

Abstract
Understanding the genetic effects on non-coding RNA (ncRNA) expression facilitates functional characterization of disease-associated genetic loci. Among several classes of ncRNAs, microRNAs (miRNAs) are key post-transcriptional gene regulators. Despite its biological importance, previous studies on the genetic architecture of miRNA expression focused mostly on the European individuals, underrepresented in other populations. Here, we mapped miRNA expression quantitative trait loci (miRNA-eQTL) for 343 miRNAs in 141 Japanese using small RNA sequencing and whole-genome sequencing, identifying 1275 cis-miRNA-eQTL variants for 40 miRNAs (false discovery rate < 0.2). Of these, 25 miRNAs having eQTL were unreported in the European studies, including 5 miRNAs with their lead variant monomorphic in the European populations, which demonstrates the value of miRNA-eQTL analysis in diverse ancestral populations. MiRNAs with eQTL effect showed allele-specific expression (ASE; e.g. miR-146a-3p), and ASE analysis further detected cis-regulatory variants not captured by the conventional miRNA-eQTL mapping (e.g. miR-933). We identified a copy number variation associated with miRNA expression (e.g. miR-570-3p, P = 7.2 × 10−6), which contributes to a more comprehensive landscape of miRNA-eQTLs. To elucidate a post-transcriptional modification in miRNAs, we created a catalog of miRNA-editing sites, including 10 canonical and 6 non-canonical sites. Finally, by integrating the miRNA-eQTLs and Japanese genome-wide association studies of 25 complex traits (mean n = 192 833), we conducted a transcriptome-wide association study, identifying miR-1908-5p as a potential mediator for adult height, colorectal cancer and type 2 diabetes (P < 9.1 × 10−5). Our study broadens the population diversity in ncRNA-eQTL studies and contributes to functional annotation of disease-associated loci found in non-European populations.
Graphical Abstract
Graphical Abstract.

Introduction
Functional characterization of genetic variants is an important challenge in elucidating the mechanisms underlying the genetics of complex diseases. Although genome-wide association studies (GWASs) have identified tens of thousands of disease-associated genetic loci, most of them are located on the non-coding regions and still remain to be functionally characterized. One of the promising approaches to tackle this challenge is identifying molecular quantitative trait loci (QTLs), where genetic loci are associated with intermediate molecular phenotypes, such as gene expression (eQTL), protein levels (pQTL) and DNA methylation (meQTL). Of these, eQTL mapping helps interpret disease-associated genetic variants with their effect on gene expression and prioritize the causal genes.
When compared with eQTL mapping of messenger RNA (mRNA), eQTL of non-coding RNA (ncRNA) has been understudied despite its importance. Among several classes of ncRNA, microRNA (miRNA), small ncRNA of 21–25 nucleotides, is known as a major post-transcriptional regulator of gene expression (1–3). MiRNAs are involved in the pathophysiology of various diseases, such as cancers (4–7) and immune-related diseases (8–11). MiRNA expressions are also heritable traits and exhibit significant associations with genetic variants [miRNA-eQTLs; (12–18)]. Previous studies reported miRNA-eQTLs associated with complex human traits (12,13). Although these lines of evidence firmly support the importance of miRNA-eQTLs in interpreting functionally uncharacterized disease-associated loci, previous studies were mainly conducted in individuals of European ancestry (12,14–18). In particular, the genetic drivers of miRNA expression variation in Asian populations are unknown. Therefore, comprehensive miRNA-eQTL studies in Asian populations are warranted.
Here, we report the first miRNA-eQTL mapping in the Asian population. We conducted small RNA-sequencing (sRNA-seq) of peripheral blood mononuclear cells (PBMCs) and whole-genome sequencing (WGS) of the 141 Japanese individuals, investigating the association between miRNA expressions and genetic variations. By leveraging the sequenced variants, we further performed (i) identification of the copy number variations (CNVs) that were associated with miRNA expression and (ii) detection of miRNA-editing events. Finally, by combining the miRNA expression data with the large-scale GWASs in Japanese individuals (mean n > 190 000), we performed a transcriptome-wide association study (TWAS) with diverse complex diseases and traits. Our results demonstrate the value of population-specific miRNA-eQTL analysis for functional characterization of disease-associated genetic loci and identification of the potential miRNAs mediating the disease biology.
Results
MiRNA-eQTLs in the Japanese population
We performed sRNA-seq on PBMCs from the 141 unrelated Japanese individuals, which allowed us to capture all types of the miRNA transcripts in principle. We quantified 343 autosomal mature miRNAs that were expressed in > 70 individuals. We used those miRNA expressions for subsequent miRNA-eQTL mapping. We performed WGS of the study participants and identified 12 171 854 autosomal genetic variants [11 396 461 single-nucleotide variants (SNVs) and 775 393 short insertions and deletions (indels)] after quality control. Of the 12 171 854 variants, we used the 7 170 825 variants with a minor allele frequency (MAF) ≥ 0.01 for the miRNA-eQTL mapping. We performed cis-miRNA-eQTL mapping using linear regression and permutation-based multiple testing correction. We defined the cis-window as 1 Mb up- and down-stream of the mature miRNA coding region. We defined ‘eMiRNAs’ as miRNAs with at least one significant eQTL variant at false discovery rate (FDR) < 0.2 (18). We report the number of cis-miRNA-eQTL variants as the number of all variant–eMiRNA pairs with FDR significance. We identified 1275 cis-miRNA-eQTL variants for 40 eMiRNAs at FDR < 0.2 (Fig. 1 and Table 1). Of these, 1011 cis-miRNA-eQTL variants for 25 miRNAs were significant at FDR < 0.1. The most significant association was between miR-146a-3p and rs2910164 (P = 2.4 × 10−16, explained variance = 38%). In agreement with previous studies on cis-eQTL (19–21), we observed strong enrichment of eQTL signals around transcription start sites (TSSs) when using the most significantly associated eQTL variant per eMiRNA (Fig. 2A) as well as when using all the eQTL variants retained after linkage disequilibrium (LD)-based clumping (r2 < 0.7; Supplementary Material, Fig. S1). PBMCs consist of several cell types, such as T/NK cells, B cells and monocytes, and each exhibits a distinct miRNA expression profile (22). To assess whether the variation in the cell type proportions in PBMCs significantly affects the miRNA-eQTL mapping results, we compared miRNA-eQTL effect estimates with and without adjustment for cell type proportions estimated by CIBERSORTx (23). We did not find apparent evidence that cell type compositions in PBMCs affected the miRNA-eQTLs (Supplementary Material, Fig. S2).
Figure 1.

Manhattan plot of cis-miRNA-eQTLs. P-values are shown for cis-windows of the 343 miRNAs examined. The 40 significant cis-miRNA-eQTLs are labeled and colored in pink. The eMiRNAs harboring genetic variants within the mature miRNA sequences are shown in green, all of which showed significant allele-specific expression (see Fig. 3). The eMiRNAs harboring genetic variants within the primary miRNA sequences but not the mature miRNA sequences are shown in blue. Diamonds indicate lead variants. Note that statistical significance was evaluated using the permutation procedures (see Materials and Methods) and that the significance cutoff P-values were different depending on the miRNA.
Table 1.
Summary association results for 40 cis-miRNA-eQTLs
| eMiRNA | Lead variant | Chr | Position | P-value | MAF (in study) | MAF (EAS) | MAF (EUR) |
|---|---|---|---|---|---|---|---|
| miR-92b-3p | rs16836028 | 1 | 154 447 505 | 2.0 × 10−5 | 0.011 | 0.016 | 0.000 |
| miR-556-3p | rs6427665 | 1 | 162 214 762 | 9.7 × 10−6 | 0.48 | 0.48 | 0.26 |
| miR-181a-5p | rs16844101 | 1 | 199 011 539 | 9.5 × 10−6 | 0.18 | 0.18 | 0.18 |
| miR-26b-5p | rs62182125 | 2 | 219 274 142 | 1.6 × 10−5 | 0.23 | 0.27 | 0.45 |
| miR-16-2-3p | rs148824756 | 3 | 160 119 084 | 3.6 × 10−5 | 0.021 | 0.024 | 0.002 |
| miR-574-3p | rs6531685 | 4 | 38 960 263 | 3.9 × 10−7 | 0.33 | 0.31 | 0.39 |
| miR-146a-3p | rs2910164 | 5 | 159 912 418 | 2.4 × 10−16 | 0.38 | 0.38 | 0.23 |
| miR-93-3p | rs375968286 | 7 | 100 389 520 | 4.3 × 10−5 | 0.028 | 0.007 | 0.000 |
| miR-335-5p | rs6947476 | 7 | 130 076 606 | 9.5 × 10−6 | 0.18 | 0.20 | 0.32 |
| miR-335-3p | rs12706931 | 7 | 130 078 291 | 6.0 × 10−7 | 0.38 | 0.39 | 0.49 |
| let-7d-3p | rs200404962 | 9 | 96 893 141 | 2.4 × 10−5 | 0.27 | 0.26 | 0.15 |
| miR-23b-3p | rs1564234 | 9 | 97 781 594 | 1.1 × 10−14 | 0.17 | 0.27 | 0.11 |
| miR-27b-3p | rs10993464 | 9 | 97 808 086 | 8.7 × 10−7 | 0.18 | 0.28 | 0.11 |
| miR-3074-3p | rs117435548 | 9 | 97 995 871 | 2.1 × 10−7 | 0.018 | 0.007 | 0.000 |
| miR-23b-5p | rs7047000 | 9 | 98 426 006 | 2.9 × 10−5 | 0.45 | 0.37 | 0.47 |
| miR-1307-3p | rs35435808 | 10 | 105 180 910 | 7.7 × 10−8 | 0.30 | 0.34 | 0.37 |
| miR-130a-3p | rs731384 | 11 | 57 408 382 | 1.0 × 10−6 | 0.096 | 0.11 | 0.29 |
| miR-1908-5p | rs174578 | 11 | 61 605 499 | 1.3 × 10−14 | 0.39 | 0.45 | 0.35 |
| miR-20a-5p | rs76518987 | 13 | 92 717 757 | 2.0 × 10−5 | 0.011 | 0.011 | 0.001 |
| miR-3173-5p | rs147808964 | 14 | 95 078 061 | 4.9 × 10−7 | 0.050 | 0.027 | 0.000 |
| miR-342-3p | rs75416067 | 14 | 100 421 429 | 5.2 × 10−7 | 0.36 | 0.37 | 0.23 |
| miR-496 | rs549766505 | 14 | 101 024 724 | 6.6 × 10−6 | 0.021 | 0.007 | 0.000 |
| miR-323b-3p | rs28366562 | 14 | 101 522 321 | 3.9 × 10−6 | 0.19 | 0.23 | 0.21 |
| miR-627-5p | rs7181577 | 15 | 42 485 444 | 3.3 × 10−15 | 0.060 | 0.084 | 0.082 |
| miR-190a-3p | rs2940333 | 15 | 62 757 544 | 1.6 × 10−8 | 0.39 | 0.46 | 0.20 |
| miR-190a-5p | rs12441323 | 15 | 62 816 101 | 6.9 × 10−8 | 0.46 | 0.43 | 0.45 |
| miR-195-5p | rs76819872 | 17 | 6 836 143 | 4.5 × 10−6 | 0.071 | 0.10 | 0.070 |
| miR-144-5p | rs10853129 | 17 | 27 191 960 | 7.8 × 10−11 | 0.45 | 0.31 | 0.30 |
| miR-144-3p | rs7214973 | 17 | 27 222 745 | 4.5 × 10−5 | 0.34 | 0.30 | 0.36 |
| miR-152-3p | rs145242009 | 17 | 46 396 778 | 5.2 × 10−6 | 0.018 | 0.002 | 0.000 |
| miR-301a-3p | rs2191245 | 17 | 56 242 752 | 5.7 × 10−5 | 0.13 | 0.13 | 0.21 |
| miR-21-5p | rs10853015 | 17 | 57 778 339 | 1.2 × 10−5 | 0.15 | 0.17 | 0.25 |
| miR-3940-3p | rs62106647 | 19 | 6 361 345 | 1.6 × 10−5 | 0.17 | 0.18 | 0.048 |
| miR-641 | rs746775867 | 19 | 40 788 494 | 2.0 × 10−5 | 0.011 | 0.009 | 0.000 |
| let-7e-5p | rs11670586 | 19 | 52 184 544 | 2.8 × 10−10 | 0.50 | 0.44 | 0.094 |
| miR-125a-5p | rs11670586 | 19 | 52 184 544 | 4.8 × 10−10 | 0.50 | 0.44 | 0.094 |
| miR-99b-5p | rs11670586 | 19 | 52 184 544 | 7.2 × 10−8 | 0.50 | 0.44 | 0.094 |
| miR-1-3p | rs60640728 | 20 | 61 145 810 | 2.4 × 10−14 | 0.21 | 0.22 | 0.21 |
| miR-130b-5p | rs425046 | 22 | 22 033 626 | 1.9 × 10−5 | 0.13 | 0.092 | 0.017 |
| miR-548j-5p | rs4822733 | 22 | 26 930 156 | 9.0 × 10−11 | 0.21 | 0.22 | 0.031 |
Novel eMiRNAs are highlighted in bold. MAF (in study), minor allele frequency in the study participants; MAF (EAS), minor allele frequency in the gnomAD (24) East Asian populations and MAF (EUR), minor allele frequency in the gnomAD non-Finnish European populations.
Figure 2.

Characteristics of cis-miRNA-eQTLs in the Japanese population. (A) The number (the upper panel) and P-values (the lower panel) of the lead variants of eMiRNAs are shown against the distance from each TSS. When more than one variant showed the minimum P-value for an eMiRNA, the median distance across them is used. (B) Effect sizes in the previous miRNA-eQTL studies [(12,17); x-axis; Europeans] are compared with those in our study (y-axis; Japanese). Pink dashed lines represent regression lines. The previously reported miRNA-eQTL variants that showed nominal P-value < 0.01 in our study are shown. (C) MAFs in the East Asian (EAS) and the European populations (EUR) are separately compared for the lead variants of the known (n = 15) and novel (n = 25) eMiRNAs. P-values are calculated using the Wilcoxon rank-sum test. Boxplots represent the interquartile range (IQR), and ends of whiskers represent the minimum and maximum values within 1.5 × IQR. The P-value was calculated using the Wilcoxon rank-sum test.
To confirm that our eQTL mapping results are consistent with previous studies in European populations, we compared the effect sizes between the previously reported miRNA-eQTLs (12,17) and our results (Fig. 2B) and found high correlations (Spearman’s correlation = 0.87 and 0.74; P = 4.0 × 10−179 and 7.1 × 10−67, respectively). Of the 40 eMiRNAs, we identified, 25 (63%) were unreported in the two previous studies on Europeans. The lead variants of the novel eMiRNAs showed significantly lower MAFs in the European populations than those in the East Asian populations of the Genome Aggregation Database [gnomAD; (24); P = 8.3 × 10−3, the Wilcoxon rank-sum test], but this was not the case for the known eMiRNAs (P = 0.38; Fig. 2C). In particular, the lead variants of the five novel eMiRNAs were monomorphic in the European populations (Table 1). These findings demonstrate that our analysis successfully identified miRNA-eQTLs common in the East Asian populations, including population-specific polymorphisms.
Next, to comprehensively delineate cis-regulatory genetic effects on miRNAs, we leveraged sRNA-seq reads covering heterozygous sites and analyzed allele-specific expression (ASE) of miRNAs. ASE is the relative expression difference between the paternal and maternal alleles within a given individual. Individuals who are heterozygous for a cis-regulatory genetic variant may exhibit ASE in which one allele is more highly expressed than the other allele. Since trans-acting or environmental factors equally influence both alleles, ASE analysis is robust to such factors and an orthogonal measure to detect cis-regulatory effects within an individual, in contrast to the conventional eQTL detection between individuals. Of the three variant–miRNA pairs that passed the quality-control criteria (see Materials and Methods), two variant–miRNA pairs showed significant ASE in at least one individual (P < 0.05/3 = 0.017); rs2910164 within miR-146a-3p and rs2620381 within miR-627-5p (Fig. 3A). These variants were also identified as significant eQTL variants in the conventional miRNA-eQTL mapping. Rs2910164 is the lead variant of the miR-146a-3p eQTL. Rs2620381 is in strong LD with the lead variant of the miR-627-5p eQTL [r2 = 0.95 in the East Asian populations of 1000 Genomes Project Phase 3 (1KG Phase 3)]. The allele with fewer read counts observed in ASE analysis consistently decreased expression (Fig. 3B).
Figure 3.

Overlap of allele-specific expression and eQTL effect of miRNA. (A) The read counts of reference and alternative alleles in a given individual are shown separately for each genotype of the variant within mature miRNA sequences. RefHom, homozygous reference; Het, heterozygote; AltHom, homozygous alternate. (B) The normalized expression of miRNAs is shown separately for each genotype of the variant within mature miRNA sequences. Each dot represents a normalized expression in a given individual. Boxplots represent IQR, and ends of whiskers represent the minimum and maximum values within 1.5 × IQR.
To improve statistical power for detecting cis-regulatory effects, we performed ASE analysis with reads combined across all the individuals heterozygous at the variant. Sixteen variants were covered by at least one sRNA-seq read from the individuals heterozygous at the variant. This combined analysis identified another variant–miRNA pair with significant ASE, rs79402775 within miR-933 (P < 0.05/24 = 2.1 × 10−3), which was not identified as significant in the miRNA-eQTL mapping. Our results demonstrate that ASE analysis expands a list of candidate cis-regulatory variants altering miRNA expression.
Influence of CNVs on miRNA expression
Previous miRNA-eQTL studies have focused on associating SNVs or short indels with miRNA expression. However, mRNA expression is not only correlated with SNVs and short indels but also CNVs (25,26). Moreover, the study using the GTEx dataset reported that nearly half of structural variants were poorly tagged by nearby SNVs or short indels and that WGS-based structural variants analysis increased the power of eQTL mapping (27). These insights motivated us to conduct miRNA-eQTL analysis using CNVs to have a more comprehensive view of the genetic regulation on miRNA expression.
We performed genome-wide CNV calling using the WGS data, identifying 7912 autosomal CNVs (3686 deletions, 2502 duplications and 1724 mixed deletions/duplications). We tested associations between miRNA expression and CNVs within 1 Mb up- and down-stream of each miRNA (27), identifying one significant miRNA–CNV association at FDR < 0.2; miR-570-3p and the CNV chr3:195 418 040–195 433 076 (P = 7.2 × 10−6, explained variance = 14%, Fig. 4). The CNV chr3:19 541 804–195 433 076 completely overlaps with a previously reported CNV chr3: 195 417 835–195 445 776 in the 1KG Phase 3 (26). Both CNV regions fully encompassed the miR-570-3p sequence. The copy number of the CNV chr3:195 418 040–195 433 076 ranged from 2 to 8, and the copy number and the expression of miR-570-3p were positively correlated, although the exact copy number of the miR-570 gene for each individual was undetermined. Notably, miR-570-3p did not exhibit any significant eQTL through an analysis focusing solely on SNVs and short indels (minimum q-value = 0.67 with rs12490110). Our results provide an evidence that examining CNVs makes our understanding of the genetic architecture of miRNA expression more comprehensive.
Figure 4.
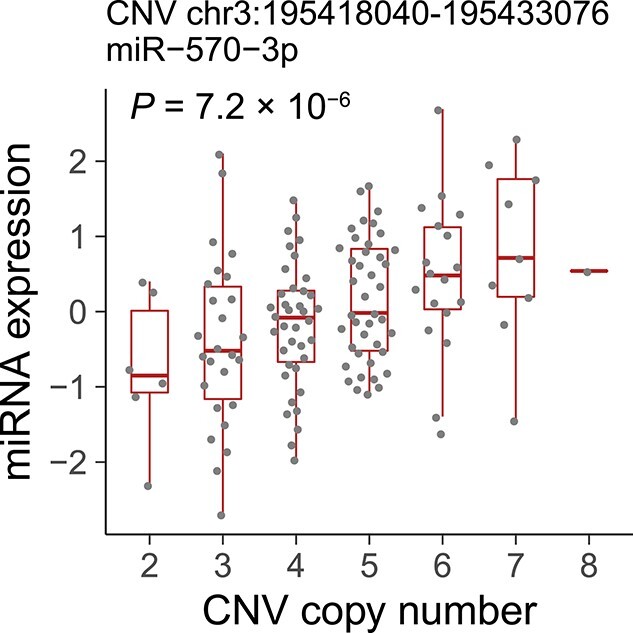
Positive correlation between genomic copy number and miRNA expression. The significant association between the genomic copy number of the CNV chr3:195418040–195 433 076 (x-axis) and the normalized expression of miR-570-3p (y-axis). Each dot represents a normalized expression in a given individual. Boxplots represent IQR, and ends of whiskers represent the minimum and maximum values within 1.5 × IQR.
MiRNA-editing detection with WGS-based genetic variants
RNA-editing is a widespread post-transcriptional modification of RNA molecules (28,29). MiRNA plays the regulatory role using partial base pairing with the target mRNA sequences (30). The bases 2–8 from the 5′-end of the mature miRNA are the main determinants of target recognition and are known as ‘seed sequence’. If an editing event occurs in the seed sequence of a miRNA, it can alter binding affinity to the target genes and thus change the set of target genes (31). To detect high-confidence RNA-editing events, it is important to rule out the possibility of falsely annotating an alternative allele of germline variants as an editing event. Leveraging the WGS-based germline variants present in the study participants, we identified 16 miRNA-editing sites while excluding false detection caused by the germline variants (Table 2). Of the 16 miRNA-editing sites, 11 (69%) were novel sites, and 6 (38%) were situated within the seed sequences and expected to alter the binding affinity to the target genes. We further validated the 16 miRNA-editing sites using an independently generated sRNA-seq dataset of the part of the study participants (n = 79). We confirmed that all the 16 sites were replicated (FDR < 0.05; Supplementary Material, Table S1).
Table 2.
Summary of the 16 canonical and non-canonical miRNA-editing events
| Editing class | miRNA | Cytoband | Position in mature miRNA | Editing in seed | Base substitution | Adjusted P | # detected individuals | Mean-editing level |
|---|---|---|---|---|---|---|---|---|
| A-to-I | miR-589-3p | 7p22.1 | 6 | Yes | A → G | 5.4 × 10−12 | 0† | 0.046 |
| miR-6503-3p | 11q12.2 | 7 | Yes | A → G | < 1 × 10−300 | 25 | 0.36 | |
| miR-411-5p | 14q32.31 | 5 | Yes | A → G | 1.0 × 10−55 | 3 | 0.033 | |
| miR-381-3p | 14q32.31 | 4 | Yes | A → G | < 1 × 10−300 | 21 | 0.17 | |
| miR-421 | Xq13.2 | 14 | No | A → G | 5.9 × 10−4 | 0† | 0.0035 | |
| miR-652-5p | Xq23 | 11 | No | A → G | 2.1 × 10−11 | 0† | 0.011 | |
| miR-505-5p | Xq27.1 | 4 | Yes | A → G | 0.037 | 0† | 0.0032 | |
| C-to-U | miR-425-5p | 3p21.31 | 14 | No | C → U | < 1 × 10−300 | 86 | 0.026 |
| miR-487b-3p | 14q32.31 | 14 | No | C → U | 3.4 × 10−5 | 0† | 0.0034 | |
| miR-652-3p | Xq23 | 11 | No | C → U | 1.9 × 10−18 | 1 | 0.013 | |
| Non-canonical | miR-1843 | 1q25.1 | 16 | No | C → A | 2.6 × 10−7 | 0† | 0.012 |
| miR-30d-5p | 8q24.22 | 9 | No | U → C | 6.4 × 10−7 | 0† | 0.0052 | |
| miR-379-3p | 14q32.31 | 11 | No | G → A | 1.6 × 10−7 | 1 | 0.017 | |
| miR-324-5p | 17p13.1 | 5 | Yes | U → C | 7.3 × 10−4 | 0† | 0.0025 | |
| miR-144-5p | 17q11.2 | 15 | No | A → C | 1.4 × 10−51 | 3 | 0.0023 | |
| miR-223-5p | Xq12 | 14 | No | A → C | 2.2 × 10−12 | 1 | 0.0030 |
Novel editing sites are highlighted in bold.
Adjusted P, P-values calculated with sRNA-seq reads combined across all individuals and adjusted by the Benjamini–Hochberg method.
# detected individuals, the number of individuals in which a given miRNA-editing event was significantly detected (see Materials and Methods).
†The editing event was detected only when sRNA-seq reads were combined across all individuals.
RNA-editing is classified into ‘canonical’ and ‘non-canonical’ editing. Canonical editing consists of two forms of well-characterized RNA-editing, adenosine-to-inosine (A-to-I) editing (32) and cytidine-to-uridine (C-to-U) editing (33). A-to-I editing is regarded as the most common form of RNA-editing in mammals. Non-canonical editing refers to the other forms of RNA-editing. We observed an over-representation of adenosine-to-guanosine (A-to-G) base substitution, which reflects A-to-I editing (7 of the 16 editing sites; Fig. 5A). The fraction of A-to-G in all the miRNA-editing sites (44%) is similar to the previously reported values (34–36).
Figure 5.

Over-representation of the two types of canonical RNA-editing. (A) The number of detected miRNA-editing sites is shown for each type of base substitution. (B) Mean-editing levels across all individuals are shown for each type of base substitution. Pink markers indicate the previously reported editing sites.
Although most previous studies on miRNA-editing focused on A-to-I editing (34–37), we also identified non-A-to-I editing events, including C-to-U and non-canonical editing. C-to-U editing showed a moderate over-representation [3 of the 16 editing sites (19%); Fig. 5A]. All the non-A-to-I editing sites detected in our analysis were novel. We calculated the RNA-editing level for each editing site as the ratio of reads with the substituted base to the total reads covering the site [(38,39); see Materials and Methods]. The novel editing sites exhibited significantly lower editing levels than the known sites (P = 0.013, the Wilcoxon rank-sum test; Fig. 5B), suggesting that non-A-to-I editing occurs at low editing levels and thus requires more sRNA-seq reads than A-to-I editing for detection. Previous studies reported that genetic variation affects mRNA-editing efficiency (38,39), known as editing QTLs (edQTLs). We investigated such edQTLs with our miRNA transcriptome data but did not identify significant associations (minimum q-value = 0.36 between G-to-A editing of the 11th base of miR-379-3p and rs61628376 at 14q32.3; Supplementary Material, Fig. S3, Supplementary Material, Table S2).
MiRNA transcriptome-wide association study
To identify disease-associated miRNAs, we performed a TWAS using the miRNA expression data and the summary statistics of the 25 large-scale GWASs of diverse human complex traits in Japanese individuals [Supplementary Material, Table S3; (40–42)]. MiRNA expressions were modeled via the elastic net method using genetic variants within 2 Mb cis-window as explanatory variables. Among the 343 autosomal miRNAs in our data, prediction models for the 22 miRNAs were successfully trained (see Materials and Methods). Based on the prediction models, we evaluated the association between genetically regulated miRNA expression and complex human traits. The miRNA TWAS identified 11 significant miRNA–complex trait associations (P < 0.05/22 = 2.3 × 10−3; Supplementary Material, Table S4), of which five fulfilled the study-wide significance threshold [P < 0.05/(22 × 25) = 9.1 × 10−5; the most significant association was between miR-1908-5p and adult height (P = 1.9 × 10−12); Fig. 6A]. TWAS associations can be spuriously detected because of LD-contamination (43), in which genetic variants used in the expression prediction models and trait-causal variants are different but in LD. To avoid capturing such false links, we performed colocalization analysis to assess whether the GWAS and eQTL associations share the same causal variants or whether the associations are due to distinct causal variants in linkage. We found that three out of five study-wide significant miRNA–complex trait associations exhibited high colocalization probability (PP4 by COLOC > 0.5); miR-1908-5p and adult height, colorectal cancer and type 2 diabetes.
Figure 6.

MiRNA TWAS for complex diseases and anthropometric traits. (A) P-values are shown for associations between genetically regulated miRNA expression and complex traits. Each diamond represents the P-value for a given miRNA–trait pair. The y-axis indicates −log10(P) with the sign of the effect sizes to represent the direction of the miRNA effects. Pink-dashed lines indicate the transcriptome-wide significance threshold via the Bonferroni correction based on the number of tested miRNAs. The miRNA–trait pairs meeting the significance threshold are labeled. The miRNA–trait pairs with COLOC PP4 > 0.5 are highlighted in pink. (B) Regional plots of colorectal cancer GWAS and miR-1908 eQTL are shown. Markers are colored by LD (r2) with the lead variant of the GWAS (rs509360). (C) The pre-miRNA mir-1908 expressions in normal and tumor tissues from colorectal cancer patients are shown (45). Each dot represents a normalized expression of mir-1908 in a given specimen sample. Boxplots represent IQR, and ends of whiskers represent the minimum and maximum values within 1.5 × IQR.
The miR-1908-5p eQTL and a colorectal cancer GWAS signal showed high colocalization probability (PP4 = 0.95). The lead GWAS variant rs509360 and its proxy variants showed strong associations with the expression of miR-1908-5p (Fig. 6B). Rs509360 was previously reported as an eQTL of four coding genes (FADS1, FADS2, TMEM258 and RAB3IL1) in the GTEx project (44), although the GTEx dataset predominantly consists of individuals of European ancestry. We examined a publicly available gene expression profile dataset of colorectal cancer clinical specimens (45), revealing that the corresponding pre-miRNA mir-1908 was differentially expressed between normal and tumor tissues (P = 3.5 × 10−5, the Wilcoxon rank-sum test; Fig. 6C). Interestingly, although TWAS analysis revealed that miR-1908-5p is protective against colorectal cancer (i.e. the negative effect size in TWAS), the expression in the clinical specimens was higher in the tumor tissues. Several miRNAs are up-regulated by the downstream signaling pathway of the target genes and form a negative regulatory feedback loop (46–48). The aberrantly activated oncogenic pathway in tumor tissues, which miR-1908-5p represses under physiological conditions, may induce the miR-1908-5p overexpression.
Discussion
In this study, we performed the first miRNA-eQTL analysis in the Asian population and identified 40 eMiRNAs, including 25 novel eMiRNAs unreported in the European populations. This resource will facilitate linking GWAS-identified loci to variations in miRNA expression in Asian populations. We demonstrated that integration of the miRNA expression and large-scale GWASs in Japanese individuals identified disease-associated miRNAs.
Our study reports several novel findings elucidating miRNA regulatory mechanisms. First, we performed an ASE analysis of miRNAs, an orthogonal approach for cis-regulatory variant detection. The detected ASE showed consistent results with eQTL mapping, confirming the validity of miRNA ASE analysis. Moreover, the analysis, which combined the reads across study participants identified the ASE of the miRNA that was not detected by the conventional eQTL mapping. Second, genomic CNVs also exhibited cis-regulatory effects on the expression of miRNAs. This is, to our knowledge, the first analysis utilizing WGS data and sRNA-seq to correlate CNVs with miRNA expression. MiR-570-3p showed significant association with an overlapping CNV but was undetected as an eMiRNA when only SNVs and short indels were used for eQTL mapping. This result indicates that eQTL mapping with CNVs makes our understanding of the genetic architecture of miRNA expression more comprehensive. In the study of protein-coding gene eQTL, duplication variants overlapping with coding regions were reported to increase gene expression, which was explained as exon duplications (27). The positive correlation between the miR-570-3p expression and the copy number of the surrounding CNV implies that duplications of the miRNA genes may increase the miRNA expression analogously to the exon duplications. Third, we created a catalog of canonical and non-canonical miRNA-editing events, while distinguishing the editing sites from an alternative allele of a germline variant using WGS data. Non-A-to-I editing showed low RNA-editing levels compared with A-to-I editing, potentially making it difficult to detect. Finally, the miRNA TWAS utilizing large-scale GWASs in Japanese individuals revealed significant miRNA–disease associations, which prioritized the causal role of the identified miRNAs. Our results provide unique insights into the pathogenic or protective role of miRNAs, which would be difficult to be obtained from differential miRNA expression profiling alone. We identified the significant associations between miR-1908-5p and adult height, colorectal cancer and type 2 diabetes. The eQTL variants (rs102275 and rs968567) of miR-1908-5p were also reported to be associated with Crohn’s disease (CD) and rheumatoid arthritis (RA) by Wohlers et al. (49) using the miRNA-eQTL and GWAS data of the European populations (rs102275 for CD; rs968567 for RA). In our TWAS, the association between miR-1908-5p and RA was not detected (P = 0.67). One potential explanation for the different result is the difference in the methods used. Although Wohlers et al. associated miR-1908-5p with RA using the joint likelihood mapping method, we performed TWAS and colocalization analysis. Further study is warranted to validate the association of miR-1908-5p with RA.
The potential caveat of this study is the limited replication analyses for miRNA-eQTLs and TWAS findings. Although the systematic comparison with the previous miRNA-eQTL studies in the European populations confirmed the overall validity of our miRNA-eQTL mapping (Fig. 2B), further replication analyses in the East Asian populations would be desirable as future work. Constructing another miRNA-eQTL dataset in the East Asian populations would also facilitate the replication of TWAS using an independent dataset.
In conclusion, by integrating sRNA-seq and WGS, our study comprehensively elucidated the genetic architecture of miRNA expression and identified potential miRNAs important for disease biology in the Japanese population.
Materials and Methods
Study populations
We enrolled 141 participants of Japanese ancestry for the WGS and the sRNA-seq. All participants signed a written informed consent form, as approved by the ethical committee of Osaka University.
WGS data processing
DNA samples isolated from whole blood were sequenced at Macrogen Japan Corporation. DNA quantity was measured by Picogreen, and degradation of DNA was assessed by gel electrophoresis. All libraries were constructed using the TruSeq DNA PCR-Free Library Preparation Kit according to the manufacturer’s protocols. Libraries were sequenced on HiSeqX (Illumina, San Diego, CA, USA), producing paired-end reads of length 2 × 150 bp, with a mean insert size of 488 bp and a mean coverage of 16.5×. Sequenced reads were aligned against the reference human genome with the decoy sequence (GRCh37, human_g1k_v37_decoy) using BWA-MEM (version 0.7.13). Duplicated reads were removed using Picard MarkDuplicates (version 2.10.10). After Base-quality score recalibration implemented in GATK (versions 3.8–0), We generated individual variant call results using HaplotypeCaller and performed multi-sample joint-calling of the variants via GenotypeGVCFs. We set genotypes satisfying any of the following criteria as missing: (i) DP < 5, (ii) GQ < 20 or (iii) DP > 60 and GQ < 95, then removed variants with low genotyping call rates (< 0.90). We performed Variant Quality Score Recalibration for SNVs and short indels according to the GATK Best Practice recommendations and adopted the variants, which passed the QC criteria. We further removed the variants (i) located in the low complexity regions, (ii) with ExcessHet > 60 or (iii) with Hardy–Weinberg P-value < 1.0 × 10−10. We kept only those presenting a non-significant difference in allele frequency (P > 1.0 × 10−10 provided by chi-square test) in the following representative reference datasets of Japanese ancestry: the combined reference panel of 1KG Phase 3 version 5 genotype (n = 2504) and Japanese WGS data [n = 1037; (41,50)], and the allele frequency panel of Tohoku Medical Megabank Project (51). Genotype refinement was performed using Beagle [version 5.1; (52)]. Also, we genotyped all the individuals using Infinium Asian Screening Array (Illumina). Comparison between WGS-based and SNP array-based genotypes showed high concordance rates (all individuals > 99.95%).
Total RNA extraction and small RNA sequencing
sRNA-seq library preparation was performed as described elsewhere (11). PBMCs were isolated from leukocyte concentrates by Ficoll–Paque density gradient centrifugation. Total RNA from PBMCs was extracted using the miRNeasy Micro Kit (Qiagen, Duesseldorf, Germany). Libraries for sRNA-seq were prepared using the SMARTer smRNA-Seq Kit (Takara, Tokyo, Japan) following the manufacturer’s instructions. Sequencing was conducted on HiSeq 2500 (Illumina, read length of 100 bp, single-end).
MiRNA expression quantification
For the QC of the sRNA-seq data, we performed adapter trimming using Cutadapt v1.8 (53) and removed reads with a low quality score (Phred quality score < 20 in >20% of total bases) using fastp (54). Also, we removed reads with a length of >29 bp or <15 bp, which are not expected to be mature miRNAs. Because of their short length, the alignment of miRNAs is known to be affected by cross-mapping, in which reads originated from one miRNA are improperly mapped to other loci with similar sequences (55). To address this issue, we adopted a stringent criterion. Specifically, we aligned the remaining reads to the human reference genome using bowtie (version 1.2.3) allowing for one mismatch (−v 1) and considered only uniquely mapped reads as valid alignment (−m 1). Some mature miRNAs are known to be encoded by multiple loci with the same sequence (56), and such miRNAs are inevitably removed by this criterion, even if they are properly aligned. To avoid such unnecessary read removal, we masked regions of the reference genome encoding the same mature miRNA according to the annotation by miRBase v22 (57) from the mapping except for a single representative locus. We counted reads mapped to mature miRNA sequences annotated by miRBase v22 using featureCounts (58) with at least 90% overlap. We obtained a median of 1.9 × 105 reads aligned to miRNAs for each individual (Supplementary Material, Fig. S4). Mature miRNAs detected with ≥1 read in at least half of the individuals were included in the subsequent analysis. We computed size factors associated with each library and normalized miRNA counts using DESeq2 (59). The normalized counts plus a pseudo-count of 1 were log2-transformed to stabilize the variance of the expression values.
cis-miRNA-eQTL analysis
A principal component analysis with the samples of HapMap project (60) confirmed that all the study participants were East Asian (Supplementary Material, Fig. S5). The obtained miRNA expression matrix was normalized using PEER (61) accounting for 15 unobserved confounders as well as the known confounders, such as library preparation batch, disease status, sex, age, number of total mapped reads, type of blood collection tube and five genotype-based principal components. The residuals for each miRNA were transformed into a standard normal distribution based on rank. We analyzed the association between genetic variants (SNVs and short indels) with MAF ≥ 0.01 within a cis-window around each miRNA and normalized expression values with MatrixEQTL (62) using linear regression with an additive effect model. The cis-window was defined as 1 Mb up- and down-stream of the mature miRNA [± 1 Mb; (12)]. If a miRNA is encoded by multiple genomic loci, we defined the cis-windows for every encoding locus and tested all the variants within either of them. To correct for multiple testing effects, we applied a permutation procedure as in previous studies (63,64). The minimal P-value per miRNA was used as the test statistic. We randomized the sample IDs of the expression data while retaining the sample IDs of the genotype data. A 5000 permutations were applied to obtain the null distribution of the minimal P-value. Thus, we derived empirical P-values for every miRNA and calculated permutation q-values using Storey’s q-value method (65) and a q-value threshold of < 0.2 was applied. To identify the list of all significant variant–miRNA pairs associated with eMiRNAs, variants with a nominal P-value below the gene-level threshold were considered significant. To evaluate the effects of cell type composition in PBMCs, we also performed cis-miRNA-eQTL mapping using expression values adjusted for cell type proportions. The cell type proportions in PBMCs were estimated by CIBERSORTx (23) with the default parameters using publicly available cell type-specific miRNA transcriptome data of the following cell types: cytotoxic T cells, T helper cells, B cells, monocytes and NK cells (22). Note that the estimation of the cell type proportions relied solely on the miRNA expression profiles and could be affected by the technical factors, such as the sample preparation batches or the quality of the reference expression data.
Evaluating relative distances of cis-miRNA-eQTL variants from TSSs
We examined the positional distribution of cis-miRNA-eQTL variants relative to TSS with the following two criteria for selecting eQTL variants:
(i) We used the single most significantly associated eQTL variant per eMiRNA. When multiple eQTL variants exhibited the smallest P-value for an eMiRNA, we used the median distance of such variants to TSS.
(ii) For each eMiRNA, we pruned the list of cis-eQTL variants to retain the most significant variants with pairwise LD (r2) < 0.7 for the study population using the clump function of PLINK.
The chromosomal coordinates of miRNA TSSs were obtained from the FANTOM5 project (66).
Allele-specific expression analysis
We kept the sRNA-seq reads aligned to mature miRNA sequences with mapping quality ≥ 10 and filtered out reads that exhibited mapping bias at heterozygous sites using the WASP mapping pipeline (67). For quantifying allele counts at heterozygous sites, we used GATK ASEReadCounter (version 4.1.7.0). The following two approaches were used to evaluate the allelic imbalance between reference and alternative alleles:
(i) We tested the ASE using the binomial test for each individual–heterozygous site pair with total coverage ≥8. To filter out potential genotyping errors, the sites were excluded where both alleles were not observed by sRNA-seq. Following the quality control, we obtained 23 individual–heterozygous site pairs composed of three SNP sites corresponding to three miRNAs (miR-146a-3p, miR-11 400 and miR-627-5p). We set a significance threshold at the level of P = 0.017 (= 0.05/3) by applying the Bonferroni correction based on the number of tested sites.
(ii) To improve statistical power, we combined reads across all individuals for each heterozygous site. After that, we used the binomial test for estimating the ASE. We set a significance threshold at the level of P = 2.1 × 10−3 (= 0.05/24) by applying the Bonferroni correction based on the number of tested sites.
CNV calling using the WGS data
We ran the Genome STRiP CNVDiscovery (25) pipeline (version 2.00.1982) to identify and genotype large deletions, duplications and mixed deletions/duplications. Considering the sequencing depth of our WGS dataset (16.5×), we set the parameters as follows: a window size of 2000 bp, a window overlap of 1000 bp, a reference gap length of 2000 bp, a boundary precision of 200 bp and a minimum refined length of 1000 bp. To estimate the FDR of the CNV call set, we performed an intensity rank-sum (IRS) test for in silico CNV validation using the intensity data of Infinium Asian Screening Array. We obtained the log-transformed R ratio of intensity values from Illumina GenomeStudio (version 2.0.4). The intensity matrix was adjusted for the effects of plates by linear regression and then used as IRS input. We tested 8373 of 23 192 autosomal CNVs and computed IRS FDR as in the 1KG Phase 3 (26). We found that a threshold of GSCNQUAL ≥1 for deletions and mixed deletions/duplications, and a threshold of GSCNQUAL ≥2 for duplications corresponded to an FDR of 0.1. CNVs fulfilling these criteria were used for downstream analysis. We excluded the individuals with excessive variation from the dataset based on the number of calls per individual exceeding the median plus three median absolute deviations. We defined MAF as the fraction of individuals that deviated from the mode copy number value in the population.
CNV cis-eQTL analysis
We selected CNV calls with MAF ≥ 0.01 for eQTL analysis. We assessed the association between the CNV copy number and normalized miRNA expression, which was used in the aforementioned eQTL analysis with SNVs and short indels. For each miRNA, we defined a cis-window as 1 Mb up- and down-stream of the mature miRNA [± 1 Mb; (27)] and tested the CNVs that overlapped with the cis-window using MatrixEQTL. Empirical P-values were calculated using 5000 permutations as described in ‘cis-miRNA-eQTL analysis’, and the miRNA-level multiple testing correction was performed using Storey’s q-value method. We set a q-value < 0.2 as a significance threshold.
MiRNA-editing calling
MiRNA-editing calling was performed using the script from previous studies (68,69). The adaptor-trimmed and length-filtered reads as described previously were further trimmed by 2 nt at the 3′-end. The resulting reads were aligned to the human reference genome (build GRCh37) using bowtie, allowing at most one mismatch per read, the best alignment and no cross-mapping. We filtered out mismatches with a base-quality score < 30. For the genomic regions of pre-miRNAs annotated in miRBase v22, mismatch calling was performed based on the binomial test. We detected candidate miRNA-editing events with all individuals combined as well as separately for each individual. To filter out genetic variants and unreliable base editing events, we applied stringent quality control criteria. Specifically, we excluded the base editing events that met the following criteria: (i) by our WGS analysis, the base editing site is called as the genetic variant, (ii) the base editing site is found in external reference datasets of genetic variants of individuals of Japanese ancestry or (iii) the base editing is called at 5′-end of the mature miRNA. We further excluded the base editing events where the aligned reads were revealed to imperfectly overlap mature miRNA sequences by visual inspection using the Integrative Genomics Viewer. Besides, we examined the bam files generated during WGS variant calling to confirm that the detected base substitutions were not observed in the aligned WGS reads.
Replication analysis of the detected miRNA-editing sites
Using the total RNA of the 79 individuals in the study participants, we independently prepared another sRNA-seq library. The sRNA-seq library was sequenced on NovaSeq 6000 (Illumina, read length of 100 bp, single-end). The sequenced reads were processed and aligned to the human reference genome as described in ‘MiRNA-editing calling’, and we counted the bases aligned to the 16 detected miRNA-editing sites. We tested base mismatches using the binomial test.
MiRNA-edQTL mapping
For each individual, we quantified miRNA-editing levels as the ratio of the number of edited reads at a specific miRNA-editing site to the number of all reads covering the site (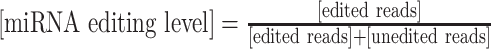 ). The obtained miRNA-editing levels were adjusted for the known confounders described previously by the linear regression model. Then, the resulting residuals were transformed into a standard normal based on rank. We tested the association between variants within a cis-window of 200 kb around each miRNA and normalized miRNA levels with MatrixEQTL using linear regression with an additive effect model.
). The obtained miRNA-editing levels were adjusted for the known confounders described previously by the linear regression model. Then, the resulting residuals were transformed into a standard normal based on rank. We tested the association between variants within a cis-window of 200 kb around each miRNA and normalized miRNA levels with MatrixEQTL using linear regression with an additive effect model.
GWAS summary statistics
We downloaded the summary statistics of Japanese GWASs for diseases (42) and anthropometric traits (40,41). Only the diseases for which harmonic means of the cases and controls were >5000 were used for downstream analysis (Supplementary Material, Table S3).
MiRNA TWAS
We performed the TWAS using the S-PrediXcan (43) software. For each miRNA quantified in our dataset, we trained a prediction model using the normalized expression values and the genotype data within 1 Mb up- and down-stream of the mature miRNA [± 1 Mb; (43)]. The prediction models were trained on the basis of the nested cross-validated elastic-net procedure (R script ‘gtex_v7_nested_cv_elnet.R’). We retained the prediction models with rho_avg > 0.1 and zscore_pval < 0.05. Then, we evaluated miRNA expression–trait associations using the summary statistics of the Japanese GWASs. We set a Bonferroni-corrected significance threshold at the level of P = 2.3 × 10−3 (= 0.05/22) based on the number of tested miRNAs. We set a study-wide significance threshold at the level of P = 9.1 × 10−5 [= 0.05/(22 × 25)] by applying the Bonferroni correction based on the number of tested miRNAs and traits.
Colocalization of miRNA-eQTLs and GWAS loci
We performed colocalization analysis via COLOC (70). The approximate Bayes factor test of COLOC estimates whether the association signals of two phenotypes share common causal variants in a given genomic region and computes posterior probabilities for the five hypotheses as follows: H0, neither trait exhibits a genetic association; H1/H2, only one trait exhibits a genetic association; H3, both traits are associated but with independent causal variants and H4, both traits are associated with a single causal variant. We ran COLOC with the default parameters and evaluated the evidence of colocalization based on the posterior probability of H4.
URLs
1000 Genomes Project, http://www.1000genomes.org/.
gnomAD, https://gnomad.broadinstitute.org/.
BWA-MEM, http://bio-bwa.sourceforge.net/.
GATK, https://software.broadinstitute.org/gatk/.
Beagle, https://faculty.washington.edu/browning/beagle/beagle.html.
PLINK, https://www.cog-genomics.org/plink2.
Cutadapt, http://cutadapt.readthedocs.io/en/stable/index.html.
fastp, https://github.com/OpenGene/fastp.
bowtie, http://bowtie-bio.sourceforge.net/index.shtml.
miRBase, http://www.mirbase.org/.
featureCounts, http://subread.sourceforge.net/.
PEER, http://www.sanger.ac.uk/science/tools/peer.
EIGENSTRAT, https://www.hsph.harvard.edu/alkes-price/software/.
MatrixEQTL, http://www.bios.unc.edu/research/genomic_software/Matrix_eQTL/.
WASP, https://github.com/bmvdgeijn/WASP.
Genome STRiP, http://software.broadinstitute.org/software/genomestrip/.
S-PrediXcan, https://github.com/hakyimlab/MetaXcan.
PredictDB Pipeline, https://github.com/hakyimlab/PredictDB_Pipeline_GTEx_v7.
R, https://www.r-project.org/.
NBDC Human Database, https://humandbs.biosciencedbc.jp/en/.
COLOC, https://cran.r-project.org/web/packages/coloc/index.html.
Acknowledgement
We thank Professor Yoichiro Kamatani at RIKEN Center for Integrative Medical Sciences for supporting the study.
Conflict of Interest statement. The authors declare that no conflicts of interest exist.
Contributor Information
Kyuto Sonehara, Department of Statistical Genetics, Osaka University Graduate School of Medicine, Suita 565-0871, Japan; Integrated Frontier Research for Medical Science Division, Institute for Open and Transdisciplinary Research Initiatives (OTRI), Osaka University, Suita 565-0871, Japan.
Saori Sakaue, Department of Statistical Genetics, Osaka University Graduate School of Medicine, Suita 565-0871, Japan; Center for Data Sciences, Harvard Medical School, Boston, MA 02114, USA; Divisions of Genetics and Rheumatology, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA; Program in Medical and Population Genetics, Broad Institute of Harvard and MIT, Cambridge, MA 02142, USA; Laboratory for Statistical Analysis, RIKEN Center for Integrative Medical Sciences, Yokohama 230-0045, Japan.
Yuichi Maeda, Integrated Frontier Research for Medical Science Division, Institute for Open and Transdisciplinary Research Initiatives (OTRI), Osaka University, Suita 565-0871, Japan; Department of Respiratory Medicine and Clinical Immunology, Osaka University Graduate School of Medicine, Suita 565-0871, Japan; Laboratory of Immune Regulation, Department of Microbiology and Immunology, Osaka University Graduate School of Medicine, Suita 565-0871, Japan.
Jun Hirata, Department of Statistical Genetics, Osaka University Graduate School of Medicine, Suita 565-0871, Japan; Pharmaceutical Discovery Research Laboratories, Teijin Pharma Limited, Hino 191-8512, Japan.
Toshihiro Kishikawa, Department of Statistical Genetics, Osaka University Graduate School of Medicine, Suita 565-0871, Japan; Department of Otorhinolaryngology - Head and Neck Surgery, Osaka University Graduate School of Medicine, Suita 565-0871, Japan; Department of Head and Neck Surgery, Aichi Cancer Center Hospital, Nagoya 464-8681, Japan.
Kenichi Yamamoto, Department of Statistical Genetics, Osaka University Graduate School of Medicine, Suita 565-0871, Japan; Department of Pediatrics, Osaka University Graduate School of Medicine, Suita 565-0871, Japan; Laboratory of Statistical Immunology, Immunology Frontier Research Center (WPI-IFReC), Osaka University, Suita 565-0871, Japan.
Hidetoshi Matsuoka, Rheumatology and Allergology, NHO Osaka Minami Medical Center, Kawachinagano 586-8521, Japan.
Maiko Yoshimura, Rheumatology and Allergology, NHO Osaka Minami Medical Center, Kawachinagano 586-8521, Japan.
Takuro Nii, Department of Respiratory Medicine and Clinical Immunology, Osaka University Graduate School of Medicine, Suita 565-0871, Japan; Laboratory of Immune Regulation, Department of Microbiology and Immunology, Osaka University Graduate School of Medicine, Suita 565-0871, Japan.
Shiro Ohshima, Rheumatology and Allergology, NHO Osaka Minami Medical Center, Kawachinagano 586-8521, Japan.
Atsushi Kumanogoh, Integrated Frontier Research for Medical Science Division, Institute for Open and Transdisciplinary Research Initiatives (OTRI), Osaka University, Suita 565-0871, Japan; Department of Respiratory Medicine and Clinical Immunology, Osaka University Graduate School of Medicine, Suita 565-0871, Japan; Department of Immunopathology, Immunology Frontier Research Center (WPI-IFReC), Osaka University, Suita 565-0871, Japan.
Yukinori Okada, Department of Statistical Genetics, Osaka University Graduate School of Medicine, Suita 565-0871, Japan; Integrated Frontier Research for Medical Science Division, Institute for Open and Transdisciplinary Research Initiatives (OTRI), Osaka University, Suita 565-0871, Japan; Laboratory of Statistical Immunology, Immunology Frontier Research Center (WPI-IFReC), Osaka University, Suita 565-0871, Japan; The Center for Infectious Disease Education and Research (CiDER), Osaka University, Suita 565-0871, Japan; Laboratory for Systems Genetics, RIKEN Center for Integrative Medical Sciences, Yokohama 230-0045, Japan.
Data Availability
The individual-level gene expression data and the summary statistics of eQTL analysis were deposited in the National Bioscience Database Center (NBDC) Human Database (https://humandbs.biosciencedbc.jp/en/) with the accession number of hum0197. The data are also available at our pheweb.jp website (https://pheweb.jp/).
Funding
Japan Society for the Promotion of Science (JSPS) KAKENHI (19H01021 and 20K21834); the Japan Agency for Medical Research and Development (AMED; JP21km0405211, JP21ek0109413, JP21gm4010006, JP21km0405217 and JP21ek0410075); JST Moonshot R&D (JPMJMS2021 and JPMJMS2024); Takeda Science Foundation; Bioinformatics Initiative of Osaka University Graduate School of Medicine; Clinical Investigator’s Research Project of the Osaka University Graduate School of Medicine; Grant Program for Next Generation Principal Investigators at Immunology Frontier Research Center (WPI-IFReC), Osaka University. S.S. was in part supported by the Mochida Memorial Foundation for Medical and Pharmaceutical Research; Kanae Foundation for the promotion of medical science; Astellas Foundation for Research on Metabolic Disorder and the JCR Grant for Promoting Basic Rheumatology. K.S. was supported by the Takeda Science Foundation and Integrated Frontier Research for Medical Science Division, Institute for Open and Transdisciplinary Research Initiatives (OTRI), Osaka University.
Author Contributions
K.S., S.S. and Y.O. designed the study and wrote the manuscript. K.S., S.S. and K.Y. conducted bioinformatics analysis. Y.M., J.H., T.K., K.Y., H.M., M.Y. and T.N. collected the samples. J.H. constructed the data for eQTL analysis. S.S., S.O., A.K. and Y.O. supervised the study.
References
- 1. Lee, R.C., Feinbaum, R.L. and Ambros, V. (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell, 75, 843–854. [DOI] [PubMed] [Google Scholar]
- 2. Lee, R.C. and Ambros, V. (2001) An extensive class of small RNAs in Caenorhabditis elegans. Science, 294, 862–864. [DOI] [PubMed] [Google Scholar]
- 3. Lee, R., Feinbaum, R. and Ambros, V. (2004) A short history of a short RNA. Cell, 116, S89–S92. [DOI] [PubMed] [Google Scholar]
- 4. Lu, J., Getz, G., Miska, E.A., Alvarez-Saavedra, E., Lamb, J., Peck, D., Sweet-Cordero, A., Ebert, B.L., Mak, R.H., Ferrando, A.A. et al. (2005) MicroRNA expression profiles classify human cancers. Nature, 435, 834–838. [DOI] [PubMed] [Google Scholar]
- 5. Esquela-Kerscher, A. and Slack, F.J. (2006) Oncomirs — microRNAs with a role in cancer. Nat. Rev. Cancer, 6, 259–269. [DOI] [PubMed] [Google Scholar]
- 6. Kumar, M.S., Lu, J., Mercer, K.L., Golub, T.R. and Jacks, T. (2007) Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat. Genet., 39, 673–677. [DOI] [PubMed] [Google Scholar]
- 7. Croce, C.M. (2009) Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet., 10, 704–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brest, P., Lapaquette, P., Souidi, M., Lebrigand, K., Cesaro, A., Vouret-Craviari, V., Mari, B., Barbry, P., Mosnier, J.-F., Hébuterne, X. et al. (2011) A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat. Genet., 43, 242–245. [DOI] [PubMed] [Google Scholar]
- 9. Zwiers, A., Kraal, L., Kraan, T.C.T.M.v.d.P., Wurdinger, T., Bouma, G. and Kraal, G. (2012) Cutting edge: a variant of the IL-23R gene associated with inflammatory bowel disease induces loss of MicroRNA regulation and enhanced protein production. J. Immunol., 188, 1573–1577. [DOI] [PubMed] [Google Scholar]
- 10. Okada, Y., Muramatsu, T., Suita, N., Kanai, M., Kawakami, E., Iotchkova, V., Soranzo, N., Inazawa, J. and Tanaka, T. (2016) Significant impact of miRNA–target gene networks on genetics of human complex traits. Sci. Rep., 6, 22223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sakaue, S., Hirata, J., Maeda, Y., Kawakami, E., Nii, T., Kishikawa, T., Ishigaki, K., Terao, C., Suzuki, K., Akiyama, M. et al. (2018) Integration of genetics and miRNA–target gene network identified disease biology implicated in tissue specificity. Nucleic Acids Res., 46, 11898–11909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huan, T., Rong, J., Liu, C., Zhang, X., Tanriverdi, K., Joehanes, R., Chen, B.H., Murabito, J.M., Yao, C., Courchesne, P. et al. (2015) Genome-wide identification of microRNA expression quantitative trait loci. Nat. Commun., 6, 6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li, J., Xue, Y., Amin, M.T., Yang, Y., Yang, J., Zhang, W., Yang, W., Niu, X., Zhang, H.-Y. and Gong, J. (2020) ncRNA-eQTL: a database to systematically evaluate the effects of SNPs on non-coding RNA expression across cancer types. Nucleic Acids Res., 48, D956–D963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Borel, C., Deutsch, S., Letourneau, A., Migliavacca, E., Montgomery, S.B., Dimas, A.S., Vejnar, C.E., Attar, H., Gagnebin, M., Gehrig, C. et al. (2011) Identification of cis- and trans-regulatory variation modulating microRNA expression levels in human fibroblasts. Genome Res., 21, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Parts, L., Hedman, Å.K., Keildson, S., Knights, A.J., Abreu-Goodger, C., Bunt, M. van de, Guerra-Assunção, J.A., Bartonicek, N., Dongen, S. van, Mägi, R., et al. (2012) Extent, causes, and consequences of small RNA expression variation in human adipose tissue. PLoS Genet., 8, e1002704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Civelek, M., Hagopian, R., Pan, C., Che, N., Yang, W., Kayne, P.S., Saleem, N.K., Cederberg, H., Kuusisto, J., Gargalovic, P.S. et al. (2013) Genetic regulation of human adipose microRNA expression and its consequences for metabolic traits. Hum. Mol. Genet., 22, 3023–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lappalainen, T., Sammeth, M., Friedländer, M.R., THoen, P.A.C., Monlong, J., Rivas, M.A., Gonzàlez-Porta, M., Kurbatova, N., Griebel, T., Ferreira, P.G. et al. (2013) Transcriptome and genome sequencing uncovers functional variation in humans. Nature, 501, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Siddle, K.J., Deschamps, M., Tailleux, L., Nédélec, Y., Pothlichet, J., Lugo-Villarino, G., Libri, V., Gicquel, B., Neyrolles, O., Laval, G. et al. (2014) A genomic portrait of the genetic architecture and regulatory impact of microRNA expression in response to infection. Genome Res., 24, 850–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stranger, B.E., Forrest, M.S., Dunning, M., Ingle, C.E., Beazley, C., Thorne, N., Redon, R., Bird, C.P., de Grassi, A., Lee, C. et al. (2007) Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science, 315, 848–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stranger, B.E., Nica, A.C., Forrest, M.S., Dimas, A., Bird, C.P., Beazley, C., Ingle, C.E., Dunning, M., Flicek, P., Koller, D. et al. (2007) Population genomics of human gene expression. Nat. Genet., 39, 1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dimas, A.S., Deutsch, S., Stranger, B.E., Montgomery, S.B., Borel, C., Attar-Cohen, H., Ingle, C., Beazley, C., Arcelus, M.G., Sekowska, M. et al. (2009) Common regulatory variation impacts gene expression in a cell type–dependent manner. Science, 325, 1246–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Juzenas, S., Venkatesh, G., Hübenthal, M., Hoeppner, M.P., Du, Z.G., Paulsen, M., Rosenstiel, P., Senger, P., Hofmann-Apitius, M., Keller, A. et al. (2017) A comprehensive, cell specific microRNA catalogue of human peripheral blood. Nucleic Acids Res., 45, 9290–9301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Newman, A.M., Steen, C.B., Liu, C.L., Gentles, A.J., Chaudhuri, A.A., Scherer, F., Khodadoust, M.S., Esfahani, M.S., Luca, B.A., Steiner, D. et al. (2019) Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol., 37, 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alföldi, J., Wang, Q., Collins, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P. et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Handsaker, R.E., Van Doren, V., Berman, J.R., Genovese, G., Kashin, S., Boettger, L.M. and McCarroll, S.A. (2015) Large multiallelic copy number variations in humans. Nat. Genet., 47, 296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sudmant, P.H., Rausch, T., Gardner, E.J., Handsaker, R.E., Abyzov, A., Huddleston, J., Zhang, Y., Ye, K., Jun, G., Hsi-Yang Fritz, M. et al. (2015) An integrated map of structural variation in 2,504 human genomes. Nature, 526, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chiang, C., Scott, A.J., Davis, J.R., Tsang, E.K., Li, X., Kim, Y., Hadzic, T., Damani, F.N., Ganel, L., Montgomery, S.B. et al. (2017) The impact of structural variation on human gene expression. Nat. Genet., 49, 692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Keegan, L.P., Gallo, A. and O’Connell, M.A. (2001) The many roles of an RNA editor. Nat. Rev. Genet., 2, 869–878. [DOI] [PubMed] [Google Scholar]
- 29. Farajollahi, S. and Maas, S. (2010) Molecular diversity through RNA editing: a balancing act. Trends Genet., 26, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ambros, V. (2004) The functions of animal microRNAs. Nature, 431, 350–355. [DOI] [PubMed] [Google Scholar]
- 31. Kawahara, Y., Zinshteyn, B., Sethupathy, P., Iizasa, H., Hatzigeorgiou, A.G. and Nishikura, K. (2007) Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science, 315, 1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Slotkin, W. and Nishikura, K. (2013) Adenosine-to-inosine RNA editing and human disease. Genome Med., 5, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rosenberg, B.R., Hamilton, C.E., Mwangi, M.M., Dewell, S. and Papavasiliou, F.N. (2011) Transcriptome-wide sequencing reveals numerous APOBEC1 mRNA-editing targets in transcript 3′ UTRs. Nat. Struct. Mol. Biol., 18, 230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gong, J., Wu, Y., Zhang, X., Liao, Y., Sibanda, V.L., Liu, W. and Guo, A.-Y. (2014) Comprehensive analysis of human small RNA sequencing data provides insights into expression profiles and miRNA editing. RNA Biol., 11, 1375–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zheng, Y., Ji, B., Song, R., Wang, S., Li, T., Zhang, X., Chen, K., Li, T. and Li, J. (2016) Accurate detection for a wide range of mutation and editing sites of microRNAs from small RNA high-throughput sequencing profiles. Nucleic Acids Res., 44, e123–e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paul, D., Ansari, A.H., Lal, M. and Mukhopadhyay, A. (2020) Human brain shows recurrent non-canonical MicroRNA editing events enriched for seed sequence with possible functional consequence. Non-Coding RNA, 6, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Joyce, C.E., Zhou, X., Xia, J., Ryan, C., Thrash, B., Menter, A., Zhang, W. and Bowcock, A.M. (2011) Deep sequencing of small RNAs from human skin reveals major alterations in the psoriasis miRNAome. Hum. Mol. Genet., 20, 4025–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Park, E., Guo, J., Shen, S., Demirdjian, L., Wu, Y.N., Lin, L. and Xing, Y. (2017) Population and allelic variation of A-to-I RNA editing in human transcriptomes. Genome Biol., 18, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Breen, M.S., Dobbyn, A., Li, Q., Roussos, P., Hoffman, G.E., Stahl, E., Chess, A., Sklar, P., Li, J.B., Devlin, B. et al. (2019) Global landscape and genetic regulation of RNA editing in cortical samples from individuals with schizophrenia. Nat. Neurosci., 22, 1402–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Akiyama, M., Okada, Y., Kanai, M., Takahashi, A., Momozawa, Y., Ikeda, M., Iwata, N., Ikegawa, S., Hirata, M., Matsuda, K. et al. (2017) Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat. Genet., 49, 1458–1467. [DOI] [PubMed] [Google Scholar]
- 41. Akiyama, M., Ishigaki, K., Sakaue, S., Momozawa, Y., Horikoshi, M., Hirata, M., Matsuda, K., Ikegawa, S., Takahashi, A., Kanai, M. et al. (2019) Characterizing rare and low-frequency height-associated variants in the Japanese population. Nat. Commun., 10, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ishigaki, K., Akiyama, M., Kanai, M., Takahashi, A., Kawakami, E., Sugishita, H., Sakaue, S., Matoba, N., Low, S.-K., Okada, Y. et al. (2020) Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat. Genet., 52, 669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barbeira, A.N., Dickinson, S.P., Bonazzola, R., Zheng, J., Wheeler, H.E., Torres, J.M., Torstenson, E.S., Shah, K.P., Garcia, T., Edwards, T.L. et al. (2018) Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun., 9, 1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. The GTEx Consortium (2020) The GTEx consortium atlas of genetic regulatory effects across human tissues. Science, 369, 1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Khamas, A., Ishikawa, T., Shimokawa, K., Mogushi, K., Iida, S., Ishiguro, M., Mizushima, H., Tanaka, H., Uetake, H. and Sugihara, K. (2012) Screening for epigenetically masked genes in colorectal cancer using 5-Aza-2′-deoxycytidine, microarray and gene expression profile. Cancer Genomics Proteomics, 9, 67–75. [PubMed] [Google Scholar]
- 46. Aguda, B.D., Kim, Y., Piper-Hunter, M.G., Friedman, A. and Marsh, C.B. (2008) MicroRNA regulation of a cancer network: consequences of the feedback loops involving miR-17-92, E2F, and Myc. Proc. Natl. Acad. Sci., 105, 19678–19683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Halappanavar, S., Nikota, J., Wu, D., Williams, A., Yauk, C.L. and Stampfli, M. (2013) IL-1 receptor regulates microRNA-135b expression in a negative feedback mechanism during cigarette smoke–induced inflammation. J. Immunol., 190, 3679–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Melling, G.E., Flannery, S.E., Abidin, S.A., Clemmens, H., Prajapati, P., Hinsley, E.E., Hunt, S., Catto, J.W.F., Coletta, R.D., Mellone, M. et al. (2018) A miRNA-145/TGF-β1 negative feedback loop regulates the cancer-associated fibroblast phenotype. Carcinogenesis, 39, 798–807. [DOI] [PubMed] [Google Scholar]
- 49. Wohlers, I., Bertram, L. and Lill, C.M. (2018) Evidence for a potential role of miR-1908-5p and miR-3614-5p in autoimmune disease risk using integrative bioinformatics. J. Autoimmun., 94, 83–89. [DOI] [PubMed] [Google Scholar]
- 50. Okada, Y., Momozawa, Y., Sakaue, S., Kanai, M., Ishigaki, K., Akiyama, M., Kishikawa, T., Arai, Y., Sasaki, T., Kosaki, K. et al. (2018) Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese. Nat. Commun., 9, 1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tadaka, S., Katsuoka, F., Ueki, M., Kojima, K., Makino, S., Saito, S., Otsuki, A., Gocho, C., Sakurai-Yageta, M., Danjoh, I. et al. (2019) 3.5KJPNv2: an allele frequency panel of 3552 Japanese individuals including the X chromosome. Hum. Genome Var., 6, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Browning, B.L., Zhou, Y. and Browning, S.R. (2018) A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet., 103, 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Martin, M. (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J., 17, 10–12. [Google Scholar]
- 54. Chen, S., Zhou, Y., Chen, Y. and Gu, J. (2018) fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics, 34, i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. de Hoon, M.J.L., Taft, R.J., Hashimoto, T., Kanamori-Katayama, M., Kawaji, H., Kawano, M., Kishima, M., Lassmann, T., Faulkner, G.J., Mattick, J.S. et al. (2010) Cross-mapping and the identification of editing sites in mature microRNAs in high-throughput sequencing libraries. Genome Res., 20, 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hu, H.Y., He, L., Fominykh, K., Yan, Z., Guo, S., Zhang, X., Taylor, M.S., Tang, L., Li, J., Liu, J. et al. (2012) Evolution of the human-specific microRNA miR-941. Nat. Commun., 3, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kozomara, A., Birgaoanu, M. and Griffiths-Jones, S. (2019) miRBase: from microRNA sequences to function. Nucleic Acids Res., 47, D155–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liao, Smyth, G.K. and Shi, W. (2014) Feature counts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30, 923–930. [DOI] [PubMed] [Google Scholar]
- 59. Love, M.I., Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Altshuler, D., Donnelly, P. and The International HapMap Consortium (2005) A haplotype map of the human genome. Nature, 437, 1299–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stegle, O., Parts, L., Piipari, M., Winn, J. and Durbin, R. (2012) Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc., 7, 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shabalin, A.A. (2012) Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics, 28, 1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. The GTEx Consortium, Ardlie, K.G., Deluca, D.S., Segre, A.V., Sullivan, T.J., Young, T.R., Gelfand, E.T., Trowbridge, C.A., Maller, J.B., Tukiainen, T. et al. (2015) The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science, 348, 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ishigaki, K., Kochi, Y., Suzuki, A., Tsuchida, Y., Tsuchiya, H., Sumitomo, S., Yamaguchi, K., Nagafuchi, Y., Nakachi, S., Kato, R. et al. (2017) Polygenic burdens on cell-specific pathways underlie the risk of rheumatoid arthritis. Nat. Genet., 49, 1120–1125. [DOI] [PubMed] [Google Scholar]
- 65. Storey, J.D. and Tibshirani, R. (2003) Statistical significance for genomewide studies. Proc. Natl. Acad. Sci., 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. de Rie, D., Abugessaisa, I., Alam, T., Arner, E., Arner, P., Ashoor, H., Åström, G., Babina, M., Bertin, N., Burroughs, A.M. et al. (2017) An integrated expression atlas of miRNAs and their promoters in human and mouse. Nat. Biotechnol., 35, 872–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. van de Geijn, B., McVicker, G., Gilad, Y. and Pritchard, J.K. (2015) WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat. Methods, 12, 1061–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Alon, S., Mor, E., Vigneault, F., Church, G.M., Locatelli, F., Galeano, F., Gallo, A., Shomron, N. and Eisenberg, E. (2012) Systematic identification of edited microRNAs in the human brain. Genome Res., 22, 1533–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang, Y., Xu, X., Yu, S., Jeong, K.J., Zhou, Z., Han, L., Tsang, Y.H., Li, J., Chen, H., Mangala, L.S. et al. (2017) Systematic characterization of A-to-I RNA editing hotspots in microRNAs across human cancers. Genome Res., 27, 1112–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Giambartolomei, C., Vukcevic, D., Schadt, E.E., Franke, L., Hingorani, A.D., Wallace, C. and Plagnol, V. (2014) Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet., 10, e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The individual-level gene expression data and the summary statistics of eQTL analysis were deposited in the National Bioscience Database Center (NBDC) Human Database (https://humandbs.biosciencedbc.jp/en/) with the accession number of hum0197. The data are also available at our pheweb.jp website (https://pheweb.jp/).