

Abstract
Rationale
The population of patients with pulmonary arterial hypertension (PAH) has evolved over time from predominantly young White women to an older, more racially diverse and obese population. Whether these changes are reflected in clinical trials is not known.
Objectives
To determine secular and regional trends among PAH trial participants.
Methods
We performed a pooled cohort analysis using harmonized data from phase III clinical trials of PAH therapies submitted to the U.S. Food and Drug Administration. We used mixed-effects linear and logistic regression to assess regional differences in participant age, sex, body habitus, and hemodynamics over time.
Results
A total of 6,599 participants were enrolled in 18 trials between 1998 and 2013; 78% were female. The mean age of participants in North America, Europe, and Latin America at the time of study start increased by 2.09 (95% confidence interval [CI], 0.67–3.51), 1.62 (95% CI, 0.24–3.00), and 4.75 (95% CI, 2.29–7.21) years per 5 years, respectively (P = 0.01). Body mass index at the time of study start increased by 0.72 kg/m2 per 5 years (95% CI, 0.44–0.99; P < 0.001) across all regions. Eighty-five percent of participants in early studies were non-Hispanic White, but this decreased over time to 70%. Ninety-seven percent of Asians and 74% of Hispanics in the sample were recruited from Asia and Latin America.
Conclusions
Patients enrolled in more recent PAH therapy trials are older and more obese, mirroring the changing epidemiology of observational cohorts. However, these trends varied by geographic region. PAH cohorts remain predominantly female, presenting challenges for generalizability to male patients. Although the proportion of non-White participants increased over time, this was primarily through recruitment in Asia and Latin America.
Keywords: pulmonary arterial hypertension, secular trends, pooled cohort analysis, clinical trial participants
Pulmonary arterial hypertension (PAH) is characterized by increased pulmonary vascular resistance (PVR) caused by progressive remodeling of small pulmonary arteries, leading to dyspnea, exercise limitation, and ultimately right heart failure and death. More than 25 years have passed since the completion of the first randomized clinical trial (RCT) of continuous intravenous epoprostenol therapy in PAH (1). Subsequent clinical trials have resulted in the approval of 14 therapies, some of which have improved exercise capacity and functional status and lengthened time to clinical worsening in PAH.
During this time, the PAH population has continued to evolve. In the 1980s, the National Institutes of Health Patient Registry for the Characterization of Primary Pulmonary Hypertension was the first large PAH registry and comprised predominantly young White women with idiopathic PAH (IPAH) (2). In contrast, patients with PAH enrolled in contemporary registries are now older, more racially diverse, and more obese than in the past (3, 4). Although RCTs have specific inclusion and exclusion criteria and require informed consent for randomization, demographic features such as age, sex, body habitus, and comorbidities should ideally mirror the larger population of patients with PAH to ensure generalizability of the results. Whether the demographics and anthropometrics of individuals enrolled in PAH RCTs have evolved to reflect those in population-based epidemiologic studies is not known.
Therefore, we performed a pooled cohort analysis of 18 phase III PAH RCTs that enrolled patients from 1998 to 2013. We sought to assess whether study populations have changed over time in terms of age, sex, body mass index (BMI), and disease characteristics. We also compared the characteristics of participants recruited in Asia and Latin America with those of participants from North American and European countries, including secular trends within these geographic regions. We hypothesized that age, BMI, and proportions of Black, Hispanic, and Asian participants have increased over time in PAH clinical trials and that these temporal trends have varied by geographic region.
Methods
Study Selection and Data Collection
Individual patient data from 21 RCTs of pulmonary hypertension therapies conducted between 1998 and 2013 submitted to the U.S. Food and Drug Administration (FDA) were provided to us by the FDA. These included RCTs for all current PAH therapies (5) as well as RCTs for sitaxentan, which was not approved by the FDA. RCTs that were not submitted to the FDA were unavailable for analysis (6). All trials were performed under the supervision of human ethics committees, and all patients provided informed consent. Harmonization and analysis of these data were exempted from review by the University of Pennsylvania institutional review board.
Individual demographic and clinical data, including subject start date, anthropometrics, pulmonary hypertension etiology, 6-minute walk distances, and invasive hemodynamics, were harmonized across all studies using the Study Data Tabulation Model version 1.4, as described in the Study Data Tabulation Model Implementation Guide: Human Clinical Trials, Version 3.2 (see the online supplement). Although the trials occurred over a period of 15 years (1998–2013), pulmonary hypertension etiology was characterized on the basis of the 2019 World Symposium on Pulmonary Hypertension classification (7). Of the 21 RCTs, we included phase III clinical trials and limited our analysis to individuals with PAH (group 1): IPAH, heritable PAH, drug- and toxin-induced PAH, PAH associated with connective tissue disease (CTD), human immunodeficiency virus infection, congenital heart disease (CHD), and portal hypertension. Eighteen trials were included in the final pooled cohort (see Figure E1 and Table E1 in the online supplement), and individual prerandomization invasive hemodynamic data were available for 12 trials (see Table E2).
For studies adhering to Study Data Tabulation Model guidelines, the subject reference start date (corresponding to the date on which each subject was first administered study treatment) was most consistently recorded among all studies and used as the start date. If this was not available, the date of subject randomization was used instead. Race was mapped to one of five standard race designations (Asian, Black, White, Native Hawaiian/Pacific Islander, or American Indian/Alaska Native) or categorized as unknown if race data were not collected. Hispanic/Latino ethnicity was mapped to a separate field. These were then collapsed into a combined race/ethnicity designation (non-Hispanic White, non-Hispanic Black, Asian, Hispanic, other and/or multiracial, or missing). The geographic region from which each patient was recruited was grouped into 1) Europe, Australia, and Israel; 2) United States and Canada; 3) Latin America; or 4) Asia on the basis of reported country of origin, study site, or available published data, as detailed in the online supplement. Thirty-four individuals recruited from South Africa could not be grouped into any of the four regions and were excluded from the analysis.
Baseline characteristics by treatment group assignment in each study reproduced the published papers, confirming the accuracy of the harmonization process. Dependent variables of interest included age, sex, BMI (kg/m2), obesity (BMI > 30 kg/m2) and most recent invasive hemodynamics before randomization (right atrial pressure [RAP; millimeters of mercury], mean pulmonary arterial pressure [mPAP; millimeters of mercury], pulmonary artery wedge pressure [PAWP; millimeters of mercury], cardiac index [L/min/m2], and PVR [dynes ⋅ s/cm5]).
Statistical Analysis
Continuous variables are expressed as mean (standard deviation) for approximately normally distributed variables and as medians with interquartile ranges for skewed variables. Categorical variables are expressed as frequency (percentage). For regression models, the independent variable was calculated as the time elapsed between the start date of each individual and December 11, 1998 (the date of the earliest study participant).
To evaluate trends in continuous variables over time, we performed mixed-effects linear regression to assess the associations of continuous variables with time (8). This method incorporates random effects for each study to account for clustering of participants within studies. A restricted maximum likelihood approach was used, as this has been demonstrated to introduce the least amount of bias (9). For each dependent variable of interest, we tested whether the mean value differed by region (Pregion) and whether the mean changed over time (Pslope). In addition, we assessed for significant effect modification by geographic region over time using the Wald test to compare models with and without the time × region interaction term (Pinteract). Because individuals from Latin America and Asia were recruited at a later time point compared with those in Western countries, we calculated least squares means for each region at a fixed time (January 2007) to allow comparisons among all regions. For binary dependent variables (obesity and male sex), we used mixed-effects logistic regression with random effects for each study. Similarly, we assessed for a difference in log odds by region, change in log odds over time, and time × region interaction.
Models with hemodynamics as dependent variables were adjusted for age, sex, BMI, pulmonary hypertension etiology, and whether other concurrent vasodilator therapy was permitted on a per-study basis. To assess for whether changes in age or BMI were due to regional differences in pulmonary hypertension etiology, we performed an additional sensitivity analysis with adjustment for pulmonary hypertension etiology as a covariate. P values less than 0.05 were considered to indicate statistical significance. All analyses were conducted using Stata/MP 17.0 (StataCorp).
Results
Among 7,604 individuals in 21 RCTs, we included 6,599 patients with PAH from 18 phase III RCTs (see Figure E1 and Table E1) recruited between 1998 and 2013 (see Figure E2). The pooled cohort had a mean age of 49.2 years (standard deviation = 15.4 years) at the time of enrollment and was 78% female; 66.5% were non-Hispanic White, 3.9% non-Hispanic Black, 14.4% Asian, and 10.5% Hispanic. Additional characteristics are summarized in Table 1. Trials were based predominantly in Western countries before 2005 (Figure 1A). Nearly all (approximately 85%) participants in early studies were non-Hispanic White, but this proportion decreased over time to 70% with the inclusion of study sites outside Western countries (Figure 1B). The proportion of male patients was lowest among early trials but subsequently has remained stable at 22% (Figure 1C). RCTs included predominantly patients with IPAH, CTD-associated PAH (CTD-PAH), and PAH secondary to CHD (Figure 1D).
Table 1.
Baseline characteristics by region
| Europe, Australia, and Israel (n = 2,954) | United States and Canada (n = 2,366) | Latin America (n = 463) | Asia (n = 816) | Overall (N = 6,599) | |
|---|---|---|---|---|---|
| Age, yr | 51.8 (15.3) | 51.5 (14.2) | 40.9 (13.9) | 38.2 (13.4) | 49.2 (15.4) |
| Female sex | 2,209 (74.8) | 1,951 (82.5) | 395 (85.3) | 619 (75.9) | 5,174 (78.4) |
| Race/ethnicity | |||||
| Non-Hispanic White | 2,622 (88.8) | 1,642 (69.4) | 97 (21.0) | 27 (3.3) | 4,388 (66.5) |
| Non-Hispanic Black | 22 (0.7) | 231 (9.8) | 3 (0.6) | 0 (0.0) | 256 (3.9) |
| Asian | 92 (3.1) | 67 (2.8) | 3 (0.6) | 789 (96.7) | 951 (14.4) |
| Hispanic | 110 (3.7) | 242 (10.2) | 341 (73.7) | 0 (0.0) | 693 (10.5) |
| Other | 10 (0.3) | 12 (0.5) | 19 (4.1) | 0 (0.0) | 41 (0.6) |
| Unknown | 98 (3.3) | 172 (7.3) | 0 (0.0) | 0 (0.0) | 270 (4.1) |
| BMI, kg/m2 (n = 5,831) | 25.9 (22.7–30.1) | 28.1 (24.1–33.3) | 25.3 (22.3–28.4) | 21.8 (19.5–24.2) | 25.8 (22.3–30.3) |
| Obese (BMI ⩾ 30 kg/m2) | 700 (25.9) | 727 (39.4) | 68 (16.3) | 30 (3.8) | 1,525 (26.5) |
| PAH etiology | |||||
| Idiopathic | 1,863 (63.1) | 1,307 (55.3) | 300 (64.8) | 486 (59.6) | 3,956 (59.9) |
| Heritable/familial | 40 (1.4) | 15 (0.6) | 5 (1.1) | 9 (1.1) | 69 (1.0) |
| Drug and toxin induced | 47 (1.6) | 68 (2.9) | 4 (0.9) | 1 (0.1) | 120 (1.8) |
| Connective tissue disease | 759 (25.7) | 757 (32.0) | 94 (20.3) | 233 (28.6) | 1,843 (27.9) |
| Congenital heart disease | 199 (6.7) | 179 (7.6) | 54 (11.7) | 84 (10.3) | 516 (7.8) |
| HIV associated | 23 (0.8) | 33 (1.4) | 6 (1.3) | 1 (0.1) | 63 (1.0) |
| Portopulmonary | 9 (0.3) | 2 (0.1) | 0 (0.0) | 2 (0.2) | 13 (0.2) |
| Other | 11 (0.3) | 2 (0.1) | 0 (0.0) | 0 (0.0) | 13 (0.2) |
| 6MWD, m (n = 6,592) | 364 (295–415) | 357 (288–401) | 353 (285–410) | 375 (319–411) | 361 (295–410) |
| WHO FC | |||||
| I | 17 (0.6) | 11 (0.5) | 6 (1.3) | 15 (1.8) | 49 (0.7) |
| II | 856 (29.0) | 619 (26.2) | 291 (62.9) | 449 (55.0) | 2,215 (33.6) |
| III | 1,959 (66.4) | 1,665 (70.5) | 150 (32.4) | 348 (42.6) | 4,122 (62.5) |
| IV | 120 (4.1) | 67 (2.8) | 16 (3.5) | 4 (0.5) | 207 (3.1) |
| Mean right atrial pressure, mm Hg (n = 4,348) | 7.0 (5.0–11.0) | 8.0 (5.0–12.0) | 10.0 (6.0–13.0) | 7.0 (5.0–11.0) | 8.0 (5.0–11.0) |
| Mean pulmonary arterial pressure, mm Hg (n = 4,679) | 50.0 (40.0–60.0) | 50.0 (40.0–60.0) | 58.0 (47.0–66.0) | 54.0 (45.0–65.0) | 51.0 (41.7–61.0) |
| Pulmonary capillary wedge pressure, mm Hg (n = 4,496) | 9.0 (6.0–12.0) | 9.0 (7.0–12.0) | 10.0 (8.0–12.0) | 9.0 (7.0–12.0) | 9.0 (7.0–12.0) |
| Cardiac output, L/min (n = 4,565) | 4.1 (3.4–5.0) | 4.2 (3.4–5.2) | 4.2 (3.2–5.1) | 3.7 (2.8–4.6) | 4.1 (3.3–5.0) |
| Cardiac index, L/min/m2 (n = 4,572) | 2.3 (1.9–2.8) | 2.3 (1.9–2.8) | 2.5 (2.0–3.1) | 2.3 (1.8–2.9) | 2.3 (1.9–2.8) |
| Pulmonary vascular resistance, dyne ⋅ s/cm5 (n = 4,545) | 778 (522–1,120) | 770 (518–1,088) | 904 (596–1,320) | 1,022 (686–1,488) | 818 (544–1,164) |
Definition of abbreviations: 6MWD = 6-minute walk distance; BMI = body mass index; FC = functional class; HIV = human immunodeficiency virus; PAH = pulmonary arterial hypertension; WHO = World Health Organization.
Data are shown as n (%) or median (interquartile range).
Figure 1.
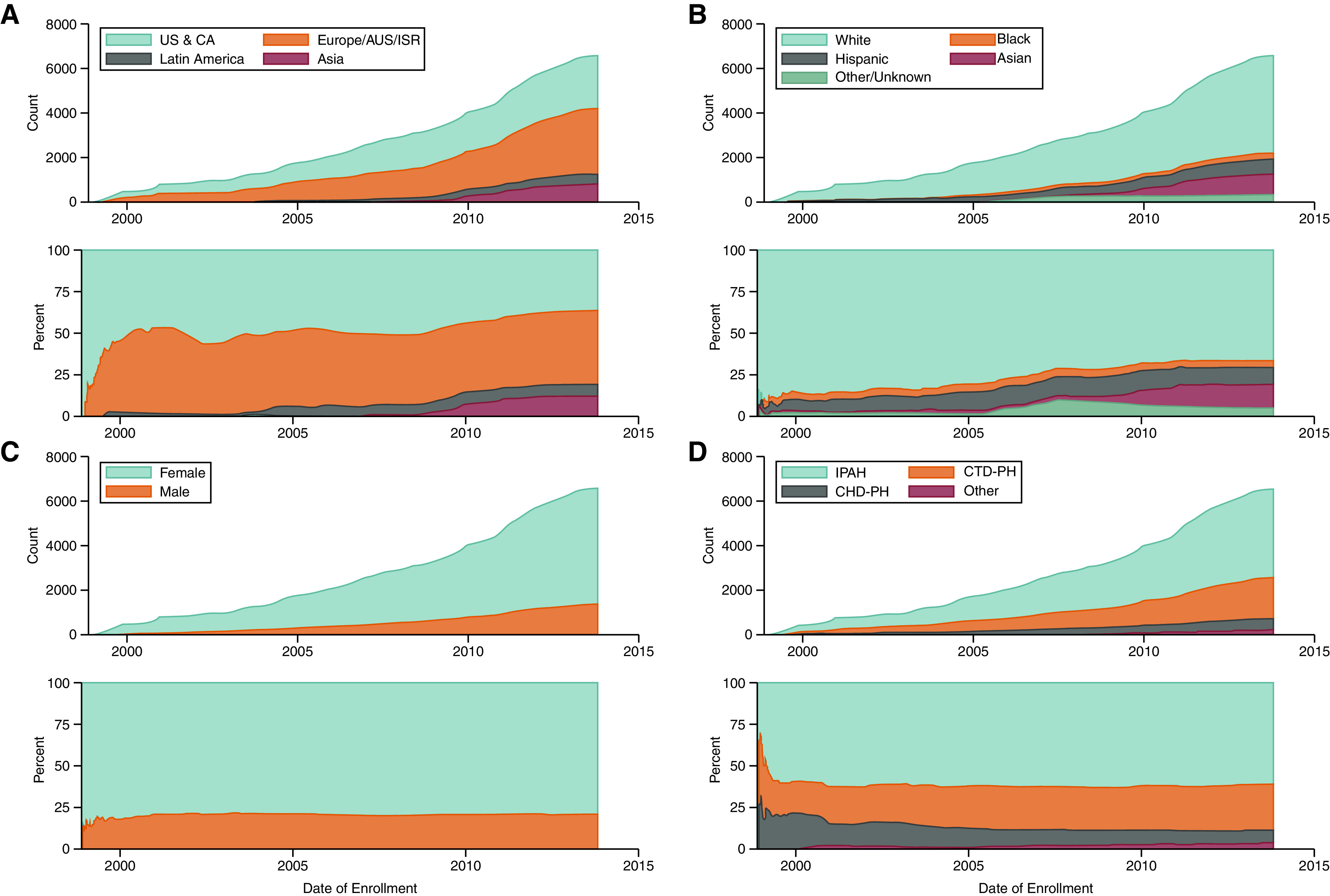
Participant (A) geographic region, (B) race, (C) sex, and (D) disease etiology over time. Data are displayed as cumulative counts (top) and overall percentage of participants (bottom) over time. AUS = Australia; CA = Canada; CHD-PH = pulmonary arterial hypertension associated with congenital heart disease; CTD-PH = pulmonary arterial hypertension associated with connective tissue disease; IPAH = idiopathic pulmonary arterial hypertension; ISR = Israel; Other = includes all other group I etiologies; US = United States.
There were significant differences in age among study participants by geographic region. The mean age of participants from Western countries was a decade greater compared with those from Latin America or Asia (Table 2). Moreover, the age of participants at recruitment increased from early studies to later studies across all regions except for Asia (Figure 2). Earlier studies from Europe and Asia had relatively higher proportions of male participants, whereas those from the United States, Canada, and Latin America had predominantly female populations (Figure 3). BMI was highest for participants from the United States and Canada and lowest for those from Asia and increased over time by 0.72 kg/m2 (95% confidence interval, 0.44–0.99; P < 0.001) in all regions (Table 2 and Figure 4A). These differences did not change after adjustment for pulmonary hypertension etiology (see Table E3). Similarly, participants from the United States and Canada had the greatest probability of being obese, followed by those from Europe and Latin America (Figure 4B).
Table 2.
Mixed-effects linear regression models
| Least Squares Mean (95% CI) | P region | Change per 5 yr (95% CI) | Pslope and Pinteract | |
|---|---|---|---|---|
| Age, yr | ||||
| US, CA | 52.0 (50.7 to 53.3) | <0.001 | 2.09 (0.67 to 3.51) | Pinteract = 0.01 |
| Europe, AUS, ISR | 51.5 (50.3 to 52.8) | 1.62 (0.24 to 3.00) | ||
| Latin America | 40.5 (38.6 to 42.4) | 4.75 (2.29 to 7.21) | ||
| Asia | 39.5 (37.0 to 42.1) | −0.81 (−4.16 to 2.54) | ||
| Body mass index, kg/m2 | ||||
| US, CA | 29.4 (29.1 to 29.8) | <0.001 | 0.72 (0.44 to 0.99) |
Pslope < 0.001 Pinteract = 0.9 |
| Europe, AUS, ISR | 26.7 (26.4 to 26.9) | |||
| Latin America | 25.6 (24.9 to 26.2) | |||
| Asia | 21.7 (21.2 to 22.2) | |||
| Right atrial pressure, mm Hg (n = 4,336) | ||||
| US, CA | 8.4 (7.9 to 8.9) | 0.03 | −0.9 (−1.4 to −0.4) | Pinteract = 0.02 |
| Europe, AUS, ISR | 8.3 (7.8 to 8.7) | −0.9 (−1.3 to −0.4) | ||
| Latin America | 9.7 (8.9 to 10.5) | 0.2 (−1.0 to 1.0) | ||
| Asia | 10.1 (9.2 to 11.1) | −2.3 (−3.6 to −1.0) | ||
| Mean pulmonary arterial pressure, mm Hg (n = 4,666) | ||||
| US, CA | 51.3 (49.8 to 52.8) | <0.001 | −2.7 (−4.0 to −1.4) |
Pslope < 0.001 Pinteract = 0.06 |
| Europe, AUS, ISR | 52.0 (50.6 to 53.3) | |||
| Latin America | 56.0 (54.0 to 57.9) | |||
| Asia | 54.0 (52.2 to 55.7) | |||
| Pulmonary artery wedge pressure, mm Hg (n = 4,483) | ||||
| US, CA | 9.1 (8.7 to 9.5) | <0.001 | −0.1 (−0.4 to 0.2) |
Pslope = 0.6 Pinteract = 0.2 |
| Europe, AUS, ISR | 9.0 (8.6 to 9.3) | |||
| Latin America | 10.1 (9.6 to 10.6) | |||
| Asia | 9.4 (9.0 to 9.9) | |||
| Cardiac index, L/min/m2 (n = 4,561) | ||||
| US, CA | 2.43 (2.36 to 2.49) | <0.001 | 0.09 (0.03 to 0.15) | Pinteract = 0.002 |
| Europe, AUS, ISR | 2.43 (2.38 to 2.49) | 0.06 (0.01 to 0.12) | ||
| Latin America | 2.73 (2.62 to 2.83) | −0.14 (−0.27 to 0.00) | ||
| Asia | 2.22 (2.08 to 2.35) | 0.23 (0.05 to 0.40) | ||
| Pulmonary vascular resistance, dynes ⋅ s/cm5 (n = 4,532) | ||||
| US, CA | 895 (845 to 944) | <0.001 | −86 (−128 to −44) |
Pslope < 0.001 Pinteract = 0.4 |
| Europe, AUS, ISR | 895 (850 to 939) | |||
| Latin America | 961 (894 to 1,029) | |||
| Asia | 1,102 (1,042 to 1,162) |
Definition of abbreviations: AUS = Australia; CA = Canada; CI = confidence interval; ISR = Israel; Pinteract = P value for region × time interaction; Pregion = P value for differences in mean hemodynamic parameter by region; Pslope = P value for change in mean hemodynamic parameter over time; US = United States.
Calculated using mixed-effects linear regression to assess the associations of continuous variables with time with clustering by trial. Least squares means were calculated at a fixed time (January 2007) to allow comparisons by region at a time point at which all regions were represented. Models of hemodynamic parameters were further adjusted for age, sex, body mass index, pulmonary arterial hypertension etiology, and whether background therapy was allowed.
Figure 2.

Participant age at study enrollment over time. Data were calculated using mixed-effects linear regression. AUS = Australia; CA = Canada; ISR = Israel; Pinteract = P value for region × time interaction; Pregion = P value for differences by region; US = United States.
Figure 3.

Probability of male sex among participants over time. Data were calculated using mixed-effects logistic regression. AUS = Australia; CA = Canada; ISR = Israel; Pinteract = P value for region × time interaction; Pregion = P value for differences by region; US = United States.
Figure 4.

Participant (A) body mass index and (B) probability of obesity over time. Data were calculated using mixed-effects (A) linear and (B) logistic regression. AUS = Australia; CA = Canada; ISR = Israel; Pregion = P value for differences by region; Pslope = P value for change over time; US = United States.
Among patients with prerandomization invasive hemodynamic measurements (n = 4,675), there were significant differences by geographic region after adjusting for age, sex, BMI, PAH etiology, and whether concurrent vasodilator therapy was allowed (Table 2). In general, participants from Latin America had the highest RAP, mPAP, and PAWP (Figure 5). RAP, mPAP, and PVR decreased over time for all regions (Figures 5A, 5B, and 5E), whereas PAWP remained unchanged (Figure 5C). Cardiac index decreased over time among patients from Latin America and increased over time for all other regions (Figure 5D).
Figure 5.
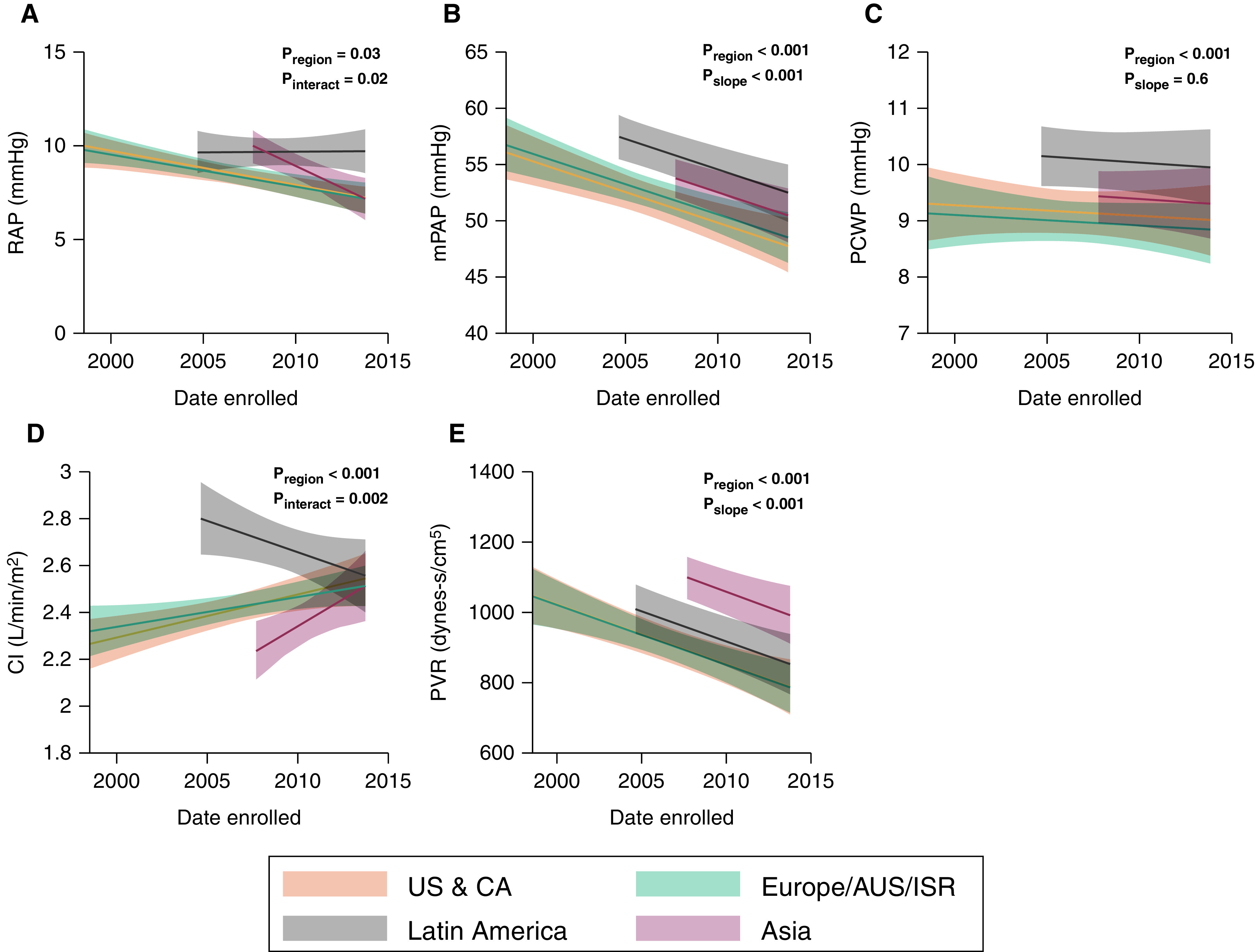
Entry hemodynamic parameters over time. (A) RAP, (B) mPAP, (C) PCWP, (D) CI, and (E) PVR. Data were calculated using mixed-effects linear regression with multivariable adjustment for age, sex, body mass index, pulmonary arterial hypertension etiology, and whether background therapy was allowed. AUS = Australia; CA = Canada; CI = cardiac index; ISR = Israel; mPAP = mean pulmonary arterial pressure; PCWP = pulmonary capillary wedge pressure; Pinteract = P value for region × time interaction; Pregion = P value for differences in mean hemodynamic parameter by region; Pslope = P value for change in mean hemodynamic parameter over time; PVR = pulmonary vascular resistance; RAP = right atrial pressure; US = United States.
Discussion
We performed pooled cohort analysis of individual patient data from phase III PAH RCTs submitted to the FDA for regulatory approval in the past two decades. There were significant differences in age, body habitus, and invasive hemodynamic measures over time, which varied by geographic region. Our findings mirrored some demographic patterns described in regional PAH registries (4, 10), whereas sex and hemodynamic trends, particularly by region, may have been unexpected. These findings have potential implications in terms of the validity of applying RCT results in a population with changing demographics and disease characteristics, as well as toward the design and conduction of future PAH therapy trials.
Changes in Age, Sex, Body Habitus, and Disease Etiology in PAH Trial Populations
The mean age of participants in PAH observational cohorts has increased from 36 years in the U.S. National Institutes of Health Patient Registry for Primary Pulmonary Hypertension (1981–1988) to 50–65 years in modern U.S. and European registries (4, 10). In particular, participants enrolled in the European Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA) were significantly older, with a mean age of 65 years (4). Within our cohort, the average baseline age of participants from Asia (comprising mostly individuals of Chinese nationality) and Latin America was more than a decade lower compared with Western cohorts. The average age of participants in all regions except Asia increased over time. Although some RCTs limited the age of participants (including the Prostacyclin [PGI2] Receptor Agonist in Pulmonary Arterial Hypertension [GRIPHON] and the Ambrisentan and Tadalafil in Patients with Pulmonary Arterial Hypertension [AMBITION] trials [12]), these restrictions would not explain the overall trends in our analyses, nor would they explain differences by region. Whether this represents differences in disease characteristics or in recognition and availability of testing among older individuals remains unknown, though our findings were unchanged in sensitivity analyses adjusting for pulmonary hypertension etiology. The increasing proportion of more elderly patients with PAH is of particular concern, as observational data have shown that patients older than age 50 at the time of diagnosis were far more likely to die (3) and may be less likely to derive benefit from PAH therapy (13).
PAH remains a disease that affects predominantly women. The proportion of women was highest in earlier studies but stabilized at 78% overall. Men were slightly underrepresented in RCTs compared with observational cohorts from similar regions. For example, 25% of patients from Europe, Australia, and Israel were male, compared with 40% in COMPERA; 17% of patients from the United States and Canada were male, compared with 20% in the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL) and 25% in the Pulmonary Hypertension Association Registry (PHAR) (4, 14). This difference between the trial population and observational data was counter to the gender bias described in RCTs for other diseases, for which men tend to be overrepresented (15–17). Epidemiologic studies have indicated that men with PAH have poorer survival compared with women (18), possibly related to the effects of sex hormones on the right ventricle and pulmonary vasculature (19). In addition, men with PAH have worse functional class and less favorable hemodynamics compared with women (20). This difference in recruited versus observed populations may be due to survival or selection bias favoring the recruitment of women or to the inclusion of individuals with mixed disease in observational cohorts, which have less strict inclusion and exclusion criteria.
Patients from Western countries, particularly from the United States and Canada, had significantly higher BMI, and 39% were obese at the time of recruitment, reflecting the obesity epidemic. In addition, the mean BMI of participants increased over time regardless of geographic region. We have previously demonstrated that obese patients had significantly worse quality of life and functional capacity compared with normal-weight patients with PAH despite the initiation of pulmonary hypertension–directed therapy in the United States (14). Furthermore, obesity itself may affect disease outcomes through direct effects on the right ventricle and pulmonary vasculature, as well as through comorbid conditions such as obstructive sleep apnea and heart failure with preserved ejection fraction (21). These trends highlight a currently unmet need to further study the role of weight loss or metabolic interventions as adjuncts to vasodilator therapy in obese patients with PAH, who tend to have more comorbidities and may have attenuated responses to vasodilator therapy and increased drug adverse events.
IPAH, CTD-PAH, and PAH secondary to CHD were the most common subtypes of PAH in our cohort, mirroring European and American registry cohorts (4). Patients with portopulmonary hypertension (PoPH) constituted only 0.2% of our study cohort, compared with 5–10% of registry populations (4). Patients with PoPH have been excluded from nearly all PAH RCTs despite being a vulnerable population with worse social determinants of health (22, 23). These patients have often been excluded because of concerns regarding the impact of liver disease on the metabolism of the study drug as well the concern for a greater adverse event rate because of thrombocytopenia, liver function abnormalities, hypotension, or peripheral edema. However, these concerns, and their magnification of the impact of social determinants of health in PoPH, should be reexamined.
Hemodynamic Trends
Preenrollment hemodynamics of patients in RCTs have improved over time, even after accounting for pulmonary hypertension etiology and the presence of concurrent vasodilator therapy. Patients who enrolled more recently had lower RAP and mPAP, higher cardiac index, and lower PVR, which may reflect earlier diagnosis or may reflect an evolving disease phenotype. The PAWP of participants did not change over time, because nearly all trials placed strict exclusion criteria for PAWP. Interestingly, mPAP at the time the study began decreased by 2.8 mm Hg for every 5 years. Participants from Latin America had the highest RAP and mPAP, while those from Asia had the lowest cardiac index and highest PVR. These differences may reflect variability in the timing of patient diagnosis. Hemodynamic trends were likely influenced by inclusion criteria for the SERAPHIN (Study with an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome) (24), GRIPHON (11), and AMBITION (12) studies, large international RCTs that enrolled a total of 2,508 patients and accounted for the majority of patients recruited after 2008. Specifically, 98% of participants in SERAPHIN and GRIPHON had World Health Organization functional class II or III symptoms, while AMBITION recruited only those with functional class II or III symptoms. This resulted in a more limited range of hemodynamic indices, potentially excluding those with mild or severe disease.
Race/Ethnicity and Geographic Region
Most study participants in our sample were non-Hispanic White. The lack of racial and ethnic diversity in RCTs is not unique to PAH (17) and has been attributed to multiple systemic barriers to participation, such as mistrust of the medical system, time and resource constraints, and lack of awareness and/or access to trial centers among people of color (25). Observational studies have suggested that Black, Asian, and Hispanic patients with PAH may have more severe disease and worse outcomes compared with White patients (26). Differences in social determinants of health and access remain major factors (26).
The majority (87%) of non-Hispanic Black patients in the sample were recruited from the United States or Canada, representing approximately 10% of patients from those regions. Although this is similar to the 12% that has been reported in U.S.-based PAH registries (25, 26), Black patients with PAH constituted only 4% of the overall cohort after accounting for other geographic sites. This significantly limits the interpretability of RCT benefits and adverse events among Black patients with PAH and suggests that targeted efforts to recruit Black patients with PAH in RCTs are warranted to improve generalizability. Eighty-two percent of Asian and 49% of Hispanic patients were recruited from Asia and Latin America, respectively. These patients may have different exposures and risk factors compared with their counterparts in the United States or Europe, limiting generalizability for patients of similar race or ethnicity residing in Western countries. We lacked information into disease subtypes, particularly among patients with CTD-PAH, which have been shown to differ by race and geographic region (26).
Although more recent RCTs have improved the overall racial diversity of study participants, several important issues remain. First, race and ethnicity data were sometimes assigned by study coordinators or not collected at all. Established common data elements for racial and ethnic self-identification exist and should become the standard for all epidemiologic studies and clinical trials in PAH (27). Active efforts in the design of clinical trials in PAH need to specifically target the barriers to inclusion and diversity of minority patients, including local champions/spokespeople, identifying race/ethnic-concordant research staff members, creation of targeted recruitment study materials, and active tracking of screening, approach, and consent rates among minority patients with PAH. Further studies are necessary to identify the factors that affect outcomes among racial minorities and demonstrate that treatment effect and adverse event profiles are consistent in this population.
Strengths and Limitations
This is the largest pooled cohort study of individual patient data from PAH RCTs to date examining trends in patient characteristics over time. We presented a comprehensive analysis using mixed-effects models to characterize these changes, which are robust in accounting for clustering of patients by study. In addition, the harmonized patient characteristics allowed robust comparison of multivariable-adjusted estimates to account for confounders. As such, our findings are highly applicable to the interpretation of current RCTs as well as the design and administration of future studies.
Our study also had several limitations. We had access only to trials submitted for regulatory approval, so there is likely bias from the exclusion of trials that were not submitted to the FDA, including those that showed a null effect, were not completed, were terminated, or were for drugs for which regulatory approval was not being sought. It is unknown how the inclusion of these trials would affect our findings, though they tended to be limited in scope (6). Although the prevalence of obesity has increased globally, regional trends have continued to change; therefore, extrapolation beyond the study period (i.e., 2013) should be cautioned. Nevertheless, a recent secular trends analysis within COMPERA demonstrated a rising prevalence of obesity from 2010 to 2019 (28). We lacked the information to uniquely identify individuals across RCTs, and therefore it is possible that individuals in our sample could have participated in multiple RCTs. RCTs often imposed strict criteria for inclusion on the basis of either 6-minute walk distance or World Health Organization functional class and thus represent a sample that excludes those with very mild or very severe disease. As noted in our discussion, our findings identified areas in which the cohort may differ from patient registries, limiting generalizability, particularly for subjects who would have otherwise been excluded from RCTs because of disease etiology, severity, or other factors. Analyses of RCTs in other fields have shown limitations in generalizing results to real-world populations, demonstrating the need for ongoing analysis of real-world data (29). Last, although local variations in PAH etiology are well described (e.g., methamphetamine-related PAH in the western United States [30] or schistosomiasis-associated PAH in Brazil [31]), we did not have region-specific data to study these differences.
Conclusions
Our findings suggest that PAH study patients are evolving, with increasing age at the time of study enrollment and a greater proportion of overweight and obese patients, particularly in Western countries. Pretreatment hemodynamics appear to be less severely affected over time, possibly reflecting earlier disease recognition and wider availability of vasodilator therapy or evolution of patients diagnosed with PAH. These trends differ on the basis of geographic region, particularly for patients in Latin America and Asia; there is a need for high-quality, prospective, multicenter registries in these regions to study changes in PAH epidemiology and outcomes. Similarly, there is a need to better understand how sex differences contribute to differences in clinical trial enrollment and outcomes. Finally, although racial and ethnic diversity has overall improved in PAH RCTs, this was due mostly to the inclusion of study sites in Asia and Latin America, limiting generalizability to patients of similar race or ethnicity residing in Western countries.
Footnotes
Supported by the Cardiovascular Medical Research and Education Fund Grant (S.M.K.), National Institutes of Health (NIH) grant K24HL103844 (S.M.K.), NIH grant K23HL141584 (N.A.-N.), the Aldrighetti Research Award for Young Investigators from the Pulmonary Hypertension Association (N.A.-N.), and NIH grant T32HL007891 (J. Min and J. Minhas).
Author Contributions: J. Min was responsible for the conception and design of the work. J. Min, D.H.A., R.L.M., J.M.S., and N.A.-N. were responsible for the analysis of the data. J. Min, D.H.A., R.L.M., J. Minhas, J.H.H., R.J.U., J.M.S., N.A.-N., and S.M.K. made contributions to the acquisition and harmonization of the data. All authors made contributions to drafting and revising the manuscript for important intellectual content and have given final approval for its publication.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, et al. Primary Pulmonary Hypertension Study Group A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med . 1996;334:296–301. doi: 10.1056/NEJM199602013340504. [DOI] [PubMed] [Google Scholar]
- 2. Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension: a national prospective study. Ann Intern Med . 1987;107:216–223. doi: 10.7326/0003-4819-107-2-216. [DOI] [PubMed] [Google Scholar]
- 3. Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JSR, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med . 2012;186:790–796. doi: 10.1164/rccm.201203-0383OC. [DOI] [PubMed] [Google Scholar]
- 4. Hoeper MM, Gibbs JSR. The changing landscape of pulmonary arterial hypertension and implications for patient care. Eur Respir Rev . 2014;23:450–457. doi: 10.1183/09059180.00007814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sitbon O, Gomberg-Maitland M, Granton J, Lewis MI, Mathai SC, Rainisio M, et al. Clinical trial design and new therapies for pulmonary arterial hypertension. Eur Respir J . 2019;53:1801908. doi: 10.1183/13993003.01908-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu H-L, Chen X-Y, Li J-R, Su S-W, Ding T, Shi C-X, et al. Efficacy and safety of pulmonary arterial hypertension-specific therapy in pulmonary arterial hypertension: a meta-analysis of randomized controlled trials. Chest . 2016;150:353–366. doi: 10.1016/j.chest.2016.03.031. [DOI] [PubMed] [Google Scholar]
- 7. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J . 2019;53:1801913. doi: 10.1183/13993003.01913-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Riley RD, Lambert PC, Abo-Zaid G. Meta-analysis of individual participant data: rationale, conduct, and reporting. BMJ . 2010;340:c221. doi: 10.1136/bmj.c221. [DOI] [PubMed] [Google Scholar]
- 9. Legha A, Riley RD, Ensor J, Snell KIE, Morris TP, Burke DL. Individual participant data meta-analysis of continuous outcomes: a comparison of approaches for specifying and estimating one-stage models. Stat Med . 2018;37:4404–4420. doi: 10.1002/sim.7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Badlam JB, Badesch DB, Austin ED, Benza RL, Chung WK, Farber HW, et al. USPHSR Investigators United States Pulmonary Hypertension Scientific Registry: baseline characteristics. Chest . 2021;159:311–327. doi: 10.1016/j.chest.2020.07.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sitbon O, Channick R, Chin KM, Frey A, Gaine S, Galiè N, et al. GRIPHON Investigators Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med . 2015;373:2522–2533. doi: 10.1056/NEJMoa1503184. [DOI] [PubMed] [Google Scholar]
- 12. Galiè N, Barberà JA, Frost AE, Ghofrani H-A, Hoeper MM, McLaughlin VV, et al. AMBITION Investigators Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med . 2015;373:834–844. doi: 10.1056/NEJMoa1413687. [DOI] [PubMed] [Google Scholar]
- 13. Hoeper MM, Boucly A, Sitbon O. Age, risk and outcomes in idiopathic pulmonary arterial hypertension. Eur Respir J . 2018;51:1800629. doi: 10.1183/13993003.00629-2018. [DOI] [PubMed] [Google Scholar]
- 14. Min J, Feng R, Badesch D, Berman-Rosenzweig E, Burger C, Chakinala M, et al. PHAR Investigators Obesity in pulmonary arterial hypertension (PAH): the Pulmonary Hypertension Association Registry (PHAR) Ann Am Thorac Soc . 2020;18:229–237. doi: 10.1513/AnnalsATS.202006-612OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim ESH, Carrigan TP, Menon V. Enrollment of women in National Heart, Lung, and Blood Institute-funded cardiovascular randomized controlled trials fails to meet current federal mandates for inclusion. J Am Coll Cardiol . 2008;52:672–673. doi: 10.1016/j.jacc.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 16. Khan SU, Khan MZ, Raghu Subramanian C, Riaz H, Khan MU, Lone AN, et al. Participation of women and older participants in randomized clinical trials of lipid-lowering therapies: a systematic review. JAMA Netw Open . 2020;3:e205202. doi: 10.1001/jamanetworkopen.2020.5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Khan MS, Shahid I, Siddiqi TJ, Khan SU, Warraich HJ, Greene SJ, et al. Ten-year trends in enrollment of women and minorities in pivotal trials supporting recent US Food and Drug Administration approval of novel cardiometabolic drugs. J Am Heart Assoc . 2020;9:e015594. doi: 10.1161/JAHA.119.015594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation . 2010;122:156–163. doi: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- 19. Ventetuolo CE, Baird GL, Barr RG, Bluemke DA, Fritz JS, Hill NS, et al. Higher estradiol and lower dehydroepiandrosterone-sulfate levels are associated with pulmonary arterial hypertension in men. Am J Respir Crit Care Med . 2016;193:1168–1175. doi: 10.1164/rccm.201509-1785OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ventetuolo CE, Praestgaard A, Palevsky HI, Klinger JR, Halpern SD, Kawut SM. Sex and haemodynamics in pulmonary arterial hypertension. Eur Respir J . 2014;43:523–530. doi: 10.1183/09031936.00027613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mair KM, Gaw R, MacLean MR. Obesity, estrogens and adipose tissue dysfunction—implications for pulmonary arterial hypertension. Pulm Circ . 2020;10:2045894020952019. doi: 10.1177/2045894020952023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Galiè N, Humbert M, Vachiery J-L, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J . 2015;46:903–975. doi: 10.1183/13993003.01032-2015. [DOI] [PubMed] [Google Scholar]
- 23. DuBrock HM, Burger CD, Bartolome SD, Feldman JP, Ivy DD, Rosenzweig EB, et al. Health disparities and treatment approaches in portopulmonary hypertension and idiopathic pulmonary arterial hypertension: an analysis of the Pulmonary Hypertension Association Registry. Pulm Circ . 2021;11:20458940211020913. doi: 10.1177/20458940211020913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pulido T, Adzerikho I, Channick RN, Delcroix M, Galiè N, Ghofrani H-A, et al. SERAPHIN Investigators Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med . 2013;369:809–818. doi: 10.1056/NEJMoa1213917. [DOI] [PubMed] [Google Scholar]
- 25. Clark LT, Watkins L, Piña IL, Elmer M, Akinboboye O, Gorham M, et al. Increasing diversity in clinical trials: overcoming critical barriers. Curr Probl Cardiol . 2019;44:148–172. doi: 10.1016/j.cpcardiol.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 26. Medrek SK, Sahay S. Ethnicity in pulmonary arterial hypertension: possibilities for novel phenotypes in the age of personalized medicine. Chest . 2018;153:310–320. doi: 10.1016/j.chest.2017.08.1159. [DOI] [PubMed] [Google Scholar]
- 27. Gray MP, Kawut SM. The Pulmonary Hypertension Association Registry: rationale, design, and role in quality improvement. Adv Pulm Hypertens . 2018;16:185–188. [Google Scholar]
- 28. Hoeper MM, Pausch C, Grünig E, Staehler G, Huscher D, Pittrow D, et al. Temporal trends in pulmonary arterial hypertension: results from the COMPERA registry. Eur Respir J . 2021 doi: 10.1183/13993003.02024-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Averitt AJ, Weng C, Ryan P, Perotte A. Translating evidence into practice: eligibility criteria fail to eliminate clinically significant differences between real-world and study populations. npj Digit Med . 2020;3:67. doi: 10.1038/s41746-020-0277-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kolaitis NA, Zamanian RT, de Jesus Perez VA, Badesch DB, Benza RL, Burger CD, et al. Pulmonary Hypertension Association Registry Investigators Clinical differences and outcomes between methamphetamine-associated and idiopathic pulmonary arterial hypertension in the Pulmonary Hypertension Association Registry. Ann Am Thorac Soc . 2021;18:613–622. doi: 10.1513/AnnalsATS.202007-774OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Valverde AB, Soares JM, Viana KP, Gomes B, Soares C, Souza R. Pulmonary arterial hypertension in Latin America: epidemiological data from local studies. BMC Pulm Med . 2018;18:106. doi: 10.1186/s12890-018-0667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]