

Abstract
Background:
Quinoline is considered to be a privileged heterocyclic ring owing to its presence in diverse scaffolds endowed with promising activity profiles. In particular, quinoline containing compounds have exhibited substantial antiproliferative effects through diverse mechanism of actions which indicates that the heteroaryl unit is flexible as well as accessible to subtle structural changes that enables its inclusion in chemically distinct antitumor constructs.
Methods:
Herein, we describe a medicinal chemistry perspective on quinolines as anticancer agents by digging into the peer reviewed literature as well as patents published in the past few years.
Results:
This review will serve as a guiding tool for medicinal chemists and chemical biologist to gain insights about the benefits of quinoline ring installation to tune the chemical architectures for inducing potent anticancer effects.
Conclusion:
Quinoline ring containing anticancer agents presents enough optimism and promise in the field of drug discovery to motivate the researchers towards the continued explorations on such scaffolds. It is highly likely that adequate efforts in this direction might yield some potential cancer therapeutics in future.
Keywords: Quinoline, anticancer, medicinal, cytotoxic, scaffold, heterocycle, cell line
1. INTRODUCTION
N-heterocycles have been a subject of significant explorations for the design and synthesis of anticancer therapeutics in the recent past. Numerous structural engineering attempts have been made towards the tactic inclusion of nitrogen containing heterocycles in the chemical architecture of drug-like molecules. The significant attention paid towards the utilization of N-heterocycles in drug discovery process could be attributed to the ability of the nitrogen atom to accommodate the positive charge and act as a hydrogen bond acceptor or donor that strongly influences the interaction between the molecule and its target [1–6]. Among the heterocyclic compounds, quinoline is considered to be a privileged scaffold due to its presence in many naturally occurring alkaloids endowed with promising biological activity profiles (Figure 1). The application of this bicyclic heteroaryl ring has specifically been expanded towards the anticancer drug discovery field [7] and quinoline containing constructs have exhibited extremely striking antitumor effects through diverse mechanism of action such as inhibition of histone deacetylase (HDAC), topoisomerase, tubulin, phosphoionositol-3-kinase (PI3K), epidermal growth factor receptor (EGFR) kinase, extracellular signal-regulated kinases (ERK) and other miscellaneous enzyme. In light of these varied mechanisms, it can be concluded that the quinoline ring is flexible as well as accessible to subtle structural changes that enable its inclusion in diverse antitumor scaffolds.
Fig. 1.
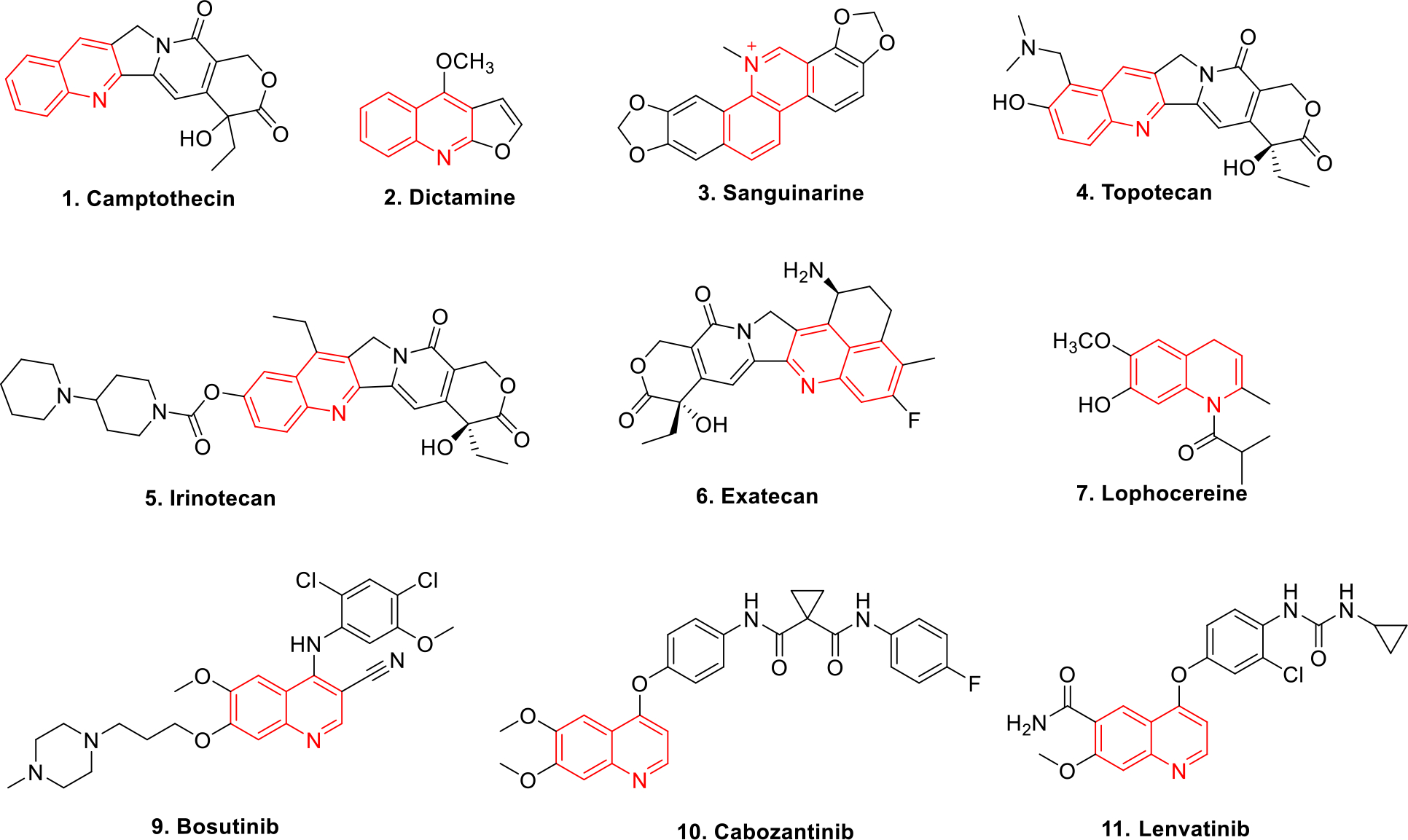
Quinoline containing anticancer drugs
It is well known that the advent of camptothecin in 1966 revolutionized the anticancer drug discovery field and transposed the focus of the chemist towards the quinoline motif for the development of antitumor constructs. The synthetic analogs of campthothecin (1) namely topotecan (4), irinotecan (5) and exatecan (6) further drove the progress of quinoline ring containing anticancer chemical architectures. As a result, explorations on quinolines escalated at a brisk rate and the subsequent preliminary success led to the advancement of several quinoline based drugs towards the clinical stage investigations. Other than the conventional anticancer therapeutics, the recent studies reporting quinoline based chemical probe inhibiting autophagy related cysteine protease ATG4B [7] and quinoline based theranostic ligands [8] represents the versatility in the application of this privileged heterocycle. These optimistic findings coupled with the applications of quinoline constructs in medicinal, synthetic organic chemistry as well as in the field of industrial chemistry has triggered the initiation of several campaigns in pursuit of establishing synthetic protocols for functionalized quinolines. In view of societal expectations influenced by the tight legislation on maintaining greenness in chemical processes, emphasis has also been laid on green and clean synthetic methodologies for the synthesis of quinoline derivatives. Thus reactions employing recyclable catalysts, one-pot reaction, solvent-free reaction conditions, using ionic liquids, ultrasound promoted synthesis and photocatalytic synthesis (UV radiation) have been extensively employed [9].
This review presents the versatility of quinoline core via compilation of recent reports on anticancer quinoline derivatives with a mention of their mechanism of action, key structural attributes and other relevant details. It is quite hopeful that this review could serve as a platform for the medicinal chemists to expand the utility of quinolines in the design of anticancer therapeutics.
2. RESEARCH ARTICLE REVIEW
This section covers the recent literature reports on quinoline based anticancer scaffolds that have displayed promising antitumor effects via diverse mechanism of actions.
2.1. HDAC inhibitors
This subsection presents the selected studies on the design of HDAC inhibitors employing quinoline ring as an integral part of the chemical architecture. Researchers have tactically synthesized compounds that can be categorized as pan-HDAC inhibitors as well as selective inhibitors of the HDAC isoforms. The studies covered in this part includes the rational design of 2-(phenylsulfonyl)quinoline N-hydroxyacrylamides, quinoline-based N-hydroxy cinnamamides and N-hydroxy benzamides, (N-Hydroxycarbonylbenzylamino) quinolines as HDAC inhibitors. The strategies employed in these studies might be useful to generate fused heteroaryl ring based HDAC inhibitors in the near future.
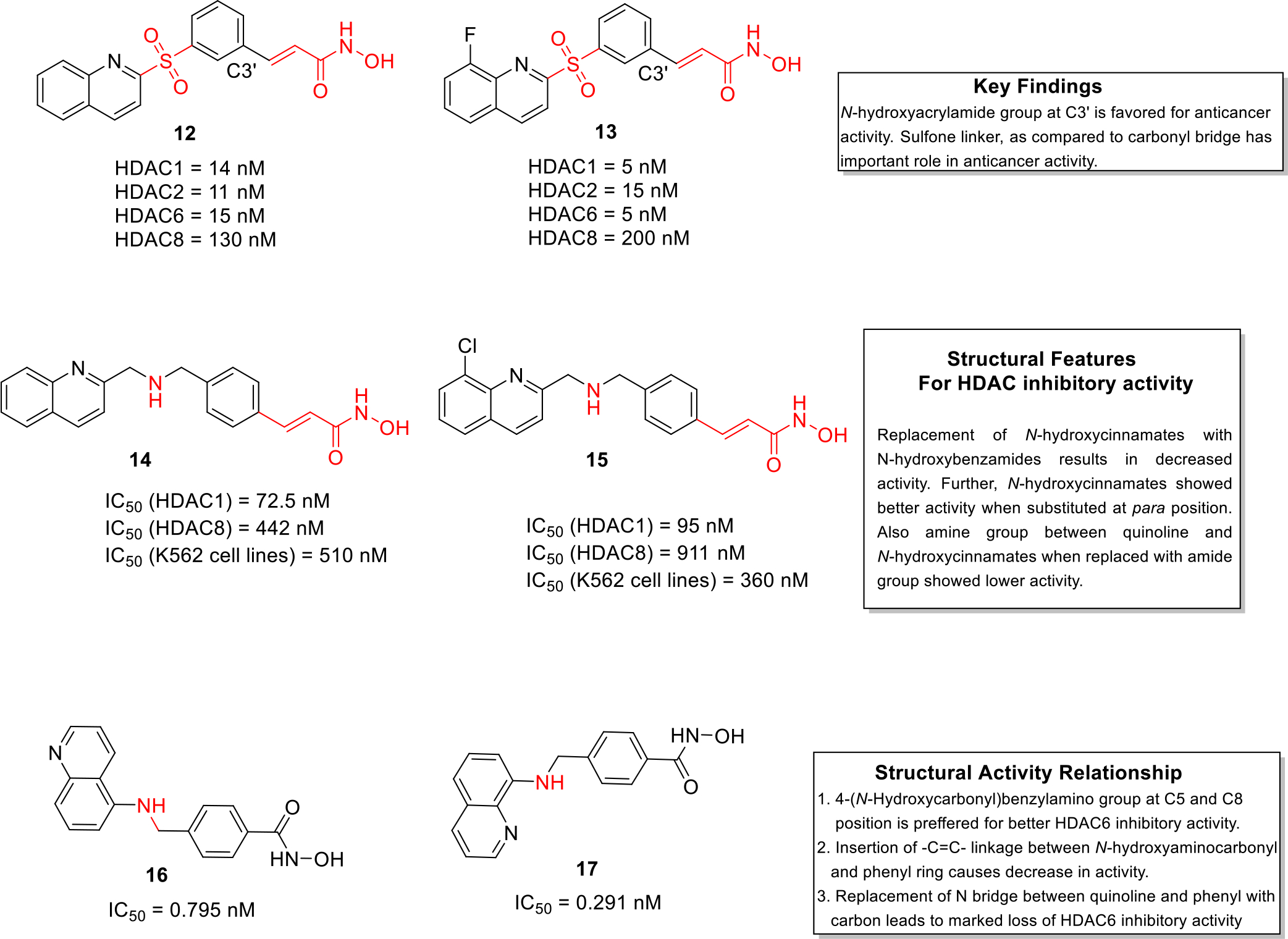
HDAC enzyme plays immense role in transcription process in cell cycle [10]. It has been observed that its overexpression is responsible for inducing various types of cancer [11]. In the recent past, significant research has been conducted to develop potent HDAC inhibitors that could be utilized in anticancer therapy [12]. In view of these revelations, Lee et al designed and synthesized 2-(phenylsulfonyl)quinoline N-hydroxyacrylamides compounds as HDAC inhibitors employing a rational approach of logically fusing the heteroaryl ring and the acrylamide unit. The lead compounds 12 and 13 showed potent inhibition of HDAC isoforms with IC50 values in the nanomolar range (Figure 2). Compounds 12 and 13 also displayed potent cell growth inhibition against HL-60, HCT116, PC-3 and A549 cell lines and the effects were more pronounced than the standard compound SAHA and PXD101. Both the candidates were further tested (in vivo) in human colon cancer xenografts in nude mice. Compound 12 showed dose dependent inhibition of tumor growth by 58.8% at a dose of 100 mg/kg. The structure-activity relationships (SAR) study determined that N-hydroxyacrylamide group is preferred at C3’ position whereas sulfone bridge played significant role in anticancer activity as compared to carbonyl bridge [13].
Fig. 2.
HDAC inhibitors
Working on similar lines, Chen et al. synthesized a series of quinoline based compounds (quinoline-based N-hydroxy cinnamamides and N-hydroxy benzamides) that showed selectivity for HDAC class I compared to HDAC class II enzyme. The synthetics were evaluated for anticancer effects against a panel of cell lines as well as HDAC inhibitory potential. The results of the biological evaluation led to the identification of compound 14 and 15 as potent inhibitors of HDAC1 and HDAC 8 isoforms. Both the compounds were endowed with striking cell growth inhibitory effects against myelogenous leukemia cell lines (Figure 2). Moreover, flow cytometry studies revealed that these two leads exerted stronger apoptotic effects as compared to standard compound vorinostat in K562 cells. In addition, molecular docking studies depicted that 14 chelated with zinc atom and demonstrated hydrogen bonding interactions with histidine and aspartate residues of class I HDAC. Overall, the results of the biological evaluation culminated in the identification of construct 14 as a promising inhibitor of class I HDACs. [14].
A team of researchers led by Liou et al designed and synthesized (N-Hydroxycarbonylbenzylamino) quinolines as selective HDAC 6 enzyme inhibitors. The HDAC isoform inhibitory assay revealed that compound 16 and 17 were potent inhibitors of the HDAC6 isoform and were endowed with IC50 values in the picomolar range. It was tempting to observe the selective HDAC isoform inhibitory profile of compound 17 which was approximately 376–12,500 times more selective towards HDAC6 as compared to the other isoforms. Moreover, in vivo studies were carried in human multiple myeloma RPMI 8226 xenograft model and the results demonstrated that compound 17 exerted total growth inhibition (TGI) of 60.4% at a dose of 30 mg/kg. In addition, the kinase selectivity analysis (KINOMEscan Kinase assay) indicated that 17 did not demonstrate high binding affinity against a panel of 97 kinases (Figure 2). Overall, the outcome of this study led to the identification of hydroxamic acid 17 as a potent antiproliferative agent for multiple myeloma [15].
2.2. Topoisomerase inhibitors
This subsection presents the logical utilization of the quinoline ring to assemble adducts that can target topoisomerase I and II, thereby exerting potent cytotoxic effects. The studies included in this part presents the insights about the activity profile of Indeno[1,2-c]quinoline derivatives, phosphorous substituted quinoline and camptothecin based topoisomerase inhibitors. It is quite hopeful that the strategies employed in these studies will spur the researchers to initiate medicinal chemistry campaigns for the design of new topoisomerase inhibitors.
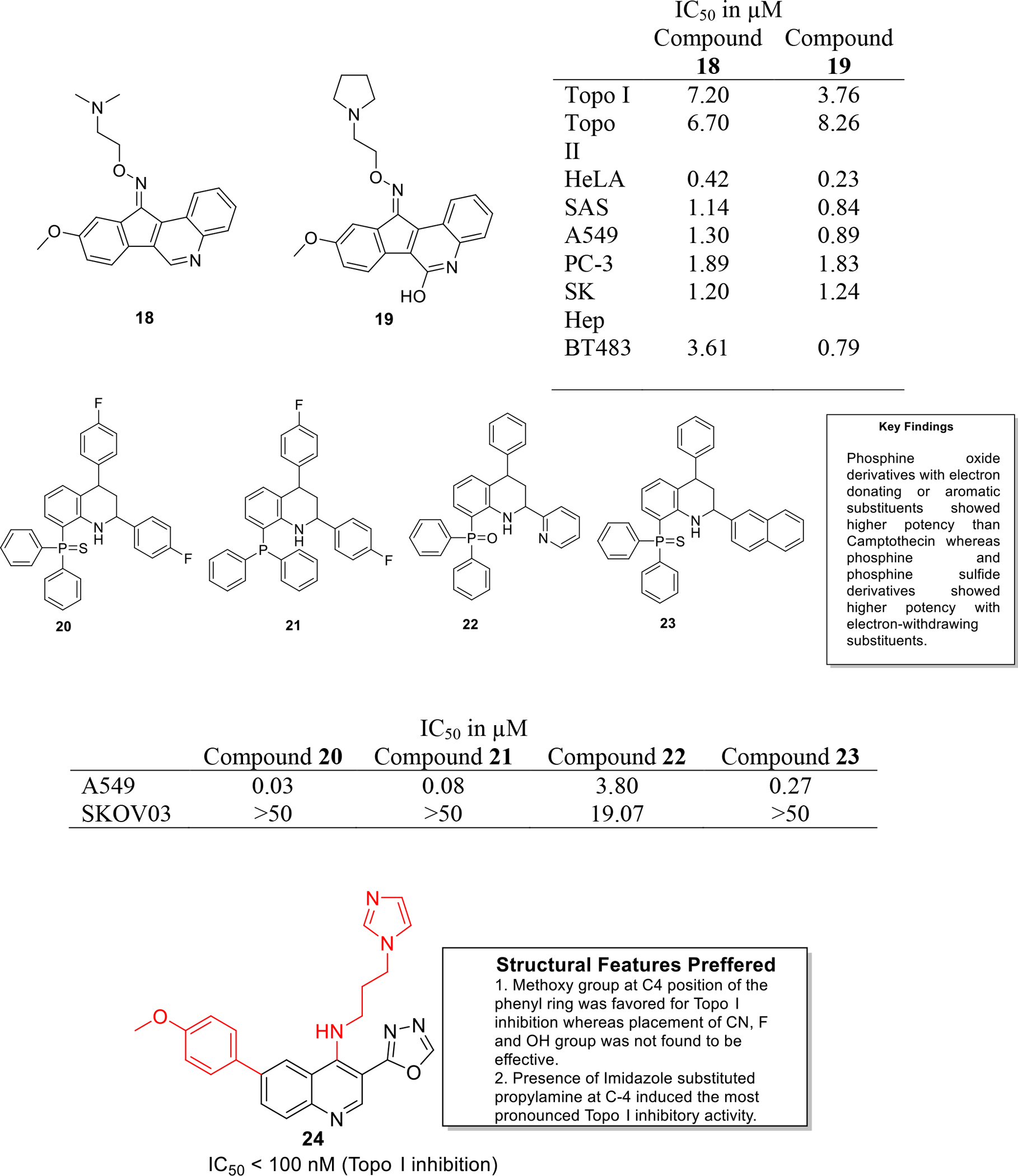
Topoisomerases are the major enzymes that are responsible for cell division. They participate in overwinding or underwinding of DNA and thus solve the topological problems of DNA [16]. In total, DNA topoisomerase assist the replication and transcription process of DNA. In humans, topoisomerases are categorized into two types as DNA Topo I and Topo II. These are overexpressed in majority of cancer cells [17]. In this context, several quinoline derivatives have been targeted against topoisomerases to furnish anticancer constructs. Tseng et al. designed and synthesized Indeno[1,2-c]quinoline derivatives as topoisomerase inhibitors that were found to exhibit dual topo I & II inhibition (Figure 3). In-vitro cytotoxicity studies indicated that compound 18 was more active as well as selective against SAS and A549 cancer cells as compared to the standard camptothecin. It was also observed that compound 19 showed strong inhibition of the growth of HeLa cells, SAS, A549 and BT483 with GI50 of 0.23, 0.84, 0.89 and 0.79 μM. Structure activity relationship was also established that highlights the favorable trends observed in the antiproliferative activity evaluation attained with placement of tertiary amine functionality (a terminal aminoalkyl side chain or a five-membered pyrrolidino ring) and confirmed that the replacement of the aforementioned with six membered rings such as morpholino or piperidine groups leads to decrease in antiproliferative activity. Among all the inhibitors tested, compound 18 and 19 were identified as the most potent topoisomerase inhibitors as evident from the results of enzymatic assays. Furthermore, the results of flow cytometric investigation revealed that 19 significantly inhibited cell cycle by exhibiting DNA fragmentation in sub-G1 phase in BT 483 cells, ultimately leading to cell death. Xenograft studies were also conducted in tumor bearing mice and it was found that tumor mass was effectively reduced and the survival rate was increased upon treatment with compound 19 at a dose of 20 mg/kg [18].
Fig. 3.
Topoisomerase inhibitors
Palacios and team synthesized a series of novel phosphorous substituted quinoline as topo I inhibitors. Some of the synthesized candidates exhibited significant inhibitory effects against topo I mediated relaxation, comparable to camptothecin (Figure 3). It was further observed that the fluorinated derivatives were substantially cytotoxic (20–23) and among them, compound 20 displayed the most potent effects against A549 cell lines (IC50 value = 0.03 μM). Moreover, the compounds were found to be selectively cytotoxic against cancer cells over non-cancerous cells [19].
Kundu et al designed quinoline derivatives via fusion of the chemical architectures of camptothecin and indenoisoquinolines (eg. indimitecan & indotecan) and analyzed them for human Topo I inhibition in plasmid DNA relaxation assay. The result of the assay indicated that compound 24 was found to be significantly potent with an activity index comparable to that of camptothecin and topotecan. Plasmid DNA cleavage assay was performed to confirm mechanism of Topo enzyme inhibition which confirmed that compound 24 is a Topo I poison. Detailed investigations of compound 24 indicated that it did not act as a DNA intercalator as per the results of Topo I unwinding and Ethidium bromide displacement assays. Furthermore, the series of hybrids was evaluated for anticancer potential using different cell lines which revealed the compound 24 exerts marked anticancer effects against different cancer cell lines including MCF7, HeLa and HCT116 with IC50 values of 2.74, 2.61 and 2.34 μM (Figure 3) [20].
2.3. Tubulin inhibitors
Literature survey indicates that the heteroaryl ring based tubulin inhibitors have demonstrated pronounced anticancer effects in the in-vitro as well as in-vivo studies. The ongoing wave in the tubulin inhibitory field is heavily inclined towards the use of 6 : 6 and 6 : 5 fused heterocycles for constructing new scaffolds. Spurred by these trends, several studies were conducted in the last decade to generate aroylquinoline-5,8-diones, quinoline analogues of flavones, quinoline-chalcone hybrids and chalcone fused quinolines. It is anticipated that the preliminary promise demonstrated by the aforementioned compounds gets replicated in the detailed preclinical studies that might render a potent compounds worthy of clinical investigation.
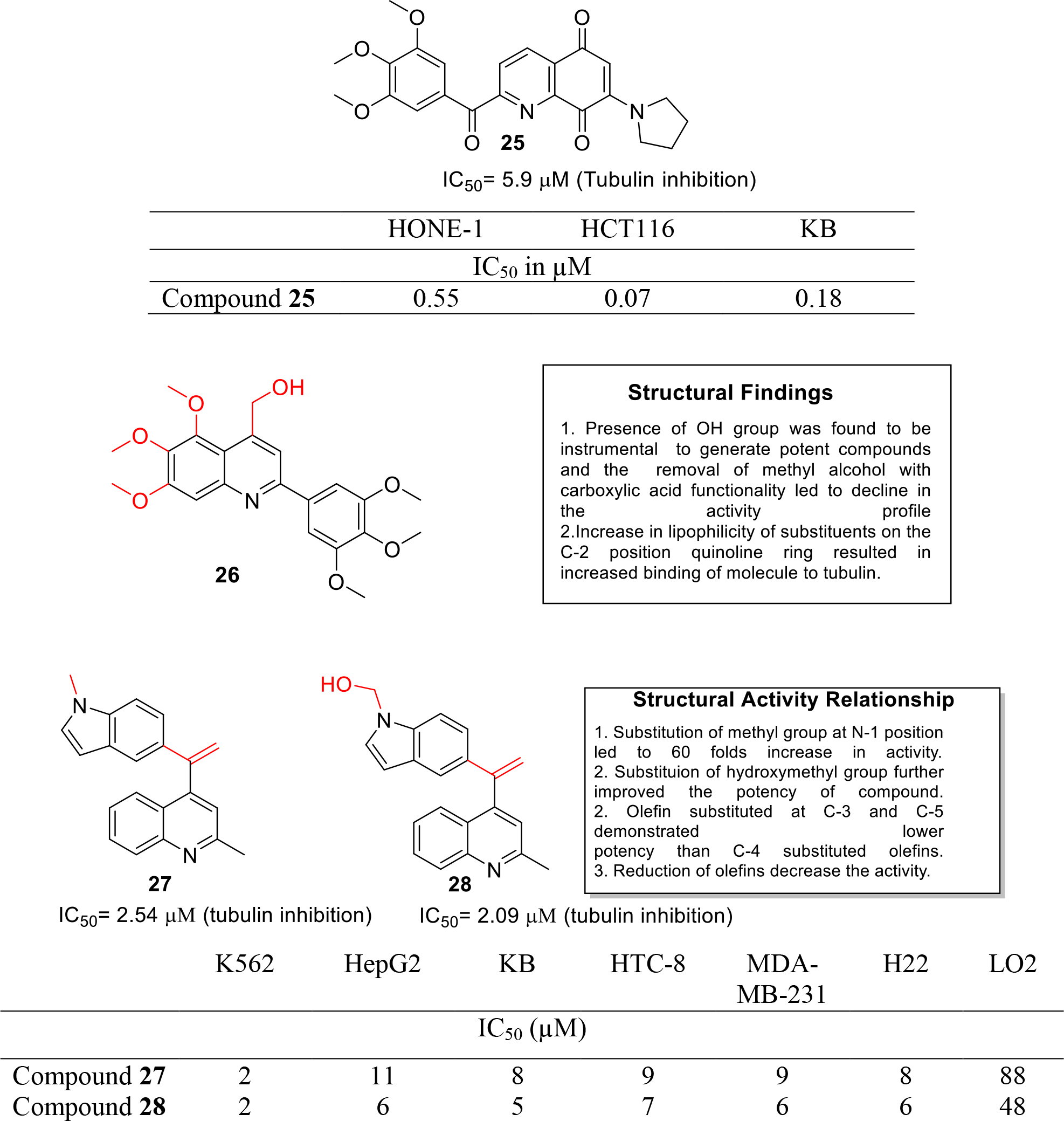
Microtubules represents a logical strategy for designing anticancer compounds owing to its involvement in important cellular activities. In light of the role of tubulin in mitosis, Liou and team synthesized 2-Aroylquinoline-5,8-diones derivatives as dual inhibitors of tubulin and HSP90 inhibitors endowed with antiproliferative effects. In vitro antitumor assay was performed using cancer cell lines HCT116, KB cells and nasopharyngeal carcinoma HONE-1. Among all the compounds, compound 25 exhibited potent inhibition of HCT116 and KB cells growth with IC50 of 0.07 and 0.18 μM (Figure 4). Compound 25 also disrupted tubulin assembly in a concentration dependent manner and inhibited polymerization of MAP-rich tubulins with IC50 value of 5.9 μM. In addition, treatment with construct 25 resulted in the induction of HSP70 along with the degradation of Akt in a dose dependent manner [21]. Overall, the results of the study culminated in the identification of compound 25 as dual HSP90/Tubulin inhibitor exerting potent antitumor effects.
Fig. 4.
Tubulin inhibitors
Shobeiri et al reported the synthesis of quinoline analogues of flavones as tubulin polymerization inhibitors (Figure 4). In vitro MTT assay was performed on four human cancer cell lines- MCF 7, MCF7/MX, A2780 and A-2780/RCIS which indicated that derivatives bearing a hydroxyl group were more potent (IC50= 7.98–60 μM) as compared to carboxylic acid derivatives (IC50 > 100 μM). The results led to the identification of a potent compound 26 that induced cell cycle arrest at G2/M phase as evidenced by flow cytometric study. The compound 26 was further subjected to molecular modeling studies and was docked into colchicine binding site where it was found to be involved in hydrophobic interactions [22].
Li et al. designed and synthesized quinoline-indole derivatives and targeted them at colchicine binding site of tubulin. The tubulin inhibitory assay demonstrated that 27 and 28 significantly inhibited the tubulin polymerization (Figure 4). It was also found that compound 28 inhibited microtubule polymerization by binding effectively to the colchicine site of tubulin. Furthermore, anti-microtubule efficacy was evaluated in K562 cells using immunofluorescent assay and the results showed that 28 induced dose-dependent disruption of microtubule formation. Flow cytometric analysis and annexin V-FITC/PI assay indicated that 28 blocked cell cycle at G2/M phase and could induce apoptosis in a dose dependent manner. Besides the in vitro studies, in vivo antitumor evaluation in liver cancer xenograft mouse model demonstrated that 27 and 28 inhibited tumor growth by 63.7% and 57.3% at doses of 20 mg/kg/day [23].
Li et al synthesized novel Quinoline-Chalcone hybrids as microtubule polymerization inhibitor and evaluated them against four cancer cell lines HepG2, KB, HCT-8 and MDA-MB-231 by MTT assay. The results demonstrated that compound 29 was the most potent with IC50 value in the low micromolar range (Figure 5). It was also observed that compound 29 arrested the cell cycle (K562) at the G2/M phase. Results of in vitro tubulin polymerization assay demonstrated the potential of compound 29 as a microtubule depolymerization agent that exerted its effects in a concentration dependent manner. Moreover, compound 29 induced apoptosis with upregulation of Bax, Bcl-Xl and Bad, and downregulation of Bcl-2. In the in vivo studies, compound 29 and its hydrochloride salt inhibited the tumor growth in mouse liver cancer xenograft model in a dose dependent fashion, with reduction in tumor volume as high as 69%. In addition, acute toxicity studies were performed in mice and median lethal dose value was found to be 665.62 mg/kg which indicated that the compound possess low toxicity and can be investigated in detail for development as cancer therapeutic [24].
Fig. 5.
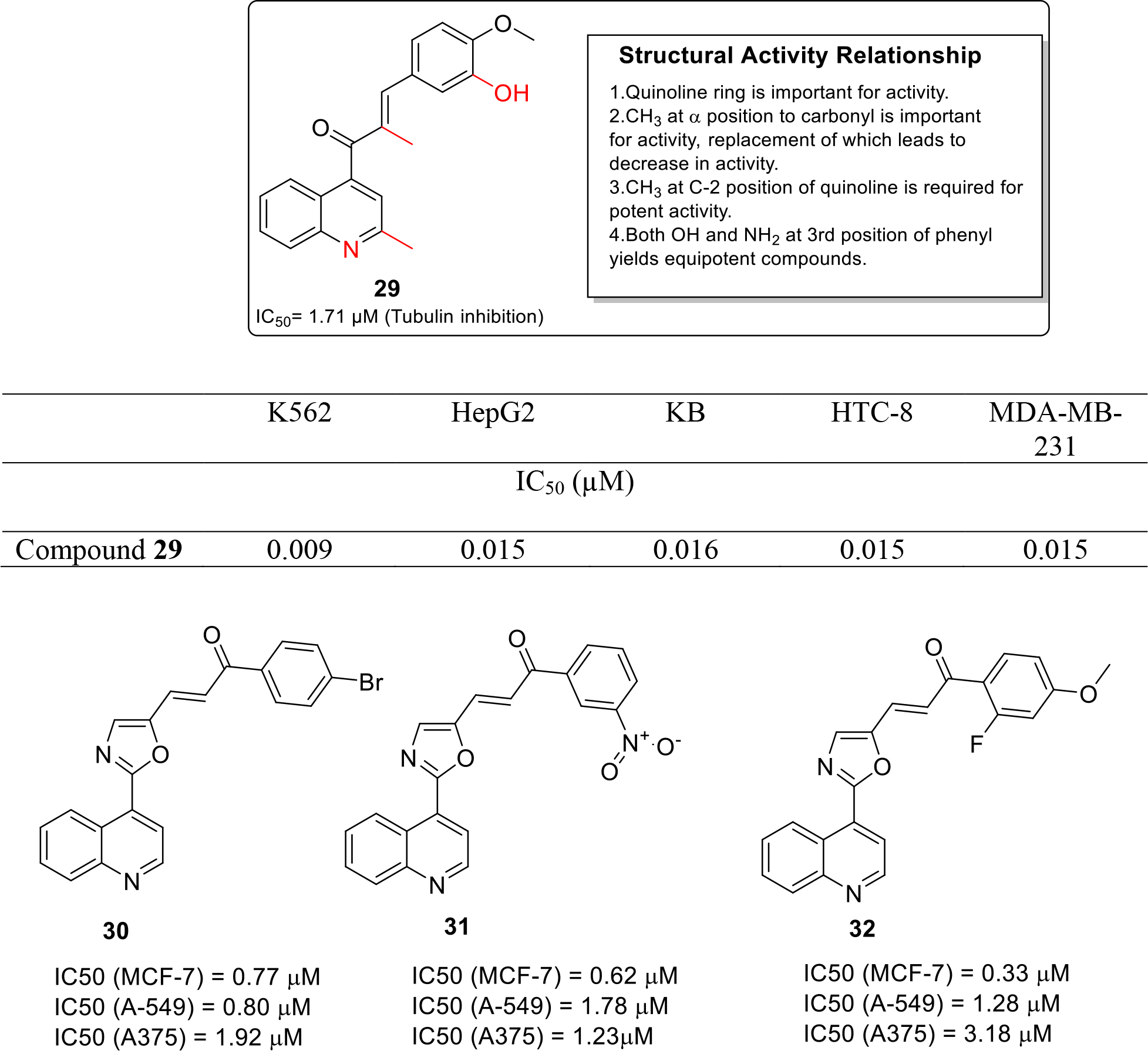
Tubulin inhibitors
Venkatarao et al. synthesized and evaluated a series of chalcone fused quinoline molecules as anticancer drug. These fused hybrids (30, 31, 32) demonstrated significant cytotoxic potential in the in vitro cytotoxicity assay studies (Figure 5) [25].
2.4. Kinase inhibitors
The subsection presents different approaches that have been utilized by the medicinal chemist to target diverse kinases for extracting antitumor effects. Specifically, investigation on -cyclopropyl-3-ethynyl-4-(4-fluorophenyl)quinolones, quinoline-3-carboxamides, 3H-pyrazolo[4,3-f]quinoline, naphthofuro[3,2-c]quinoline-6,7,12-triones and pyrano[3,2-c]quinoline-6,7,8,13-tetraones based analogues have been covered. It is quite astonishing to observe that quinoline ring demonstrates utmost flexibility to target different kinase and exert cell killing effects against various cell lines.
PI3K enzymes are related to intracellular signal transducers kinases and are involved in numerous cellular processes including cell growth and survival [26]. PI3K enzyme has been found to be overexpressed in several different types of cancers and significant attention was paid towards the development of novel PI3K inhibitor as anticancer agents in the last decade. In view of this valuable information, Manikandan and coworkers synthesized novel 2-cyclopropyl-3-ethynyl-4-(4-fluorophenyl)quinoline based molecules as PI3-kinase inhibitors. The results of the in-vitro enzymatic assay led to the identification of compound 33 that exerted maximum percentage inhibition of 72.57 % and was found to be endowed with IC50 of 0.45 μM (Figure 6). Molecular docking studies revealed that compound 33 demonstrated non-covalent and hydrogen bonding interactions with PI3K enzyme [27].
Fig. 6.
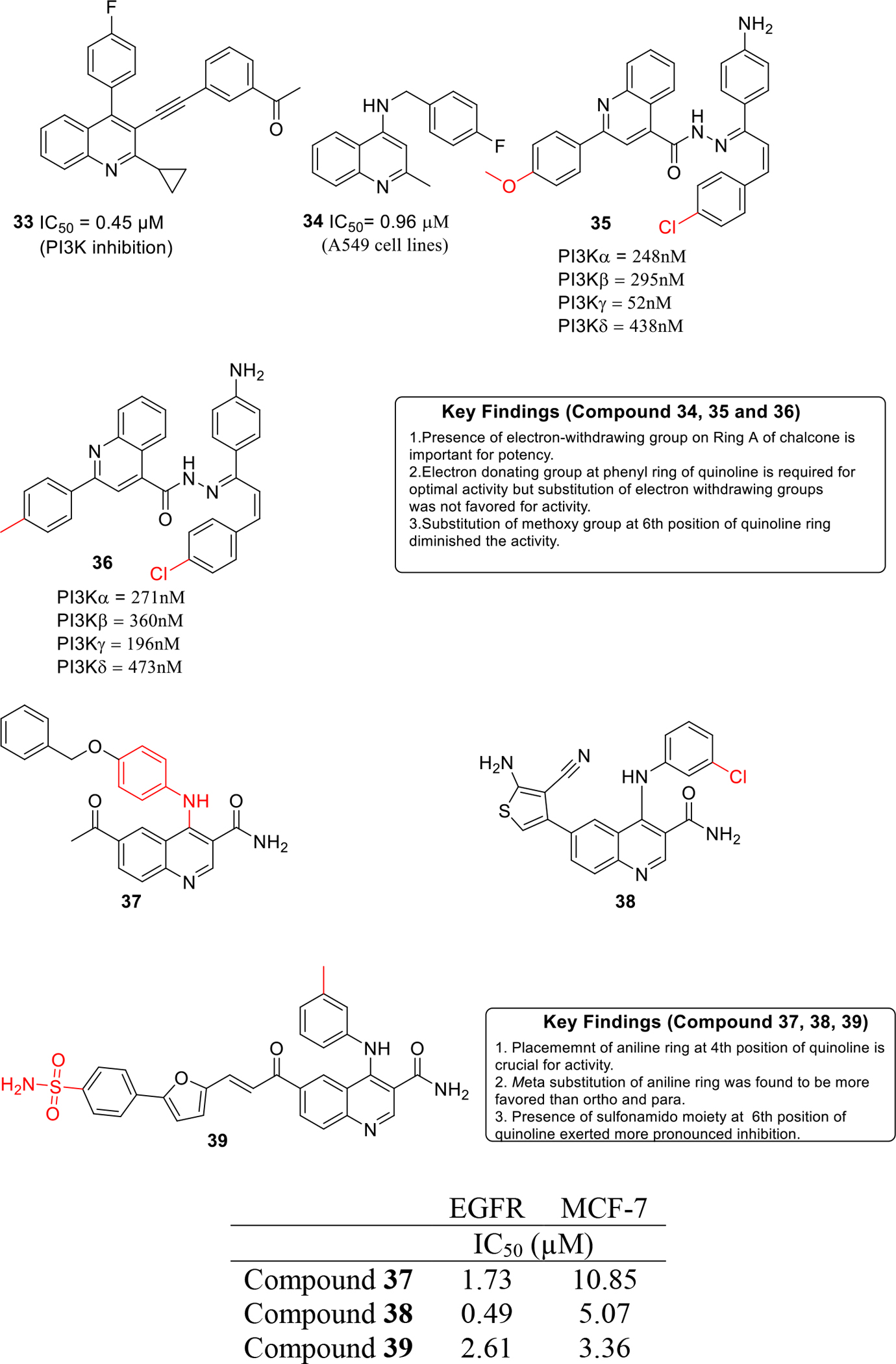
Kinase inhibitors
Working on similar lines, Vennila and others designed 4-substituted quinolines as PDK1 enzyme inhibitors. In vitro studies were carried out against A549 cells that demonstrated the potential of compound 34 as the most potent from the series with IC50 value of 0.96 μM (Figure 6) [28].
Abbas et al. designed and synthesized new hybrids of quinoline/chalcone and evaluated them as PI3K enzyme inhibitors (Figure 6). The synthesized candidates were screened against NCI60 cell lines and candidates 35 and 36 exhibited GI50 values in the range of 0.3–52 μM. Flow cytometric studies further indicated that 35 and 36 arrests the growth in the G2/M phase in A549 and K-562 cells. To further elucidate the molecular basis of induction of G2/M cell cycle arrest and apoptosis, western blotting analysis was carried out which revealed that 35 and 36 mitigated PI3K/Akt/mTOR signaling in A549 and K562 cells [29].
EGFR kinase is a transmembrane protein that is involved in cell growth and proliferation [30]. Numerous cancers are associated with amplification of EGFR kinase expression. In this context, development of EGFR kinase inhibitors is a crucial step towards cancer inhibition [31]. Capitalizing on these revelations, Ella and team designed and synthesized quinoline-3-carboxamides derivatives and evaluated them as EGFR inhibitors (Figure 6). The results of the biological evaluation demonstrated that compound 37–39 were substantially potent inhibitors of the enzyme with 38 displaying the best activity (IC50 = 0.49 μM). In-vitro cytotoxicity studies revealed that the carboxamide 39 exerted striking cell growth inhibitory potential against MCF-7 cancer cell line with an IC50 value of 3.35 μM. It is highly anticipated that these compounds can emerge as a lead structures for further optimization in order to generate a more potent EGFR inhibitors as anti-breast cancer agents [32].
George et al. synthesized quinoline based chalcones and 4,5-dihydropyrazoles as EGFR inhibitors (Figure 7). EGFR inhibitory assay revealed that compound 40 displayed striking inhibition of the enzyme at nanomolar concentration (IC50 = 37 nM). Anti-proliferative activity of the synthesized adducts was evaluated against three human cancer cell lines MCF-7, DLD1 and HeLa and the results indicated that the substitution of pyrazole NH with 4-arylthiazolyl moiety and placement of 4-p-tolyl ring at the pyrazole ring improved the cell growth inhibitory activity. Compound 41 (4-p-tolyl derivative) was found to be a potent cytotoxic agent against the cell lines employed and displayed showed IC50 values in the nanomolar to low micromolar range [33].
Fig. 7.
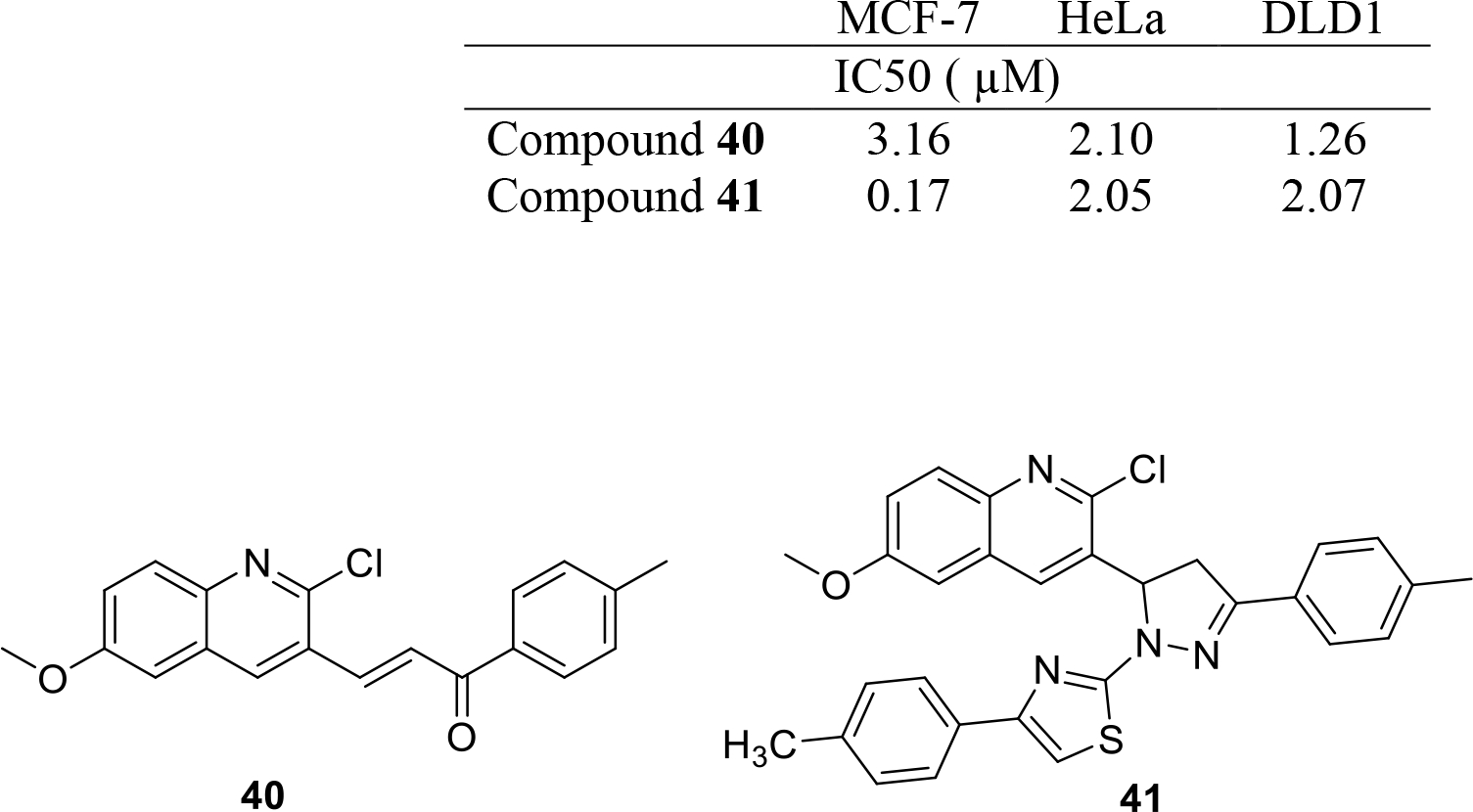
Kinase inhibitors
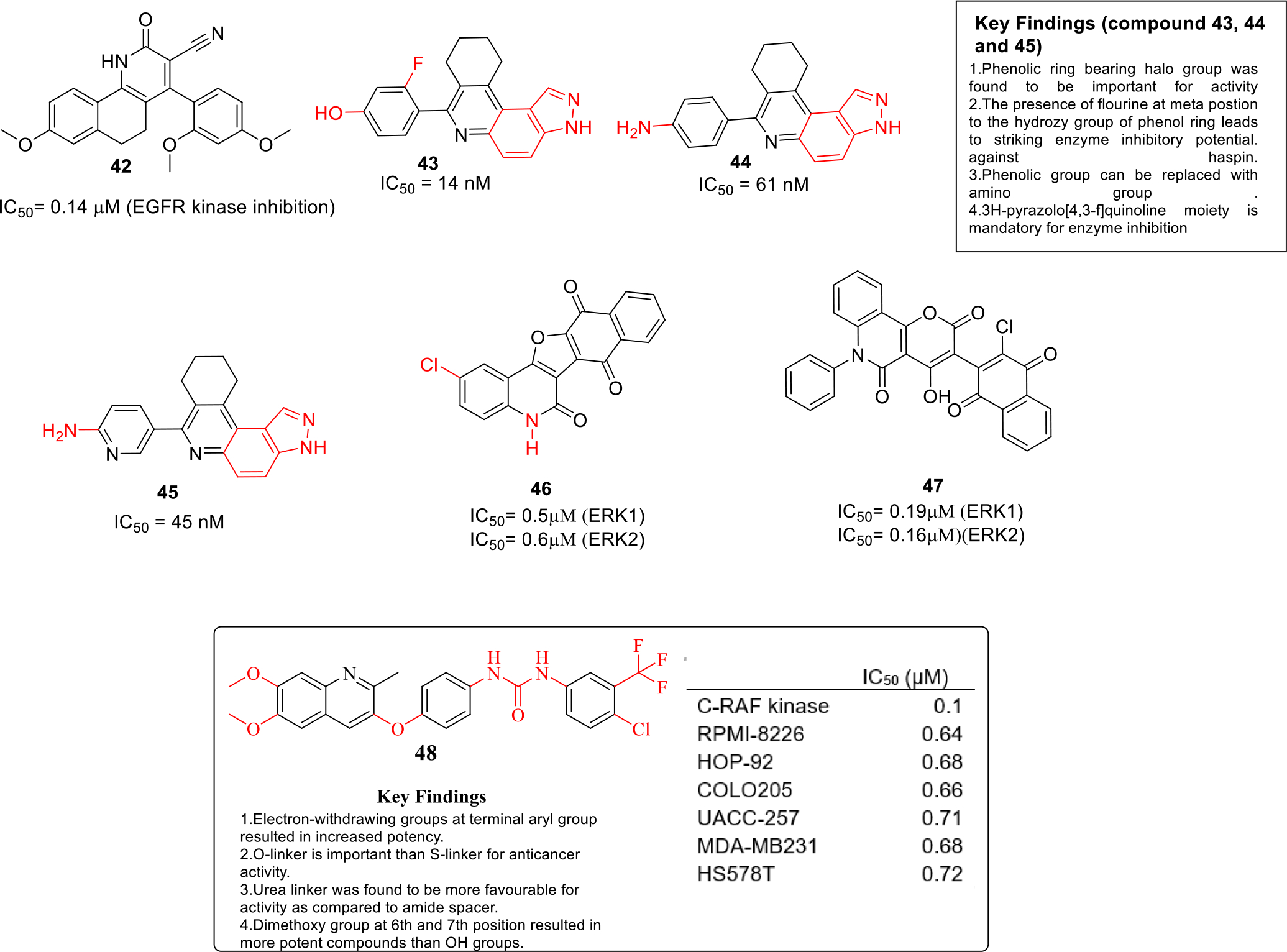
Abdelsalam et al designed and synthesized fused quinolines, quinazoline and indazole based compounds as EGFR inhibitors and the compounds were evaluated for in vitro anticancer activity against four cancerous cell lines HepG2, MCF-7, HCT116 and Caco-2. Among all the compounds, compound 42 displayed significant potency against MCF-7 cell line with IC50 value of 7.21 μM. Moreover, compound 42 was also selective against MCF-7 cell lines over healthy fibroblast cell lines (Figure 7). In addition, the results of the EGFR kinase assay also revealed that compound 42 was a potent inhibitor of the enzyme with potency equivalent to that of Erlotinib (IC50= 0.14μM). Molecular docking studies were carried out and it was predicted that 42 fits well into EGFR active site cavity in almost similar manner as Erlotinib [34].
Haspin is a protein kinase which is known for phosphorylating histone H3 that ultimately progresses mitosis process in cells [35]. Inhibition of Haspin is a new target for ceasing rapid proliferation of cancer cells. In quest to capitalize on these revelations, Sintim and team synthesized 3H-pyrazolo[4,3-f]quinoline based compounds as haspin kinase inhibitors. In total, 21 compounds were synthesized and the results of biological evaluation demonstrated that three compounds 43, 44 and 45 exhibited marked enzyme inhibition with IC50 values in nanomolar range. (Figure 7). Furthermore, molecular docking studies were performed and it was found that the interaction pattern of 43 with Haspin at hinge region matched with the haspin-ADP interaction. Moreover, flow cytometry and caspase 3/7 activity assays indicated that 43 induced apoptosis in HCT116 cells. Overall, the outcome of the study present compound 43–45 as ideal lead compounds to develop cancer therapeutics [36].
Aly et al synthesized naphthofuro[3,2-c]quinoline-6,7,12-triones and pyrano[3,2-c]quinoline-6,7,8,13-tetraones based analogues as ERK inhibitors. The results of the biological evaluation revealed that compound 46 and 47 were endowed with high selectivity towards ERK1 & ERK2 enzyme (Figure 7). In vitro anticancer studies were also performed against NCI-60 panel of tumor cell lines and compound 46 and 47 were found to be significantly potent anticancer agents. In addition, apoptosis induction assay data suggested that 46 and 47 activated caspase 3/7 in A375 cell lines and the enzyme kinetic studies indicated that 46 and 47 are ATP competitive type of inhibitors [37].
Rapidly accelerated fibrosarcoma (RAF) kinases are involved in many cellular processes including cell cycle regulation and organ development. Overactivation of RAF leads to the development of proliferative diseases including cancers [38]. El-Gamal et al designed and synthesized new quinoline derivatives as RAF kinases inhibitors. Most of the compounds were found to be highly potent and compound 48 was identified as the most active with IC50 value of 0.10 μM (Figure 7). Moreover, compound 48 was selective against RAF kinase over 12 different kinases that were tested. In addition, the docking studies indicated that the binding of compound 48 to RAF kinase was attributed to three hydrogen bonding interactions [39].
2.5. Miscellaneous
This subsection basically covers the quinoline based anticancer agents that have been majorly evaluated only for in-vitro cytotoxic effects. It is noteworthy to mention that exhaustive evaluation of these compounds (mechanistic studies, toxicity studies, in-vivo investigation in xenograft models along with pharmacokinetic studies) might render new dimensions to the activity profile of these preliminarily active compounds that might end up turning to clinical candidates in the longer run. In addition, detailed investigation of these compounds will further validate the ability of quinoline based chemical architectures to act in mechanistically diverse ways.
Arafa et al synthesized novel quinoline based derivatives as antiproliferative agents and evaluated them against two cancer cell lines HT29 and MDA-MB231 by MTT assay. The most potent compound of the series 49 exhibited strong anticancer activity against both the cell lines (Figure 8). Moreover, the SAR studies showed that the N’-benzylideneacetohydrazides were more potent than the N-phenylhydrazine-(thio)carboxamides and N’-(1-phenylethylidene)acetohydrazides [40].
Fig. 8.
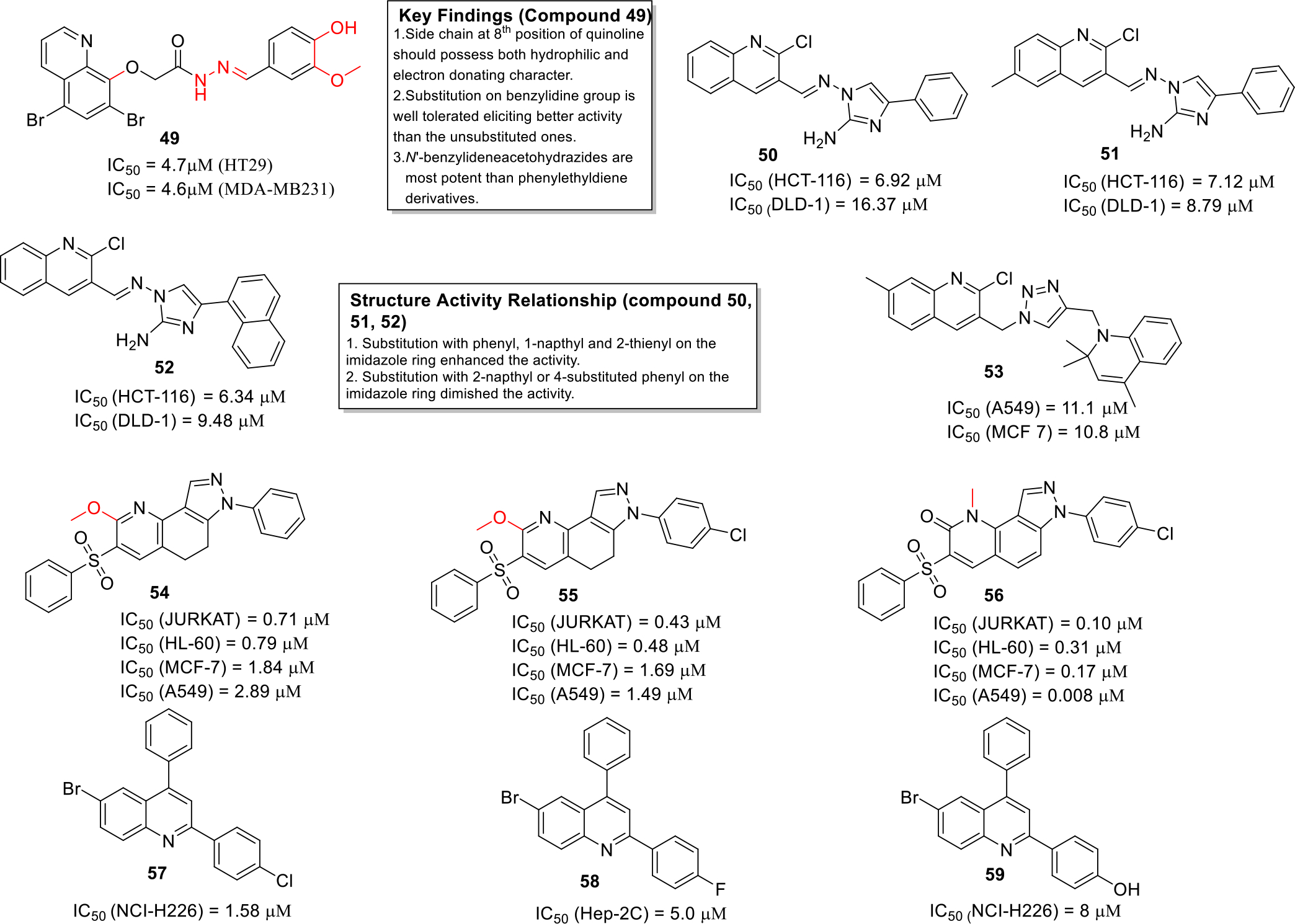
Anticancer quinolines
Singh et al synthesized a series of 2-aminoimidazoleequinoline hybrids and evaluated them against two human colon cancer cell lines HCT-116 and DLD-1. The results of the biological evaluation demonstrated that compounds 50–52 displayed strong anticancer activity (Figure 8) and were also found to be non-toxic to normal cell lines. Structure activity relationship studies indicated that the replacement of the phenyl ring at position 4 of imidazole with classical ring equivalent bioisosters leads to more effective compounds, however, substitutions on phenyl ring led to decrease in selective cytotoxic action towards cancer cell and increase in cytotoxicity towards the normal cells [41].
Pal and coworkers designed and synthesized a series of new hybrid compounds by linking quinoline, triazole and dihydroquinoline pharmacophores. The compounds exhibited potential cytotoxic effects against A549 and MCF 7 cancer cell lines (Figure 8). Furthermore, the synthesized candidates were screened by molecular docking studies and the results indicated that compound 53 showed minimum docking score and it also utilized the conserved binding pocket of PDE4 enzyme [42].
Spano et al synthesized pyrazolo[3,4-h]quinolines as photosensitizing agents. Some of the compounds (54–56) exhibited significant potency against various cancer cell lines including T lymphocyte cell line JURKAT, human leukemia cell line HL-60, MCF-7 and A549 cell lines (Figure 8). The compounds were also evaluated by colony formation assay using A-549 cells which depicted that 56 reduced colony formation at a lower concentration as compared to 55. Moreover, it was observed that compound 56 induced apoptosis with large lysosomal and mitochondrial involvement. Furthermore, the SAR studies lead to the fact that presence of chlorine on phenyl group attached to the pyrazole NH enhances the cytotoxic potential [43].
Liberto et al. synthesized a library of 28 quinoline derivatives and evaluated them by MTT assay against human cancer cell lines namely TOV-21G (ovary), NCI-H226 (lung) and Hep-2c (Hela contaminant). Compound 57–59 were reported to be most potent antiprolifertaive agents (Figure 8). Attempts to delineate the structural features required for the activity indicated that substitution at the 4th position of phenyl with groups such as fluorine and nitro favored the anticancer activity [44].
Alegaon et al designed and synthesized quinoline-azetidinone hybrids and evaluated them in vitro against human liver carcinoma cell line HEP G2 and human hepatoma cell line HEP 3B. Compounds 60 and 61 exhibited potent antiproliferative activity with IC50 value in the mid-nanomolar to low micromolar range. Moreover, the compounds were able to induce apoptosis in these cancer cell lines. Furthermore, SAR was established which revealed that the presence of strong electron donating group like OH at para position of phenyl leads to significant activity whereas a decline in activity was observed with electron withdrawing groups (Figure 9). It was also found that a phenyl at C-4 of azetidinone is crucial for activity and could not be replaced by other 5-membered rings. The compounds synthesized in this study followed the Lipinski rule of 5 [45].
Fig. 9.
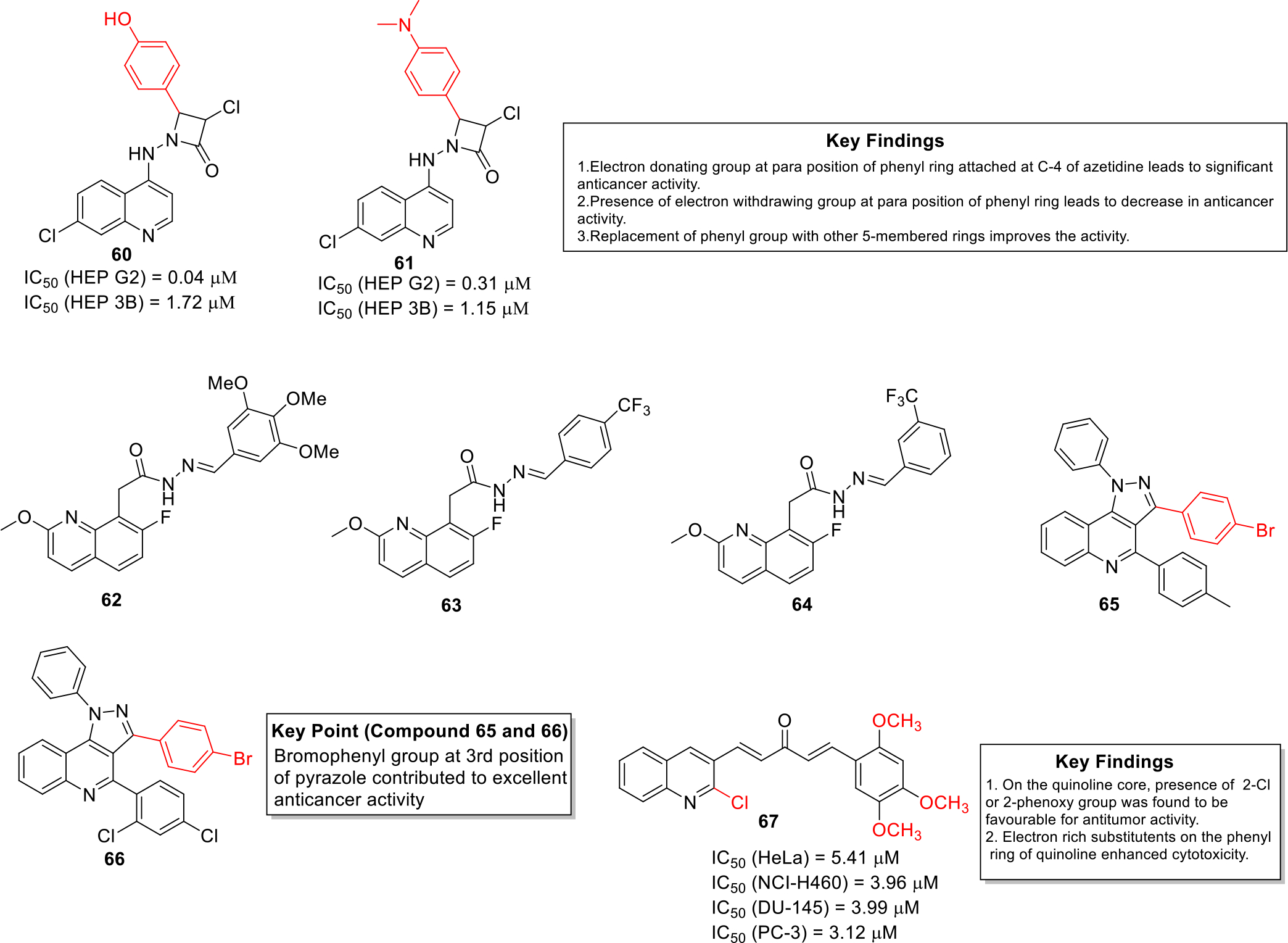
Anticancer quinolines
Reddy et al synthesized novel quinoline acetohydrazide derivatives and tested against MCF 7 and skin cancer cell lines G-361. The results of the in vitro cytotoxicity studies indicated that the potent compounds 62, 63 and 64 were significantly active in comparison to that of standard drug doxorubicin (IC50 = 2.444 μM) (Figure 9). SAR studies revealed that the trifluoro derivatives were substantially potent and displayed excellent binding properties in the docking studies against topoisomerase I. [46].
Kasaboina et al synthesized novel pyrazolo-quinoline based compounds and evaluated them against various cancer cell lines including lung, breast and other cancer cell lines. The research group identified that the compounds 65 and 66 (Figure 9) showed IC50 values of 2.43 and 6.01 μM, against A549 and MCF7 cancer cell lines respectively [47].
Ramya et al designed and synthesized quinoline-curcumin hybrid analogues as potential anticancer agents. The synthesized compounds exhibited better aqueous solubility in comparison to curcumin. Moreover, compound 67 was found to be more efficacious than curcumin against cancer cell lines such as human cervical cancer cell line HeLa, NCI-H460, human prostate cancer cell lines DU-145 and PC-3 (Figure 9). The detailed investigation revealed that compound 67 induced G2/M cell cycle arrest and induced apoptosis in PC-3 cells [48].
Li et al designed and synthesized quinoline based compounds and evaluated them against a panel of seven cancer cell lines (human laryngeal carcinoma (HCT-116, RKO and DLD1), gastric carcinoma (BGC-823), non-small-cell lung carcinoma (NCI-H1650), liver carcinoma (HepG2), and ovarian carcinoma (SK-OV-3). Among all the compounds, compound 68 demonstrated potent antitumor effects against all the employed cell lines (Figure 10). In vivo experiments were performed using colorectal cancer xenograft model in nude mice and it was observed that compound 68 exerted significant tumor growth inhibition at a dose of 80 mg/kg. Detailed studies also revealed that 68 induced p53/Bax-dependent colorectal cancer cell apoptosis via triggering p53 transcriptional activity [49].
Fig. 10.
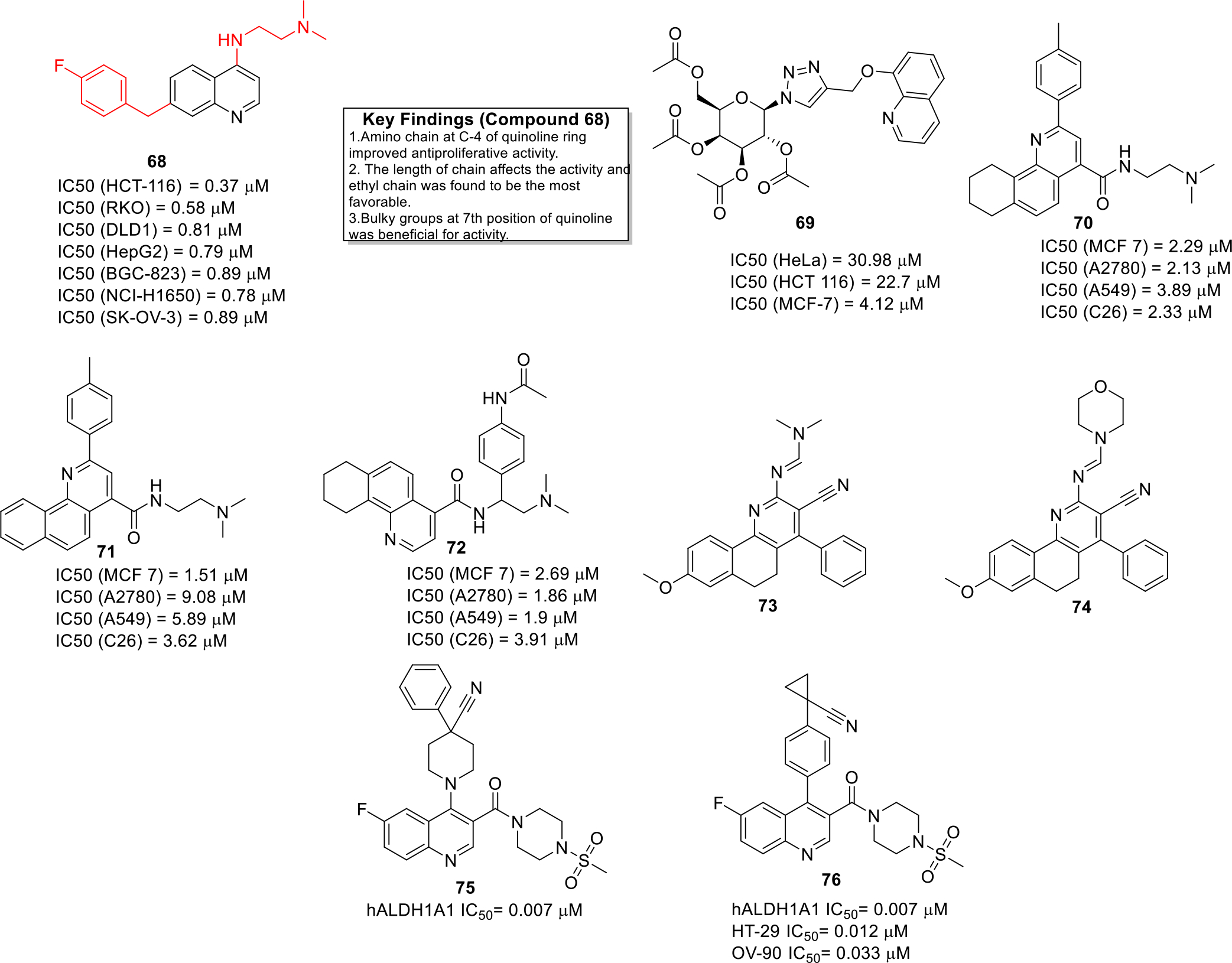
Anticancer quinolines
Krawczyk et al synthesized 8-hydroxyquinoline glycoconjugates and explored the compounds for their anti-cancer properties. The prepared glycoconjugates were screened against various cancer lines and compound 69 was identified as a hit (Figure 10). The glycoconjugation strategy resulted in both the improvement of bioavailability as well as the activity of 8-hydroxyquinoline derivatives [50].
Jafari et al developed novel benzo- and tetrahydrobenzo-[h]quinoline derivatives and tested them against various cancer cell lines. The novel compounds depicted IC50 values in the low micromolar range (Figure 10) against various cancer cell lines. Detailed investigation indicated that the compound 71 acted as a DNA intercalating agent. Furthermore, the docking studies also confirmed that these compounds interacted and intercalated with DNA. Annexin V-FITC/Propidium iodide staining assay demonstrated that 70, 71 and 72 induced apoptosis in A549 cells [51].
Haiba et al. synthesized novel Benzo[h]chromene and Benzo[h]quinoline derivatives (73 and 74) and the entire library was found to be endowed with cytotoxicity activities in the mid micromolar range against human cancer cell lines HepG-2 and MCF-7. (Figure 10) [52].
Overexpression of ALDH1A1 type of aldehyde dehydrogenase serves as a biomarker in cancer cells [53]. In quest to capitalize on this useful information, Yang and others developed quinoline containing ALDH1A1 inhibitors. Among all the compounds, compound 75 & 76 were found to be the most potent ALDH1A1 inhibitors with IC50 value of 0.007 μM (Figure 10). The pharmacokinetic parameters were also studied for these potent candidates and it was found that these compounds were suitable for in vivo drug administration. In particular, compound 76 was endowed with substantial antiproliferative effects against HT-29 and OV-90 cell lines and also demonstrated low drug clearance as well as long half-life period [54].
Human dihydroorotate dehydrogenase (hDHODH) enzyme plays its key role in pyrimidine biosynthesis [55]. In this context, significant attention has been paid towards the design of hDHODH inhibitors as anticancer drugs in recent times. Working on similar lines, Vyas et al designed and synthesized substituted quinoline derivatives as hDHODH inhibitor [56]. The compounds were designed on the basis of 3D quantitative SAR studies and the designed compounds docked well in the binding site of the enzyme with good docking scores. In vitro screening was performed including enzyme inhibition assays and MTT assays against HT-29 and MDA-MB-231 cancer cell lines that led to the identification of 77 and 78 as potent hDHODH possessing anticancer potential (Figure 11) [57].
Fig. 11.
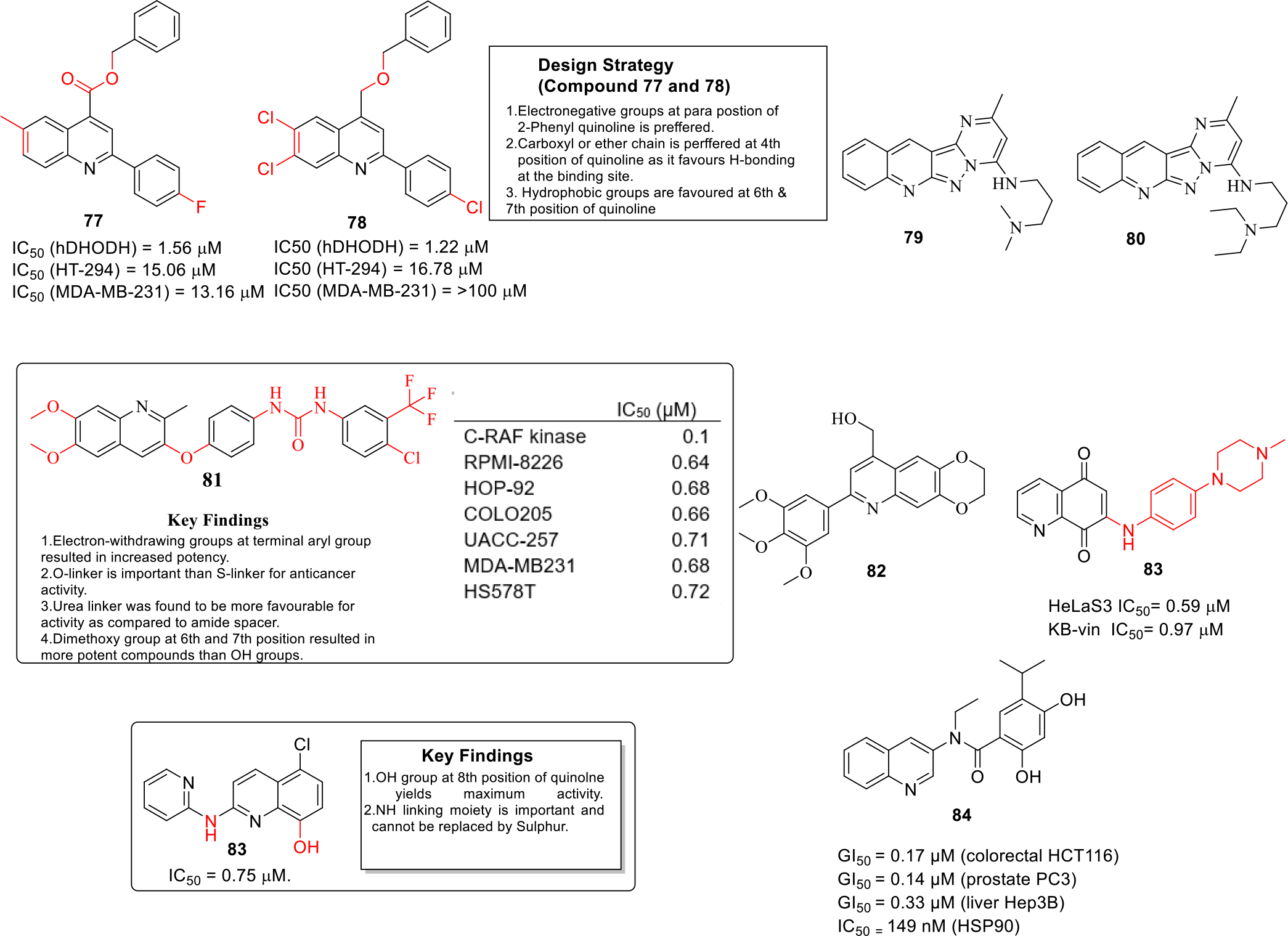
Anticancer quinolines
ATP-binding cassette transporters are involved in important cellular processes whereas overexpression of a particular ATP-binding cassette transporter (ABC) subfamily G2 is responsible for multidrug resistance which is a major cause for cancer chemotherapy failure [58]. Karthikeyan et al. developed novel fused Pyrazolo-quinolines compounds as ABCG2 inhibitors (Figure 11) that could be utilized as MDR reversal agents. From the enzymatic assays, compound 79 and 80 were found to be most potent ABCG2 inhibitors. Docking studies indicated that the 80 binds to ABCG2 cavity with high affinity and was involved in key interactions [59]
Heat shock protein 90 (HSP90) is a cellular protein that is involved in protection of cell against heat stress and other processes [60] and is found to be elevated in several malignancies [61]. Emphasizing on these revelations, Malayeri and team designed 7-(aryl)-2,3-dihydro-[1,4]dioxino[2,3-g]quinoline derivatives as HSP90 inhibitors and screened them against three cancer cell lines MCF-7, A549 and DU145 by MTT assay. The results of the MTT assay demonstrated that compound 81 exerted significant anticancer effects against MCF-7 and A549 cancer cell lines with IC50 value of 45.3 and 64.15 μM (Figure 11). The results of experimental assay were rationalized with the help of computational studies as the molecular docking studies revealed that compound 81 was involved in adenine type interactions with N-terminal ATP-binding site of HSP90 [62].
The levels of NAD(P)H:quinone oxidoreductase (NQO1) has been observed to be overexpressed in cancer cells as compared to normal cells [63] and thus NQO1 has been validated as an anticancer target [64]. Ling and others synthesized novel amino-quinoline-5,8-dione derivatives as NQO1 inhibitors. The results of the biological evaluation revealed that compound 82 possessed NQO1-dependent antiproliferative activity with IC50 values within 0.59 and 1.52 μM (Figure 11). Detailed investigation indicated that these compounds cause lethal mitochondrial dysfunction in a dose dependent manner in cancer cells. Apoptotic studies further suggested that compound 82 downregulated the anti-apoptotic protein levels of Bcl-2 and up-regulated the pro-apoptotic Bax proteins and ultimately cleaved caspase-3 in a concentration dependent manner in HeLa S3 cells. [65].
Proviral insertion site in moloney murine leukemia virus (PIM) proteins family is involved in biological processes including mainly cell proliferation and cell survival. Elevated levels of PIM-1 is detected in solid malignancies. In light of these revelations, Li and others synthesized quinolines derivatives as Pim-1 kinase inhibitors that could be utilized as anti-prostate cancer agents. SAR studies led to the identification of compound 83 as the lead compound (Figure 11) that was endowed with the potential to to induce apoptosis [66].
In quest to extract the benefits of HSP90 inhibition, nepali et al employed a molecular hybridization strategy to construct amide tethered quinoline-resorcinol conjugates. Among all the conjugates, hybrid 84 exerted significant cell growth inhibitory effects against PC-3 prostate cancer cell lines and also triggered degradation of HSP90 client proteins. Furthermore, compound 84 was found to be endowed with apoptosis inducing ability, caused G2M phase cell cycle arrest in PC-3 cells and demonstrated key interactions with the amino acid residues of the HSP90 chaperone protein (Fig. 11) [67].
3. PATENT REVIEW
Numerous quinoline based inhibitors with anticancer potential patented during year 2014–2019 have been discussed in this section.
Knight et al published a patent on the synthesis of new quinoline derivatives as PI3K alpha enzyme inhibitors. The compounds were evaluated by PI3K alpha Leadseeker SPA assay and it was found that the synthesized candidates were potent inhibitors of PI3 kinase alpha enzymes and were endowed with IC50 values in the picomolar to low nanomolar range (Figure 12) [68].
Fig. 12.
Quinoline derivatives as PI3K alpha enzyme inhibitors
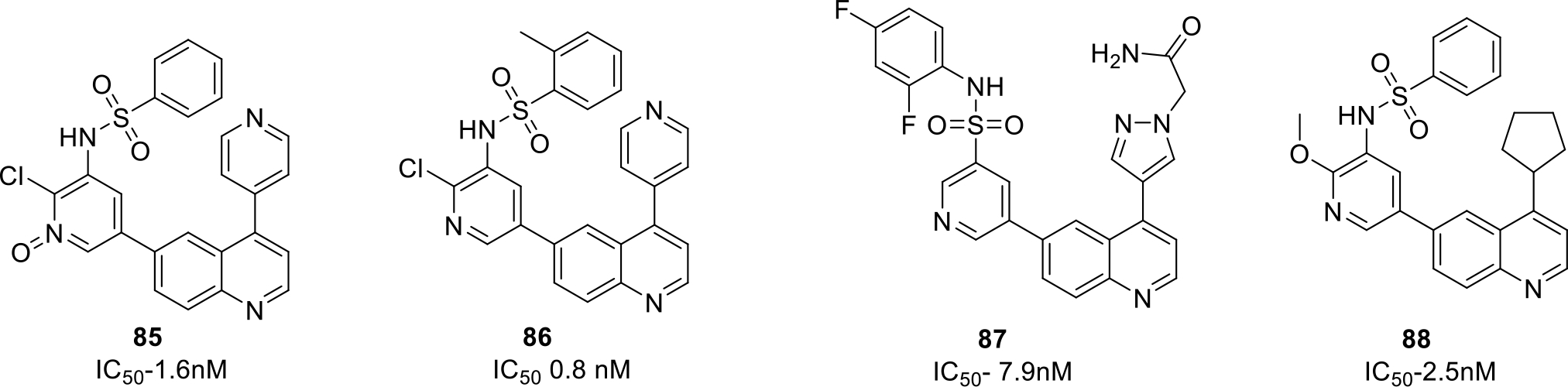
Kumar et al., filed a patent for the discovery of Imidazo[4,5-c]quinoline derivatives as antitumor agents. The compounds were evaluated by performing various cellular and enzymatic assays. The result of the cytotoxicity assay demonstrated that the compounds 89-93 were endowed with significant cell growth inhibitory effects (IC50 < 1 μM). Among them, compound 90 and 92 exerted a % inhibition > 50 against PI3K α enzyme and mTOR kinase (Figure 13) [69].
Fig. 13.
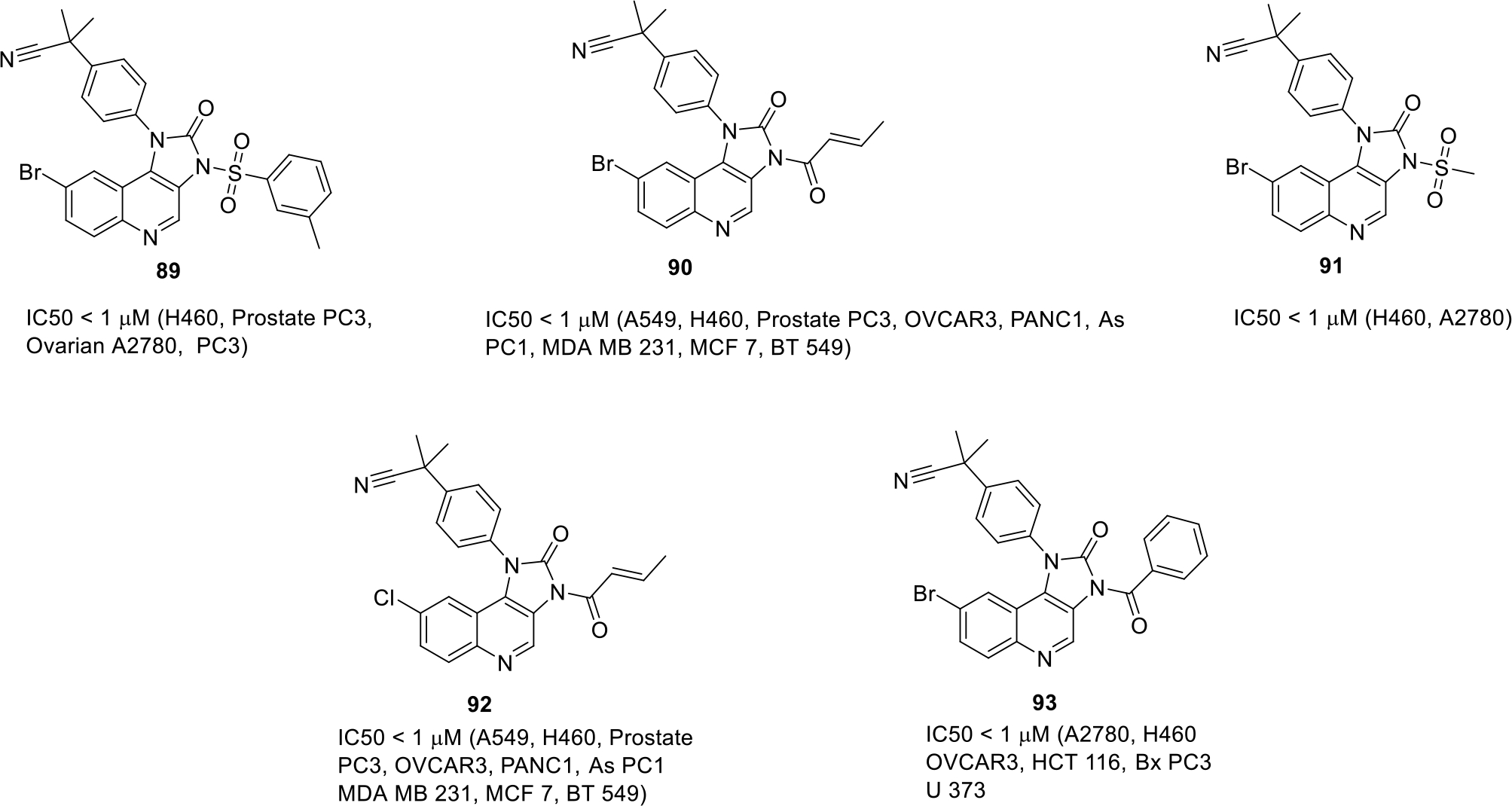
Imidazo[4,5-c]quinoline derivatives with cancer cell lines for which IC50 value is less than 1μM
Liu and team designed quinoline compounds as a potent human methionine aminopeptidase enzyme (hMetAP) inhibitors. The synthesized compounds (94–99) showed more selectivity towards hMetAP2 isoform of enzyme as compared to hMetAP1. Among them, compound 96 was strikingly potent and exhibited the maximum potency with IC50 of 0.03 μM for hMetAP2 and 11.9 μM for hMetAP1. The structures of compound 100 along with other potent compounds are shown in Figure 14 [70].
Fig. 14.
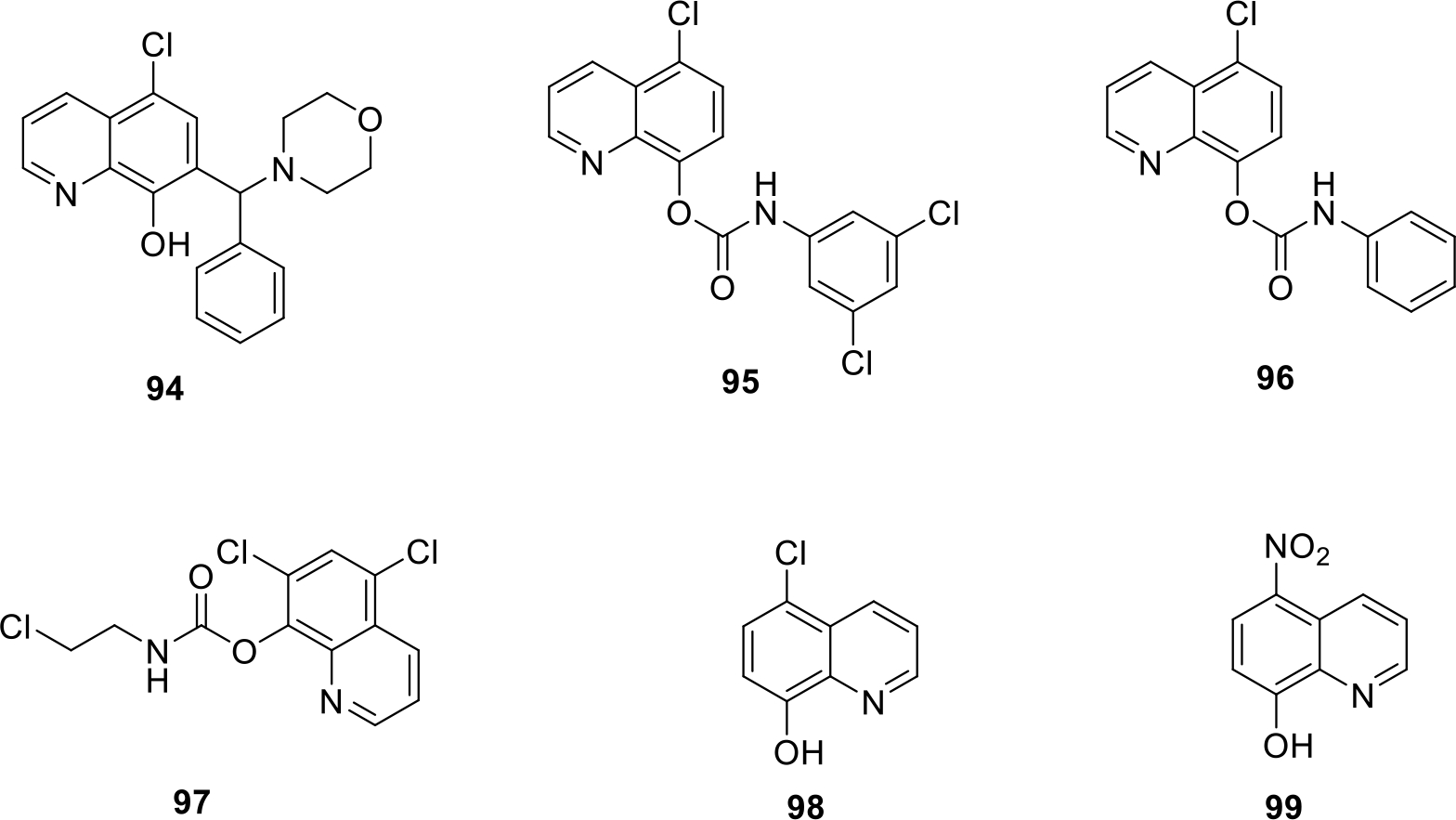
Quinoline compounds as hMetAP enzyme inhibitors with IC50 values (in μM)
Furet and team filed a patent for new quinoline carboxamide derivatives as Fibroblast growth factor receptor 3 (FGFR3) inhibitor. The synthesized compounds (100–103) displayed significant inhibitory potential at nanomolar concentration. Among all the compounds, compound 100 was found to be the most potent (IC50 value = 9 nM). The structure of compound 100 along with other potent compounds are shown in Figure 15 [71].
Fig. 15.
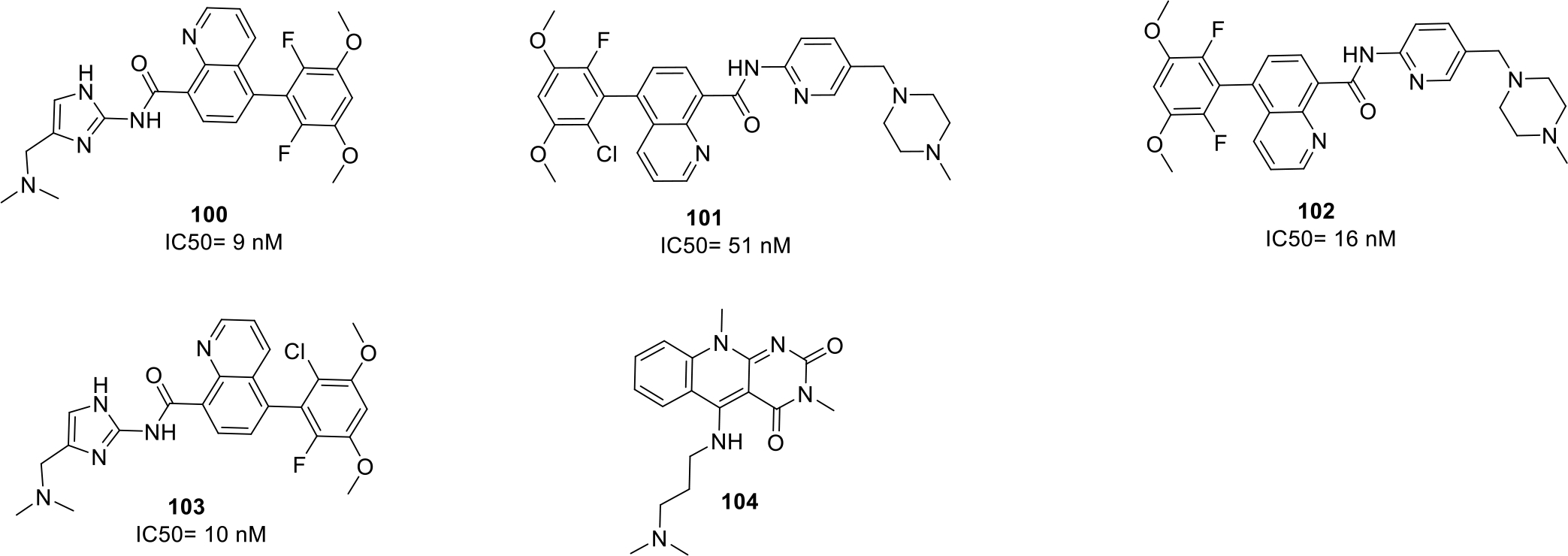
Anticancer quinolines
Literature reports indicates that HDM2 protein has been found to be overexpressed in tumor cells that leads to inhibition of an important p53 protein responsible for the induction of tumor cell apoptosis. Therefore, HDM2 inhibition presents a rational approach for the development of novel anticancer agents. In light of these revelations, Weissman et. al., patented pyrimidodione-quinoline compounds as a selective human double minute 2 protein (HDM2) inhibitors. The structure of the most potent compound 104 that displayed selective HDM2 inhibitory activity with high solubility is shown in Figure 15 [72].
Tzeng et al., filed a patent on 4-Anilinofuro[2,3-b]quinoline derivatives as anticancer agents. The synthesized compounds were tested for their anticancer activity by MTT assay using cancer cell lines breast cancer cell lines MCF 7 and lung cancer cell lines NCI-H460. The results of the assay indicated that the compounds (105 – 108) displayed substantial cell growth inhibitory effects. (Figure 16) [73].
Fig. 16.
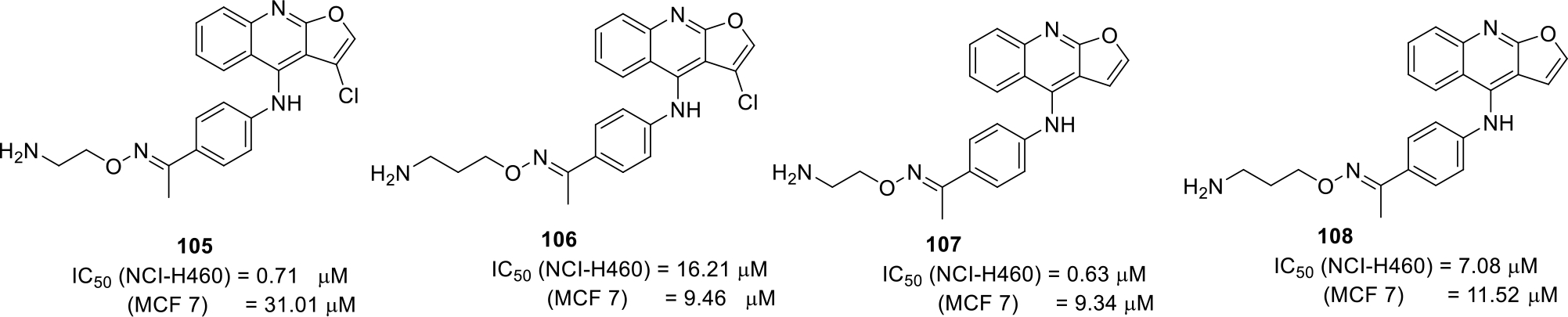
4-Anilinofuro[2,3-b]quinoline derivatives
Xu published a patent quinoline derivatives reporting them as c-Met kinase inhibitors. The results of the enzymatic assay led to the identification of several potent inhibitors with IC50 values ranging from 0.009 to 8.2 μg/mL. All the compounds were also evaluated against human cancer cell lines including Non-small cell lung carcinoma H460, human colon cancer HT-29 and human gastric cancer MKN-45. Most potent compounds (109 – 112) were found to be endowed with IC50 value between 0.0002–0.57 μg/mL as shown in Figure 17 [74].
Fig. 17.
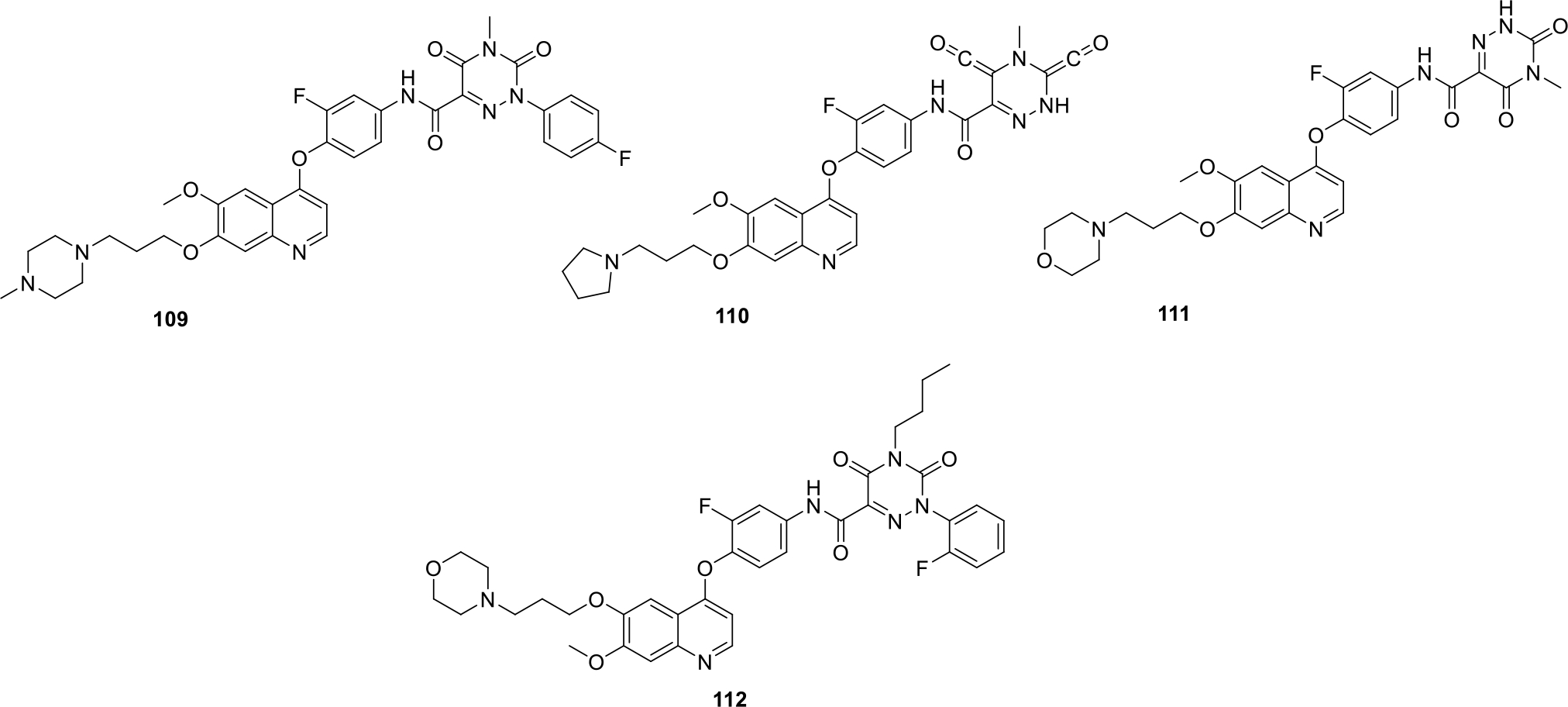
Quinoline derivatives as c-Met kinase inhibitors
Fuchss et. al., filed a patent on Imidazo[4,5-c]quinolines compounds as DNA-protein kinase (DNA-PK) inhibitor. The synthesized series of compounds were analyzed by DNA-PK Biochemical assay. Most of the synthesized compounds (113 – 116) were found to be potent as DNA-PK inhibitor with IC50 value < 0.1μM. Some of the potent structures are shown in Figure 18 [75].
Fig. 18.
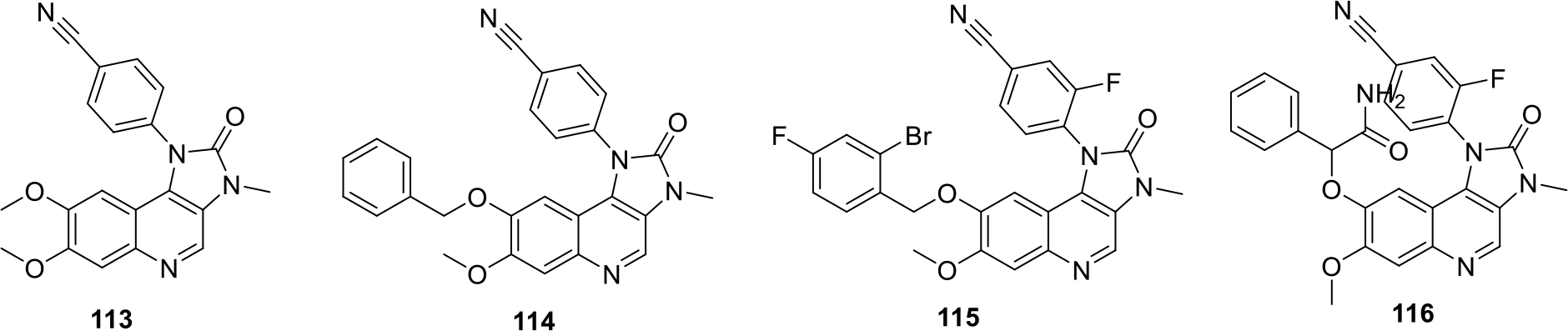
Imidazo[4,5-c]quinoline compounds as DNA-protein kinase inhibitors
Kuo and team filed three patents for 2-Selenophene-4-quinolones, their hydrophilic derivatives and 2-phenyl-4-quinolines as antiproliferative agents. The structures of the promising compounds are shown in Figure 19. The most potent compound 117 was further tested against five more cancer cell lines HCT-116, Hep G2, NCI-H226, A549 and A498 and it was found that 117 was endowed with significant anticancer potential against HCT-116 cell lines with IC50 of 0.9 μM [76–78].
Fig. 19.
2-Selenophene-4-quinolones derivatives with IC50 (μM) values
Xi patented series of substituted quinolines compounds as kinase inhibitors. The compounds were targeted against c-Met kinase, Kinase insert domain receptor (KDR) and AxL receptor. The results of the biological evaluation led to the identification of potent inhibitors 121–124 with IC50 values in the nanomolar range (Figure 20). Among them, compound 122 showed strong IC50 value of 5 nM in cellular AXL phosphorylation assay whereas compound 124 inhibits KDR with 23 nM during VEGF-R2 phosphorylation assays [79].
Fig. 20.
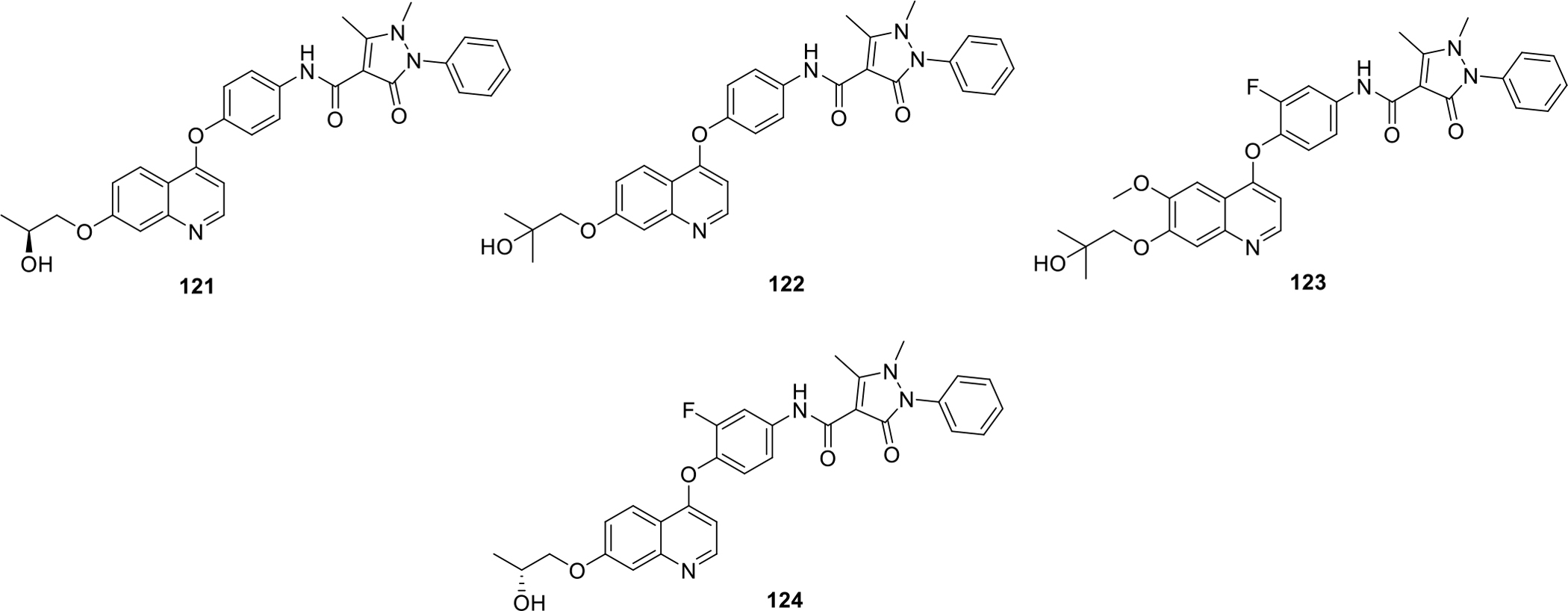
Substituted quinoline compounds as c-Met kinase inhibitors
LSF factor is considered as an oncogene found to be overexpressed in cancer cells. As such, LSF factor upregulates metalloproteinase-9 which is responsible for inducing angiogenesis in tumors and is mainly involved in hepatocellular carcinoma. In this context, Schaus et al. filed a patent for [1,3]Dioxolo[4,5-G][1,2,4]triazolo[1,5-a]quinoline derivatives as Late SV40 factor (LSF) inhibitor and evaluated them against hepatocellular carcinoma. The potent triazole and sulphur containing compounds (125 – 127) from the patent are shown in Figure 21 [80].
Fig. 21.
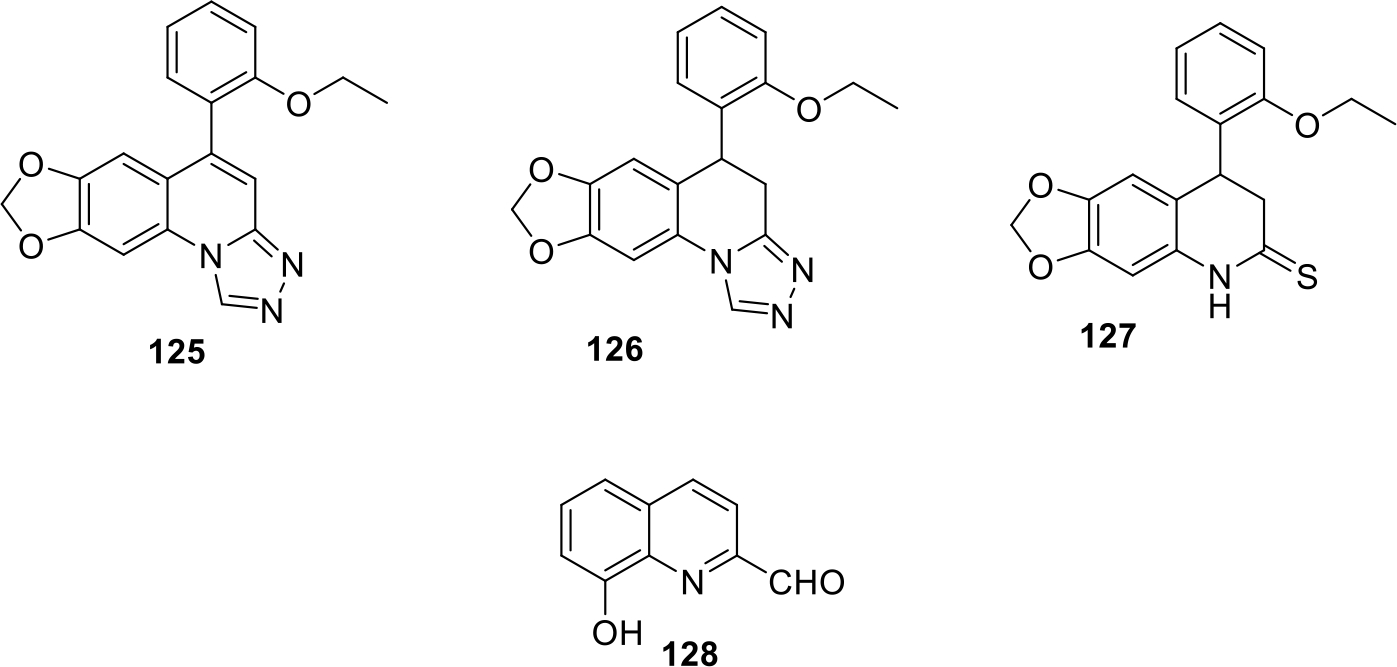
Anticancer quinolines
Chan et al patented quinoline derivatives (Figure 21) as potent anticancer agents. The compounds were evaluated against human breast cancer cell lines Hs578t, T47D and chronic myelogenous leukemia cell lines K562. Among all the compounds, compound 128 was most potent with IC50 value of 8.37μg/ml in T-47D cell lines and 7.00μg/ml in K562 cell lines. Candidate 128 was further explored by in vitro morphological studies for evaluation of its cytotoxic effects and the results demonstrated that compound showed cell shrinkage on all three cell lines at 25 μg/ml concentration whereas the breast cancer cell lines lost its adherent property. In vitro colony forming assay revealed that the anchorage dependent cologenicity ability of T47D was inhibited effectively by 128 in a dose dependent pattern [81].
It has been well evidenced that Hedgehog pathway plays its role in embryogenesis and also in adult stem cells repair. Overactivation of this pathway is known to increase angioproteins which in turn leads to development of various cancers in body. Capitalizing on the information, Tao and team filed a patent on quinolines and other related derivatives and evaluated them for their Hedgehog signaling inhibitory activity. The structure of the most potent quinoline compound 129 is shown Figure 22 [82].
Fig. 22.
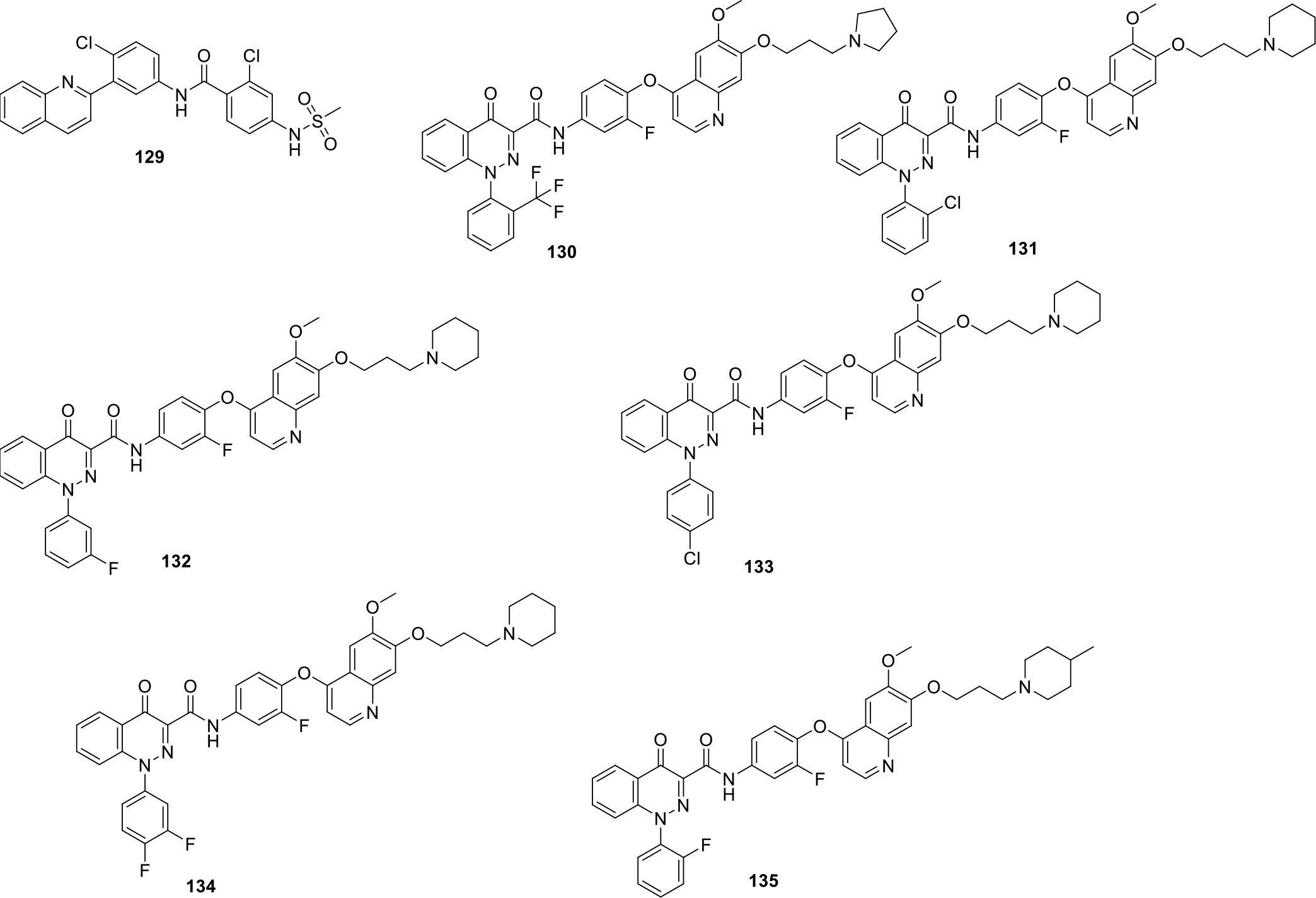
Anticancer quinolines
Gong et al., patented a series of novel quinoline and cinnoline derivatives as c-Met kinase enzyme inhibitors. A series of seventy compounds was synthesized and tested against various cancer cell lines including human lung cancer H460, human malignant glioblastoma cells U87MG, colon cancer cell HT-29, human gastric carcinoma MKN-45 and hepatoma cell SMMC-7721. Among them, compound 132 exhibited the most significant inhibitory potential against SMMC-7721 and H460 cell lines with IC50 of 0.007 μg/mL. The structures of the most potent compounds (130–135) are shown in Figure 22 and the activity profile is presented in Table 1 [83].
Table 1.
Activity profile of quinoline derivatives
| Compound | H460 IC50(μg/mL) |
U87MG IC50(μg/mL) |
HT-29 IC50(μg/mL) |
MKN-45 IC50(μg/mL) |
SMMC IC50(μg/mL) |
c-Met kinase IC50(μg/mL) |
|---|---|---|---|---|---|---|
| 130 | 0.04 | 0.18 | 0.18 | 0.27 | 0.57 | 0.05 |
| 131 | 0.052 | 0.35 | 0.19 | 0.007 | 0.3 | 0.03 |
| 132 | 0.007 | 1.2 | 0.17 | 0.0075 | 0.65 | 0.3 |
| 133 | 0.06 | 0.86 | 0.42 | 0.087 | 1 | 0.01 |
| 134 | 0.06 | 3.5 | 0.38 | 0.12 | 0.63 | 0.03 |
| 135 | 0.47 | 0.15 | 0.31 | 0.45 | 1.1 | 0.05 |
Berdini et al filed a patent on new quinoline compounds as Fibroblast growth factor receptor (FGFR) inhibitor I in view of the role of FGFR in cell differentiation and proliferation. Some of the potent compounds (136 – 139) are shown in Figure 23 [84].
Fig. 23.
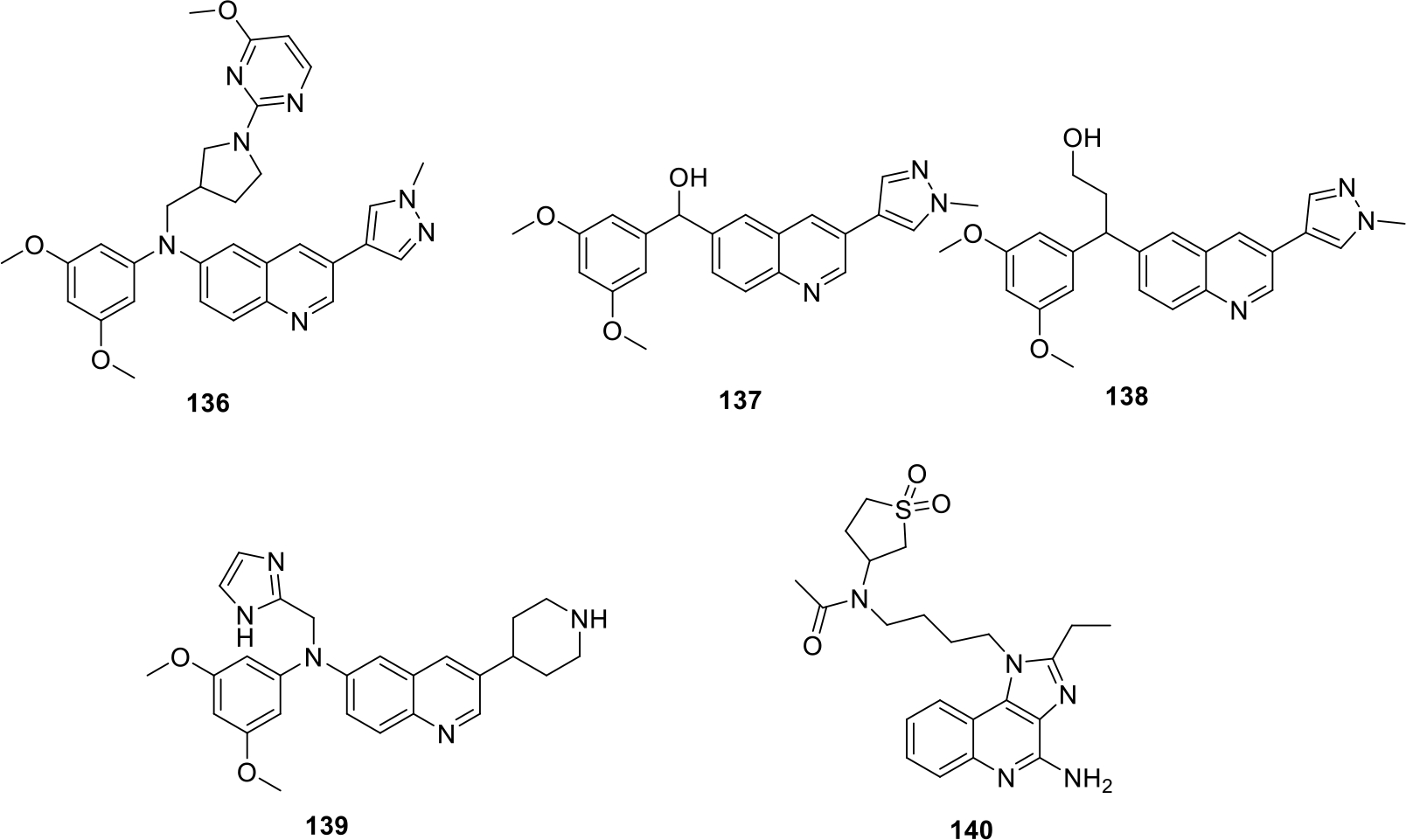
Anticancer quinolines
Gekeler et al. synthesized substituted Imidazoquinolines derivatives as Toll like receptor 7 (TLR7) activators. In total seven compounds were synthesized, and the anticancer evaluation of compounds were conducted using rat models. The results of the evaluation demonstrated that molecule 140 was found to be most potent having strong anti-metastatic effect (Figure 23) [85].
Tang et al. published a patent on quinoline derivatives as potent anticancer agents. The compounds were evaluated by MTS assay on various cancer cell lines involving human hepatoma cell lines Hep3B, adenocarcinoma human alveolar cell lines A549, human esophageal cancer cell lines HKESC-4 and others. The structures of the most potent compounds (141–145) are presented in Figure 24. Among all the compounds, compound 145 being optically active was screened for separately for both its (+) and (−) isomer. Both isomers showed significant anticancer effect with MTS50 ranging from 4 to 10 μg/mL. In vitro studies showed that (+)-145 exhibited 2 folds higher cytotoxicity against KYSE150 cancer cell lines as compared to Cisplatin whereas, both the isomers exhibited similar cytotoxic activity on Hep3B, A549 and HKESC-4 cell lines. Moreover, in vivo antitumor efficacy evaluation of pure enantiomer (−)-145 on mice demonstrated effective suppression of KYSE150 xenograft tumors in nude mice [86].
Fig. 24.
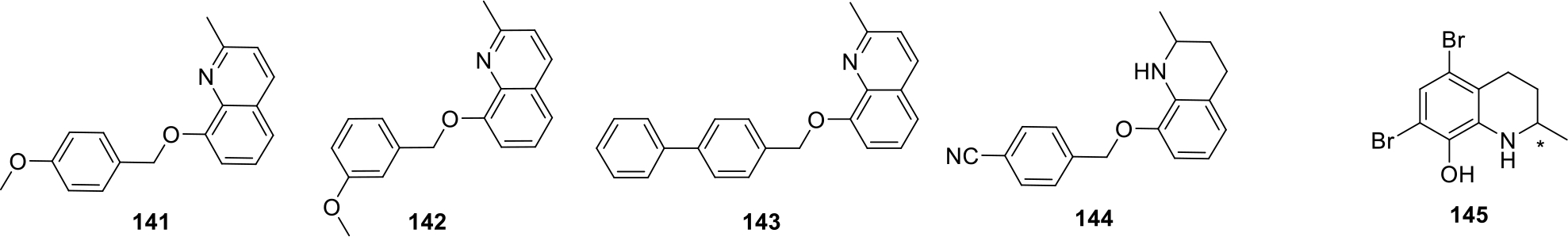
Quinoline derivatives with relative MTS activity at 50 μg/ml concentration
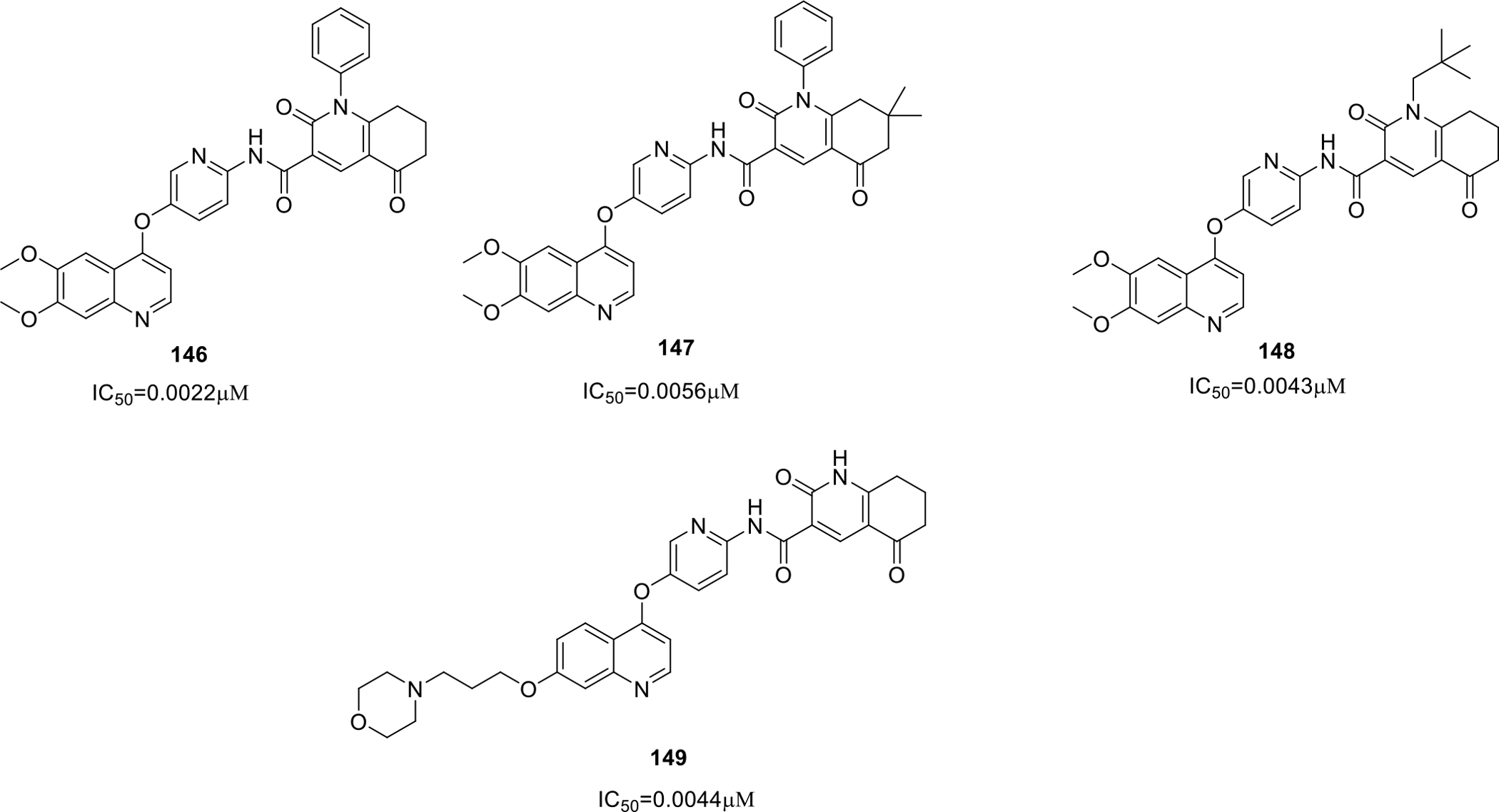
Inukai and team patented quinoline derivatives as AXL inhibitors. The synthesized compounds (146 – 149) were found to have potent AXL inhibitory action with IC50 ranging from 0.0011–0.0056 μM (Figure 25). Moreover, kinase selectivity evaluation in vitro test was also conducted to test the selectivity of the synthesized candidates towards AXL over KDR. Compound 147 showed approximately 1800 times selective AXL inhibition effect over KDR. The results of the study were quite optimistic as inhibition of KDR may lead to high blood pressure and selective AXL inhibition demonstrated by 147 can lead to its emergence as a potent anticancer agent with less side effects [87].
Fig. 25.
Quinoline derivatives as AXL inhibitor
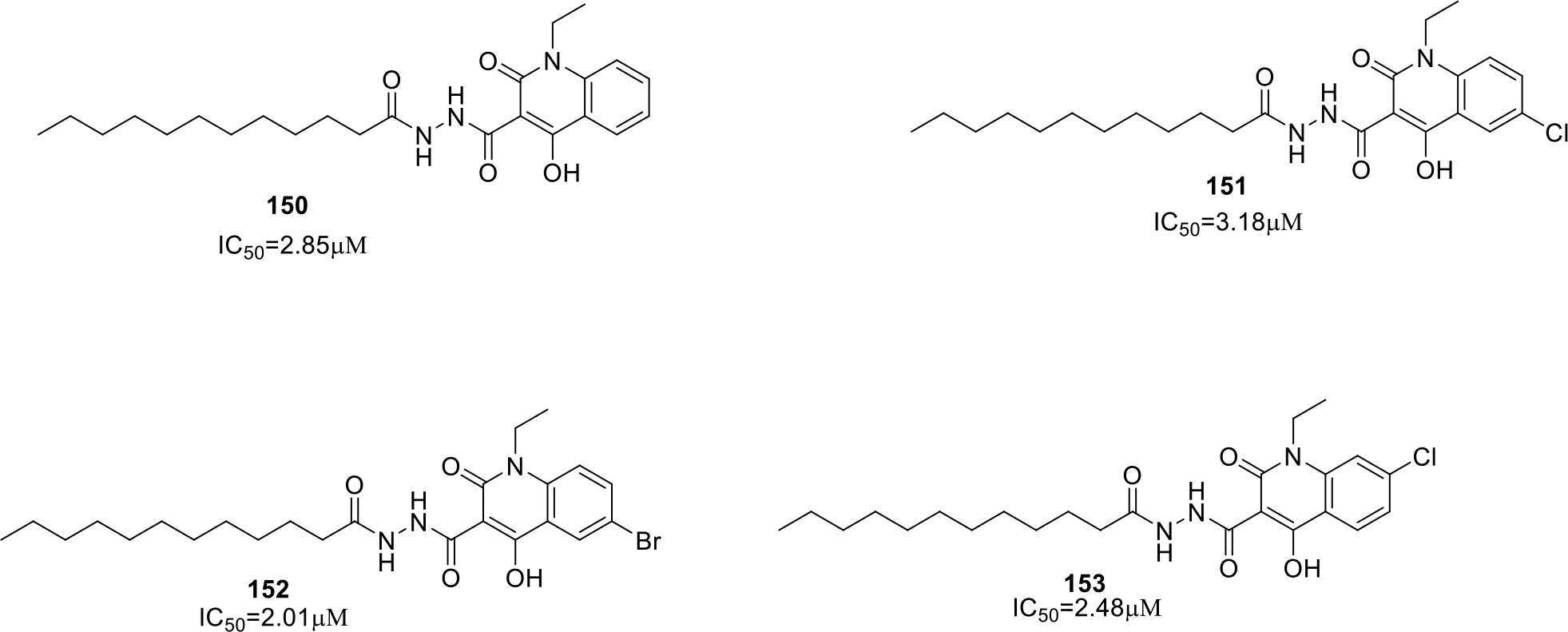
Gil et al filed a patent on quinoline derivatives with heterocyclic substitutions as glycogen synthase kinase-3 (GSK-3) inhibitors. The structures of the active compounds (150 – 153) are presented in Figure 26. Among the compounds synthesized, compound 150 was also studied to determine the binding mode of the compound with the kinase and the results demonstrated that 150 acts as a non-competitive inhibitor of GSK-3 enzyme [88].
Fig. 26.
Heterocyclic quinoline compounds as Glycogen Synthase kinase-3 (GSK-3) inhibitors
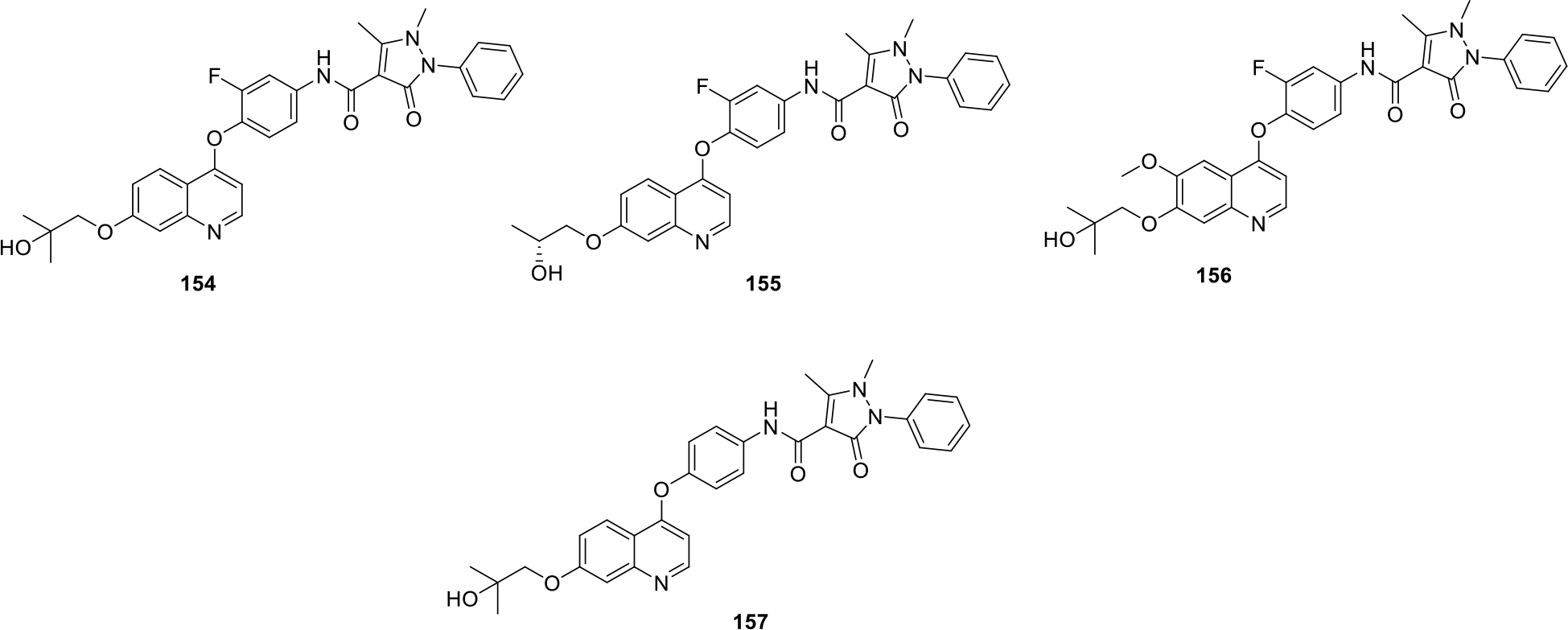
Xi patented substituted quinoline compounds as c-Met kinase, Kinase insert domain receptor (KDR) and Axl inhibitor. The synthesized compounds (154 – 157) showed potent inhibitory effects on kinase enzymes (IC50 values in nanomolar range) are shown in Figure 27. Tumor xenograft model studies were also performed and it was observed that compound 154 exerted significant reduction in mean tumor volume at doses of 10, 20 and 40 mg/kg [89].
Fig. 27.
Substituted quinoline compounds with IC50 values in nM range
APE-1 is a multifunctional enzyme involved in DNA repairing and is found to be overexpressed in tumors. Gold et al. filed a patent for the synthesis of 4-aminoquinoline derivatives as AP endonuclease-1 (APE-1) enzyme inhibitor that can be used as effective anticancer agents. Compounds 158–161 were found to be effective APE-1 inhibitors with Ki values of 0.12–0.22 μM as shown in Figure 28 [90].
Fig. 28.
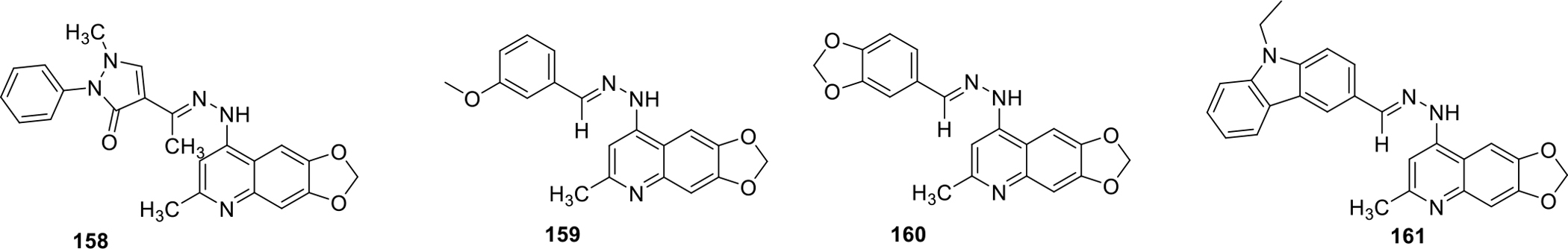
4-Aminoquinoline derivatives as APE-1 enzyme inhibitors
Scott and team patented substituted 2,3-dihydroimidazo[1,2-c]quinoline derivatives as PI3 kinase enzyme inhibitors. The patented compounds (162, 163, 164) showed potent PI3K enzyme inhibition and their structures are shown in Figure 29 [91].
Fig. 29.
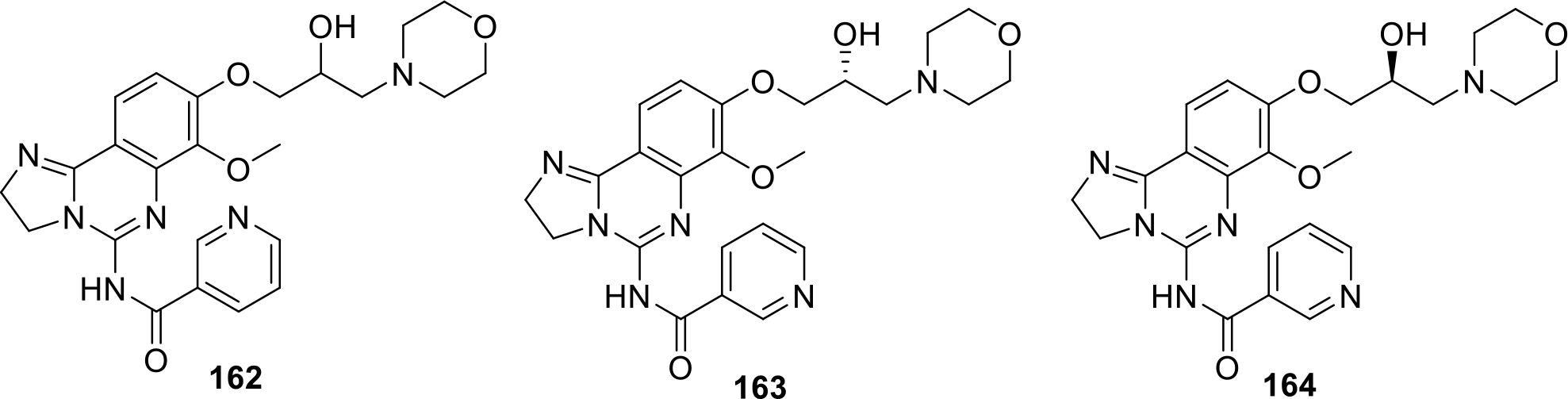
Substituted 2,3-dihydroimidazo[1,2-c]quinoline derivatives as PI3 kinase enzyme inhibitors
Castro et al. published trisubstituted bicyclic quinoline derivatives as kinase inhibitors. The biological assay results indicated potent PI3K inhibitory action of compounds (165–167) with IC50 values ranging from 1 to 10000 nM as shown in Figure 30. Compound 167 showed strong PI3Kγ isoform inhibitory action at 100 to <500 nanomolar concentration. Moreover the selectivity test between the two isoforms PI3K δ and γ was also performed for the synthesized compounds in which 167 showed more selectivity towards the δ isoform with IC50 selectivity of 10 to <50 [92].
Fig. 30.
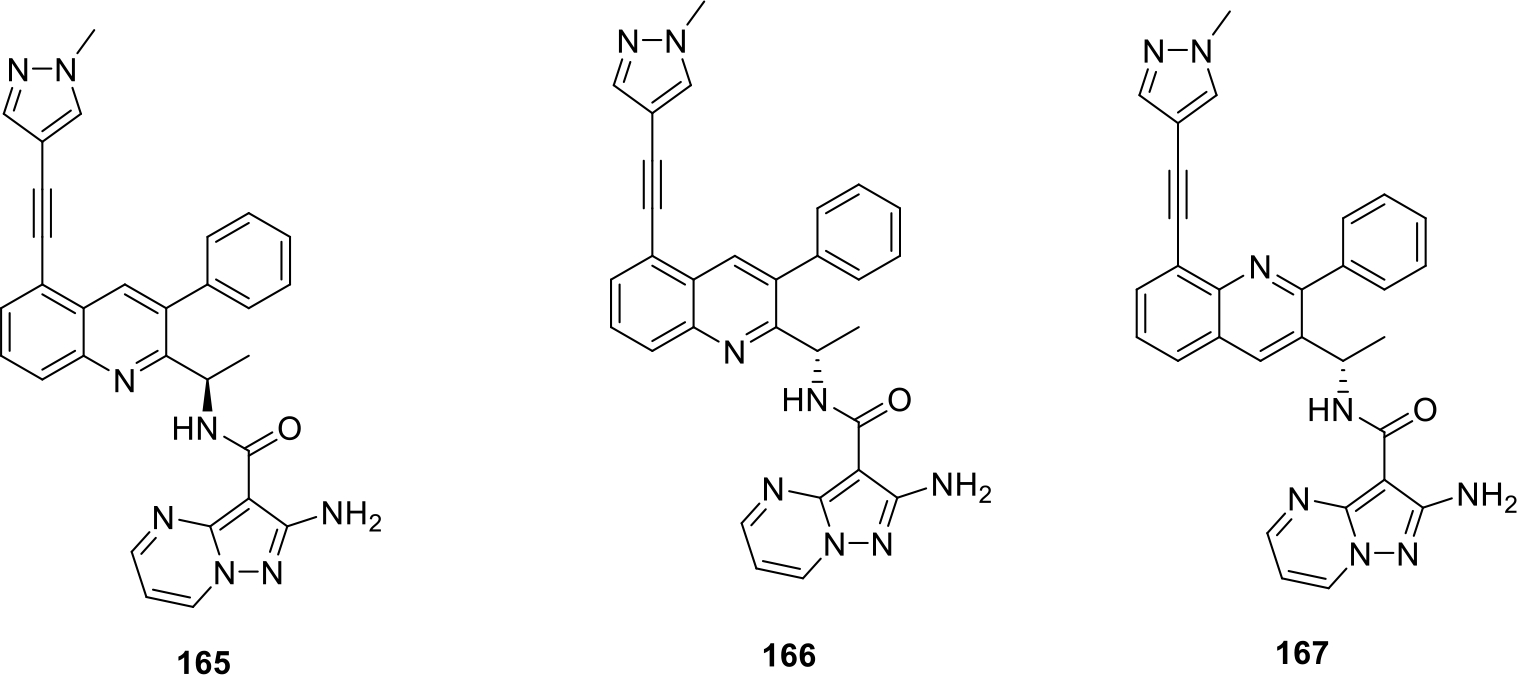
Trisubstituted bicyclic quinoline derivatives as kinase inhibitors
Chu et al patented aryl-quinoline based compounds as vasculogenic mimicry (VM) inhibitors. The results of the biological evaluation indicated that compound 168 (Figure 31) showed maximal reduction in number of cell junctions (43%) at 5 nM concentrations. The compound also inhibited the vasculogenic mimicry network in melanoma cells. Detailed mechanistic studies revealed that compound 168 reduced VM network by inhibition of mRNA expression of Notch4 intracellular domain and CDHE5. Moreover, compound 168 also decreased protein level expression of pro-Nodal, a precursor of nodal. In vivo anticancer studies were performed in mice by HCT-116 orthotopic human colon cancer xenograft model and it was concluded that 168 significantly reduced tumor volume by 49% at a dose of 50mg/kg [93].
Fig. 31.
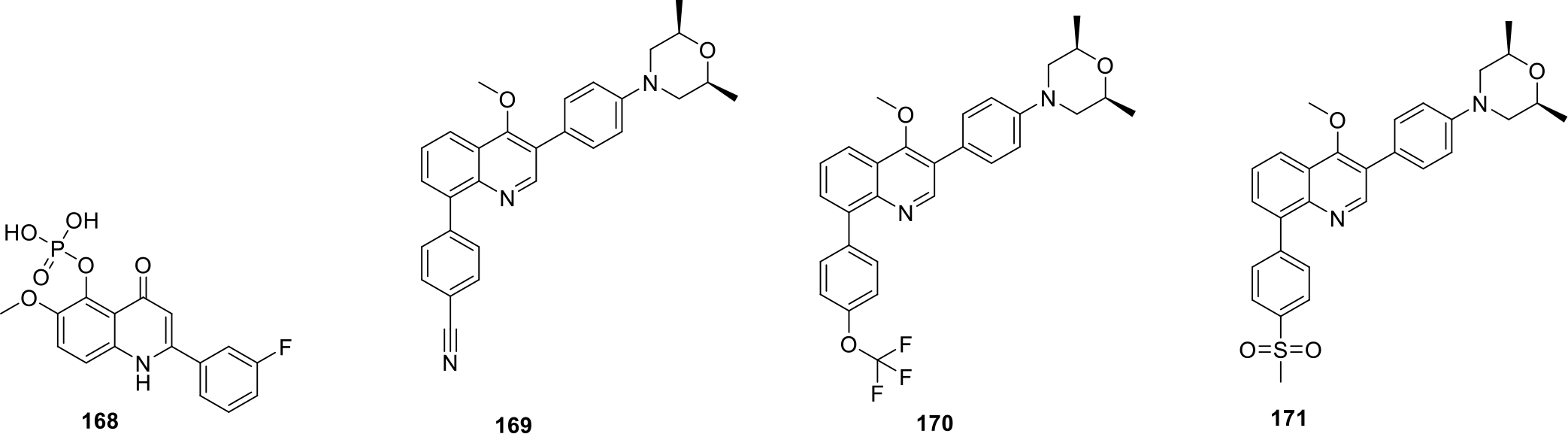
Anticancer quinolines
Wu and team patented quinoline derivatives as GPCRs-like receptors oncogene smoothened (SMO) inhibitors. The potent compounds (169–171) (as shown in Figure 31) showed IC50 values < 50 nM for hedgehog pathway inhibition [94].
Lee et al synthesized quinoline sulfonyl derivatives and evaluated them for cell growth inhibitory effects. The cellular assays were done on cancer cell lines including breast cancer cell lines MDA-MB231, MCF7 and MDA-MB468. The structure of the potent compounds 172–177 are presented in Fig. 32. Compound 172 was found to be most potent that inhibited angiogenesis and prevented spread of tumor cells to other tissues or organs in ATH490 mice engrafted with MDA-MB 231 cancer cells. Moreover, compound 172 also slowed down cell cycle progression from late G1 phase to S phase (Figure 32 and Table 2) [95].
Fig. 32.
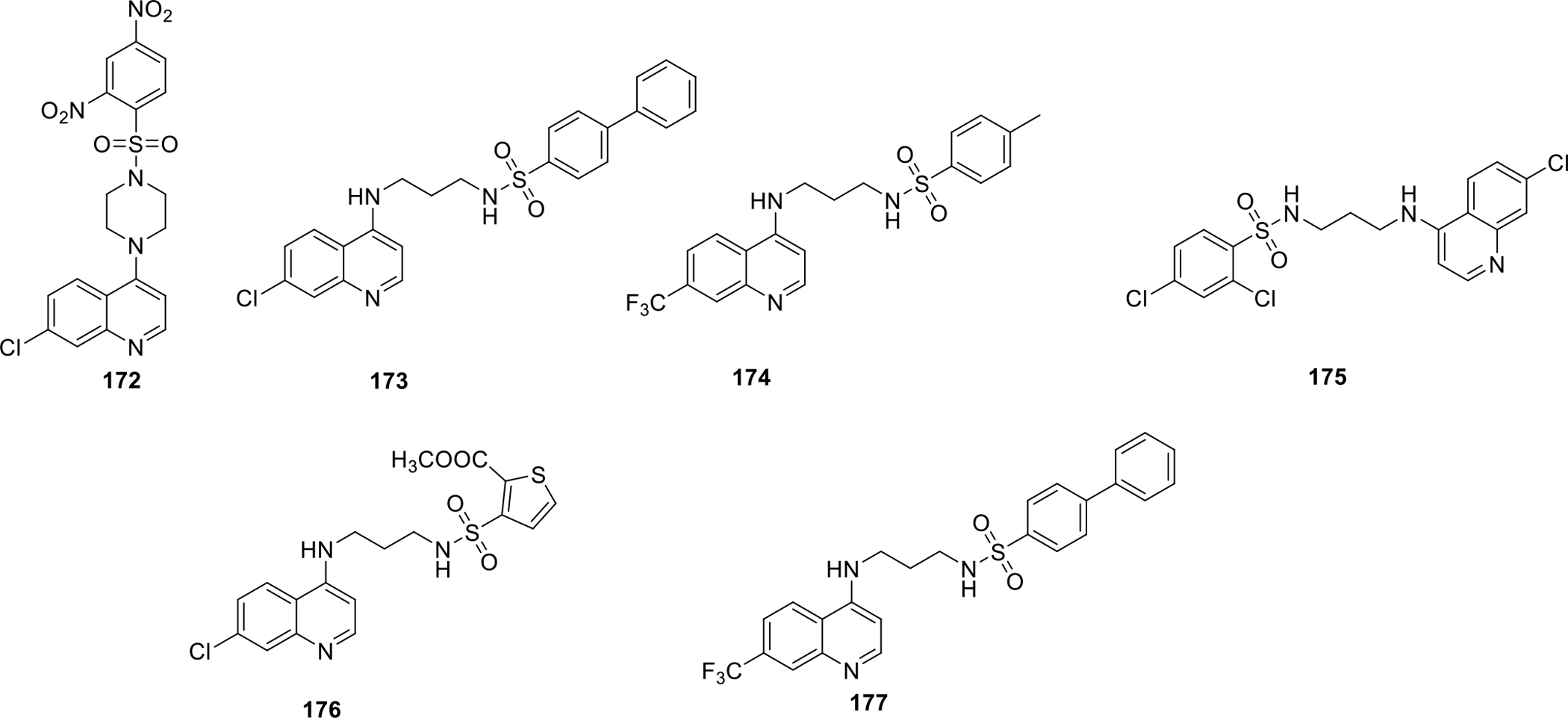
Quinoline sulfonyl derivatives
Table 2.
Activity profile of quinoline sulfonyl derivatives
| Compounds | Cancer cell lines (GI50, μM) | |||
|---|---|---|---|---|
| MDA-MB-231 | MDA-MB-468 | MCF7 | 184B5 | |
| 172 | 3.41 | 0.7 | 2.32 | 8.99 |
| 173 | 4.63 | 4.46 | 2.47 | 2.41 |
| 174 | 6.36 | 6.21 | 6.90 | 12.21 |
| 175 | 6.76 | 3.61 | 4.04 | 3.55 |
| 176 | 5.97 | 4.18 | 4.22 | 15.71 |
| 177 | 6.03 | 4.19 | 3.60 | 2.82 |
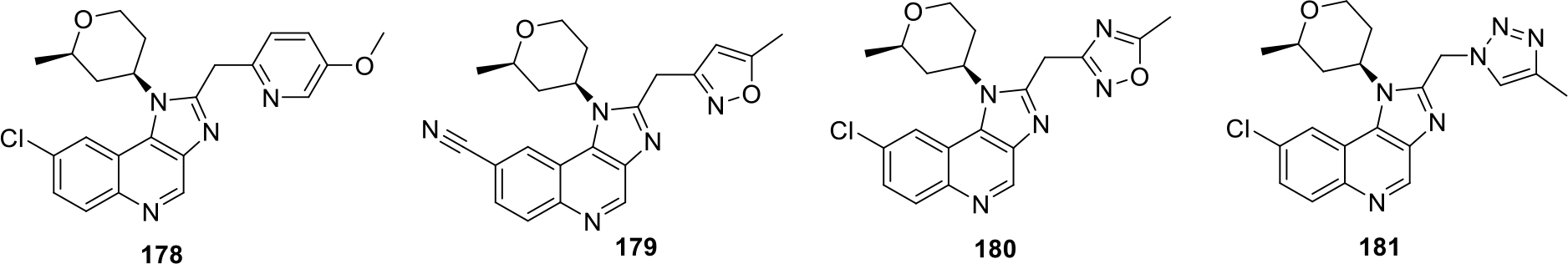
Coffman and others filed a patent for synthesizing imidazo[4,5-c]quinoline derivatives as Leucine rich receptor kinase 2 (LRRK2) inhibitors. In total 250 compounds were synthesized and among them, compounds 180 and 181 were found to be most potent with IC50 value of 2.7 nM. Other potent compound 178–181 with IC50 values in nanomolar range as shown in Figure 33 [96].
Fig. 33.
Imidazo[4,5-c]quinoline derivatives as LRRK2 inhibitors
Xie and others patented quinoline derivatives as potent anticancer compounds (Figure 34). The compounds were screened using various cancer cell lines including A549 lung cancer cell lines, DU145 prostate cancer cells, KB nasopharyngeal cancer cells and KB-VIN drug-resistant nasopharyngeal cancer cells. Compound 183 was found to be more potent than the standard drug used i.e. paclitaxel. The structure of the potent compounds (182–184) are shown in Figure 34 and the activity profile is presented in Table 3 [97].
Fig. 34.
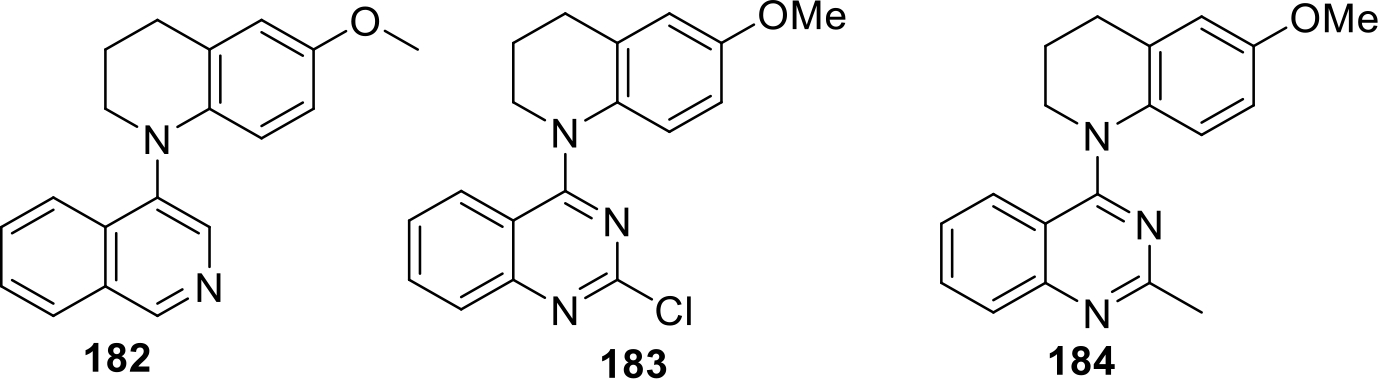
Quinoline derivatives as anticancer compounds
Table 3.
Activity profile of quinoline derivatives
| S. NO | Compound | Cancer cell lines GI50( in μg/ml) | |||
|---|---|---|---|---|---|
| A549 | DU145 | KB | KB-VIN | ||
| 1 | 182 | 0.0063 | 0.0072 | 0.0056 | 0.0082 |
| 2 | 183 | 0.0006 | 0.0005 | 0.0006 | 0.0006 |
| 3 | 184 | 0.0056 | 0.0046 | 0.0034 | 0.0055 |
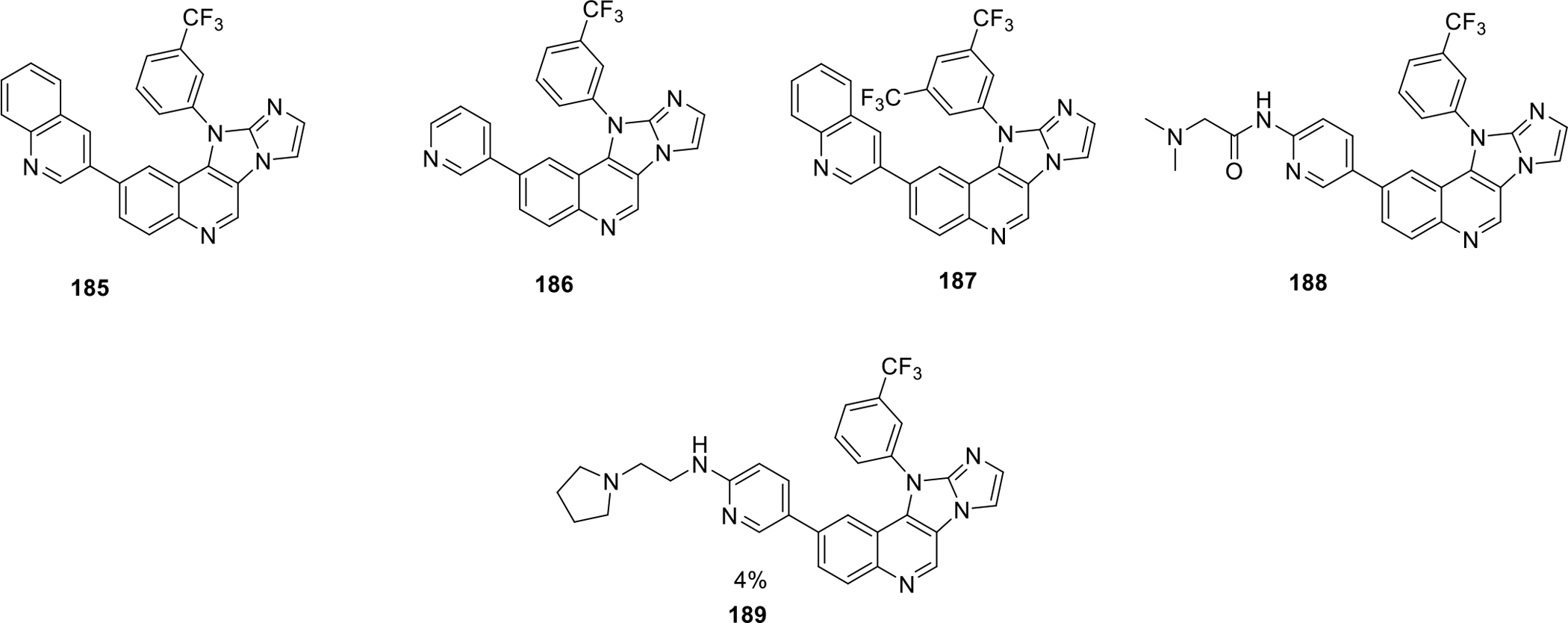
Chen et al. synthesized fused quinoline compounds for their mammalian target of rapamycin (mTOR) inhibitory activity. From the synthesized compounds, 185–189 was found to be most potent with percentage MTOR activity of −5 to 7% at 0.03 μM (Figure 35). The most potent compound 185 showed −5% MTOR activity at 0.03 μM concentration [98].
Fig. 35.
Fused quinoline compounds as mTOR inhibitory activity
Fuchss and others filed a patent for the synthesis of arylquinazoline compounds as DNA-protein kinase inhibitors. A total of approximately 479 compounds were synthesized and evaluated for both cellular as well as biochemical DNA PK phosphorylation assays. The structures of the potent compounds (190–195) that showed IC50 value less than 3 nM in the enzymatic assay are shown in Figure 36 [99].
Fig. 36.
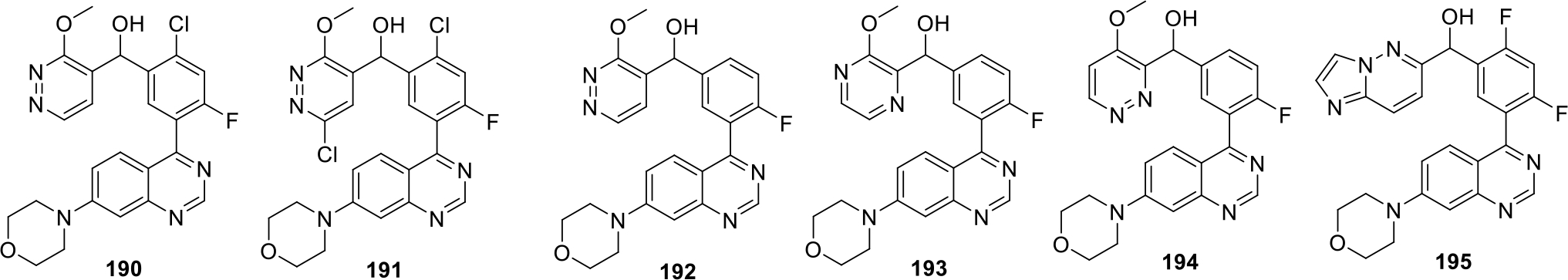
Arylquinazoline compounds as DNA-protein kinase inhibitors
Courcambeck and team filed a patent on novel quinoline derivatives. The compounds were screened against three different cancer cell lines including human malignant melanoma cell lines A375, human colon cancer cell lines HCT-116, acute monocytic leukemia cell lines MOLM-14. The compounds 196–198 were found to possess potent anticancer activity by showing cell viability percentage of less than 20%. (Figure 37) [100].
Fig. 37.
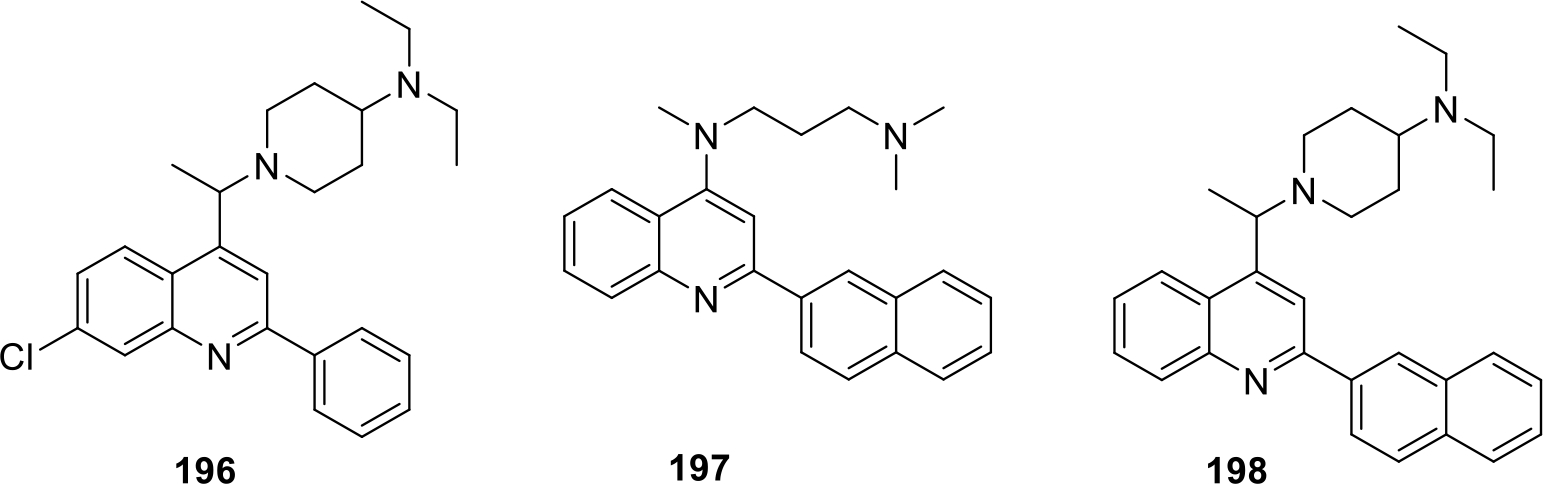
Novel quinoline derivatives
Inukai et al synthesized quinoline derivatives as Axl inhibitor. The compounds were screened by enzymatic assays and the best compounds (199–202) with IC50 values were shown in Figure 38 [101].
Fig. 38.
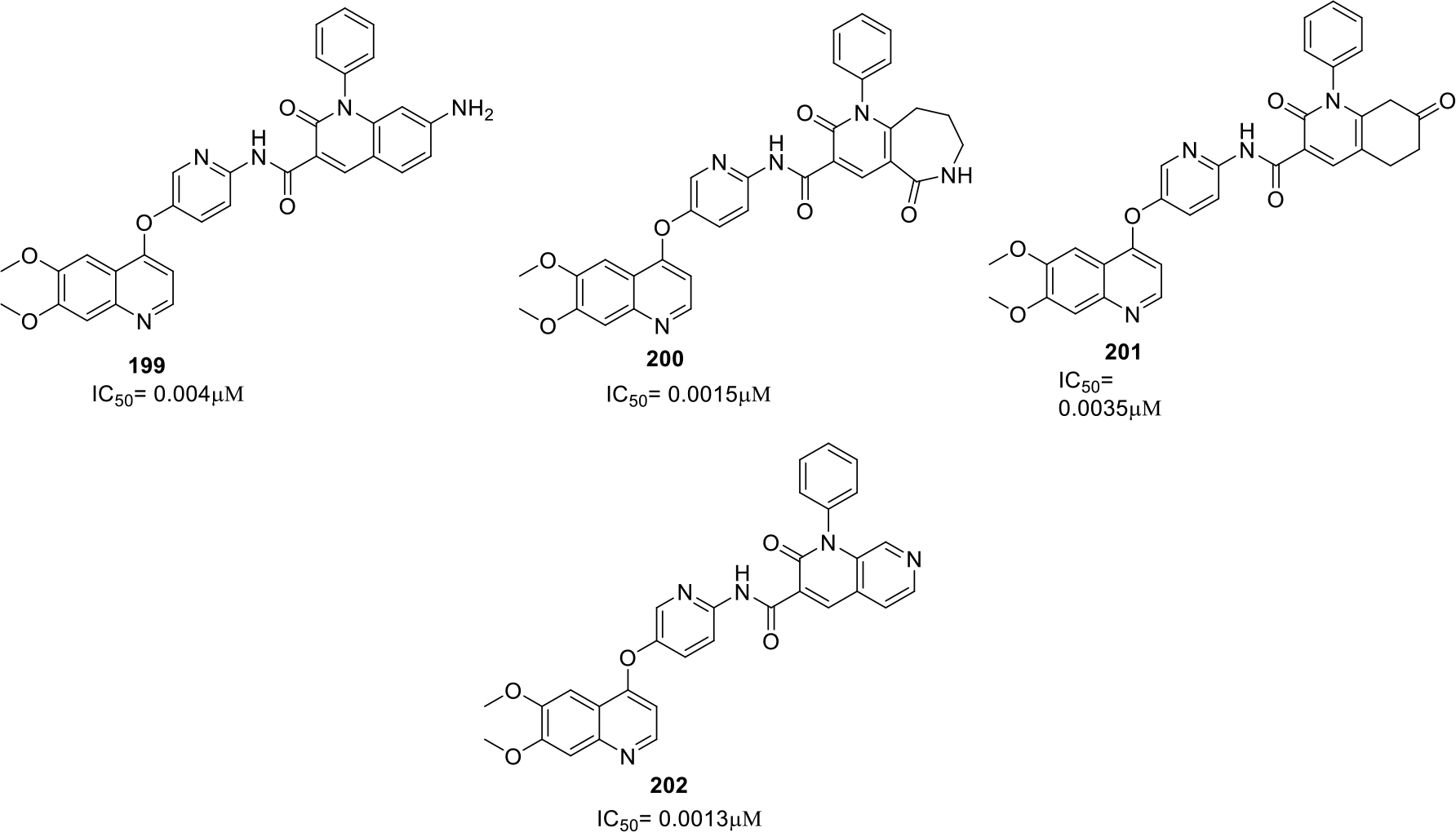
Quinoline derivatives as Axl inhibitor
Amaravadi et al., synthesized bisaminoquinolines with different linkers and evaluated them by using A375P melanoma cell lines and the potent compounds 203–206 showed logIC50 value of −6.57 to −6.39 (Figure 39). It was concluded that increasing linker length within quinolone rings in dimeric chloroquines resulted in improved cytotoxicity action as well as improved ROS production [102].
Fig. 39.
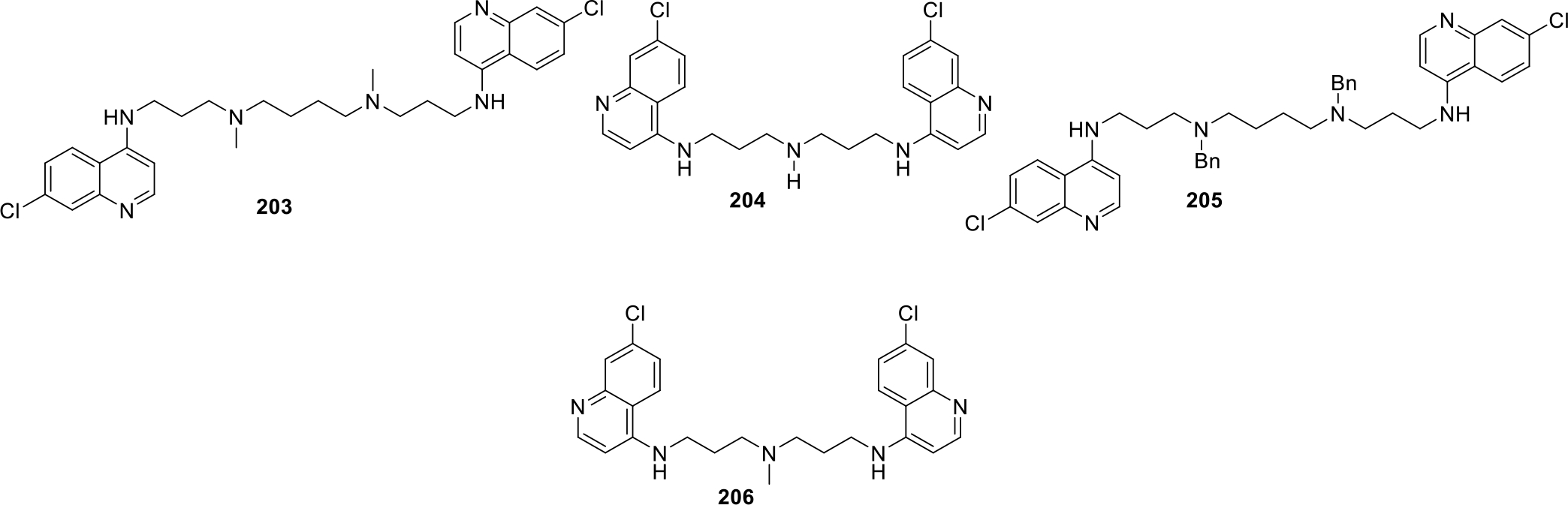
Bisaminoquinoline derivatives
4. CURRENT & FUTURE DEVELOPMENTS
Literature reports and the patents covered in this compilation clearly validates the potential of quinoline ring as a privileged scaffold for constructing antitumor chemical architectures. This review makes it evident that the preliminary pipeline is flooded with numerous quinoline based scaffolds that can be employed as leads for further structural alterations to develop cancer therapeutics.
Though the majority of the preliminary/preclinical studies covered in this review appears to be preponderant, there are some specific investigations that demonstrates utmost urgency to conduct detailed preclinical or clinical stage investigation. It is noteworthy to mention that remarkable antiproliferative effects demonstrated by a quinoline based selective HDAC6 inhibitor against multiple myeloma is one of the most optimistic and promising findings attained recently in the field of HDAC inhibitor based anticancer drug discovery. The HDAC6 inhibitor was evaluated by kinome assay to confirm its off target effects and it was found to be highly selective for the HDAC6 isoform [15]. These results are quite inspiring and can act as a spur for other researchers to initiate a program centered at the identification of selective HDAC6 inhibitors bearing different heteroaryl rings to exert anticancer effects. Likewise, some studies covered in this article that presents quinoline based topoisomerase, tubulin and kinase inhibitors [18–25, 27–29, 32–34, 36–37] endowed with striking potential to exert cytotoxic effects against various human cancer cell lines are also promising enough to garner the attention of the chemist as well as the cancer biologist. Overall, the compounds listed in this review have majorly exhibited substantial antitumor effects against colon cancer, multiple myeloma, myelogenous leukemia, lung cancer, breast cancer, colon cancer and liver cancer. In light of the evidenced preliminary/preclinical results of the scaffolds presented in section 2 and 3, it is extremely imperative to conduct detailed investigations of the most potent compounds from the aforementioned studies to establish their candidature as anticancer compounds. To accomplish this, evaluations should be conducted in varied directions beginning with the testing of their cytotoxic potential against normal cells that will perspicuously establish their profile as anticancer compounds and not just as cytotoxic compounds. Profiling of physicochemical and in vitro ADME properties, solubility and permeability assessments, PK assessments in rats should be prioritized. Attempts should majorly focus at the synthesis of new compounds via logical structural modification of compounds (covered in this review) as leads to reduce the off-target activities, amplify the potency and tune the pharmacokinetic profile to ensure their therapeutic growth as drug candidates. The structure activity relationship presented in this paper can act as a valuable tool for the medicinal chemist to conduct structural engineering attempts on leads. As such, the experience and intuition of the medicinal chemist will be of utmost importance to synthesize more efficacious derivatives. Furthermore, evaluations such as Ames test, high-dose pharmacology, PK studies (in vivo), dose linearity, toxicological and chemical screens should be performed for the optimized structures in future. Appropriate efforts should also be made to the assess the tumor growth suppression potential of the potent compounds in the animal models that mimic human conditions, such as knockouts or genetically-modified mice, to ascertain the safety of drugs for clinical stage investigations.
In addition, preliminary evaluation of the potent compounds (presented in section 2 and 3) in combination with approved anticancer agents shall be carried out to fully explore their anticancer potential. The results of the preliminary combination therapy might render us with ample scope to further evaluate the specific combinations in detailed preclinical studies. Moreover, this study will also assist the researchers to conceive the idea of constructing hybrid drugs in pursuit of attaining balanced modulation of biochemically correlated targets, thereby extracting enhanced anticancer potential. Another way to maximize the utilization of the encouraging results of the studies covered in this review would be application of the PROTAC model to construct new scaffolds as degraders of the protein. The potent compounds could be employed as ligands for the protein of interest (required to be degraded for extracting antitumor effects) and can be linked to E3 ligase ligands via appropriate linkers to generate PROTACS. Design of PROTAC is an emerging approach of drug discovery and application of PROTAC model could give new dimensions to the activity profile of the quinoline based chemical architectures covered in this compilation.
CONCLUSION
In a nut shell, quinoline ring containing anticancer agents presents enough optimism and promise in the field of drug discovery to motivate the researchers towards the continued explorations on such scaffolds. It is highly likely that adequate efforts in this direction might yield some potential cancer therapeutics in future.
Acknowledgements:
Kunal Nepali is supported by grants fromTaipei Medical University, Taiwan (Grant No. TMU107-AE1-B33).
Funding:
Jing Ping Liou is supported by the Ministry of Science and Technology, Taiwan (grant no. MOST 107–2113-M-038–001). Kunal Nepali is supported by grants from Ministry of Science and Technology, Taiwan [Grant no. MOST108–2320-B-038–010-MY2 (2–1)]
Footnotes
Conflict of Interest - The authors declare no conflict of interest
Consent for Publication: All the authors give their consent for this publication of this manuscript in the journal “Anticancer Agents in Medicinal Chemistry”
REFERENCES
- [1].Afzal O; Kumar S; Haider MR; Ali MR; Kumar R; Jaggi M; Bawa S A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem, 2015, 97, 871–910. [DOI] [PubMed] [Google Scholar]
- [2].Salat K; Moniczewski A; Librowski T Nitrogen, oxygen or sulfur containing heterocyclic compounds as analgesic drugs used as modulators of the nitroxidative stress. Mini Rev. Med. Chem, 2013, 13, 335–352. [PubMed] [Google Scholar]
- [3].Kinarivala N; Patel R; Boustany RM; Al-Ahmad A; Trippier PC Discovery of aromatic carbamates that confer neuroprotective activity by enhancing autophagy and inducing the anti-apoptotic protein B-cell lymphoma 2 (Bcl-2). J. Med. Chem, 2017, 60, 9739–9756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Makoukji J; Saadeh JF; Mansour KA; El‐Sitt S; Al Ali J; Kinarivala N; Trippier PC; Boustany RM Flupirtine derivatives as potential treatment for the neuronal ceroid lipofuscinoses. Ann. Clin. Transl. Neurol, 2018, 5, 1089–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Singh H; Singh JV; Bhagat K; Gulati HK; Sanduja M; Kumar N; Kinarivala N; Sharma S Rational approaches, design strategies, structure activity relationship and mechanistic insights for therapeutic coumarin hybrids. Biorg. Med. Chem, 2019, 27, 3477–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Singh H; Kinarivala N; Sharma S Multi-Targeting Anticancer Agents: Rational Approaches, Synthetic Routes and Structure Activity Relationship. Anti-Cancer Agents Med. Chem, 2019, 19, 842–874. [DOI] [PubMed] [Google Scholar]; b) Bhagat K; Singh JV; Pagare PP; Kumar N; Sharma A; Kaur G; Kinarivala N;Gandu S; Singh H; Sharma S; Bedi PMS Rational approaches for the design of various GABA modulators and their clinical progression. Mol. Divers, 2020, DOI: 10.1007/s11030-020-10068-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jain S; Chandra V; Jain PK; Pathak K; Pathak D; Vaidya A Comprehensive review on current developments of quinoline-based anticancer agents. Arab. J. Chem, 2019, 12, 4920–4946. [Google Scholar]
- [8].Lindner T; Loktev A; Altmann A; Giesel F; Kratochwil C; Debus J; Jäger D; Mier W; Haberkorn U Development of quinoline-based theranostic ligands for the targeting of fibroblast activation protein. J. Nucl. Med, 2018, 59, 1415–1422. [DOI] [PubMed] [Google Scholar]
- [9].Prajapati SM; Patel KD; Vekariya RH; Panchal SN; Patel HD Recent advances in the synthesis of quinolines: a review. Rsc Advances, 2014, 4, 24463–24476. [Google Scholar]
- [10].Kouzarides T Histone acetylases and deacetylases in cell proliferation. Curr. Opin. Genet. Dev, 1999, 9, 40–48. [DOI] [PubMed] [Google Scholar]
- [11].Ropero S; Esteller M The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol, 2007, 1, 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Khan O; La Thangue NB HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol. Cell Biol, 2012, 90, 85–94. [DOI] [PubMed] [Google Scholar]
- [13].Lee HY; Chang CY; Su CJ; Huang HL; Mehndiratta S; Chao YH; Hsu CM; Kumar S; Sung TY; Huang YZ 2-(Phenylsulfonyl) quinoline N-hydroxyacrylamides as potent anticancer agents inhibiting histone deacetylase. Eur. J. Med. Chem, 2016, 122, 92–101. [DOI] [PubMed] [Google Scholar]
- [14].Chen C; Hou X; Wang G; Pan W; Yang X; Zhang Y; Fang H Design, synthesis and biological evaluation of quinoline derivatives as HDAC class I inhibitors. Eur. J. Med. Chem, 2017, 133, 11–23. [DOI] [PubMed] [Google Scholar]
- [15].Lee HY; Nepali K; Huang FI; Chang CY; Lai MJ; Li YH; Huang HL; Yang CR; Liou JP (N-hydroxycarbonylbenylamino) quinolines as selective histone deacetylase 6 inhibitors suppress growth of multiple myeloma in vitro and in vivo. J. Med. Chem, 2018, 61, 905–917. [DOI] [PubMed] [Google Scholar]
- [16].Wang JC Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol, 2002, 3, 430. [DOI] [PubMed] [Google Scholar]
- [17].Järvinen T; Kononen J; Pelto-Huikko M; Isola J Expression of topoisomerase IIalpha is associated with rapid cell proliferation, aneuploidy, and c-erbB2 overexpression in breast cancer. Am. J. Pathol, 1996, 148, 2073. [PMC free article] [PubMed] [Google Scholar]
- [18].Tseng CH; Tzeng CC; Yang CL; Lu PJ; Chen HL; Li HY; Chuang YC; Yang CN; Chen YL Synthesis and antiproliferative evaluation of certain indeno [1, 2-c] quinoline derivatives. Part 2. J. Med. Chem, 2010, 53, 6164–6179. [DOI] [PubMed] [Google Scholar]
- [19].Alonso C; Fuertes M; Martín-Encinas E; Selas A; Rubiales G; Tesauro C; Knudssen BK; Palacios F Novel topoisomerase I inhibitors. Syntheses and biological evaluation of phosphorus substituted quinoline derivates with antiproliferative activity. Eur. J. Med. Chem, 2018, 149, 225–237. [DOI] [PubMed] [Google Scholar]
- [20].Kundu B; Das SK; Chowdhuri SP; Pal S; Sarkar D; Ghosh A; Mukherjee A; Bhattacharya D; Das BB; Talukdar A Discovery and Mechanistic Study of Tailor-Made Quinoline Derivatives as Topoisomerase 1 Poisons with Potent Anticancer Activity. J. Med. Chem, 2019, 62, 3428–3446. [DOI] [PubMed] [Google Scholar]
- [21].Nepali K; Kumar S; Huang HL; Kuo FC; Lee CH; Kuo CC; Yeh TK; Li YH; Chang JY; Liou JP 2-Aroylquinoline-5, 8-diones as potent anticancer agents displaying tubulin and heat shock protein 90 (HSP90) inhibition. Org. Biomol. Chem, 2016, 14, 716–723. [DOI] [PubMed] [Google Scholar]
- [22].Shobeiri N; Rashedi M; Mosaffa F; Zarghi A; Ghandadi M; Ghasemi A; Ghodsi R Synthesis and biological evaluation of quinoline analogues of flavones as potential anticancer agents and tubulin polymerization inhibitors. Eur. J. Med. Chem, 2016, 114, 14–23. [DOI] [PubMed] [Google Scholar]
- [23].Li W; Xu F; Shuai W; Sun H; Yao H; Ma C; Xu S; Yao H; Zhu Z; Yang DH Discovery of Novel Quinoline–Chalcone Derivatives as Potent Antitumor Agents with Microtubule Polymerization Inhibitory Activity. J. Med. Chem, 2018, 62, 993–1013. [DOI] [PubMed] [Google Scholar]
- [24].Li W; Shuai W; Sun H; Xu F; Bi Y; Xu J; Ma C; Yao H; Zhu Z; Xu S Design, synthesis and biological evaluation of quinoline-indole derivatives as anti-tubulin agents targeting the colchicine binding site. Eur. J. Med. Chem, 2019, 163, 428–442. [DOI] [PubMed] [Google Scholar]
- [25].Kumar L; Jha A; Sridhar G Synthesis and Biological Evaluation of Chalcone fused Quinoline Derivatives as Anticancer Agents. Chem. Data Collect, 2019, 22, 100236. [Google Scholar]
- [26].Cantley LC The phosphoinositide 3-kinase pathway. Science, 2002, 296, 1655–1657. [DOI] [PubMed] [Google Scholar]
- [27].Thangarasu P; Selvi ST; Manikandan A Unveiling novel 2-cyclopropyl-3-ethynyl-4-(4-fluorophenyl) quinolines as GPCR ligands via PI3-kinase/PAR-1 antagonism and platelet aggregation valuations; development of a new class of anticancer drugs with thrombolytic effects. Bioorg. Chem, 2018, 81, 468–480. [DOI] [PubMed] [Google Scholar]
- [28].Vennila K; Sunny D; Madhuri S; Ciattini S; Chelazzi L; Elango KP Design, synthesis, crystal structures and anticancer activity of 4-substituted quinolines to target PDK1. Bioorg. Chem, 2018, 81, 184–190. [DOI] [PubMed] [Google Scholar]
- [29].Abbas SH, El-Hafeez AAA; Shoman ME; Montano MM; Hassan HA New quinoline/chalcone hybrids as anti-cancer agents: Design, synthesis, and evaluations of cytotoxicity and PI3K inhibitory activity. Bioorg. Chem, 2019, 82, 360–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schlessinger J Cell signaling by receptor tyrosine kinases. Cell, 2000, 103, 211–225. [DOI] [PubMed] [Google Scholar]
- [31].Arora A; Scholar EM Role of tyrosine kinase inhibitors in cancer therapy. J. Pharmacol. Exp. Ther, 2015, 315, 971–979. [DOI] [PubMed] [Google Scholar]
- [32].Aly RM; Serya RA; El-Motwally AM; Esmat A; Abbas S; El Ella DAA Novel quinoline-3-carboxamides (Part 2): Design, optimization and synthesis of quinoline based scaffold as EGFR inhibitors with potent anticancer activity. Bioorg. Chem, 2017, 75, 368–392. [DOI] [PubMed] [Google Scholar]
- [33].George RF; Samir EM; Abdelhamed MN; Abdel-Aziz HA; Abbas SE Synthesis and anti-proliferative activity of some new quinoline based 4, 5-dihydropyrazoles and their thiazole hybrids as EGFR inhibitors. Bioorg. Chem, 2019, 83,186–197. [DOI] [PubMed] [Google Scholar]
- [34].Abdelsalam EA; Zaghary WA; Amin KM; Taleb NAAA; Mekawey AA; Eldehna WM, W.M.; Abdel-Aziz HA; Hammad SF Synthesis and in vitro anticancer evaluation of some fused indazoles, quinazolines and quinolines as potential EGFR inhibitors. Bioorg. Chem, 2019, 89, 102985. [DOI] [PubMed] [Google Scholar]
- [35].Maiolica A; de Medina-Redondo M; Schoof EM; Chaikuad A; Villa F; Gatti M; Jeganathan S; Lou HJ; Novy K; Hauri S Modulation of the chromatin phosphoproteome by the Haspin protein kinase. Mol. Cell Proteomics, 2014, 13, 1724–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Opoku-Temeng C; Dayal N; Sooreshjani MA; Sintim HO 3H-pyrazolo [4, 3-f] quinoline haspin kinase inhibitors and anticancer properties. Bioorg. Chem, 2018, 78, 418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Aly AA; El-Sheref EM; Bakheet ME; Mourad MA; Bräse S; Ibrahim MA; Nieger M; Garvalov BK; Dalby KN; Kaoud TS Design, synthesis and biological evaluation of fused naphthofuro [3, 2-c] quinoline-6, 7, 12-triones and pyrano [3, 2-c] quinoline-6, 7, 8, 13-tetraones derivatives as ERK inhibitors with efficacy in BRAF-mutant melanoma. Bioorg. Chem, 2019, 82, 290–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Leicht DT; Balan V; Kaplun A; Singh-Gupta V; Kaplun L; Dobson M; Tzivion G Raf kinases: function, regulation and role in human cancer. Bba.-Mol. Cell. Res, 2007, 1773, 1196–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].El-Gamal MI; Khan MA; Tarazi H; Abdel-Maksoud MS; El-Din MMG; Yoo KH; Oh C-H Design and synthesis of new RAF kinase-inhibiting antiproliferative quinoline derivatives. Part 2: Diarylurea derivatives. Eur. J. Med. Chem, 2017, 127, 413–423. [DOI] [PubMed] [Google Scholar]
- [40].Arafa RK; Hegazy GH; Piazza GA; Abadi A,Synthesis H and in vitro antiproliferative effect of novel quinoline-based potential anticancer agents. Eur. J. Med. Chem, 2013, 63, 826–832. [DOI] [PubMed] [Google Scholar]
- [41].Kuldeep S; Vikas V; Kavita Y; Vedagopuram S; Devinder K; Avinash B; Vinod K Design, regioselective synthesis and cytotoxic evaluation of 2-aminoimidazoleequinoline hybrids against cancer and primary endothelial cells. Eur. J. Med. Chem, 2014, 87, 150–158. [DOI] [PubMed] [Google Scholar]
- [42].Praveena KSS; Ramarao EVVS; Murthy NYS; Akkenapally S; Kumar CG; Kapavarapu R; Pal S Design of new hybrid template by linking quinoline, triazole and dihydroquinoline pharmacophoric groups: A greener approach to novel polyazaheterocycles as cytotoxic agents. Bioorg. Med. Chem. Lett, 2015, 25, 1057–1063. [DOI] [PubMed] [Google Scholar]
- [43].Spano V; Parrino B; Carbone A; Montalbano A; Salvador A; Brun P; Vedaldi D; Diana P; Cirrincione G; Barraja P Pyrazolo [3, 4-h] quinolines promising photosensitizing agents in the treatment of cancer. Eur. J. Med. Chem, 2015, 102, 334–351. [DOI] [PubMed] [Google Scholar]
- [44].Liberto NA; Simões JB; de Paiva Silva S; da Silva CJ; Modolo LV; de Fátima Â; Silva LM; Derita M; Zacchino S; Zuñiga OMP Quinolines: Microwave-assisted synthesis and their antifungal, anticancer and radical scavenger properties. Biorg. Med. Chem, 2017, 25, 1153–1162. [DOI] [PubMed] [Google Scholar]
- [45].Alegaon SG; Parchure P; Araujo L; Salve P; Alagawadi K; Jalalpure S; Kumbar V Quinoline-azetidinone hybrids: Synthesis and in vitro antiproliferation activity against Hep G2 and Hep 3B human cell lines. Bioorg. Med. Chem. Lett, 2017, 27, 1566–1571. [DOI] [PubMed] [Google Scholar]
- [46].Manohar CS; Manikandan A; Sridhar P; Sivakumar A; Kumar BS; Reddy SR Drug repurposing of novel quinoline acetohydrazide derivatives as potent COX-2 inhibitors and anti-cancer agents. J. Mol. Struct, 2018, 1154, 437–444. [Google Scholar]
- [47].Kasaboina S; Ramineni V; Banu S; Bandi Y; Nagarapu L; Dumala N; Grover P Iodine mediated pyrazolo-quinoline derivatives as potent anti-proliferative agents. Bioorg. Med. Chem. Lett, 2018, 28, 664–667. [DOI] [PubMed] [Google Scholar]
- [48].Ramya PS; Guntuku L; Angapelly S; Karri S; Digwal CS; Babu BN; Naidu V; Kamal A Curcumin inspired 2-chloro/phenoxy quinoline analogues: synthesis and biological evaluation as potential anticancer agents. Bioorg. Med. Chem. Lett, 2018, 28, 892–898. [DOI] [PubMed] [Google Scholar]
- [49].Li S; Hu L; Li J; Zhu J; Zeng F; Huang Q; Qiu L; Du R; Cao R Design, synthesis, structure-activity relationships and mechanism of action of new quinoline derivatives as potential antitumor agents. Eur. J. Med. Chem, 2019, 162, 666–678. [DOI] [PubMed] [Google Scholar]
- [50].Krawczyk M; Pastuch-Gawolek G; Mrozek-Wilczkiewicz A; Kuczak M; Skonieczna M; Musiol R Synthesis of 8-hydroxyquinoline glycoconjugates and preliminary assay of their β1, 4-GalT inhibitory and anti-cancer properties. Bioorg. Chem, 2019, 84, 326–338. [DOI] [PubMed] [Google Scholar]
- [51].Jafari F; Baghayi H; Lavaee P; Hadizadeh F; Soltani F; Moallemzadeh H; Mirzaei S; Aboutorabzadeh SM; Ghodsi R Design, synthesis and biological evaluation of novel benzo-and tetrahydrobenzo-[h] quinoline derivatives as potential DNA-intercalating antitumor agents. Eur. J. Med. Chem, 2019, 164, 292–303. [DOI] [PubMed] [Google Scholar]
- [52].Haiba ME; Al-Abdullah ES; Ahmed NS; Ghabbour HA; Awad HM Efficient and easy synthesis of new Benzo [h] chromene and Benzo [h] quinoline derivatives as a new class of cytotoxic agents. J. Mol. Struct, 2019, 701–711. [Google Scholar]
- [53].Ucar D; Cogle CR; Zucali JR; Ostmark B; Scott EW; Zori R; Gray BA; Moreb JS Aldehyde dehydrogenase activity as a functional marker for lung cancer. Chem. Biol. Interact, 2009, 178, 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yang S-M; Martinez NJ; Yasgar A; Danchik C; Johansson C; Wang Y; Baljinnyam B; Wang AQ; Xu X; Shah P Discovery of orally bioavailable, quinoline-based aldehyde dehydrogenase 1A1 (ALDH1A1) inhibitors with potent cellular activity. J. Med. Chem, 2018, 61, 4883–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Löffler M; Fairbanks LD; Zameitat E; Marinaki AM; Simmonds HA Pyrimidine pathways in health and disease. Trends Mol. Med, 2005, 11, 430–437. [DOI] [PubMed] [Google Scholar]
- [56].Vyas VK; Qureshi G; Oza D; Patel H; Parmar K; Patel P; Ghate MD Synthesis of 2-, 4,−6-, and/or 7-substituted quinoline derivatives as human dihydroorotate dehydrogenase (hDHODH) inhibitors and anticancer agents: 3D QSAR-assisted design. Bioorg. Med. Chem. Lett, 2019, 29, 917–922. [DOI] [PubMed] [Google Scholar]
- [57].Lolli ML; Sainas S; Pippione AC; Giorgis M; Boschi D; Dosio F Use of human dihydroorotate dehydrogenase (hDHODH) inhibitors in autoimmune diseases and new perspectives in cancer therapy. Recent pat. anti-Canc, 2018, 13, 86–105. [DOI] [PubMed] [Google Scholar]
- [58].Leonessa F; Clarke R ATP binding cassette transporters and drug resistance in breast cancer, Endocr.-Relat. Cancer, 2003, 10, 43–73. [DOI] [PubMed] [Google Scholar]
- [59].Karthikeyan C; Malla R; Ashby CR Jr; Amawi H; Abbott KL; Moore J; Chen J; Balch C; Lee C; Flannery PC Pyrimido [1 ″, 2 ″: 1, 5] pyrazolo [3, 4-b] quinolines: Novel compounds that reverse ABCG2-mediated resistance in cancer cells. Cancer Lett, 2016, 376, 118–126. [DOI] [PubMed] [Google Scholar]
- [60].Pearl LH; Prodromou C Structure and in vivo function of Hsp90. Curr. Opin. Struct. Biol, 2000, 10, 46–51. [DOI] [PubMed] [Google Scholar]
- [61].Bagatell R; Whitesell L Altered Hsp90 function in cancer: a unique therapeutic opportunity. Mol. Cancer Ther, 2004, 3, 1021–1030. [PubMed] [Google Scholar]
- [62].Malayeri SO; Abnous K; Arab A; Akaberi M; Mehri S; Zarghi A; Ghodsi R Design, synthesis and biological evaluation of 7-(aryl)-2, 3-dihydro-[1, 4] dioxino [2, 3-g] quinoline derivatives as potential Hsp90 inhibitors and anticancer agents. Biorg. Med. Chem, 2017, 25, 1294–1302. [DOI] [PubMed] [Google Scholar]
- [63].Oh E-T; Park HJ; Implications of NQO1 in cancer therapy. BMB reports, 2015, 48, 609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Scott KA; Barnes J; Whitehead RC; Stratford IJ; Nolan KA Inhibitors of NQO1: identification of compounds more potent than dicoumarol without associated off-target effects. Biochem. Pharmacol, 2011, 81, 355–363. [DOI] [PubMed] [Google Scholar]
- [65].Ling Y; Yang Q-X; Teng Y-N; Chen S; Gao W-J; Guo J; Hsu P-L; Liu Y; Morris-Natschke SL; Hung C-C Development of novel amino-quinoline-5, 8-dione derivatives as NAD (P) H: quinone oxidoreductase 1 (NQO1) inhibitors with potent antiproliferative activities. Eur. J. Med. Chem, 2018, 154, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Li K; Li Y; Zhou D; Fan Y; Guo H; Ma T; Wen J; Liu D; Zhao L Synthesis and biological evaluation of quinoline derivatives as potential anti-prostate cancer agents and Pim-1 kinase inhibitors. Biorg. Med. Chem, 2016, 24, 1889–18. [DOI] [PubMed] [Google Scholar]
- [67].Nepali K; Lin MH, Chao MW; Penga SJ; Hsu KC; Lin TE; Chen MC; Mei-Jung Lai, Pan MJ, Jing-Ping SL Liou JP Amide-tethered quinoline-resorcinol conjugates as a new class of HSP90 inhibitors suppressing the growth of prostate cancer cells. Bioorg. Chem, 2019, 91, 103119. [DOI] [PubMed] [Google Scholar]
- [68].Knight SD; Schmidt SJ Quinoline derivatives as PI3 kinase inhibitors. US8633187B2, January 21, 2014. [Google Scholar]
- [69].Kumar S; Vishwakarma R; Mundada R; Deore V; Kumar P; Sharma S Imidazo[4, 5-c]quinoline derivatives and their use in the treatment of tumors and/or inflammation. US8637670B2, January 28, 2014. [Google Scholar]
- [70].Liu JO; Shim JS; Chong CR; Bhat S Quinoline compounds as inhibitors of angiogenesis, human methionine aminopeptidase, and sirt1, and methods of treating disorders. US8729097B2, May 20, 2014. [Google Scholar]
- [71].Furet P; Porta DG; Guagnano V Quinoline carboxamide derivatives as protein tyrosine kinase inhibitors. US8815901B2, August 26, 2014. [Google Scholar]
- [72].Weissman AM; Yang Y Highly soluble pyrimido-dione-quinoline compounds and their use in the treatment of cancer. US8877765B2, November 4, 2014. [Google Scholar]
- [73].Tzeng C-C; Chen Y-L; Chen Y-W; Lu P-J 4-anilinofuro [2, 3-b] quinoline derivatives, their preparation processes, and pharmaceutical compositions comprising the same. US8952033B2, Febuary 10, 2015. [Google Scholar]
- [74].Xu H Quinoline compound composing 1, 2, 4-triazine-dione and use thereof. US8993566B2, March 31, 2015. [Google Scholar]
- [75].Fuchss T; Mederski W; Zenke F Imidazo [4, 5-c] quinolines as DNA-PK inhibitors. US9000153B2, April 7, 2015. [Google Scholar]
- [76].Kuo S-C; Teng C-M; Lee K-H; Huang L-J; Chou L-C; Chang C-S; Sun C-M; Wu T-S; Pan S-L; Way T-D 2-selenophene-4-quinolones as anticancer agents. US9023866B2, May 5, 2015. [Google Scholar]
- [77].Kuo S-C; Teng C-M; Lee K-H; Huang L-J; Chou L-C; Chang C-S; Sun C-M; Wu T-S; Pan S-L; Way T-D Hydrophilic derivatives of 2-Selenophene-4-quinolones as anticancer agents. US9023867B2, May 5, 2015. [Google Scholar]
- [78].Kuo S-C; Teng C-M; Lee K-H; Huang L-J; Chou L-C; Chang C-S; Sun C-M; Wu T-S; Pan S-L; Way T-D 2-Phenyl-4-quinolines as anticancer agents. US9029394B2, May 12, 2015. [Google Scholar]
- [79].Xi N Substituted quinoline compounds and methods of use, US9133162B2 September 15, 2015. [Google Scholar]
- [80].Schaus SE; Hansen U; Bishop JA [1, 3] dioxolo [4, 5-g][1, 2, 4] triazolo [1, 5-a] quinoline derivatives as inhibitors of the late SV40 factor (LSF) for use in treating cancer. US9175001B2, November 3, 2015. [Google Scholar]
- [81].Chan AS-C; Tang J.C.-o.; Lam K-H; Chui C-H; Kok SH-L; Chan SH; Cheung F; Gambari R; Cheng CH Method of making and administering quinoline derivatives as anti-cancer agents. US9321730B2, April 26, 2016. [Google Scholar]
- [82].Tao C; Sun X; Han H; Koroniak L; Desai N Isoquinoline, quinoline, and quinazoline derivatives as inhibitors of hedgehog signaling. US9345699B2, May 24, 2016. [Google Scholar]
- [83].Gong P; Zhao Y; Liu Y; Zhai X; Li S; Zhu W; Qin M Quinoline and cinnoline derivatives and their applications. US9382232B2, July 5, 2016 [Google Scholar]
- [84].Berdini V; Angibaud PR; Woodhead SJ; Saxty G Quinolines as FGFR kinase modulators. US9439896B2, September 13, 2016. [Google Scholar]
- [85].Gekeler V; Maier T; Zimmermann A; Hofmann H-P; Kulkarni SA; Jagtap AP; Chaure GS Substituted imidazoquinolines. US9446040B2, September 20, 2016. [Google Scholar]
- [86].Tang JC-O; Chan ASC; Lam KH; Chan SH Quinoline derivatives as anti-cancer agents. US9493419B2, November 15, 2016. [Google Scholar]
- [87].Inukai T; Takeuchi J; Yasuhiro T Quinoline derivative. US9573935B2, Febuary 21, 2017. [Google Scholar]
- [88].Gil AM; Ayuso-Gontan CG; Ruiz VP; Martin CP; Fernandez DIP; Rodriguez JAR Heterocyclic GSK-3 allosteric modulators. US9585879B2, March 7, 2017. [Google Scholar]
- [89].Xi N Substituted quinoline compounds and methods of use. US9598400B2, March 21, 2017. [Google Scholar]
- [90].Gold BI; Xie X; Srinivasan A; Wang L Compounds and methods for inhibition of AP endonuclease-1/redox factor-1 (HAPE1) activity. US9624235B2, April 18, 2017. [Google Scholar]
- [91].Scott WJ; Möwes M; Liu N; Mönning U; Bömer U Alkoxy-substituted 2, 3-dihydroimidazo [1, 2-C] quinazolines. US9675616B2, June 13, 2017. [Google Scholar]
- [92].Castro AC; Evans CA; Tremblay MR Trisubstituted bicyclic heterocyclic compounds with kinase activities and uses thereof. US9708348B2, July 18, 2017. [Google Scholar]
- [93].Chu Y-W; Chien D-S Use of aryl-quinolin derivatives as inhibitors of vasculogenic mimicry. US9717721B2, August 1, 2017. [Google Scholar]
- [94].Wu H; Chaofeng L; Lin J; Chen X; Yunhui L; Zhuowei L; Changqing W; Chen L; Chen S Quinoline derivatives as SMO inhibitors. US9938292B2, April 10, 2018. [Google Scholar]
- [95].Lee H; Solomon VR; Pundir S Quinoline sulfonyl derivatives and uses thereof. US9975852B2, May 22, 2018. [Google Scholar]
- [96].Coffman KJ; Galatsis P; Garnsey MR; Henderson JL; Kormos B.Lyn.; Kurumbail R;G; Pettersson MY; Reese MR; Stepan AF; Verhoest PR; Wager TT Imidazo[4,5-c]quinoline and Imidazo[4,5- C]naphthyridine derivatives as LLRK2 inhibitors. US10039753B2, August 7, 2018. [Google Scholar]
- [97].Xie L; Wang X; Lee K-H N-aryl unsaturated fused ring tertiary amine compounds, preparation method and anti-tumor applications thereof. EP2857393B1, September 12, 2018. [Google Scholar]
- [98].Chen PG; Yan C; Reale M; Chen M Fused quinoline compunds as PI3K, mTOR inhibitors. US10112945B2, October 30, 2018. [Google Scholar]
- [99].Fuchss T; Emde U; Buchstaller H-P; Mederski W Arylquinazolines. US10172859B2, January 8, 2019. [Google Scholar]
- [100].Sonia B; Beret A; Bassissi F; Halfon P; Courcambeck J Substituted 2, 4-diamino-quinoline derivatives for use in the treatment of proliferative diseases. US10179770B2, January 15, 2019. [Google Scholar]
- [101].Inukai T; Takeuchi J; Yasuhiro T Quinoline Derivative. US10208034B2, Febuary 19, 2019. [Google Scholar]
- [102].Amaravadi RK; Winkler F Dimeric quinacrine derivatives as autophagy inhibitors for cancer therapy. US10221140B2, March 5, 2019. [Google Scholar]