

Abstract
Phospholipases A2 (PLA2s) are a diverse family of lipolytic enzymes which hydrolyze the acyl bond at the sn-2 position of glycerophospholipids to produce free fatty acids and lysophospholipids. These products are precursors of bioactive eicosanoids and platelet-activating factor which have been implicated in pathological states of numerous acute and chronic neurological disorders. To date, more than 27 isoforms of PLA2 have been found in the mammalian system which can be classified into four major categories: secretory PLA2, cytosolic PLA2, Ca2+-independent PLA2, and platelet-activating factor acetylhydrolases. Multiple isoforms of PLA2 are found in the mammalian spinal cord. Under physiological conditions, PLA2s are involved in diverse cellular responses, including phospholipid digestion and metabolism, host defense, and signal transduction. However, under pathological situations, increased PLA2 activity, excessive production of free fatty acids and their metabolites may lead to the loss of membrane integrity, inflammation, oxidative stress, and subsequent neuronal injury. There is emerging evidence that PLA2 plays a key role in the secondary injury process after traumatic spinal cord injury. This review outlines the current knowledge of the PLA2 in the spinal cord with an emphasis being placed on the possible roles of PLA2 in mediating the secondary SCI.
Keywords: Phospholipases A2, Spinal cord injury, Inflammation, Oxidation, Apoptosis
Introduction
Phospholipases A2 (PLA2s) are a diverse family of lipolytic enzymes which hydrolyze the acyl bond at the sn-2 position of glycerophospholipids to produce free fatty acids and lysophospholipids (Fig. 1) [1–3]. These products are precursors of bioactive eicosanoids and platelet-activating factor (PAF) which are well-known mediators of inflammation and tissue damage implicated in pathological states of numerous acute and chronic neurological disorders including spinal cord injury (SCI) [3–6]. The hydrolysis of membrane phospholipids by PLA2 is a rate-limiting step for generation of eicosanoids and PAF [3, 7]. Stimulation of PLA2 is thought to be an important event in production of lipid inflammatory mediators. Under physiological conditions, PLA2s are involved in diverse cellular responses, including phospholipid digestion and metabolism, host defense, and signal transduction. However, in pathological situations, increased PLA2 activity and excessive production of free fatty acids such as arachidonic acid (AA) and pro-inflammatory mediators such as eicosanoids and PAF, may lead to the loss of membrane integrity, inflammation, oxidative stress, and subsequent neuronal injury [2, 3, 5, 8, 9]. This review outlines the current knowledge of the PLA2 in the spinal cord with an emphasis being placed on the possible role of PLA2 in mediating the secondary injury after an initial trauma.
Fig. 1.
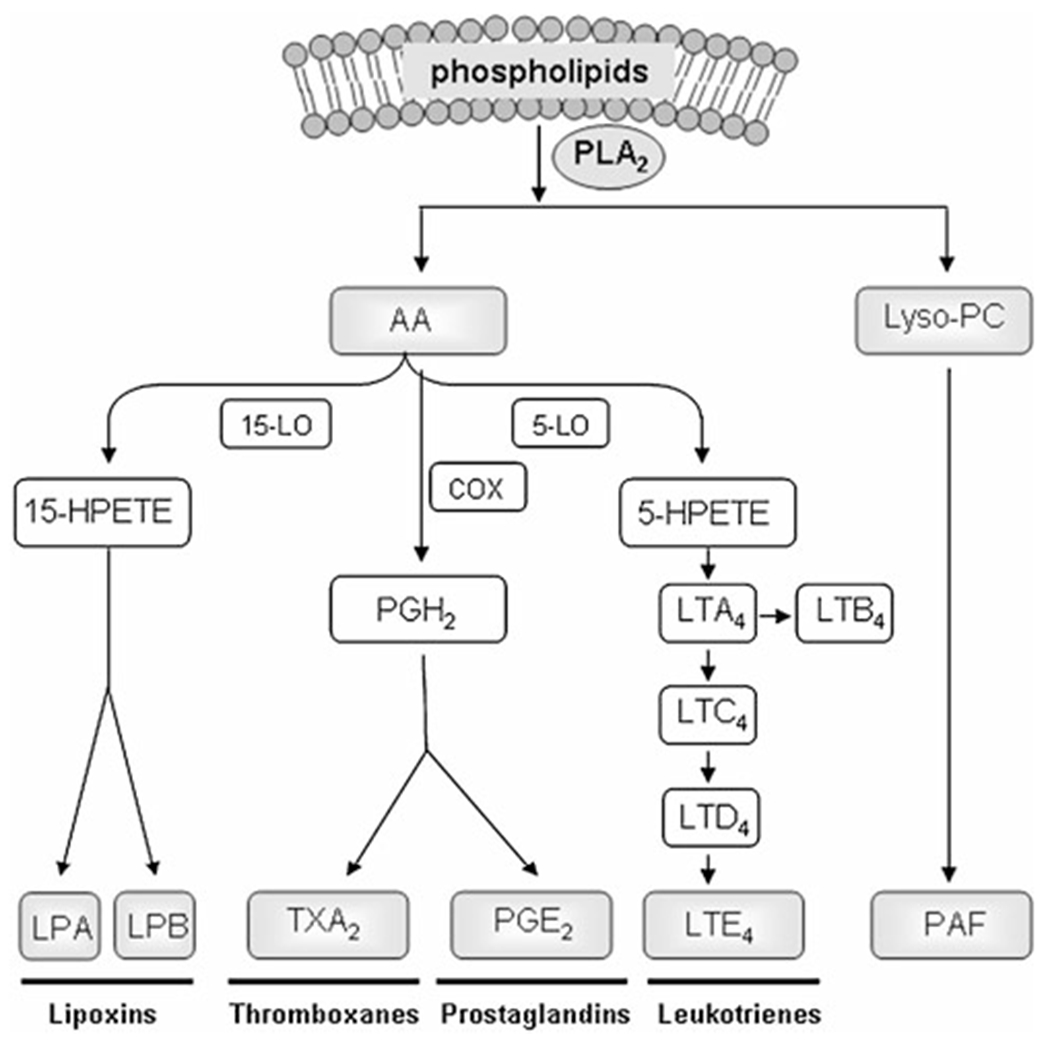
PLA2 mediates the production of lipid mediators. PLA2 hydrolyzes the membrane phospholipids to produce a free fatty acid such as arachidonic acid (AA) and a lysophospholipid such as lysophosphatidylcholine (Lyso-PC). AA can give rise to eicosanoids via cyclooxygenases (COX), 5-lipoxygenase (5-LO), and 15-lipoxygenase (15-LO) enzymes. Eicosanoids such as thromboxanes (TX), prostaglandins (PG), and leukotrienes (LT) are potent mediators of inflammation which can increase vascular permeability and induce chemotaxis of immune cells. Lipoxin is also an AA-derived lipid mediator. However, in contrast to above eicosanoids, it has an anti-inflammatory effect. In addition, platelet-activating factor (PAF) is biosynthesized from Lyso-PC and acetyl CoA by enzyme Lyso-PC acetyltransferase. Lyso-PC is a myelinolytic agent and can act as a chemoattractant for immune cells. PAF is a potent phospholipid activator and a mediator of many leukocyte functions, including platelet aggregation, inflammation, and anaphylaxis. 5-HPETE 5-hydroperoxyeicosatetraenoic acid, PGH2 prostaglandin H2
Classification, Structure, and Properties of PLA2
To date, more than 27 isoforms of PLA2s have been found in the mammalian system which can be classified into four major categories: secretory PLA2 (sPLA2), cytosolic PLA2 (cPLA2), Ca2+-independent PLA2 (iPLA2), and platelet-activating factor acetylhydrolases (PAF-AH; Table 1) [1, 2, 10, 11]. sPLA2s, in which ten isozymes have been identified, have a low molecular mass of about 14–18 kD and require the presence of submillimolar to millimolar concentrations of Ca2+ for effective hydrolysis of a substrate phospholipid without any fatty acid selectivity [11–14]. They are synthesized intracellularly and then secreted into the extracellular space and can act extracellularly [15, 16]. sPLA2 binds to two types of cell surface receptors, namely the N type, identified in neurons, and the M type, identified in skeletal muscles, of sPLA2 receptors although this nomenclature is merely academic since neither receptor is limited to these tissues and the expression has been shown widely for both types [17]. Members of the cPLA2 have a higher molecular mass (85–110 kD), selectively hydrolyze phospholipids containing AA, and require a submicromolar concentration of Ca2+ for optimal activity [2, 13, 18]. cPLA2s consist of 6 isoforms, among which cPLA2α plays an essential role in the initiation of AA metabolism. Intracellular activation of cPLA2α is tightly regulated by Ca2+ and phosphorylation [11, 18]. iPLA2s, containing seven enzymes, are intracellular enzymes with higher molecular mass ranging from 28 to 91 kD that shows no Ca2+ requiring for its activity. iPLA2 is generally considered as a housekeeping enzyme for the maintenance of membrane phospholipids [2, 10, 11]. Recent evidence, however, suggests that iPLA2 may also be involved in the pathogenesis of childhood neurological disorders [19]. PAF-AH family represents a unique group of PLA2 that contains four enzymes exhibiting unusual substrate specificity toward PAF and/or oxidized phospholipids [2, 10, 11].
Table 1.
A summary of mammalian PLA2 enzymes
| Family | Group | Other name | Size (kD) | Ca2+ requirement | Catalytic site | sn-2 FA Preference | Spinal cord localization | Human chromosome |
|---|---|---|---|---|---|---|---|---|
| sPLA2 | IB | Pancreatic PLA2 | 14 | mM | His/Asp dyad | No | + | 12q23-24 |
| IIA | Synovial PLA2 | 14 | mM | His/Asp dyad | No | + | 1p34-36 | |
| IIC | 15 | mM | His/Asp dyad | No | + | 1p34-36 | ||
| IID | 14 | mM | His/Asp dyad | No | NRA | 1p34-36 | ||
| IIE | 14 | mM | His/Asp dyad | No | + | 1p34-36 | ||
| IIF | 16 | mM | His/Asp dyad | No | NRA | 1p34-36 | ||
| III | 55 | mM | His/Asp dyad | No | NRA | 22q | ||
| V | 14 | mM | His/Asp dyad | No | + | 1p34-36 | ||
| X | 14 | mM | His/Asp dyad | No | + | 16p12-13 | ||
| XII | 19 | mM | His/Asp dyad | No | + | 4q25 | ||
| cPLA2 | IVA | cPLA2α | 85 | μM | Ser/Asp dyad | AA | + | 1q25 |
| IVB | cPLA2β | 110 | μM | Ser/Asp dyad | To be confirmed | NRA | 15 | |
| IVC | cPLA2γ | 60 | None | Ser/Asp dyad | To be confirmed | NRA | 19 | |
| IVD | cPLA2δ | 92-93 | μM | Ser/Asp dyad | To be confirmed | NRA | 15 | |
| IVE | cPLA2ε | 100 | μM | Ser/Asp dyad | To be confirmed | NRA | 15 | |
| IVF | cPLA2ξ | 96 | μM | Ser/Asp dyad | To be confirmed | NRA | 15 | |
| iPLA2 | VIA1 | iPLA2 | 84-85 | None | Ser/His/Asp triad | No | + | 22q13.1 |
| VIA2 | iPLA2β | 88-90 | None | Ser/His/Asp triad | No | + | 22q13.1 | |
| VIB | iPLA2γ | 88-91 | None | Ser/His/Asp triad | No | NRA | 7q31 | |
| VIC | iPLA2δ | 146 | None | Ser/His/Asp triad | No | NRA | NRA | |
| VID | iPLA2ε | 53 | None | Ser/His/Asp triad | No | NRA | NRA | |
| VIE | iPLA2ξ | 57 | None | Ser/His/Asp triad | No | NRA | NRA | |
| VIF | iPLA2η | 28 | None | Ser/His/Asp triad | No | NRA | NRA | |
| PAF-AH | VIIA | Plasma PAF-AH | 45 | None | Ser/His/Asp triad | Acetate | NRA | N.D. |
| VIIB | PAF-AH II | 40 | None | Ser/His/Asp triad | Acetate | NRA | N.D. | |
| VIIIA | PAF-AH-Iα1 (Ib) | 26 | None | Ser/His/Asp triad | Acetate | NRA | N.D. | |
| VIIIB | PAF-AH-Iα2 (Ib) | 26 | None | Ser/His/Asp triad | No | NRA | 11q23 |
ND not determined, NRA no report available, SC spinal cord. Adopted from Liu et al. [83] with modifications
PLA2 Isozymes in the Normal Spinal Cord
Multiple isoforms of PLA2s have been found in the mammalian spinal cord. sPLA2 activity was detected in the normal rat spinal cord homogenate [20]. Western blot analysis revealed the presence of sPLA2 IIA and V in the normal rat spinal cord [20]. mRNAs of sPLA2s IB, IIA, IIC, and V were also detected in the normal rat spinal cord [21]. We have recently shown that mRNAs for sPLA2-IB, IIA, IIC, IIE, V, X, and XIIA were all expressed in the normal rat spinal cord [22]. Immunohistochemistry and Western blot analysis confirmed the expression of sPLA2-IB and IIA in the spinal cord at the protein level [22]. Immunofluorescence double staining revealed that sPLA2-IB and IIA were localized to neurons, axons, astrocytes, and oligodendrocytes [22].
cPLA2 activity was also detected in the cytosolic fraction of the rat spinal cord [23]. cPLA2 immunoreactivity was found in the dorsal horn and motor neurons of the rat and monkey [24] and in the rat spinal cord shown by Western blot [25]. Lucas et al. [21] reported that cPLA2 activity was present in the normal rat spinal cord homogenates and demonstrated that both the mRNA and protein of cPLA2 are expressed in the normal rat spinal cord. Recently, we confirmed cPLA2 expression in the normal adult rat spinal cord and defined its cellular localization in neurons and oligodendrocytes but not in astrocytes [26].
iPLA2 mRNA is constitutively expressed in the human [27] and rat spinal cords [28]. Recently, Lucas et al. [21] demonstrated the activity as well as protein and mRNA expressions of iPLA2 in the normal rat spinal cord using iPLA2 activity assay, RT-PCR, and Western blot analyses.
PLA2 Isozymes in the Injured Spinal Cord
Acute SCI triggers a secondary injury by multiple biological processes [29–32]. One such a critical process is the activation of PLA2 which can result in the hydrolysis of membrane phospholipids, releasing free fatty acid, generation of oxygen free radicals, formation of eicosanoids, and ultimately leading to neuronal death [9, 33, 34].
SCI Induces Increases of PLA2 Metabolites including Free Fatty Acids, Eicosanoids, and Lipid Peroxides
It has been demonstrated in several experimental SCI models that the degradation of membrane phospholipids, along with the generation of free fatty acids, eicosanoids, and lipid peroxides, increased following SCI [35–39], suggesting that PLA2 activity increased following SCI. Phospholipids are main components of the cell membrane that play important roles in maintaining the structure and function of cell membranes. SCI resulted in an immediate decrease of total phospholipid content [37]. During the first minute of compression trauma to the spinal cord, 10% of the plasmenylethanolamine (PlsEtn) was lost with an overall loss of 18% found at 30 min after the injury [40].
One of the first pathophysiological events occurring following SCI is the release of free fatty acids due to the activation of PLA2. Within the first few minutes after SCI, the level of free fatty acids has increased in the gray matter and later in the white matter to a lesser extent [35, 41]. A time course study showed that there existed biphasic increases in the free fatty acid level in the spinal cord following injury [37]. The first increase occurred within 5 min after SCI which was declined at 30 min. The second increase occurred at 1 h after SCI, peaked at 24 h, and remained significantly high at 7 days after SCI [37].
Hydrolysis of membrane phospholipids by PLA2 is a rate-limiting step for generation of pro-inflammatory eicosanoids and PAF [3, 7]. Following SCI, eicosanoids such as thromboxane A2 and prostaglandin E2 (PGE2), metabolites of PLA2, increased in the injured cord tissue within a few minutes after SCI, and persisted at significantly high levels for 72 h after injury [42, 43]. These eicosanoids and PAF are key mediators of tissue damage and cell death following SCI.
SCI Induces Increased PLA2 Activity and Expression
Following a contusive SCI in adult rats, PLA2 activity increased significantly which was peaked at 4 h post-injury and remained at a significant high level at 7 days [26]. The expression of cPLA2, an important PLA2 isotype, was also increased and peaked at 3 and 7 days post-injury [26]. Immunohistochemical studies revealed that cPLA2 immunoreactivity was markedly increased in both the injured gray and white matter at 7 days after injury [26]. Immunofluorescence double labeling demonstrated that increased level of cPLA2 was localized in neurons, swollen axons, oligodendrocytes, and a subpopulation of microglia [26].
In addition to cPLA2, increased expression of a subset of sPLA2 was found following SCI [22]. In this study, expression of ten sPLA2 mRNAs including sPLA2-IB, IIA, IIC, IID, IIE, IIF, III, V, X, and XIIA at 4 h after SCI was studied. Among them, seven were detected in naïve and spinal cord contused animals (sPLA2-IB, IIA, IIC, IIE, V, X, and XIIA). Among these expressed sPLA2 isoforms, sPLA2-IIA showed the most dramatic increase after SCI. Increased expression of sPLA2-IIE mRNA was also found in the injured cord in a similar pattern. In contrast, sPLA2-X showed a decrease in signal intensity. No significant difference was found in expression patterns of sPLA2-IB, IIC, V, and XIIA mRNAs between the sham and SCI groups. Real-time qRT-PCR revealed that sPLA2-IIA mRNA expression had a significant fourfold increase at 1 h following contusion and remained highly elevated at 4 h after SCI [22]. Weston blot confirmed an increase in the expression of sPLA2-IIA and IIE after SCI [22]. Immunohistochemistry showed that significantly more immunoreactivity of IIA and IIE was found in the injured cord at 4 h after SCI as compared to the sham control [22]. Finally, immunofluorescence double labeling revealed that sPLA2-IIA was localized in neurons, axons, oligodendrocytes, astrocytes, and some myelin rings [22]. Thus, sPLA2 isoforms are differentially expressed following SCI and are localized in both neurons and glial cells. Localization of specific sPLA2 isoforms such as sPLA2IIA in neurons and oligodendrocytes indicate that these molecules may play important roles in mediating neuronal and oligodendrocyte cell death following spinal cord injury.
PLA2 Activation is Induced by Several Toxic Factors including Inflammatory Cytokines, Free Radicals, and Excitatory Amino Acids Generated in the Injured Spinal Cord
Although increased activity and expression of PLA2s after SCI was observed, the mechanism(s) by which they increase remains unclear. Recent studies showed that PLA2 activity and/or expression could be induced by several toxic factors such as inflammatory cytokines [2, 44], free radicals [45, 46], and excitatory amino acids [47–49]. All of these injury mediators were demonstrated to increase in the spinal cord following injury. It is therefore possible that PLA2 may serve as a converging molecule that mediates the pathogenesis of these different injury mechanisms associated with spinal cord secondary injury [29–31].
Increased PLA2 Results in Spinal Cord Tissue Damage and Behavioral Impairment
Increasing evidence suggests that PLA2 and their metabolites may mediate inflammation, oxidation, and neurotoxicity following SCI. In vitro experiments showed that both PLA2 and melittin, an activator of endogenous PLA2, induced spinal neuronal death in a dose-dependent manner, an effect that could be substantially reversed by mepacrine, a PLA2 inhibitor [26]. When PLA2 was directly microinjected into the normal rat spinal cord, it induced tissue damage, demyelination, and sustained impairment in motor function. Such PLA2-induced demyelination, however, could be effectively attenuated with mepacrine, a PLA2 inhibitor [26]. Injections of PLA2 also induced the expression of inflammatory cytokines TNF-α and IL-1β, as well as 4-HNE, a product of lipid peroxidation and a marker for oxygen free radical-mediated membrane injury [26]. PLA2 has also been reported to mediate myelin breakdown and axonal degeneration [50].
In vitro experiments also showed that sPLA2 induced spinal oligodendrocyte death in a dose-dependent manner [22]. Low levels of exogenously added sPLA2-IIA (0.01 and 0.1 μM) result in a loss of processes extending from the soma and at higher dose (2 μM) triggers a complete loss of process and cell death. In contrast, 2 μM of sPLA2-IIA had no effect on cultured Schwann cells and astrocytes, suggesting a specific sensitivity of oligodendrocytes to sPLA2-IIA. In addition, sPLA2-IIA mediates H2O2, IL-1β, and TNFα-induced oligodendrocyte cell death [22].
Annexin A1 (ANXA1) is an endogenous nonselective inhibitor of PLA2. Our experiment showed that administration of ANXA1 inhibited SCI-induced increases in PLA2 and myeloperoxidase activities [51]. In addition, ANXA1 administration reduced the expression of interleukin-1β and activated caspase-3 at 24 h post-injury and glial fibrillary acidic protein at 4 weeks post-injury [51]. Furthermore, ANXA1 administration significantly reversed PLA2-induced spinal cord neuronal death in vitro and reduced tissue damage and increased white matter sparing in vivo, compared to the vehicle-treated controls [51]. Fluoro-Gold retrograde tracing showed that ANXA1 administration protected axons of long descending pathways at 6 weeks post-SCI [51]. ANXA1 administration also significantly increased the number of animals that responded to transcranial magnetic motor-evoked potentials [51].
A recent study showed that intravenous administration of arachidonyl trifluoromethyl ketone (AACOCF3; 7.13 mg/kg), a cPLA2 inhibitor, at 30 min after SCI significantly increased the number of surviving neurons and oligodendrocytes at 7 days post-SCI as well as improved behavior recovery [52]. This finding suggests that PLA2 activation plays a critical role in mediating secondary SCI.
Increased Metabolites of PLA2 Result in Neurotoxicity and Demyelination
Metabolism of free fatty acid represents a source of reactive oxygen species (ROS). Generation of free fatty acids in SCI is closely associated with increases in free radical formation observed in the lesioned region of injured spinal cord [53, 54]. Several studies showed that free fatty acids activated NADPH oxidase, a key enzyme mediating ROS production [46, 55–57]. Application of pathophysiological concentrations of free fatty acids has been demonstrated in vitro to induce oxidative injury to spinal cord cell cultures [58]. Low concentrations of free fatty acid, AA, support cultured neurons to survive whereas higher concentrations are neurotoxic [59]. Neurotoxic effects of AA have also been observed in hippocampal neurons and cortical neurons [59, 60] as well as oligodendrocytes [61].
The bioactive eicosanoids such as thromboxanes, prostaglandins, and leukotrienes from AA, induced by PLA2, have been implicated as mediators of secondary injury via a host of mechanisms [62, 63]. For example, thromboxane A2 stimulates platelet aggregation and vasoconstriction. PGE2 and leukotrienes B4 (LTB4) increases vascular permeability. LTB4 also is a potent stimulator of polymorphonuclear leukocyte chemokinesis and chemotaxis.
In contrast to the above-mentioned eicosanoids, another AA-derived lipid mediator, lipoxin, has been shown to have an anti-inflammatory effect through modulating key steps in leukocyte trafficking [64]. However, changes of lipoxins and its possible role following SCI remain to be determined.
Lysophosphatidylcholine (Lyso-PC) and PAF are also metabolic products mediated by PLA2. Injection of Lyso-PC into the spinal cord causes demyelination as well as expression of a number of chemokines and cytokines [65, 66], which occurred in the injured cord after SCI. PAF levels have been shown to increase 20-fold after spinal cord injury induced by stroke [67]. Intrathecal administration of PAF leads to reduced spinal cord blood flow and motor deficits, an effect which can be blocked by the PAF receptor antagonist, WEB 2170 [68]. Treatment with WEB 2170 after acute spinal cord contusion resulted in significant increases in white matter sparing as well as decreases in pro-inflammatory cytokine mRNA levels within the lesion epicenter [69, 70]. Treatment of a PAF receptor antagonist BN52021 also improves behavioral recovery after SCI [71]. In vitro experiments showed that low concentrations of PAF resulted in neuronal differentiation and sprouting, while higher concentrations were neurotoxic [72]. PAF not only induced neuronal death in a dose-dependent manner in vitro [73, 74] but also death of oligodendrocytes and astrocytes [69].
PLA2 Mediates Excytotoxic Neuronal Death and Tissue Damage
It has been shown that the release of high levels of excitatory amino acids (EAA) such as glutamate and aspartate in experimental SCI is an important mechanism of secondary injury [30]. Growing evidence suggests that PLA2 mediates EAA-induced neuronal death and tissue damage. Marked increases in PLA2 activity and AA release have been reported after treatments of neuronal cultures with glutamate, N-methyl-D-aspartate (NMDA), and kainic acid (KA) [47, 75, 76]. This increased PLA2 activity can be inhibited by a PLA2 inhibitor, mepacrine as well as a KA/AMPA receptor antagonist, CNQX [76]. Recently, phosphorylated cPLA2 expression and AA release have been reported to increase in cultured primary neurons after NMDA stimulation [77]. The NMDA-induced AA release was inhibited by a cPLA2 inhibitor methyl arachidonyl fluorophosphonate and a NADPH oxidase-ROS pathway has been demonstrated to mediate NMDA-induced cPLA2 phosphorylation [77]. It has been hypothesized that the glutamate release triggers a sequence of events including NMDA receptor activation, increases of intracellular Ca2+, activation of PLA2, and eventually neuronal death [49]. It has been shown that glutamate release in the spinal cord can be suppressed by PLA2 inhibitors such as indomethacin by 40%, AACOCF3 by 45%, and 4-bromophenacyl bromide by 36%, suggesting that PLA2-induced EAA release mediates the pathogenesis of secondary injury in a positive feedback manner [78]. Thus, excessive stimulation of NMDA receptors, as occurs in the spinal cord trauma, may result in stimulation of PLA2 activity leading to alterations in membrane composition, permeability, and fluidity which could cause neuronal and glial death. Indeed, in vivo and in vitro experiments showed that exogenous administration of PLA2 induced neuronal death and tissue damage [79–81].
Possible Mechanisms of PLA2 Action after Spinal Cord Injury
To date, mechanisms underlying PLA2-mediated SCI remain unclear. Several hypotheses, however, have been proposed to interpret PLA2-mediated injury. These possible mechanisms include the effect of PLA2 on membrane damage, release of pro-inflammatory mediators, generation of free radicals, release of excitotoxic neurotransmitters, and enhancement of apoptosis (Fig. 2) [3, 49, 63, 82, 83].
Fig. 2.
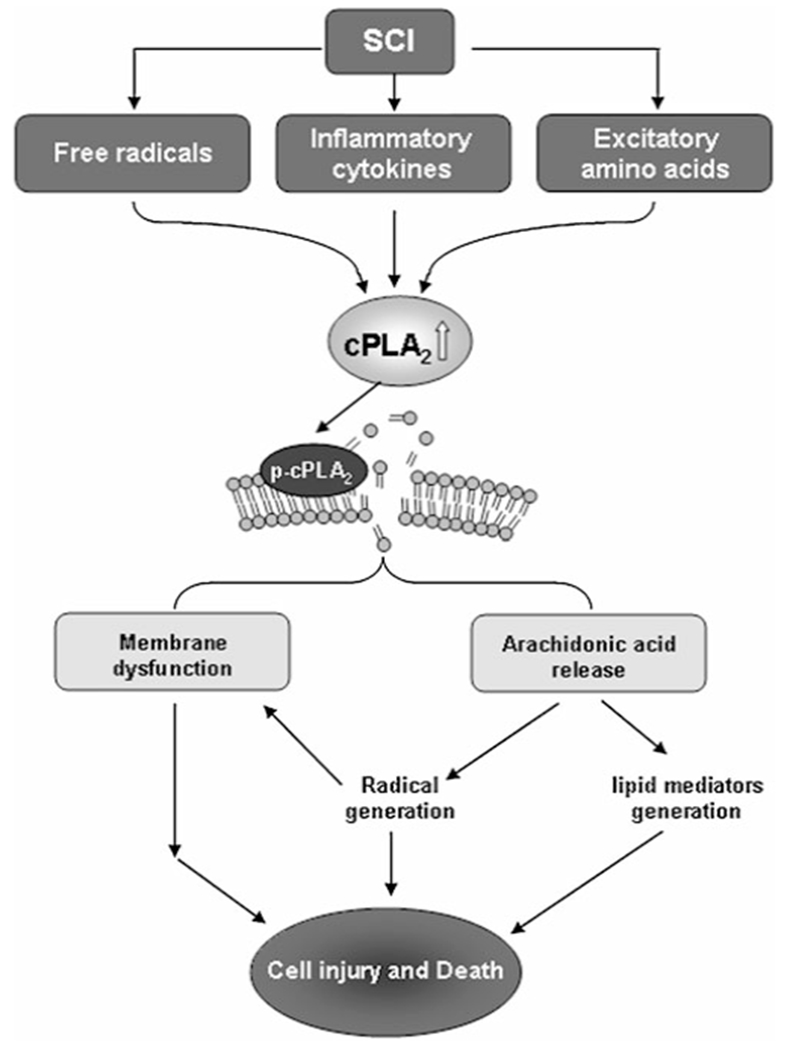
Possible mechanisms underlying PLA2-mediated secondary spinal cord injury. Acute traumatic SCI triggers a secondary injury process mediated by multiple injury inducers including inflammatory cytokines, free radicals, and excitatory amino acids; all these inducers may activate PLA2. Over-activation of PLA2 may enhance membrane phospholipid hydrolysis, arachidonic acid release, oxygen free radical generation, eicosanoids production, and ultimately lead to neuronal death
Injury to the neural membrane can be a result of PLA2’s direct action. Phospholipids are the main components of a neural cell bi-layer membrane. They not only constitute the backbone of neural membrane, but also provide the membrane with suitable environment, fluidity, and ion permeability, which are required for the proper function of integral membrane proteins, receptors, and ion channels. PLA2 activation induces phospholipid degradation and membrane breakdown directly through hydrolysis of neural membrane phospholipids, resulting in alteration of membrane function such as fluidity and permeability, behavior of transporters and receptors, ion homeostasis, and eventually leading to functional failure of excitable membranes [3, 49, 84]. Once the neural membranes are destroyed, the functional loss may become irreversible.
Induction of abnormally high levels of PLA2 metabolites could be another indirect mechanism of PLA2-mediated injury. PLA2 metabolites mediate inflammation, oxidation, and neurotoxicity following SCI. For example, metabolism of free fatty acids represents a source of ROS. A number of studies showed that free fatty acids activated NADPH oxidase, a key enzyme of ROS production [46, 55–57]. Application of pathophysiological concentrations of free fatty acids has been demonstrated to induce oxidative injury to spinal cord cell cultures [58]. In addition, several well-known bioactive mediators of inflammation such as eicosanoids (prostaglandins, thromboxanes, leukotrienes, and lipoxins) and PAF induced by PLA2 have been implicated as mediators of secondary injury [62, 63, 68–71].
PLA2 could also mediate damage induced by EAA. Application of PLA2 to the rat ischemic cerebral cortex resulted in a significant increase in EAA levels and a PLA2 inhibitor mepacrine significantly decreased the ischemia-evoked efflux of EAA into cortical superfusates, suggesting the involvement of PLA2 in EAA release [82]. The release of high levels of EAAs is an important mechanism of secondary injury after acute SCI [30].
Increasing evidence suggests the involvement of multiple isoforms of PLA2 in apoptosis, which has been identified as an important mechanism of cell death in many neurological disorders including SCI [85–87]
In summary, PLA2 can be activated by several key injury mediators such as inflammatory cytokines, free radicals, and excitatory amino acids that have been shown to increase following traumatic SCI. Increased PLA2 activity can in turn hydrolyze neural membrane and further increase inflammation, oxidation, and excitatory amino acid release. This indicates that PLA2 activation may play a central role in this positive feedback loop triggered by traumatic SCI. Such activation may eventually lead to neuronal and glial cell death, tissue damage, and electrophysiological and behavioral impairments. Thus, PLA2 may act as a convergence molecule that mediates multiple injury mechanisms after SCI and blocking PLA2 action may represent a novel and efficient strategy to block multiple injury pathways that occur following SCI.
Acknowledgment
This work was supported by NIH NS036350, NS052290, NS050243, NS059622, Indiana Spinal Cord and Brain Injury Research Board (no. 91910), Mari Hulman George Endowment, and the Paralysis Project of America.
Contributor Information
Nai-Kui Liu, Spinal Cord and Brain Injury Research Group, Stark Neurosciences Research Institute, Indiana University School of Medicine, 950 W. Walnut St., R-2 Building, Room 402, Indianapolis, IN 46202, USA; Department of Neurological Surgery, Indiana University School of Medicine, 950 W. Walnut St., R-2 Building, Room 402, Indianapolis, IN 46202, USA.
Xiao-Ming Xu, Spinal Cord and Brain Injury Research Group, Stark Neurosciences Research Institute, Indiana University School of Medicine, 950 W. Walnut St., R-2 Building, Room 402, Indianapolis, IN 46202, USA; Department of Neurological Surgery, Indiana University School of Medicine, 950 W. Walnut St., R2 Building, Room 402, Indianapolis, IN 46202, USA.
References
- 1.Kudo I, Murakami M (2002) Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat 68–69:3–58 [DOI] [PubMed] [Google Scholar]
- 2.Murakami M, Nakatani Y, Atsumi G, Inoue K, Kudo I (1997) Regulatory functions of phospholipase A2. Crit Rev Immunol 17:225–283 [DOI] [PubMed] [Google Scholar]
- 3.Farooqui AA, Yang HC, Rosenberger TA, Horrocks LA (1997) Phospholipase A2 and its role in brain tissue. J Neurochem 69:889–901 [DOI] [PubMed] [Google Scholar]
- 4.Bonventre JV (1996) Roles of phospholipases A2 in brain cell and tissue injury associated with ischemia and excitotoxicity. J Lipid Mediat Cell Signal 14:15–23 [DOI] [PubMed] [Google Scholar]
- 5.Farooqui AA, Ong WY, Horrocks LA (2006) Inhibitors of brain phospholipase A2 activity: their neuropharmacological effects and therapeutic importance for the treatment of neurologic disorders. Pharmacol Rev 58:591–620 [DOI] [PubMed] [Google Scholar]
- 6.Yedgar S, Cohen Y, Shoseyov D (2006) Control of phospholipase A2 activities for the treatment of inflammatory conditions. Biochim Biophys Acta 1761:1373–1382 [DOI] [PubMed] [Google Scholar]
- 7.Farooqui AA, Litsky ML, Farooqui T, Horrocks LA (1999) Inhibitors of intracellular phospholipase A2 activity: their neurochemical effects and therapeutical importance for neurological disorders. Brain Res Bull 49:139–153 [DOI] [PubMed] [Google Scholar]
- 8.Bazan NG, Rodriguez de Turco EB, Allan G (1995) Mediators of injury in neurotrauma: intracellular signal transduction and gene expression. J Neurotrauma 12:791–814 [DOI] [PubMed] [Google Scholar]
- 9.Phillis JW, O’Regan MH (2004) A potentially critical role of phospholipases in central nervous system ischemic, traumatic, and neurodegenerative disorders. Brain Res Brain Res Rev 44:13–47 [DOI] [PubMed] [Google Scholar]
- 10.Murakami M, Kudo I (2002) Phospholipase A2. J Biochem (Tokyo) 131:285–292 [DOI] [PubMed] [Google Scholar]
- 11.Schaloske RH, Dennis EA (2006) The phospholipase A2 superfamily and its group numbering system. Biochim Biophys Acta 1761:1246–1259 [DOI] [PubMed] [Google Scholar]
- 12.Mayer RJ, Marshall LA (1993) New insights on mammalian phospholipase A2(s): comparison of arachidonoyl-selective and - nonselective enzymes. Faseb J 7:339–348 [DOI] [PubMed] [Google Scholar]
- 13.Dennis EA (1994) Diversity of group types, regulation, and function of phospholipase A2. J Biol Chem 269:13057–13060 [PubMed] [Google Scholar]
- 14.Murakami M, Kudo I, Inoue K (1995) Secretory phospholipases A2. J Lipid Mediat Cell Signal 12:119–130 [DOI] [PubMed] [Google Scholar]
- 15.Murakami M, Kambe T, Shimbara S, Yamamoto S, Kuwata H, Kudo I (1999) Functional association of type IIA secretory phospholipase A(2) with the glycosylphosphatidylinositol-anchored heparan sulfate proteoglycan in the cyclooxygenase-2-mediated delayed prostanoid-biosynthetic pathway. J Biol Chem 274:29927–29936 [DOI] [PubMed] [Google Scholar]
- 16.Murakami M, Koduri RS, Enomoto A, Shimbara S, Seki M, Yoshihara K, Singer A, Valentin E, Ghomashchi F, Lambeau G, Gelb MH, Kudo I (2001) Distinct arachidonate-releasing functions of mammalian secreted phospholipase A2s in human embryonic kidney 293 and rat mastocytoma RBL-2H3 cells through heparan sulfate shuttling and external plasma membrane mechanisms. J Biol Chem 276:10083–10096 [DOI] [PubMed] [Google Scholar]
- 17.Hanasaki K, Arita H (2002) Phospholipase A2 receptor: a regulator of biological functions of secretory phospholipase A2. Prostaglandins Other Lipid Mediat 68–69:71–82 [DOI] [PubMed] [Google Scholar]
- 18.Clark JD, Schievella AR, Nalefski EA, Lin LL (1995) Cytosolic phospholipase A2. J Lipid Mediat Cell Signal 12:83–117 [DOI] [PubMed] [Google Scholar]
- 19.Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S, Cangul H, Coryell J, Canham N, Nardocci N, Zorzi G, Pasha S, Rodriguez D, Desguerre I, Mubaidin A, Bertini E, Trembath RC, Simonati A, Schanen C, Johnson CA, Levinson B, Woods CG, Wilmot B, Kramer P, Gitschier J, Maher ER, Hayflick SJ (2006) PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet 38:752–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Svensson CI, Lucas KK, Hua XY, Powell HC, Dennis EA, Yaksh TL (2005) Spinal phospholipase A2 in inflammatory hyperalgesia: role of the small, secretory phospholipase A2. Neuroscience 133:543–553 [DOI] [PubMed] [Google Scholar]
- 21.Lucas KK, Svensson CI, Hua XY, Yaksh TL, Dennis EA (2005) Spinal phospholipase A2 in inflammatory hyperalgesia: role of group IVA cPLA2. Br J Pharmacol 144:940–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Titsworth WL, Cheng X, Ke Y, Deng L, Burckardt KA, Pendleton C, Liu NK, Shao H, Cao QL, Xu XM (2009) Differential expression of sPLA(2) following spinal cord injury and a functional role for sPLA(2)-IIA in mediating oligodendrocyte death. Glia 57(14):1521–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farooqui AA, Ong WY, Horrocks LA, Farooqui T (2000) Brain cystosolic phospholipase A2: localization, role, and involvement in neurological diseases. Neuroscientist 6:169–180 [Google Scholar]
- 24.Ong WY, Horrocks LA, Farooqui AA (1999) Immunocytochemical localization of cPLA2 in rat and monkey spinal cord. J Mol Neurosci 12:123–130 [DOI] [PubMed] [Google Scholar]
- 25.Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ (2001) Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 410:471–475 [DOI] [PubMed] [Google Scholar]
- 26.Liu NK, Zhang YP, Titsworth WL, Jiang X, Han S, Lu PH, Shields CB, Xu XM (2006) A novel role of phospholipase A2 in mediating spinal cord secondary injury. Ann Neurol 59:606–619 [DOI] [PubMed] [Google Scholar]
- 27.Larsson Forsell PK, Kennedy BP, Claesson HE (1999) The human calcium-independent phospholipase A2 gene multiple enzymes with distinct properties from a single gene. Eur J Biochem 262:575–585 [DOI] [PubMed] [Google Scholar]
- 28.Svensson CI, Yaksh TL (2002) The spinal phospholipase-cyclooxygenase-prostanoid cascade in nociceptive processing. Annu Rev Pharmacol Toxicol 42:553–583 [DOI] [PubMed] [Google Scholar]
- 29.Young W (1993) Secondary injury mechanisms in acute spinal cord injury. J Emerg Med 11(Suppl 1):13–22 [PubMed] [Google Scholar]
- 30.Park E, Velumian AA, Fehlings MG (2004) The role of excitotoxicity in secondary mechanisms of spinal cord injury: a review with an emphasis on the implications for white matter degeneration. J Neurotrauma 21:754–774 [DOI] [PubMed] [Google Scholar]
- 31.Hall ED, Braughler JM (1986) Role of lipid peroxidation in post-traumatic spinal cord degeneration: a review. Cent Nerv Syst Trauma 3:281–294 [DOI] [PubMed] [Google Scholar]
- 32.Buki A, Okonkwo DO, Wang KK, Povlishock JT (2000) Cytochrome c release and caspase activation in traumatic axonal injury. J Neurosci 20:2825–2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakano S, Kogure K, Abe K, Yae T (1990) Ischemia-induced alterations in lipid metabolism of the gerbil cerebral cortex: I. Changes in free fatty acid liberation. J Neurochem 54:1911–1916 [DOI] [PubMed] [Google Scholar]
- 34.Muralikrishna Adibhatla R, Hatcher JF (2006) Phospholipase A2, reactive oxygen species, and lipid peroxidation in cerebral ischemia. Free Radic Biol Med 40:376–387 [DOI] [PubMed] [Google Scholar]
- 35.Demediuk P, Saunders RD, Anderson DK, Means ED, Horrocks LA (1985) Membrane lipid changes in laminectomized and traumatized cat spinal cord. Proc Natl Acad Sci USA 82:7071–7075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy EJ, Behrmann D, Bates CM, Horrocks LA (1994) Lipid alterations following impact spinal cord injury in the rat. Mol Chem Neuropathol 23:13–26 [DOI] [PubMed] [Google Scholar]
- 37.Demediuk P, Daly MP, Faden AI (1989) Changes in free fatty acids, phospholipids, and cholesterol following impact injury to the rat spinal cord. J Neurosci Res 23:95–106 [DOI] [PubMed] [Google Scholar]
- 38.Cherian L, Kuruvilla A, Chandy MJ, Abraham J (1996) Changes in phospholipids and acetylcholinesterase during early phase of injury to spinal cord—an experimental study in rats. Indian J Physiol Pharmacol 40:134–138 [PubMed] [Google Scholar]
- 39.Lukacova N, Halat G, Chavko M, Marsala J (1996) Ischemia-reperfusion injury in the spinal cord of rabbits strongly enhances lipid peroxidation and modifies phospholipid profiles. Neurochem Res 21:869–873 [DOI] [PubMed] [Google Scholar]
- 40.Horrocks LA, Demediuk P, Saunders RD, Dugan L, Clendenon NR, Means ED, Anderson DK (1985) The degradation of phospholipids, formation of metabolites of arachidonic acid, and demyelination following experimental spinal cord injury. Cent Nerv Syst Trauma 2:115–120 [DOI] [PubMed] [Google Scholar]
- 41.Faden AI, Chan PH, Longar S (1987) Alterations in lipid metabolism, Na+, K+-ATPase activity, and tissue water content of spinal cord following experimental traumatic injury. J Neurochem 48:1809–1816 [DOI] [PubMed] [Google Scholar]
- 42.Tonai T, Taketani Y, Ueda N, Nishisho T, Ohmoto Y, Sakata Y, Muraguchi M, Wada K, Yamamoto S (1999) Possible involvement of interleukin-1 in cyclooxygenase-2 induction after spinal cord injury in rats. J Neurochem 72:302–309 [DOI] [PubMed] [Google Scholar]
- 43.Resnick DK, Nguyen P, Cechvala CF (2001) Regional and temporal changes in prostaglandin E2 and thromboxane B2 concentrations after spinal cord injury. Spine J 1:432–436 [DOI] [PubMed] [Google Scholar]
- 44.Beck S, Lambeau G, Scholz-Pedretti K, Gelb MH, Janssen MJ, Edwards SH, Wilton DC, Pfeilschifter J, Kaszkin M (2003) Potentiation of tumor necrosis factor alpha-induced secreted phospholipase A2 (sPLA2)-IIA expression in mesangial cells by an autocrine loop involving sPLA2 and peroxisome proliferator-activated receptor alpha activation. J Biol Chem 278:29799–29812 [DOI] [PubMed] [Google Scholar]
- 45.van Rossum GS, Drummen GP, Verkleij AJ, Post JA, Boonstra J (2004) Activation of cytosolic phospholipase A2 in Her14 fibroblasts by hydrogen peroxide: a p42/44(MAPK)-dependent and phosphorylation-independent mechanism. Biochim Biophys Acta 1636:183–195 [DOI] [PubMed] [Google Scholar]
- 46.Sun GY, Horrocks LA, Farooqui AA (2007) The roles of NADPH oxidase and phospholipases A2 in oxidative and inflammatory responses in neurodegenerative diseases. J Neurochem 103:1–16 [DOI] [PubMed] [Google Scholar]
- 47.Kim DK, Rordorf G, Nemenoff RA, Koroshetz WJ, Bonventre JV (1995) Glutamate stably enhances the activity of two cytosolic forms of phospholipase A2 in brain cortical cultures. Biochem J 310(Pt 1):83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sandhya TL, Ong WY, Horrocks LA, Farooqui AA (1998) A light and electron microscopic study of cytoplasmic phospholipase A2 and cyclooxygenase-2 in the hippocampus after kainate lesions. Brain Res 788:223–231 [DOI] [PubMed] [Google Scholar]
- 49.Klein J (2000) Membrane breakdown in acute and chronic neurodegeneration: focus on choline-containing phospholipids. J Neural Transm 107:1027–1063 [DOI] [PubMed] [Google Scholar]
- 50.De S, Trigueros MA, Kalyvas A, David S (2003) Phospholipase A2 plays an important role in myelin breakdown and phagocytosis during Wallerian degeneration. Mol Cell Neurosci 24:753–765 [DOI] [PubMed] [Google Scholar]
- 51.Liu NK, Zhang YP, Han S, Pei J, Xu LY, Lu PH, Shields CB, Xu XM (2007) Annexin A1 reduces inflammatory reaction and tissue damage through inhibition of phospholipase A2 activation in adult rats following spinal cord injury. J Neuropathol Exp Neurol 66:932–943 [DOI] [PubMed] [Google Scholar]
- 52.Huang W, Bhavsar A, Ward RE, Hall JC, Priestley JV, Michael-Titus AT (2009) Arachidonyl trifluoromethyl ketone is neuroprotective after spinal cord injury. J Neurotrauma 26:1429–1434 [DOI] [PubMed] [Google Scholar]
- 53.Hamada Y, Ikata T, Katoh S, Tsuchiya K, Niwa M, Tsutsumishita Y, Fukuzawa K (1996) Roles of nitric oxide in compression injury of rat spinal cord. Free Radic Biol Med 20:1–9 [DOI] [PubMed] [Google Scholar]
- 54.Azbill RD, Mu X, Bruce-Keller AJ, Mattson MP, Springer JE (1997) Impaired mitochondrial function, oxidative stress and altered antioxidant enzyme activities following traumatic spinal cord injury. Brain Res 765:283–290 [DOI] [PubMed] [Google Scholar]
- 55.Daniels I, Lindsay MA, Keany CI, Burden RP, Fletcher J, Haynes AP (1998) Role of arachidonic acid and its metabolites in the priming of NADPH oxidase in human polymorphonuclear leukocytes by peritoneal dialysis effluent. Clin Diagn Lab Immunol 5:683–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hartfield PJ, Robinson JM (1998) Arachidonic acid activates NADPH oxidase by a direct, calmodulin-regulated mechanism. Prostaglandins Other Lipid Mediat 56:1–6 [DOI] [PubMed] [Google Scholar]
- 57.Kerkhoff C, Nacken W, Benedyk M, Dagher MC, Sopalla C, Doussiere J (2005) The arachidonic acid-binding protein S100A8/A9 promotes NADPH oxidase activation by interaction with p67phox and Rac-2. FASEB J 19:467–469 [DOI] [PubMed] [Google Scholar]
- 58.Toborek M, Malecki A, Garrido R, Mattson MP, Hennig B, Young B (1999) Arachidonic acid-induced oxidative injury to cultured spinal cord neurons. J Neurochem 73:684–692 [DOI] [PubMed] [Google Scholar]
- 59.Okuda S, Saito H, Katsuki H (1994) Arachidonic acid: toxic and trophic effects on cultured hippocampal neurons. Neuroscience 63:691–699 [DOI] [PubMed] [Google Scholar]
- 60.Li Y, Maher P, Schubert D (1997) A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron 19:453–463 [DOI] [PubMed] [Google Scholar]
- 61.Wang H, Li J, Follett PL, Zhang Y, Cotanche DA, Jensen FE, Volpe JJ, Rosenberg PA (2004) 12-Lipoxygenase plays a key role in cell death caused by glutathione depletion and arachidonic acid in rat oligodendrocytes. Eur J Neurosci 20:2049–2058 [DOI] [PubMed] [Google Scholar]
- 62.Anderson DK, Demediuk P, Saunders RD, Dugan LL, Means ED, Horrocks LA (1985) Spinal cord injury and protection. Ann Emerg Med 14:816–821 [DOI] [PubMed] [Google Scholar]
- 63.Sapirstein A, Bonventre JV (2000) Phospholipases A2 in ischemic and toxic brain injury. Neurochem Res 25:745–753 [DOI] [PubMed] [Google Scholar]
- 64.Serhan CN (2005) Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot Essent Fatty Acids 73:141–162 [DOI] [PubMed] [Google Scholar]
- 65.Ousman SS, David S (2000) Lysophosphatidylcholine induces rapid recruitment and activation of macrophages in the adult mouse spinal cord. Glia 30:92–104 [PubMed] [Google Scholar]
- 66.Ousman SS, David S (2001) MIP-1alpha, MCP-1, GM-CSF, and TNF-alpha control the immune cell response that mediates rapid phagocytosis of myelin from the adult mouse spinal cord. J Neurosci 21:4649–4656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lindsberg PJ, Yue TL, Frerichs KU, Hallenbeck JM, Feuerstein G (1990) Evidence for platelet-activating factor as a novel mediator in experimental stroke in rabbits. Stroke 21:1452–1457 [DOI] [PubMed] [Google Scholar]
- 68.Faden AI, Halt P (1992) Platelet-activating factor reduces spinal cord blood flow and causes behavioral deficits after intrathecal administration in rats through a specific receptor mechanism. J Pharmacol Exp Ther 261:1064–1070 [PubMed] [Google Scholar]
- 69.Hostettler ME, Knapp PE, Carlson SL (2002) Platelet-activating factor induces cell death in cultured astrocytes and oligodendrocytes: involvement of caspase-3. Glia 38:228–239 [DOI] [PubMed] [Google Scholar]
- 70.Hostettler ME, Carlson SL (2002) PAF antagonist treatment reduces pro-inflammatory cytokine mRNA after spinal cord injury. Neuroreport 13:21–24 [DOI] [PubMed] [Google Scholar]
- 71.Xiao J, Zhao D, Hou T, Wu K, Zeng H (1998) Synergetic protective effects of combined blockade by two kinds of autolesion mediator receptor on neurological function after cervical cord injury. Chin Med J (Engl) 111:443–446 [PubMed] [Google Scholar]
- 72.Komecki E, Ehrlich YH (1988) Neuroregulatory and neuropathological actions of the ether-phospholipid platelet-activating factor. Science 240:1792–1794 [DOI] [PubMed] [Google Scholar]
- 73.Xu Y, Tao YX (2004) Involvement of the NMDA receptor/nitric oxide signal pathway in platelet-activating factor-induced neurotoxicity. Neuroreport 15:263–266 [DOI] [PubMed] [Google Scholar]
- 74.Bate C, Rumbold L, Williams A (2007) Cholesterol synthesis inhibitors protect against platelet-activating factor-induced neuronal damage. J Neuroinflammation 4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dumuis A, Sebben M, Haynes L, Pin JP, Bockaert J (1988) NMDA receptors activate the arachidonic acid cascade system in striatal neurons. Nature 336:68–70 [DOI] [PubMed] [Google Scholar]
- 76.Farooqui AA, Yi Ong W, Lu XR, Halliwell B, Horrocks LA (2001) Neurochemical consequences of kainate-induced toxicity in brain: involvement of arachidonic acid release and prevention of toxicity by phospholipase A(2) inhibitors. Brain Res Brain Res Rev 38:61–78 [DOI] [PubMed] [Google Scholar]
- 77.Shelat PB, Chalimoniuk M, Wang JH, Strosznajder JB, Lee JC, Sun AY, Simonyi A, Sun GY (2008) Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J Neurochem 106:45–55 [DOI] [PubMed] [Google Scholar]
- 78.Sundstrom E, Mo LL (2002) Mechanisms of glutamate release in the rat spinal cord slices during metabolic inhibition. J Neurotrauma 19:257–266 [DOI] [PubMed] [Google Scholar]
- 79.Clapp LE, Klette KL, DeCoster MA, Bernton E, Petras JM, Dave JR, Laskosky MS, Smallridge RC, Tortella FC (1995) Phospholipase A2-induced neurotoxicity in vitro and in vivo in rats. Brain Res 693:101–111 [DOI] [PubMed] [Google Scholar]
- 80.Kolko M, Bruhn T, Christensen T, Lazdunski M, Lambeau G, Bazan NG, Diemer NH (1999) Secretory phospholipase A2 potentiates glutamate-induced rat striatal neuronal cell death in vivo. Neurosci Lett 274:167–170 [DOI] [PubMed] [Google Scholar]
- 81.Kolko M, DeCoster MA, de Turco EB, Bazan NG (1996) Synergy by secretory phospholipase A2 and glutamate on inducing cell death and sustained arachidonic acid metabolic changes in primary cortical neuronal cultures. J Biol Chem 271:32722–32728 [DOI] [PubMed] [Google Scholar]
- 82.O’Regan MH, Smith-Barbour M, Perkins LM, Phillis JW (1995) A possible role for phospholipases in the release of neurotransmitter amino acids from ischemic rat cerebral cortex. Neurosci Lett 185:191–194 [DOI] [PubMed] [Google Scholar]
- 83.Liu NK, Titsworth WL, Xu XM (2009) Phospholipase A2 in CNS disorders: implication on traumatic spinal cord and brain injuries. In: Banik N, Ray S (Eds.), Handbook of neurochemistry and molecular neurobiology, Springer, New York, pp. 321–341 [Google Scholar]
- 84.Farooqui AA, Ong WY, Horrocks LA (2004) Biochemical aspects of neurodegeneration in human brain: involvement of neural membrane phospholipids and phospholipases A2. Neurochem Res 29:1961–1977 [DOI] [PubMed] [Google Scholar]
- 85.Beattie MS, Farooqui AA, Bresnahan JC (2000) Review of current evidence for apoptosis after spinal cord injury. J Neurotrauma 17:915–925 [DOI] [PubMed] [Google Scholar]
- 86.Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW, Dong HX, Wu YJ, Fan GS, Jacquin MF, Hsu CY, Choi DW (1997) Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci 17:5395–5406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS (1997) Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med 3:73–76 [DOI] [PubMed] [Google Scholar]