

Conspectus
With complex molecular architectures, intriguing oxidation patterns, and wide-ranging biological activities, diterpene natural products have greatly impacted research in organic chemistry and drug discovery. Our laboratory has completed total syntheses of several highly oxidized diterpenes, including the ent-kauranoids maoecrystal Z, trichorabdal A, and longikaurin E; the antibiotic pleuromutilin; and the insecticides ryanodol, ryanodine and perseanol. In this Account, we show how analysis of oxidation patterns and inherent functional group relationships can inform key C–C bond disconnections that greatly simplify the complexity of polycyclic structures and streamline their total syntheses. In articulating these concepts, we draw heavily from the approaches to synthetic strategy that were codified by Evans, Corey, Seebach, and others, based on the formalism that heteroatoms impose an alternating acceptor and donor reactivity pattern upon a carbon skeleton. We find these ideas particularly useful when considering oxidized diterpenes as synthetic targets.
In the first part of the Account, we describe the use of reductive cyclizations as strategic tactics for building polycyclic systems with γ-hydroxyketone motifs. We have leveraged Sm-ketyl radical cyclizations as ‘reactivity umpolungs’ to generate γ-hydroxyketones in our total syntheses of the Isodon ent-kauranoid diterpenes (−)-maoecrystal Z, (−)-longikaurin E, and (−)-trichorabdal A. Following this work, we identified the same γ-hydroxyketone pattern in the diterpene antibiotic (+)-pleuromutilin, which again inspired the use of a SmI2-mediated reductive cyclization, this time to construct a bridging eight-membered ring. This collection of four total syntheses highlights how reductive cyclizations are particularly effective umpolung tactics when used to simultaneously form rings and introduce 1,4-dioxygenation patterns.
In the second part of the Account, we detail the syntheses of the complex and highly oxidized ryanodane and isoryanodane diterpenes, and present the oxidation pattern analysis that guided our synthetic designs. We first discuss our 15-step total synthesis of (+)-ryanodol, which incorporated five of the eight oxygen atoms in just two transformations: a dihydroxylation of (S)-pulegone and a SeO2-mediated trioxidation of the A-ring cyclopentenone. This latter transformation gave rise to an independent investigation of SeO2-mediated peroxidations of simple bicyclic cyclopent-2-en-1-ones. The syntheses of (+)-ryanodine and (+)-20-deoxyspiganthine are also presented, which required modified end-game strategies to selectively incorporate the key pyrrole-2-carboxylate ester. Finally, we describe our fragment coupling approach to prepare the isoryanodane diterpene (+)-perseanol. Using a similar oxidation pattern analysis to that developed in the synthesis of ryanodol, we again identified a two-stage strategy to install the five hydroxyl groups. This strategy was enabled by a Pd-mediated carbopalladation/carbonylation cascade and leveraged unexpected, emergent reactivity to sequence a series of late-stage oxidations.
While each of the diterpene natural products discussed in this Account present unique synthetic questions, we hope that through their collective discussion, we provide a conceptual framework that condenses and summarizes the chemical knowledge we have learned, and inspires future discourse and innovations in strategy design and methodology development.
Graphical Abstract
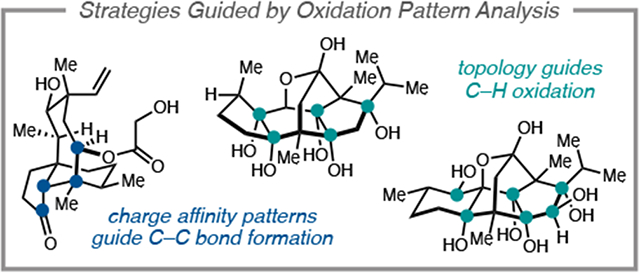
1. Introduction
Over the past decade, we have been engaged in efforts to synthesize complex and highly oxygenated diterpenes using functional group-guided topological strategies.5 We are far from alone in our interests: diterpenes have long drawn the attention of synthetic chemists due to their diverse and fascinating molecular architectures, which are coupled to a wide range of biological activities. Promising anticancer, antibacterial, and antimalarial activities have inspired multimillion-dollar campaigns to develop complex diterpenes, such as paclitaxel, pleuromutilin, and artemisinin, as therapeutics.6 These efforts highlight how the preparation of diterpenes by chemical- or semi- synthesis can transform our ability to use such molecules and their synthetic derivatives as biological probes or as lead compounds for the development of new medicines. Nonetheless, the elaborate polycyclic systems and intricate oxidation patterns that characterize many diterpenes can still hinder traditional medicinal chemistry studies of their biological activity.7 New advances in the science of chemical synthesis, driven both by the invention of new reactions and by innovations in strategy, are required to provide flexible and modular access to this important class of natural products.
In this Account, we review our syntheses of several oxidized diterpenes (Figure 1a). The syntheses can be roughly grouped into two types of strategies, both of which were guided by consideration of the oxidation pattern of the target. The first group includes the Isodon diterpenoids maoecrystal Z (1), trichorabdal A (2), and longikaurin E (3), and the antibiotic pleuromutilin (4); each possesses a key γ-hydroxyketone motif embedded within its polycyclic framework. The γ-hydroxyketone is an example of what Evans classified a ‘dissonant’ charge affinity pattern: the polarization imparted along the carbon chain between the oxygen functional groups is mismatched (Figure 1b).5b, 5d Carbon–carbon bond formation within this dissonant path requires a polarity inversion, or as Seebach defined, ‘reactivity umpolung’.5d The position of this dissonant charge affinity pattern in each structure inspired synthetic strategies involving ring closure by Sm-mediated addition of an aldehyde-derived ketyl radical to an α,β-unsaturated cabonyl.8,9 These syntheses highlight how this tactic is particularly effective when used to simultaneously form rings and introduce 1,4-dioxygenation patterns.
Figure 1.

(a) Oxidized diterpene targets completed by the Reisman laboratory. (b) Difunctional relationships guide key C–C bond constructions. (c) Carbon framework topology guides early vs. late stage C–H oxidation.
The second group includes the targets ryanodine (5) and perseanol (6) and required a different strategy to address their high degree of oxidation (Figure 1c). In these systems, the oxidation was introduced in two stages: 1) in the first few steps of the synthesis, prior to construction of the primary carbon framework; and 2) in the late stage, through strategic deployment of C–H oxidation reactions. In both syntheses, nearly all oxidation is introduced in the required oxidation state, with the correct stereochemistry, avoiding lateral functional group manipulations.
Taken together, the syntheses described in this Account demonstrate two approaches to diterpene synthesis that we anticipate can be extended to new contexts and syntheses of complex and highly oxidized natural products.
2. Reductive Cyclization Strategies: Addressing Dissonant Charge Affinity Patterns
2a. ent-Kauranoid Diterpenes
Plants of the Isodon genus are often used in Chinese traditional medicine and are the source of over 600 ent-kauranoid diterpenes.10,11 The parent compound, ent-kaurene, is the first cyclic diterpene produced as part of the gibberellin biosynthetic pathway in plants and fungi.12 One large subfamily of ent-kauranoids bears oxidation at C20. Examples of these natural products include (−)-longikaurin E (3), the 6,7-seco-ent-kauranoid (−)-trichorabdal A (2), and the rearranged 6,7-seco-ent-kauranoid (−)-maoecrystal Z (1).11a,13 Recognizing that these ent-kauranoids share oxidation at C7, C11, and C20, we envisioned that a unified strategy might provide access to all three targets. In the biosynthetic pathway, ent-kaurene is oxidized and rearranges to 1, 2, and 3; we sought to develop an abiotic approach, incorporating key oxidized functional groups in spiro-lactone 8, then forming the C6–C7 and C9–C11 bonds through reductive cyclizations.
Our general strategy was guided by identification of the γ-hydroxyketone motif spanning C7 to C11 (Figure 2).14 This dissonant charge affinity pattern keyed the use of a SmI2-mediated reductive cyclization to generate an umpole at the C11 position. In the context of preparing maoecrystal Z, use of a dialdehyde (8a) was envisioned to enable a cascade process;8,9 initial formation of the C9–C11 bond followed by single electron reduction would give the corresponding enolate, which could undergo an intramolecular aldol reaction to close the central five-membered ring.15 Alternatively, SmI2-mediated monocyclization of 8b, followed by subsequent oxidative cyclization to form the requisite bicyclo[3.2.1]octane, could enable elaboration to trichorabdal A (3) and longikaurin E (4).
Figure 2.

Retrosynthetic analysis of ent-kauranoid diterpenes.
With (−)-1 as our first target,1 we initially investigated the TiIII-mediated reductive coupling of 12 with a variety of acrylates, hoping to incorporate the requisite side chain and minimize steps after lactone formation (Scheme 1). Whereas the use of simple acrylates, such as methyl acrylate, gave 14 as the product in varying yield, acrylates with α-substituents failed to undergo the desired coupling. Fortunately, use of 2,2,2-trifluoroethyl acrylate as an electrophilic partner under modified Gansäuer conditions16 led to the isolation of lactone 14 as a single diastereomer in good yield. The reaction presumably proceeds through radical intermediate 13, in which approach of the acrylate from opposite the large siloxymethylene group affords the desired C10 stereochemistry. Alkylation of the lactone followed by desaturation, desilylation, and oxidation gave dialdehyde 8a.
Scheme 1.

12-step total synthesis of (−)-maoecrystal Z (1).
In the key SmI2-mediated reductive cyclization, lithium salts were found to be critical for desired reactivity. Treatment of dialdehyde 8a with SmI2 in the presence of LiBr and tBuOH as a proton source thus provided the tetracyclic diol, which was isolated as a single diastereomer. This transformation builds two rings and introduces two secondary carbinols in a single step. In fact, the cascade reaction was critical to forming the central five-membered ring; attempts to form the two rings sequentially, in a non-cascade process, failed to identify conditions that engage the C6 aldehyde derived from tricycle 9 (see Scheme 2) in the intramolecular aldol. Diacetylation, ozonolysis, and methenylation gave enal 18, which upon mono-deacetylation delivered (−)-maoecrystal Z (1) in 12 linear steps from (−)-γ-cyclogeraniol (11).1
Scheme 2.

Total synthesis of (−)-longikaurin E (3) and (−)-trichorabdal A (2).
In order to access trichorabdal A and longikaurin E,17 we returned to the key spiro-lactone, executing selective deprotection/oxidation of 17 to prepare 8b (Scheme 2). Fortunately, subjection of monoaldehyde 8b to our previously developed reductive cyclization conditions gave tricycle 9 as a single diastereomer, which was protected as the methoxymethyl ether. Taking inspiration from Pd-catalyzed oxidative cyclizations of silyl enol ethers,18 we envisioned that a similar aerobic transformation of a silyl ketene acetal could establish the final carbocyclic ring and leverage the pendant alkene without need for further functionalization. Indeed, following extensive reaction development, subjection of 20 to stoichiometric Pd(OAc)2 in DMSO provided tetracycle 21.
At this stage, intermediate 21 was diverted to targets (−)-3 and (−)-2 through slightly different reaction sequences. Global deprotection of 21 followed by selective oxidation at C6 and acetylation at C11 provided aldehyde 23. A SmI2-mediated intramolecular aldehyde–lactone pinacol coupling afforded lactol 10 as a single diastereomer, another example of the strategic use of SmI2 to introduce dissonant oxidation patterns via C–C bond formation. Finally, ozonolysis and methenylation gave (−)-3 in 17 steps (longest linear sequence) from 11. Alternatively, 21 could be advanced to 25 through ozonolyis and methenylation. Acidic deprotection and oxidation then revealed the aldehyde of (−)-2.17
Our unified strategy enabled the first total syntheses of three architecturally distinct ent-kauranoids from a common intermediate and illustrates the utility of single-electron reduction chemistry for the preparation of congested polycyclic systems bearing dissonant oxidation patterns.
2b. Pleuromutilin
The fungal diterpene (+)-pleuromutilin (4), first isolated in 1951, inhibits bacterial protein synthesis,19 and semi-synthetic analogues have been advanced into the clinic as antibiotics.20 Importantly, amine-containing 12-epi-4 derivatives have been reported as efficacious against gram-negative pathogens.21 With the goal of providing enabling chemistry for the synthesis of new mutilin-based antibacterial agents, we sought to develop concise and flexible syntheses of both 4 and its C12 epimer.2 Although there were three prior syntheses of 4 when we initiated our studies,22 these approaches all built the eight-membered ring relatively early in the synthesis, requiring lengthy sequences to then introduce the C10-C11-C12 stereotriad. In our synthetic planning, we envisioned building the eight-membered ring by annulation with a functionalized C10–C14 fragment in order to minimize functional group manipulations after the tricyclic core was formed (Figure 3).
Figure 3.

Retrosynthetic analysis of (+)-pleuromutilin (4) and 12-epi-4.
Similar to our approach to the Isodon ent-kauranoids, we identified the dissonant γ-hydroxyketone pattern from C3 to C14 as a keying element for a SmI2-mediated reductive cyclization to form the C5–C14 bond. Although Procter had previously employed a SmI2-mediated cascade cyclization for the synthesis of 4,22c,d the design of that cascade required an ester at C15 to induce a ketyl radical conjugate addition. As a result, the Procter approach required several additional steps to adjust the oxidation states at C3 and C15. In contrast, a SmI2-mediated cyclization of 28 was expected to provide 26 with C3, C14, and C15 in the correct oxidation states.
Having identified this reductive cyclization tactic for forming the eight-membered ring, we devised a synthetic plan that would allow modular construction of aldehyde 28, so that both (+)-4 and 12-epi-4 could be prepared. It was anticipated that 28 could arise from the bifunctional hydrindanone fragment 30 by crotylation with either Z- or E-boronic acid 31.23 Hydrindanone enal 30 was mapped back to cyclohexenone 32, available in one step from (+)-trans-dihydrocarvone.24
In the forward direction, ketone 34, bearing a C9 quaternary center, was prepared via sequential conjugate additions (Scheme 3). Allylic chlorination of the iso-propenyl group of 34 was performed prior to intramolecular aldol condensation in order to avoid a competing, undesired Prins-type cyclization. Ketone 35 could be elaborated to enal 30 in three additional steps.
Scheme 3.

18-step total synthesis of (+)-pleuromutilin (4).
To complete the first of two key bond-forming steps, selective syn-crotylation with boronic acid Z-31 gave desired diastereomer 29a after chromatographic separation. Unfortunately, despite the use of a chiral BINOL catalyst,23 we were not able obtain high diastereoface selectivity for addition to the aldehyde. Nonetheless, the required diastereomer (29a) could be elaborated in three steps to aldehyde 28. To form the eight-membered ring, a freshly prepared Sml2 solution was added dropwise to 28; quenching of presumed Sm-enolate 38 with trimethylsilyl chloride then delivered tricycle 26 as a separable 23:1 mixture of diastereomers. Substantial optimization determined that rigorously anaerobic conditions were critical for minimizing byproduct formation and that addition of water was important for high diastereoselectivity.
At this stage, completion of the synthesis hinged upon selective hydrogenation of the C10 exo-methylene in the presence of the C19 vinyl group. It was hypothesized that metal-catalyzed hydrogen atom transfer (MHAT) reduction might provide a solution, wherein the thermodynamic preference for formation of a tertiary carbon-centered radical could be leveraged. Indeed, chemoselective reduction of the exocyclic C10–C17 olefin was achieved; however, this reduction was accompanied by unanticipated C14 oxidation. Deuterium labeling experiments revealed that this reaction proceeds by a transannular [1,5]-HAT redox relay, thermodynamically driven by formation of the C14 ketone. By protecting the C3 ketone of 26 as a silyl enol ether prior to the MHAT redox relay, the C14 ketone of 41 could be reduced using dissolving metal conditions while preserving the ketone oxidation state at C3. Finally, acylation followed by global deprotection under acidic conditions afforded (+)-pleuromutilin (4) in 18 steps in the longest linear sequence.2
An advantage of this modular approach was the ability to readily prepare the 12-epi-4 framework by varying the relative configuration of cyclization substrates at C12 (Scheme 4). To this end, enal 30 was crotylated with E-31 to deliver 29c, which was elaborated to 12-epi-28 without difficulty. Exposure of 12-epi-28 to the optimal SmI2 cyclization conditions furnished 12-epi-26, then advanced via the previously developed 4-step sequence to complete the synthesis of 12-epi-4.
Scheme 4.

18-step total synthesis of (+)-12-epi-pleuromutilin (12-epi-4).
We anticipate that the brevity and modularity of these syntheses, enabled by the unique ability of radical cyclizations to overcome the challenges that dissonant charge affinity patterns pose to standard bond constructions, will facilitate preparation of new pleuromutilin antibiotics.
3. Complex Oxidation Topology
3a. Ryanodane Diterpenes
A classic example of how natural products can impact our understanding of biology is the story of (+)-ryanodine. (+)-Ryanodine (5) is a highly oxidized diterpene isolated from the tropical shrub Ryania speciose Vahl.25 Investigation of ryanodine’s insecticidal properties ultimately led to the purification and characterization of intracellular Ca2+ ion channels now known as the ryanodine receptors (RyRs),26,27 which are involved in a number of signal transduction processes. Whereas both 5 and its hydrolysis product (+)-ryanodol (43) bind insect RyRs,28 43 exhibits significantly lower affinity for mammalian receptors.29 In addition, structurally related diterpenes that vary in peripheral oxidation, including (+)-20-deoxyspiganthine (44), have also been identified as RyR modulators.30 Due to their biological importance, ryanodanes have been the focus of total synthesis31,32 and medicinal chemistry30,33 efforts for several decades. However, at the outset of our studies, there were no completed total syntheses of ryanodine, due in part to the challenges associated with introduction of the pyrrole-2-carboxylate at the C3 alcohol of ryanodol (43). With the long-term goal of developing new probes of RyR function, we devised a synthetic platform for the preparation of (+)-5 and related congeners (i.e., (+)-44).
Our initial efforts focused on the preparation of (+)-43 envisioning that the synthetic strategy could be applied to (+)-5 and other ryanodanes (Figure 4).3,34 We drew inspiration from early relay studies by Deslongchamps and coworkers, which established that the C1–C15 bond of the bridging E ring could be formed via epoxidation of 45 followed by reductive cyclization. 32 With (+)-anhydroryanodol (45) as a slightly simpler target, our strategy was directed by the dense oxidation of the tetracyclic core. We hypothesized that an A-ring cyclopentenone, such as that in 49, could undergo sequential oxidation in the final steps of the synthesis to install the C3 alcohol and the syn-C4–C12-diol with the appropriate stereochemistry. In contrast, we sought to incorporate the C-ring oxidation early in the synthesis while assembling the carbon framework. With this goal in mind, an intramolecular Pauson–Khand reaction (PKR)35 was identified as a key tactic to prepare cyclopentenone 49. We mapped the requisite enyne (50) back to (S)-(−)-pulegone (51), expecting that sequential oxidation α to the ketone could be used to install the C6 and C1O alcohols.
Figure 4.

Retrosynthetic analysis of ryanodane diterpenes.
We began our studies with the oxidation of (S)-(−)-pulegone (51, Scheme 5). Although we initially envisioned a multistep sequence to 53 via known pulegone oxide, we ultimately found that treatment of 51 with excess KHMDS followed by oxaziridine 52 resulted in dihydroxylation to form 53 as the major diastereomer. Although further mechanistic investigations are required, preliminary studies suggest that the reaction proceeds first by generation of the thermodynamic potassium dienolate, which upon reaction with 52 gives rise to the C6 tertiary alkoxide as a mixture of diastereomers.36 A second enolization and oxidative trapping gives rise to α,α’-diol 53 in 40–50% yield following silica gel chromatography. In this single transformation, we access a key building block with the complete C-ring functionality found in ryanodol and ryanodine. Following protection of the alcohols, the D ring was constructed by a four-step sequence involving 1,2-addition of prop-1-yn-1-y1 magnesium bromide followed by ozonolytic olefin cleavage to afford ketone 55. A second 1,2-addition and subsequent Ag-catalyzed cyclization/elimination cascade37 then gave α,β-unsaturated lactone 56. The critical all-carbon quaternary center was formed as a single diastereomer by 1,4-addition of a vinyl cuprate reagent, giving 50. Pauson–Khand cyclization with catalytic [RhC1(CO)2]2 under an atmosphere of carbon monoxide (CO) provided cyclopentenone 49 as a single diastereomer.38
Scheme 5.

15-step total synthesis of (+)-ryanodol (43).
Having identified a concise route to tetracycle 49, we faced the challenging task of introducing the alcohols at C3, C4, and C12, with the C4 alcohol expected to be the most challenging of the three. We initially investigated conditions for allylic oxidation at C4; however, this proved unfruitful and led us to pursue SeO2-mediated α-oxidation of the C2 ketone.39 To our surprise, treatment of 49 with SeO2 and K3PO4 in refluxing dioxane resulted in the isolation of hydroxyenone 58, wherein the presumed intermediate enediketone underwent hydration at C12. Careful analysis of the reaction mixtures indicated the presence of a minor side-product assigned the structure of 57, a compound which had undergone further oxidation at C4. Although the yield was very low (<5%), we recognized that this remarkable transformation installs the oxygen atoms at C3, C4, and C12, as well as a functional group handle for incorporation of the C2-iso-propyl group, all in a single step. As result, we set out to improve the yield, ultimately finding that heating 49 with excess SeO2 in rigorously anhydrous 1,4-dioxane gave increased amounts of 57, which could be isolated as enol triflate 48 in 28% yield over the two steps. Alternatively, oxidation of 49 with SeO2 and 10 equiv water in 1,4-dioxane, followed by triflation, gives 47 in 56% yield.
To complete the synthesis of (+)-43, enol triflate 48 was submitted to Pd-catalyzed Stifle cross-coupling, ketone reduction, and hydrogenation to deliver (+)-anyhdyroryanodol (45). In slight modification of the reported protocol,32 treatment of 45 with freshly prepared trifluoroperacetic acid cleanly afforded the epi-anhydroryanodol epoxide, which could be subjected to Li in NH3 to effect reductive cyclization to (+)-ryanodol (43). 3 With only 15 steps, this synthesis represents a ca. 50% reduction in length compared to previous syntheses.32, 33
Given the known challenge associated with preparing ryanodine by site-selective C2 acylation of 43, our synthetic strategy to 5 called for departure from the ryanodol end game, such that the pyrrole 2-carboxalate could be introduced selectively without extensive additional protecting group manipulations. 34, 40 Moreover, we anticipated that dioxidation product 47 could be used to prepare the related natural product 20-deoxyspiganthine. Although there was some uncertainty as to whether the pyrrole 2-carboxalate could survive the final reductive cyclization, we nonetheless identified anhydroryanodol derivatives 61 (Scheme 6) and 68 (Scheme 7) as potential candidates for acylation.
Scheme 6.

18-step total synthesis of (+)-ryanodine (5).
Scheme 7.

19-step total synthesis of (+)-20-deoxyspiganthine (44).
The synthesis of (+)-5 commenced with diol 59, which was strategically protected as a dioxaborinane (Scheme 6). Reduction of the C3-ketone furnished 61, bearing a single unprotected alcohol. A survey of standard acylating reagents derived from pyrrole-2-carboxylic acid failed to deliver pyrrole ester 63; however, deprotonation of alcohol 61 before addition of trichloroketone 62 successfully provided 63. This reaction is noteworthy: efforts to perform the acylation on a compound bearing an iso-propyl, instead of iso-propenyl, substituent at C2 were unsuccessful. Subsequent deprotection, hydrogenation, and hydroxyl-directed epoxidation revealed 66. Whereas the use of Li/NH3, the conditions employed for the synthesis of ryanodol, resulted in reductive cyclization with concomitant deacylation, the use of lithium di-tert-butylbiphenylide (LiDBB) in THF effected C–C bond formation and alcohol debenzylation to provide (+)-ryanodine (5) in 65% yield.35
A similar sequence was used to elaborate alcohol 67 to 20-deoxyspiganthine (44, Scheme 7). In this case, the C12 alcohol was protected as a TMS ether prior to reduction of the C3 ketone. Despite the similarity of 68 to 61, this acylation required further optimization of the reaction conditions in order to minimize non-productive translactonization. Ultimately, 68 was treated with excess KHMDS to enolize the lactone and prevent translactonization; while this enabled acylation of the C3 alcohol with pyrrole 69, α-chlorination of the lactone was also observed. Radical dechlorination of 70 afforded lactone 71, which was uneventfully advanced to (+)-20-deoxyspiganthine (44) by hydrogenation, epoxidation, and reductive cyclization.35 This represents the first total synthesis of a spiganthine-type natural product.
Taken together, our ryanodane studies highlight how managing formation of C–C and C–0 bonds to introduce functionality at the correct oxidation level can minimize protecting group adjustments and lead to concise syntheses.
3b. SeO2-Mediated Oxidative Transpositions
The successful syntheses of the ryanodane natural products discussed above were greatly enabled by the serendipitous discovery of SeO2-mediated oxidation of tetracyclic enone 49 (see Scheme 5). Whereas the oxidation of ketones to α-diketones by SeO2, known as the Riley oxidation,40 is well established, applications of this reaction to cyclopent-2-en-1-ones had not been systematically studied. We recognized that the SeO2-mediated di- and trioxidations of simple bicyclic cyclopent-2-en-1-ones, prepared by intramolecular Pauson–Khand reactions, enables the transposition of the enone functionality to give highly oxidized cyclopentenone products that are difficult to access by other methods. Given the ability of these di- and trioxidations to construct functional group relationships elusive via canonical PKRs, we pursued further studies of this interesting transformation (Scheme 8).41
Scheme 8.

(a) Dioxidation of bicyclic cyclopent-2-en-1-ones with aqueous SeO2. (b) Trioxidation of bicyclic cyclopent-2-en-1-ones with anhydrous SeO2.
Investigations of a series of bicyclic enones accessible via PKR revealed that increasing the amount of water relative to our previously reported conditions3,35 significantly improved dioxidation yields. Consistent with our earlier studies, we found omitting water gives the best selectivity for trioxidation products. While we were unable to improve the trioxidation yields beyond ~20–30%, this reaction nonetheless accomplishes the stereospecific incorporation of three oxygen atoms in a single reaction. Both the di- and trioxidations were found to tolerate a range of bicyclopentenones, featuring saturated and unsaturated heterocycles and bulky quaternary centers, although 5,5-fused bicyclic enones were uniquely reactive under these conditions. Additional investigation of reaction kinetics and enantiospecificity implicated competitive mechanistic pathways: water-dependent sequencing of Riley oxidation and allylic C–H oxidation steps appear responsible for product selectivity. The diosphenol products could be readily converted to enol triflates, enabling further elaboration through cross-coupling to access diverse, fully substituted cyclopentenones pertinent to the synthesis of complex natural products.
3c. Isoryanodane Diterpenes
Having established a synthetic strategy to prepare several ryanodane natural products, we turned our attention to a related family of targets, the isoryanodanes. The isoryanodanes possess a 5-6-5 ABC ring system instead of the 5-5-6 system found in the ryanodanes but bear a similar, dense oxidation pattern. Although it has not been conclusively established, it seems likely that these compounds are biosynthetically related. One interesting divergence is that none of the isoryanodanes isolated to date possess a pyrrole-2-carboxylate ester like that found in ryanodine, although many retain potent insecticidal properties. For example, (+)-perseanol (6)42 displays potent antifeedant activity with low toxicity to mammalian cells,43 as do related isoryanodane diterpenes.44 This has raised the question as to whether the target of these compounds is indeed the insect RyR or whether they act through a distinct mode-of-action. Perseanol is decorated by six free hydroxyl groups around a highly caged pentacyclic core that includes a bridging 7-membered lactol. Prior to our studies, there was a single report describing an approach to the complex framework but no completed syntheses.45 Inspired by the potential to further investigate isoryanodane mode-of-action, we developed a convergent synthesis of (+)-6.4
Guided by our approach to ryanodine, we envisioned that a late-stage reductive cyclization could be used to form the C1–C15 bond, leading us to retrosynthetically simplify 6 to a structure we termed anhydroperseanol (87, Figure 5). Although we initially pursued a Pauson–Khand approach to prepare the carbon framework of anhydroperseanol, we encountered challenges in forming the 7-membered lactone-containing enyne. These challenges led us to revise our synthetic strategy. Analysis of the oxidation patterns in 87 suggested that the A–B ring fusion diol could be accessed from tetracyclic alkene 88, whereas the B–C ring fusion diol might be introduced early in the synthesis. Tetracyclic lactone 88 was dissected into two fragments that could be joined in two C–C bond-forming steps. First, we envisioned 1,2-addition of a metallated fragment derived from 91 into aldehyde 92. Second, the resulting alkenyl bromide (90) could undergo an intramolecular Pd-catalyzed carbopalladation/carbonylation cascade to forge the central 7-membered lactone. To access the enantioenriched fragments required for a convergent fragment coupling, we expected to prepare enal 92 from (R)-(+)-pulegone (51) and hoped to prepare vicinal dihalide 91 from commercially available 3-ethoxycyclopent-2-en-1-one (99).
Figure 5.

Retrosynthetic analysis of (+)-perseanol (6).
In the forward sense, the C-ring fragment (92) was prepared by known Favorskii rearrangement46 of 51 followed by diastereoconvergent α-hydroxylation to yield 94 (Scheme 9a). Hydroxyl-directed epoxidation and isomerization then provided syn-diol 96. Through this four-step sequence, the B–C ring fusion diol was efficiently introduced at the beginning of the synthesis with the correct relative stereochemistry. Diol 96 could be elaborated in two additional steps to alkenyl aldehyde 92.
Scheme 9.

(a) Preparation of the C-ring fragment (92). (b) Preparation of the A-ring fragment (91).
Synthesis of the A-ring fragment (91) began with alkylation of the zinc enolate of vinylogous ester 99 (Scheme 9b). An iodination/hydrolysis sequence provided diketone 102, which was immediately brominated to yield bromoiodocyclopentenone 103. Finally, an enantioselective Corey–Bakshi–Shibata (CBS) reduction47 of (±)-103 resulted in a kinetic resolution that provided alcohol (−)-106 in 91% enantiomeric excess, which was protected to furnish dihalide fragment 91.
With both fragments in hand, we investigated a two-step annulation strategy to forge the tetracyclic ring system. Selective lithiation of iodide 91 and addition to aldehyde 92 provided secondary alcohol 90 in good yield but modest diastereoselectivity (Scheme 10). To close the central B ring, a carbopalladation/carbonylation cascade was investigated, with the goal of forging two C–C bonds and setting the all-carbon quaternary center in a single step. This reaction required extensive optimization to identify conditions that favored formation of the seven-membered lactone in preference to the five-membered lactone accessible via competitive direct carbonylation of the alkenyl bromide. In particular, it was important to maintain a low concentration of CO in solution to avoid inhibition of alkenyl bromide oxidative addition and to favor alkene insertion prior to carbonylation. Ultimately, use of N-formylsaccharin with KF as an activator48 and 50 mol% Pd(PPh3)4 proved optimal; under these conditions, the bridging lactone 88 could be formed in 57% yield as a single diastereomer.
Scheme 10.

16-step total synthesis of (+)-perseanol (6).
To complete the synthesis, the tetrasubstituted allylic alcohol needed to be transformed into the A–B ring fusion diol. To this end, PMB deprotection of 88 followed by treatment with dimethyl dioxirane (DMDO) was found to deliver the C15 ketone, in addition to unexpected benzylidene acetal oxidation. This transformation had not been a part of our initial plan; we nonetheless continued with efforts to elaborate towards 6. Following 1,2-addition of methyl Grignard to hydroxybenzoate 108, it was discovered that diol 109 could be converted to orthobenzoate 111 by treating with trifluoroacetic acid, presumably proceeding through dioxolenium ion 110. This three-step sequence, although outside of the original synthetic design, allowed us to repurpose the benzylidene acetal from a diol protecting group to an anchimeric directing group. Having enabled installation of the C4 tertiary alcohol, it resumed the role as an alcohol protecting group, now in the form of an orthobenzoate. This series of transformations illustrates how some of the most strategic reaction sequences arise from leveraging the emergent reactivity of the molecule, rather than strictly adhering to a preconceived synthetic design.
Tetracyclic alkene 111 was found to undergo selective allylic oxidation of the tertiary C–H bond at C2 to give allylic alcohol 112. Again, we were faced with an opportunity to revise our synthetic plan. Although we had initially envisioned epoxidizing anhydroperseanol (87) in order to execute a reductive cyclization analogous to those employed in the syntheses of ryanodine and ryanodol, we recognized that the synthesis could be streamlined by instead preparing the isomeric epoxide 113. This type of reductive cyclization was unprecedented; however, given that 112 could be stereoselectively converted to 113 in good yield, it seemed worthwhile to pursue. Extensive investigation of a variety of reductants revealed that lithium 2-phenylnaphthalenide in THF/benzene effected the reductive cyclization of 113 to construct the C1–C15 bond and deliver 115 in 25% yield. Interestingly, C2-des-iso-propyl-113 undergoes reductive cyclization in >50% yield (not shown), indicating that the steric bulk of the iso-propyl substituent likely slows down the rate of C–C bond formation. Final hydrogenolysis of the orthobenzoate afforded (+)-perseanol (6). 4 The successful first total synthesis of (+)-6 in 16 linear steps testifies to the power of convergent fragment couplings; additionally, the opportunistic formation of the A–B ring fusion diol illustrates that strategic redox planning can enable fortuitous discoveries to drive synthetic efficiency.
4. Concluding Remarks
In summary, we have presented insight into several total syntheses of diterpene natural products. Our synthetic planning was guided by target structures, with bond-forming tactics chosen to leverage inherent functional group relationships and their charge affinity patterns in order to minimize redundant changes to carbon oxidation states. In addition, careful choreography of C–C and C–O bond construction allowed strategic introduction of oxidation, thereby reducing synthetic manipulations ancillary to the direct assembly of the natural product.
At the broader level, our work is among that developed by the synthetic community to utilize distinct conceptual frameworks to fill gaps in biosynthetic and chemical knowledge. We are compelled to implement these intellectual programs as practical synthetic sequences by the growing need for significant quantities of natural products to answer interesting biological questions. Just as we have been inspired to solve the unique challenges presented by each target diterpene, we encourage continued innovation in design strategies and chemical methods for the synthesis of natural products of ever-increasing complexity.
Acknowledgements
We would like to gratefully acknowledge the past and present members of the Reisman laboratory who have contributed to the science that is captured in this Account. Fellowship support was provided by the NSF (S. E. D., Grant No. DGE-1144469). S.E.R. is a Heritage Medical Research Institute Investigator. Financial support from the NIH (R35GM118191) is acknowledged.
Biographies
Sara E. Dibrell received a B.S. in Biochemistry from The University of Texas at San Antonio, conducting research under Prof. Doug E. Frantz. She is currently a graduate student in the Reisman group, investigating reductive cross-coupling methods and natural product synthesis.
Yujia Tao received a B.S. in Chemistry from The University of California, Berkeley, conducting research under Prof. Thomas Maimone. She is currently a graduate student in the Reisman group, studying natural product synthesis.
Sarah E. Reisman earned a B.A. in Chemistry from Connecticut College and then a Ph.D. in Organic Chemistry from Yale University under the direction of John L. Wood. Following post-doctoral studies with Eric N. Jacobsen at Harvard University, Sarah began her independent career at Caltech in 2008, where she is presently the Bren Professor of Chemistry.
References
- 1. Cha JY; Yeoman JTS; Reisman SE A Concise Total Synthesis of (−)-Maoecrystal Z. J. Am. Chem. Soc 2011, 133 (38), 14964–14967. The synthesis of ent-kauranoid diterpene maoecrystal Z leveraged a SmI2-mediated reductive cascade cyclization to quickly forge the tricyclic core from a spiro-lactone intermediate accessed via reductive epoxide coupling
- 2. Farney EP;‡ Feng SS; Schäfers F;‡ Reisman SE Total Synthesis of (+)-Pleuromutilin. J. Am. Chem. Soc 2018, 140 (4), 1267–1270. Access to the central eight-membered ring of pleuromutilin was enabled by a SmI2-mediated reductive cyclization of a bifunctional hydrindane fragment, formed via a modular crotylation that also allowed synthesis of natural product analogs.
- 3. Chuang KV;‡ Xu C;‡ Reisman SE A 15-Step Synthesis of (+)-Ryanodol. Science 2016, 353 (6302), 912–915. The oxidative complexity of ryanodol guided its expedient synthesis, which hinged on a strategic Pauson–Khand reaction of a pulegone-derived intermediate and unique SeO2-mediated oxidative transposition to quickly install challenging patterns of oxidation.
- 4. Han A; Tao Y; Reisman SE A 16-Step Synthesis of the Isoryanodane Diterpene (+)-Perseanol. Nature 2019, 573 (7775), 563–567. In the convergent synthesis of the isoryanodane skeleton, a key two-step sequence of 1,2-addition and intramolecular Pd-catalyzed carbopalladation/carbonylation cascade coupled complex aldehyde and bromoiodocyclopentene fragments, followed by a reductive cyclization that furnished perseanol.
- 5.(a) Corey EJ; Cheng X-M The Logic of Chemical Synthesis; Wiley, 1989. [Google Scholar]; (b) Evans DA; Andrews GC Allylic Suloxides: Useful intermediates in orgnic Synthesis. Acc. Chem. Res 1971, 7, 147–155. [Google Scholar]; (c) Evans DA An Organizational Format for the Classification of Functional Groups. Applications to the Construction of Difunctional Relationships. [Google Scholar]; (d) Seebach D Methods of Reactivity Umpolung. Angew. Chem. Int. Ed 1979, 18(4), 239–258. [Google Scholar]; (e) Hendrickson JB Systematic Characterization of Structures and Reactions for Use in Organic Synthesis. J. Chem. Soc 1971, 93 (25), 6847–6854. [Google Scholar]
- 6.Breitmaier E Terpenes: Flavors, Fragrances, Pharmaca, Pheromones; John Wiley & Sons, 2006. [Google Scholar]
- 7.Koehn FE; Carter GT The Evolving Role of Natural Products in Drug Discovery. Nat. Rev. Drug Discov 2005, 4 (3), 206–220. [DOI] [PubMed] [Google Scholar]
- 8.(a) Molander GA; Harris CR Sequencing Reactions with Samarium(II) Iodide. Chem. Rev 1996, 96 (1), 307–338. [DOI] [PubMed] [Google Scholar]; (b) Edmonds DJ; Johnston D; Procter DJ Samarium(II)-Iodide-Mediated Cyclizations in Natural Product Synthesis. Chem. Rev 2004, 104 (7), 3371–3404. [DOI] [PubMed] [Google Scholar]
- 9.(a) Fukuzawa S-I; Nakanishi A; Fujinami T; Sakai S Reductive Coupling of Ketones or Aldehydes with Electron-deficient Alkenes Promoted by Samarium di-iodide. J Chem. Commun 1986, 624–625. [Google Scholar]; (b) Enholm EJ; Trivellas T Lanthanide Induced Intramolecular Coupling of Aldehydes and Ketones with Electron-Deficient Olefins. Tetrahedron Lett. 1989, 30, 1063–1066. [Google Scholar]; (c) Helm MD; Sucunza D; Da Silva M; Helliwell M; Procter DJ SmI2-Mediated Dialdehyde Cyclization Cascades. Tetrahedron Lett. 2009, 50, 3224–3226. [Google Scholar]
- 10.Sun H-D; Huang S-X; Han Q-B Diterpenoids from Isodon species and their biological activities. Nat. Prod. Rep 2006, 23, 673–698. [DOI] [PubMed] [Google Scholar]
- 11.(a) Han Q-B; Cheung S; Tai J; Qiao C-F; Song J-Z; Tso T-F; Sun H-D; Xu H-X Maoecrystal Z, a Cytotoxic Diterpene from Isodon eriocalyx with a Unique Skeleton. Org. Lett 2006, 8 (21), 4727–4730. [DOI] [PubMed] [Google Scholar]; (b) Node M; Sai M; Fuji K; Fujita E; Shingu T; Watson WH; Grossie D Three New Anti-Tumor Diterpenoids, Trichorabdals A, C, And D. Chem. Lett 1982, 11 (12), 2023–2026. [Google Scholar]; (c) Fujita T; Takeda Y; Shingu T Longikaurin C, D, E and F; New Antibacterial Diterpenoids from Rabdosia longituba. Heterocycles 1981, 16 (92), 227–230. [Google Scholar]
- 12.Kawaide H; Imai R; Sassa T; Kamiya Y ent-Kaurene Synthase from the Fungus Phaeosphaeria sp. L487 cDNA Isolation, Characterization, and Bacterial Expression of a Bifunctional Diterpene Cyclase in Fungal Gibberellin Biosynthesis. J. Biol. Chem 1997, 272 (35), 21706–21712. [DOI] [PubMed] [Google Scholar]
- 13.(a) Fuji K; Node M; Sai M; Fujita E; Takeda S; Unemi N Terpenoids L III.: Antitumor Activity of Trichorabdals and Related Compounds. Chem. Pharm. Bull 1989, 37 (6), 1472–1476. [DOI] [PubMed] [Google Scholar]; (b) Zhao W; Pu J-X; Du X; Su J; Li X-N; Yang J-H; Xue Y-B; Li Y; Xiao W-L; Sun H-D Structure and Cytotoxicity of Diterpenoids from Isodon adenolomus. J. Nat. Prod 2011, 74 (5), 1213–1220. [DOI] [PubMed] [Google Scholar]
- 14.Yeoman JTS; Cha JY; Mak VW; Reisman SE A Unified Strategy for the Synthesis of (−)-Maoecrystal Z, (−)-Trichorabdal A, and (−)-Longikaurin E. Tetrahedron 2014, 70 (27), 4070–4088. [Google Scholar]
- 15.Fujita E; Fuji K; Sai M; Node M; Watson WH; Zabel V The structure of trichorabdal B and its transformation into a novel skeleton; X-ray crystal structures. J Chem. Soc. Chem. Commun 1981, 17, 899–900. [Google Scholar]
- 16.(a) RajanBabu TV; Nugent WA J Am. Chem. Soc 1989, 111, 4525. [Google Scholar]; (b) RajanBabu TV; Nugent WA J Am. Chem. Soc 1994, 116, 986. [Google Scholar]; (c) Gansäauer A; Pierobon M; Bluhm H Catalytic, Highly Regio- and Chemoselective Generation of Radicals from Epoxides: Titanocene Dichloride as an Electron Transfer Catalyst in Transition Metal Catalyzed Radical Reactions. Angew. Chem. Int. Ed 1998, 37 (1), 101–103. [Google Scholar]; (d) Gansäuer A; Bluhm H; Rinker B; Narayan S; Schick M; Lauterbach T; Pierobon M Reagent-Controlled Stereoselectivity in Titanocene-Catalyzed Epoxide Openings: Reductions and Intermolecular Additions to α,β-Unsaturated Carbonyl Compounds. Chem. Eur. J 2003, 9 (2), 531–542. [DOI] [PubMed] [Google Scholar]
- 17.Yeoman JTS; Mak VW; Reisman SE A Unified Strategy to Ent-Kauranoid Natural Products: Total Syntheses of (−)-Trichorabdal A and (−)-Longikaurin E. J Am. Chem. Soc 2013, 135 (32), 11764–11767. [DOI] [PubMed] [Google Scholar]
- 18.(a) Ito Y; Aoyama H; Hirao T; Mochizuki A; Saegusa T Cyclization reactions via oxo-.pi.-allylpalladium(II) intermediates. J Am. Chem. Soc 1979, 101 (2), 494–496. [Google Scholar]; (b) Kende AS; Roth B; Sanfilippo PJ Facile, palladium(II)-mediated synthesis of bridged and spirocyclic bicycloalkenones. J. Am. Chem. Soc 1982, 104, 1784–1785. [Google Scholar]
- 19.(a) Kavanagh F; Hervey A; Robbins WJ Proc. Natl. Acad. Sci. U. S. A 1951, 37, 570. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Birch AJ; Holzapfel CW; Rickards RW The structure and some aspects of the biosynthesis of pleuromutilin. Tetrahedron 1966, 22 (Suppl. 8), 359–387. [Google Scholar]; (c) Poulsen SM; Karlsson M; Johansson LB; Vester B The pleuromutilin drugs tiamulin and valnemulin bind to the RNA at the peptidyl transferase centre on the ribosome. Mol. Microbiol 2001, 41 (5), 1091–1099. [DOI] [PubMed] [Google Scholar]
- 20.(a) Brooks G; Burgess W; Colthurst D; Hinks JD; Hunt E; Pearson MJ; Shea B; Takle AK; Wilson JM; Woodnutt G Pleuromutilins. Part 1: The Identification of Novel Mutilin 14- Carbamates. Bioorg. Med. Chem 2001, 9 (5), 1221–1231. [DOI] [PubMed] [Google Scholar]; (b) Rittenhouse S; Biswas S; Broskey J; McCloskey L; Moore T; Vasey S; West J; Zalacain M; Zonis R; Payne D Selection of Retapamulin, a Novel Pleuromutilin for Topical Use. Antimicrob. Agents Chemother 2006, 50 (11), 3882–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thirring K; Heilmayer W; Riedl R; Kollmann H; Ivezic- Schoenfeld Z; Wicha W; Paukner S; Strickmann D WO2015110481A1, July 30, 2015. [Google Scholar]
- 22.(a) Gibbons EG Total synthesis of (+)-pleuromutilin. J. Am. Chem. Soc 1982, 104 (6), 1767–1769. [Google Scholar]; (b) Boeckman RK Jr.; Springer DM; Alessi TR Synthetic studies directed toward naturally occurring cyclooctanoids. 2. A stereocontrolled assembly of (+)-pleuromutilin via a remarkable sterically demanding oxy-Cope rearrangement. J. Am. Chem. Soc 1989, 111 (21), 8284–8286. [Google Scholar]; (c) Helm MD; Da Silva M; Sucunza D; Findley TJK; Procter DJ A dialdehyde cyclization cascade: An approach to pleuromutilin. Angew. Chem. Int. Ed 2009, 48 (49), 9315–9317. [DOI] [PubMed] [Google Scholar]; (d) Fazakerley NJ; Helm MD; Procter DJ Total Synthesis of (+)-Pleuromutilin. Chem. Eur. J 2013, 19 (21), 6718–6723. [DOI] [PubMed] [Google Scholar]; (e) Murphy SK; Zeng M; Herzon SB A modular and enantioselective synthesis of the pleuromutilin antibiotics. Science 2017, 356 (6341), 956–959. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zeng M; Murphy SK; Herzon SB Development of a Modular Synthetic Route to (+)-Pleuromutilin, (+)−12-epi-Mutilins, and Related Structures. J. Am. Chem. Soc 2017, 139 (45), 16377–16388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Lou S; Moquist PN; Schaus SE Asymmetric allylboration of ketones catalyzed by chiral diols. J. Am. Chem. Soc 2006, 128 (39), 12660–12661. [DOI] [PubMed] [Google Scholar]; (b) Alam R; Vollgraff T; Eriksson L; Szabo KL Synthesis of adjacent quaternary stereocenters by catalytic asymmetric allylboration. J. Am. Chem. Soc 2015, 137 (35), 11262–11265. [DOI] [PubMed] [Google Scholar]
- 24.White JD; Grether UM; Lee CS (R)-(+)-3, 4-Dimethylcyclohex-2-en-1-one: ((R)-(+)-3, 4-Dimethyl-2-cyclohexen-1-one). Org. Synth 2005, 82, 108–114. [Google Scholar]
- 25.(a) Rogers EF; Koniuszy FR; Shavel J; Folkers K Plant Insecticides. I. Ryanodine, A New Alkaloid from Ryania Speciosa Vahl. J. Am. Chem. Soc 1948, 70 (9), 3086–3088. [DOI] [PubMed] [Google Scholar]; (b) Wiesner K; Valenta Z; Findlay JA The structure of ryanodine. Tetrahedron Lett. 1967, 8 (3), 221–223. [Google Scholar]
- 26.(a) Pessah IN; Waterhouse AL; Casida JE The calcium-ryanodine receptor complex of skeletal and cardiac muscle. Biochem. Biophys. Res. Commun 1985, 128 (1), 449–456. [DOI] [PubMed] [Google Scholar]; (b) Inui M; Saito A; Fleischer S solation of the ryanodine receptor from cardiac sarcoplasmic reticulum and identity with the feet structures. J. Biol. Chem 1987, 262 (32), 15637–15642. [PubMed] [Google Scholar]
- 27.(a) Ryanodine Receptors: Structure, Function and Dysfunction in Clinical Disease; Wehrens XHT, Marks AR, Eds.; Developments in Cardiovascular Medicine; Springer US, 2005. [Google Scholar]; (b) Lanner JT Ryanodine Receptor Physiology and Its Role in Disease. In Calcium Signaling; Islam MS, Ed.; Advances in Experimental Medicine and Biology; Springer: Dordrecht, The Netherlands, 2012. [DOI] [PubMed] [Google Scholar]
- 28.(a) Meissner G Ryanodine activation and inhibition of the Ca2+ release channel of sarcoplasmic reticulum. J. Biol. Chem 1986, 261 (14), 6300–6306. [PubMed] [Google Scholar]; (b) Meissner G; El-Hashem A Ryanodine as a functional probe of the skeletal muscle sarcoplasmic reticulum Ca 2+ release channel. Mol. Cell. Biochem 1992, 114 (1), 119–123. [DOI] [PubMed] [Google Scholar]
- 29.Sutko JL; Airey JA; Welch W; Ruest L The pharmacology of ryanodine and related compounds. Pharmacol. Rev 1997, 49 (1), 53–98. [PubMed] [Google Scholar]
- 30.(a) Achenbach H; Hübner H; Vierling W; Brandt W; Reiter M Spiganthine, the cardioactive principle of Spigelia anthelmia. J. Nat. Prod 1995, 58 (7), 1092–1096. [DOI] [PubMed] [Google Scholar]; (b) Hübner H; Vierling W; Brandt W; Reiter M; Achenbach H Minor constituents of Spigelia anthelmia and their cardiac activities. Phytochemistry 2001, 57 (2), 285–296. [DOI] [PubMed] [Google Scholar]
- 31.(a) Beĺanger A; Berney DJF; Borschberg HJ; Brousseau R; Doutheau A; Durand R; Katayama H; Lapalme R; Leturc DM; Liao CC; MacLachlan FN; Maffrand JP; Marazza F; Martino R; Moreau C; Saint-Laurent L; Saintonge R; Soucy P; Ruest L; Deslongchamps P Total Synthesis of Ryanodol. Can. J. Chem 1979, 57 (24), 3348–3354. [Google Scholar]; (b) Deslongchamps P; Beĺanger A; Bemey DJF; Borschberg HJ; Brousseau R; Doutheau A; Durand R; Katayama H; Lapalme R; Leturc DM; Liao CC; Maclachlan FN; Maffrand JP; Marazza F; Martino R; Moreau C; Ruest L; Saint-Laurent L; Saintonge R; Soucy P The Total Synthesis of (+)-Ryanodol. 1. General Strategy and Search for a Convenient Diene for the Construction of a Key Tricyclic Intermediate. Can. J. Chem 1990, 68 (1), 115–126. [Google Scholar]; (c) Deslongchamps P; Beĺanger A; Berney DJF; Borschberg HJ; Brousseau R; Doutheau A; Durand R; Katayama H; Lapalme R; Leturc DM; Liao CC; Maclachlan FN; Maffrand JP; Marazza F; Martino R; Moreau C; Ruest L; Saint-Laurent L; Saintonge R; Soucy P The Total Synthesis of (+)-Ryanodol. 2. Model Studies for Ring B and Ring C of (+)-Anhydroryanodol. Preparation of a Key Pentacyclic Intermediate. Can. J. Chem 1990, 68 (1), 127–152. [Google Scholar]; (d) Deslongchamps P; Beĺanger A; Bemey DJF; Borschberg HJ; Brousseau R; Doutheau A; Durand R; Katayama H; Lapalme R; Leturc DM; Liao CC; Maclachlan FN; Maffrand JP; Marazza F; Martino R; Moreau C; Ruest L; Saint-Laurent L; Saintonge R; Soucy P The Total Synthesis of (+)-Ryanodol. 3. Preparation of (+)-Anhydroryanodol from a Key Pentacyclic Intermediate. Can. J. Chem 1990, 68 (1), 153–185. [Google Scholar]; (e) Deslongchamps P; Beianger A; Bemey DJF; Borschberg HJ; Brousseau R; Doutheau A; Durand R; Katayama H; Lapalme R; Leturc DM; Liao CC; Maclachlan FN; Maffrand JP; Marazza F; Martino R; Moreau C; Ruest L; Saint-Laurent L; Saintonge R; Soucy P The Total Synthesis of (+)-Ryanodol. 4. Preparation of (+)-Ryanodol from (+)-Anhydroryanodol. Can. J. Chem 1990, 68 (1), 186–192. [Google Scholar]
- 32.(a) Nagatomo M; Koshimizu M; Masuda K; Tabuchi T; Urabe D; Inoue M Total Synthesis of Ryanodol. J. Am. Chem. Soc 2014, 136 (16), 5916–5919. [DOI] [PubMed] [Google Scholar]; (b) Nagatomo M; Hagiwara K; Masuda K; Koshimizu M; Kawamata T; Matsui Y; Urabe D; Inoue M Symmetry-Driven Strategy for the Assembly of the Core Tetracycle of (+)-Ryanodine: Synthetic Utility of a Cobalt-Catalyzed Olefin Oxidation and α-Alkoxy Bridgehead Radical Reaction. Chem. Eur. J 2016, 22 (1), 222–229. [DOI] [PubMed] [Google Scholar]; (c) Masuda K; Koshimizu M; Nagatomo M; Inoue M Asymmetric Total Synthesis of (+)-Ryanodol and (+)-Ryanodine. Chem. Eur. J 2016, 22 (1), 230–236. [DOI] [PubMed] [Google Scholar]; (d) Koshimizu M; Nagatomo M; Inoue M Unified Total Synthesis of 3-epi-Ryanodol, Cinnzeylanol, Cinncassiols A and B, and Structural Revision of Natural Ryanodol and Cinnacasol. Angew. Chem. Int. Ed 2016, 55 (7), 2493–2497. [DOI] [PubMed] [Google Scholar]; (e) Nagatomo M; Hagiwara K; Masuda K; Koshimizu M; Kawamata T; Matsui Y; Urabe D; Inoue M Symmetry-Driven Strategy for the Assembly of the Core Tetracycle of (+)-Ryanodine: Synthetic Utility of a Cobalt-Catalyzed Olefin Oxidation and α-Alkoxy Bridgehead Radical Reaction. Chem. Eur. J 2016, 22 (1), 222–229. [DOI] [PubMed] [Google Scholar]; (f) Masuda K; Koshimizu M; Nagatomo M; Inoue M Asymmetric Total Synthesis of (+)-Ryanodol and (+)-Ryanodine. Chem. Eur. J 2016, 22 (1),230–236. [DOI] [PubMed] [Google Scholar]
- 33.(a) Waterhouse AL; Pessah IN; Francini AO; Casida JE; Structural aspects of ryanodine action and selectivity. J. Med. Chem 1987, 30 (4), 710–716. [DOI] [PubMed] [Google Scholar]; (b) Welch W; Ahmad S; Airey JA; Gerzon K; Humerickhouse RA; Besch HRJ; Ruest L; Deslongchamps P; Sutko JL Structural determinants of high-affinity binding of ryanoids to the vertebrate skeletal muscle ryanodine receptor: a comparative molecular field analysis. Biochemistry 1994, 33 (20), 6074–6085. [DOI] [PubMed] [Google Scholar]
- 34.Xu C; Han A; Reisman SE An Oxidative Dearomatization Approach To Prepare the Pentacyclic Core of Ryanodol. Org. Lett 2018, 20 (13), 3793–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khand IU; Knox GR; Pauson PL; Watts WE; Foreman MI Organocobalt complexes. Part II. Reaction of acetylenehexacarbonyldicobalt complexes, (R1C2R2)Co2(CO)6, with norbornene and its derivatives. J. Chem. Soc. Perkin Trans 1 1973, 977–981. [Google Scholar]
- 36.Chuang KV A Total Synthesis of (+)-Ryanodol. PhD Thesis, California Institute of Technology: Pasadena, CA, 2016. DOI: 10.7907/Z95X26ZV [DOI] [Google Scholar]
- 37.Egi M, Ota Y; Nishimura Y; Shimizu K; Azechi K; Akai S Efficient intramolecular cyclizations of phenoxyethynyl diols into multisubstituted α, β-unsaturated lactones. Org. Lett 2013, 15(16), 4150–4153. [DOI] [PubMed] [Google Scholar]
- 38.Koga Y; Kobayashi T; Narasaka K Rhodium-catalyzed intramolecular Pauson-Khand reaction. Chem. Lett 1998, 27 (3), 249–250. [Google Scholar]
- 39.Riley HL; Morley JF; Friend NAC; 255. Selenium dioxide, a new oxidising agent. Part I. Its reaction with aldehydes and ketones. J. Chem. Soc. Res 1932, 1875–1883. [Google Scholar]
- 40.Masuda K; Nagatomo M; Inoue M Chemical Conversion of Ryanodol to Ryanodine. Chem. Pharm. Bull 2016, 64, 874–879. [DOI] [PubMed] [Google Scholar]
- 41.Dibrell SE;‡ Maser MR;Reisman SE SeO2-Mediated Oxidative Transposition of Pauson–Khand Products. J. Am. Chem. Soc 2020, 142 (14), 6483–6487. [DOI] [PubMed] [Google Scholar]
- 42.González-Coloma A; Terrero D; Perales A; Escoubas P; Fraga BM Insect Antifeedant Ryanodane Diterpenes from Persea Indica. J. Agric. Food Chem 1996, 44 (1), 296–300. [Google Scholar]
- 43.Ling S-Q; Xu Y-N; Gu Y-P; Liu S-Y; Tang W-W Toxicity and Biochemical Effects of Itol A on the Brown Planthopper, Nilaparvata Lugens (Stål) (Hemiptera: Delphacidae). Pestic. Biochem. Physiol 2018, 152, 90–97. [DOI] [PubMed] [Google Scholar]
- 44.(a) Nohara T; Kashiwada Y; Tomimatsu T; Kido M; Tokubuchi N; Nishioka I Cinncassiol D1 and Its Glucoside, Novel Pentacyclic Diterpenes from Cinnamomi Cortex. Tetrahedron Lett. 1980, 21 (27), 2647–2648. [Google Scholar]; (b) Chai X-Y; Bai C-C; Shi H-M; Xu Z-R; Ren H-Y; Li F-F; Lu Y-N; Song Y-L; Tu P-F Six Insecticidal Isoryanodane Diterpenoids from the Bark and Twigs of Itoa Orientalis. Tetrahedron 2008, 64 (24), 5743–5747. [Google Scholar]; (c) Zeng J; Xue Y; Shu P; Qian H; Sa R; Xiang M; Li X-N; Luo Z; Yao G; Zhang Y Diterpenoids with Immunosuppressive Activities from Cinnamomum Cassia. J. Nat. Prod 2014, 77 (8), 1948–1954. [DOI] [PubMed] [Google Scholar]
- 45.Koshimizu M; Nagatomo M; Inoue M Construction of a Pentacyclic Ring System of Isoryanodane Diterpenoids by SmI2-Mediated Transannular Cyclization. Tetrahedron 2018, 74 (26), 3384–3390. [Google Scholar]
- 46.Shen Y; Li L; Pan Z; Wang Y; Li J; Wang K; Wang X; Zhang Y; Hu T; Zhang Y Protecting-group-free total synthesis of (−)-jiadifenolide: development of a [4 + 1] annulation toward multisubstituted tetrahydrofurans. Org. Lett 2015, 17 (21), 5480–5483. [DOI] [PubMed] [Google Scholar]
- 47.(a) Denmark SE; Beutner GL Lewis Base Catalysis in Organic Synthesis. Angew. Chem. Int. Ed 2008, 47 (9), 1560–1638. [DOI] [PubMed] [Google Scholar]; (b) Corey EJ; Link JO A new chiral catalyst for the enantioselective synthesis of secondary alcohols and deuterated primary alcohols by carbonyl reduction. Tetrahedron Lett. 1989, 30 (46), 6275–6278. [Google Scholar]
- 48.Ueda T; Konishi H; Manabe K Palladium-catalyzed fluorocarbonylation using N-formylsaccharin as CO source: general access to carboxylic acid derivatives. Org. Lett 2013, 15 (20), 5370–5373. [DOI] [PubMed] [Google Scholar]