

Abstract
KCNT1 encodes the sodium-activated potassium channel KNa1.1, expressed preferentially in the frontal cortex, hippocampus, cerebellum, and brainstem. Pathogenic missense variants in KCNT1 are associated with intractable epilepsy, namely epilepsy of infancy with migrating focal seizures (EIMFS), and sleep-related hypermotor epilepsy (SHE). In vitro studies of pathogenic KCNT1 variants support predominantly a gain-of-function molecular mechanism, but how these variants behave in a neuron or ultimately drive formation of an epileptogenic circuit is an important and timely question. Using CRISPR/Cas9 gene editing, we introduced a gain-of-function variant into the endogenous mouse Kcnt1 gene. Compared to wild-type (WT) littermates, heterozygous and homozygous knock-in mice displayed greater seizure susceptibility to the chemoconvulsants kainate and pentylenetetrazole (PTZ), but not to flurothyl. Using acute slice electrophysiology in heterozygous and homozygous Kcnt1 knock-in and WT littermates, we demonstrated that CA1 hippocampal pyramidal neurons exhibit greater amplitude of miniature inhibitory postsynaptic currents in mutant mice with no difference in frequency, suggesting greater inhibitory tone associated with the Kcnt1 mutation. To address alterations in GABAergic signaling, we bred Kcnt1 knock-in mice to a parvalbumin-tdTomato reporter line, and found that parvalbumin-expressing (PV+) interneurons failed to fire repetitively with large amplitude current injections and were more prone to depolarization block. These alterations in firing can be recapitulated by direct application of the KNa1.1 channel activator loxapine in WT but are occluded in knock-in littermates, supporting a direct channel gain-of-function mechanism. Taken together, these results suggest that KNa1.1 gain-of-function dampens interneuron excitability to a greater extent than it impacts pyramidal neuron excitability, driving seizure susceptibility in a mouse model of KCNT1-associated epilepsy.
Keywords: Epilepsy, Potassium channel, KCNT1, Interneuron, Mouse
1. Introduction
KCNT1 encodes the sodium-activated potassium channel KNa1.1 (Yuan et al., 2003). Early work describing KNa1.1 in neurons struggled to identify whether the channel could contribute to cellular excitability given the necessity of supraphysiologic intracellular sodium concentrations for channel activation. However, it is now thought that sub-cellular localization of the channel near microdomains of elevated sodium, maintained by voltage-gated sodium channels and the sodium-potassium pump, allows KNa1.1 to impact neuronal excitability as a delayed rectifier conductance, contributing to the resting membrane potential, afterhyperpolarization current, and action potential threshold (Gray and Johnston, 2021; Wallen et al., 2007). In particular, the frequency of action potentials during high-frequency stimulation is fine-tuned by the level of KNa1.1 activity (Yang et al., 2006).
Since the first reported association with epilepsy pathogenesis in 2012 (Barcia et al., 2012), pathogenic variants in KCNT1 are increasingly reported with a broad phenotypic spectrum from early-onset developmental and epileptic encephalopathies including epilepsy of infancy with migrating focal seizures (EIMFS) to later-onset focal epilepsy and sleep-related hypermotor epilepsy (SHE) (Borlot et al., 2020; Heron et al., 2012; Ishii et al., 2013; Kingwell, 2012; Lim et al., 2016; McTague et al., 2013; McTague et al., 2018; Ohba et al., 2015).
Functional studies performed in heterologous expression systems have demonstrated for nearly all pathogenic variants (with the exception of p.Ile924Phe (Evely et al., 2017; Vanderver et al., 2014) an enhanced potassium conductance mediated by larger current amplitudes, more hyperpolarized voltage dependence of activation, and more rapid activation kinetics compared to WT channels (Kim et al., 2014; McTague et al., 2018; Milligan et al., 2014; Tang et al., 2016). Yet, our understanding of how this enhanced potassium channel function serves as a circuit-level substrate for epileptogenesis is incomplete. The first described Kcnt1 knock-in mouse model (p.R455H, homologous to the recurrent human p.R474H variant) exhibits increased seizure susceptibility to PTZ and interictal discharges on EEG, but this study did not elucidate a cellular mechanism for these findings (Quraishi et al., 2020). A second Kcnt1 knock-in mouse model homologous to the human p. Y796H variant similarly has spontaneous tonic and generalized tonic-clonic seizures only in the homozygous state, attributed primarily to reduced excitability of ‘non-fast spiking’ GABAergic neurons’ and enhanced, homotypic connectivity in both excitatory and inhibitory neurons (Shore et al., 2020). To further explore the cell-type specific implications of pathogenic KNa1.1 gain-of-function, we investigated a novel Kcnt1 knock-in mouse and tested the hypothesis that channel gain-of-function dampens the excitability of PV+ interneurons in epileptogenic regions such as the hippocampus, leading to loss of inhibitory regulation and seizure susceptibility.
2. Materials and methods
2.1. Mice
All experiments were performed in accord with the Northwestern University Animal Care and Use Committee in compliance with the NIH Guide for the Care and Use of Laboratory Animals and consistent with the ARRIVE guidelines. PV-tdTomato BAC (#027395) and C57BL/6 J (#000664) mice were obtained from Jackson Laboratory. Mice were group-housed in a 14:10 h light-dark cycle. Food and water were provided ad libitum. Both male and female mice were used in this study, compared, and reported as a group if not significantly different.
2.2. KNa1.1 expression in CHO cells
As previously described (Gertler et al., 2019), a recombinant human KNa1.1 cDNA (RefSeq NM_020822.2) subcloned into a pCMV-IRES-eGFP reporter was transfected into CHO-K1 cells maintained in DMEM/F12 at <70% confluence. The L456F variant was introduced by site-directed mutagenesis using Q5 DNA polymerase (New England Biolabs, Ips-wich, MA) and verified with Sanger sequencing. Manual patch-clamp recording of cells with moderate GFP expression after a 48-h incubation period was performed with a K-based internal solution containing (in mM): 100 KCl, 40 NaCl, 5 HEPES-K, 5 EGTA-K in a standard artificial cerebrospinal fluid (aCSF) external solution containing (in mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2.0 CaCl2, 1.0 MgCl2, 25 NaHCO3, and 12.5 glucose, bubbled continuously with carbogen (95% O2 and 5% CO2). Whole-cell capacitance was measured in voltage clamp with a 10 mV hyperpolarizing step from a holding potential of −60 mV immediately following rupture with a 10 kHz Bessel filter, prior to measuring currents using a 2 s voltage step from −100 mV to +100 mV with a 1 kHz Bessel filter. Cells with an access resistance greater than 15 MΩ were discarded, and liquid junction potential was not corrected. Instantaneous current and steady state current are defined as peak currents during the first 100 msec and last 100 msec of the voltage step, respectively. Comparisons between WT and variant channels were made using a two-tailed unpaired t-test.
2.3. Generation of mutant Kcnt1 knock-in mice
A Cas9 guide RNA (GACCTCATTCCTGCTGCTG) was designed complementary to the coding strand of the mouse Kcnt1 gene (NM_175462.4) to promote a double-strand break in exon 14 (encoding the RCK1 domain) and high-fidelity, homology-directed repair of c.1309C > T to create the murine equivalent of human p.L456F (Kcnt1 p. L457F), which itself is not a known disease-causing variant. Primers spanning intron-exon boundaries outside the repair oligonucleotide (tctcctgacctcccggggaggcctctgggtccttagcca-cactcagcagctgctgctctgcagGATGGACAACGGA-GAGGCCTGCTTTATCTtcAGCAGCAGGAATGAGGTGGACCGCACAG; intron sequence shown as lower case) were used to recognize on-target insertions/deletions resulting from non-homologous end joining. A Cas9 targeting vector (Addgene #62988, based on pX459) (Ran et al., 2013) with the previously mentioned guide sequence inserted at the BbsI site was electroporated into ND7/23 cells to test targeting and cleavage efficiency. A transactivating RNA complexed to crRNA with target site homology was commercially obtained (Integrated DNA Technologies, Coralville, IA) and injected into super-ovulated C57BL/6 J zygotes. All potential founders were genotyped by PCR of tail DNA, with subsequent cloning into pCR4-Blunt (Invitrogen) and Sanger sequencing of subcloned amplicons from potential founders. Next-generation sequencing of founders was employed to further genotype and verify chimeric founders.
Three independent lines were outbred to C57BL/6 J mice from the Jackson laboratory. All N1 mice were similarly genotyped by direct Sanger sequencing of PCR products and verified by next-generation sequencing prior to outbreeding of two independent N1 founders with the Kcnt1-L437F knock-in allele. The target site was sequenced up to 1 kb in either direction to exclude the possibility of concatameric insertion. Expression of the Kcnt1 transcript was verified by real time-quantitative PCR (qPCR) using probes located 5′ and 3′ to the predicted gRNA binding site. Mice were backcrossed with C57BL/6 J for at least three generations prior to experimental use, and were routinely genotyped using digital droplet PCR or real time-qPCR to identify each allele. All mice were reared and housed with WT littermates.
2.4. Immunofluorescence
Six week-old PV-tdTomato transgenic mice were perfused with 4% paraformaldehyde (PFA), after which 40–60 μm sections were cut on a vibrating microtome (Leica). Sections were blocked with 10% Normal goat serum prior to overnight incubation at 4 °C with primary antibodies for mouse Kcnt1 (1:400, Neuromab #75–051) and PV (1:1000, Synaptic Systems #195–004). The Kcnt1 antibody used for these experiments is directed against the cytoplasmic C-terminus and was raised against an immunogen comprised of rat KNa1.1 amino acids 1168–1237, which is 98% identical to the mouse KNa1.1 in this region. Because Kcnt1 alternative splicing occurs with N-terminal exons, this C-terminal antibody is expected to bind to all isoforms present in permeabilized cells. Alexa-488 and 594-conjugated secondary antibodies (1:1000, Life Technologies) were incubated for 1 h at room temperature prior to PBS washing and mounting with Prolong antifade (Life Technologies). Images were captured on a confocal microscope (Fluoview FV1000; Olympus).
2.5. Fluorescence in situ hybridization
Fluorescence in situ hybridization was performed using the RNA-scope multiplex fluorescent system (Advanced Cell Diagnostics). Brains extracted from 8 to 10 week old WT and PV-tdTomato C57BL/6 J mice were rapidly embedded in OCT and snap frozen in a dry ice/ethanol bath prior to storage at −80 °C in airtight containers. Sections (10 μm) were cut on a cryostat (Leica) cleaned with RNase away and mounted prior to fixation with 4% PFA. The remainder of the protocol was per manufacturer’s instructions using a custom Kcnt1 antisense probe directed against the longest transcript variant, isoform 1 (NCBI RefSeq NM_175462.4), as well as a tdTomato probe, and a three-channel positive and three-channel negative control probe in sections from the same tissue block.
2.6. Behavioral phenotyping
Rotarod, zero maze, open field, and home cage analyses were performed on P42-P54 mice between 1:00 pm and 6:00 pm in a standard lit room. For rotarod testing, all mice were acclimated to the equipment for 5 min with no acceleration prior to three trials on an accelerating ramp program, spaced at least 15 min between trials. For the zero maze test, Limelight software (Actimetrics) was used to video record each trial, track the position of the mouse, and calculate distance traveled and relative position in the arena. Trials where mice jumped off the maze were excluded from the analysis. Open-field boxes (28 cm × 28 cm) were cleaned with 70% ethanol prior to experimentation and before subsequent tests. All mice were acclimated to the testing space with ambient white noise for 1 h prior to a 20 min trial, recorded and analyzed using EthoVision XT (Noldus).
2.7. Seizure susceptibility testing
Wild-type and knock-in littermates were tested at 3 and 6–8 weeks of age as noted by experimenters blinded to genotype. Separate cohorts were used to test susceptibility to seizures induced by the chemoconvulsant flurothyl (Bis(2,2,2-trifluoroethyl ether, Sigma-Aldrich, St. Louis, MO), which was quantified by measuring latency to first myoclonic jerk and to generalized convulsions with loss of posture, following introduction of liquid flurothyl by a syringe pump into a clear, plexiglass chamber at a rate of 20 μl/min. Kainate (Tocris) dissolved in sterile saline was injected intraperitoneally at 30 mg/kg; Pentylenetetrazole (PTZ) dissolved in sterile saline was injected intraperitoneally at 60 mg/kg. Kainate and PTZ drugs were prepared immediately prior to test injections at their final concentrations. Responses were videotaped and scored on a modified Racine scale (Van Erum et al., 2019) by an observer blinded to genotype.
2.8. Visualized recording in ex vivo brain slices
Mice aged postnatal day 42–60 were anesthetized with a ketamine-xylazine mixture and perfused transcardially with ice-cold aCSF containing (in mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2.0 CaCl2, 1.0 MgCl2, 25 NaHCO3, and 12.5 glucose, bubbled continuously with carbogen (95% O2 and 5% CO2). The brains were rapidly removed, glued to the stage of a vibrating microtome (Leica Instrument), and immersed in ice-cold aCSF. Coronal ex vivo slices containing the dorsal hippocampus were cut at a thickness of 300 μm and transferred to a holding chamber, where they were submerged in aCSF at 35 °C for 30 min, and returned to room temperature before recording.
Ex vivo slices were then transferred to a small volume (~0.5 ml) Delrin recording chamber that was mounted on a fixed-stage, upright microscope (Olympus). Neurons were visualized using differential interference contrast optics (Olympus), illuminated at 735 nm (Thorlabs), imaged with a 60× 1.0 NA water-immersion objective (Olympus) and a CCD camera (QImaging). PV+ neurons were identified by the presence of somatic tdTomato fluorescence examined under epifluorescence microscopy with a daylight (6500 K) LED (Thorlabs) and an appropriate filter cube (Semrock).
Recordings were made at room temperature (20–22 °C) with patch electrodes fabricated from capillary glass (Sutter Instruments) pulled on a Flaming-Brown puller (Sutter Instruments) and fire-polished with a microforge (Narishige) immediately before use. Pipette resistance was ~3–4 MΩ. Internal solution for voltage-clamp recordings of inhibitory postsynaptic currents contained (in mM): 120 CsCl, 10 Na2-phosphocreatine, 5 HEPES, 5 tetraethylammonium-Cl, 2 Mg2ATP, 1 QX314-Cl, 0.5 Na3GTP, 0.5 CaCl2, 0.25 EGTA, and 0.2% (WT/vol) biocytin, pH adjusted to 7.25–7.30 with CsOH. This internal solution had an osmolarity of ~290 mOsm. Somatic whole-cell patch-clamp recordings were obtained with a 700B amplifier (Molecular Devices). The signal for voltage-clamp recordings was filtered at 1 kHz and digitized at 10 kHz with a digitizer (Molecular Devices). Series resistance was measured but not compensated for, and data were discarded if series resistance increased by 20% during recordings. Data acquisition was performed using pClamp (Molecular Devices). All mIPSC recordings were made in the presence of TTX (1 μM), R-CPP (10 μM) and NBQX (5 μM) to prevent confounding spontaneous glutamatergic inputs; all recorded events were able to be blocked by addition of SR95531 (10 μM). Internal solution for current-clamp recordings contained (in mM): 135 KMeSO4, 5 KCl, 0.5 CaCl2, 5 HEPES-K, 5 EGTA-K, 10 Na2-phosphocreatine, 2 Mg2-ATP, 0.5 Na3GTP, 0.2% (w/v) biocytin without synaptic blockers present. pH was adjusted to 7.25 with KOH; osmolarity was adjusted to 290 mOsm. The liquid junction potential for this internal solution was ~6 mV and was not corrected. Off-line data analyses were performed with ClampFit (Molecular Devices), MATLAB (MathWorks), and Wdetecta (Marion-Poll and Tobin, 1991).
2.9. Drugs
For seizure susceptibility testing, flurothyl (Bis(2,2,2-trifluoroethyl ether) and pentylenetetrazole (PTZ) were obtained from Sigma-Aldrich, and kainate was obtained from Tocris. All drugs were prepared immediately prior to test injections at their final concentrations and shielded from light. For use in acute slice electrophysiology experiments, R-CPP and NBQX disodium salt were obtained from Tocris Bioscience. Na3GTP and TTX were from Roche and Alomone Laboratories, respectively. Drugs were dissolved as stock solutions in either water or DMSO and aliquoted and frozen at −30 °C before use. Each drug was diluted to the appropriate concentrations by adding to the perfusate immediately before the experiment. The final concentration of DMSO in the perfusate was 0.1%.
2.10. Statistical analyses
Statistical analyses were performed with Prism (GraphPad). Scatter dot plots are used for graphic representation (Krzywinski and Altman, 2014; Streit and Gehlenborg, 2014) where the central line represents the mean and error bars represent SEM. Sample size (n value) is defined by the number of observations (i.e. cells or mice). No statistical method was used to predetermine sample size. Comparisons for unrelated samples were performed using a one-way ANOVA with the Holm-Sidak post hoc correction test at a significance level (α) of 0.05. Unless <0.0001 or >0.99, exact P values (two-tailed) are reported. Differences in frequency-intensity (F–I) curves were determined using the Kolmogorov-Smirnov test.
3. Results
Pathogenic KCNT1-encoded KNa1.1 variants associated with epilepsy (including predominantly EIMFS and SHE) are clustered within the S5 transmembrane segment, RCK1 and RCK2 cytoplasmic domains of the channel protein. We directly compared previously reported KCNT1 variants classified as disease-causing in the Human Gene Mutation Database (HGMD) (Stenson et al., 2014) to population single-nucleotide variants contained within the genome aggregation database (gnomAD) (Lek et al., 2016) (Fig. 1A–C). Whereas gnomAD variants are distributed throughout the gene, disease-causing variants were disproportionately more common in the S5 segment (12% HGMD vs 1.6% gnomAD), RCK1 and RCK2 domains (RCK1: 24.5% HGMD vs 18.2% in gnomAD; RCK2: 36% in HGMD vs 13.8% in HGMD).
Fig. 1.
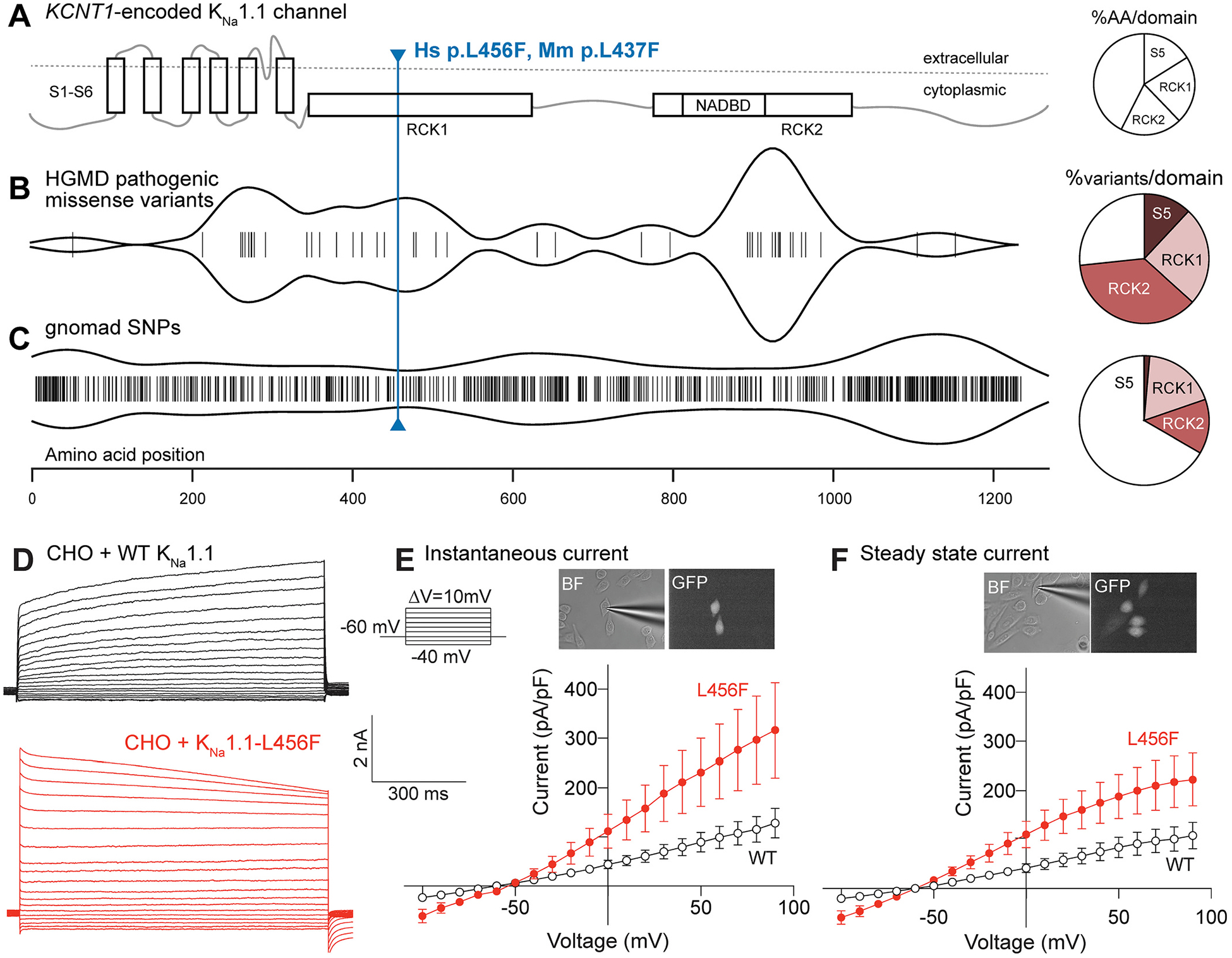
KCNT1-L456F is a novel, predicted pathogenic variant. (A) Schematic showing structural domains of the KCNT1-encoded KNa1.1 channel. Pie chart illustrates the percentage of amino acid residues within S5, RCK1, RCK2 and the rest of the protein (unlabeled). (B) HGMD pathogenic missense variants cluster most notably in the RCK2 domain of the KNa1.1 channel, as shown by violin plot. Pie chart shows the number of variants in the indicated domain, normalized by the number of amino acids in the same domain. (C) Genome aggregation database (gnomAD) shows single nucleotide polymorphisms (SNPs) for KNa1.1 that approach a random distribution, with some clustering in the N- and C- termini. Pie chart shows the number of variants in the indicate domain, normalized by the number of amino acids in the same domain. (D) Whole-cell currents recorded from a holding potential of −60 mV from CHO cells transfected with WT KNa1.1 (top, black traces) and L456F (bottom, red traces). (E) Current density-voltage response curve for an instantaneous current component (first 100 msec, L456F, red, n = 18; WT, black, n = 12) showing a significant difference between CHO cells expressing KCNT1-L456F (M = 327.4 pA/pF, SEM = 96.7 pA/pF) and those expressing KCNT1-WT (M = 125.4 pA/pF, SEM = 27.8 pA/pF) by two-tailed unpaired t-test analysis, where t(38) = 2.66, p = 0.011. Inset shows bright field (BF) and green fluorescent protein (GFP) images (60× magnification) of recorded CHO cells. Data shown are mean ± SEM. (F) Current density-voltage responses for steady-state current (last 100 msec, L456F, red, n = 18; WT, black, n = 12) showing a significant difference between CHO cells expressing KCNT1-L456F (M = 222.7 pA/pF, SEM = 53.7 pA/pF) and those expressing KCNT1-WT (M = 107.1 pA/pF, SEM = 24.8 pA/pF) by two-tailed unpaired t-test analysis, where t(38) = 2.26, p = 0.029. Inset shows 60× BF and GFP images of recorded CHO cells. Data shown are mean ± SEM. Differences in current kinetics were not secondary to differences in series resistance errors as there was no difference in access resistance between the two groups (WT: 14.7 ± 1.5 MΩ, n = 12; L456F: 11.6 ± 1.1 MΩ, n = 18, p = 0.09), and no correlation between access resistance and peak current density measured at 90 mV.
Based on these findings, we engineered the human missense KCNT1 variant, L456F, which represents a non-conservative amino acid substitution (SIFT4G, predicted damaging, score = 0) (Vaser et al., 2016) of a highly conserved leucine residue (Likelihood ratio test, predicted damaging, score = 0.2 × 10−5) (Chun and Fay, 2009) within RCK1. This variant is in close proximity to clusters of known pathogenic variants and is also predicted to be pathogenic by in silico variant classification algorithms (Fig. S1), but is not a known disease-associated variant. To determine the functional impact of the channel variant, we compared the current generated by heterologous expression of the consensus human KNa1.1 long isoform to that generated by the L456F variant (Fig. 1D–F). Similar to previously reported pathogenic missense variants (Ishii et al., 2013; Milligan et al., 2014; Rizzo et al., 2016), cells expressing L456F exhibited significantly larger amplitude currents than WT expressing cells across the entire voltage range upon channel activation (instantaneous current; p = 0.011) and during sustained depolarization (current measured at quasi-steady state; p = 0.029) as determined by unpaired t-test. These findings support human KCNT1-L456F as a gain-of-function variant.
3.1. Hippocampal expression of KNa1.1
To understand the specific cell types most likely to be impacted by a change in KNa1.1 channel function, we surveyed the literature delineating transcriptional and translational expression patterns of Kcnt1 mRNA and the KNa1.1 channel protein. Previous in situ hybridization studies suggested that Kcnt1 mRNA is robustly expressed in neurons throughout the CNS, including the cortex, hippocampus, and brainstem, whereas immunohistochemical studies confirmed its presence in brainstem nuclei, yet show sparse cellular positivity in hippocampus and frontal cortex (Rizzi et al., 2016). Consistent with an interneuron expression pattern, a knockout-validated KNa1.1 antibody identified sparse somatic labeling in the stratum lacunosum-moleculare of the hippocampus and the dentate gyrus (Bhattacharjee et al., 2002; Martinez-Espinosa et al., 2015). We additionally surveyed previously published single-cell RNA sequencing data for cortical or hippocampal mouse neurons and found that Kcnt1 expression appears robust in parvalbumin-positive (PV+) and somatostatin-positive (SOM+) interneuron subsets, near-absent in other interneuron subsets (e.g. vasoactive intestinal peptide-positive), and with intermediate expression in excitatory pyramidal neurons (Fig. S2) (Cembrowski et al., 2016; Ero et al., 2018).
In our experiments, immunohistochemistry and in situ hybridization demonstrated cell-type specific Kcnt1 expression. We performed in situ hybridization in transgenic mouse lines where tdTomato is expressed under the control of the parvalbumin (PV) or somatostatin (SOM) promoters. Gene-specific oligonucleotide probes for the long isoform of Kcnt1 were enriched in PV+ interneurons of the hippocampus with limited expression in SOM+ interneurons, and undetectable expression in the CA1-CA3 pyramidal layer (Fig. 2A–C). As Kcnt1 expression is highly expressed in the cerebellum in both granule cells and PV+ basket cells, we used the cerebellum as a positive control to demonstrate binding of both probes to each sagittal section. Using an anti- KNa1.1 cytoplasmic, C-terminal antibody, channel expression was readily identified in the CA2 region and subiculum, and noted to co-localize with PV expression throughout the hippocampus, though there are a minority of KNa1.1+ PV− cells in CA1–3 (presumably SOM+) and a large number of KNa1.1+ PV− neurons in the subiculum (Fig. 2D,E).
Fig. 2.
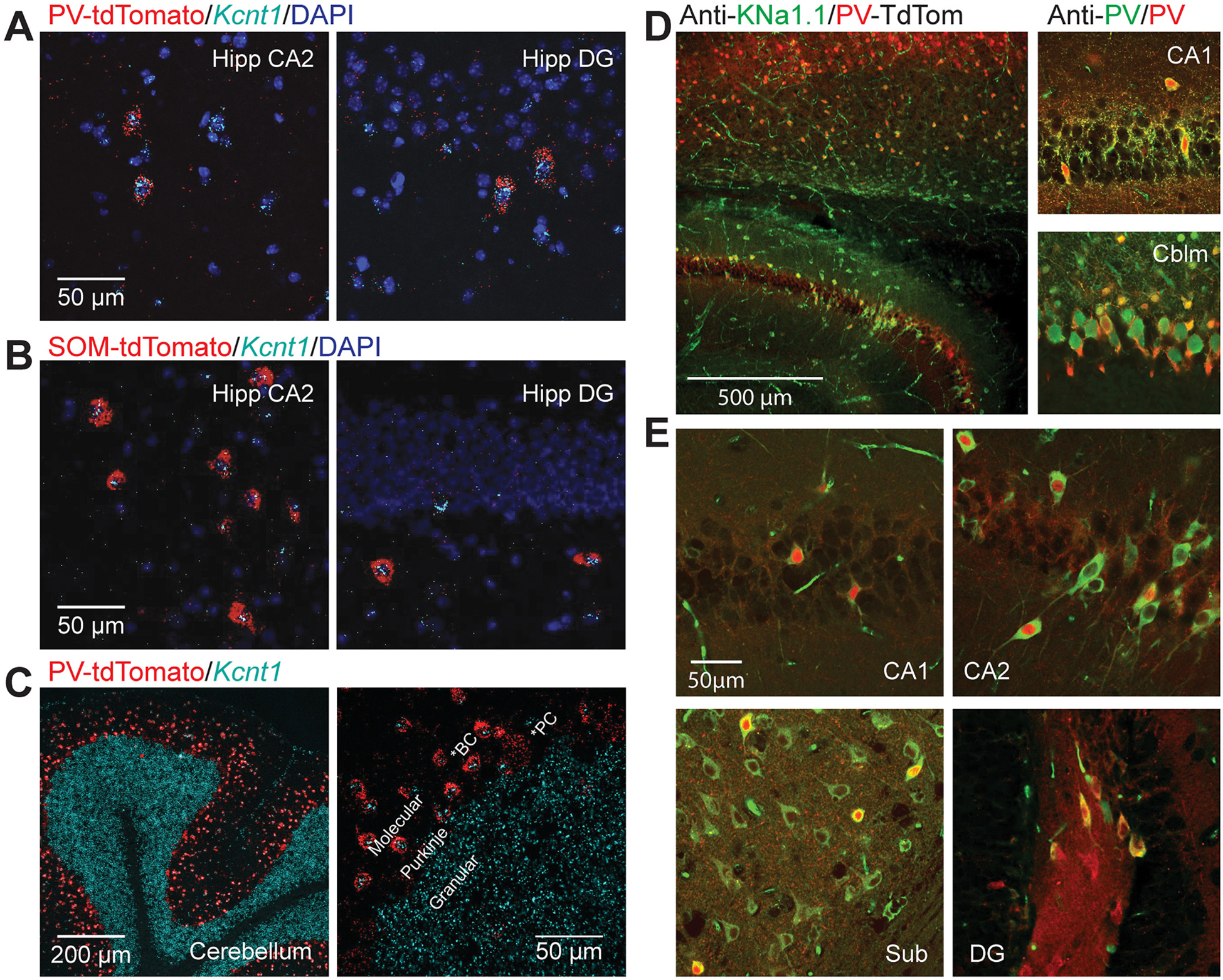
KCNT1 is predominantly expressed and translated in PV+ interneurons of the hippocampus. (A) In situ hybridization showing co-localization of tdTomato driven by the native PV promoter (red) and the mouse Kcnt1 probe (cyan) in CA2 and dentate gyrus. (B) In situ hybridization showing co-localization of tdTomato driven by the native SOM promoter (red) and the Kcnt1 probe (cyan) in CA2 and dentate gyrus. (C) Control in situ hybridization demonstrating correct binding of the Kcnt1 probe (cyan) in granule cells and tdTomato driven by the native PV promoter (red) in basket cells of the cerebellum. (D) Immunocytochemistry showing antibody labeling of KNa1.1 in a PV-tdTomato reporter mouse line, with (inset) verification of PV localization using a PV antibody in the hippocampus and cerebellum. (E) Immunocytochemistry showing co-labeling of PV in a subset of KNa1.1-labeled neurons in CA1, CA2, the subiculum, and the dentate gyrus.
3.2. Kcnt1-L437F mice exhibit seizure susceptibility
We used CRISPR/Cas9 gene editing to introduce the human KCNT1 variant p.L456F into the orthologous mouse gene (Kcnt1-L437F; Fig. S3A). Backcrossing of two independent founder lines of knock-in mice to the isogenic strain C57BL/6 J yielded two colonies without any genotype-dependent differences in birth rates or survival. Whole brain Kcnt1 transcript levels were not significantly different among wild-type (WT), heterozygous (KI/+) and homozygous (KI/KI) knock-in mutant mice (Fig. S3B,C).
Mice were tested in a battery of behavioral tasks to measure motor activity, coordination, anxiety, and fear, and showed no significant differences among genotypes (Fig. 3A–C). No readily observable, spontaneous seizures were evident in heterozygous or homozygous knock-in mice from birth through 12 weeks. To assess seizure susceptibility, we introduced animals to flurothyl, a GABAA receptor antagonist that provokes generalized and recurrent seizures after a quantifiable time latency as a model of seizure susceptibility (Baraban and Schwartzkroin, 1996); we also separately introduced the non-competitive GABA receptor antagonist PTZ (Mandhane et al., 2007). We then measured latency to first myoclonic jerk and generalized tonic-clonic seizure. Neither KI/+ nor KI/KI littermates showed a difference in susceptibility to flurothyl compared with WT littermates (Fig. S3D). By contrast, we observed a more prolonged seizure duration in KI/+ mice upon PTZ exposure, and a more severe clinical seizure correlate in KI/KI mice as evidenced by significant differences in the distribution of Racine scores (Fig. 3D). We also examined the response to injection of the glutamate receptor agonist kainate and observed that WT mice either had no response or exhibited brief focal seizures without loss of posture (Fig. 3E). By contrast, KI/+ mice exhibited frequent focal seizures with occasional progression to generalized seizure activity, whereas KI/KI mice invariably proceeded to status epilepticus and death. These data indicate that KI/+ and KI/KI mice have enhanced seizure susceptibility.
Fig. 3.
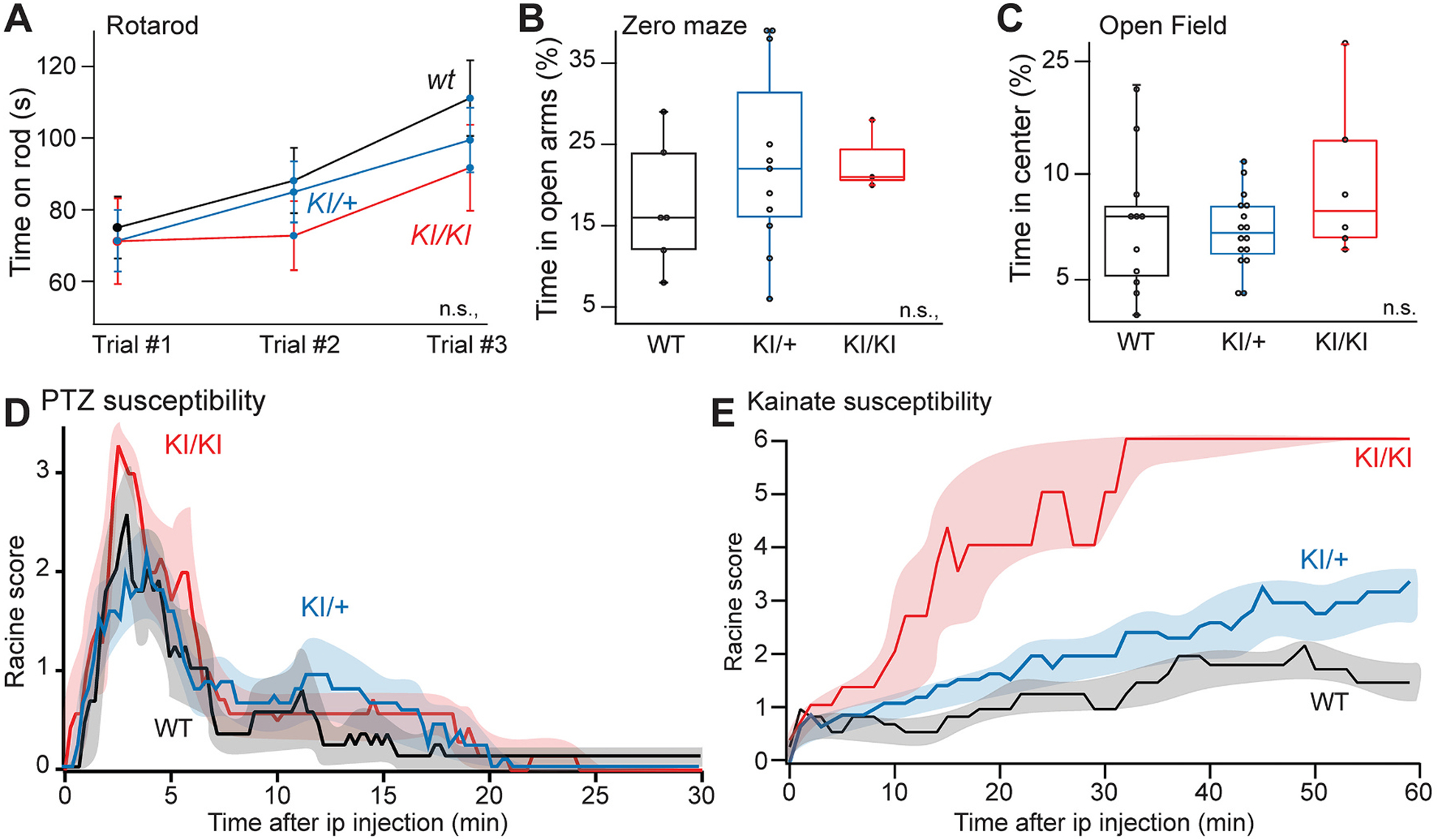
Enhanced seizure susceptibility in L437F knock-in mice. (A) Rotarod, (B) zero maze, and (C) open field behavioral testing was performed for each mouse line but did not reveal significant differences. (D) Seizure severity and duration following intraperitoneal injection of PTZ for WT, KI/+ and KI/KI littermates. Differences were significant by Kolmogorov-Smirnov test when either KI/+ or KI/KI were compared to WT mice (WT, n = 9 vs KI/+, n = 14: ks = 2.5, p < 0.0001; WT vs KI/KI, n = 7: ks = 3.0, p < 0.0001). (E) Seizure severity and duration following intraperitoneal injection of kainate (30 mg/kg) between WT and KI/+ (WT, n = 9 vs KI/+, n = 12: D = 0.72, ks = 3.9, p < 0.0001) and between WT and KI/KI (WT, n = 6 vs KI/KI, n = 6: D = 0.80, ks = 4.4, p < 0.0001).
3.3. Impact of Kcnt1-L437F on neuronal excitability
We investigated the impact of Kcnt1-L437F on neuronal excitability. As pyramidal neurons predominate in the CA1 hippocampal subfield and are heavily innervated by interneurons (Inan et al., 2013), we examined the intrinsic excitability of visually-identified neurons from the hippocampus by performing current-clamp recordings in WT, KI/+, and KI/KI littermates (Fig. 4A; Table S1). With depolarizing current steps, we did not observe significant differences in the firing activity to current intensity (F–I) relationship of either KI/+ or KI/KI neurons (Fig. 4B,C). The whole-cell capacitance of neurons of each genotype did not differ (Table S1). There was no statistical difference among neurons of each genotype with respect to rheobase current, action potential threshold, or maximum firing rate (Fig. 4D).
Fig. 4.
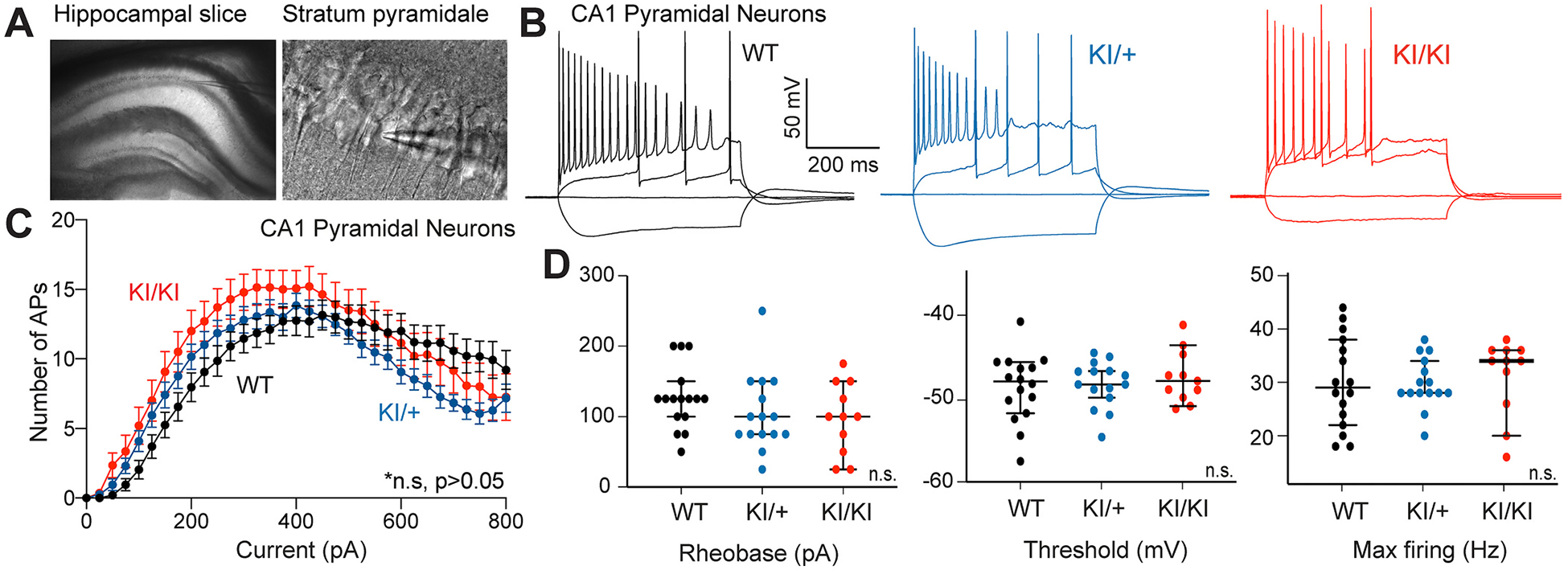
KNa1.1 gain-of-function dampens intrinsic excitability of CA1 pyramidal neurons. (A) Image (4× magnification) showing location of patch pipette used to record from acute hippocampal slice (left) and 60× image of recorded CA1 pyramidal neuron in stratum pyramidale (right). (B) Sample traces from whole cell current clamp recordings from CA1 pyramidal neurons in response to current step injections from WT (black), KI/+ (blue) and KI/KI (red) mice. Image (4× magnification) showing location of patch pipette used to record from an acute hippocampal slice (top) and 60× image of recorded CA1 pyramidal neuron in stratum pyramidale (bottom). (C) Number of spikes-injected current curves plotted from WT, KI/+ and KI/KI neurons. Plot shows slight shift to the left and reduction in number of spikes beyond 400 pA for KI/+ and KI//KI compared to WT (WT, n = 20; KI/+, n = 20; KI/KI, n = 15). The F–I relationships for heterozygous or the homozygous cells are not significantly different from WT (p = 0.12; p = 0.07, respectively; Kolmogorov-Smirnov test). (D) Rheobase current, threshold potential, and maximum (Max) firing of recorded cells from WT, KI/+ and KI/KI mice. Table S1 provides a summary of intrinsic electrophysiological properties of these neurons. (WT, n = 19; KI/+, n = 20; KI/KI, n = 14).
We next recorded miniature inhibitory postsynaptic currents (mIPSCs) in pyramidal neurons to quantify inhibitory input (Fig. 5A). CA1 pyramidal neurons were visually identified for electrophysiological recording using a cesium-based internal solution. Glutamatergic antagonists and TTX were used to inhibit glutamate transmission and action-potential dependent release, respectively. While there was no difference in the frequency of mIPSCs recorded between WT and KI/+ littermates (Fig. 5B), the mIPSC amplitude was significantly larger in KI/+ neurons (Fig. 5C).
Fig. 5.
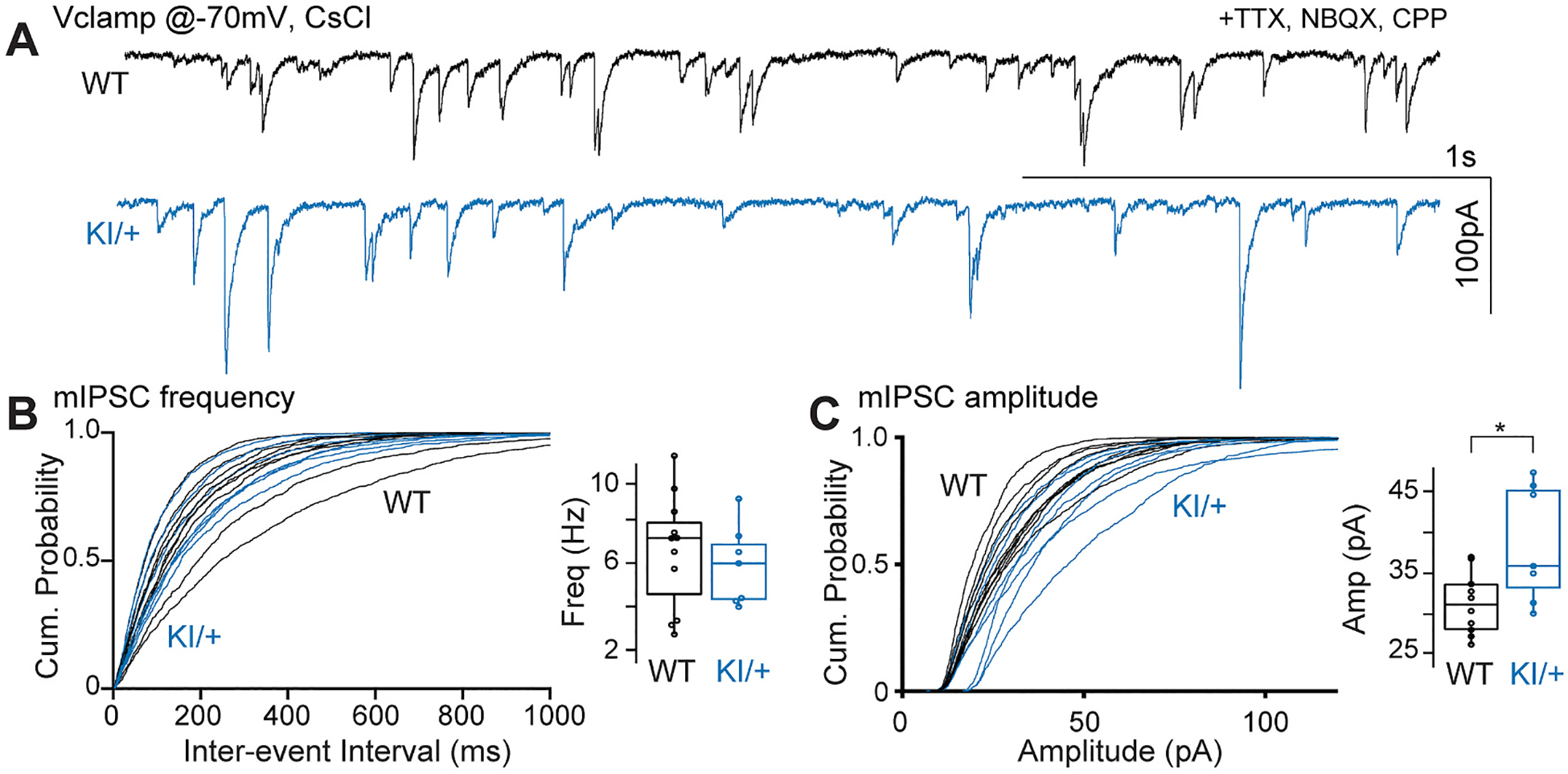
KNa1.1 gain-of-function increases mIPSC amplitude in CA1 pyramidal neurons. (A) Sample traces showing whole cell voltage clamp recordings of mIPSCs using a CsCl internal solution at −70 mV in the presence of TTX, CPP and NBQX from CA1 pyramidal neurons from WT and KI/+ acute hippocampal slices. Cumulative probability plots comparing frequency (B) and amplitude (C) of mIPSCs from WT and KI/+ mice. Insets in B and C show box plots comparing the two groups (WT, n = 12; KI/+, n = 7; * indicates p < 0.05).
Given the pattern of interneuron-predominant Kcnt1 and increased inhibitory neurotransmission without significant change in excitatory neuron excitability, we were motivated to investigate the intrinsic properties of interneurons. To specifically assay intrinsic properties of PV+ interneurons given their relatively higher Kcnt1 expression, we crossed Kcnt1-L437F mice with the PV-tdTomato reporter mouse line maintained on the same C57BL/6 J background and compared the electrophysiological properties of PV+ neurons identified by fluorescence in an acute slice between WT and KI/+ littermates (Fig. 6A,B). In the current clamp configuration, we observed a failure of KI/+ neurons to fire repetitively at high amplitude current injections due to depolarization block (Fig. 6B), and significantly lower firing of KI/+ neurons throughout the range of current inputs (Fig. 6C). However, there were no differences in average maximum firing frequency, threshold voltage, and rheobase current (Fig. 6D). There was no difference in the whole-cell capacitance to suggest either sampling bias for a different subset of PV+ interneurons or differences in morphology to explain these differences. These findings indicate a significant blunting of PV+ interneuron excitability in KI/+ mice.
Fig. 6.
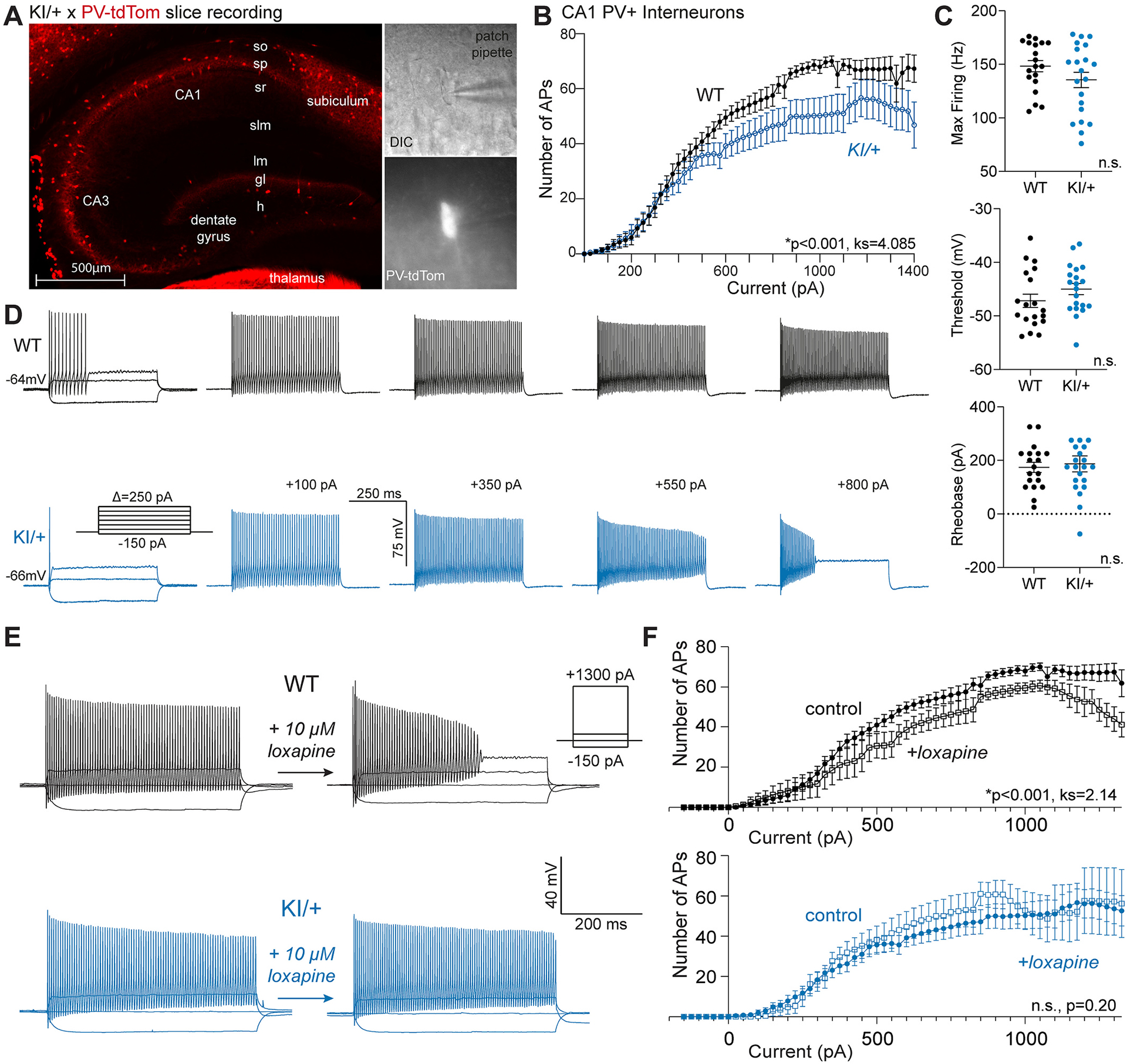
KNa1.1 gain-of-function dampens intrinsic excitability of CA1 PV+ interneurons. (A) Image showing PV+ interneurons (red) in a hippocampal slice from a KI/+ X PV-tdTomato mouse (left). Bright field image (60× magnification) of recorded PV+ interneuron (top, right) and tdTomato-positive image of the same cell (60×, bottom right). (B) Plot showing number of action potentials vs current injected comparing PV+ interneurons from different genotypes (WT, n = 19; KI/+, n = 21). The F–I relationships are significantly different (ks = 4.085, p < 0.001); Kolmogorov-Smirnov test). (C) Plots comparing maximum (Max) firing frequency, threshold potential, and rheobase current for PV+ interneurons between WT and KI/+ mice (WT, n = 19; KI/+, n = 21). (D) Current clamp recordings from single PV+ interneurons in acute hippocampal slices from WT (top, black) or KI/+ (bottom, blue) mice. Table S2 provides a summary of intrinsic electrophysiological properties of these neurons. Data shown are mean ± SEM. (E) Representative current clamp recordings from WT and KI/+ CA1 PV+ interneurons before and after exposure to 10 μM loxapine. Insets illustrate the voltage protocol and scale bars. (F) Comparisons of the number of action potentials evoked by varying injected current between control and loxapine treated CA1 PV+ interneurons from WT (control, n = 19; loxapine, n = 7) and KI/+ (control, n = 21; loxapine, n = 8) mice. Statistical differences between control and loxapine treated groups were determined by the Kolmogorov-Smirnov test and p-values are included in panels. Tables S1 and S2provide summaries of intrinsic electrophysiological properties of neurons measured in the absence and presence of loxapine.
3.4. Direct channel activation phenocopies genetic KNa1.1 gain-of-function
Mechanistically, we reasoned that altered intrinsic neuronal excitability was a direct result of KNa1.1 gain-of-function. We tested if exposure of neurons to loxapine, a KNa1.1 channel activator (Biton et al., 2012), was sufficient to replicate the effect of genetic KNa1.1 gain-of-function. We applied loxapine to acute hippocampal slices from WT, KI/+, and KI/KI PV-tdTomato mice. Loxapine (10 μM) impeded repetitive firing at higher current injection in WT and KI/+ pyramidal neurons (Fig. S4A,B; Table S1) but not KI/KI neurons (Fig. S4C). In PV+ interneurons from WT mice, acute loxapine exposure evoked depolarization block and lower action potential firings (Fig. 6E,F), which phenocopies behavior of PV+ neurons from KI/+ mice (Fig. 6B,D). In contrast, loxapine had no significant effect on action potential firing of PV+ interneurons from KI/+ mice (Fig. 6E,F; Table S2), which suggested that the Kcnt1-L437F variant occludes the effect of loxapine.
4. Discussion
KCNT1 was identified as an epilepsy-associated gene in 2012, and our understanding of the mechanisms responsible for KCNT1-associated epilepsy has progressed from a broadening of the clinical phenotype in individuals with pathogenic variants to heterologous channel expression demonstrating gain-of-function in KNa1.1 and initial characterization of mouse models. Our study expands upon previous work by investigating the neurophysiological impact of a novel gain-of-function variant in a genomic hotspot where previously reported pathogenic variants are clustered within a channel domain essential for sodium-gated pore opening.
Cohort studies of KCNT1-associated epilepsy have demonstrated enrichment of pathogenic variants within the S5 and RCK KNa1.1 channel domains, suggesting that the binding of sodium and conformational changes of the channel pore are central to its aberrant conductance (Hite and MacKinnon, 2017; Hite et al., 2015), in contrast to altered voltage sensor properties in other well-known potassium channelopathies (Niday and Tzingounis, 2018). In a heterologous expression system, the p.L456F variant we studied behaves similarly to other recurrent pathogenic variants, conducting a potassium current with an instantaneously active conductance of greater current amplitude than WT channels.
In our study of the novel Kcnt1-L437F mouse model, we did not see overt behavioral differences between WT and mutant littermates, but did observe a genotype-dependent response to specific proconvulsant compounds indicating enhanced seizure susceptibility. To pursue the cellular mechanisms responsible for enhanced seizure susceptibility, we used whole-cell patch clamp electrophysiology in acute brain slice preparations to demonstrate a key physiological function of KNa1.1 in sustaining high-frequency, repetitive firing. As fast-spiking PV+ interneurons are uniquely predisposed to depolarization block, we hypothesize that decreasing their maximum achievable firing rate leads to a circuit-wide reduction in inhibitory tone, and can thus be considered proconvulsant. These effects can be phenocopied in WT mice upon exposure to the KNa1.1 activator loxapine, but are occluded by heterozygous expression of the L437F variant in PV+ interneurons. We noticed that loxapine had little impact on pyramidal neuron excitability in KI/KI homozygous pyramidal neurons despite reduced firing in WT and KI/+ neurons; whether this effect is the result of changes in sodium channel expression or trafficking of a hyperexcitable KNa1.1 requires additional investigation. Differentiating direct channel effects from systems-level changes is critically important as development of novel therapeutic strategies to reduce channel expression move to preclinical stages.
There are limited data to quantify the impact of KNa1.1 channel activators on channels expressing pathogenic variants. Although activation of protein kinase C (PKC) with 2-O-tetradecanoylphorbol-13-acetate (TPA) has been demonstrated to increase KNa1.1 current amplitudes, heterologous expression of p.R409Q or p.A913T variants occludes this effect (Barcia et al., 2012). In contrast, application of the KNa1.1 activator bithionol does increase current amplitude of channels with pathogenic variants, however, the downstream effect of its binding, as measured by presumed dissociation of the protein Phactr1 from the membrane, was absent in channels with these same pathogenic variants (Fleming et al., 2016), suggesting altered channel activity. Consistent with a state of maximal activation in comparison to the WT channel, the KNa1.1 channel activator loxapine similarly has little impact on the p. L437F pathogenic variant in vitro (Gertler et al., 2019).
Other genetically engineered mice with phenotypes attributable to variant Kcnt1 expression have been reported recently (Quraishi et al., 2020; Shore et al., 2020). Kcnt1 knock-out mice exhibit a lower seizure susceptibility than WT mice, as well as deficits in spontaneous motor activity, the ability to learn and to extinguish a motor task, and an enhanced peripheral sensitivity to neuropathic pain (Martinez-Espinosa et al., 2015; Quraishi et al., 2020). In the Kcnt1-R455H mouse model (analogous to the recurrent human variant KCNT1-R474H), homozygous knock-in mice were stillborn while heterozygous knock-in mice showed interictal discharges and electrographic seizures on video EEG, and enhanced susceptibility to the proconvulsant compound PTZ (Quraishi et al., 2020); a mechanistic basis for these findings was not explored. In the Kcnt1-Y777H mouse model (corresponding to the recurrent human variant KCNT1-Y796H), heterozygous mice were not significantly different from WT mice in terms of spontaneous seizures detected on video EEG, prompting the use of homozygous mice throughout the study; as measured by widefield calcium imaging and in vivo electrocorticography, frontal seizures predominate (Shore et al., 2020). To explain these findings, cultured primary neurons were used to demonstrate reduced excitability of non-fast spiking interneurons whereas non-fast spiking layer 2/3 neurons recorded in acute slices were shown to have lower input resistances and increased rheobase currents contributing to reduced overall excitability. In comparison, our model shows a relatively mild impact of KNa1.1 gain-of-function compared to phenotypic human EIMFS or the Kcnt1-R455H mouse model, yet heterozygous and homozygous mice were more susceptible to PTZ and kainate. Similar susceptibility could not be demonstrated with flurothyl, a GABA-A receptor antagonist. Interestingly, an increase in amplitude of mIPSCs in heterozygous mice suggests an increase in compensatory postsynaptic GABA-A receptor expression.
A notable unifying feature of all mouse models reported thus far is the relatively mild impact of gain-of-function channel conductance in heterozygous mice as compared to children with KCNT1-associated EIMFS. Reasons for this include inherent limitations of gain-of-function mouse models and the contribution of unknown genetic modifiers in the human and mouse genomes. There may also be clues in unexplored areas such as KCNT1 alternative mRNA splicing and species-specific redundancy among potassium channels involved in membrane repolarization during sustained action potential firing. Despite these limitations, mouse models remain valuable for the acquisition of pre-clinical data as different avenues for precision medicine approaches to KCNT1-associated epilepsy appear on the horizon, including KNa1.1 channel blockers (Spitznagel et al., 2020) and antisense oligonucleotides, as has recently been introduced for other epileptic channelopathies (Han et al., 2020; Lenk et al., 2020).
5. Conclusions
Our work demonstrated that mice heterozygous or homozygous for a novel KNa1.1 gain-of-function mutation exhibit enhanced seizure susceptibility, and that parvalbumin-positive hippocampal interneurons, a predominant KNa1.1-expressing neuron subtype, exhibit blunted evoked action potential firing consistent with impaired inhibitory neurotransmission. Our findings implicate blunted excitability of a specific subset of interneurons as a pathophysiological mechanism responsible for a severe childhood genetic epilepsy.
Supplementary Material
Acknowledgments and funding sources.
This work was supported by NIH grants NS104237 (TSG) and NS108874 (ALG, JAK), a research grant from the Child Neurology Foundation and Pediatric Epilepsy Research Foundation (TSG), a gift from the Davee Foundation (ALG), and the Northwestern Transgenic and Targeted Mutagenesis Laboratory.
Declaration of Competing Interest
T.S.G. received consulting fees from Biogen. J.A.K. received research funding from Pfizer, Praxis, Ovid and Takeda, received consulting fees from Neurocycle Therapeutics, and received royalty income from GW Pharmaceuticals, Novartis, Stoke Therapeutics, StrideBio, Pfizer, Lund-beck, Bright Minds Biosciences, Xenon Pharmaceuticals, Noema Pharma, and Encoded Genomics. A.L.G. received research funding from Tevard Biosciences and Praxis Precision Medicines, and received consulting fees from Neurocrine Biosciences.
Footnotes
CRediT authorship contribution statement
Tracy S. Gertler: Conceptualization, Methodology, Formal analysis, Writing – original draft, Funding acquisition. Suraj Cherian: Methodology. Jean-Marc DeKeyser: Conceptualization, Methodology. Jennifer A. Kearney: Supervision, Writing – review & editing. Alfred L. George: Supervision, Writing – review & editing, Funding acquisition.
Appendix A. Supplementary data.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2022.105713.
References
- Baraban SC, Schwartzkroin PA, 1996. Flurothyl seizure susceptibility in rats following prenatal methylazoxymethanol treatment. Epilepsy Res 23, 189–194. [DOI] [PubMed] [Google Scholar]
- Barcia G, et al. , 2012. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat. Genet 44, 1255–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee A, et al. , 2002. Localization of the slack potassium channel in the rat central nervous system. J. Comp. Neurol 454, 241–254. [DOI] [PubMed] [Google Scholar]
- Biton B, et al. , 2012. The antipsychotic drug loxapine is an opener of the sodium-activated potassium channel slack (Slo2.2). J. Pharmacol. Exp. Ther 340, 706–715. [DOI] [PubMed] [Google Scholar]
- Borlot F, et al. , 2020. KCNT1-related epilepsy: an international multicenter cohort of 27 pediatric cases. Epilepsia. 61, 679–692. [DOI] [PubMed] [Google Scholar]
- Cembrowski MS, et al. , 2016. Hipposeq: a comprehensive RNA-seq database of gene expression in hippocampal principal neurons. Elife. 5, e14997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S, Fay JC, 2009. Identification of deleterious mutations within three human genomes. Genome Res 19, 1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ero C, et al. , 2018. A cell atlas for the mouse brain. Front. Neuroinform 12, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evely KM, et al. , 2017. The Phe932Ile mutation in KCNT1 channels associated with severe epilepsy, delayed myelination and leukoencephalopathy produces a loss-of-function channel phenotype. Neuroscience. 351, 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming MR, et al. , 2016. Stimulation of slack K(+) channels alters mass at the plasma membrane by triggering dissociation of a phosphatase-regulatory complex. Cell Rep 16, 2281–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler TS, et al. , 2019. Functional consequences of a KCNT1 variant associated with status dystonicus and early-onset infantile encephalopathy. Ann. Clin. Transl. Neurol 6, 1606–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray R, Johnston D, 2021. Sodium sensitivity of KNa channels in mouse CA1 neurons. J. Neurophysiol 125, 1690–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, et al. , 2020. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci. Transl. Med 12. [DOI] [PubMed] [Google Scholar]
- Heron SE, et al. , 2012. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet 44, 1188–1190. [DOI] [PubMed] [Google Scholar]
- Hite RK, MacKinnon R, 2017. Structural titration of Slo2.2, a Na(+)-dependent K(+). Channel. Cell 168, 390–399 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hite RK, et al. , 2015. Cryo-electron microscopy structure of the Slo2.2 Na(+)-activated K(+) channel. Nature. 527, 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inan M, et al. , 2013. Dense and overlapping innervation of pyramidal neurons by chandelier cells. J. Neurosci 33, 1907–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, et al. , 2013. A recurrent KCNT1 mutation in two sporadic cases with malignant migrating partial seizures in infancy. Gene 531, 467–471. [DOI] [PubMed] [Google Scholar]
- Kim GE, et al. , 2014. Human slack potassium channel mutations increase positive cooperativity between individual channels. Cell Rep 9, 1661–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingwell K, 2012. Genetics: mutations in potassium channel KCNT1-a novel driver of epilepsy pathogenesis. Nat. Rev. Neurol 8, 658. [DOI] [PubMed] [Google Scholar]
- Krzywinski M, Altman N, 2014. Visualizing samples with box plots. Nat. Methods 11, 119–120. [DOI] [PubMed] [Google Scholar]
- Lek M, et al. , 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenk GM, et al. , 2020. Scn8a antisense oligonucleotide is protective in mouse models of SCN8A encephalopathy and Dravet syndrome. Ann. Neurol 87, 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CX, et al. , 2016. KCNT1 mutations in seizure disorders: the phenotypic spectrum and functional effects. J. Med. Genet 53, 217–225. [DOI] [PubMed] [Google Scholar]
- Mandhane SN, et al. , 2007. Timed pentylenetetrazol infusion test: a comparative analysis with s.c.PTZ and MES models of anticonvulsant screening in mice. Seizure. 16, 636–644. [DOI] [PubMed] [Google Scholar]
- Marion-Poll F, Tobin TR, 1991. Software filter for detecting spikes superimposed on a fluctuating baseline. J. Neurosci. Methods 37, 1–6. [DOI] [PubMed] [Google Scholar]
- Martinez-Espinosa PL, et al. , 2015. Knockout of Slo2.2 enhances itch, abolishes KNa current, and increases action potential firing frequency in DRG neurons. Elife. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTague A, et al. , 2013. Migrating partial seizures of infancy: expansion of the electroclinical, radiological and pathological disease spectrum. Brain. 136, 1578–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTague A, et al. , 2018. Clinical and molecular characterization of KCNT1-related severe early-onset epilepsy. Neurology. 90, e55–e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan CJ, et al. , 2014. KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann. Neurol 75, 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niday Z, Tzingounis AV, 2018. Potassium Channel gain of function in epilepsy: an unresolved paradox. Neuroscientist. 24, 368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba C, et al. , 2015. De novo KCNT1 mutations in early-onset epileptic encephalopathy. Epilepsia. 56, e121–e128. [DOI] [PubMed] [Google Scholar]
- Quraishi IH, et al. , 2020. Impaired motor skill learning and altered seizure susceptibility in mice with loss or gain of function of the Kcnt1 gene encoding slack (KNa1.1) Na(+)-activated K(+) channels. Sci. Rep 10, 3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, et al. , 2013. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzi S, et al. , 2016. Differential distribution of the sodium-activated potassium channels slick and slack in mouse brain. J. Comp. Neurol 524, 2093–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo F, et al. , 2016. Characterization of two de novoKCNT1 mutations in children with malignant migrating partial seizures in infancy. Mol. Cell. Neurosci 72, 54–63. [DOI] [PubMed] [Google Scholar]
- Shore AN, et al. , 2020. Reduced GABAergic neuron excitability, altered synaptic connectivity, and seizures in a KCNT1 gain-of-function mouse model of childhood epilepsy. Cell Rep 33, 108303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitznagel BD, et al. , 2020. VU0606170, a selective slack channels inhibitor, decreases calcium oscillations in cultured cortical neurons. ACS Chem. Neurosci 11, 3658–3671. [DOI] [PubMed] [Google Scholar]
- Stenson PD, et al. , 2014. The human gene mutation database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet 133, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit M, Gehlenborg N, 2014. Bar charts and box plots. Nat. Methods 11, 117. [DOI] [PubMed] [Google Scholar]
- Tang QY, et al. , 2016. Epilepsy-related Slack Channel mutants Lead to channel over-activity by two different mechanisms. Cell Rep 14, 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Erum J, et al. , 2019. PTZ-induced seizures in mice require a revised Racine scale. Epilepsy Behav 95, 51–55. [DOI] [PubMed] [Google Scholar]
- Vanderver A, et al. , 2014. Identification of a novel de novo p.Phe932Ile KCNT1 mutation in a patient with leukoencephalopathy and severe epilepsy. Pediatr. Neurol 50, 112–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaser R, et al. , 2016. SIFT missense predictions for genomes. Nat. Protoc 11, 1–9. [DOI] [PubMed] [Google Scholar]
- Wallen P, et al. , 2007. Sodium-dependent potassium channels of a slack-like subtype contribute to the slow afterhyperpolarization in lamprey spinal neurons. J. Physiol 585, 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, et al. , 2006. Pharmacological activation and inhibition of slack (Slo2.2) channels. Neuropharmacology. 51, 896–906. [DOI] [PubMed] [Google Scholar]
- Yuan A, et al. , 2003. The sodium-activated potassium channel is encoded by a member of the Slo gene family. Neuron 37, 765–773. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.