

Abstract
The varicella-zoster virus (VZV) genome, as with all other human herpesviruses, comprises both unique and repeated regions. In addition, the genome includes six sequences of tandemly repeating short elements termed reiteration regions, designated R1 to R5. The R4 element is duplicated, with one copy each in the terminal repeat short (TRs) and the internal repeat short (IRs) regions. The reiteration regions (R) are an understudied feature of the VZV genome even though three of them (R1, R2 and R3) reside inside the coding regions of important viral genes. R1 is located in ORF11, a tegument protein and the homoolog to herpes simplex virus UL47. R2 is located in ORF 14, which encodes glycoprotein C. R3 is located in ORF22, a component of the viral tegument and the largest gene product encoded by the virus. It is the homolog to herpes simplex virus UL36. R4A is located in the IRs and is duplicated, usually in inverted orientation, as R4B in the TRs. R5 is located in a non-coding region between ORF60 and 61. We developed primers to amplify and Sanger sequence all six reiteration regions, including primers that independently amplify the two copies of R4. We sequenced the reiteration regions from more than 80 genomes for viruses selected from the Kaiser Permanente Herpes Zoster Study, each of which was sampled from patients with well-characterized cases of herpes zoster. Samples were selected to provide a broad representation of VZV clades. The remainder of these genomes were sequenced using the Illumina MiSeq platform.
Introduction
Varicella Zoster Virus (VZV) is an alphaherpesvirus of the family Herpesviridae. Primary infection causes varicella (chickenpox), an acute and typically uncomplicated febrile pruritic rash illness. During acute varicella, VZV establishes a permanent latent infection in the cranial and dorsal root ganglia. The virus can reactivate, most frequently after the age of 50, to cause herpes zoster (HZ), also called shingles, a rash illness that is often associated with severe pain. About 10% of HZ cases progress to postherpetic neuralgia, which is characterized by continued intense local pain that persists for months to years after skin lesions have resolved.
Varicella-zoster virus is a double-stranded DNA virus with an average genome size of 125 Kb, encoding at least 71 open reading frames. The genome is subdivided into unique and repeat regions and has analogous genomic architecture to herpes simplex virus. Thus, the terminal repeat long (TRL) region of VZV is shorter than the terminal repeat short (TRS), a carry-over of designations from the similar architecture of the HSV genome (1, 2). The regions of the genome are (from left to right) designated the TRL, unique long (UL), internal repeat long (IRL), internal repeat short (IRS), unique short (US) and TRS. The unique long (UL) is flanked by the TRL and the IRL and at ~100Kb contains most of the open reading frames of VZV. The unique short (US) region (5.2 Kb) is flanked by the IRS region (7.3 Kb) and TRS region (7.3 Kb); this complex can exist in two isomeric forms that nearly always attach to the 3’ end of the UL region (3). The R regions are clusters of short repeating elements located in 6 positions on the VZV genome. The R regions each vary among strains in the number of copies of repeating elements present, and thus are variable in length.
R1 is located within the coding region of ORF11, which encodes a tegument protein involved in RNA binding and may be required for VZV pathogenesis in skin (4). R1 comprises a mixture of three repeating elements of 6, 15 and 18 base pairs (bp), respectively. The number of elements in R1 regions ranges from 12 to 20, and terminates with the 3bp sequence, GGA.
R2 is located in the coding region of VZV glycoprotein C (gC), which is involved in viral attachment; it also binds to complement component C3b, preventing the generation of C5 convertase and thereby blocking the complement cascade (5). R2 has a 42bp repeat element and terminates in a 32bp element that also varies in sequence.
R3 is situated in the coding region of the large tegument protein , and is involved in capsid transport and tegumentation and is essential for VZV growth in cell culture (6). The repeat element is only 9 bp in length, terminating in a 4 bp partial element, GCCC. R3 has previously been observed to have the greatest variation in size of all of the reiteration regions and has been the most problematic for determining a clean sequence (3, 7–9).
R4 is located in a non-coding region in close proximity to the origins of replication (ORI). R4 consists of a 27 bp repeating unit terminating with a partial 11 bp sequence; it has not been reported to exhibit sequence variation within the element. R4 is present in the genome in two copies, in the IRS (located between ORF62 and ORF63) and the TRS (between ORF70 and ORF71). Since our study was designed to independently sequence each of the two R4 regions, we have designated them as R4A (IRS) and R4B (TRS).
R5 is situated in a non-coding region between ORF 60 and 61. It comprises two elements of 88 bp and 24 bp, typically beginning and ending with a copy of the longer element. The elements also generally alternate. Some variability was observed in the 88-mer, but not in the 24-mer.
We determined full genome sequences for 73 of 84 viral samples obtained from well-characterized cases of HZ, using a combination of Illumina and Sanger sequencing. Most or all of the R regions were determined for 84 samples. Our evaluation of the six R regions from each of these viruses revealed a number of new findings, including: 1) a number of previously unreported variant elements (several in R4); 2) the clade-specific association of some variant elements and motifs of elements for several of the R regions; 3) the observation of within-strain mixtures for several of the R regions; 4) and within-strain length variation between R4A and R4B in nearly half of the viruses studied.
Methods
Study samples:
Samples of previously extracted and genotyped viral DNA were selected from the 1033 confirmed HZ case patients enrolled in the Kaiser Permanente Herpes Zoster Study (KPHZ) (13, 14). This cohort study was conducted among Kaiser Permanente of Southern California members aged 60 and older. Cohorts of HZ vaccinated and unvaccinated persons with well-characterized cases of HZ were matched by age and sex; DNA from cases with herpes zoster were selected to provide a broad cross-section of VZV genotypes, with bias toward genotypes for which fewer complete genome sequences have been published (Clades 2, 4, 5 and 6). Clade designations were determined from complete genome sequences applying the nomenclature system proposed by Breuer, et al (15). For 11 samples that could not be completely sequenced using MiSeq, genotype was established using the revised SNP-based scheme published by Jensen, et al (16).
Sequencing of Repeat Regions:
Two sets of primers each were developed for PCR amplification of R1, R3, R4A and R4B. Only one set of primers each was designed to amplify R2 and R5. With the exception of R2, for which the same primer set could also be used for sequencing, additional primers were developed for that purpose. The PCR reaction volumes for R1, R2 and R5 were 10μl total volume with 0.5μl of template. For R4A and R4B a 20μl reaction was used with 2.0μl of template. R3 PCR was performed in two rounds with 2.0μl of original DNA template used in a 20μl reaction for the first round and 2.0μl of the first round PCR used as the template in the second semi-nested round of PCR with a total volume of 20μl. The PCR enzyme extensor 2X master mix was used (ThermoFisher, Waltham, MA). Each reaction required 0.4μM of each primer. After the first ten cycles, elongation time was increased by 10 seconds in each of 25 cycles. The starting elongation time and annealing temperatures as well as the primer names and positions (Dumas reference strain, accession #NC_001348.1) are listed in Table 1. Sanger sequencing was performed on an ABI 3730 Genetic analyzer using Big Dye V1.1 (ThermoFisher Scientific).
Table 1.
Primers and conditions used for PCR and sequencing. Nucleotide positions are based on the reference strain Dumas (Clade 1, GenBank accession # NC_001348.1). Extensor 2X master mix was used per manufacturer’s instructions; the elongation time was increased by 10 seconds per cycle starting at cycle 11.
| Region | Annealing Temp | Elongation Time | Forward Primer | Reverse Primer |
|---|---|---|---|---|
| R1 Option 1 | 55°C | 2 minutes | 1iiF 13459-13480 | 2CR 14696-14673 |
| R1 Option 2 | 58°C | 1 minute | 2AF 13809-13830 | New2BR 14337-14318 |
| R2 | 55°C | 1 minute | R2Fpcr 20593-20617 | R2Rpcr 21143-21126 |
| R5 | 61°C | 2 minutes | 8VF 101650-101669 | 8XR 102573-102553 |
| R4A Option 1 | 61°C | 6 minutes | 9IF 108433-108454 | 9UR 113719-113698 |
| R4A Option 2 | 61°C | 6 minutes | 9IF 108433-108454 | 9TR 113268-113247 |
| R4B Option 1 | 55°C | 5 minutes | 9iF2 116319-116336 | 10FR2 120700- 120685 |
| R4B Option 2 | 61°C | 5 minutes | 9iiF 116781-116801 | 10FR 120704- 120683 |
| R3 is semi-nested but follows the same PCR protocol apart from having two rounds of amplification prior to sequencing | ||||
| R3 semi-nested Option 1 | ||||
| Round 1 | 55°C | 8 minutes | 3861F 37914-37931 | 4MR 44413-44392 |
| Round 2 | 58°C | 8 minutes | 3861F 37914-37931 | 4LR 44123-44103 |
| R3 semi-nested Option 2 | ||||
| Round 1 | 58°C | 8 minutes | 2139 37799-37816 | 4LR 44123-44103 |
| Round 2 | 58°C | 8 minutes | 3861F 37914-37931 | 4LR 44123-44103 |
| Sequencing Primers | ||||
|---|---|---|---|---|
| Region | Forward | Reverse | ||
| R1 | 2AF 13809-13830 | New 2BR 14337-14318 | ||
| R2 | R2Fpcr 20593-20617 | R2Rpcr 21143-21126 | ||
| R5 | R5Fseq 101825-101839 | R5Rseq 102335-102316 | ||
| R4A | R4Aseq 109628-109647 | R4AR 110016-110000 | ||
| R4B | R4Bseq 119862-119879 | R4BRseq 120268-120250 | ||
| R3 | R3Fpcr 41286-41303 | R3R-JB 41806-41784 | ||
Ruling out PCR induced artefacts in R region mixtures:
PCR products were cloned from Ellen strain DNA for R1, R2 (TOPO TA cloning kit, Invitrogen, Carlsbad, CA), R3, R4A and R4B (TOPO XL PCR cloning kit, Invitrogen). PCR was completed using the Option 1 conditions for R1, R4A and R4B (Table 1). Conditions for R3 were modified as indicated below to produce a shorter fragment for cloning:
R3 round 1 59°C 8 minutes 2139 37799–37816 4LR 44123–44103
R3 round 2 59°C 3 minutes R3F3 40604–40623 4JR 44310–43288
Colonies were picked and plasmid DNA prepared for a single clone from each R region. This approach provided sufficient concentrations of DNA to sequence it directly, with no requirement for additional PCR. The number of copies of the element was determined from the sequence. In addition, 12 separate PCR reactions were set up for each set of primers. Both sets of primers were used for R1, R3, R4A and R4B and the single set of primers for R2. All of the PCR products were then sequenced and evaluated for the number of R region elements in each sample. The forward and reverse sequences were aligned using Sequencher 5.4.6 (Gene Codes Corp., Ann Arbor, MI).
Evaluation of number of R region variants in mixtures:
Topo TA cloning was performed according to the manufacturer’s specifications (see previous section).
PCR products for R4A and R4B from a single sample were cloned via TOPO XL PCR cloning kit per the manufacturer’s instructions. One hundred colonies were picked for both R4A and R4B, miniprep DNA was prepared and sequenced. This was repeated for an additional sample and 100 colonies were picked and sequenced. To determine whether or not the PacBio platform (Pacific Biosciences of California, Inc., Menlo Park, CA), which can read long DNA fragments, could reproduce results from Sanger sequencing, two runs on the same sample used for Sanger sequencing R4A were performed using different input concentrations of DNA. PacBio was performed in accordance with manufacturer’s instructions in the CDC Biotechnology Core Facility.
Sequencing of the remainder of the genome:
The remainder of these genomes was determined using the Miseq platform (Illumina, Inc., San Diego, CA) as previously described (17, 18).
Sequence analysis and assembly of R regions:
The pattern of reiteration region was analyzed by Geneious Version R11 (Biomatter, New Zealand). Unique repeat elements were identified and annotated alphabetically, starting with previously described elements (8). Identical reiteration regions across different samples were analyzed by ElimDupes (https://hcv.lanl.gov/content/sequence/ELIMDUPES/elimdupes.html).
Results
R4B DNA extracted from the same shingles specimen and amplified using two different primer sets revealed the presence of sequences with differences in the number of elements present in individual R4B regions. Figure 1 shows the sequences derived using two distinct primer sets on the same extracted DNA sample. The top two sequences (primer set 1) differ from the bottom two sequences (primer set 2) by 27bp – the length of a single repeat element in the R4 reiteration region.
Figure 1.
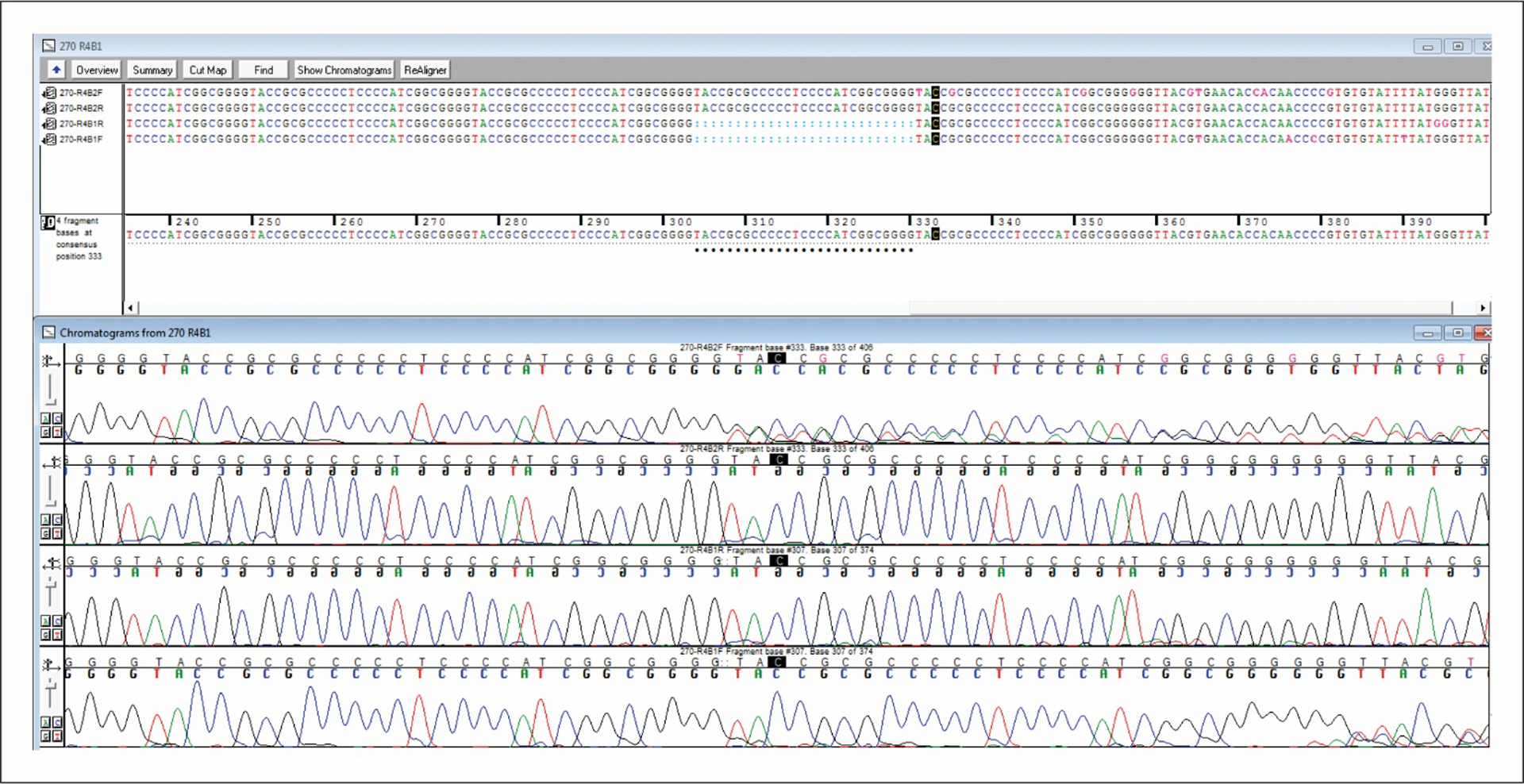
Mismatches are indicated by black dots. The top two sequences represent one pair of R4B primers and the bottom two sequences the other primer pair with the same sample. The addition of 27bp (length of a single repeating element for R4) brings the two sequences into alignment indicating there was one less copy of the reiteration region element in the second primer pair.
We considered whether a PCR artefact might introduce length polymorphisms, although the mechanism by which a single discrete 27bp element might be inserted or eliminated was unclear. To address this possibility, we cloned the PCR product from Ellen DNA for R1, R2, R3, R4A and R4B and prepared plasmid DNA from one clone for each of five regions (R5 was not evaluated because it has so little variation in element copy number). The DNA concentration was sufficient to sequence the DNA without PCR amplification. Using the plasmid DNA, we performed 12 different PCR reactions for each primer pair for each R region cloned. The two primer sets were used for R1, R3, R4A and R4B and a single primer set was used for R2. The results are shown in Table 2. There was no variation in copy number for any of the regions when compared to the unamplified plasmid DNA. Thus, there was every indication that PCR was not responsible for introducing the discrepancy in copy number seen with R4B primer sets 1 and 2.
Table 2.
Comparison of number of copies of the element for the R region PCR products with the number of copies from the original cloned DNA.
| R1 Clone 31 | R2 Clone 1 | R3 Clone 230 | R4A Clone 4 | R4B Clone 5 | |||||
|---|---|---|---|---|---|---|---|---|---|
| Tube # | Primer Set A | Primer Set B | Primer Set A | Primer Set A | Primer Set B | Primer Set A | Primer Set B | Primer Set A | Primer Set B |
| 1 | 14 | 14 | 7 | 4 | 4 | 7 | 7 | 8 | 8 |
| 2 | 14 | 14 | 7 | 4 | 4 | 7 | 7 | 8 | 8 |
| 3 | 14 | 14 | 7 | 4 | 4 | 7 | 7 | 8 | 8 |
| 4 | 14 | 14 | 7 | 4 | 4 | 7 | 7 | 8 | 8 |
| 5 | 14 | 14 | 7 | 4 | 4 | 7 | 7 | 8 | 8 |
| 6 | 14 | 14 | 7 | 4 | 4 | 7 | 7 | 8 | 8 |
| 7 | 14 | 14 | 7 | 4 | 4 | 7 | 7 | 8 | 8 |
| 8 | 14 | 14 | 7 | 4 | 4 | 7 | 7 | 8 | 8 |
| 9 | 14 | 14 | 7 | 4 | 4 | 7 | 7 | 8 | 8 |
| 10 | 14 | 14 | 7 | 4 | 4 | 7 | 7 | 8 | 8 |
| 11 | 14 | 14 | 7 | 4 | 4 | 7 | 7 | 8 | 8 |
| 12 | 14 | 14 | 7 | 4 | 4 | 7 | 7 | 8 | 8 |
| Copies in original clone | 14 | 14 | 7 | 4 | 4 | 7 | 7 | 8 | 8 |
The absence of errors introduced by the PCR protocols confirmed, we wanted to evaluate the number of variant R regions present in samples detected as mixtures. To this end, DNA obtained from a VZV sample with evidence of R region mixtures was cloned using the TOPO XL PCR cloning kit, and DNA from 100 clones each was sequenced for R4A and R4B. Analysis of the copy number for each of the clones in the one pair revealed the predominant copy number in both R4A and R4B was 9, but the range for R4A was 6–10 and for R4B was 8–11 (Figure 2). R4A for the second pair had a predominant copy number of 12 repeating elements, while R4B had two roughly equivalent predominant number of elements of 6 and 7.
Figure 2.
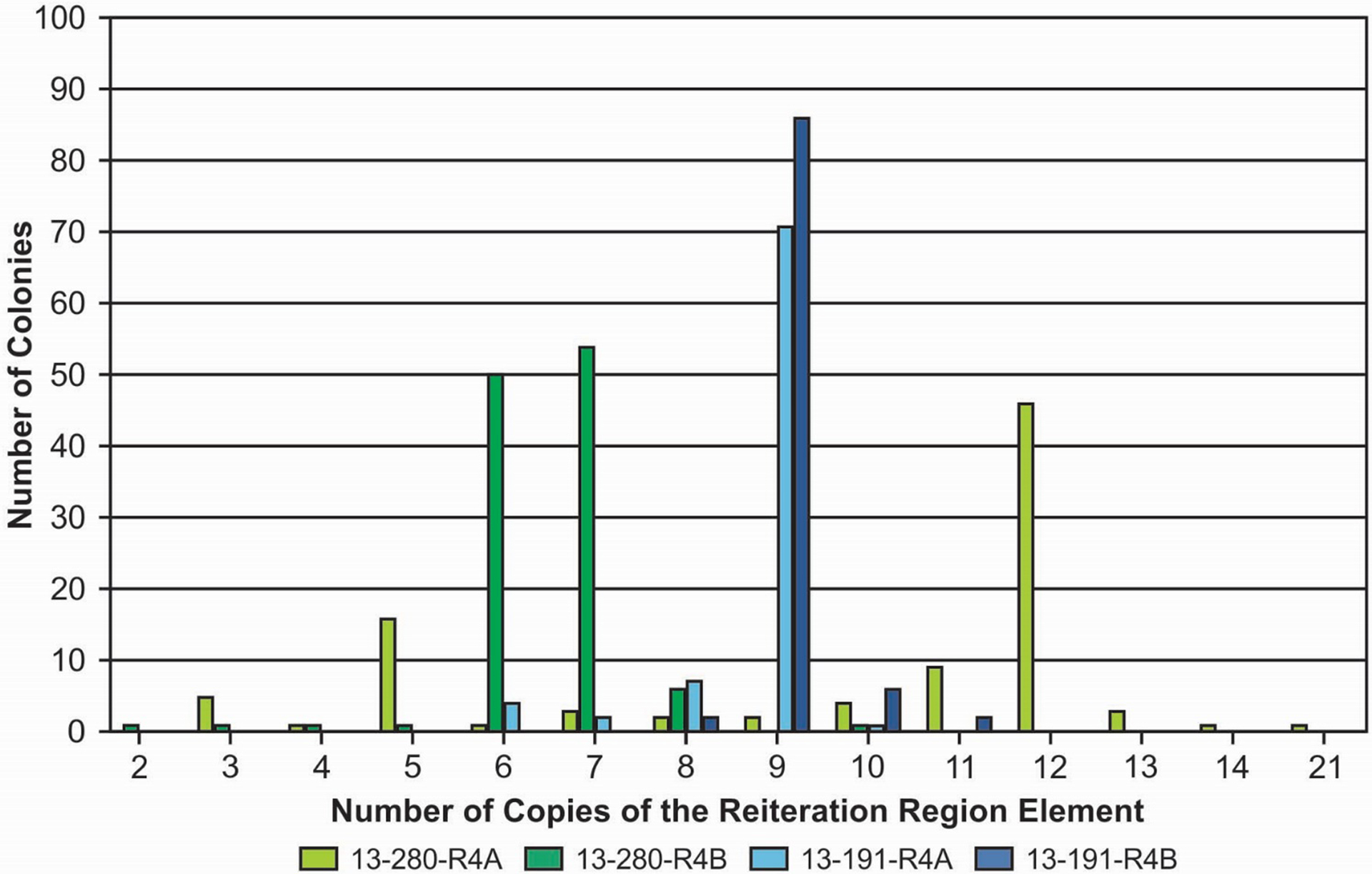
Evaluation of the number of copies of the element in R4A and R4B by TA cloning. In sample 13–191 both the R4A and the R4B had the same predominant copy number of 9 copies in the reiteration region. In 13–280, R4A had a predominant copy number of 12 while R4B was split almost evenly between 6 and 7 copies of the reiteration region.
Since TA cloning is impractical for evaluating large numbers of mixed R region sequences, we evaluated the PacBio platform, which is capable of reading long fragments of DNA, for its suitability for determining the relative frequency of R region variants in a mixture. The results of two separate PacBio runs are displayed in Figure 3. We also cloned this sample and picked colonies for sequencing to compare the results. By TA cloning, the predominant number of element copies was 12 (47%), followed by 5 copies (19%). The remaining 11 variants were present at levels of 10% or less. In contrast, both PacBio runs identified 5 copies as the predominant R region variant, with run 2 detecting a smaller peak at 12 copies.
Figure 3.
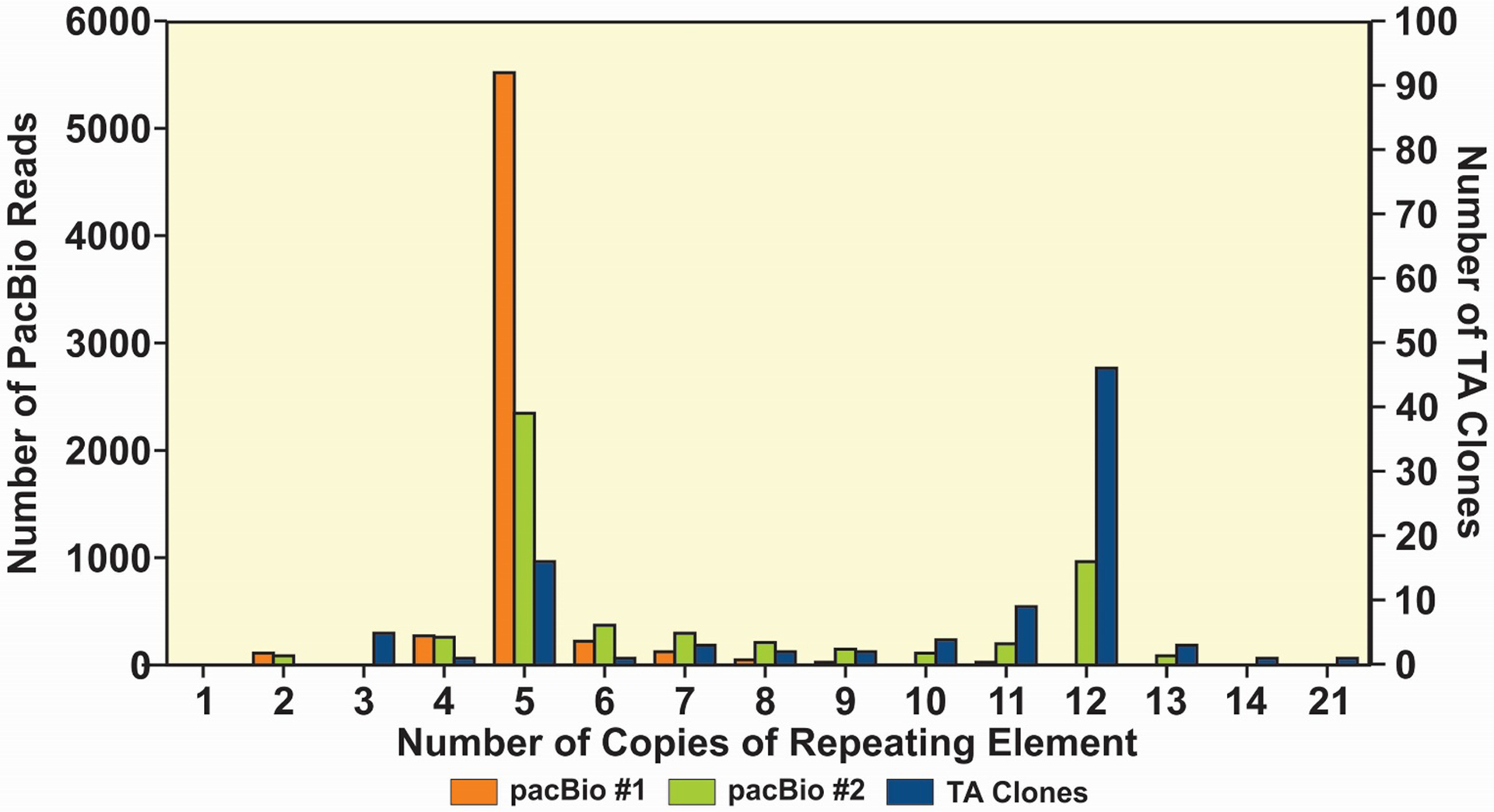
Comparison of copy number of elements from an R4A region by TA cloning and PacBio sequencing for the same sample. The TA cloning showed a predominant copy number of 12 with a smaller peak at 5 copies, while PacBio run #1 showed a predominant copy number of 5. PacBio run #2 also showed a predominant copy number of 5 with a smaller peak at 12 copies.
Whole genome sequencing of this clinical sample confirmed that it was not, apart from the variation observed in 3 of the reiteration regions (R3, R4A, R4B), a mixture of strains. We were able to determine complete genome sequences for 73 of the 84 KPHZ study samples using a combination of Illumina MiSeq and Sanger sequencing. The phylogenetic distribution of these specimens is shown in Figure 4. Note that the analysis of these samples has led to the identification of 2 potential new VZV clades; these will be addressed in future studies.
Figure 4.
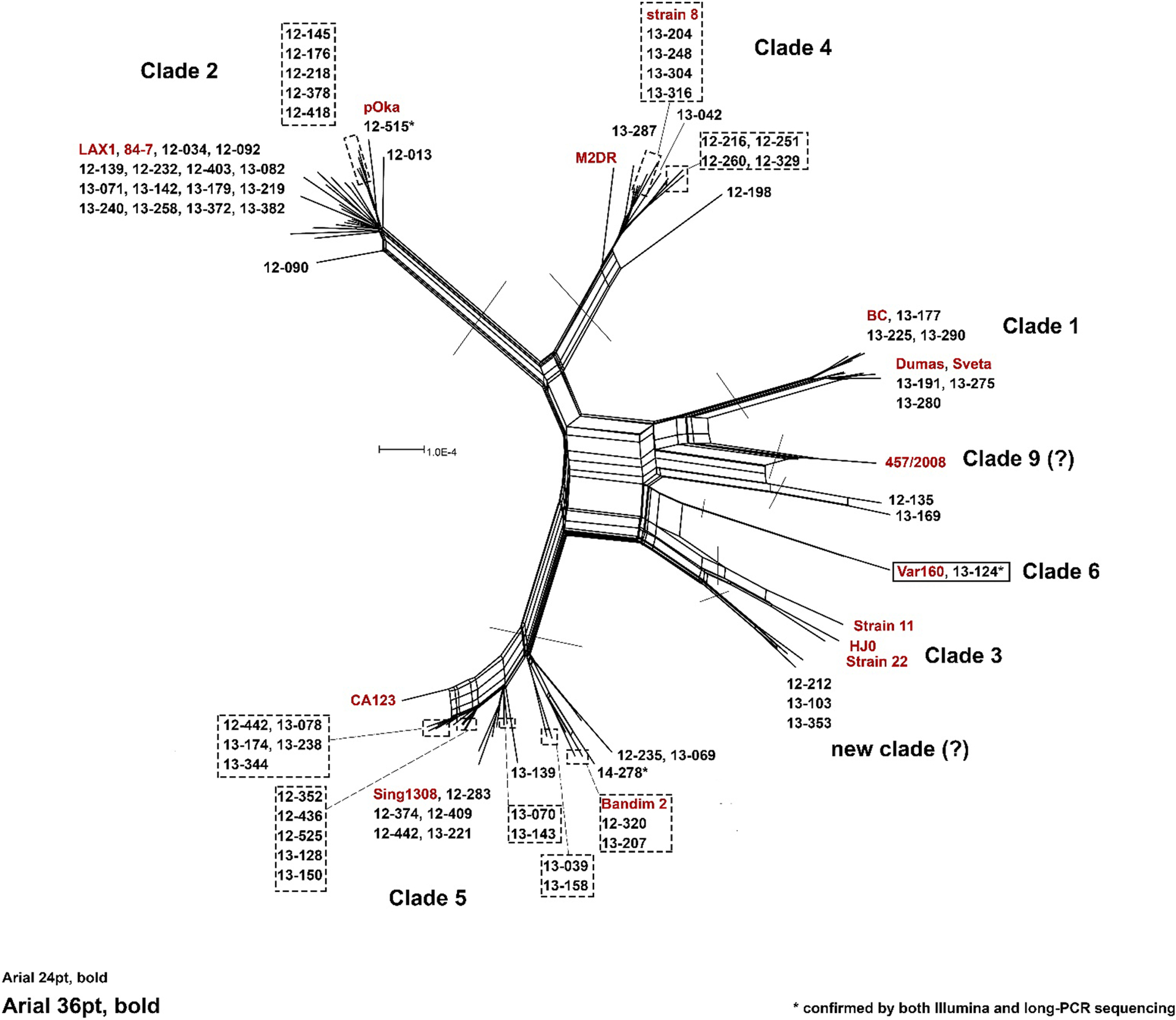
Phylogenetic network reveals clade distribution of study samples. A phylogenetic network was constructed using SplitsTree4 () and represents the alignment of all VZV genomes sequenced in this study and sixteen representative sequences (red text) chosen to highlight the six major established VZV clades (REF2). Note that repeat elements within the genomes (R1-R5 and the terminal repeat region) were masked prior to network construction.
Analyses of the six reiteration regions from these 84 specimens are displayed in Figures 5–9. Roughly 40% of the genomes revealed the presence of element copy number mixtures in at least one region; mixtures were identified only in R3, R4A and R4B, with a single exception in R1.
Figure 5.
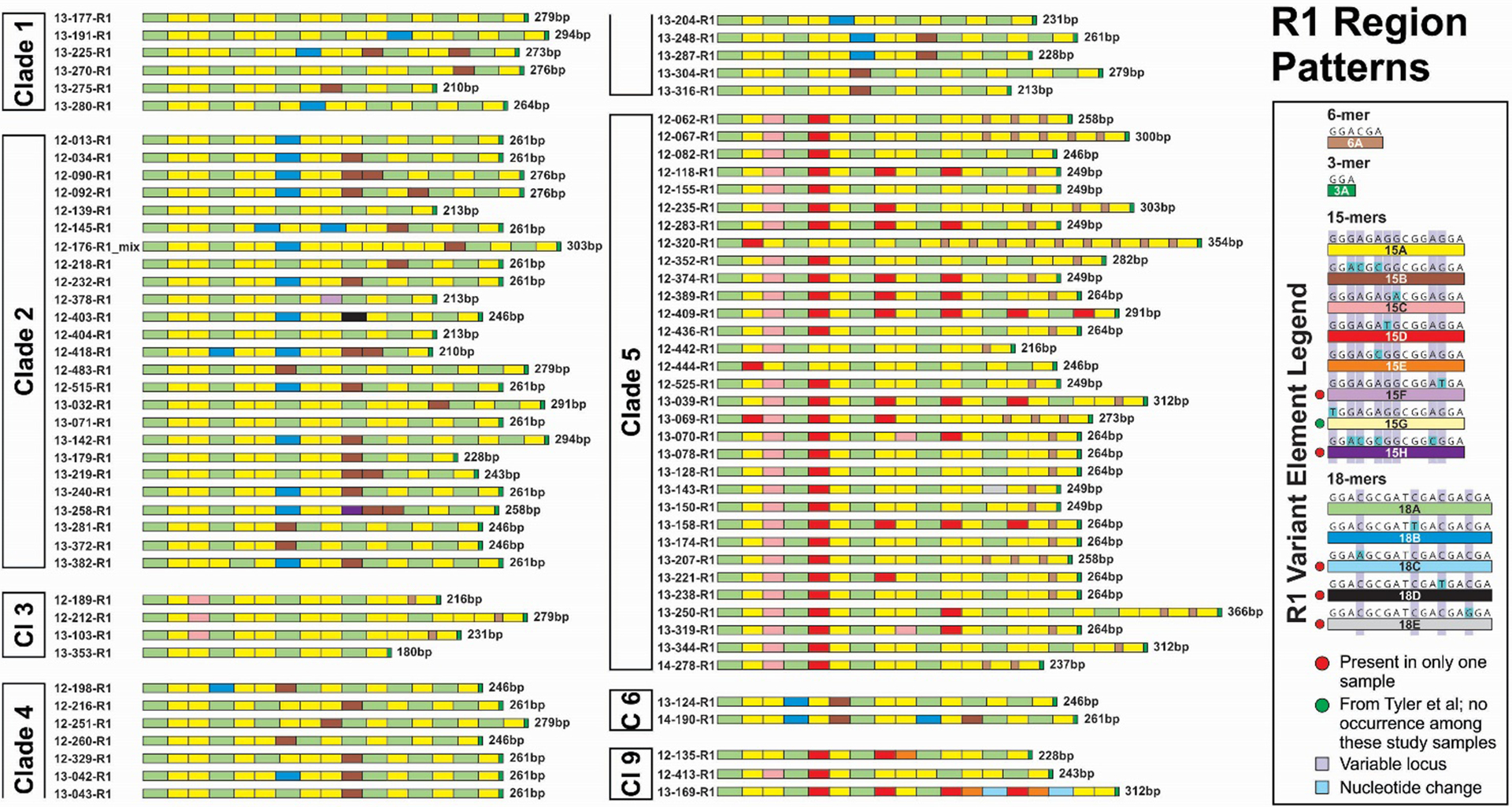
R1 variant element patterns for study samples grouped by VZV clade. Representations of all elements are scaled to reflect the relative actual length in base pairs.
Figure 9.
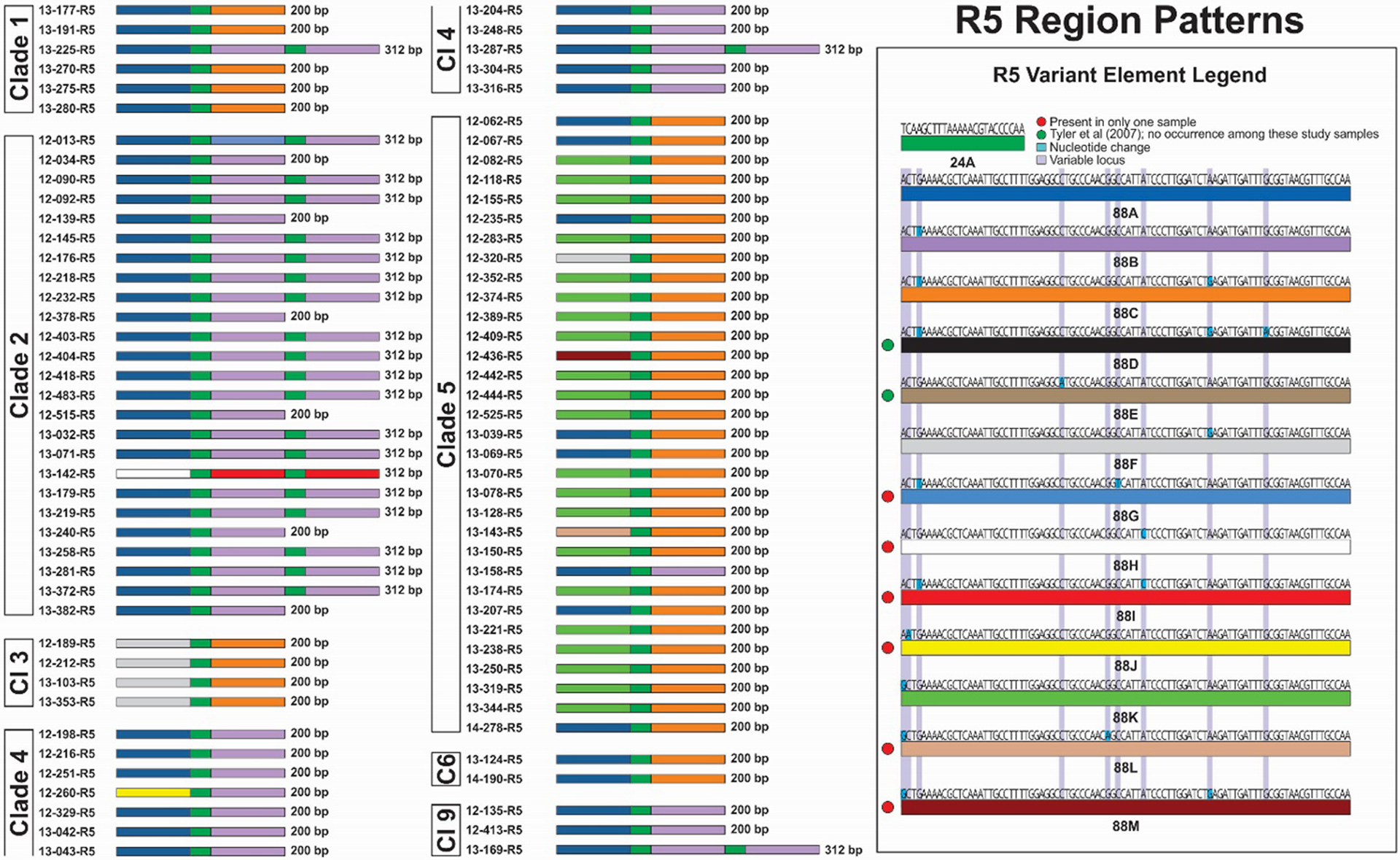
R5 variant element patterns for study samples grouped by VZV clade. Representations of all elements are scaled to reflect the relative actual length in base pairs.
The R1 element patterns are shown in Figure 5. The R1 regions comprise combinations of 15-mer, 18-mer, and 6-mer elements, always terminating with a GGA 3-mer. Including the variant elements observed by Tyler et al (8), a total of eight 15-mers and five 18-mers have now been identified. We provided a large text summary of DNA sequences for all of the R region variant elements in Supplementary Table 2. While no single element appears to be VZV clade-specific, Clades 1, 2, 4 and 6 were found to contain both 15B and 18B elements, but no 15C or 15D elements. For Clade 5 viruses and Clade 9 viruses, 15C and 15D elements were consistently present, but not 15B or 18B. Variability in R1 region length was substantially less than for R3, ranging from 180bp to 366bp, a little over 2-fold. We observed the recently identified novel 6bp element (6A, GGACGA) in R1 exclusively in Clade 3 and Clade 5 viruses. The 6A element was present as a single copy in 3 of 4 (75%) Clade 3 R1 sequences, and in 29 of 32 (91%) Clade 5 sequences. Among Clade 5 viruses, 6A was present in between 1 and 9 copies (mean = 2). Only a single genome (12–176, Clade 2) was found to contain a mixture of variant R1 sequences. A leading motif of R1 elements, 18A-15A-15C-18A-15D-15A-18A, is common to 28 of 32 Clade 5 samples and is found only in one other sample (12–413) which is a Clade 9 virus. Another leading element motif (18A-15A-15A-18A-15A-15A-18B-15A-15A) occurred in 11 of 25 (44%) Clade 2 viruses and otherwise only in three other genomes (13–042, 13–248 and 13–287, Clade 4). The 15D R1 element was found only in Clade 5 viruses and in three Clade 9 viruses.
We identified ten new variant elements in R2 region sequences; R2 sequences were determined for all 84 strains evaluated for this study (Figure 6). The length variation in R2 ranged from 200bp to 494bp, or about 2.5-fold. The variant elements 42A through G have been described previously, although 42D, 42F and 42G were not observed among the strains sequenced here. The association of specific elements and signature series motifs of elements are less pronounced for R2, but some clear trends are apparent. All but one of Clade 4 and Clade 9 viruses comprise combinations of 42A and 42B with a terminal 32A element. Seventeen of 25 (68%) Clade 2 viruses have a terminal 32C element, elsewhere found in only a single Clade 1 virus (13–225). Terminal 32B elements are dominant for Clade 5 viruses, but were also found in scattered genomes from other VZV clades. Element 42E occurred in all but one Clade 2 virus, usually in 2–4 copies; it was otherwise present as a single copy in one Clade 1 and two Clade 5 viruses and 3 of the 4 Clade 3 viruses.
Figure 6.

R2 variant element patterns for study samples grouped by VZV clade. Representations of all elements are scaled to reflect the relative actual length in base pairs.
Seventy-nine of the R3 regions from these VZV samples were successfully sequenced; of these, 33 (42%) had evidence for mixtures of variant element copy number (Figure 7). A total of 8 variants of R3 elements have now been identified, 7 of which were observed among these samples. The elements ranged from 2 copies of the 9-mer element (22bp total) to 54 copies (447bp total). Both the shortest and longest copy number variants were observed as pure R3 sequences (not mixtures). Given that this region is contained within the coding region of ORF22, the large tegument protein, a 20-fold difference in element length might be expected to impact protein function. The occurrence of clade-specific element motifs is less consistent for R3 than for some of the other R regions, but for the Clade 1 viruses in this study, all but one had the 9G element in the first position, followed by the 9A element (Figure 7). The remaining sequence (13–275) had the 9G element followed by a 9C element. We were unable to sequence an R3 region PCR product for 5 (6%) of the genomes.
Figure 7.

R3 variant element patterns for study samples grouped by VZV clade. Representations of all elements are scaled to reflect the relative actual length in base pairs. For R regions with mixtures the sequences with the strongest chromatogram signal are represented in the figure.
R4 sequences were obtained for 83 of 84 VZV samples (Figure 8); the R4A and R4B variant sequences with the strongest chromatogram profiles for these samples are displayed pairwise, side by side for ease of comparison. R4 regions that contained mixtures of sequences varying in copy number were detected as follows: 50 of 83 (60%) were found in either R4A, R4B or both; 11 had mixtures in R4A only (13%); 14 in R4B only (17%); and 25 had mixtures in R4A and B (30%). Altogether, 36 of 83 samples had mixtures in R4A (43%) and 39 of 83 had mixtures in R4B (47%). In addition, 37/83 (45%) of the samples revealed a length difference between predominant variant for R4A and R4B. The remainder were identical in both copies. R4A and R4B displayed the lowest number of variant elements compared with any of the other R regions (4 variant elements total).
Figure 8.
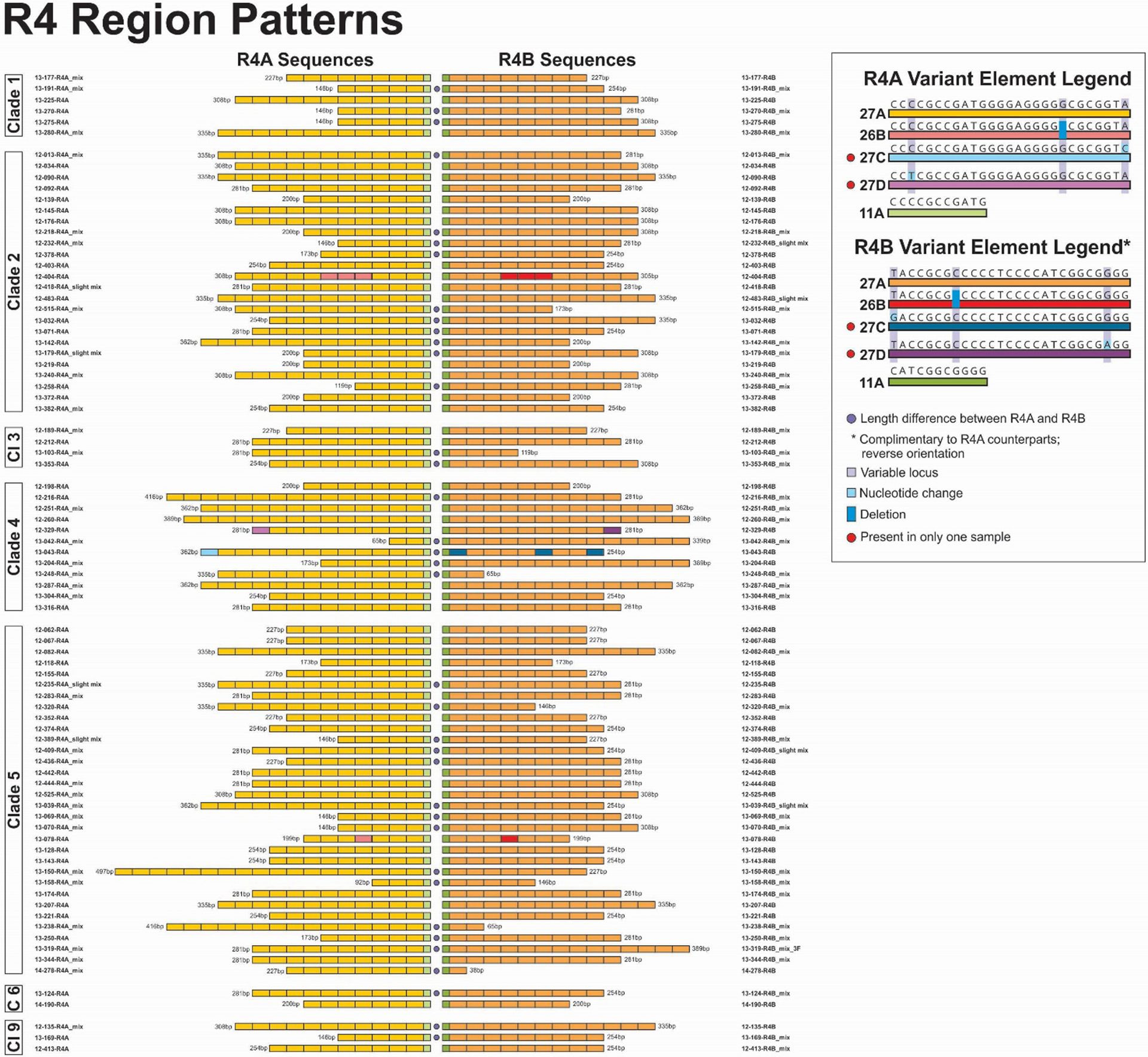
Pairwise comparison of R4A and R4B variant element patterns for study samples grouped by VZV clade. Representations of all elements are scaled to reflect the relative actual length in base pairs. For R regions with mixtures the sequences with the strongest chromatogram signal are represented in the figure.
The R5 region patterns are displayed in Figure 9. In terms of length and organization, R5 is the most consistent of all the R regions. There are two lengths and configurations of R5 regions: 1) 200 bp, comprising a 24bp element (24A) flanked on both ends by 88bp elements; and 2) 312 bp, which comprises three 88-mer elements alternating with two 24-mer elements (88-24-88-24-88). We have observed no sequence variation in the 24-mer element but, together with those reported by Tyler et al (8), a total of 13 alleles of the 88-mer elements have now been identified. Two of those previously reported, 88D and 88E were not present in any of the viruses evaluated in this report. Moreso than any of the other R regions, the element patterns of R5 regions were associated by VZV clade. Five of six (83%) Clade 1 viruses had the pattern 88A-24A-88C. Seventeen of 25 (68%) Clade 2 viruses had the pattern 88A-24A-88B-24A-88B. An additional 6 (24%) Clade 2 viruses had a short version of the same sequence (88A-24A-88B). Clade 4 viruses displayed the same pattern as Clade 2 viruses, but with the short version predominating. The Clade 3 viruses all had the pattern 88F-24A-88C. Clade 5 viruses displayed six patterns two of which were dominant: 88K-24A-88C (21 of 32 viruses, 66%) and 88A-24A-88C (7 of 32 viruses, 22%). Clade 6 viruses displayed the same pattern as Clade 1 viruses, and Clade 9 viruses displayed the same patterns as Clade 2 and 4 viruses. Of additional interest, the 88B and 88C elements are only found in the 2nd or 3rd 88-mer position and, with only 2 exceptions, 88G and 88I found in 12–013 and 13–142, Clade 2 are the only two 88-mer elements found in those positions.
A summary of samples that shared identical R regions sequences with at least one other sample are shown in Supplementary Figure 1. For R1, one sequence was shared by six samples, one with five samples, three sequences were shared among three samples each, and six pairs of samples had identical R1 sequences. In instances where an R1 region was identical in samples representing more than one VZV clade, Clades 2 and 4, and Clades 3 and 5 always grouped together. For R2, one sequence was shared by three samples (all Clade 1), and six pairs of samples had identical R2 region sequences. R3 and R5 region sequences, due to their reduced complexity and limited number of variant elements, included larger numbers of samples with shared sequences (Supplementary Figure 1). The R4 regions are constructed almost entirely from a single element sequence, and as such were not displayed in this figure. Supplementary Table 1 shows the GenBank accession numbers and summary data for the study samples.
Among the R regions characterized, a total of 16 allelic elements were detected in only a single sample. Five of these single occurrence elements were in R1 regions (15F, 15H, 18C, 18D and 18E), 6 in R2 regions (42J, 42L, 42M, 42O, 42P and 42Q), and 5 in R5 regions (88F, 88H, 88I, 88J and 88L). The stable circulation of these variant elements will need to be confirmed in broader studies. Numbers were limited for this study, and no apparent associations between HZ severity and R region element motif patterns were observed.
Discussion
In this study of the six VZV R regions in 84 clinical samples from cases of well-characterized HZ, we have confirmed and expanded on previous observations (3, 7, 8, 12, 19–21), and uncovered new properties of these unusual regions of the VZV genome.
Sanger sequencing of VZV DNA from the HZ samples using two different primer sets revealed that several of the R regions within a strain comprise mixtures with varying numbers of the repeat elements. This was a common feature for R3 and R4A and R4B. One mixture in R1 was observed. No evidence of mixtures was observed for either R2 or R5, but we cannot yet rule out whether they occur. Complete genome sequences for an additional 1,100 clinical HZ specimens are now being analyzed and are expected to provide definitive answers, not just for the R regions but for features across the entire VZV genome. Plasmid DNA preparations containing R1, R2, R3, R4A and R4B region DNA were sequenced directly without amplification and compared with PCR products from the same template. The number of copies of the variant element was identical for both materials, providing convincing evidence that the PCR protocols themselves were not introducing any errors in the element copy number. TOPO TA cloning of the R4A and R4B PCR products from strains with evidence of an R4 mixture detected four to twelve R4 variants, with one or two variants clearly dominating. As previously observed by Tyler and co-workers (8), using Sanger sequencing and shotgun cloning of the unique regions of the virus indicated that these viruses were clonal apart from the mixture of R region sequences. Davison and Scott (3) noted a size disparity between the native virion and the clone containing the R3 region in their report of the first complete genome sequence of VZV (Dumas strain). This was attributed to the instability of the repetitive structure of R3 in E. coli. In the current study variability in size and element copy number was also observed for R4 clones using E. coli. However, the same variability was present in samples sequenced using the PacBio sequencing platform, which involves no E. coli step. In addition, considering more recent experience with, e.g., the generation of infectious herpesvirus clones via BAC, it seems less likely that E. coli is responsible for this variation (22). As such, we conclude that, for several of the R regions, variability in the size and copy number commonly occur in VZV samples that are otherwise clonal. Among the HZ VZV samples evaluated here, 33/79 (42%) had evident mixtures in R3, 36/83 (43%) in R4A and 39/83 (47%) in R4B. We were able to determine complete genome sequences for 73 of 84 evaluated in this study using a combination of MiSeq Illumina and Sanger sequencing; all 73 viruses were clonal except for some of the R regions. In this study and in previous work (8), R3 exhibits the most variation in length and sequence; we observed a 20-fold difference between the shortest and the longest R3 region (22bp versus 447bp). A report on the complete genome sequencing of the Oka vaccine strain and its parental strain mentions they were unable to sequence R3 directly from the PCR product (7). They succeeded only in reading to just before the 4th repeat element using the sense primer and the last 3 elements with the antisense primer. We have also experienced difficulty in reading all of the copies of the R3 region in some samples. Careful examination often reveals the presence of an additional copy/ies in the smaller peaks in the chromatogram. Since there may be two or more variant R region sequences in some strains, the sense primer may be reading one element of the reiteration region while the antisense primer may be aligning that copy with different element from the reverse direction. This can lead to what look like mismatches and minor peaks. The mixture in the number of variants making up the R3 may account for some of the minor bases and mixed bases that have been reported (8, 9). Since R3 lies within a coding region, the length and sequence variation conceivably could result in significant biological consequences for the virus or may simply be tolerated in the protein structure (3). Resolution of that question will require studying variably lengthed R3 regions incorporated into BAC VZV viruses. VZV ORF 22 is the homolog of HSV-1 UL36; the protein is a component of the tegument and is thought to play a role in VZV assembly (6, 23–25). More studies will be required to determine if the R1 and R2 regions, also located in coding regions, are commonly present in mixtures in otherwise clonal strains. In this study, only a single mixed R1 region was found in these clinical samples, and no R2 regions were mixed.
The 6-mer element in R1 has previously been observed only in Clade 5 viruses and was present in 29 of 32 (91%) of these Clade 5 viruses in 1 to 9 copies. The 6-mer element is always found 3’ of a 15-mer element. Thus far, this has also invariably been a 15A element. In this study a single copy of the 6-mer was found near the terminus of three out of four Clade 3 viruses in this study. Three of the Clade 3 viruses (12–212, 13–103 and 13–353) appeared to diverge from the reference sequences for Clade 3 and may represent Clade3/5 recombinants. One of the four Clade 3 viruses (12–189) failed to yield sufficient template for deep sequencing and, as such, was not included in the phylogenetic analysis of the study samples. Genotyping for 12–189 and 10 other samples that could not be sequenced on MiSeq was done using the revised genotyping scheme published by Jensen et al (16). All R1 regions terminated in a GGA 3-mer as previously reported (3, 7, 26).
We determined that more than 40% of the R3, R4A and R4B regions in our sample comprise “allelic” mixtures of variant R region repeating elements. Given the limit of detection for Sanger sequencing, it is quite possible that we are underestimating the frequency of this property, particularly given our results using TOPO TA cloning, where all but one or two of the variant R region sequences in R4 would have gone undetected by Sanger. We evaluated the PacBio next gen platform as a practical alternative to TA cloning, for which analyzing large numbers of genomes would be prohibitive. PacBio identified a different dominant copy number, with the shorter sequences more common than the longer ones. This discrepancy may have been a consequence of the PCR step, where the random amplification in early cycles of some R region variants may produce misleading results with respect to the predominant copy number. PacBio and similar long-read technologies may be more useful than Sanger for determining how frequently mixtures occur, and what the number and composition of elements for each variant are. While R4 is located in a non-coding region, it is proximal to the origins of replication and contains a palindrome postulated to play a role in replication (3); as such, it is plausible that length variation in R4 could have biological consequences.
We identified 28 new variant elements among these VZV genomes: R1, two new 15-mers and three 18-mers; R2, one 32-mer and ten 42-mers; R3, one 9-mer; R4 three 27-mers; and R5, eight 88-mers. We adopted a revised nomenclature for these variant elements, consisting of the length of the element followed by a letter designation.
Twenty-four of the 28 (86%) newly described variant elements were found only in a single VZV sample. Since the variant elements often differ by only 1 or 2 base pairs, these elements will need to be confirmed in a broader evaluation of VZV genomes. That said, eight of these elements (R1 18C, R2 42H, 42K, 42N, R3 9H, R4 27C and 27D, and R5 88I were present in more than one copy in the same genome, suggesting that these variant elements were not attributable to sequencing error. In addition, in R2, variable loci for 7 of the 10 new elements had been previously observed to vary and some of the bases observed in these new elements were also observed previously. It is in the combination of variable bases that these new elements differ from those determined earlier. Eight new variable loci were identified for R2 elements. In R5, we identified six variable loci in the 88-mer element that had not been previously observed. Altogether, nine loci in the 88-mer are now known to vary (4 were previously described); of these, 6 had the substituted base in only a single variant element. Among the five 88-mer elements reported previously (8), two (88D and 88E) were not observed in these samples. Of eight elements identified for the first time in this study, five were present only a single time in a single sample (88G, 88H, 88J, 88L, 88M) and one (88I) was present twice in a single sample. In both of the R4 variant elements present in only a single genome, the same variant element was present in both R4A and R4B. More interestingly, in three of these samples (12–404, 12–329, and 13–078) the respective R4A and R4B copies were identical, including the placement of the variant R4 element/s. In the case of sample 12–404, three adjacent copies of element 26B were separated from the 11A terminus by three 27A elements. This may suggest a tendency to maintain identity between R4A and R4B, driven by homologous recombination. Possibly this could also be driven by the proximity of R4A and R4B to the origins of replication. Among variant elements previously reported (8) we did not identify the following in the genomes evaluated here: R1, 15G; R2, 42D, 42F 42G; R3, 9F; and R5, 18D and 18E.
Beyond the work reported here, we also sequenced samples obtained from varicella lesions and observed the same presence of mixtures in R3, R4A and R4B, indicating that this property is inherent to the virus and not a consequence of viral modification during latency (data not shown).
Most of the nucleotide changes to variant elements in R1 to R3 conferred no changes in amino acid sequence, but some non-synonymous changes were introduced. Detailed discussion of these mutations will be deferred to a subsequent publication.
Further studies in the context of viable, genetically modified viruses will be needed to determine the impact of length variation on virus phenotype. The most obvious region to begin such studies is R3, for which the impact is obscured by the frequent presence of mixtures of differently sized reiteration regions. Evaluation of differently sized R1 and R2 regions will also be of interest. The potential impact of variation in R4 and R5 is less clear; both are located in non-coding regions but are also located in close proximity to both open reading frames and, in the case of R4, to the origins of replication.
This represents the most extensive study of the VZV reiteration regions thus far, and has revealed a level of diversity, both in size and in the number of variant elements comprising those regions, than was previously observed. Some of the variability observed here, including length and sequence variation, warrants further study in the context of viable bacterial artificial chromosome (BAC) viruses for potential impacts on VZV phenotype. More advanced methods of DNA sequencing in addition to assessing the impact of length and sequence variation on protein function and virus phenotype in viable viruses should help to resolve outstanding questions about these unusual features of VZV.
Supplementary Material
Supplementary Figure 1. Repeating element patterns common to two or more study samples.
Importance.
This analysis of the R regions has revealed a number of newly described characteristics, including a total of 28 variant elements – at least one for each of the R regions. Some of these variant elements introduce non-synonymous changes to their respective proteins. We also identified substantial length heterogeneity, notably for the three regions located within coding regions. Finally, we observed considerable within-strain length heterogeneity for the R3 region and the R4 regions; this included both the occurrence of mixtures of variably sized R regions and length heterogeneity between the two copies of R4.
Footnotes
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Diseases Control and Prevention (CDC).
References
- 1.Pellett PE, Roizman B. 2013. Herpesviridae, p 1802–22. In Knipe DM. Howley PM (ed) Fields Virology. Lippincott, Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Arvin AM, Gilden D. Varicella-Zoster Virus. 2013. Herpesviridae, p 1802–22. In Knipe DM. Howley PM (ed) Fields Virology. Lippincott, Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Davison AJ, Scott JE 1986. The Complete DNA Sequence of Varicella-Zoster Virus. J Gen Virol 67:1759–1816. [DOI] [PubMed] [Google Scholar]
- 4.Che X, Oliver SL, Sommer MH, Rajamani J, Reichelt M, Arvin AM. 2011. Identification and functional characterization of the varicella-zoster virus OR11 gene product. Virology 412:156–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grose C, Carpenter JE, Jackson W, Duus KM. 2010. Overview of varicella-zoster virus glycoproteins gC, gH and gL. In (Abendroth A, Arvin AM, Moffat JF (ed) Varicella-Zoster Virus. Curr Topics in Microbiol Immunol 342:114–27. [DOI] [PubMed] [Google Scholar]
- 6.Lebrun M, Thelen N, Thiry M, Riva L, Ote I, Conde C, Vandevenne P, Di Valentin E, Bontems S, Sadzot-Delvaux C. 2014. Varicella-zoster virus induces the formation of dynamic nuclear capsid aggregates. Virology 454–455:311–27. [DOI] [PubMed] [Google Scholar]
- 7.Gomi Y, Sunamachi H, Mori Y, Nagaike K, Takahashi M, Yamanishi K. 2002. Comparison of the Complete Sequence of the Oka Varicella Vaccine and Its Parental Virus. J Virol 76:11447–11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tyler SD, Peters GA, Grose C, Severini A, Gray MJ, Upton C, Tipples GA. 2007. Genomic cartography of varicella-zoster virus: A complete genome-based analysis of strain variability with implications for attenuation and phenotypic differences. Virology 359:447–458. [DOI] [PubMed] [Google Scholar]
- 9.Won YH, Kim JI, Kim YY, Lee CH. 2014. Characterization of the repeat Sequences of Varicella-Zoster Virus. J Bacteriol Virol 44:326–335. [Google Scholar]
- 10.Hondo R, Togo Y. 1988. Strain variation of R5 Direct repeats in the Right-Hand Portion of the Long Unique Segment of Varicella-Zoster Virus DNA. J Virol 68: 2916–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takada M, Suzutani T, Yoshida I, Matoba M, Azuma M. 1995. Identification of Varicella-Zoster Virus by PCR Analysis of Three Repeat Elements and a PstI-Site-Less Region. J Clin Microbiol 33: 658–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshida M, Tamura T. 1999. An analytical method for r5 repeated structure in varicella-zoster virus DNA by polymerase chain reaction. J Virol Meth 80: 213–215. [DOI] [PubMed] [Google Scholar]
- 13.Tseng HF, Chi M, Hung P, Harpaz R, Schmid DS, LaRussa P, Sy LS, Luo Y, Holnquist K, Takhar H, Jacobsen SJ. 2018. Family history of zoster and risk of developing herpes zoster. Int J Infect Dis 66:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tseng HF, Schmid DS, Harpaz R, LaRussa P, Jensen NJ, Rivailler P, Radford K, Folster J, Jacobsen SJ. 2014. Herpes Zoster caused by Vaccine-Strain Varicella zoster Virus in an Immunocompetent Recipient of Zoster Vaccine. Clin Infect Dis 58:1125–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Breuer J, Grose C, Norberg P, Tipples G, Schmid DS. 2010. A proposal for a common nomenclature for viral clades that form the species varicella-zoster virus: summary of VZV Nomenclature Meeting 2008, Barts and the London School of Medicine and Dentistry, 24–25 July 2008. J Gen Vir 91:821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jensen NJ, Rivailler P, Tseng HF, Quinlivan ML, Radford K, Folster J, Harpaz R, LaRussa P, Jacobsen S, Schmid DS. 2017. Revisiting the genotyping scheme for varicella-zoster viruses based on whole genome comparisons. J Gen Virol 98:1434–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Depledge DP, Kundu S, Jensen NJ, Gray E, Jones M, Steinberg S, Gershon A, Kinchington PR, Schmid DS, Balloux F, Nichols RA, Breuer J. 2013. Deep Sequencing of Viral Genomes Provides Insight into the Evolution and Pathogenesis of Varicella Zoster Virus and Its Vaccine in Humans. Mol Biol Evol. 31:397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Norberg P, Depledge DP, Kundu S, Atkinson C, Brown J, Haque T, Hussaini Y, MacMahon E, Molyneaux P, Papaevangelou V, Sengupta N, Koay ESC, Tang JW, Underhill GS, Grahn A, Studahl M, Breuer J, Bergström T. 2015. Recombination of globally circulating varicella-zoster virus. J Virol 89:7133–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takayama M, Takayama N, 2004. New method of differentiating wild-type varicella virus (VZV) strains from Oka varicella vaccine strain by VZV ORF6-based PCR and restriction fragment length polymorphism analysis. J Clin Virol 29:113–9. [DOI] [PubMed] [Google Scholar]
- 20.Ida M, Kageyama S, Sato H, Kamiyama T, Toyomoto T, Ozaki T, Kajita Y, Morohashi M, Shiraki K. 2000. Characterization of acyclovir susceptibility and genetic stability of varicella-zoster viruses isolated during acyclovir therapy. J Dermatol Sci 23:63–72. [DOI] [PubMed] [Google Scholar]
- 21.Yoshida M, Tamura T, Shimizu A, Ohashi N, Itoh M. 2003. Analysis of numbers of repeated units in R2 region among varicella-zoster virus strains. J Dermatol Sci 31:129–33. [DOI] [PubMed] [Google Scholar]
- 22.Tischer BK, Kaufer BB. 2012. Viral bacterial artificial chromosomes: generation, mutagenesis, and removal of mini-F sequences. J Biomed Biotechnol 2012:472537. doi: 10.1155/2012/472537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McNabb DS, Courtney RJ. 1992. Characterization of the large tegument protein ICP1/2 of herpes simplex virus type 1. Virology 190:221–32. [DOI] [PubMed] [Google Scholar]
- 24.Morrison EE, Stevenson AJ, Wang YF, Meredity DM. 1998. Differences in the intracellular localization and fate of herpes simplex virus tegument proteins early in the infection of Vero cells. J Gen Virol 79: 2517–28. [DOI] [PubMed] [Google Scholar]
- 25.Bucks MA, O’Regan KJ, Murphy MA, Wills JW, Courtney RJ. 2007. Herpes simplex virus type 1 tegument proteins VP1/2 and UL37 are associated with intranuclear capsids. Virology 361: 316–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abe T, Sato M, Tamai M. 2000. Variable R1 region in varicella zoster virus in fulminant type of acute retinal necrosis syndrome. Br. J Opthhalmol 84:193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Repeating element patterns common to two or more study samples.