

Abstract
Angiotensin-(1-7) [Ang-(1-7)] is an endogenous seven-amino acid peptide hormone of the renin-angiotensin system that has anti-proliferative properties. In this study, Ang-(1-7) inhibited the growth of cancer-associated fibroblasts (CAFs) and reduced fibrosis in the tumor microenvironment. A marked decrease in tumor volume and weight was observed in orthotopic human BT-474 or ZR-75–1 breast tumors positive for the estrogen receptor and HER2 following Ang-(1-7) administration to athymic mice. Ang-(1-7) concomitantly reduced interstitial fibrosis in association with a significant decrease in collagen I deposition, along with a similar reduction in perivascular fibrosis. In CAFs isolated from orthotopic breast tumors, the heptapeptide markedly attenuated in vitro growth as well as reduced fibronectin, transforming growth factor-β (TGF-β), and ERK1/2 kinase activity. An associated increase in the MAPK phosphatase DUSP1 following treatment with Ang-(1-7) suggested a potential mechanism by which the heptapeptide reduced MAPK signaling. Consistent with these in vitro observations, immunohistochemical analysis of Ang-(1-7)-treated orthotopic breast tumors revealed reduced TGF-β and increased DUSP1. Together, our findings indicate that Ang-(1-7) targets the tumor microenvironment to inhibit CAF growth and tumor fibrosis.
Keywords: angiotensin-(1-7), fibrosis, breast cancer, tumor-associated fibroblasts, DUSP1, fibronectin, transforming growth factor
INTRODUCTION
Crosstalk between cancer cells and surrounding tissue is essential for the development and progression of tumors. The interaction between cancerous cells and the adjacent microenvironment transforms the stroma into an abnormal phenotype, altering normal function (1, 2). The altered stromal microenvironment impacts tissue architecture, cellular morphology, and extracellular matrix (ECM)-cell interactions that directly contribute to formation of the neoplasia (1). Solid tumors, in particular breast tumors, are characterized by pathological desmoplasia, resulting in increased fibrosis and ECM deposition (1, 2). About 80% of reactive stroma associated with breast carcinoma is composed of activated myofibroblasts (3) which secrete ECM proteins, resulting in desmoplasia and breast tumor progression (4).
Activated myofibroblasts play a vital role in tumor initiation, growth, and metastases. Tumor stroma myofibroblasts or “cancer-associated fibroblasts” (CAF) are characterized by their expression profile and are distinguished from normal fibroblasts by the expression of α-smooth muscle actin and vimentin (5). Moreover, the presence of α-smooth muscle actin-positive myofibroblasts around non-invasive epithelium in breast ductal carcinoma in situ is strongly correlated with the onset of tumor invasion and poor prognosis (5, 6). Inflammation and cytokine secretion by cancer cells results in the recruitment of CAF to the tumor site. Activated myofibroblasts are transformed by cytokines such as transforming growth factor-β (TGF-β) and secrete tumor promoting growth factors including hepatocyte growth factor (HGF), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), and TGF-α and -β (5), which leads to the production of ECM components including collagens, tenacin-C, and fibronectin (7).
CAF play an important role in the initiation and progression of cancer; however, cancer therapies that target the tumor stroma are limited (5, 8). In this study, we assessed whether angiotensin-(1-7) [Ang-(1-7)], a seven-amino acid peptide hormone of the renin-angiotensin system with vasodilatory, anti-proliferative, and anti-fibrotic properties (9), alters the tumor microenvironment to reduce tumor growth and fibrosis. Ang-(1-7) exerts its biological activity through a unique G protein-coupled receptor, mas (9, 10). The heptapeptide decreased mitogen-stimulated growth of vascular smooth muscle cells (VSMCs) in vitro (11) and attenuated neointima formation in a balloon catheter injury model of the rat carotid artery, with no effect on the underlying media layer, indicating that the anti-growth properties of the heptapeptide are limited to proliferating cells (12). In a retrospective study of hypertensive patients treated with angiotensin converting enzyme (ACE) inhibitors which increase Ang-(1-7), ACE inhibitor treatment reduced the risk of cancer, particularly lung and breast cancer (13). More importantly, we showed that Ang-(1-7) significantly decreased the proliferation of human lung cancer cells in vitro by reducing mitogen-activated protein (MAP) kinase activity (14). Ang-(1-7) infusion decreased human A549 lung adenocarcinoma xenograft growth with a corresponding reduction in cyclooxygenase 2 (COX-2) (15). Lung tumors from mice injected with the heptapeptide had reduced vessel density with an associated decrease in VEGF, indicating that Ang-(1-7) inhibited tumor angiogenesis (16). These data suggested that Ang-(1-7) also regulates the tumor microenvironment to inhibit cancer growth.
Fibrosis correlates with the progression and invasion of breast cancer. The increased ECM deposition and secretion of growth factors by myofibroblasts directly contribute to breast tumor growth by stimulating tumor cell proliferation, increasing angiogenesis, and promoting invasion (5). The purpose of this study was to determine whether Ang-(1-7) could serve as an anti-fibrotic agent that targets the tumor microenvironment to reduce breast cancer tumor growth and fibrosis.
MATERIALS AND METHODS
Materials:
The following materials were purchased from the companies in the parentheses. Ang-(1-7) and [D-Alanine7]-angiotensin-(1-7) or [D-Ala7]-Ang-(1-7) (Bachem, King of Prussia, PA); [D-Proline7]-angiotensin-(1-7) or [D-Pro7]-Ang-(1-7) (GenScript Corporation, Piscataway, NJ); penicillin, RPMI-1640, DMEM/F12, streptomycin, fetal bovine serum (FBS), and Hypoxanthine-Aminopterin-Thymidine (HAT) supplement (Gibco Invitrogen BRL, Carlsbad, CA); TGF-β (Calbiochem, San Diego, CA); Matrigel (BD Biosciences, Bedford, MA); picric acid (Sigma-Aldrich, St. Louis, MO); saturated picric acid (LabChem Inc., Pittsburgh PA); and collagenase, trypsin, and soybean trypsin inhibitor (Worthington Biochemical, Lakewood, NJ). Antibodies were obtained from the following sources: Collagen I and TGF-β for immunohistrochemistry (Abcam, Cambridge, MA); phosphorylated ERK 1/2 and TGF-β for Western blot hybridization (Cell Signaling Technology, Danvers, MA); MKP-1 for Western blot hybridization (Upstate Biotech, Lake Placid, NY) and for immunohistochemistry (Santa Cruz Biotechnology, Santa Cruz, CA); fibronectin, vimentin, α-smooth muscle actin, and β-actin (Sigma-Aldrich, St. Louis, MO); Cy2 FITC-coupled donkey anti-rabbit, Cy3 FITC-coupled donkey anti-mouse (Jackson Laboratories, West Grove, PA); and polyclonal and HRP-conjugated secondary antibodies (GE Health care, Buckinghamshire, UK).
Cell Culture:
ZR-75–1 breast ductal carcinoma cells (ATCC CRL-1500), derived from a 63 year-old Caucasian female, and BT-474 breast carcinoma cells (ATCC HTB-20), derived from a 60 year-old Caucasian female, were grown in RPMI media containing 10% FBS, 100 μg/mL penicillin, 100 units/mL streptomycin and 10 nM Hepes. Isolated tumoral fibroblasts were grown in DMEM/F12 media containing 10% FBS, 100 μg/mL penicillin, 100 units/mL streptomycin and 10 nM Hepes. Cells were grown at 37°C in a humidified atmosphere of 5% CO2:95% room air.
Orthotopic Model of Human Breast Cancer:
Female athymic mice (15–20 g, 4–6 weeks of age; Charles River Laboratory, Wilmington, MA) were housed in cages with HEPA-filtered air (12-h light dark cycle) and provided ad libitum access to food and autoclaved water. All procedures complied with the policies of the Wake Forest University Animal Care and Use Committee. Female athymic mice were ovariectomized and supplemented with 0.18 mg 17β-estradiol 90-day time release pellets (Innovative America, Saratoga, FL) to prevent cycling of hormones (17, 18). Actively growing ZR-75–1 cells (2 ×106) or BT-474 cells (5 × 106) at 75% confluence were suspended in 50% PBS:50% Matrigel and injected into the inguinal mammary fat pad (19, 20). Tumor size was measured every other day using a caliper and tumor volume was calculated using the formula V= [(Length × Width2)/2]. When tumors reached a volume of 100 mm3 (ZR-75–1) or 200 mm3 (BT-474), the mice were implanted with osmotic minipumps for subcutaneous release of saline or 24 μg/kg/h Ang-(1-7). After 18 days of treatment, the animals were sacrificed and tumors were excised.
Immunohistochemistry:
Paraformaldehye-fixed paraffin embedded tumors were cut into five micron thick sections and stained with hematoxylin and eosin (H&E) to determine morphology. Interstitial and perivascular fibrosis was measured by picrosirius red histochemical staining (21). Immunostaining was performed with an antibody to collagen I (1:100) using the streptavidin-biotin method (22). Immunostaining for TGF-β (1:100), DUSP1 (1:200), and vimentin (1:100) was performed using FITC-coupled Cy2 or Cy3 as secondary antibodies and counterstained with 4’,6-diamidino-2-phenylindole (DAPI). Stained sections were visualized with a Leica DM 4000 microscope (Amax = 492 nm Cy2, Amax = 550 nm Cy3 for fluorescence) and photographed with a QImaging Retiga 1300R camera. A computer-assisted counting technique with a pixel counter to select stained fibers was used to quantify picrosirius red and collagen I staining. Interstital fibrosis was expressed as a percentage of reactive fibers/field (four fields/tumor section/mouse), while perivascular fibrosis is expressed as a percentage of reactive fibers/blood vessel (four vessels/tumor section/mouse).
Tumor Fibroblast Isolation:
Orthotopic ZR-75–1 breast tumors (200 mm3) were excised from mammary fat pads. Minced tumors were digested overnight at 4°C with trypsin (50 μg/mL) and soybean trypsin inhibitor (100 μg/mL) was added to stop the reaction. The minced tissue, harvested by centrifugation, was further digested with collagenase (85 U/mL) at 37°C for 30 min. Undigested tumor tissue was removed with a cell strainer and tumor fibroblasts were isolated by differential plating as previously described (23).
Quantification of Cell Number:
Isolated tumor fibroblasts (1 × 104 cells/mL) in DMEM/F12 containing 0.5% FBS and 10 ng/mL TGF-β were treated with PBS or 100 nM Ang-(1-7) in PBS, added daily due to degradation of the heptapeptide. Cell number was determined using a hemocytometer.
Immunofluorescence:
Fibroblasts were fixed with paraformaldehyde, permeabilized with 0.1% Triton X-100, and incubated overnight at 4°C with antibodies to fibronectin, vimentin, α-smooth muscle actin, or collagen I (1:100). Following a 30 min incubation with Cy2- or Cy3–labeled antibodies (1:100), the cells were counter-stained with DAPI.
Western Blot Hybridization:
Treated cell monolayers were solubilized in lysis buffer (14) and protein was measured by a modification of the Lowry method (24). Proteins were separated by polyacrylamide gel electrophoresis and transferred to hydrophobic polyvinylidene diflouride membrane. Non-specific binding was blocked by incubation with Blotto (Tris-buffered saline with 5% powdered milk and 4% Triton X-100). Membranes were incubated overnight at 4°C with primary antibodies specific to DUSP1 (MKP1, 1:1000), fibronectin (1:5000), pERK1/2 or TGF-β (1:1000) followed by a 1 h incubation with polyclonal horseradish peroxidase (HRP)-conjugated secondary antibodies (1:2000) at room temperature. Immunoreactive products were visualized by chemiluminescence (SuperSignal Femto or Pico West, Pierce Biotechnology, Rockford, IL) and quantified by densitometry using MCID digital densitometry software (Cambridge, UK). Protein loading was visualized by incubation of stripped membranes with a monoclonal antibody to β-actin (1:2000).
Statistics:
All data are presented as the mean±standard error of the mean (SEM). Statistical differences were evaluated by Student’s t test or one way analysis of variance (ANOVA) followed by Dunnett’s post hoc test. The criterion for statistical significance was set at p < 0.05.
RESULTS
Inhibition of Orthotopic Human Breast Tumors by Ang-(1-7)
Aythmic mice bearing human ZR-75–1 or BT-474 orthotopic breast tumors were administered saline or 24 μg/kg/h Ang-(1-7) via osmotic minipump. The ZR-75–1 tumor volume in the two treatment groups was not statistically different at the beginning of treatment [117.3±4.3 mm³ in the saline-treated group when compared to 123.8±7.6 mm³ in the Ang-(1-7)-treated group]. Tumor volume of saline-medicated mice continued to increase, while the growth of tumors in Ang-(1-7)-treated mice was significantly inhibited beginning at day 4 of treatment [Figure 1A, day 18 tumor volumes: 287.4±22.2% change in the saline-treated mice compared to 59.7±9.8% change in the Ang-(1-7)-treated animals when compared to original tumor volume; p<0.001]. By the end of the study, the volume of ZR-75–1 tumors from Ang-(1-7)-medicated mice was reduced 25% compared to the initial tumor volume, resulting in a %T/C of 22.3%. Due to the diminished size of the tumors from mice administered Ang-(1-7), the study was terminated at 18 days to provide sufficient tumor tissue for mechanistic studies.
Figure 1.
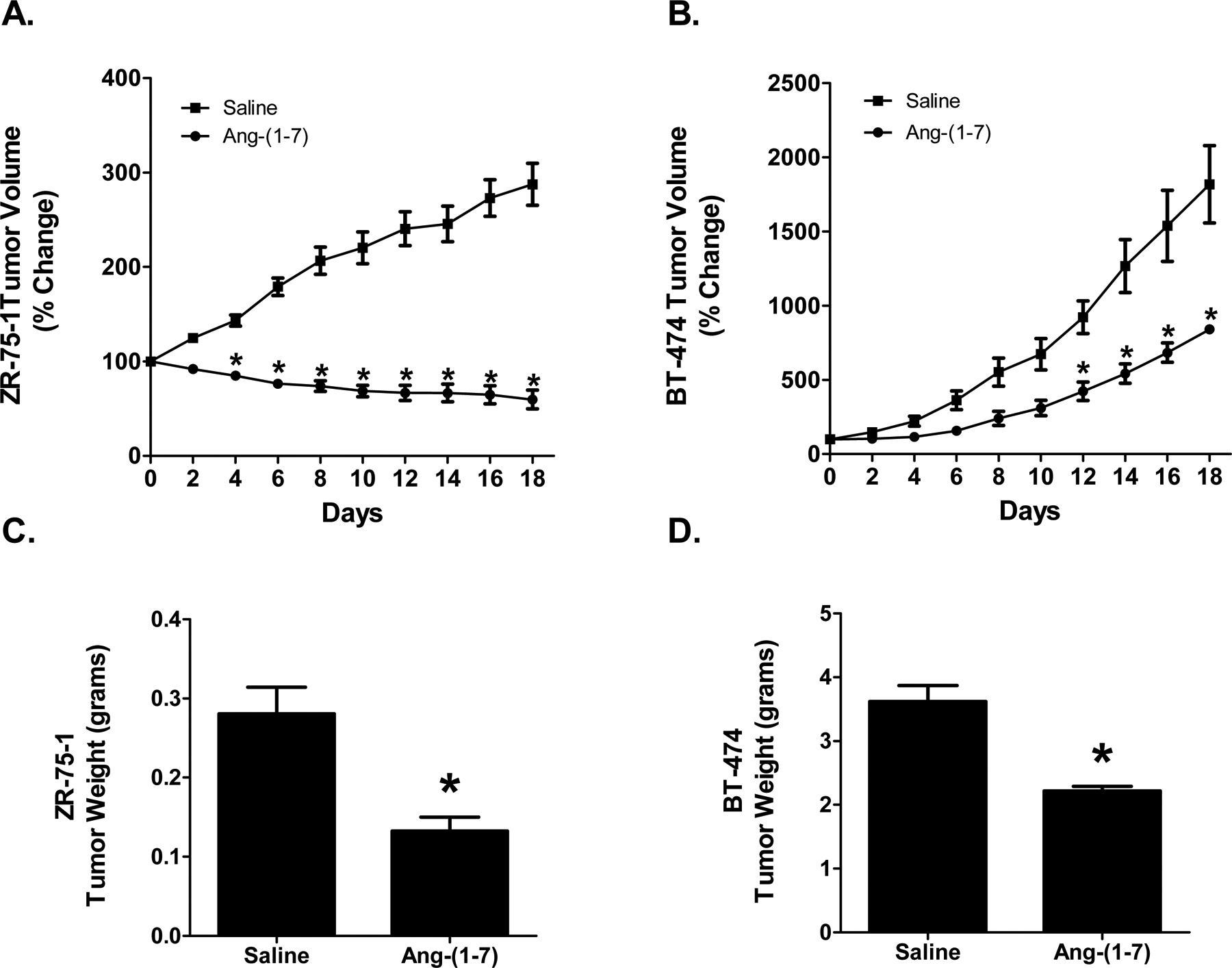
Effect of Ang-(1–7) on orthotopic human breast cancer growth. The volumes of human ZR-75–1 (Panel A) or BT-474 (Panel B) orthotopic breast tumors from mice treated with saline or Ang-(1–7) were measured using a caliper, calculated as V=½(L x W²), and presented as the percent change compared to initial tumor volume. ZR-75–1 (Panel C) or BT-474 (Panel D) tumor weight from mice infused with saline or Ang-(1–7) was determined at time of sacrifice. * denotes p<0.05; n=5–6.
The tumor volume of the two treatment groups of athymic mice injected with human BT-474 breast cancer cells was not statistically different at the beginning of treatment [327.0±72.5 mm³ in the saline-treated group compared to 197.7±15.9 mm³ in the Ang-(1-7)-administered group]. The volume of the tumors in saline-treated mice continued to increase, while the growth of tumors from the heptapeptide-medicated animals was inhibited significantly beginning at day 12 [Figure 1B, final tumor volumes: 1818±260.2% change in the saline-treated animals compared to 841.2±24.9% change in the Ang-(1-7)-treated animals when compared to initial tumor volumes; p<0.001]. Ang-(1-7) administration of BT-474 orthotopic human breast tumors resulted in a %T/C of 29.9%. The study was terminated at 18 days for health concerns due to the large tumor burden of the saline-treated mice.
No gross pathological side effects were observed with heptapeptide administration; there was no change in motor function, body weight, and food and water intake. No metastases were observed in mice with AR-75–1 or BT-474 tumors, due to the short time of the study. Upon completion of the study, the mice were euthanized and tumors were excised and weighed. The ZR-75–1 tumors from mice treated with Ang-(1-7) weighed approximately 50% less than the tumors from the saline control animals [0.29±0.03 g versus 0.14±0.02 g] (Figure 1C). Similarly, the BT-474 tumors from mice that were medicated with Ang-(1-7) weighed approximately 40% less than the tumors from the mice treated with saline [3.62±0.25 g versus 2.22±0.07 g] (Figure 1D).
Inhibition of Tumor Fibrosis by Ang-(1-7)
Interstitial tumoral fibrosis was quantified in orthotopic breast tumor sections stained with picrosirius red, a non-specific collagen stain. ZR-75–1 tumors and BT-474 tumors from saline-treated mice displayed heavy deposits of collagen which were reduced by Ang-(1-7) administration (Figures 2A and 2B). The relative amount of picrosirius red staining was quantified and the amount of collagen within the tumors was expressed as percent fibrosis per field. Treatment with the heptapeptide reduced interstitial fibrosis by 64% in the ZR-75–1 tumors [23.3±2.4% versus 8.3±0.8% fibrosis per field] (Figure 2A) and by 75% in the BT-474 tumors [4.9±1.0% versus 1.2±0.2% fibrosis per field] (Figure 2B), indicating that Ang-(1-7) reduces tumor fibrosis in orthotopic breast tumors. ZR-75–1 tumors from saline-treated mice had 3-fold more interstitial fibrosis when compared to BT-474 tumors from saline-treated mice, demonstrating a differential deposition of collagen between the two types of breast tumors. The amount of picrosirius red staining surrounding blood vessels in BT-474 tumors was also quantified, to measure perivascular fibrosis. It was not possible to quantify perivascular fibrosis in ZR-75–1 tumors due to pervasive interstitial fibrosis throughout the tumor. Heavy deposits of collagen were visualized around blood vessels in BT-474 tumors from saline-treated animals while tumor sections from Ang-(1-7)-medicated animals had reduced perivascular fibrosis. The heptapeptide decreased collagen deposition around the blood vessels by 73% [49.3±3.2% versus 13.4±2.2% fibrosis per blood vessel] (Figure 2C), suggesting that Ang-(1-7) reduces both interstitial and perivascular fibrosis in breast tumors.
Figure 2.
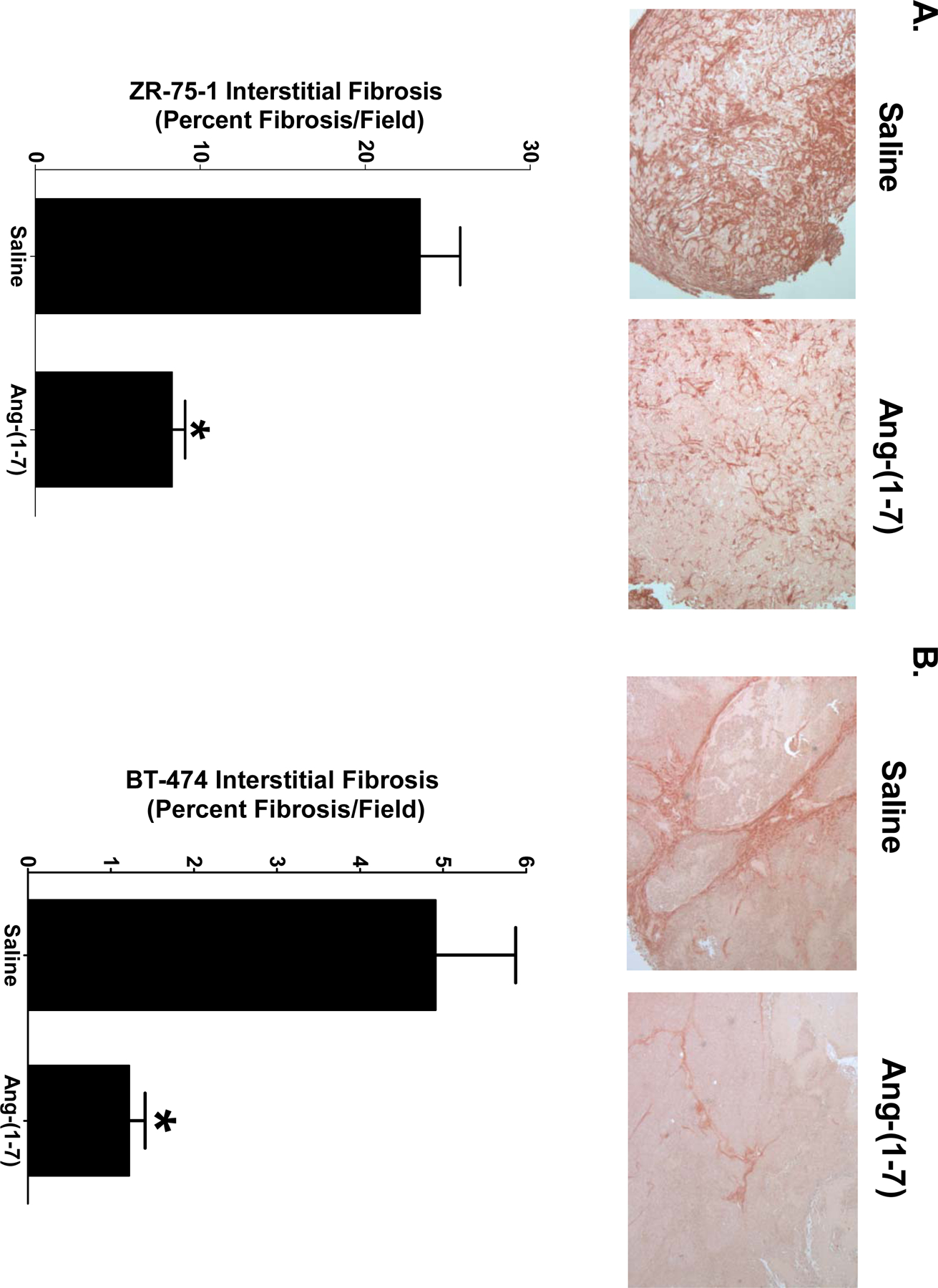
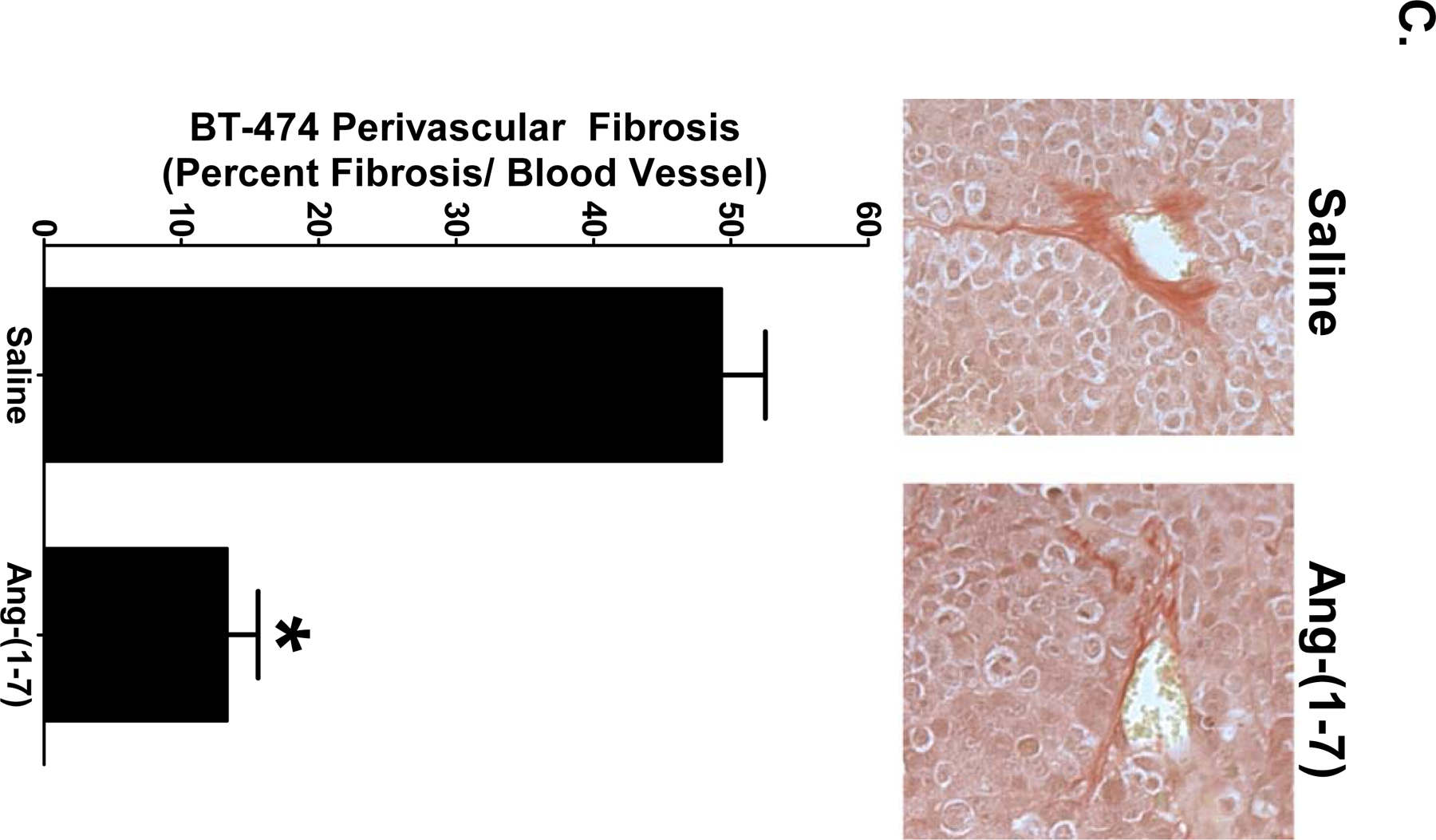
Inhibition of breast tumor fibrosis by Ang-(1–7). Representative pictures of picrosirius red-stained sections of ZR-75–1 (Panel A) and BT-474 tumors (Panel B) from mice treated with saline or Ang-(1–7) are shown at x200 magnification. Interstitial fibrosis was calculated as the percent fibrosis per field in sections of ZR-75–1 or BT-474 tumors. Representative pictures of picrosirius red-stained blood vessels from BT-474 tumors (Panel C) at x200 magnification. Perivascular fibrosis was calculated as percent fibrosis per vessel. * denotes p<0.05; n=5–6.
Interstitial fibrosis in orthotopic breast tumors was further characterized by the immunoreactivity of collagen I, one of the main isoforms of collagen found in fibrosis breast tissue. Representative pictures of collagen I immunoreactivity in ZR-75–1 and BT-474 tumor sections are shown in Figures 3A and 3B, respectively. Ang-(1-7) reduced collagen I deposition by 80% in ZR-75–1 orthotopic breast tumors [16.2±1.7% per field in the tumors of saline-treated mice versus 3.3±0.9% per field in tumors from Ang-(1-7)-treated mice], while a 78% decrease in collagen I deposition was observed in the BT-474 tumors [3.3±0.7% per field in the tumors of saline-treated animals when compared to 0.7±0.1% per field in tumors from Ang-(1-7)-medicated animals].
Figure 3.
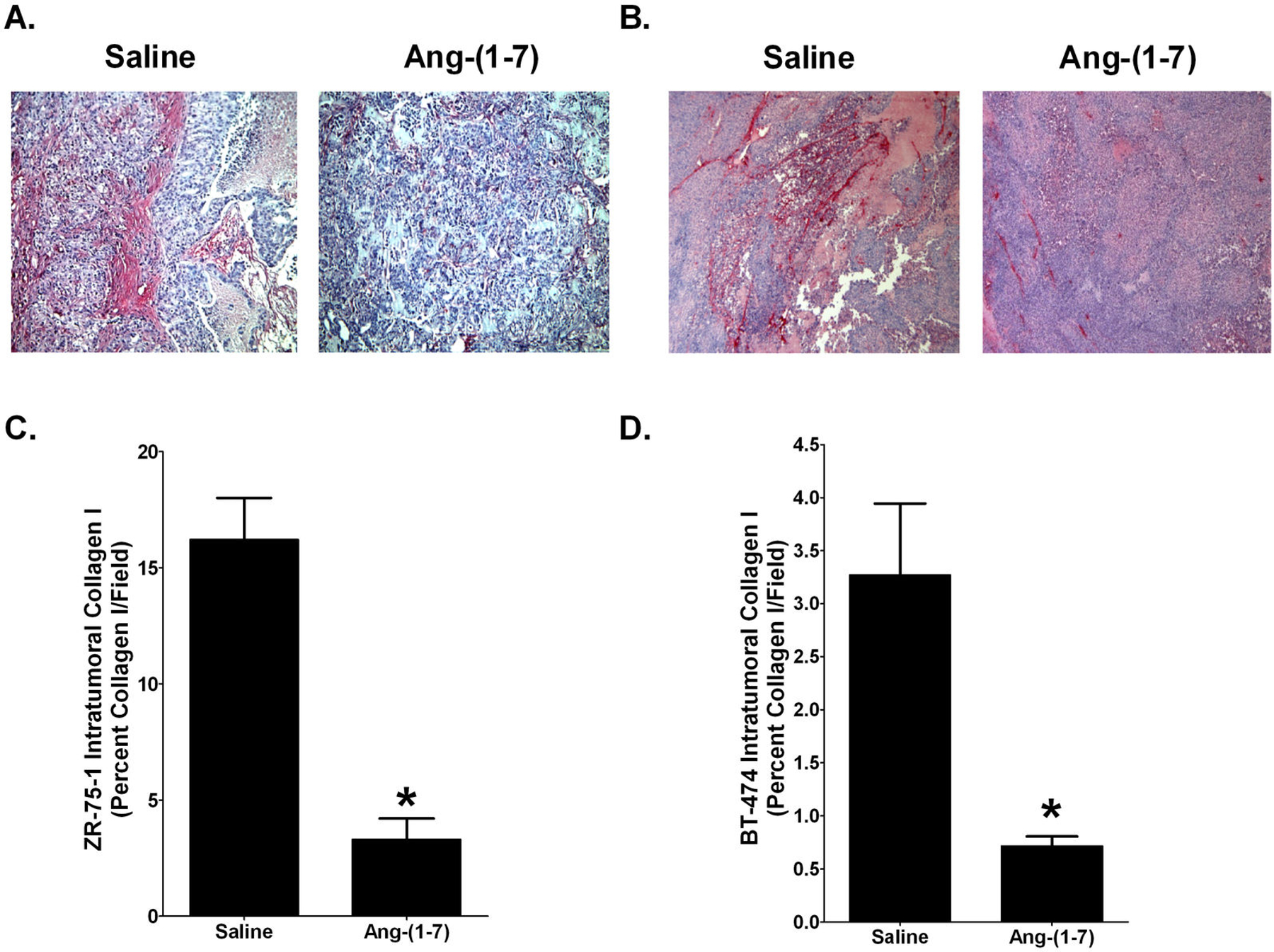
Ang-(1–7) reduction in collagen I deposition in breast tumors. ZR-75–1 (Panel A) or BT-474 tumors (Panel B) from mice treated with saline of Ang-(1–7) were incubated with an antibody to collagen I and representative pictures are shown at x200 magnification. Collagen deposition was quantified as the percent collagen I per field in ZR-75–1 (Panel C) and BT-474 tumors (Panel D). * denotes p<0.05; n=5–6.
Inhibition of Tumor-Associated Fibroblast Growth by Ang-(1-7)
Fibroblasts were isolated from ZR-75–1 tumors to identify the molecular mechanism for the Ang-(1-7)-mediated reduction in tumor fibrosis. Tumor-associated fibroblasts, isolated by differential plating, were characterized as myofibroblasts by positive immunoreactivity to fibronectin, vimentin, collagen I, and α-smooth muscle actin (Figure 4A). The percent of cells which showed positive immunoreactivity for fibronectin, vimentin, and α-smooth muscle actin was determined in sequentially passaged tumoral fibroblasts, to determine whether the cells maintain their phenotype as activated myofibroblasts with time in culture. As shown in Figure 4B, fibroblasts isolated from orthotopic tumors retained the activated myofibroblast phenotype until passage 5; therefore, only cells from passages 2–4 were used for in vitro experiments.
Figure 4.
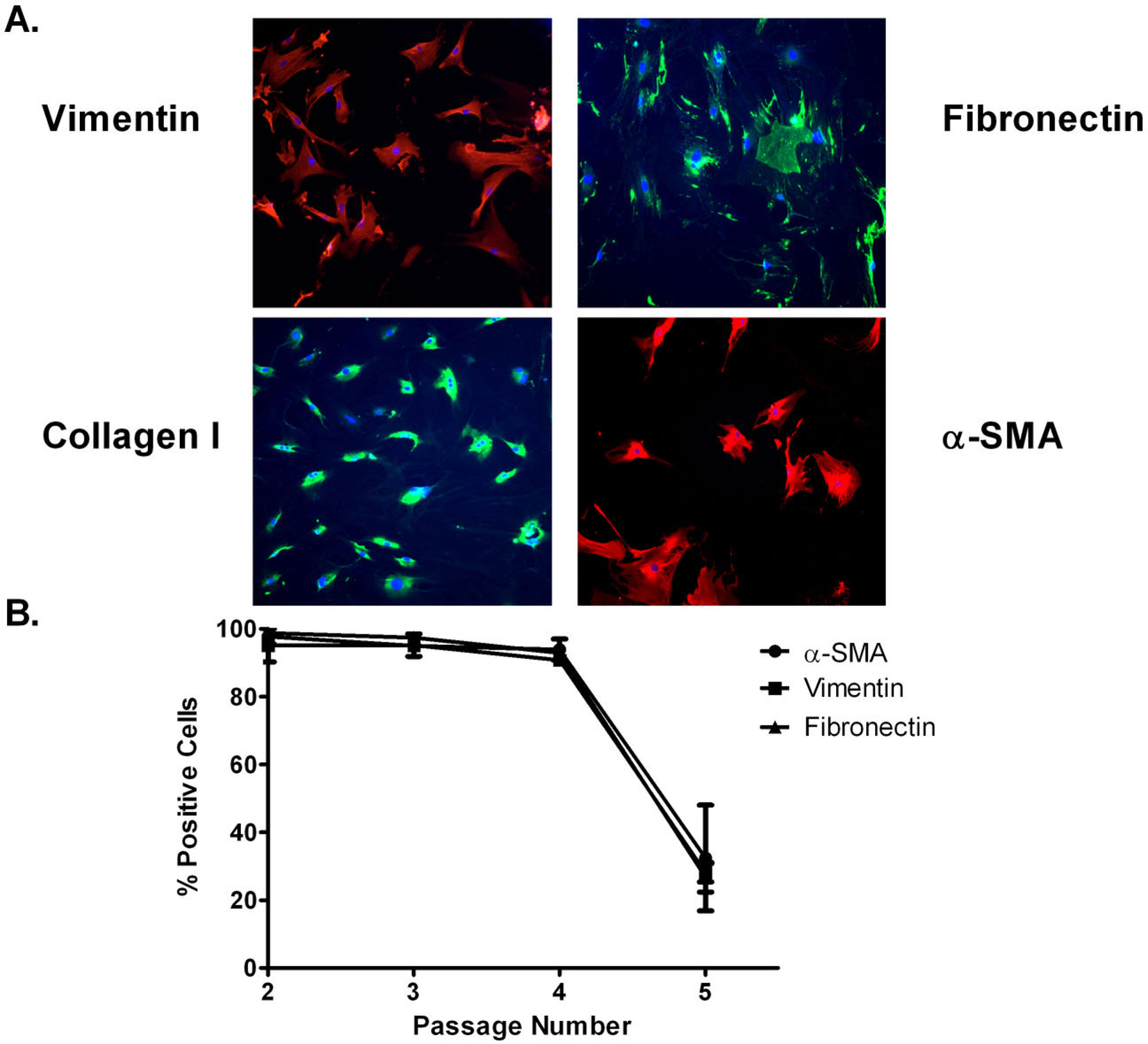
Characterization of isolated tumoral fibroblasts. A: Isolated tumoral fibroblasts were incubated with antibodies to vimentin, fibronectin, collagen I, and α-smooth muscle actin (α-SMA). B: The percent of cells which showed positive immunoreactivity for fibronectin, vimentin, and α-SMA was determined in sequentially passaged tumoral fibroblasts. n=2–3.
Isolated tumoral fibroblasts were treated with PBS or 100 nM Ang-(1-7) daily for 10 days and the cells were counted using a hemocytometer, as a measure of cell proliferation. Ang-(1-7) significantly reduced the growth of cultured myofibroblasts isolated from orthotopic breast tumors at days 4, 7 and 10, with a 33% reduction in cell growth at day 10 [10,700±400 PBS-treated myofibroblasts versus 7000±200 Ang-(1-7)-treated myofibroblasts] (Figure 5A).
Figure 5.
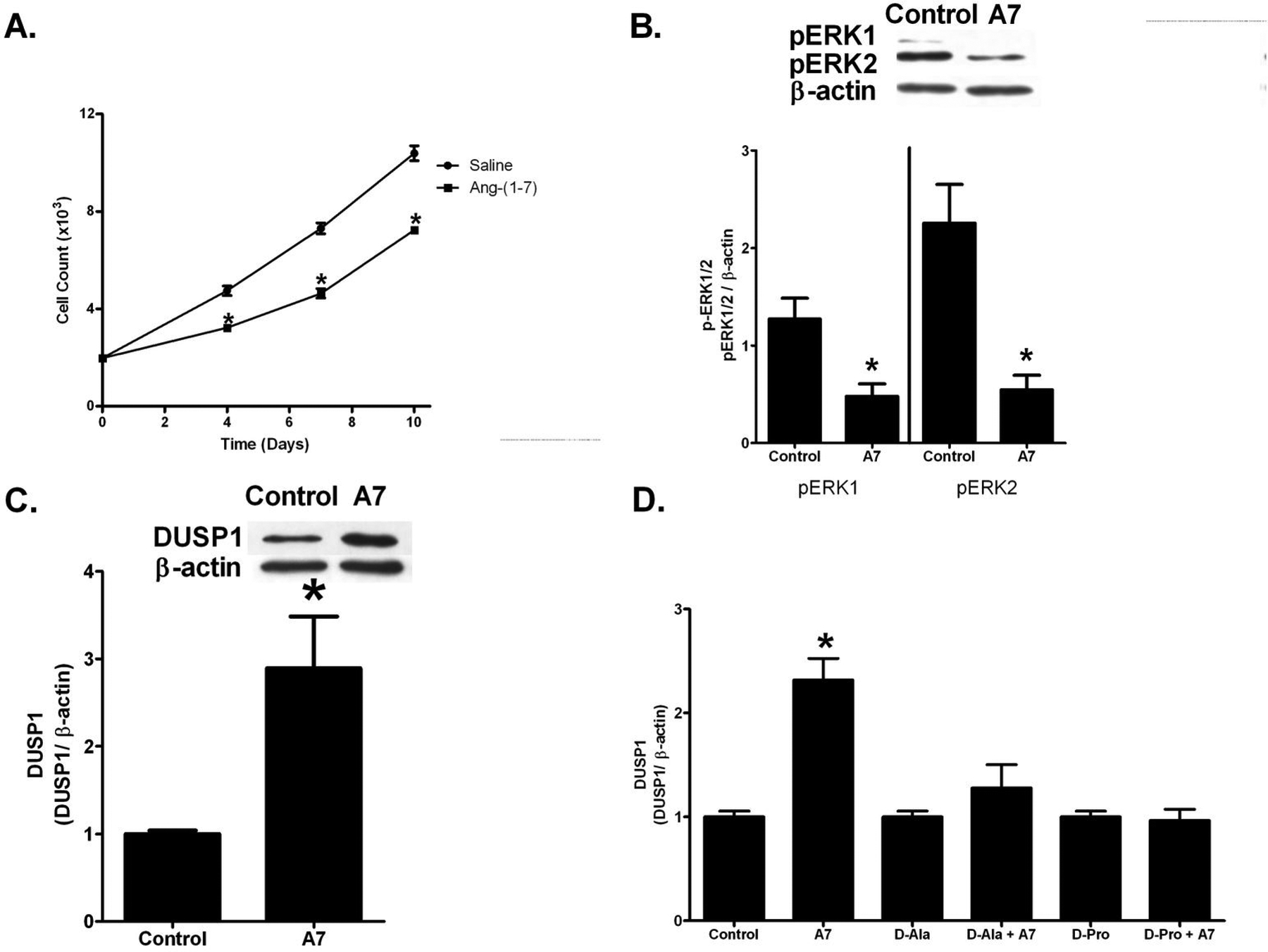
Ang-(1–7) inhibition of isolated tumor myofibroblast growth. A: Tumoral fibroblasts were treated with PBS or 100 nM Ang-(1–7) and 2% FBS and cell number was determined on days 4, 7 and 10, using a hemocytometer. B: Tumoral fibroblasts were serum starved overnight, pre-treated with PBS (control) or 100 nM Ang-(1–7) for four h and stimulated with 10 ng/mL TGF-β1 for 2 h. pERK1/2 was assessed by Western blot hybridization and is presented as a function of β-actin. C: Tumoral fibroblasts were serum starved overnight and treated with PBS (control) or 100 nM Ang-(1–7) for four h. DUSP1 was assessed by Western blot hybridization and presented as a function of β-actin. D: Tumoral fibroblasts were incubated for 4 h with PBS (Control), 100 nM Ang-(1–7) (A7), 1 µM D-[Ala7]-Ang-(1–7) (D-Ala), 100 nM Ang-(1–7) and 1 µM D-[Ala7]-Ang-(1–7) (D-Ala + A7), 1 µmol/L D-[Pro7]-Ang-(1–7) (D-Pro), and 100 nM Ang-(1–7) and 1 µmol/L D-[Pro7]-Ang-(1–7) (D-Pro + A7). DUSP1 was assessed by Western blot hybridization. * denotes p<0.05; n=3–4 of cells from different passage numbers isolated from 3 different tumors.
Ang-(1-7) Reduces MAP Kinase Activity by Up-regulation of a MAP Kinase Phosphatase
Phosphorylated-extracellular regulated kinases (pERK1/2) are potent mitogenic signaling proteins implicated in cell survival, growth, and proliferation. pERK1/2 was measured by Western blot hybridization in protein homogenates from myofibroblasts stimulated with 10 ng/mL TGF-β and treated with PBS or 100 nM Ang-(1-7), to determine if the heptapeptide regulates phosphorylated MAP kinases. Ang-(1-7) decreased pERK1 by 47% and pERK2 by 63% [1.13±0.23 relative density units (RDU) in PBS-treated cells versus 0.60±0.06 RDU in Ang-(1-7)-treated cells for pERK1, and 2.27±0.56 RDU in PBS-treated cells versus 0.84±0.08 RDU in Ang-(1-7)-treated cells for pERK2] (Figure 5B), indicating that the Ang-(1-7)-mediated anti-proliferative effect may be due to a reduction in phosphorylated ERK1/2.
MAP kinases are phosphorylated and activated by MAP kinases kinases (MEKs) and dephosphorylated and inactivated by MAP kinase phosphatases. Dual specificity phosphatase 1 (DUSP1), a MAP kinase phosphatase that dephosphorylates and inactivates ERK1/2, was up-regulated 2.52±0.29-fold by Ang-(1-7) in tumor-associated fibroblasts, suggesting that the heptapeptide may reduce pERK1/2 by up-regulation of the MAP kinase phosphatase DUSP1 (Figure 5C). Pretreatment with the Ang-(1-7) receptor (AT(1–7)R) antagonist D-[Ala7]-Ang-(1-7) or D-[Pro7]-Ang-(1-7) (100 nM) completely blocked the Ang-(1-7)-mediated increase in DUSP1 (Figure 5D), whereas the antagonists alone had no effect, indicating that Ang-(1-7) activated an AT(1–7) receptor to increase DUSP1 in tumor-associated fibroblasts.
Ang-(1-7) Reduces TGF-β1 and Fibronectin in Tumoral Fibroblasts
TGF-β1 is a potent stimulator of fibroblast activation that transforms fibroblasts to secreting myofibroblasts. TGF-β1 was quantified by Western blot hybridization in protein homogenates from myofibroblasts isolated from orthotopic breast tumors and treated with PBS or 100 nm Ang-(1-7) for 24 h. Ang-(1-7) reduced TGF-β1 by 45.4±11.7% in myofibroblasts compared to controls (Figure 6A). Since TGF-β1 stimulates myofibroblasts to synthesize and deposit ECM proteins, myofibroblasts were stimulated with 10 ng/mL TGF-β1 and treated with either PBS or 100 nM Ang-(1-7) for 24 h, to determine if the heptapeptide regulated fibronectin synthesis. Ang-(1-7) administration decreased fibronectin by 37.5±12.0% compared to the control (Figure 6B), suggesting that Ang-(1-7) reduces fibronectin synthesis to attenuate ECM protein deposition, fibrosis, and fibronectin signaling.
Figure 6.
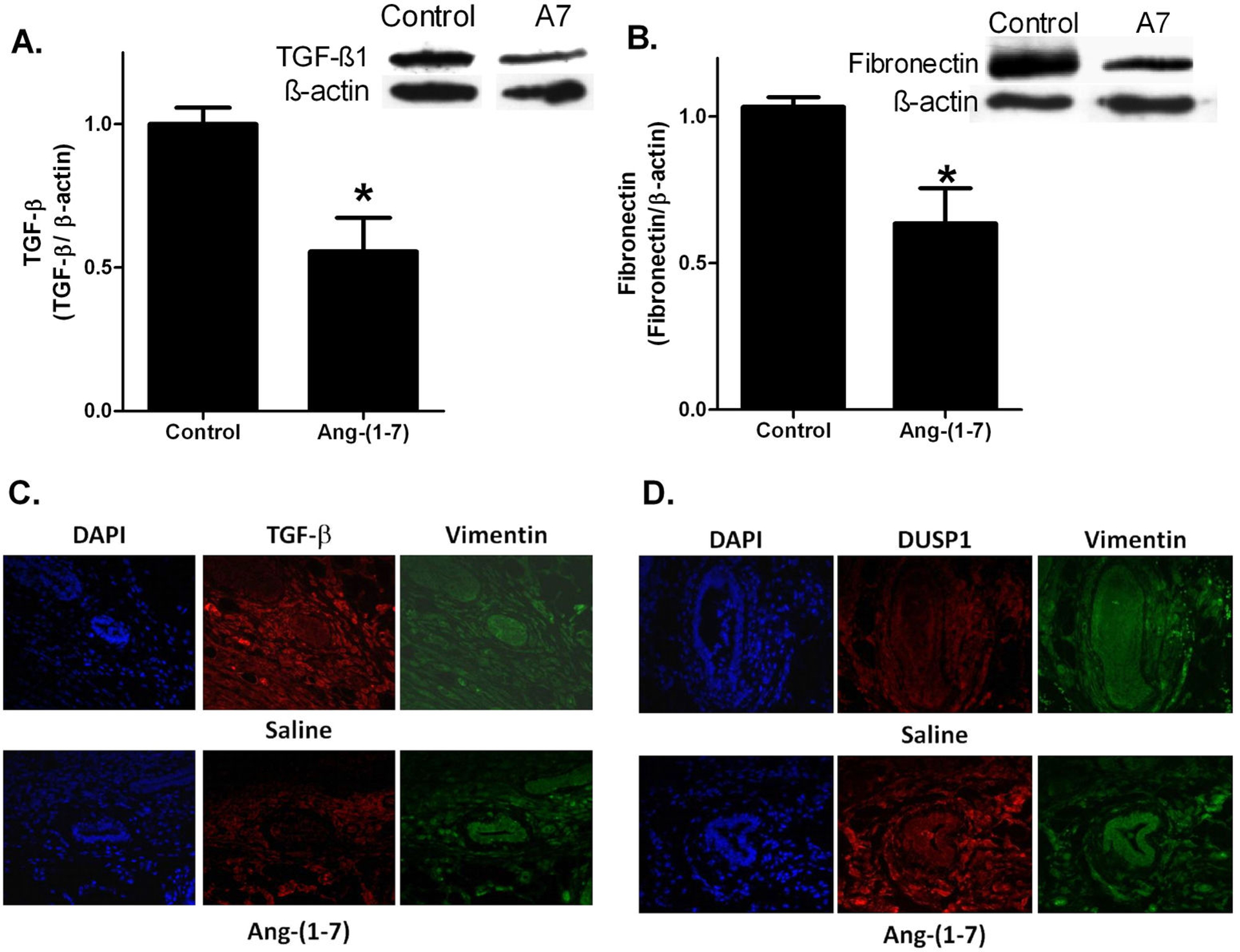
Effect of Ang-(1–7) on TGF-β, fibronectin, and DUSP1 in isolated tumor myofibroblasts and tumor sections. A: Isolated tumoral fibroblasts were serum starved overnight and incubated with 100 nM Ang-(1–7) [A7] for 24 h. TGF-β was assessed by Western blot hybridization and quantified as a function of β-actin. B: Isolated tumoral fibroblasts were serum starved overnight and treated with 100 nM Ang-(1–7) [A7] and 10 ng/mL TGF-β1 for 24 h. Fibronectin was assessed by Western blot hybridization and quantified as a function of β-actin. * denotes p<0.05; n=3–4. C: ZR-75–1 tumors from mice treated with saline or Ang-(1–7) were incubated with antibodies to DUSP1 and vimentin and counterstained with DAPI. Representative pictures are shown at x200 magnification. D: ZR-75–1 tumors from mice treated with saline or Ang-(1–7) were incubated with antibodies to TGF-β and vimentin and counterstained with DAPI. Representative pictures are shown at x200 magnification.
Sections from ZR-75–1 tumors in mice treated with saline or Ang-(1-7) were incubated with antibodies to TGF-β1 (Figure 6, Panel C) or DUSP1 (Figure 6, Panel D) and vimentin. TGF-β1 immunoreactivity was reduced and DUSP1 immunoreactivity was increased in perivascular fibroblasts identified by positive immunoreactivity to the vimentin antibody and by vascular morphology in mice treated with Ang-(1-7) compared to saline, in agreement with studies in isolated tumor-associated fibroblasts.
DISCUSSION
The link between fibrosis and breast cancer is well established. Collagen I deposition in the breast leads to increased mammographic density, which correlates with breast cancer risk (25). Increased ECM remodeling and stiffening by collagen and fibronectin enhances tumor cell survival and proliferation (26). Moreover, the secretion of growth factors, cytokines, and pro-angiogenic peptides by myofibroblasts promotes breast tumor growth (5). The present study is the first to show that Ang-(1-7) reduces the growth of human breast orthotopic tumors with a corresponding decrease in tumoral fibrosis. The reduction in interstitial and perivascular fibrosis was associated with a decrease in collagen I deposition. The heptapeptide reduced the serum-stimulated proliferation of isolated tumoral fibroblasts, suggesting that Ang-(1-7) inhibits myofibroblast growth to reduce tumor fibrosis. The reduction in fibroblast proliferation by Ang-(1-7) was associated with increased DUSP1 and a corresponding decrease in phosphorylated ERK1/2. The up-regulation of DUSP1 was blocked by the AT(1–7) receptor antagonist, indicating that DUSP1 induction by Ang-(1-7) was a receptor-mediated process. Ang-(1-7) also reduced production of active TGF-β and synthesis of fibronectin in the isolated tumoral fibroblasts. This ability of Ang-(1-7) to prevent production of TGF-β1 as well as decrease TGF-β1-stimulated ERK1/2 activation and fibronectin synthesis indicate that the heptapeptide is an antagonist of TGF-β1-mediated fibrosis in breast cancer. Of note, Ang-(1-7) also reduced the proliferation of breast tumor cell in vitro; the molecular mechanisms involved in the inhibition of breast cancer cell proliferation are detailed in a separate manuscript (in preparation).
Collagen I deposition was significantly decreased by Ang-(1-7), to reduce tumor fibrosis. However, the inhibition of collagen synthesis by the heptapeptide may also participate in the reduction in breast tumor growth. Collagen I forms a tissue scaffold to promote tumor growth and stimulates expression of genes associated with cellular metabolism, transcription and translation, including eukaryotic translation initiation factor 4B, glutamyl-prolyl-tRNA polymerase, and Poly (a) polymerase, playing a role in both tumor structure as well as growth and progression (27). Intratumoral collagen I modulates e-cadherin-mediated cell-to-cell contact, to increase tumor invasiveness and metastases (28). This suggests that the Ang-(1-7)-mediated decrease in collagen I production may also participate in the reduction in tumor proliferation as well as the inhibition of tumor metastases.
Fibronectin, another component of the ECM secreted by activated myofibroblasts, is involved in cell-matrix cell-cell adhesions, cell migration, and oncogenic transformation (29). The deposition of fibronectin in breast tumor stroma is positively correlated with tumor grade, size, and lymph node invasion (30). Fibronectin stimulates tumor cells through activation of integrin signaling to activate both focal adhesion kinases (FAKs) and MAP kinases, increasing cell proliferation, survival, and angiogenesis (31). We observed a 40% decrease in fibronectin in Ang-(1-7)-treated isolated tumoral fibroblasts, suggesting that the heptapeptide may attenuate ECM deposition, tumor invasion and proliferation by reducing fibronectin.
TGF-β binds to its receptors on fibroblasts to activate both MAP kinase and Smad signaling pathways and stimulate cell proliferation and fibrosis. Activation of the Smad signaling pathway by TGF-β results in ECM synthesis and deposition, tenascin-C production as well as its own production, creating an autocrine cycle of fibroblast activation and ECM deposition (5). TGF-β1 expression in breast cancer biopsies positively correlated with the rate of disease progression, independent of node status, tumor stage, age, and ER status, suggesting its role in tumor progression (32). Since Ang-(1-7) reduced TGF-β1 in isolated tumoral fibroblasts, the heptapeptide may attenuate myofibroblast activation by reducing TGF-β1 synthesis and signaling.
The exact molecular mechanism for the Ang-(1-7) mediated down-regulation of TGF-β1 is not known. However, we previously showed that Ang-(1-7) increased cAMP in vascular smooth muscle cells; (33) since an increase in cAMP inhibited TGF-β-stimulated collagen synthesis by ERK1/2 signaling in cardiac fibroblasts, (34) Ang-(1-7) may increase cAMP in tumoral fibroblasts to reduce TGF-β. In addition, the DUSP1 promoter contains a cAMP-responsive element (35), suggesting that Ang-(1-7) may increase cAMP in tumoral fibroblasts to up-regulate DUSP1, reduce MAP kinase activities, and inhibit fibrosis.
The amount of interstitial fibrosis was 3-fold higher in ZR-75–1 tumors than in BT-474 tumors, as assessed by picrosirius red staining in saline-treated mice. Several factors could account for this difference. BT-474 cells express the c-Met receptor, while ZR-75–1 cells do not. Since activation of the c-Met receptor by HGF reduces fibrosis, (36, 37) HGF signaling through the c-Met receptor may be responsible for the decreased total interstitial fibrosis observed in the BT-474 tumors (38, 39). On the other hand, HER2 signaling increases the expression of the Wilms’ tumor suppressor gene (WT1) product through activation of protein kinase B (Akt); (40) WT1 is implicated in tumor suppression and progression as well as inhibition of TGF-β signaling, which may reduce interstitial fibrosis (41, 42). The over-expression of HER2 in BT-474 cells and increased WT1 signaling could also contribute to reduced interstitial fibrosis. Further investigation is warranted to determine the molecular mechanism for the differences in interstitial fibrosis in the ZR-75–1 and BT-474 tumors.
We observed a significant reduction in perivascular fibrosis in BT-474 tumors from Ang-(1-7)-medicated mice when compared to tumors from saline-treated mice. It was not possible to quantify perivascular fibrosis in the ZR-75–1 tumors due to pervasive interstitial fibrosis. The heptapeptide-mediated reduction in perivascular fibrosis in BT-474 tumors may lead to an overall decrease in the tumoral interstitial fluid pressure. High interstitial fluid pressure, found in breast tumors as well as other types of tumors, is associated with vessel leakiness, lymph vessel abnormalities, and perivascular fibrosis, leading to matrix rigidity and fibroblast contractility and resulting in increased fiber tension (43, 44). This results in decreased transcapillary transport, limiting chemotherapeutic drug delivery to the tumor. These results suggest that Ang-(1-7) may enhance the delivery of chemotherapeutic agents when administered in combination with other therapeutic drugs (43).
The inhibition of tumor fibrosis by Ang-(1-7) is supported by previous studies demonstrating the anti-fibrotic effect of Ang-(1-7) in cardiac and renal cells and tissues. Ang-(1-7) infusion prevented cardiac fibrosis in deoxycorticosterone acetate salt-induced hypertension with a significant decrease in left ventricular wall fibrosis and reduced perivascular fibrosis, (45) in agreement with studies showing that the heptapeptide reduced collagen formation and TGF-β in rat cardiac fibroblasts (46). Studies by our group showed that Ang-(1-7) infusion reduced cardiac fibrosis in Ang II-treated rats, further illustrating the role of Ang-(1-7) as an anti-fibrotic agent (47). Mice with ablated mas, the Ang-(1-7) receptor, have impaired cardiac function with increased cardiac collagen I, collagen III, and fibronectin deposition, (48) while mas deletion increased collagen III, collagen IV, and fibronectin in the renal cortex and medulla (49). Our results demonstrate that Ang-(1–7), through activation of mas, also inhibits tumoral fibrosis.
In a recently reported Phase I clinical trial assessing Ang-(1-7) as a chemotherapeutic agent, we showed that the heptapeptide reduced plasma placental growth factor (PlGF) in patients with clinical benefit, (50) in agreement with our preclinical studies demonstrating that Ang-(1-7) inhibits angiogenesis (16). Since activated myofibroblasts secrete cytokines which stimulate blood vessel formation, (5) Ang-(1-7) may reduce angiogenesis by inhibiting the growth of tumor-associated fibroblasts. Taken together, these results suggest that Ang-(1-7) has pleiotropic effects on the tumor microenvironment, to decrease tumor fibrosis, inhibit angiogenesis, and reduce tumor growth.
Acknowledgements
We thank L. Tenille Howard, R. Lanning, and M. Landrum for technical assistance.
Grant Support
This work was supported in part by Grant HL-51952 and HL-56973 from the National Institutes of Health as well as a grant from the Susan G. Komen Breast Cancer Research Foundation (PEG and EAT). The authors gratefully acknowledge support provided by Unifi, Inc., Greensboro, NC, Golfers Against Cancer, Greensboro, NC, and Farley-Hudson Foundation, Jacksonville, NC. KLC was supported by a Department of Defense Breast Cancer Research Program Predoctoral Traineeship.
Footnotes
Disclosure of Potential Conflict of Interest
Drs. Gallagher and Tallant hold a patent for the treatment of cancer with Ang-(1-7).
REFERENCES
- (1).Sheffer Y, Leon O, Pinthus JH, Nagler A, Mor Y, Genin O, et al. Inhibition of fibroblast to myofibroblast transition by halofuginone contributes to the chemotherapy-mediated antitumoral effect. Mol Cancer Ther 2007;6:570–7. [DOI] [PubMed] [Google Scholar]
- (2).Anton K, Glod J. Targeting the tumor stroma in cancer therapy. Curr Pharm Biotechnol 2009;10:185–91. [DOI] [PubMed] [Google Scholar]
- (3).Sappino AP, Skalli O, Jackson B, Schurch W, Gabbiani G. Smooth-muscle differentiation in stromal cells of malignant and non-malignant breast tissues. Int J Cancer 1988;41:707–12. [DOI] [PubMed] [Google Scholar]
- (4).Cardone A, Tolino A, Zarcone R, Borruto CG, Tartaglia E. Prognostic value of desmoplastic reaction and lymphocytic infiltration in the management of breast cancer. Panminerva Med 1997;39:174–7. [PubMed] [Google Scholar]
- (5).Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006;6:392–401. [DOI] [PubMed] [Google Scholar]
- (6).Hasebe T, Sasaki S, Imoto S, Ochiai A. Highly proliferative fibroblasts forming fibrotic focus govern metastasis of invasive ductal carcinoma of the breast. Mod Pathol 2001;14:325–37. [DOI] [PubMed] [Google Scholar]
- (7).De WO, Demetter P, Mareel M, Bracke M. Stromal myofibroblasts are drivers of invasive cancer growth. Int J Cancer 2008;123:2229–38. [DOI] [PubMed] [Google Scholar]
- (8).Micke P, Ostman A. Tumour-stroma interaction: cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer 2004;45 Suppl 2:S163–S175. [DOI] [PubMed] [Google Scholar]
- (9).Santos RA, Ferreira AJ, Simoes E Silva AC. Recent advances in the angiotensin-converting enzyme 2-angiotensin(1–7)-Mas axis. Exp Physiol 2008;93:519–27. [DOI] [PubMed] [Google Scholar]
- (10).Santos RA, Simoes E Silva AC, Maric C, Silva DM, Machado RP, de B,I, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A 2003;100:8258–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Freeman EJ, Chisolm GM, Ferrario CM, Tallant EA. Angiotensin-(1-7) inhibits vascular smooth muscle cell growth. Hypertension 1996;28:104–8. [DOI] [PubMed] [Google Scholar]
- (12).Strawn WB, Ferrario CM, Tallant EA. Angiotensin-(1-7) reduces smooth muscle growth after vascular injury. Hypertension 1999;33:207–11. [DOI] [PubMed] [Google Scholar]
- (13).Lever AF, Hole DJ, Gillis CR, McCallum IR, McInnes GT, MacKinnon PL, et al. Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer? Lancet 1998;352:179–84. [DOI] [PubMed] [Google Scholar]
- (14).Gallagher PE, Tallant EA. Inhibition of human lung cancer cell growth by angiotensin-(1-7). Carcinogenesis 2004;25:2045–52. [DOI] [PubMed] [Google Scholar]
- (15).Menon J, Soto-Pantoja DR, Callahan MF, Cline JM, Ferrario CM, Tallant EA, et al. Angiotensin-(1-7) inhibits growth of human lung adenocarcinoma xenografts in nude mice through a reduction in cyclooxygenase-2. Cancer Res 2007;67:2809–15. [DOI] [PubMed] [Google Scholar]
- (16).Soto-Pantoja DR, Menon J, Gallagher PE, Tallant EA. Angiotensin-(1-7) inhibits tumor angiogenesis in human lung cancer xenografts with a reduction in vascular endothelial growth factor. Mol Cancer Ther 2009;8:1676–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Moran AL, Nelson SA, Landisch RM, Warren GL, Lowe DA. Estradiol replacement reverses ovariectomy-induced muscle contractile and myosin dysfunction in mature female mice. J Appl Physiol 2007;102:1387–93. [DOI] [PubMed] [Google Scholar]
- (18).Nelson JF, Felicio LS, Osterburg HH, Finch CE. Altered profiles of estradiol and progesterone associated with prolonged estrous cycles and persistent vaginal cornification in aging C57BL/6J mice. Biol Reprod 1981;24:784–94. [DOI] [PubMed] [Google Scholar]
- (19).Arpino G, Gutierrez C, Weiss H, Rimawi M, Massarweh S, Bharwani L, et al. Treatment of human epidermal growth factor receptor 2-overexpressing breast cancer xenografts with multiagent HER-targeted therapy. J Natl Cancer Inst 2007;99:694–705. [DOI] [PubMed] [Google Scholar]
- (20).Couillard S, Gutman M, Labrie C, Belanger A, Candas B, Labrie F. Comparison of the effects of the antiestrogens EM-800 and tamoxifen on the growth of human breast ZR-75–1 cancer xenografts in nude mice. Cancer Res 1998;58:60–4. [PubMed] [Google Scholar]
- (21).Young M, Fullerton M, Dilley R, Funder J. Mineralocorticoids, hypertension, and cardiac fibrosis. J Clin Invest 1994;93:2578–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Floyd HS, Farnsworth CL, Kock ND, Mizesko MC, Little JL, Dance ST, et al. Conditional expression of the mutant Ki-rasG12C allele results in formation of benign lung adenomas: development of a novel mouse lung tumor model. Carcinogenesis 2005;26:2196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Tallant EA, Ferrario CM, Gallagher PE. Angiotensin-(1-7) inhibits growth of cardiac myocytes through activation of the mas receptor. Am J Physiol Heart Circ Physiol 2005;289:H1560–H1566. [DOI] [PubMed] [Google Scholar]
- (24).Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 1951;193:265–75. [PubMed] [Google Scholar]
- (25).Martin LJ, Boyd NF. Mammographic density. Potential mechanisms of breast cancer risk associated with mammographic density: hypotheses based on epidemiological evidence. Breast Cancer Res 2008;10:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009;139:891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kiefer J, Alexander A, Farach-Carson MC. Type I collagen-mediated changes in gene expression and function of prostate cancer cells. Cancer Treat Res 2004;118:101–24. [DOI] [PubMed] [Google Scholar]
- (28).Koenig A, Mueller C, Hasel C, Adler G, Menke A. Collagen type I induces disruption of E-cadherin-mediated cell-cell contacts and promotes proliferation of pancreatic carcinoma cells. Cancer Res 2006;66:4662–71. [DOI] [PubMed] [Google Scholar]
- (29).Gould VE, Koukoulis GK, Virtanen I. Extracellular matrix proteins and their receptors in the normal, hyperplastic and neoplastic breast. Cell Differ Dev 1990;32:409–16. [DOI] [PubMed] [Google Scholar]
- (30).Ioachim E, Charchanti A, Briasoulis E, Karavasilis V, Tsanou H, Arvanitis DL, et al. Immunohistochemical expression of extracellular matrix components tenascin, fibronectin, collagen type IV and laminin in breast cancer: their prognostic value and role in tumour invasion and progression. Eur J Cancer 2002;38:2362–70. [DOI] [PubMed] [Google Scholar]
- (31).Meng XN, Jin Y, Yu Y, Bai J, Liu GY, Zhu J, et al. Characterisation of fibronectin-mediated FAK signalling pathways in lung cancer cell migration and invasion. Br J Cancer 2009;101:327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Gorsch SM, Memoli VA, Stukel TA, Gold LI, Arrick BA. Immunohistochemical staining for transforming growth factor beta 1 associates with disease progression in human breast cancer. Cancer Res 1992;52:6949–52. [PubMed] [Google Scholar]
- (33).Tallant EA, Clark MA. Molecular mechanisms of inhibition of vascular growth by angiotensin-(1-7). Hypertension 2003;42:574–9. [DOI] [PubMed] [Google Scholar]
- (34).Liu X, Sun SQ, Hassid A, Ostrom RS. cAMP inhibits transforming growth factor-beta-stimulated collagen synthesis via inhibition of extracellular signal-regulated kinase 1/2 and Smad signaling in cardiac fibroblasts. Mol Pharmacol 2006;70:1992–2003. [DOI] [PubMed] [Google Scholar]
- (35).Boutros T, Chevet E, Metrakos P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol Rev 2008;60:261–310. [DOI] [PubMed] [Google Scholar]
- (36).Mizuno S, Matsumoto K, Nakamura T. HGF as a renotrophic and anti-fibrotic regulator in chronic renal disease. Front Biosci 2008;13:7072–86. [DOI] [PubMed] [Google Scholar]
- (37).Schievenbusch S, Strack I, Scheffler M, Wennhold K, Maurer J, Nischt R, et al. Profiling of anti-fibrotic signaling by hepatocyte growth factor in renal fibroblasts. Biochem Biophys Res Commun 2009;385:55–61. [DOI] [PubMed] [Google Scholar]
- (38).Lee WY, Chen HH, Chow NH, Su WC, Lin PW, Guo HR. Prognostic significance of co-expression of RON and MET receptors in node-negative breast cancer patients. Clin Cancer Res 2005;11:2222–8. [DOI] [PubMed] [Google Scholar]
- (39).Shattuck DL, Miller JK, Carraway KL, III, Sweeney C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res 2008;68:1471–7. [DOI] [PubMed] [Google Scholar]
- (40).Tuna M, Chavez-Reyes A, Tari AM. HER2/neu increases the expression of Wilms’ Tumor 1 (WT1) protein to stimulate S-phase proliferation and inhibit apoptosis in breast cancer cells. Oncogene 2005;24:1648–52. [DOI] [PubMed] [Google Scholar]
- (41).Dey BR, Sukhatme VP, Roberts AB, Sporn MB, Rauscher FJ, III, Kim SJ. Repression of the transforming growth factor-beta 1 gene by the Wilms’ tumor suppressor WT1 gene product. Mol Endocrinol 1994;8:595–602. [DOI] [PubMed] [Google Scholar]
- (42).Loeb DM, Sukumar S. The role of WT1 in oncogenesis: tumor suppressor or oncogene? Int J Hematol 2002;76:117–26. [DOI] [PubMed] [Google Scholar]
- (43).Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure - an obstacle in cancer therapy. Nat Rev Cancer 2004;4:806–13. [DOI] [PubMed] [Google Scholar]
- (44).Radisky DC, Kenny PA, Bissell MJ. Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem 2007;101:830–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Grobe JL, Mecca AP, Mao H, Katovich MJ. Chronic angiotensin-(1-7) prevents cardiac fibrosis in DOCA-salt model of hypertension. Am J Physiol Heart Circ Physiol 2006;290:H2417–H2423. [DOI] [PubMed] [Google Scholar]
- (46).Iwata M, Cowling RT, Gurantz D, Moore C, Zhang S, Yuan JX, et al. Angiotensin-(1-7) binds to specific receptors on cardiac fibroblasts to initiate antifibrotic and antitrophic effects. Am J Physiol Heart Circ Physiol 2005;289:H2356–H2363. [DOI] [PubMed] [Google Scholar]
- (47).McCollum LT, Gallagher E, Tallant EA. Angiotensin-(1-7) Attenuates Cardiac Fibrosis and Vascular Hypertrophy in a Model of Angiotensin II-Dependent Hypertension. Hypertension 2008;52:e34. [Google Scholar]
- (48).Santos RA, Castro CH, Gava E, Pinheiro SV, Almeida AP, Paula RD, et al. Impairment of in vitro and in vivo heart function in angiotensin-(1-7) receptor MAS knockout mice. Hypertension 2006;47:996–1002. [DOI] [PubMed] [Google Scholar]
- (49).Pinheiro SV, Ferreira AJ, Kitten GT, da Silveira KD, da Silva DA, Santos SH, et al. Genetic deletion of the angiotensin-(1-7) receptor Mas leads to glomerular hyperfiltration and microalbuminuria. Kidney Int 2009;75:1184–93. [DOI] [PubMed] [Google Scholar]
- (50).Petty WJ, Miller AA, McCoy TP, Gallagher PE, Tallant EA, Torti FM. Phase I and pharmacokinetic study of angiotensin-(1-7), an endogenous antiangiogenic hormone. Clin Cancer Res 2009;15:7398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]