the pathogenesis of CVD. This was evidenced by a study that demonstrated that precise Ca2+ transport from the ER to the mitochondria regulates the cardiac contraction cycle. 90 Moreover, mitochondrial Ca2+ fluctuations and Ca2+ oscillation triggered by ER are present during cardiomyocyte beating. 91
Among the proteins involved in the maintenance of MAM, Mfn1/2 seems to be the most relevant one in the pathogenesis of CVD. It was confirmed that adult hearts deleted for both mitofusins showed compromised cardiac function, augmented left ventricular end‐diastolic volume, and reduced fractional shortening. This is supported by the fact that transgenic Mfn‐2−/− mice exhibited reduced contact length between these organelles resulting in a reduction of ER–mitochondrial Ca2+ transfer, and increased production of ROS that activate the NLRP3 inflammasome. 85
Finally, it has been reported that specific proteins conserving the ER–mitochondria interface are involved in ischemia/reperfusion (I/R). For example, OPA1 deficiency was associated with increased sensitivity to I/R, whereas the inhibition of Fis1 and DRP1 function was reported to be cardioprotective. 92
3.3.2. MAM and cardiovascular diseases
Missiroli et al. briefly summarized in their review that excess ROS production and subsequent NLRP3 activation are frequently found in CVD. In the presence of excess cholesterol deposition in the arterial wall, it forms crystals that induce inflammatory injury. This can be supported by the finding that macrophages can internalize these crystals and promote NLRP3 inflammasome activation in a process involving leakage of cathepsin B and L into the cytoplasm. This, in turn, causes the excessive formation of mitochondrial ROS and lowering in potassium concentrations. 19
The important role of inflammasomes was confirmed in atherosclerosis using ApoE−/− mice deletion of IL‐1β gene reduced the size of atherosclerotic lesions by up to 30%. 93 Moreover, the deletion of the IL‐ 18 receptors (IL‐18R−/−) decreased the size of the lesions. 94 Despite this, NLRP3 may not be the only source of pro‐inflammatory cytokines in atherosclerosis. Transgenic mice ApoE−/− crossed with mice deleted for different components of the NLRP3 such as (Nlrp3−/−, ASC−/−, or caspase‐1−/−) exhibited no differences in atherosclerotic lesions and plaques when compared to the double knockout and control mice. 95
NLRP3 inflammasome recruitment and the appropriate MAM composition also have an important role during I/R. Notably, IL‐1β and IL‐18 are primary mediators of I/R‐induced human myocardial injury through the inhibition of caspase‐1 activity that reduces the depression in contractile force after I/R. 96 Similarly, in ASC−/− mice the level of inflammatory cytokines was reduced and this results in a significant reduction of injuries such as the development of infarctions, myocardial fibrosis, and dysfunction in myocardial I/R injury compared to wild‐type controls. 97
Additionally, Shengnan and Ming‐Hui demonstrated that FUNDC1 also participates in MAM formation in cardiomyocytes by binding to IP3R2. This is because FUNDC1 deletion causes an 80% reduction in ER and mitochondria contact sites resulting in the decrease of Ca2+ transfer from ER to mitochondria resulting in elevation of ROS generation which induces chronic inflammation. 1
3.4. The role of MAM in the onset and progression of cancer
3.4.1. Alteration of MAM composition in breast cancer
In breast cancers, the expression of the stress‐activated Sig1R was found to be higher in metastatic potential cancer cells than in normal tissue. Under basal conditions, Sig1R binds the MAM chaperone GRP78; however, upon activation of IP3R3, Sig1R dissociates from GRP78 and binds the receptor, thereby stabilizing it at the MAM and enhancing IP3R3‐mediated Ca2+ fluxes to the mitochondria. However, during conditions of chronic ER stress involving prolonged ER Ca2+ depletion, Sig1R translocates from MAM to the peripheral ER and attenuates cellular damage, thereby preventing cell death. Sig1R also regulates Ca2+ homeostasis by forming a functional molecular platform with the calcium‐activated K+ channels, thus driving Ca2+ influx and favoring the migration of cancer cells. This implicates protumorigenic functions of this protein as stated in a current review by Morciano et al. 11
3.4.2. Alteration of MAM in hepatocellular cancer
Alteration of Mnfs or OPA1 function leads to decreased mitochondrial fusion, shifting the balance of mitochondrial dynamics to over‐fragmentation. This phenomenon was observed in experimental settings, aimed to investigate cancer biology. 57 For instance, a study demonstrated that MFN1 loss‐of‐function triggered the epithelial‐to‐mesenchymal transition of hepatocellular carcinoma favoring its metastasis. 98 Another study demonstrated that knockdown of Mfn‐1 and OPA1 inhibited mitochondrial fusion in experimental settings, leading to reduced cell growth and tumor formation. This implicates the antitumor effect of OPA1 and Mfn‐1 by silencing the induction of proapoptotic mechanisms, inhibition of oxidative metabolism, and ATP production. 99
4. CONCLUSION
MAM, a tiny membrane contact site, serves a far more important physiological function than most people realize. Based on the physiological function of multiple MAM resident proteins, there are still many unanswered questions about these contact sites. Apparently, Ca2+ homeostasis, mitochondrial dynamics, inflammasome formation and activation, cellular autophagy, and apoptosis are all affected when this membrane contact site is disrupted. The cumulative effect of its disruption is strongly associated with inflammatory‐mediated metabolic diseases, and it has a dramatic impact on health. MAM, on the other hand, plays an important role in innate immune cell response to ER stress and serves as a site of NLRP3 inflammasome activation under stress conditions, implying that MAM could serve as a novel potential therapeutic target for inflammatory‐related metabolic diseases. However, the nonspecific alteration of MAM makes it so difficult to use it as a target to treat some of these diseases.
AUTHOR CONTRIBUTIONS
Sisay Teka Degechisa wrote the manuscript draft. Yosef Tsegaye Dabi and Solomon Tebeje Gizaw contributed to the gathering of data, draft reviewing, and editing of the manuscript. All authors revised the manuscript and approved the final version of the manuscript before submission.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
We thank Dr. Sisay Addisu, Dr. Tibebe Antonios, Dr. Maria Degef, and Dr. Tsehayney Kelemu for providing critical comments after reading and evaluating a Ph.D. seminar from which this review manuscript was drafted.
Degechisa ST, Dabi YT, Gizaw ST. The mitochondrial associated endoplasmic reticulum membranes: a platform for the pathogenesis of inflammation‐mediated metabolic diseases. Immun Inflamm Dis. 2022;10:e647. 10.1002/iid3.647
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no data sets were generated or analyzed during the current study.
REFERENCES
- 1. Shengnan W, Ming‐Hui Z. Mitochondria‐associated endoplasmic reticulum membranes in the heart. Arch Biochem Biophys. 2019;662:201‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lackner LL, Voeltz GK. The mechanisms and functions of inter‐organelle interactions. Mol Biol Cell. 2017;28(6):703‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Scorrano L, De Matteis MA, Emr S, et al. Coming together to define membrane contact sites. Nat Commun. 2019;10(1):1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Islinger M, Godinho LF, Costello J, Schrader M. The different facets of organelle interplay—an overview of organelle interactions. Front Cell Dev Biol. 2015;3:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bernhard W, Haguenau F, Gautier A, Oberling C. Submicroscopical structure of cytoplasmic basophils in the liver, pancreas and salivary gland; study of ultrafine slices by electron microscope. Z Zellforsch Mikrosk Anat. 1952;37(3):281‐300. [PubMed] [Google Scholar]
- 6. Bernhard W, Rouiller C. Close topographical relationship between mitochondria and elastoplast of liver cells in a definite phase of cellular activity. J Cell Biol. 1956;2(4):73‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Copeland DE, Dalton A. An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J Cell Biol. 1959;5(3):393‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vance JE. Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem. 1990;265(13):7248‐7256. [PubMed] [Google Scholar]
- 9. Rusinol AE, Cui Z, Chen MH, Vance JE. A unique mitochondria‐associated membrane fraction from the rat liver has a high capacity for lipid synthesis and contains pre‐Golgi secretory proteins including nascent lipoproteins. J Biol Chem. 1994;269(44):27494‐27502. [PubMed] [Google Scholar]
- 10. Csordás G, Renken C, Várnai P, et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006;174(7):915‐921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morciano G, Marchi S, Morganti C, et al. Role of mitochondria‐associated ER membranes in calcium regulation in cancer‐specific settings. Neoplasia. 2018;20(5):510‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Naon D, Scorrano L. At the right distance: ER‐mitochondria juxtaposition in cell life and death. BBA Mol Cell Res. 2014;1843(10):2184‐2194. [DOI] [PubMed] [Google Scholar]
- 13. Yang M, Li C, Yang S, et al. Mitochondria‐associated ER membranes—the origin site of autophagy. Front Cell Dev Biol. 2020;8:595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tubbs E, Rieusset J. Metabolic signaling functions of ER‐mitochondria contact sites: role in metabolic diseases. J Mol Endocrinol. 2017;58(2):R87‐R106. [DOI] [PubMed] [Google Scholar]
- 15. Bernhardt D, Müller M, Reichert AS, Osiewacz HD. Simultaneous impairment of mitochondrial fission and fusion reduces mitophagy and shortens replicative lifespan. Sci Rep. 2015;5(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rieusset J. The role of endoplasmic reticulum‐mitochondria contact sites in the control of glucose homeostasis: an update. Cell Death Dis. 2018;9(3):1‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thoudam T, Jeon JH, Ha CM, Lee IK. Role of mitochondria‐associated endoplasmic reticulum membrane in inflammation‐mediated metabolic diseases. Mediat Inflamm. 2016;2016:1851420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. López‐Crisosto C, Bravo‐Sagua R, Rodriguez‐Peña M, et al. ER‐to‐mitochondria miscommunication and metabolic diseases. Biochim Biophys Acta. 2015;1852(10):2096‐2105. [DOI] [PubMed] [Google Scholar]
- 19. Missiroli S, Patergnani S, Caroccia N, et al. Mitochondria‐associated membranes and inflammation. Cell Death Dis. 2018;9(3):1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perrone M, Caroccia N, Genovese I, et al. The role of mitochondria‐associated membranes in cellular homeostasis and diseases. Int Rev Cell Mol Biol. 2020;350:119‐196. [DOI] [PubMed] [Google Scholar]
- 21. Rizzuto R, Pinton P, Carrington W, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280(5370):1763‐1766. [DOI] [PubMed] [Google Scholar]
- 22. Leal S, João N. The Interplay Between Mitochondria‐Endoplasmic Reticulum Contacts and Alzheimer's Disease. Thesis; 2019.
- 23. Gómez‐Suaga P, Bravo‐San Pedro JM, González‐Polo RA, Fuentes JM, Niso‐Santano M. ER–mitochondria signaling in Parkinson's disease. Cell Death Dis. 2018;9(3):1‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rowland AA, Voeltz GK. Endoplasmic reticulum‐mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol. 2012;13(10):607‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper. Trends Cell Biol. 2009;19(2):81‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang A, Williamson CD, Wong DS, et al. Quantitative proteomic analyses of human cytomegalovirus‐induced restructuring of endoplasmic reticulum‐mitochondrial contacts at late times of infection. Mol Cell Proteomics. 2011;10(10):111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu Y, Zhu X. Endoplasmic reticulum‐mitochondria tethering in neurodegenerative diseases. Transl Neurodegener. 2017;6(1):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reane DV, Rizzuto R, Raffaello A. The ER‐mitochondria tether at the hub of Ca2+ signaling. Curr Opin Physiol. 2020;17:261‐268. [Google Scholar]
- 29. De Vos KJ, Mórotz GM, Stoica R, et al. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Gen. 2012;21(6):1299‐1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stoica R, De Vos KJ, Paillusson S, et al. ER‐mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD‐associated TDP‐43. Nat Commun. 2014;5(1):1‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chandra D, Choy G, Deng X, Bhatia B, Daniel P, Tang DG. Association of active caspase 8 with the mitochondrial membrane during apoptosis: potential roles in cleaving BAP31 and caspase 3 and mediating mitochondrion‐endoplasmic reticulum cross‐talk in etoposide‐induced cell death. Mol Cell Biol. 2004;24(15):6592‐6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC. A caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol. 2003;160(7):1115‐1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Iwasawa R, Mahul‐Mellier AL, Datler C, Pazarentzos E, Grimm S. Fis1 and Bap31 bridge the mitochondria–ER interface to establish a platform for apoptosis induction. EMBO J. 2011;30(3):556‐568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Simmen T, Aslan JE, Blagoveshchenskaya AD, et al. PACS‐2 controls endoplasmic reticulum–mitochondria communication and Bid‐mediated apoptosis. EMBO J. 2005;24(4):717‐729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Betz C, Stracka D, Prescianotto‐Baschong C, Frieden M, Demaurex N, Hall MN. mTOR complex 2‐Akt signaling at mitochondria‐associated endoplasmic reticulum membranes regulates mitochondrial physiology. Proc Natl Acad Sci USA. 2013;110(31):12526‐12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paillusson S, Stoica R, Gomez‐Suaga P, et al. There's something wrong with my MAM; the ER‐mitochondria axis and neurodegenerative diseases. Trends Neurosci. 2016;39(3):146‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Phillips MJ, Voeltz GK. Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol. 2016;17(2):69‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu H, Carvalho P, Voeltz GK. Here, there, and everywhere: The importance of ER membrane contact sites. Science. 2018;361(6401). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Checchetto V, Teardo E, De Stefani D, et al. Electrophysiological characterization of the activity and regulation of the mitochondrial calcium uniporter. Biophys J. 2014;106(2):760. [Google Scholar]
- 40. Kerkhofs M, Bittremieux M, Morciano G, et al. Emerging molecular mechanisms in chemotherapy: Ca2+ signaling at the mitochondria‐associated endoplasmic reticulum membranes. Cell Death Dis. 2018;9(3):1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vance JE. Newly‐made phosphatidylserine and phosphatidylethanolamine are preferentially translocated between rat liver mitochondria and endoplasmic reticulum. J Biol Chem. 1991;266(1):89‐97. [PubMed] [Google Scholar]
- 42. Van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9(2):112‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Prasad M, Kaur J, Pawlak KJ, Bose M, Whittal RM, Bose HS. Mitochondria‐associated endoplasmic reticulum membrane (MAM) regulates steroidogenic activity via steroidogenic acute regulatory protein‐voltage‐dependent anion channel 2 (VDAC2) interactions. J Biol Chem. 2015;290(5):2604‐2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lewin TM, Kim JH, Granger DA, Vance JE, Coleman RA. Acyl‐CoA synthetase isoforms 1, 4, and 5 are present in different subcellular membranes in the rat liver and can be inhibited independently. J Biol Chem. 2001;276(27):24674‐24679. [DOI] [PubMed] [Google Scholar]
- 45. Csordás G, Thomas AP, Hajnóczky G. Quasi‐synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J. 1999;18(1):96‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marchi S, Rimessi A, Giorgi C, et al. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+‐dependent apoptotic stimuli. Biochem Biophys Res Commun. 2008;375(4):501‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell. 2011;144(5):757‐768. [DOI] [PubMed] [Google Scholar]
- 48. Bononi A, Bonora M, Marchi S, et al. Identification of PTEN at the ER and MAMs and its regulation of Ca 2+ signaling and apoptosis in a protein phosphatase‐dependent manner. Cell Death Differ. 2013;20(12):1631‐1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Giorgi C, Ito K, Lin HK, et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science. 2010;330(6008):1247‐1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Del Campo A, Parra V, Vásquez‐Trincado C, et al. Mitochondrial fragmentation impairs insulin‐dependent glucose uptake by modulating Akt activity through mitochondrial Ca2+ uptake. Am J Physiol Endocrinol Metab. 2014;306(1):E1‐E3. [DOI] [PubMed] [Google Scholar]
- 51. Gutiérrez T, Parra V, Troncoso R, et al. Alteration in mitochondrial. Ca 2+ uptake disrupts insulin signaling in hypertrophic cardiomyocytes. Cell Commun Signal. 2014;12(1):1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Scrivo A, Bourdenx M, Pampliega O, Cuervo AM. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018;17(9):802‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2018;14(2):207‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER‐mitochondria contact sites. Nature. 2013;495(7441):389‐393. [DOI] [PubMed] [Google Scholar]
- 55. Senft D, Ze'ev AR. UPR, autophagy, and mitochondria crosstalk underlie the ER stress response. Trends Biochem Sci. 2015;40(3):141‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Axe EL, Walker SA, Manifava M, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3‐phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182(4):685‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gomez‐Suaga P, Paillusson S, Stoica R, Noble W, Hanger DP, Miller CCJ. The ER‐mitochondria tethering complex VAPB‐PTPIP51 regulates autophagy. Curr Biol. 2017;27(3):371‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87(1):99‐163. [DOI] [PubMed] [Google Scholar]
- 59. Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER‐mitochondria Ca 2+ transfer in the control of apoptosis. Oncogene. 2008;27(50):6407‐6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria‐associated membranes in cellular signaling. BBA Mol Cell Res. 2014;1843(10):2253‐2262. [DOI] [PubMed] [Google Scholar]
- 61. Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18(6):716‐731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Morris G, Puri BK, Walder K, et al. The endoplasmic reticulum stress response in neurodegenerative diseases: emerging pathophysiological role and translational implications. Mol Neurobiol. 2018;55(12):8765‐8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bernales S, Morales Soto MA, McCullagh E. Unfolded protein stress in the endoplasmic reticulum and mitochondria: a role in neurodegeneration. Front Aging Neurosci. 2012;4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21(7):677‐687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20(13):3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang T, Zhao J, Liu T, et al. A novel mechanism for NLRP3 inflammasome activation. Metab Open. 2022;13:100166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pereira AC, De Pascale J, Resende R, et al. ER‐mitochondria communication is involved in NLRP3 inflammasome activation under stress conditions in the innate immune system. Cell Mol Life Sci. 2022;79(4):1‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dugger BN, Dickson DW. Pathology of neurodegenerative diseases. Cold Spring Harbor Perspect Biol. 2017;9(7):a028035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of α‐synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson's disease brain. J Biol Chem. 2008;283(14):9089‐9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fogal B, Li J, Lobner D, McCullough LD, Hewett SJ, System XC. ‐activity and astrocytes are necessary for interleukin‐1β‐mediated hypoxic neuronal injury. J Neurosci. 2007;27(38):10094‐10105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Area‐Gomez E, Schon EA. Mitochondria‐associated ER‐membranes and Alzheimer's disease. Curr Opin Genet Dev. 2016;38:90‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Area‐Gomez E, de Groof A, Bonilla E, et al. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer's disease. Cell Death Dis. 2018;9(3):335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Area‐Gomez E, de Groof AJ, Boldogh I, et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol. 2009;175(5):1810‐1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yu W, Jin H, Huang Y. Mitochondria‐associated membranes: a potential therapeutic target for treating Alzheimer's disease. Clin Sci. 2021;135(1):109‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Del Prete D, Suski JM, Oulès B, et al. Localization and processing of the amyloid‐β protein precursor in mitochondria‐associated membranes. J Alzheimer's Dis. 2017;55(4):1549‐1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nakanishi A, Kaneko N, Takeda H, et al. Amyloid β directly interacts with NLRP3 to initiate inflammasome activation: identification of an intrinsic NLRP3 ligand in a cell‐free system. Inflamm Regen. 2018;38(1):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Franchi L, Eigenbrod T, Muñoz‐Planillo R, Nuñez G. The inflammasome: a caspase‐1‐activation platform that regulates immune responses and disease pathogenesis. Nature Immunol. 2009;10(3):241‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Guardia‐Laguarta C, Area‐Gomez E, Rüb C, et al. α‐Synuclein is localized to mitochondria‐associated ER membranes. J Neurosci. 2014;34(1):249‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gustin A, Kirchmeyer M, Koncina E, et al. NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS One. 2015;10(6):e0130624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Codolo G, Plotegher N, Pozzobon T, et al. Triggering of the inflammasome by aggregated α–synuclein, an inflammatory response in synucleinopathies. PLoS One. 2013;8(1):55375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yan Y, Jiang W, Liu L, et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell. 2015;160(1‐2):62‐73. [DOI] [PubMed] [Google Scholar]
- 83. Lee S, Min KT. The interface between ER and mitochondria: molecular compositions and functions. Mol Cells. 2018;41(12):1000‐1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Deora V, Lee JD, Albornoz EA, et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia. 2020;68(2):407‐421. [DOI] [PubMed] [Google Scholar]
- 85. Dorn II, GW , Song M, Walsh K. Functional implications of mitofusin‐2 mediated mitochondrial‐SR tethering. J Mol Cell Cardiol. 2015;78:123‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sebastián D, Hernández‐Alvarez MI, Segalés J, et al. Mitofusin‐2 links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA. 2012;109(14):5523‐5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Johann S, Heitzer M, Kanagaratnam M, et al. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia. 2015;63(12):2260‐2273. [DOI] [PubMed] [Google Scholar]
- 88. Rutter GA, Pinton P. Mitochondria‐associated endoplasmic reticulum membranes in insulin signaling. Diabetes. 2014;63(10):3163‐3165. [DOI] [PubMed] [Google Scholar]
- 89. Tubbs E, Theurey P, Vial G, et al. Mitochondria‐associated endoplasmic reticulum membrane integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes. 2014;63(10):3279‐3294. [DOI] [PubMed] [Google Scholar]
- 90. Bassani RA, Bassani JW, Bers DM. Mitochondrial and sarcolemmal Ca2+ transport reduce [Ca2+] ion during caffeine contractures in rabbit cardiac myocytes. J Physiol. 1992;453(1):591‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Robert V, Gurlini P, Tosello V, et al. Beat‐to‐beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J. 2001;20(17):4998‐5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Papanicolaou KN, Khairallah RJ, Ngoh GA, et al. Mfn‐2 maintains the mitochondrial structure and contributes to stress‐induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31(6):1309‐1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kirii H, Niwa T, Yamada Y, et al. Lack of interleukin‐1β decreases the severity of atherosclerosis in ApoE‐deficient mice. Arterioscler Thromb Vasc Biol. 2003;23(4):656‐660. [DOI] [PubMed] [Google Scholar]
- 94. Mallat Z, Corbaz A, Scoazec A, et al. Interleukin‐18/interleukin‐18 binding protein signaling modulates atherosclerotic lesion development and stability. Circ Res. 2001;28 89(7):41‐45. [DOI] [PubMed] [Google Scholar]
- 95. Menu P, Pellegrin M, Aubert JF, et al. Atherosclerosis in ApoE‐deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2011;2(3):137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kawaguchi M, Takahashi M, Hata T, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123(6):594‐604. [DOI] [PubMed] [Google Scholar]
- 97. Le Page S, Niro M, Fauconnier J, et al. Increase in cardiac ischemia‐reperfusion injuries in Opa1+/‐ mouse model. PLoS One. 2016;11(10):0164066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zhang Z, Li TE, Chen M, et al. Mfn‐1‐dependent alteration of mitochondrial dynamics drives hepatocellular carcinoma metastasis by glucose metabolic reprogramming. Br J Cancer. 2020;122(2):209‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Li M, Wang L, Wang Y, et al. Mitochondrial fusion via OPA1 and Mfn‐1 supports liver tumor cell metabolism and growth. Cells. 2020;9(1):121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no data sets were generated or analyzed during the current study.