

Abstract
Cutaneous lupus erythematosus (CLE) is a disfiguring and poorly understood condition frequently associated with systemic lupus. Previous studies suggest that non-lesional keratinocytes play a role in disease predisposition, but this has not been investigated in a comprehensive manner or in the context of other cell populations. To investigate CLE immunopathogenesis, normal-appearing skin, lesional skin, and circulating immune cells from lupus patients were analyzed via integrated single-cell RNA-sequencing and spatial RNA-sequencing. We demonstrate that normal-appearing skin of lupus patients represents a type I interferon-rich, prelesional environment that skews gene transcription in all major skin cell types and dramatically distorts predicted cell-cell communication networks. We also show that lupus-enriched CD16+ dendritic cells undergo robust interferon education in the skin, thereby gaining pro-inflammatory phenotypes. Together, our data provide a comprehensive characterization of lesional and non-lesional skin in lupus and suggest a role for skin education of CD16+ dendritic cells in CLE pathogenesis.
ONE SENTENCE SUMMARY:
Non-lesional and lesional lupus skin share inflammatory phenotypes that drive activation of CD16+ dendritic cells.
EDITOR’S SUMMARY FOR ABN2263
Comprehending Cutaneous Lupus
Cutaneous lupus erythematosus (CLE) is disfiguring skin condition that affects most patients with systemic lupus erythematosus (SLE) and can be resistant to treatment even when systemic disease is responsive. Billi et al. analyzed analyzed CLE lesions and paired normal-appearing skin biopsies, as well as circulating immune cell subsets, to better understand changes in the skin that drive CLE pathogenesis. Using single-cell RNA sequencing and spatial RNA sequencing, they identified a type I IFN-rich signature in prelesional, normal looking skin that influenced transcription and cell-cell communication for all major skin cell types. CD16+ dendritic cells, which are associated with SLE, were also shaped by the type I IFN environment, and cells in these sites shifted toward a pro-inflammatory phenotype. Together these data provide insights into transcriptional changes in the skin that contribute to CLE pathogenesis.
INTRODUCTION
Cutaneous lupus erythematosus (CLE) is a disfiguring inflammatory skin disease that affects 70% of patients with systemic lupus erythematosus (SLE). Whereas ~50% of patients respond to SLE-directed therapies (1), many patients suffer from refractory skin lesions, even when their systemic disease is controlled. Lack of knowledge regarding the inflammatory composition of CLE and the drivers that instigate disease has delayed effective therapy development.
Intriguingly, the etiology of skin lesions in CLE may be in part found in abnormalities of non-lesional, normal-appearing skin. Recent data support a role for increased epidermal type I interferon (IFN) production (2–5) and dysfunction of Langerhans cells (6) as important for priming inflammatory and apoptotic responses. However, the role of other cells in the skin, the skewed communication networks between them, and cellular mediators of this inflammatory predisposition have not been well-defined.
In this paper, we examined the cellular composition of paired lesional and non-lesional skin samples from SLE patients with active CLE lesions to comprehensively define the cellular makeup and to characterize the principal mediators of inflammatory changes that contribute to the disease. We examined the peripheral blood of the same patients to investigate the cutaneous education of monocyte-derived CD16+ dendritic cells, which were found to be prominent in lesional and non-lesional skin. Overall, we found an IFN-rich signature and a discrete pro-inflammatory cellular communication network between stromal and inflammatory cells in lesional and non-lesional skin that supports a critical role for the skin in priming inflammatory responses in SLE patients.
RESULTS
Single-cell RNA-sequencing of lesional and non-lesional skin from patients with CLE identifies diverse skin and immune cell populations
To investigate the cellular composition and comprehensive transcriptional effects of CLE, we performed single-cell RNA-sequencing (scRNA-seq) on lesional and sun-protected non-lesional skin from 7 patients with active CLE (Table S1), 6 of whom also carried a diagnosis of SLE. Samples were analyzed in parallel with skin from 14 healthy controls from diverse anatomical sites. The final dataset comprised 46,540 cells, with an average of 2,618 genes and 11,645 transcripts per cell. Visualization using Uniform Manifold Approximation and Projection (UMAP) revealed 26 distinct cell clusters (Fig. 1a) that were annotated as 10 major cell types, each comprising cells from lesional, non-lesional, and healthy control skin biopsies (Fig. 1b–d). Conspicuous clustering by disease state was evident for many cell types, including keratinocytes (KCs) and myeloid cells. Cell composition analysis revealed an increase in the proportion of myeloid cells in both lesional and non-lesional skin relative to healthy control (Fig. 1e,f).
Figure 1. Single-cell RNA-sequencing (scRNA-seq) captures the cellular diversity within lesional and non-lesional skin of patients with cutaneous lupus erythematosus (CLE).
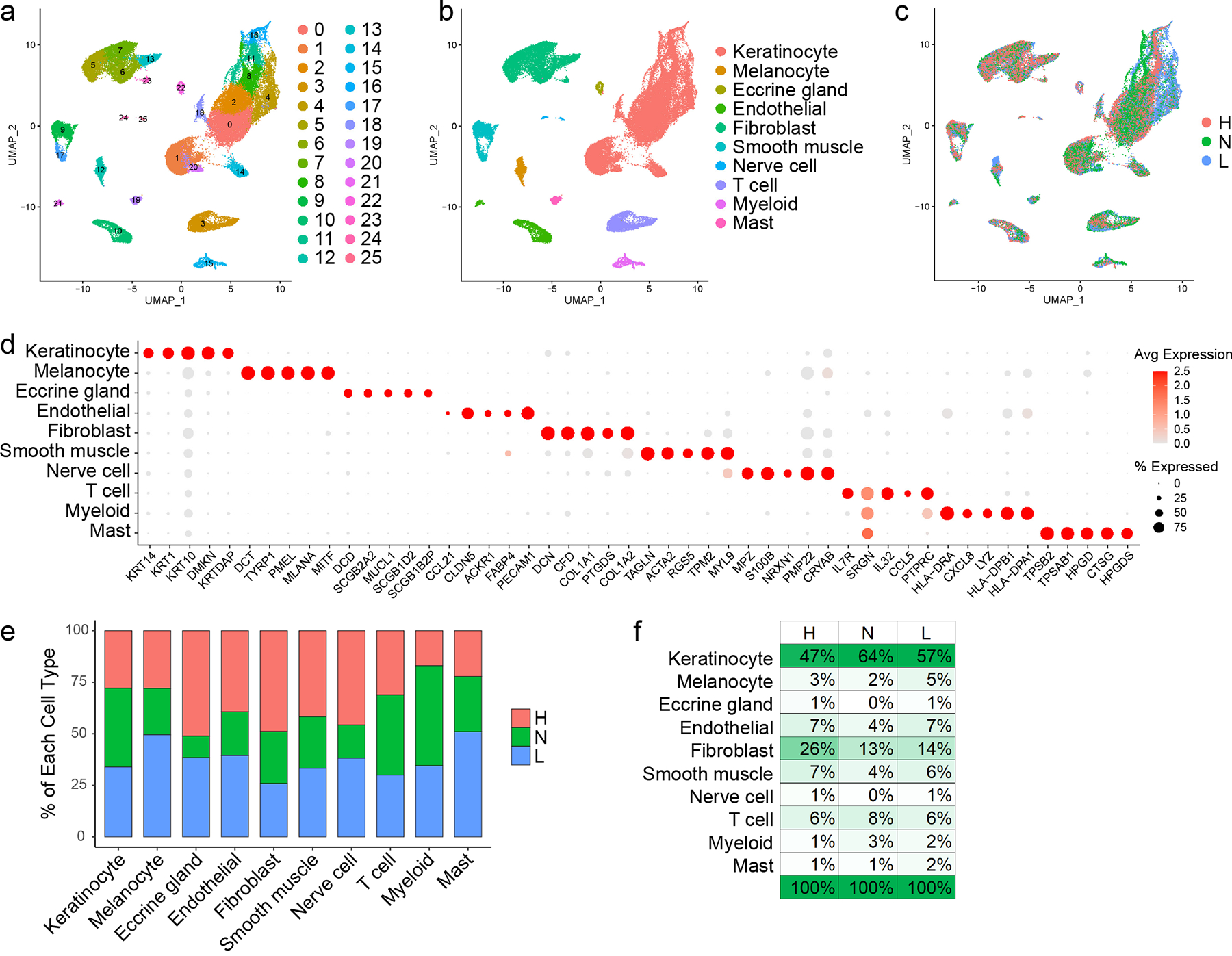
a. UMAP plot of 46,540 cells colored by cluster. b. UMAP plot of cells colored by cell type. c. UMAP plot of cells colored by disease state (H, healthy control skin; N, non-lesional lupus skin; L, lesional lupus skin). d. Dot plot of representative marker genes for each cell type. Color scale, average marker gene expression. Dot size, percentage of cells expressing marker gene. e. Bar plot showing the relative contribution of the three disease states to the total number of each cell type. Values are normalized to the total number of cells for each disease state. f. Composition of cells recovered for each disease state by cell type.
KCs from both lesional and non-lesional skin of patients with CLE exhibit a pathologic type I IFN signature
KCs constituted the majority of cells sequenced (25,675 cells). Sub-clustering analysis of KCs identified 14 sub-clusters (Fig. 2a), including several (5, 6, 8, 13) dominated by KCs from lupus patients (Fig. 2b,c). Analyses of characteristic KC subtype markers identified 5 KC states: basal, spinous, supraspinous, follicular, and cycling (Fig. 2d, Fig. S1a). Lupus-dominated sub-clusters corresponded to subpopulations within basal and spinous KC states.
Figure 2. Interferon (IFN) responses shape the transcriptomic landscape of both lesional and non-lesional keratinocytes in patients with CLE.
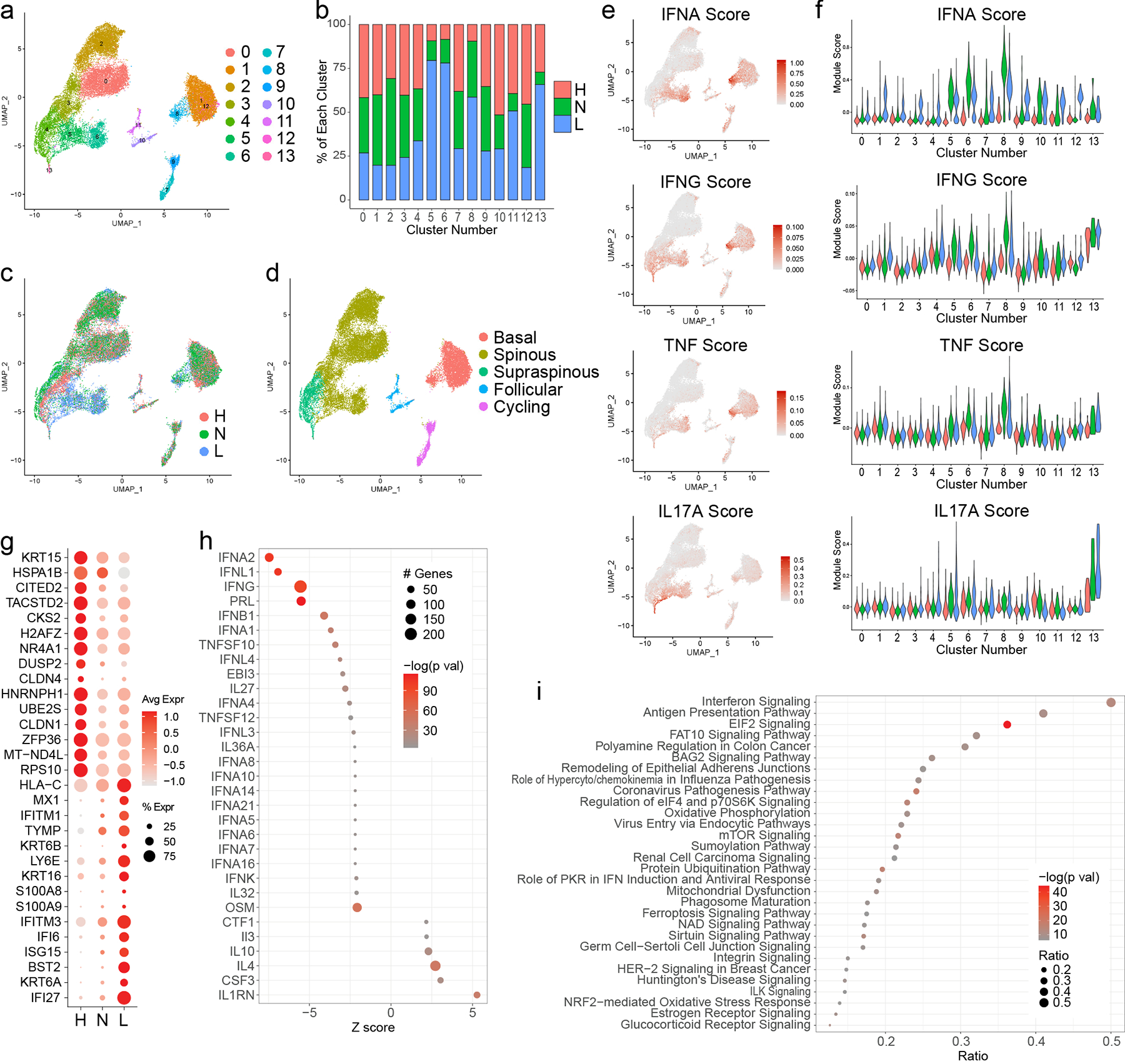
a. UMAP plot of 25,675 keratinocytes (KCs) colored by sub-cluster. b. Bar plot of each KC sub-cluster showing the relative contribution of the three disease states. Values are normalized to the total number of KCs for each disease state. c. UMAP plot of KCs colored by disease state. d. UMAP plot of KCs colored by subtype. e. Feature plots of module scores for the indicated KC cytokine modules. f. Violin plots of KC scores for the indicated cytokine modules split by disease state. g. Dot plot of the top 15 differentially expressed genes (DEGs) downregulated and upregulated in L vs. H basal KCs. Color scale, average marker gene expression. Dot size, percentage of cells expressing marker gene. h. Dot plot of the top 30 upstream regulators enriched among DEGs in L vs. H basal KCs. Color scale, −log10(p value) from the enrichment analysis. Dot size, number of DEGs corresponding to each upstream regulator. Negative Z score, enriched in L; positive, enriched in H. i. Dot plot of the top 30 canonical pathways enriched among DEGs in L vs. H basal KCs. Color scale, −log10(p value) from the enrichment analysis. Dot size, ratio of [number of pathway genes among the DEGs]/[total number of pathway genes].
The relatively shallow depth of scRNA-seq precludes direct examination of transcript levels for many cytokines implicated in CLE, particularly IFNs. To investigate whether cytokine responses were driving KC sub-clustering by disease in CLE, we used cytokine response signatures previously developed by our group that contain genes induced upon stimulation of primary human keratinocytes with a panel of cytokines (IFN-α, IFN-γ, TNF, IL-17A, IL-1B, IL-36, IL-4, IL-13, or IL-17A and TNF in combination). We plotted the cytokine module scores for each KC in our single-cell dataset (Fig. 2e, Fig. S1b). Lupus-enriched sub-clusters corresponded best with cells exhibiting high scores for type I IFN (IFN-α), type II IFN (IFN-γ), and, to a lesser degree, TNF. Our and others’ prior work has identified a critical role for type I IFN in SLE and CLE keratinocytes (2–5). Accordingly, cytokine module violin plots revealed that lupus-enriched basal (sub-cluster 8) and spinous (5 and 6) sub-clusters consisted almost entirely of KCs with high IFN-α module scores (Fig. 2f, Fig. S1c), whereas the separation was less striking for IFN-γ and TNF. Notably, cells scoring highest in these clusters originated from non-lesional biopsies. This suggests that even normal-appearing skin from patients with CLE exists in a ‘prelesional’ state, primed by heightened type I IFN signaling. Follicular KC sub-clusters (10 and 11) showed elevated IFN-α cytokine module scores in non-lesional and lesional samples relative to healthy controls as well (Fig. 2f), suggesting the follicular epithelium also represents an abnormal, IFN-rich environment in CLE. Scores were far lower for follicular than basal KCs, implicating the interfollicular epidermis more strongly than the follicular epithelium in type I IFN education of neighboring cells.
For a broader understanding of the transcriptomic differences in lesional KCs of patients with CLE, we performed differential expression analysis between the lesional CLE vs. healthy basal KCs and identified type I IFN downstream genes (e.g., MX1, IFITM1, IFITM3, IFI6, ISG15, IFI27) among the top upregulated genes in the lesional cells (Fig. 2g). We then used Ingenuity Pathway Analysis (IPA) to identify the top cytokines predicted to serve as upstream regulators for the genes induced in lesional samples, identifying primarily IFNs as upstream regulators of CLE-enriched transcripts (Fig. 2h). Corroborating this, canonical pathway analysis distinguished IFN signaling as highly enriched in lesional samples (Fig. 2i).
scRNA-seq identifies a CLE-enriched fibroblast subtype exhibiting a strong IFN response signature
We next analyzed fibroblasts (FBs), the other major stromal cell constituent of the skin. Sub-clustering analysis of 8,622 FBs identified 10 sub-clusters (Fig. 3a). Only one sub-cluster (4) was dominated by FBs from lupus patients (Fig. 3b,c). Annotation of these sub-clusters based on published dermal FB marker genes (7) revealed three subtypes as previously described (SFRP2+, COL11A1+, and SFRP4+ FBs) and a small cluster marked by expression of COL26A1 and RAMP1 (RAMP1+ FBs) (Fig. 3d, Fig. S2a). Immunohistochemistry of these key markers (Fig. S2b) confirmed that SFRP2+ FBs constituted the majority of FBs (7). The lupus-enriched sub-cluster lay within the SFRP2+ FBs and was analyzed as an independent subtype. Analysis of the top gene markers of each FB subtype indicated that these FBs were distinguished by high IFN-stimulated gene (ISG) expression (Fig. S2c, Data File S1), and thus we designated these IFN FBs. To corroborate this, we took advantage of FB cytokine response signatures generated by treating primary human FBs with a panel of cytokines (IFN-α, IFN-γ, TNF, IL-1B, IL-36, IL-4, IL-13, or TGF-β) followed by bulk RNA-seq to identify significantly induced genes as done above for KCs. Consistent with our annotation of IFN FBs, UMAP and violin plots depicting FB cytokine module scores revealed that IFN FBs were most uniquely distinguished by IFN-α and IFN-γ cytokine signatures (Fig. 3e,f, Fig. S2d,e). As in KCs (Fig. 2e), IFN cytokine module scores were highest in non-lesional FBs, reinforcing that normal-appearing skin of lupus patients represents a prelesional, IFN-primed environment. We compared the cytokine upstream regulators identified in the comparisons of non-lesional vs. healthy basal KCs and non-lesional IFN FBs vs. healthy SFRP2+ FBs. This revealed that type I IFNs, and potentially type III IFNs such as IFN-λ1, served as the top upstream regulators in both cell types (Fig. 3g). Notably, high FB IFN module scores were primarily restricted to the single sub-cluster of IFN FBs (Fig. 3f), indicating that only a specific subset of FBs in skin of patients with CLE exhibits robust IFN-induced gene expression changes. This is an interesting contrast to KCs, where non-lesional and/or lesional KCs showed elevated IFN module scores for the majority of sub-clusters (Fig. 2e).
Figure 3. Analysis of fibroblast (FB) heterogeneity in CLE patient skin identifies an IFN-responsive FB subtype present in both non-lesional and lesional skin.
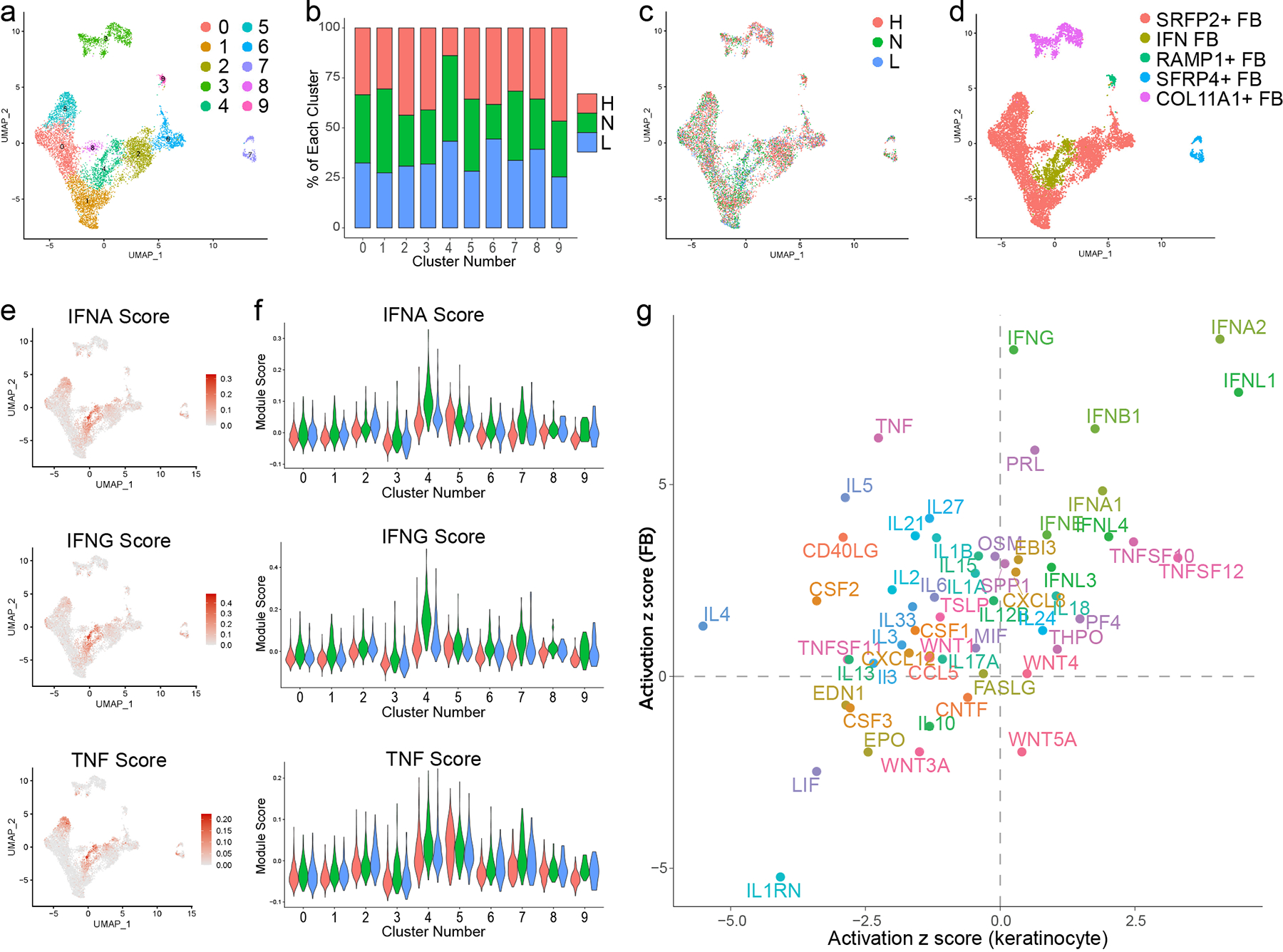
a. UMAP plot of 8,622 FBs colored by sub-cluster. b. Bar plot of each FB sub-cluster showing the relative contribution of the three disease states. Values are normalized to the total number of FBs for each disease state. c. UMAP plot of FBs colored by disease state. d. UMAP plot of FBs colored by subtype. e. Feature plots of scores for the indicated FB cytokine module scores. f. Violin plots of FB module scores for the indicated cytokine modules split by disease state. g. Scatter plot showing activation z scores for cytokine upstream regulators common to basal KCs (N vs. H) and IFN FBs (N) vs. SFRP2+ FBs (H).
T cells infiltrating lesional and non-lesional skin of patients with CLE demonstrate IFN education across multiple subsets including regulatory T cells
Having analyzed the major stromal cell types of the skin, we moved on to examination of the immune cells. Sub-clustering and annotation based on established marker genes identified nine T cell subsets (Fig. 4a–c). Cells of one sub-cluster were distinguished by expression of ISGs and were therefore designated IFN T cells (Fig. 4c). IFN T cells derived primarily from lupus samples (Fig. 4d), constituting 13% and 15% of T cells from non-lesional and lesional samples, respectively, but <1% of healthy control T cells. Probable regulatory T cells (Tregs), annotated based on FOXP3 expression, were detected in similar proportions across healthy control, non-lesional, and lesional samples (Fig. 4d). Comparison of gene expression in non-lesional vs. healthy control Tregs identified numerous ISGs, suggestive of chronic IFN stimulation (Fig. 4e). We and others have identified a role for type I IFNs in driving Treg dysfunction in SLE (8, 9). Thus, the chronic IFN stimulation of Tregs that we observe in non-lesional skin may contribute to impaired Treg ability to maintain immune homeostasis and self-tolerance in lupus. Additionally, Treg function and disease activity improve following low dose IL-2 therapy (10), further supporting a pathogenic role for IFN-mediated Treg dysfunction.
Figure 4. Skin of CLE patients exhibits an abnormal T cell infiltrate at both lesional and non-lesional sites.

a. UMAP plot of 3,030 T cells colored by disease state. b. UMAP plot of T cells colored by subset. Tcm, central memory T cells; Tem, effector memory T cells; Trm, tissue-resident memory T cells; CD161+ Tm, CD161+ memory T cells; Treg, probable regulatory T cells; Tph/Tfh, T peripheral helper and/or T follicular helper cells; IFN T, interferon T cells; CD8T, CD8+ T cells; NKC, natural killer cells. c. Dot plot of representative marker genes for each T cell subset. Color scale, average marker gene expression. Dot size, percentage of cells expressing marker gene. d. Bar plot of each T cell subset showing the relative contribution of the three disease states. Values are normalized to the total number of T cells for each disease state. e. Dot plot of the top 15 differentially expressed genes (DEGs) downregulated and upregulated in N vs. H Tregs.
One sub-cluster of T cells clustered closely with Tregs but did not express FOXP3. Rather, this sub-cluster was distinguished by expression of CXCL13 (Fig. 4c), a B cell-attracting chemokine and SLE biomarker that appears to play a pathogenic role (reviewed by Schiffer et al. (11)), as well as ICOS and PDCD1 (encoding PD-1), leading us to annotate these as T peripheral helper and/or T follicular helper (Tph/Tfh) cells. Abundance of these cells varied by disease state at 1%, 4%, and 2% of healthy control, non-lesional, and lesional T cells, respectively. Closer inspection revealed that Tph/Tfh cells from healthy control and non-lesional samples differed in expression of ISGs including CXCL13 (expressed by 0% of healthy control vs. 76% of non-lesional Tph/Tfh cells). The role of Tph/Tfh cells in skin inflammation in lupus warrants further investigation.
Altogether, T cell imbalances and the presence of IFN T cells and other IFN-educated T cell subsets including Tregs in non-lesional samples indicate the presence of an abnormal T cell infiltrate poised in the prelesional environment of normal-appearing skin of patients with CLE.
Major shifts in myeloid cell subsets are detected in lesional and non-lesional skin of CLE patients
We evaluated myeloid cells, the other major immune cell type detected in our skin samples. Sub-clustering and annotation identified nine myeloid cell subsets with largely distinct marker genes (Fig. 5a–c): classical type 1 dendritic cell (cDC1; CLEC9A, IRF8), classical type 2 dendritic cell subset A (cDC2A; LAMP3 and CD1B), classical type 2 dendritic cell subset B (cDC2B; CLEC10A, IL1B), plasmacytoid dendritic cell (pDC; GZMB, JCHAIN), CD16+ dendritic cell (CD16+ DC; FCGR3A, HES1), Langerhans cell (LC; CD207, CD1A), lipid-associated macrophage (LAM; APOE, APOC1), perivascular macrophage (PVM; CD163, SELENOP), and plasmacytoid dendritic cell-like cell (pDC-like; PPP1R14A, TRPM4) that was previously described by Villani et al. (12). Myeloid cell subsets showed far greater variability in representation among healthy control, non-lesional, and lesional samples than the above cell types (Fig. 5a,d,f).
Figure 5. Non-lesional skin of CLE patients shows major myeloid cell subset shifts including infiltration of plasmacytoid and CD16+ dendritic cells.
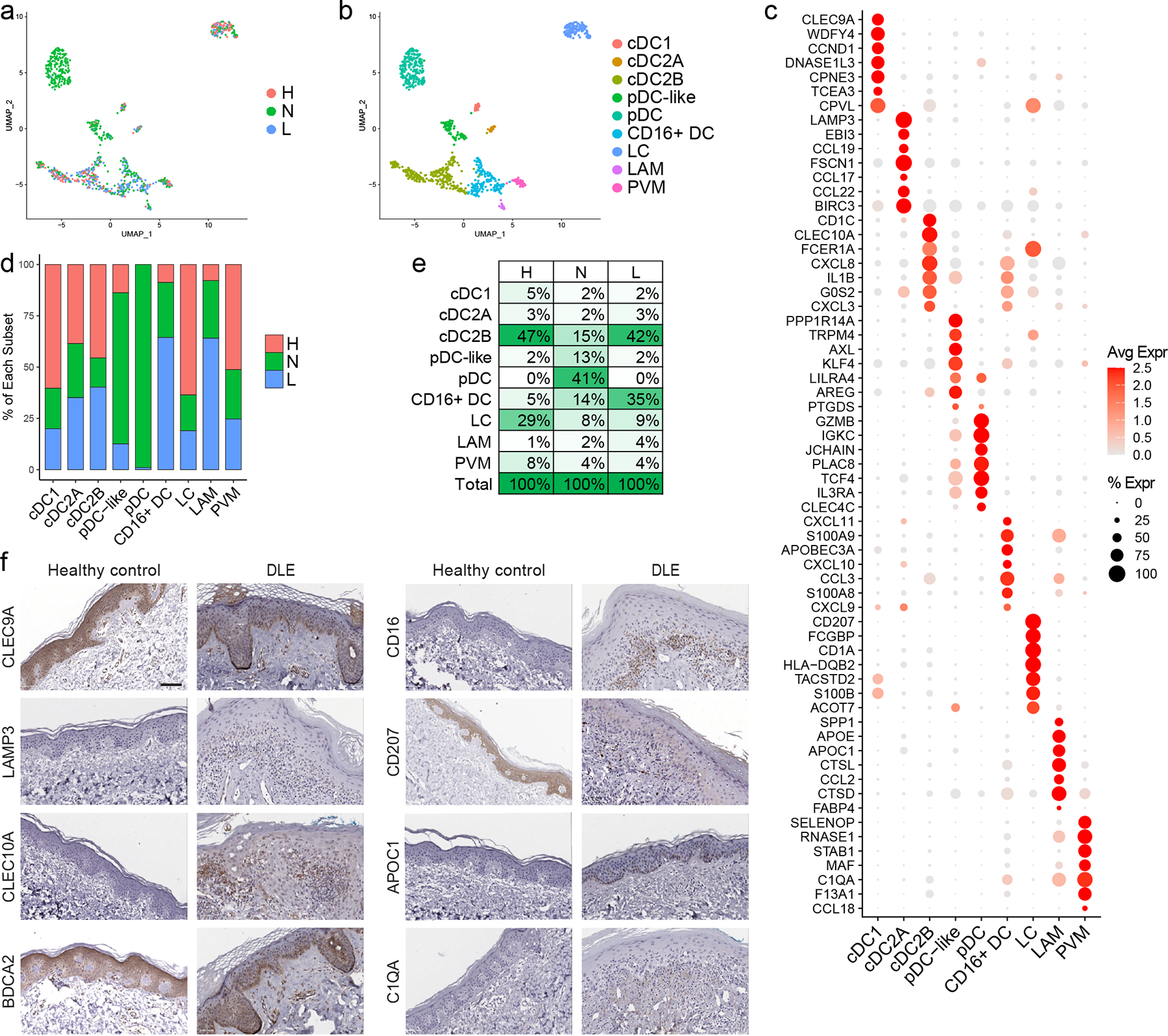
a. UMAP plot of 973 myeloid cells colored by disease state. b. UMAP plot of myeloid cells colored by subset. cDC1, classical type 1 dendritic cells (DCs); cDC2A, classical type 2 DC subset A; cDC2B, classical type 2 DC subset B; pDC, plasmacytoid DC; DC, dendritic cell; LC, Langerhans cell; LAM, lipid-associated macrophage; PVM, perivascular macrophage. c. Dot plot of representative marker genes for each myeloid cell subset. Color scale, average marker gene expression. Dot size, percentage of cells expressing marker gene. d. Bar plot of each myeloid cell subset showing the relative contribution of the three disease states. Values are normalized to the total number of myeloid cells for each disease state. e. Percentage of cells in each myeloid cell subset divided by disease state. f. Immunostaining for the indicated marker genes for each myeloid cell subset in healthy control and discoid lupus erythematosus (DLE) lesional skin. CLEC9A, cDC1; LAMP3, cDC2A; CLEC10A, cDC2B; BDCA2, pDC; CD16, CD16+ DC; CD207, LC; APOC1, LAM; and C1QA, PVM. Scale bar, 50 μm. Images are representative of 2 healthy control and 3 DLE sections.
In healthy control skin, cDC2Bs accounted for nearly half (47%) of the myeloid cells; this differed greatly from non-lesional lupus skin, where pDCs dominated (41%), although the majority were from two patients (Fig. 5e). In keeping with prior reports, LCs were decreased in lesional (13, 14) and non-lesional (6) lupus skin (Fig. 5d,e). Among the most striking differences between healthy control and lupus skin, however, was the overrepresentation of CD16+ DCs in both non-lesional and lesional CLE samples compared to healthy skin (Fig. 5d,e). While the exact identity of these cells remains somewhat in flux, CD16+ DCs are gaining recognition as a unique DC subset characterized by expression of FCGR3A/CD16a that can be detected as a transcriptomically distinct population (labeled DC4 in work by Villani et al. profiling circulating mononuclear cell populations in healthy individuals using scRNA-seq (12)). This population is thought to overlap with CD16+ DCs previously described by MacDonald et al. as expressing high levels of CD86 and CD40 and possessing potent T cell stimulatory capabilities (15). CD16+ DCs also exhibit enhanced capacity relative to cDC2Bs for secretion of inflammatory cytokines upon toll-like receptor stimulation (16), a capacity that is further enhanced in CD16+ DCs isolated from peripheral blood of patients with SLE (17). Expansion and enhanced function of CD16+ DCs in lupus patients could therefore promote pathogenesis. Based on shared surface marker expression, this subset may also overlap with 6-Sulfo LacNAc-dendritic cells (slanDCs) (18), a pro-inflammatory myeloid DC subset that has been linked to lupus immunopathogenesis (19, 20) and is increased in lesional skin of lupus patients (19).
Ligand-receptor analysis demonstrates lupus-enriched predicted cell-cell interactions prominently involving CD16+ DCs
Following identification of cellular populations, we then sought to understand how cell-cell communication differed in the skin of lupus patients. We performed ligand-receptor (L-R) analyses among all major cell populations within healthy control, non-lesional, and lesional skin samples using CellPhoneDB. Each L-R pair was then assigned to the condition in which it showed the highest interaction score, and the number of interactions for each cell type pair was plotted. Few L-R interactions were strongest in healthy control skin, and the majority of these represented KC-KC crosstalk (Fig. 6a). Non-lesional skin, in contrast, showed many more interactions (Fig. 6b). FBs represented the main ligand-expressing cell type among non-lesional-enriched pairs, but myeloid and endothelial cells (ECs) were also highly interactive. Additionally, eccrine gland cells participated in a high number of interactions in non-lesional skin as expressers of both ligands and receptors, which is of interest given that perieccrine inflammation is a hallmark of CLE. In lesional skin, however, myeloid and ECs were most prominent among cell-cell interactors (Fig. 6c).
Figure 6. Ligand-receptor (L-R) analysis indicates major shifts in cell-cell communication in CLE and identifies CD16+ DCs as the top candidate cellular interactors in non-lesional skin.
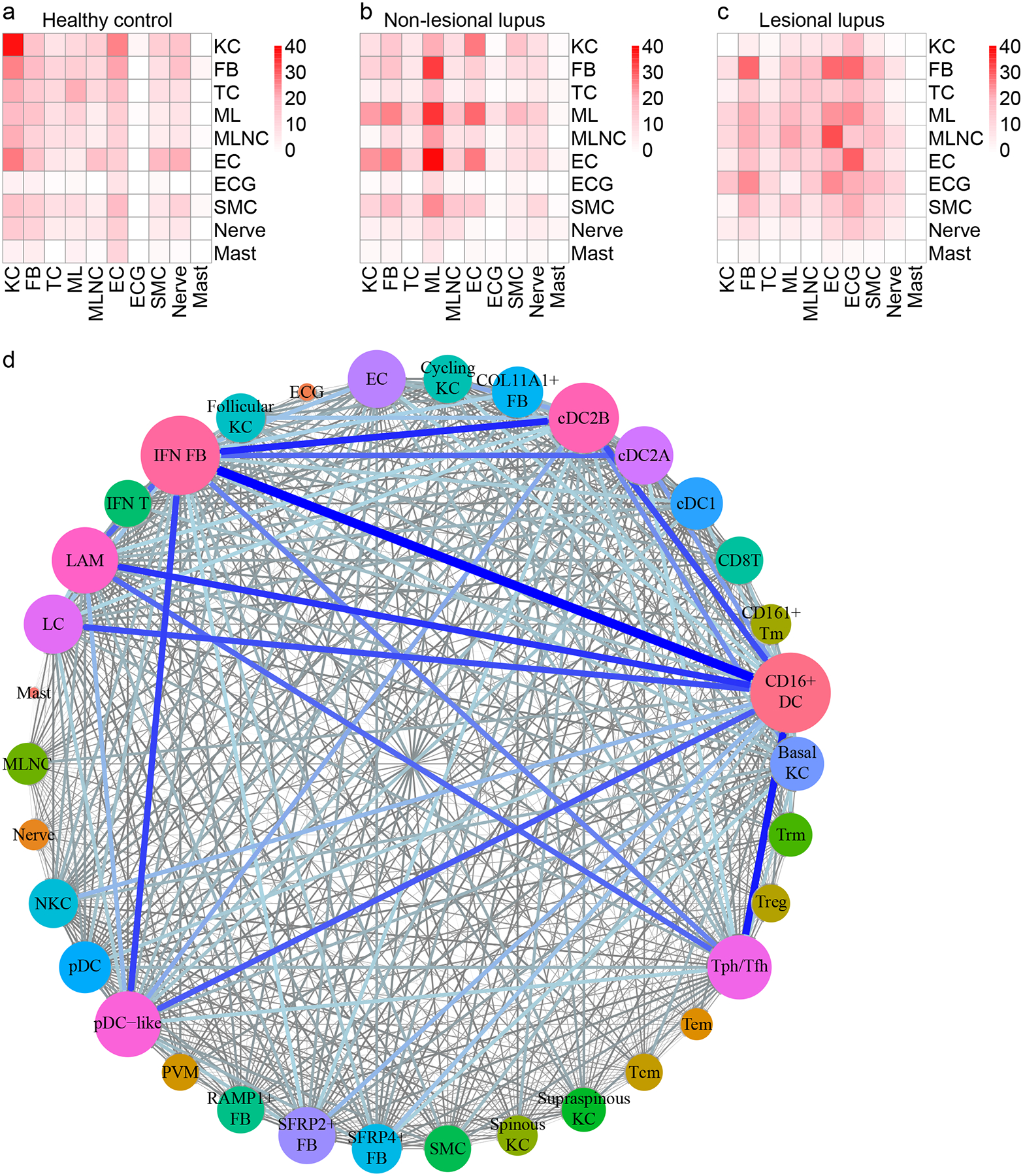
a. Heatmap depicting the number of L-R pairs with interaction scores highest in H samples divided by cell type. Row, cell type expressing the ligand; column, cell type expressing the receptor. Color scale, number of L-R pairs. TC, T cell; ML, myeloid cell; MLNC, melanocyte; EC, endothelial cell; ECG; eccrine gland cell; SMC, smooth muscle cell. b. Heatmap depicting the number of L-R pairs with interaction scores highest in N samples. c. Heatmap depicting the number of L-R pairs with interaction scores highest in L samples. d. Connectome web analysis of interacting cell types in N samples. Vertex (colored cell node) size is proportional to the number of interactions to and from that cell type, whereas the thickness of the connecting lines is proportional to the number of interactions between two nodes.
These analyses suggest a prominent role for myeloid cells in non-lesional and lesional skin. Given the functional heterogeneity within the myeloid cell population, we sought to define more precisely the myeloid and other cellular participants in these interactions. Thus, we divided KCs, FBs, T cells, and myeloid cells into their respective subsets and repeated analysis of L-R pairs. Plotting the L-R interactions in non-lesional skin revealed an even denser network of candidate cellular interactors (Fig. 6d). Regarding stromal cells, ligands expressed by KC subsets primarily signaled to receptors on ECs, suggesting a mechanism by which KCs may influence tissue infiltration by immune cells. IFN FBs were among the most active of all cell subsets. However, CD16+ DCs represented the top interactors. Expressing both ligands and receptors, CD16+ DCs showed numerous enriched interactions involving IFN FBs, Tph/Tfh cells, and cDC2B cells. pDCs were comparatively inert, rarely participating in L-R pairs (Fig. 6d), consistent with recent literature supporting a more senescent phenotype of pDCs in SLE skin(4).
Integration of spatial-seq and scRNA-seq analyses provides architectural context shaping cell-cell interactions within lupus skin
L-R analysis derives exclusively from differential gene enrichment and therefore lacks critical spatial context to substantiate putative interactions. To bolster our interaction analyses, we analyzed discoid lupus lesional skin sections using spatial sequencing on the 10x Genomics Visium platform. A section containing multiple hair follicle segments was selected for in depth analysis (Fig. 7a). We detected 632 spatially defined spots with an average of 3,704 genes and 10,176 transcripts per spot. Abundant dermal deposition of extracellular glycosaminoglycans, termed mucin, is a frequent feature of cutaneous lupus and is evident in the section as collagen fiber splaying; accordingly, many dermal areas showed very low transcript detection and were excluded during quality control.
Figure 7. Spatial sequencing reinforces the effects of IFN-producing interfollicular KCs on CD16+ DCs and FBs in the superficial dermis.
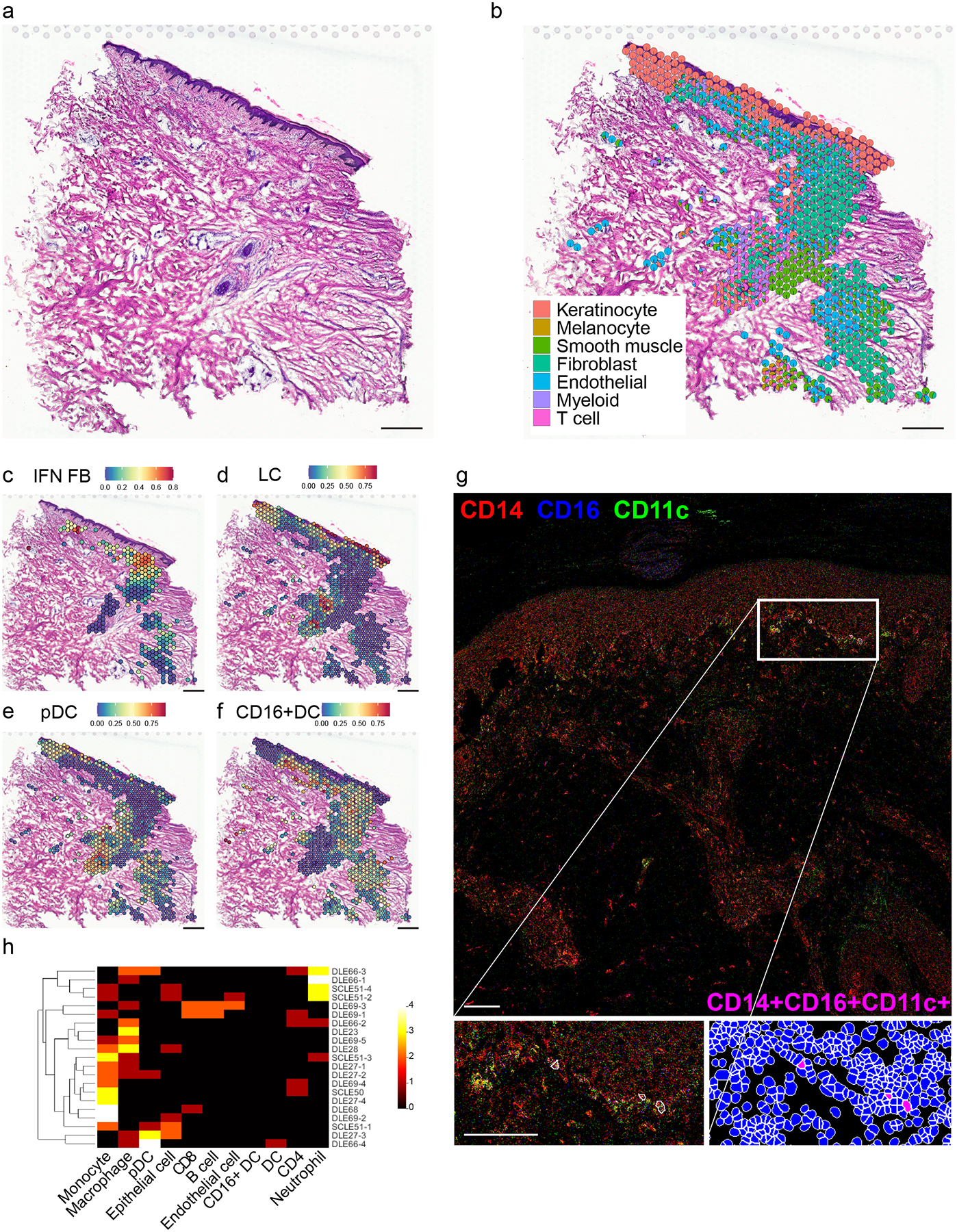
N=4 biological replicates; data are shown for the most complex sample as defined by the highest number of spots after quality control steps. a. Hematoxylin and eosin staining of DLE tissue section corresponding to spatial sequencing data below. Scale bar, 200 μm. b. Spatial scatter pie plot showing cell type composition based on detection of scRNA-seq signatures corresponding to 7 cell types. Each spot is represented as a pie chart showing relative cell type proportions. Spot coordinates correspond to tissue location. c. Spatial heatmap of the IFN FB subset gene signature. Color, scaled expression of each subset gene signature. Only spots meeting a FB prediction score threshold of 0.25 are shown. d. Spatial heatmap of the LC subset gene signature. All spots are shown. e. Spatial heatmap of the pDC subset gene signature. All spots are shown. f. Spatial heatmap of the CD16+ DC subset gene signature. All spots are shown. g. Representative image of a DLE skin section showing the localization of CD16+ DCs (as indicated by the CD14+CD11c+CD16+ immunophenotype) generated by imaging mass cytometry. Scale bar, 100 μm. Insets, subepidermal enrichment of CD16+ DCs. h. Heatmap depicting the number of the indicated cell types (columns) located within 4μm of each of the CD16+ DCs (rows) located across 6 DLE (N=16 CD16+ DCs) and 2 subacute CLE (SCLE; N=5 CD16+ DCs) sections. Color scale, number of neighboring cells.
At a diameter of 55μm, each spot may account for multiple cells of intermixed types. This was corroborated by spatial heatmaps showing overlapping expression of representative marker genes corresponding to the major cell types (Fig. S3a). Accordingly, rather than assigning a single cell type to each spot, we generated a pie chart for each spot showing the representation of the transcriptomic signature of each major cell type (Fig. 7b, Fig. S3b). This approach recapitulated the architecture visible on H&E staining, with the epidermis and follicle showing high KC signature detection and the dermis showing a mix of signatures corresponding primarily to FBs, ECs, and smooth muscle cells, which include both vascular smooth muscle and the cells of the arrector pili muscle attached to the hair follicle. The majority of spots with high immune cell signatures localize to the subepidermal and perifollicular regions (Fig. 7b), corresponding respectively to the characteristic interface dermatitis and periadnexal infiltrate of discoid lupus. Subepidermal spots showed a particularly prominent myeloid signature with a comparatively weak T cell signature, suggesting strong localization of myeloid cells to the interface, possibly as a direct effect of signaling initiated by KCs or FBs in the prelesional CLE environment.
To further understand how the KC, FB, T cell, and myeloid cell heterogeneity observed in our scRNA-seq mapped onto the architecture of the lesional tissue, we generated spatial heatmaps showing prediction scores corresponding to the subsets defined above. Spatial KC subset analysis demonstrated appropriate localization of the supraspinous, spinous, and basal KC signals to the superficial, mid, and basal epidermis, respectively (Fig. S3c). The follicular KC signal localized to the follicular epithelium and the cycling KC signal primarily to the deeper portion of the follicle, where the stem cells that give rise to the follicle are located. Consistent with the interfollicular epidermis representing the primary site of exaggerated IFN education in CLE, spatial FB subset analysis revealed prominent localization of the IFN FB signal to the superficial dermis (Fig. 7c) and detection of other FB subsets in the perifollicular dermis (Fig. S3d). Spatial T cell subset analysis also showed subset-specific localization within the tissue section (Fig. S3e).
Myeloid cell subset spatial heatmaps (Fig. S3f) complemented our immunostaining results (Fig. 5f). As expected, spots showing a strong LC gene signature were restricted to the epidermis and the follicular epithelium (Fig. 7d). Many spots in the perifollicular dermis scored highly for pDCs (Fig. 7e). Spots scoring highly for CD16+ DC clustered most densely in the superficial dermis (Fig. 7f), again suggesting these cells can be modulated in the IFN-rich environment generated by the basal KCs of the interfollicular epidermis (2).
To further dissect CD16+ DC cell-cell communication at the level of the individual cell, we performed imaging mass cytometry of lesional DLE and subacute CLE (SCLE) skin biopsies, defining CD16+ DCs as CD14+CD11c+CD16+ cells. This identified CD16+ DCs primarily concentrated in the superficial dermis directly under the dermo-epidermal junction (Fig. 7g). Enumeration of neighboring cells revealed that diverse immune cell types are detected within a 4 μm distance of CD16+ DCs, with monocytes and macrophages being more common than lymphocytes (Fig. 7h). Among stromal cell types included in the analysis, epithelial cells (here, keratinocytes), occurred more commonly in proximity to CD16+ DCs than ECs, supporting interaction between CD16+ DCs and basal KCs. Statistical analysis demonstrated significant overrepresentation of innate inflammatory cells including pDCs and monocytes in proximity to CD16+ DCs in both DLE and SCLE (Fig. S4).
Pseudotime analysis of paired circulating and skin-infiltrating myeloid cells suggests that CD16+ DCs arise from non-classical monocytes that undergo IFN education in lupus skin
Overall, our data thus far supported close communication of CD16+ DCs with stromal cells in the skin, so we next wanted to understand the origin and phenotype of CD16+ DCs that infiltrate the skin in lupus patients. Reasoning that these likely arise from circulating mononuclear cells similar to the DC4 subset identified by Villani et al. (12), we examined peripheral blood mononuclear cells (PBMCs) by scRNA-seq from the same seven lupus patients above as well as from four healthy controls. PBMC and skin cell data were aggregated for clustering, and clusters containing myeloid cells were selected for further analysis based on expression of established markers (Fig. S5). Myeloid cells from PBMCs and skin were then re-clustered together to assess for connections between circulating and skin-infiltrating subsets.
Sub-clustering of the aggregated myeloid cells revealed an apparent transition spanning PBMCs and skin (Fig. 8a). Annotation identified the bridging cells as CD14+CD16++ non-classical monocytes (ncMos), which derived exclusively from PBMCs, and CD16+ DCs from both PBMCs and skin (Fig. 8b–d). Pseudotime analysis of ncMos and CD16+ DCs performed using Monocle arranged the cells along a single trajectory. While further in vivo experimental evidence is required to substantiate this prediction, these transcriptional analyses suggest that circulating ncMos infiltrate the skin to give rise to CD16+ DCs (Fig. 8e), a process that appears to occur much more frequently in lupus than healthy control skin based on relative abundance (Fig. 8c). This increased entry into the skin could even account for the decreased proportion of ncMos and circulating CD16+ DCs observed in lupus patients relative to controls in our dataset (Fig. 8c).
Figure 8. Pseudotime analysis suggests CD16+ DCs arise from non-classical monocytes (ncMos) that migrate into skin of lupus patients and undergo IFN education.

a. UMAP plot of 6,576 myeloid cells from skin and peripheral blood mononuclear cells (PBMCs) colored by origin and disease state. H, healthy skin; N, non-lesional lupus skin; L, lesional lupus skin; HP, healthy PBMCs; LP, lupus PBMCs. b. UMAP plot of myeloid cells colored by subset. Red ellipse, bridge between circulating and skin-derived myeloid cells consisting of non-classical monocyte (ncMo) and CD16+ DC subsets. Mono, monocytes. c. Bar plot of each myeloid cell subset showing the relative contribution of H, N, L, HP, and LP samples. Values are normalized to the total number of myeloid cells for each sample type. d. Dot plot of representative marker genes for each myeloid cell subset. Color scale, average marker gene expression. Dot size, percentage of cells expressing marker gene. e. Pseudotime trajectory of ncMos and CD16+ DCs colored by origin and disease state (top), by subset (mid), and by pseudotime (bottom). X-axis, component 1; Y-axis, component 2. f. Pseudotime heatmap depicting expression of significant marker genes corresponding to 5 expression patterns that span the transition from ncMo to CD16+ DC. Color scale, scaled marker gene expression across pseudotime. g. Pseudotime heatmap depicting expression of the top 80 marker genes across the transition from ncMo to CD16+ DC. Color scale, scaled marker gene expression across pseudotime. h. Scatter plots depicting scores for the indicated upstream regulators for each cell across pseudotime. X-axis, pseudotime; Y-axis, module score. i. Dot plot of CD16+ DC gene scores for lesional skin biopsies from healthy controls (N=13) and CLE patients (N=90) analyzed by microarray. j. Dot plot presenting the same data as in i split by CLE subtype and presence vs. absence of SLE. N=28, 19, 20, and 23 for DLE −SLE, DLE +SLE, SCLE −SLE, and SCLE +SLE, respectively.
To understand the transcriptional changes that accompany this transition, we performed differential expression analysis along the pseudotime. This identified five gene expression patterns spanning the transition from ncMo to CD16+ DC (Fig. 8f). Close inspection of the top 80 most significant marker genes revealed that this transition was marked primarily by late upregulation of numerous genes encoding chemokines, which could support retention of CD16+ DCs and recruitment of other inflammatory cells into the skin of CLE patients, as well as ISGs, consistent with migration of these cells into the type I IFN-rich lupus skin environment (Fig. 8g). Broadly, these changes support our prior findings that keratinocyte-derived type I IFN can promote DC activation, enabling them to stimulate immune responses in the skin (2).
We next analyzed these five gene expression patterns for canonical pathway enrichment for insight into what cellular functions might be acquired and lost across this transition (Fig. S6). Top canonical pathways enriched among gene patterns expressed earlier in the transition (mostly periphery-associated) tended to relate to leukocyte trafficking. These included ephrin receptor signaling (p=7.44×10−3), the top pathway enriched in pattern A, as well as actin cytoskeleton signaling (p=5.57×10−7) and integrin signaling (p=4.57×10−4), the top pathways enriched in pattern B. In pattern C, encompassing genes induced mid-transition, the top pathway was autophagy (p=6.41×10−3), which has been implicated in the monocyte-to-DC transition (21). Top pathways enriched among gene patterns expressed later in the transition (mostly skin-associated) related more to cytokine signaling. The top pathway enriched in pattern D was the role of hypercytokinemia/hyperchemokinemia in the pathogenesis of influenza (p=6.79×10−6), with IFN signaling (p=2.80×10−4) also highly ranked. The top pathway enriched in pattern E was IL-6 signaling (p=1.85×10−6), in line with data supporting a role for increased production of IL-6 by lupus KCs secondary to conditioning by IFN signaling (3).
To analyze the cytokines influencing the transition from ncMo to skin-infiltrating CD16+ DC, we calculated upstream regulator scores for all cytokines included in IPA for each cell and determined the correlation of these scores with pseudotime. Scores for a panel of type I IFNs (IFN-α1/13, IFN-β, and IFN-κ) and IFN-γ (Fig. 8h) indicated very high correlation (r= 0.774, 0.880, 0.666, and 0.873, respectively) with a more pronounced rise late in pseudotime, consistent with robust IFN education representing an important terminal step in this transition. A number of upstream regulators showed more gradual induction across pseudotime and higher correlation scores, suggestive of an earlier role in the transition. These included IL-1β (r= 0.928), the top-correlated cytokine, and TNF (r= 0.903); of note, these also emerged as top significant genes in the pseudotime DEG analysis, showing late upregulation in the transition from ncMo to CD16+ DC (Fig. 8g). This is in keeping with our prior report that prolonged type I IFN exposure primes monocytes for inflammasome activation and enhances their production of IL-1β (22).
The interactions that mediate infiltration of CD16+ DCs into normal-appearing skin of patients with CLE are not yet known. To highlight L-R pairs that could promote this accumulation, we generated circos plots of all cytokine interaction pairs in which CD16+ DCs expressed either receptor (Fig. S7a) or ligand (Fig. S7b). CD16+ DCs expressed 12 cytokine receptors involved in L-R pairs (Fig. S7a). ECs and smooth muscle cells, which include vascular smooth muscle cells, expressed the highest number of interacting ligands. Next highest among stromal cells were IFN FBs. In a similar pattern, CD16+ DCs expressed 23 ligands involved in L-R pairs (Fig. S7b), with ECs and IFN FBs expressing the highest number of interacting receptors. Together, these L-R data suggest enhanced interactions with ECs and IFN FBs enable CD16+ DCs to accumulate in the skin of CLE patients, where the IFN-rich environment augments their pro-inflammatory properties and capacity for cell-cell communication.
Finally, to validate these findings, we interrogated microarray data from lesional skin of patients with CLE for transcriptional evidence of the ncMo to CD16+ DC transition. We constructed a gene signature from the top pseudotime marker genes showing induction across pseudotime (Fig. 8g) and calculated scores for a panel of skin biopsies from healthy controls (N=13) and discoid (N=47) and subacute (N=43) CLE lesions. CLE patient CD16+ DC gene scores were significantly higher than control (Fig. 8i), with minimal variation by CLE subtype or presence vs. absence of SLE (Fig. 8j). While this analysis is limited by overlapping expression of some signature gene markers with other inflammatory cell types present in CLE, the strong difference is consistent with increased CD16+ DCs in skin of all patients with CLE, regardless of subtype or association with SLE. Stratification of patients by disease severity according to Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI) activity scores (23, 24) revealed no difference (Fig. S8a); however, when CLASI damage was evaluated, patients with moderate damage scores (ranging from 10–20) showed a trend toward higher CD16+ DC gene scores (p=0.1098) (Fig. S8b).
DISCUSSION
Collectively, our data describe the cellular composition and architecture of cutaneous lupus at unprecedented resolution. We demonstrate the pervasive effects of IFN on skin stromal and immune cells alike. Most intriguingly, these effects are pronounced in non-lesional samples, suggesting that normal-appearing skin of patients with CLE exists in an immunologically primed, ‘prelesional’ state. This state skews the transcriptional programs of many of the major cell types in the skin, with dramatic effects on the capacity for cell-cell communication. Indeed, even the minor cellular constituents of the skin not examined in detail here exhibited transcriptional shifts in non-lesional CLE skin that alter their potential to engage other stromal and immune cells (Fig. 6b). The majority of this IFN education appears to occur at the interfollicular dermo-epidermal junction, where keratinocytes, a known source of type I IFN (2, 4), and myeloid cells, including pDCs, primarily localize. Which cells are producing IFN is difficult to discern in these data given the lack of depth of scRNA-seq. Indeed, the inflammatory environment of non-lesional skin may alter myeloid cell function, including TNF-mediated repression of IFN production in pDCs (25), but further investigation, using alternate modalities, is required to resolve this question.
This investigation highlighted CD16+ DCs, a myeloid cell subset increasingly implicated in lupus pathogenesis (15–17, 19, 20), as proficient intercellular communicators even in non-lesional skin of CLE patients (Fig. 6d), where they are highly abundant (Fig. 5d). Unsupervised clustering followed by pseudotime analysis of combined myeloid cells from skin and peripheral blood suggests that progenitor ncMos in circulation give rise CD16+ DCs (Fig. 8a,b,e). Clustering of DCs isolated from peripheral blood of healthy patients identified a subset of so-called DC4 cells characterized by expression of FCGR3A, encoding CD16a, with high transcriptional similarity to a monocyte subset with features of ncMos (12), inspiring some discussion that DC4 cells may in fact represent monocytes (26, 27). Here, however, utilization of single-cell technologies across blood and tissues has enabled identification of a CD16+ DC population enriched in the skin of patients with SLE and defined their probable precursors and the transcriptomic changes accompanying this transition that arm CD16+ DCs to instigate tissue inflammation. ScRNA-seq analysis of immune cells isolated from kidney biopsies of patients with lupus nephritis (LN) and healthy controls suggests that this paradigm may not be limited to the skin. Arazi et al. identified several myeloid subpopulations resembling DC4 cells (CM0, CM1, and CM4); these were highly enriched in LN biopsies and showed upregulation of IFN response scores relative to steady-state kidney macrophages and conventional DCs (28), suggesting IFN education is a characteristic feature of tissue-infiltrating CD16+ DCs that facilitates their pathogenicity in lupus. Whether and how the skin-educated CD16+ DCs characterized here may contribute to systemic disease remains to be determined, but there is precedent for systemic translation of skin inflammation (29).
Accumulation of CD16+ DCs in the skin represents a distinguishing feature of not just the lesional but also the prelesional CLE environment. In CLE patients, exit of CD16+ DCs from the circulation into non-lesional skin is likely enhanced through robust L-R interactions between CD16+ DCs and the vasculature (Fig. S7). Following tissue infiltration, CD16+ DCs may be directed to accumulate in the superficial dermis (Fig. 7g) by ligand gradients generated by subepidermal FBs that have been transformed to an IFN FB phenotype through the effects of KC-secreted IFN (2) (Fig. 3g, Fig. 7c). Upon encountering the IFN-rich non-lesional CLE environment, CD16+ DCs upregulate a panoply of ISG-encoded cytokines and chemokines (Fig. 8f,g). This endows them with the capacity for extensive cell-cell communication with diverse cell types (Fig. S7) including innate immune cells in their immediate proximity (Fig. S4), whereby they may contribute to genesis of CLE lesions and ultimately promote tissue damage (Fig. S8b). Thus, the prelesional environment of skin of CLE patients represents a collaboration between stromal and immune cells, with critical contributions from KCs, FBs, and CD16+ DCs. Further investigations directed at differences in cell-cell communication between non-lesional and lesional CLE will provide additional insight into the downstream events that precipitate clinically evident inflammation and lesion formation and provide targeted therapeutic strategies to improve treatment response in patients with this devastating disease.
Our study has several limitations. First, our CLE patient cohort includes only one patient without SLE, a bias that reflects our recruitment through a rheumatology clinic. However, CLE patients without SLE are as numerous as those with SLE (30) and merit deeper investigation. Contrast of skin and blood data from patients with and without SLE may bring to light important and clinically relevant differences in these populations. Second, isolation of skin cells for scRNA-seq requires processing that may introduce biases into cell recovery. While integration with spatial-seq and imaging mass cytometry lessens the impact of this potential bias, additional corroboration via other modalities would strengthen these conclusions. Finally, the bulk of our analyses rely on transcriptomic data. Further studies will be needed to validate these findings at the protein level and identify the consequences of these expression changes in vivo. Predictions such as cell-cell interaction networks and transition of circulating ncMos to skin-infiltrating CD16+ DCs require further evidence generated through alternate experimental approaches. These limitations notwithstanding, our findings support an integral role for non-lesional skin of lupus patients in augmenting the inflammatory capabilities of pathogenic myeloid cells.
MATERIALS AND METHODS
Study design
The pre-specified objectives of the research were to (1) examine the compositional and transcriptional differences between lesional and non-lesional skin of CLE patients and healthy controls and (2) identify circulating precursors of tissue-infiltrating myeloid cells via scRNA-seq. Sample size was determined by the two-month period selected for human sample acquisition (07/18/2019–09/18/2019). No rules for stopping data collection were defined in advance. Regarding data inclusion/exclusion criteria, all samples from all patients enrolled were included in the analysis. No outliers were excluded. The research subjects were adult patients (18–99 years of age) with active CLE in a location amenable to skin biopsy; exclusion criteria were active infection, active malignancy undergoing chemotherapy, pregnancy at enrollment, hemoglobin <8.5 g/dL, or bleeding disorder. 7 patients with active CLE (Table S1) were recruited for this study. All patients contributed lesional and non-lesional (sun-protected skin of the buttock) 6mm punch skin biopsies and whole blood for isolation of PBMCs. A diagnosis of SLE was confirmed for 6 of 7 patients via the European League Against Rheumatism/American College of Rheumatology criteria (31). 14 healthy controls were recruited for skin biopsy and 4 for whole blood. No blinding was used in the analysis. The study was approved by the University of Michigan Institutional Review Board (HUM00066116), and all patients gave written consent. The study was conducted according to the Declaration of Helsinki Principles.
Immunohistochemistry
Paraffin embedded tissue sections from punch biopsies from patients with discoid lupus erythematosus and healthy control skin were heated at 60°C for 30 minutes, de-paraffinized, and rehydrated. Slides were placed in antigen retrieval buffer at the pH indicated in Table S2 and heated at 125°C for 30 seconds in a pressure cooker water bath. After cooling, slides were treated with 3% H2O2 (5 minutes) and blocked using 10% goat serum (30 minutes). Overnight incubation was performed at 4°C with primary antibodies at the indicated concentrations (Table S2). Slides were then washed, treated with appropriate secondary antibodies, peroxidase (30 minutes), and diaminobenzidine substrate, before imaging.
Single-cell RNA-seq library preparation, sequencing, and alignment
Generation of single-cell suspensions for scRNA-seq was performed as follows: Skin biopsies were incubated overnight in 0.4% dispase (Life Technologies) in Hank’s Balanced Saline Solution (Gibco) at 4°C. Epidermis and dermis were separated. Epidermis was digested in 0.25% Trypsin-EDTA (Gibco) with 10U/mL DNase I (Thermo Scientific) for 1 hour at 37°C, quenched with FBS (Atlanta Biologicals), and strained through a 70μM mesh. Dermis was minced, digested in 0.2% Collagenase II (Life Technologies) and 0.2% Collagenase V (Sigma) in plain medium for 1.5 hours at 37°C, and strained through a 70μM mesh. Epidermal and dermal cells were combined in 1:1 ratio, and libraries were constructed by the University of Michigan Advanced Genomics Core on the 10X Chromium system with chemistry v3. Libraries were then sequenced on the Illumina NovaSeq 6000 sequencer to generate 150 bp paired end reads. Data processing including quality control, read alignment (hg38), and gene quantification was conducted using the 10X Cell Ranger software. The samples were then merged into a single expression matrix using the cellranger aggr pipeline.
Cell clustering and cell type annotation
The R package Seurat (v3.1.2) (32) was used to cluster the cells in the merged matrix. Cells with less than 500 transcripts or 100 genes or more than 10% of mitochondrial expression were first filtered out as low-quality cells. The NormalizeData function was used to normalize the expression level for each cell with default parameters. The FindVariableFeatures function was used to select variable genes with default parameters. The ScaleData function was used to scale and center the counts in the dataset. Principal component analysis (PCA) was performed on the variable genes, and the first 30 PCs were used in the RunHarmony function from the Harmony package (33) to remove potential batch effect among samples processed in different libraries. Uniform Manifold Approximation and Projection (UMAP) dimensional reduction was performed using the RunUMAP function. The clusters were obtained using the FindNeighbors and FindClusters functions with the resolution set to 0.5. The cluster marker genes were found using the FindAllMarkers function. The cell types were annotated by overlapping the cluster markers with the canonical cell type signature genes. To calculate the disease composition based on cell type, the number of cells for each cell type from each disease condition were counted. The counts were then divided by the total number of cells for each disease condition and scaled to 100 percent for each cell type. Differential expression analysis between H and N or H and L were carried out using the FindMarkers function.
Cell type sub-clustering
Sub-clustering was performed on the abundant cell types. The same functions described above were used to obtain the sub-clusters. Sub-clusters that were defined exclusively by mitochondrial gene expression, indicating low quality, were removed from further analysis. The subtypes were annotated by overlapping the marker genes for the sub-clusters with the canonical subtype signature genes. Ingenuity pathway analysis was applied to the differentially expressed genes to determine the canonical pathways and the potential upstream regulators. The upstream regulators with an activation z score ≥2 or ≤2 were considered significant. The module scores were calculated using the AddModuleScore function on the genes activated by the intended cytokine from bulk RNA-seq analysis as previously described (34).
Ligand receptor interaction analysis
CellphoneDB (v2.0.0) (35) was applied for L-R analysis. In the first analysis, the major cell type annotations were used. The cells were separated by their disease classifications (H, N, L), and a separate run was performed for each disease classification. Pairs with p value >0.05 were filtered out from further analysis. To compare among the three disease conditions, each pair was assigned to the condition in which it showed the highest interaction score. The number of interactions for each cell type pair was then calculated. In the second analysis, KCs, FBs, T cells, and myeloid cells were divided into their respective subtypes. The number of interactions between each cell type pair was calculated. The cytokine pairs for CD16+ DC was plotted in circos plots using the R package “circlize”. The connectome web was plotted using the R package “igraph”.
Pseudotime trajectory construction
Pseudotime trajectory for myeloid cell sub-clusters 6 and 8 from Fig. 8a was constructed using the R package Monocle (v2.10.1) (36). The raw counts for cells were extracted from the Seurat analysis and normalized by the estimateSizeFactors and estimateDispersions functions with the default parameters. Genes detected in >10 cells were retained for further analysis. Variable genes were determined by the differentialGeneTest function with a model against the Seurat sub-cluster identities. The orders of the cells were determined by the orderCells function, and the trajectory was constructed by the reduceDimension function with default parameters. Differential expression analysis was carried out using the differentialGeneTest function with a model against the pseudotime, and genes with an adjusted p value smaller than 0.05 were clustered into five patterns and plotted in the heatmap. Ingenuity Pathway Analysis was used to determine the upstream regulators for the genes in each expression pattern. A module score was calculated for each upstream regulator on gene targets from all five patterns. The module scores were calculated using the Seurat function AddModuleScore with default parameters. Pearson correlation was then performed between the upstream regulator module scores and the pseudotime.
Integration with keratinocyte and fibroblast cytokine signatures
As previously described by our group (37, 38), we utilized RNA-seq-based KC and FB cytokine response signatures for the following cytokines: IFN-α (5ng/ml), IFN-γ (5ng/ml), TNF (10ng/ml), IL-17A (10ng/ml, KCs only), IL-17A and TNF in combination (10ng/ml each, KCs only), IL-1β (10ng/ml), IL-36 (10ng/ml), IL-4 (10ng/ml), IL-13 (10ng/ml), or TGF-β (10ng/ml, FBs only). Briefly, primary human KCs from 50 donors were treated with a panel of cytokines as above for 8 hours and harvested for RNA isolation. Primary human FBs from 13 donors were treated with a panel of cytokines as above for 6 hours and harvested for RNA isolation. Unstimulated control KCs and FBs were cultured and harvested in parallel. Bulk RNA-seq was performed on the Illumina NovaSeq 6000 sequencer with the assistance of the University of Michigan Advanced Genomics Core. For each stimulation condition vs. unstimulated controls, differential expression analysis was performed using DESeq2 (39). Differentially expressed genes (twofold increase, false discovery rate <0.05) were used to construct response signatures for each cytokine, provided in Data File S2 (KCs) and Data File S3 (FBs).
Spatial sequencing library preparation
Skin samples were frozen in OCT medium and stored at −80°C until sectioning. Optimization of tissue permeabilization was performed on 20 μm sections using Visium Spatial Tissue Optimization Reagents Kit (10X Genomics, Pleasanton, CA, USA), which established an optimal permeabilization time to be 9 minutes. Samples were mounted onto a Gene Expression slide (10X Genomics), fixed in ice-cold methanol, stained with hematoxylin and eosin, and scanned under a microscope (Keyence, Itasca, IL, USA). Tissue permeabilization was performed to release the poly-A mRNA for capture by the poly(dT) primers that are precoated on the slide and include an Illumina TruSeq Read, spatial barcode, and unique molecular identifier (UMI). Visium Spatial Gene Expression Reagent Kit (10X Genomics) was used for reverse transcription to produce spatially barcoded full-length cDNA and for second strand synthesis followed by denaturation to allow a transfer of the cDNA from the slide into a tube for amplification and library construction. Visium Spatial Single Cell 3ʹ Gene Expression libraries consisting of Illumina paired-end sequences flanked with P5/P7 were constructed after enzymatic fragmentation, size selection, end repair, A-tailing, adaptor ligation, and PCR. Dual Index Kit TT Set A (10X Genomics) was used to add unique i7 and i5 sample indexes and generate TruSeq Read 1 for sequencing the spatial barcode and UMI and TruSeq Read 2 for sequencing the cDNA insert, respectively.
Spatial sequencing data analysis
After sequencing, the reads were aligned to the human genome (hg38), and the expression matrix was extracted using the spaceranger pipeline. Seurat was then used to analyze the expression matrix. Specifically, the SCTransform function was used to scale the data and find variable genes with default parameters. PCA and UMAP were applied for dimensional reduction. The FindTransferAnchors function was used to find a set of anchors between the spatial-seq data and scRNA-seq data, which were then transferred from the scRNA-seq to the spatial-seq data using the TransferData function. These two functions construct a weights matrix that defines the association between each query cell and each anchor. These weights sum to 1 and were used as the percentage of the cell type in the spots.
Imaging mass cytometry of tissue sections
Formalin-fixed, paraffin-embedded (FFPE) skin biopsy tissue sections from lesional skin of patients with SCLE or DLE were analyzed using the Hyperion imaging CyTOF system (Fluidigm) as previously described (34) with modifications of the antibody panel. Specifically, metal-tagged antibodies including pan-keratin (C11, Biolegend), BDCA2 (Polyclonal, R&D Systems), CD56 (123C3, ThermoFisher Scientific), HLA-DR (LN3, Biolegend), CD11c (EP1347Y, Abcam), and CD4 (EPR6855, Fluidigm) were added in this study. Markers used to annotate each cell type are listed in Table S3.
Imaging mass cytometry data analysis
Multiplexed imaging mass cytometry data were converted first to .TIFF images using MCD Viewer v1.0.560.2 (Fluidigm) and then segmented using CellProfiler v3.1.8 for single-cell analysis. The unsupervised clustering algorithm Phenograph was performed on 16 markers (CD56, CD15, CD11c, CD16, CD14, CD31, CD27, CD4, HLA-DR, CD68, CD20, CD8, BDCA2, CD138, E-cadherin, and Pan-Keratin) using histoCAT v1.75 software. To identify the rare CD16+ DCs, manual gating was performed on high CD14+CD11c+CD16+ expression. Neighborhood analysis was performed by permutation test (40) using histoCAT.
Calculation of the CD16+ DC gene score and analysis of an independent CLE cohort
The CD16+ DC gene score was calculated using the top 80 pseudotime marker genes across the transition from ncMo to CD16+ DC shown in Fig. 8g (excluding VMO1 and FCGR3A, as these showed depletion across pseudotime, and DNAJB1 and CCL3L1, as these were not detected in the microarray dataset) in the CLE cohort data previously published by Berthier et al. (41), with the algorithm described by Feng et al. (42). Statistical analysis of gene score comparisons were generated using an unpaired parametric t-test with GraphPad Prism software version 8.0.0; p-values<0.05 were considered significant and reported in all panels. All comparisons across all groups were performed and, for clarity, only the most relevant reported.
Statistical analysis
The statistical methods relevant scRNA-seq data analysis are specified above in context within subsections: Cell clustering and cell type annotation, Cell type sub-clustering, Ligand receptor interaction analysis, Pseudotime trajectory construction, Spatial sequencing data analysis, Imaging mass cytometry data analysis, and Calculation of the CD16+ DC gene score and analysis of an independent CLE cohort.
Data and materials availability:
The scRNA-seq data are available in GEO under accession number GSE186476. All reagents used in this study are commercially available. No cell lines were generated in this study.
Supplementary Material
ACKNOWLEDGEMENTS:
Funding:
This work was supported by the Taubman Institute via Innovative Program (JMK, JEG, and FW), Parfet Emerging Scholar (JMK), and Taubman Emerging Scholar (ACB) funds; the Babcock Endowment Fund (LCT, JEG); and the National Institute of Health (NIH): K08-AR078251 (ACB), R01-AR071384 (JMK), K24-AR076975 (JMK), R01-AR069071 (JEG), R01-AR073196 (JEG), P30-AR075043 (JEG), K01-AR072129 (LCT), R01-AI022553 (RLM), R01-AR040312 (RLM), R01-AR074302 (RLM), Office of the Director S10-OD020053 (FW), the National Cancer Institute P30-CA046592 (FW), and the Lupus Research Alliance (JMK). ACB is supported by the Dermatology Foundation. LCT is supported by the Dermatology Foundation, Arthritis National Research Foundation, and National Psoriasis Foundation. FW is supported by the National Science Foundation (1653611).
Competing interests:
JMK has received Grant support from Q32 Bio, Celgene/BMS, Ventus Therapeutics, ROME therapeutics and Janssen. JEG has received Grant support from Celgene/BMS, Janssen, Eli Lilly, and Almirall. JMK has served on advisory boards for AstraZeneca, Eli Lilly, GlaxoSmithKline, Gilead, Bristol Myers Squibb, Avion Pharmaceuticals, Provention Bio, Aurinia Pharmaceuticals, Ventus Therapeutics, and Boehringer Ingelheim. JEG has served on advisory boards for AstraZeneca, Sanofi, Eli Lilly, Boehringer Ingelheim, Novartis, Janssen, Almirall, BMS. All other authors have nothing to disclose.
Footnotes
LIST OF SUPPLEMENTARY MATERIALS
REFERENCES
- 1.Chasset F, Arnaud L, Costedoat-Chalumeau N, Zahr N, Bessis D, Frances C, The effect of increasing the dose of hydroxychloroquine (HCQ) in patients with refractory cutaneous lupus erythematosus (CLE): An open-label prospective pilot study. Journal of the American Academy of Dermatology, (2016). [DOI] [PubMed] [Google Scholar]
- 2.Sarkar MK, Hile GA, Tsoi LC, Xing X, Liu J, Liang Y, Berthier CC, Swindell WR, Patrick MT, Shao S, Tsou PS, Uppala R, Beamer MA, Srivastava A, Bielas SL, Harms PW, Getsios S, Elder JT, Voorhees JJ, Gudjonsson JE, Kahlenberg JM, Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann Rheum Dis 77, 1653–1664 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stannard JN, Reed TJ, Myers E, Lowe L, Sarkar MK, Xing X, Gudjonsson JE, Kahlenberg JM, Lupus Skin Is Primed for IL-6 Inflammatory Responses through a Keratinocyte-Mediated Autocrine Type I Interferon Loop. The Journal of investigative dermatology 137, 115–122 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Psarras A, Alase A, Antanaviciute A, Carr IM, Md Yusof MY, Wittmann M, Emery P, Tsokos GC, Vital EM, Functionally impaired plasmacytoid dendritic cells and non-haematopoietic sources of type I interferon characterize human autoimmunity. Nat Commun 11, 6149 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Der E, Suryawanshi H, Morozov P, Kustagi M, Goilav B, Ranabothu S, Izmirly P, Clancy R, Belmont HM, Koenigsberg M, Mokrzycki M, Rominieki H, Graham JA, Rocca JP, Bornkamp N, Jordan N, Schulte E, Wu M, Pullman J, Slowikowski K, Raychaudhuri S, Guthridge J, James J, Buyon J, Tuschl T, Putterman C, A. Accelerating Medicines Partnership Rheumatoid, C. Systemic Lupus Erythematosus, Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat Immunol 20, 915–927 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shipman WD, Chyou S, Ramanathan A, Izmirly PM, Sharma S, Pannellini T, Dasoveanu DC, Qing X, Magro CM, Granstein RD, Lowes MA, Pamer EG, Kaplan DH, Salmon JE, Mehrara BJ, Young JW, Clancy RM, Blobel CP, Lu TT, A protective Langerhans cell-keratinocyte axis that is dysfunctional in photosensitivity. Science translational medicine 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tabib T, Morse C, Wang T, Chen W, Lafyatis R, SFRP2/DPP4 and FMO1/LSP1 Define Major Fibroblast Populations in Human Skin. The Journal of investigative dermatology 138, 802–810 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolf SJ, Estadt SN, Theros J, Moore T, Ellis J, Liu J, Reed TJ, Jacob CO, Gudjonsson JE, Kahlenberg JM, Ultraviolet light induces increased T cell activation in lupus-prone mice via type I IFN-dependent inhibition of T regulatory cells. J Autoimmun 103, 102291 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan B, Ye S, Chen G, Kuang M, Shen N, Chen S, Dysfunctional CD4+,CD25+ regulatory T cells in untreated active systemic lupus erythematosus secondary to interferon-alpha-producing antigen-presenting cells. Arthritis Rheum 58, 801–812 (2008). [DOI] [PubMed] [Google Scholar]
- 10.von Spee-Mayer C, Siegert E, Abdirama D, Rose A, Klaus A, Alexander T, Enghard P, Sawitzki B, Hiepe F, Radbruch A, Burmester GR, Riemekasten G, Humrich JY, Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann Rheum Dis 75, 1407–1415 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Schiffer L, Worthmann K, Haller H, Schiffer M, CXCL13 as a new biomarker of systemic lupus erythematosus and lupus nephritis - from bench to bedside? Clin Exp Immunol 179, 85–89 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, Jardine L, Dixon D, Stephenson E, Nilsson E, Grundberg I, McDonald D, Filby A, Li W, De Jager PL, Rozenblatt-Rosen O, Lane AA, Haniffa M, Regev A, Hacohen N, Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bos JD, van Garderen ID, Krieg SR, Poulter LW, Different in situ distribution patterns of dendritic cells having Langerhans (T6+) and interdigitating (RFD1+) cell immunophenotype in psoriasis, atopic dermatitis, and other inflammatory dermatoses. The Journal of investigative dermatology 87, 358–361 (1986). [DOI] [PubMed] [Google Scholar]
- 14.Sontheimer RD, Bergstresser PR, Epidermal Langerhans cell involvement in cutaneous lupus erythematosus. The Journal of investigative dermatology 79, 237–243 (1982). [DOI] [PubMed] [Google Scholar]
- 15.MacDonald KP, Munster DJ, Clark GJ, Dzionek A, Schmitz J, Hart DN, Characterization of human blood dendritic cell subsets. Blood 100, 4512–4520 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Piccioli D, Tavarini S, Borgogni E, Steri V, Nuti S, Sammicheli C, Bardelli M, Montagna D, Locatelli F, Wack A, Functional specialization of human circulating CD16 and CD1c myeloid dendritic-cell subsets. Blood 109, 5371–5379 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Henriques A, Ines L, Carvalheiro T, Couto M, Andrade A, Pedreiro S, Laranjeira P, Morgado JM, Pais ML, da Silva JA, Paiva A, Functional characterization of peripheral blood dendritic cells and monocytes in systemic lupus erythematosus. Rheumatol Int 32, 863–869 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Rhodes JW, Tong O, Harman AN, Turville SG, Human Dendritic Cell Subsets, Ontogeny, and Impact on HIV Infection. Front Immunol 10, 1088 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hansel A, Gunther C, Baran W, Bidier M, Lorenz HM, Schmitz M, Bachmann M, Dobel T, Enk AH, Schakel K, Human 6-sulfo LacNAc (slan) dendritic cells have molecular and functional features of an important pro-inflammatory cell type in lupus erythematosus. J Autoimmun 40, 1–8 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Dobel T, Kunze A, Babatz J, Trankner K, Ludwig A, Schmitz M, Enk A, Schakel K, FcgammaRIII (CD16) equips immature 6-sulfo LacNAc-expressing dendritic cells (slanDCs) with a unique capacity to handle IgG-complexed antigens. Blood 121, 3609–3618 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Martin C, Espaillat MP, Santiago-Schwarz F, IL-10 restricts dendritic cell (DC) growth at the monocyte-to-monocyte-derived DC interface by disrupting anti-apoptotic and cytoprotective autophagic molecular machinery. Immunol Res 63, 131–143 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Berthier CC, Kahlenberg JM, Enhanced Inflammasome Activity in Systemic Lupus Erythematosus Is Mediated via Type I Interferon-Induced Up-Regulation of Interferon Regulatory Factor 1. Arthritis Rheumatol 69, 1840–1849 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein RS, Morganroth PA, Werth VP, Cutaneous lupus and the Cutaneous Lupus Erythematosus Disease Area and Severity Index instrument. Rheum Dis Clin North Am 36, 33–51, vii (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klein R, Moghadam-Kia S, LoMonico J, Okawa J, Coley C, Taylor L, Troxel AB, Werth VP, Development of the CLASI as a tool to measure disease severity and responsiveness to therapy in cutaneous lupus erythematosus. Arch Dermatol 147, 203–208 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Psarras A, Antanaviciute A, Alase A, Carr I, Wittmann M, Emery P, Tsokos GC, Vital EM, TNF-alpha Regulates Human Plasmacytoid Dendritic Cells by Suppressing IFN-alpha Production and Enhancing T Cell Activation. J Immunol 206, 785–796 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calzetti F, Tamassia N, Micheletti A, Finotti G, Bianchetto-Aguilera F, Cassatella MA, Human dendritic cell subset 4 (DC4) correlates to a subset of CD14(dim/-)CD16(++) monocytes. J Allergy Clin Immunol 141, 2276–2279 e2273 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Dutertre CA, Becht E, Irac SE, Khalilnezhad A, Narang V, Khalilnezhad S, Ng PY, van den Hoogen LL, Leong JY, Lee B, Chevrier M, Zhang XM, Yong PJA, Koh G, Lum J, Howland SW, Mok E, Chen J, Larbi A, Tan HKK, Lim TKH, Karagianni P, Tzioufas AG, Malleret B, Brody J, Albani S, van Roon J, Radstake T, Newell EW, Ginhoux F, Single-Cell Analysis of Human Mononuclear Phagocytes Reveals Subset-Defining Markers and Identifies Circulating Inflammatory Dendritic Cells. Immunity 51, 573–589 e578 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Arazi A, Rao DA, Berthier CC, Davidson A, Liu Y, Hoover PJ, Chicoine A, Eisenhaure TM, Jonsson AH, Li S, Lieb DJ, Zhang F, Slowikowski K, Browne EP, Noma A, Sutherby D, Steelman S, Smilek DE, Tosta P, Apruzzese W, Massarotti E, Dall’Era M, Park M, Kamen DL, Furie RA, Payan-Schober F, Pendergraft WF 3rd, McInnis EA, Buyon JP, Petri MA, Putterman C, Kalunian KC, Woodle ES, Lederer JA, Hildeman DA, Nusbaum C, Raychaudhuri S, Kretzler M, Anolik JH, Brenner MB, Wofsy D, Hacohen N, Diamond B, S. L. E. n. Accelerating Medicines Partnership in, The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol 20, 902–914 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skopelja-Gardner S, Tai J, Sun X, Tanaka L, Kuchenbecker JA, Snyder JM, Kubes P, Mustelin T, Elkon KB, Acute skin exposure to ultraviolet light triggers neutrophil-mediated kidney inflammation. Proc Natl Acad Sci U S A 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drenkard C, Parker S, Aspey LD, Gordon C, Helmick CG, Bao G, Lim SS, Racial Disparities in the Incidence of Primary Chronic Cutaneous Lupus Erythematosus in the Southeastern US: The Georgia Lupus Registry. Arthritis Care Res (Hoboken) 71, 95–103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, Smolen JS, Wofsy D, Boumpas DT, Kamen DL, Jayne D, Cervera R, Costedoat-Chalumeau N, Diamond B, Gladman DD, Hahn B, Hiepe F, Jacobsen S, Khanna D, Lerstrøm K, Massarotti E, McCune J, Ruiz-Irastorza G, Sanchez-Guerrero J, Schneider M, Urowitz M, Bertsias G, Hoyer BF, Leuchten N, Tani C, Tedeschi SK, Touma Z, Schmajuk G, Anic B, Assan F, Chan TM, Clarke AE, Crow MK, Czirják L, Doria A, Graninger W, Halda-Kiss B, Hasni S, Izmirly PM, Jung M, Kumánovics G, Mariette X, Padjen I, Pego-Reigosa JM, Romero-Diaz J, Rúa-Figueroa Fernández Í, Seror R, Stummvoll GH, Tanaka Y, Tektonidou MG, Vasconcelos C, Vital EM, Wallace DJ, Yavuz S, Meroni PL, Fritzler MJ, Naden R, Dörner T, Johnson SR, 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Annals of the Rheumatic Diseases 78, 1151–1159 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, Raychaudhuri S, Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gudjonsson JE, Tsoi LC, Ma F, Billi AC, van Straalen KR, Vossen A, van der Zee HH, Harms PW, Wasikowski R, Yee CM, Rizvi SM, Xing X, Xing E, Plazyo O, Zeng C, Patrick MT, Lowe MM, Burney RE, Kozlow JH, Cherry-Bukowiec JR, Jiang Y, Kirma J, Weidinger S, Cushing KC, Rosenblum MD, Berthier C, MacLeod AS, Voorhees JJ, Wen F, Kahlenberg JM, Maverakis E, Modlin RL, Prens EP, Contribution of plasma cells and B cells to hidradenitis suppurativa pathogenesis. JCI Insight 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R, CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc 15, 1484–1506 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL, The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsoi LC, Rodriguez E, Degenhardt F, Baurecht H, Wehkamp U, Volks N, Szymczak S, Swindell WR, Sarkar MK, Raja K, Shao S, Patrick M, Gao Y, Uppala R, Perez White BE, Getsios S, Harms PW, Maverakis E, Elder JT, Franke A, Gudjonsson JE, Weidinger S, Atopic Dermatitis Is an IL-13-Dominant Disease with Greater Molecular Heterogeneity Compared to Psoriasis. The Journal of investigative dermatology 139, 1480–1489 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang Y, Tsoi LC, Billi AC, Ward NL, Harms PW, Zeng C, Maverakis E, Kahlenberg JM, Gudjonsson JE, Cytokinocytes: the diverse contribution of keratinocytes to immune responses in skin. JCI Insight 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schapiro D, Jackson HW, Raghuraman S, Fischer JR, Zanotelli VRT, Schulz D, Giesen C, Catena R, Varga Z, Bodenmiller B, histoCAT: analysis of cell phenotypes and interactions in multiplex image cytometry data. Nat Methods 14, 873–876 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berthier CC, Tsoi LC, Reed TJ, Stannard JN, Myers EM, Namas R, Xing X, Lazar S, Lowe L, Kretzler M, Gudjonsson JE, Kahlenberg JM, Molecular Profiling of Cutaneous Lupus Lesions Identifies Subgroups Distinct from Clinical Phenotypes. J Clin Med 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feng X, Wu H, Grossman JM, Hanvivadhanakul P, FitzGerald JD, Park GS, Dong X, Chen W, Kim MH, Weng HH, Furst DE, Gorn A, McMahon M, Taylor M, Brahn E, Hahn BH, Tsao BP, Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheum 54, 2951–2962 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.