

Abstract
Troponin released from irreversibly injured myocytes is the gold standard biomarker for the rapid identification of an acute coronary syndrome. In acute myocardial infarction, necrotic cell death is characterized by sarcolemmal disruption in response to a critical level of energy depletion after more than 15 min of ischemia. Although troponin I and T are highly specific for cardiomyocyte death, high-sensitivity assays have demonstrated that measurable circulating levels of troponin are present in many normal subjects. In addition, transient as well as chronic elevations have been demonstrated in many disease states not clearly associated with myocardial ischemia. The latter observations have given rise to the clinical concept of myocardial injury. This review will summarize evidence supporting the notion that circulating troponin levels parallel the extent of myocyte apoptosis in normal ventricular remodeling and in pathophysiological conditions not associated with infarction or necrosis. It will review the evidence that myocyte apoptosis can be accelerated by diastolic strain from elevated ventricular preload and systolic strain from dyskinesis after brief episodes of ischemia too short to cause a critical level of myocyte energy depletion. We then show how chronic, low rates of myocyte apoptosis from endogenous myocyte turnover, repetitive ischemia, or repetitive elevations in left ventricular diastolic pressure can lead to significant myocyte loss in the absence of neurohormonal stimulation. Finally, we posit that the differential response to strain-induced injury in heart failure may determine whether progressive myocyte loss and heart failure with reduced ejection fraction or interstitial fibrosis and heart failure with preserved ejection fraction become the heart failure phenotype.
Keywords: apoptosis, HFpEF, HFrEF, ischemia, troponin
Because of the low rate of endogenous myocyte cell division, the loss of cardiomyocytes from disease leads to compensatory cellular hypertrophy of surviving myocytes and varying forms of myocardial fibrosis. When myocyte loss becomes extensive, it results in the development of heart failure with systolic and/or diastolic left ventricular dysfunction (1–3). This can develop slowly over time as in hypertensive cardiovascular disease or abruptly as it does following prolonged regional ischemia in acute myocardial infarction. During ischemia, the absence of perfusion and oxygen delivery leads to the depletion of glycogen and a rapid decline in myocardial ATP production (4, 5). After 15 min, irreversible myocyte injury begins as a wavefront of necrotic myocyte cell death that progresses from the subendocardium to the subepicardium (6, 7). Necrosis is characterized by sarcolemmal disruption allowing the release of multiple myocyte proteins into the extracellular space that rapidly enter the circulation at reperfusion. Many of these proteins have been used as biomarkers of myocyte cell death.
Cardiac troponins are now the most widely used clinical biomarkers to diagnose myocardial infarction. In addition to having a high specificity, they are extremely sensitive markers of cardiomyocyte cell death that can identify an evolving myocardial infarction at a very early stage (8). Although troponin and other cardiac enzymes are released by necrotic myocytes, the advent of high-sensitivity troponin assays has revealed that low levels of troponin I (TnI) and troponin T (TnT) can be detected in the circulation of many normal individuals (9). These assays have also identified an increasing number of circumstances where troponin is acutely as well as chronically elevated above the 99% upper reference limit (URL) for normal in the absence of clinical evidence of myocardial infarction (10). These isolated elevations of troponin have now been collectively characterized as reflecting “myocardial injury” (9). Troponin I elevation in the absence of infarction can exhibit an acute rise and fall pattern as it does following cardiac surgery (11), in myocardial injury after noncardiac surgery (MINS) (12) and microembolization with, as well as without, epicardial coronary artery disease (13). In addition, it can be persistently elevated in chronic disease states such as heart failure (9). Frequently, these troponin elevations are not associated with elevations of other cardiac biomarkers of myocardial necrosis such as creatinine kinase MB. Thus, it remains unclear whether these circumstances reflect an increased sensitivity of troponin to identify tiny numbers of irreversibly injured myocytes, troponin release as a consequence of senescence, and normal myocyte turnover or the selective degradation and release of troponins from otherwise viable myocytes. Despite these uncertainties, multiple clinical studies have demonstrated that even minor acute and chronic troponin elevations unassociated with myocardial infarction have an impact on long-term prognosis, are associated with increased cardiovascular event rates, and negatively impact survival (14).
This review will summarize preclinical experimental evidence to support the point of view that troponin elevation developing in the absence of necrosis typical of infarction reflects programmed myocyte death or apoptosis. We will review the experimental data that show transient apoptosis and troponin I release developing following brief “reversible ischemia” and transient myocyte diastolic strain associated with elevated left ventricular diastolic filling pressure. We will then review the evidence demonstrating that even low levels of chronic myocyte apoptosis from repetitive reversible ischemia or repetitive diastolic strain can lead to substantial myocyte loss when present on a chronic basis. Finally, we propose the hypothesis that myocyte strain is a common stimulus for apoptosis arising from both brief ischemia and preload elevation and discuss how this may differentially modulate patterns of myocardial remodeling seen in chronic heart failure with reduced (HFrEF) and preserved (HFpEF) ejection fraction.
CIRCULATING TROPONIN I CORRELATES WITH MYOCYTE APOPTOSIS IN NORMAL ADULTS AND CARDIAC REMODELING IN NEONATES IMMEDIATELY AFTER BIRTH
Postmortem studies in humans and preclinical studies have demonstrated adult myocyte turnover throughout life in the absence of disease or physiological stress. Myocyte apoptosis assessed by TUNEL staining has been identified at a low rate (0.013% of myocytes) in the normal human heart (15). This has been corroborated by similarly low rates of myocyte apoptosis in normal animals (e.g., in swine this averages 0.003% of myocytes) (16). This low rate of myocyte loss is counterbalanced by a low rate of myocyte cell division that has been estimated to average 0.45%–1.0% of cardiac myocytes per year by carbon-14 dating studies (17).
Although the rates of apoptosis at a single point in time are extremely low, the time frame of myocyte apoptosis in vivo is short and estimated to be between 6 and 24 h (18). As a result, even low rates can lead to chronic myocyte loss when the frequency of apoptosis exceeds that of myocyte cell division. The rate of myocyte death from apoptosis in otherwise healthy subjects without underlying cardiovascular disease is higher in men than in women while rates of myocyte cell division are similar (19). The impact of this sex-related difference in apoptosis over many years is significant with total myocyte numbers in the female heart remaining constant, whereas the male heart loses ∼1% of myocytes/yr (20). Despite a substantial loss of myocytes in the male heart, the reduction in anatomic left ventricular mass is attenuated because of compensatory myocyte cellular hypertrophy (19, 20). The sex-related differences in myocyte apoptosis in normal hearts also correlate with similar sex-related differences in circulating troponin I in subjects without known cardiovascular disease (21). Although there are no simultaneous comparisons of myocyte apoptosis with circulating troponin levels in normal subjects, high-sensitivity troponin I levels within the normal range are higher in men (2.4 ng/L) than in women (1.5 ng/L) (21) and these sex-related differences impact the 99th URL for identifying abnormal circulating troponin values (22, 23).
A correlation between elevated circulating troponin and myocyte apoptosis can also be found immediately after birth in neonates. This arises from physiological postnatal remodeling with involution of the right ventricle and fetal pulmonary circulation as right ventricular systolic pressure declines from systemic levels shortly after birth. Studies in normal neonates have demonstrated a systematic rise and fall in troponin I in the immediate postpartum period in the absence of severe hypoxia or other clinical circumstances associated with cardiac injury (24). In preclinical studies, this transition after birth is associated with marked elevations in myocyte apoptosis in the right ventricle and associated with a reduction in right ventricular mass as it adapts to the new low-pressure pulmonary circulation (25). Fetal measurements obtained in experimental animals demonstrate that myocyte apoptosis occurs throughout cardiac development but declines immediately before birth (26, 27). Developmental ventricular remodeling is not associated with myocyte necrosis as this would lead to an inflammatory response, reactive fibrosis, and scarring which apoptotic cell death avoids. Thus, the transient increases in circulating troponin levels immediately after birth support the view that troponin can be elevated in circumstances associated with “physiological” levels of myocyte apoptosis from normal myocyte turnover.
TROPONIN I PROTEOLYSIS FOLLOWING MYOCYTE STRAIN FROM ISCHEMIA AND PRELOAD ELEVATION
Brief ischemia (<15 min) is associated with the development of stunned myocardium that is considered to be completely reversible since contractile dysfunction normalizes within 24 h after reperfusion (28, 29). Ultrastructural studies have demonstrated an intact sarcolemma with neither electron microscopic nor light microscopic evidence typical of myocyte necrosis (7, 30). Although the precise mechanism responsible for reversible dysfunction in stunning remains unclear, prior studies have raised the possibility that it could be secondary to reversible enzymatic cleavage of troponin I. Studies by Gao et al. (31) in isolated Langendorff hearts demonstrated that brief global ischemia induced calpain-mediated proteolytic cleavage of troponin I. Preventing this with the μ-calpain inhibitor calpastatin or reducing perfusate calcium prevented troponin I degradation and improved global function after reperfusion. Although these results supported a potential role of troponin I proteolysis as a mechanism of myocardial stunning in the globally ischemic isolated rat heart model, subsequent studies including in vivo studies of regional ischemia in swine from our laboratory failed to identify troponin I proteolysis (32, 33). To resolve these discordant findings, Feng et al. (34) demonstrated that myocardial TnI degradation in the isolated Langendorff rat heart was related to elevations in left ventricular (LV) preload and stretch rather than ischemia. After ischemia, the isovolumic Langendorff rat heart preparation typically develops transient elevations in LV end-diastolic pressure exceeding 25 mmHg. Elevating diastolic filling pressure to 25 mmHg in the absence of ischemia produced TnI degradation as reflected by an increase in the 27-kDa degradation band by Western blot analysis (Fig. 1A). Inhibiting µ-calpain with calpeptin prevented TnI degradation after preload elevation and improved LV contractile function as assessed by developed pressure. In addition, preventing LV end-diastolic pressure elevation after reperfusion by venting the Langendorff heart preparation prevented TnI degradation after global ischemia (Fig. 1B). Thus, while TnI degradation impairs contractile function in the absence of necrosis or infarction, the mechanism appears to be related to myocyte stretch or strain rather than ischemia. Myocyte apoptosis or alternative pathways of cell death were not specifically evaluated in any of these early studies. Nevertheless, the fact that calpain activation is associated with caspase-3 activation supports this possibility (35). These observations may also explain the release of multiple proteolytic fragments of troponin I in response to a variety of myocardial stresses (36, 37). The subsequent sections will review more recent experimental evidence confirming myocyte apoptosis and troponin I release after similar pathophysiological stresses in vivo.
Figure 1.

The critical role of left ventricular (LV) preload elevation on troponin I proteolysis in the buffer-perfused Langendorff rat heart. A: Western blot analysis of two troponin I antibodies (C5 and 8I7) from normal control excised tissue (Ex) vs. isolated rat hearts subjected to 40 min of preload elevation to an LV end-diastolic pressure (LVEDP) of 25 mmHg. Preload elevation with normal perfusion (Nl) increased the 26-kDa degradation band. The increase in TnI degradation was blocked by inhibiting µ calpain with calpeptin. *P < 0.05 vs. excised. B: troponin I degradation after 20 min of ischemia-reperfusion in isolated rat hearts was prevented by venting the LV to prevent excessive increases in LVEDP during reperfusion of the Langendorff heart preparation. From Feng et al. (34) and reproduced with the permission of the American Heart Association.
TROPONIN I RELEASE AND MYOCYTE APOPTOSIS AFTER REVERSIBLE REGIONAL ISCHEMIA IN STUNNED MYOCARDIUM
Several clinical studies have demonstrated that low levels of troponin release can occur during increased myocardial workload in the absence of infarction. Examples include patients subjected to transient tachycardia by atrial pacing (38), exercise testing (39), and in clinically asymptomatic subjects after severe exercise or marathon runners (40–42). These observations raise the possibility that a transient imbalance between myocardial oxygen delivery and demand could, in some circumstances, lead to troponin release. To determine whether troponin I could be released after reversible supply-induced ischemia without the complexities of neurohormonal activation or increased myocardial oxygen consumption, we subjected closed-chest swine to 10-min left anterior descending (LAD) occlusions to produce stunned myocardium (43). Prior studies have demonstrated that this duration of ischemia is not associated with histological evidence of necrosis nor creatine kinase MB isoform (CK-MB) release (44). Coronary sinus sampling demonstrated a delayed but progressive increase in TnI as early as 30 min after reperfusion with the highest values found 24 h after brief ischemia (Fig. 2A). In a subset of animals, hearts were harvested 1 h after reperfusion to assess pathological evidence of myocyte injury. Although there was no light microscopic evidence of necrosis, TUNEL staining demonstrated an approximately sixfold increase in myocyte apoptosis in the postischemic region that returned to baseline in hearts stained at 24 h (Fig. 2B).
Figure 2.

Troponin I (cTnI) release and myocyte apoptosis after “reversible” regional ischemia in pigs with stunned myocardium. A: serum TnI measurements (log scale) in systemic and coronary venous samples (great cardiac vein) before and after a 10-min LAD occlusion. Regional LAD wall thickening became dyskinetic during the occlusion and gradually returned to normal after reperfusion consistent with stunned myocardium (data not shown). The 99th percentile upper reference limit (URL) for TnI is depicted by the dotted line. There was a delayed increase in TnI which exceeded the normal range within 1 h after reperfusion with levels after 24 h reaching ∼1,000 ng/L although regional function at this time was normal. B: myocyte apoptosis by TUNEL staining was transiently increased in the ischemic LAD region at 1 h but returned to normal 24 h after brief ischemia. Values are means ± SE. †P <0.05 vs. remote. Modified from Weil et al. (43) and reproduced with permission of the American College of Cardiology.
The translational relevance of these preclinical observations has been confirmed in humans. A previous study of patients undergoing angioplasty for single-vessel coronary artery disease found that even a 1-min coronary occlusion in humans was functionally significant and produced stunning with cumulative dysfunction after repeated transient occlusion of a stenotic vessel (45). After completing the protocol to investigate stunning, percutaneous coronary intervention (PCI) was performed. Although periprocedural troponin I measured at 12 and 24 h rose and met the definition of myocardial infarction in half of the patients, the independent role of the brief repetitive ischemia versus the procedure could not be ascertained. To address this, Arnadottir et al. (46) recently subjected patients without coronary artery disease to brief, transient ischemia using balloon occlusions of the coronary artery lasting less than 90 s (Fig. 3). Like the swine studies, these investigators demonstrated a delayed increase in circulating troponin using multiple high-sensitivity TnI and TnT assays. After only 90 s of ischemia (Fig. 3, bottom right), TnI rose eightfold above baseline levels and exceeded the 99% URL level (consistent with the current definition of myocardial infarction) in more than 20% of the patients. More than 60% of the patients exceeded the 99% URL for TnT. Troponin I and T levels also rose, albeit to slightly lower levels, after 30 s and 60 s of ischemia. Collectively, the preclinical and clinical studies indicate that a delayed rise and fall of troponin can occur after what was previously considered to be reversible ischemia and compatible with what clinicians would define as angina rather than myocardial infarction (47). The tissue analyses after brief ischemia in swine demonstrate that TnI release is accompanied by cell death because of transient myocyte apoptosis in the absence of light microscopic evidence of necrosis. Whether other forms of myocyte death (e.g., autophagy, necroptosis) contribute to the elevation of TnI after brief ischemia remains uncertain (48, 49).
Figure 3.

Troponin release after brief coronary occlusions of 30–90 s in patients. This slide summarizes serial troponin release up to 4 h following a brief angioplasty balloon occlusion in patients with normal coronary arteries or nonobstructive coronary artery disease. Box and whiskers plots depict troponin normalized to the baseline level before coronary occlusion. Top, left: relative troponin in subjects not receiving a balloon occlusion. The remaining panels summarize measurements up to 4 h after release of a 30-s occlusion (top, right), a 60-s occlusion (bottom, left), or a 90-s occlusion (bottom, right). Like the data following a 10-min occlusion in swine (Fig. 2), high-sensitivity (hs) troponin I assays (red bars and data points Siemens Centaur assay: green bars and data points Abbott ARCHITECT STAT assay) and troponin T (blue bars and data points Roche Elecsys 2010 assay) demonstrated a delayed rise above baseline values that exceeded the 99th URL in a significant number of patients (data not shown). Longer-term measurements beyond 4 h were not performed. These data further support the notion that troponin is released following brief ischemia of a duration compatible with angina. Values are presented as medians and interquartile ranges. From Arnadottir et al. (46), and reproduced with permission of the American Heart Association.
MYOCYTE APOPTOSIS AND TROPONIN I RELEASE FROM ACUTELY ELEVATED PRELOAD AND DIASTOLIC STRAIN
Myocardial injury and troponin elevation are frequently found in the absence of obstructive coronary disease and myocardial ischemia in clinical conditions including heart failure, acute hypertension, and advanced chronic kidney disease (9). Although left ventricular hypertrophy and elevated diastolic filling pressures in heart failure and hypertension can promote the development of stress-induced ischemia with normal coronary arteries (50), troponin elevation is frequently found at rest. In heart failure, this can arise from several mechanisms. For example, neurohormonal activation can enhance myocyte apoptosis because of chronic β-adrenergic activation (51, 52). In addition, acute preload elevation and diastolic strain can stimulate myocyte apoptosis in the absence of ischemia or neurohormonal activation. With regard to diastolic strain, Cheng et al. (53) subjected isolated papillary muscles to graded stretch in vitro to simulate the effects of elevated preload in vivo. They demonstrated that increased diastolic strain produced a progressive increase in myocyte apoptosis. Like brief ischemia in vivo, this was associated with oxidative stress as reflected by increased superoxide anion production. To determine whether transient elevations in preload could lead to myocyte apoptosis and troponin I release in vivo, we subjected closed-chest swine to a 1-h episode of pressure overload produced by elevating afterload with the α-agonist phenylephrine (54). Ischemia was circumvented since, unlike other species, swine have no vasoconstrictor response to intracoronary α-adrenergic agonists (55). This transiently elevated LV end-diastolic pressure to a level similar to what we had previously demonstrated to cause myocardial troponin I degradation in the isolated heart (34). Elevating LV end-diastolic pressure to 30 mmHg for 1 h had no effect on myocardial perfusion or coronary vasodilator reserve. Nevertheless, there was a marked reduction in global systolic function similar to myocardial stunning with the ejection fraction falling from 58% to 32% after phenylephrine was stopped and LV pressure normalized. Circulating troponin I progressively rose after restoration of preload to normal (Fig. 4A) and peaked at ∼1,400 ng/L 24 h later. TUNEL staining of hearts removed 3 h after preload elevation showed an approximately sixfold increase in myocyte apoptosis (Fig. 4B). Apoptosis normalized along with the ejection fraction after 24 h. Collectively, these results demonstrate that transient preload elevation in the absence of ischemia can produce transient myocyte apoptosis and reversible contractile dysfunction with increases in troponin I above the 99% URL in the normal heart. This mechanism of myocardial injury is independent of ischemia (54) and may be responsible for circulating troponin I elevations in heart failure and other fluid overload states.
Figure 4.
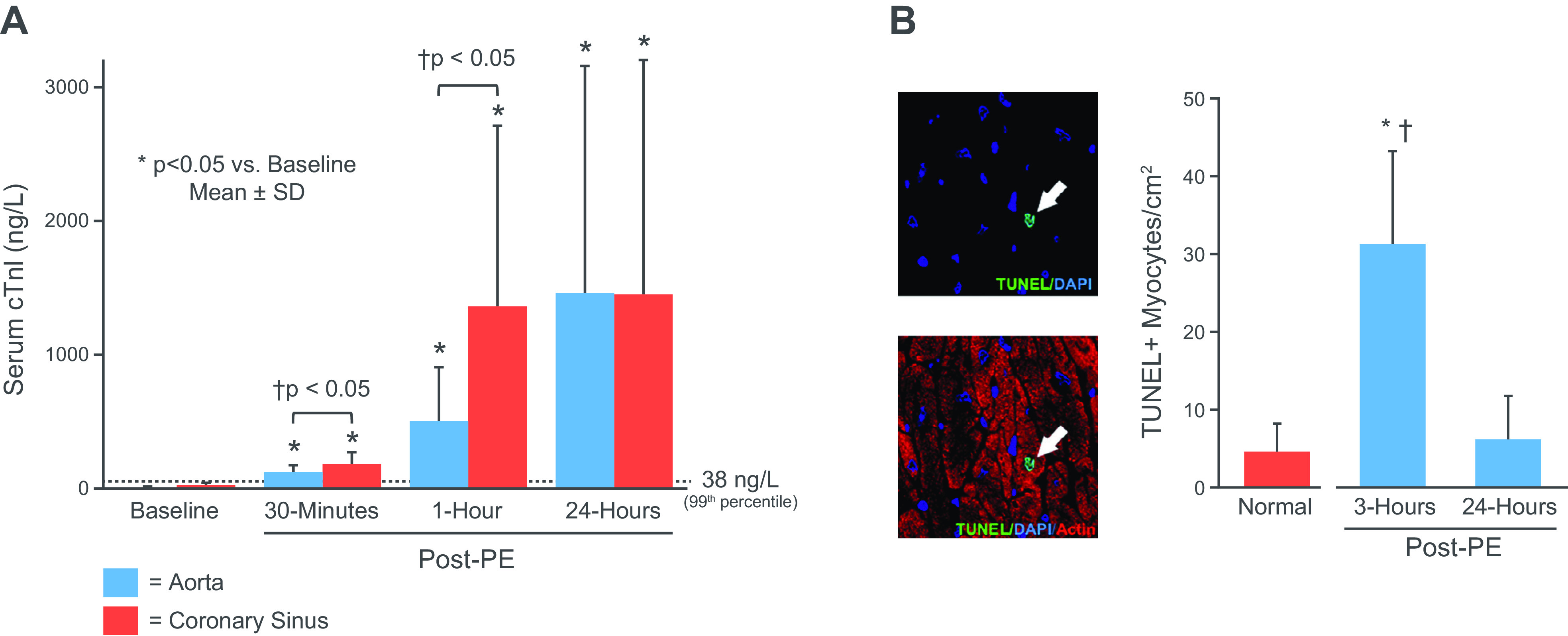
Apoptosis, troponin I release and stretch-induced stunning. A: measurements of serum TnI (aorta-blue; coronary sinus red) at baseline and selected time points following a transient 1-h elevation in left ventricular (LV) end-diastolic pressure to 35 mmHg in response to an increase in afterload with phenylephrine (PE). Troponin rose above the 99th URL within 30 min and reached ∼1,400 ng/L after 24 h. Western blot analysis of tissue at 3 h showed TnI proteolysis, light microscopy showed no evidence of necrosis or infarction, and microsphere flow measurements showed no evidence of ischemia (data not shown). B: myocyte apoptosis was increased sixfold in tissue harvested at 3 h and returned to normal levels 24 h after transient preload elevation. Values are means ± SD. *P < 0.05 vs. normal control; †P < 0.05 vs. 24-h post-PE. Adapted from Weil et al. (54) and published with permission of the American College of Cardiology.
CHRONIC APOPTOSIS LEADS TO CUMULATIVE REGIONAL MYOCYTE LOSS FROM REPETITIVE ISCHEMIA IN HIBERNATING MYOCARDIUM
Like the sex-related differences in apoptosis and differential variations in myocyte numbers over the lifespan in aging men and women (19), the impact of low levels of myocyte apoptosis occurring after repetitive “reversible” ischemia of a duration compatible with angina could become considerable since the rate of de novo myocyte formation is low. The functional impact of low rates of apoptosis was clearly demonstrated using conditional caspase activation to increase myocyte apoptosis in mice (56). In these studies, a persistent increase in myocyte apoptosis to only 0.023% (∼15-fold over normal) resulted in dilated cardiomyopathy.
Studies by our group and others have found that regional myocyte apoptosis develops in response to prolonged moderate ischemia or “short-term hibernation” (57, 58), as well as distal to chronic coronary stenosis resulting in hibernating myocardium in swine (16, 29, 59, 60). As the physiological severity of the chronic stenosis increased and coronary flow reserve during pharmacological vasodilation declined, there was regional myocyte loss and compensatory myocyte cellular hypertrophy in the hibernating region of the heart (Fig. 5, A and B). The myocyte loss reflected chronic myocyte apoptosis. In this model of chronic hibernating myocardium, regional apoptosis peaks at a rate of ∼0.02% (∼1 in 5,000 myocytes, Fig. 5C). This presumably reflected the propensity of the region to develop spontaneous demand-induced ischemia (62). Like transient coronary occlusion, transient inotropic stimulation with isoproterenol to assess contractile reserve resulted in troponin I release in association with demand-induced subendocardial ischemia (Fig. 5D; 61). Clinical studies have also demonstrated myocyte apoptosis (63) and chronic troponin elevation in humans with hibernating myocardium (64).
Figure 5.

Myocyte apoptosis and regional myocyte loss in hibernating myocardium. A: histological slides of interstitial connective tissue staining and myocyte cellular size. Chronic repetitive ischemia distal to a severe left anterior descending (LAD) stenosis resulted in hibernating myocardium. This was associated with a small increase in reticular collagen (blue) indicating interstitial fibrosis and a prominent increase in myocyte size indicating regional cellular hypertrophy. This occurred in the absence of anatomic hypertrophy and normally perfused remote myocytes were not enlarged and similar to sham controls. B: myocyte cellular hypertrophy reflected a loss of myocytes as myocyte nuclear density in the hibernating region was reduced as compared with sham controls. There was no evidence of anatomic left ventricular hypertrophy C: chronic repetitive ischemia in collateral-dependent myocardium was associated with an increase in myocyte apoptosis by fluorescence TUNEL staining (green myocyte nucleus). After 3 mo of a chronic stenosis, myocyte apoptosis increased approximately sixfold over sham controls. *P < 0.05, hibernating vs. sham. D: when swine with hibernating myocardium were subjected to β-adrenergic stimulation with isoproterenol to assess contractile reserve, heart rate increased to ∼200 beats/min for 20 min. Two hours after recovery, there was a prominent increase in troponin I. Mean TnI (first generation assay) increased from 350 ± 180 (SE) ng/L to 3,530 ± 560 ng/L (P < 0.001, n = 8). Thus, like brief supply-induced ischemia, transient increases in demand resulting in subendocardial ischemia caused troponin I release. Data in A–C adapted from Lim et al. (16) and D from Valeti et al. (61). Used with permission.
Over time, regional myocyte apoptosis rates in chronic hibernating myocardium in swine with a chronic LAD occlusion and collateral-dependent myocardium decline despite a persistent critical limitation in coronary flow reserve and stable dysfunction. The ability to prevent further apoptosis likely reflects induction of cell survival and antiapoptotic program along with intrinsic mitochondrial adaptations that reduce regional oxygen consumption and prevent a supply/demand imbalance in response to increases in external workload (58, 65–68).
SEVERE REPETITIVE REVERSIBLE ISCHEMIA, OXIDATIVE STRESS, AND FIBROSIS
Although ischemia-induced myocyte loss in chronic hibernating myocardium can occur without infarction or evidence of replacement fibrosis, there is a small increase in interstitial myocardial connective tissue staining from ∼4% of cross-sectional area in the normal heart to ∼6% in chronic hibernating myocardium. Interestingly, more severe degrees of repetitive ischemia from total coronary occlusions can lead to substantial regional fibrosis that is reversible. Dewald and colleagues (69) produced repetitive daily 15-min total coronary occlusions (resulting in dyskinesis) in mice to produce chronic repetitive stunning in the absence of evidence of infarction. The more severe ischemia in this model was associated with an acute inflammatory response that increased interstitial fibrosis within 7 days (from 5% to 20% of myocardial area via picrosirius red staining). Apoptosis and myocyte loss (myocyte nuclear density) were not reported. Interestingly, this initial inflammatory response was transient and spontaneously resolved despite continued repetitive ischemia. Furthermore, in contrast to myocardial infarction, both contractile dysfunction and fibrosis were partially reversible after cessation of ischemia.
The fibrosis from brief total coronary occlusions appears to arise from myocardial oxidative stress and reactive oxygen species. It was markedly attenuated in mice overexpressing superoxide dismutase (69). This is consistent with studies of Bolli et al. (70, 71) that demonstrated a burst of oxygen-free radicals after single as well as repetitive coronary occlusions in dogs. In contrast to mice, a shorter (5 min) duration of ischemia in dogs did not result in fibrosis but inhibiting free radical production attenuated contractile dysfunction and accelerated functional recovery. Collectively, the differences in the pathological response to repetitive ischemia in the mouse versus dog and swine models of repetitive ischemia raise the possibility that the severity of ischemia or contractile dysfunction during ischemia may be an important determinant of whether myocyte loss or interstitial fibrosis predominate. They provide several potential mechanisms through which repetitive ischemia could contribute to the development of ischemic cardiomyopathy when multivessel coronary artery disease is present (72).
REPETITIVE PRESSURE OVERLOAD LEADS TO MYOCYTE LOSS WITH THE DEVELOPMENT OF INTERSTITIAL FIBROSIS, MARKEDLY REDUCED LV COMPLIANCE AND A “HFpEF LIKE” PHENOTYPE
Although the frequency of myocyte apoptosis after elevating LV preload in vivo or subjecting papillary muscles to increased passive strain in vitro is low, the cumulative myocyte loss could become significant in the setting of repetitive episodes of stretch. This is particularly relevant to circumstances where there are transient elevations in LV end-diastolic pressure such as labile hypertension and demand-induced myocardial ischemia. Perrino et al. (73) demonstrated that the nature of systolic pressure overload was associated with marked variations in physiological and molecular cardiac remodeling. Intermittent pressure overload produced by transverse aortic constriction (iTAC) for 2 h/day in mice led to increased LV end-diastolic pressure, apoptosis, and fibrosis with a physiological phenotype consistent with diastolic dysfunction, a preserved ejection fraction, and minor anatomic hypertrophy. In this model, iTAC caused transient neurohormonal activation resulting in a downregulation of β-adrenergic signaling that could be prevented by β-blockade. Thus, the effects of iTAC on neurohormonal activation make it difficult to separate the role of transient sympathetic stimulation from stretch and elevated LV filling pressure on the responses.
We hypothesized that repetitive elevations in LV end-diastolic pressure from transient increases in afterload in swine would produce chronic apoptosis, cumulative myocyte loss, and culminate in the development of a dilated cardiomyopathy. As transient aortic constriction was not feasible in the large animal model, we subjected swine to daily infusions of the α-agonist phenylephrine to increase afterload and transiently elevate LV end-diastolic pressure to ∼30 mmHg for 2 h/day for a period of 2 wk (74). This approach avoided apoptosis from β-adrenergic stimulation yet, led to a cumulative reduction in myocyte nuclear density of ∼20% with compensatory myocyte cellular hypertrophy (Fig. 6A). Surprisingly, although there was myocyte loss and cellular hypertrophy, ejection fraction remained normal and there was concentric LV remodeling with no evidence of anatomic hypertrophy (Fig. 6B). Instead, there was a marked increase in interstitial fibrosis with a profound reduction in LV diastolic compliance (Fig. 7, A and B). In contrast to the fibrosis developing after repetitive ischemia (75), the interstitial fibrosis and reductions in diastolic distensibility were not rapidly reversible after pressure overload was stopped (76). Interestingly, once diastolic distensibility was reduced, transient pressure overload no longer caused myocardial stunning (Fig. 7C) and myocyte injury in terms of troponin I release (Fig. 7D) was markedly attenuated (74). Thus, the development of interstitial fibrosis and reductions in diastolic distensibility could in a sense be considered compensatory as they served to prevent progressive strain-induced myocyte loss and transient systolic dysfunction after acute pressure overload. The attenuation of strain-induced injury in the adapted heart may also explain the differential importance of proinflammatory macrophages and inflammatory mechanisms during acute versus sustained pressure overload (77, 78). These preclinical studies also demonstrate that substantial reductions in LV distensibility can develop without overt heart failure or abnormalities in hemodynamic parameters at rest. This may explain how some patients develop HFpEF and exercise intolerance without resting hypertension, congestive signs at rest, or structural remodeling of the heart on echocardiography. This is one of several HFpEF phenotypes and typical of patients with reduced arterial compliance and concentric inward remodeling without an increase in mass and is particularly common with aging (79, 80).
Figure 6.
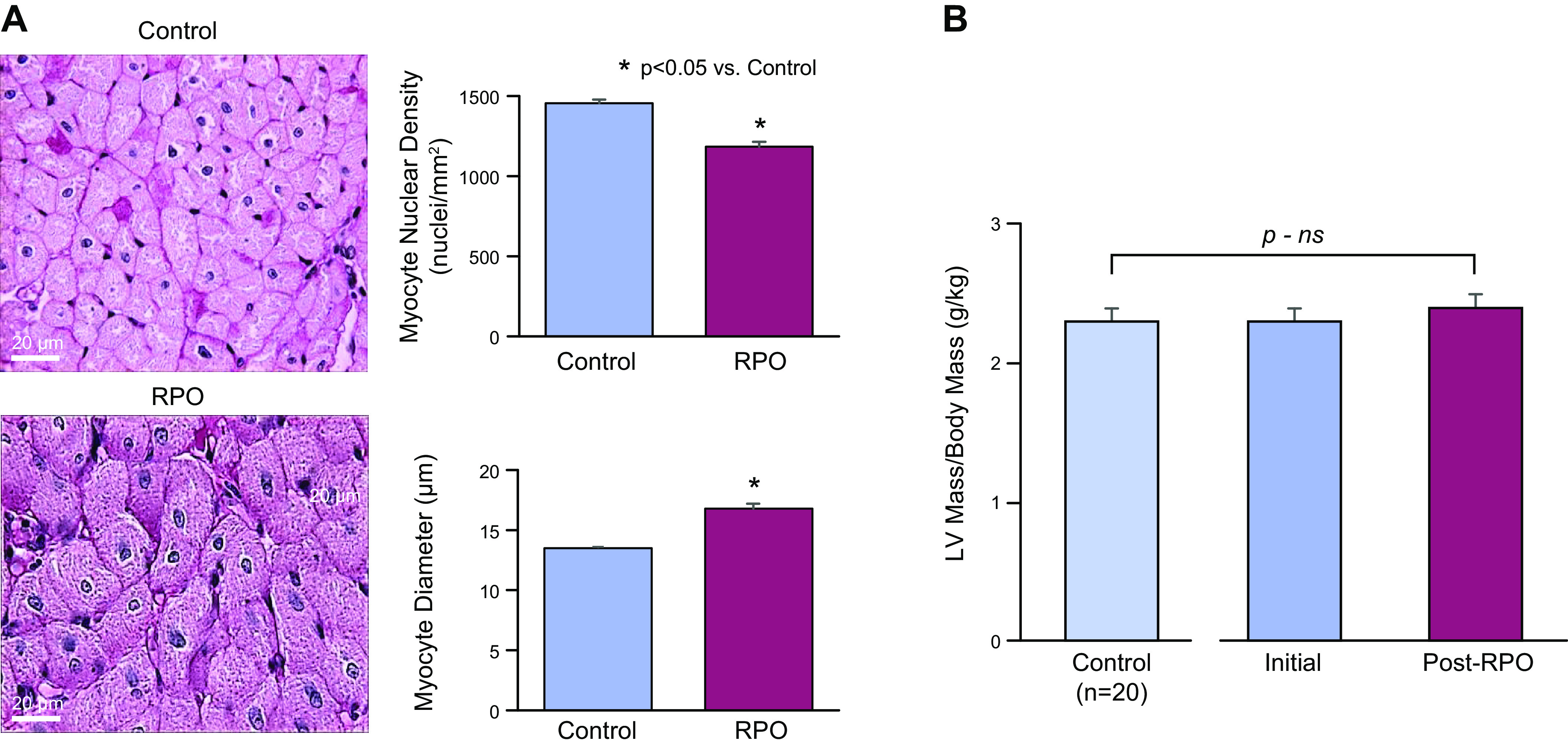
Myocyte loss and myocyte cellular hypertrophy in the absence of anatomic hypertrophy after repetitive pressure overload (RPO). A: after only 2 wk of RPO, there was an ∼20% reduction in myocyte nuclear density and a corresponding increase in myocyte diameter. *P < 0.05, RPO vs. control. B: reduction in myocyte nuclear density and cellular hypertrophy occurred in the absence of anatomic left ventricular hypertrophy as assessed by the serial left ventricular (LV) mass-to-body weight ratio from multidetector CT as well as postmortem measurements. These findings are consistent with the notion that repetitive stretch-induced apoptosis led to considerable global myocyte loss and concentric LV remodeling in this model. Adapted from Weil et al. (74) and published with permission of the American College of Cardiology.
Figure 7.
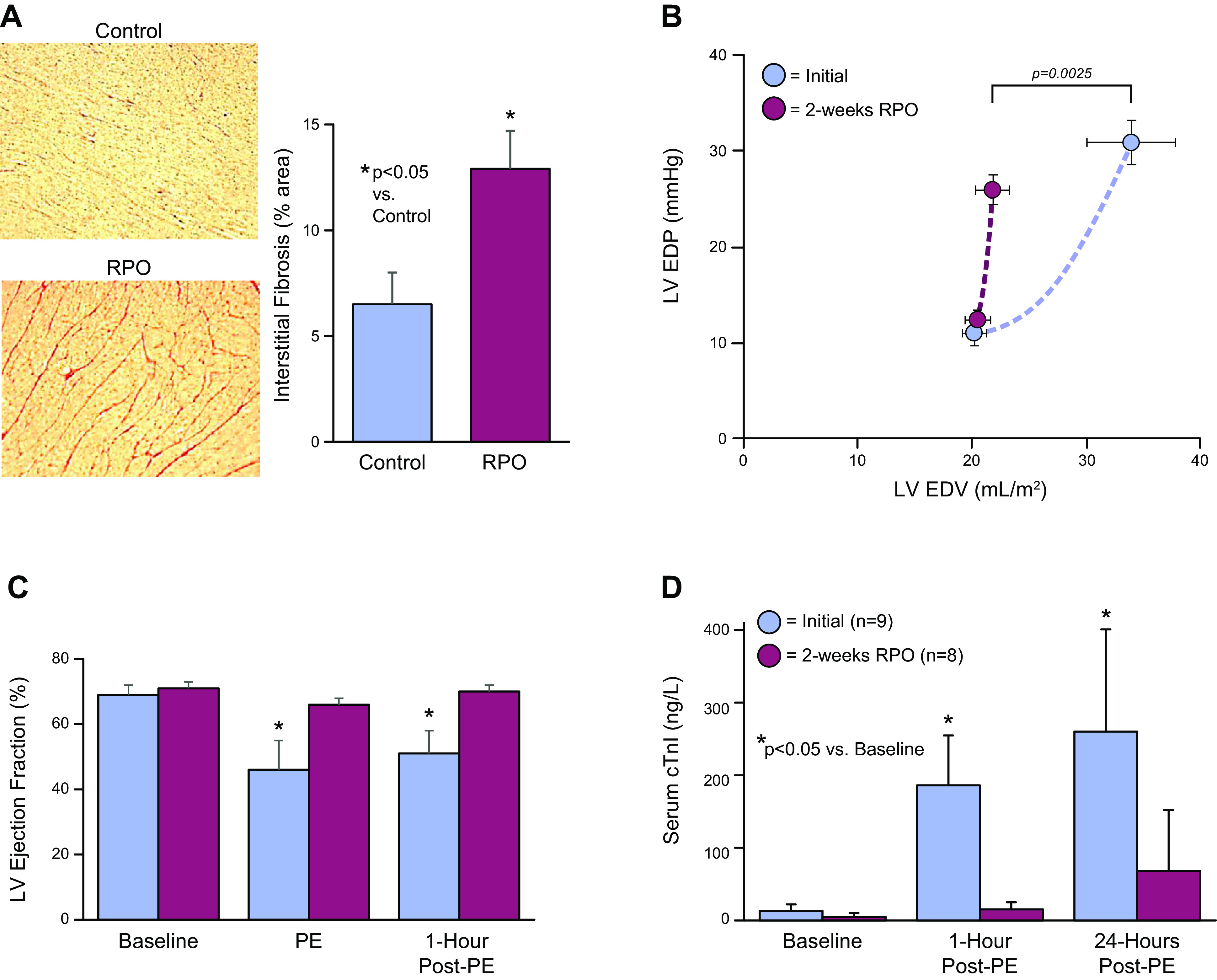
Interstitial fibrosis and reduced diastolic left ventricular (LV) compliance following 2 wk of repetitive pressure overload (RPO). Swine were subjected to a daily 2-h phenylephrine infusion to increase LV end-diastolic pressure to 30–35 mmHg. A: summarizes the increase in connective tissue found after 2 wk. Interstitial fibrosis averaged 6.5% of LV area in controls (blue) and increased to 12.5% after 2 wk of RPO (red). *P < 0.05, RPO vs. control. B: increase in connective tissue markedly reduced LV diastolic compliance after 2 wk of RPO (red) vs. initial control measurements (blue) assessed using estimates of LV end-diastolic volume (LVEDV) based on echocardiography. Directionally similar changes were obtained using LV diastolic pressure-volume relations using an admittance catheter. C: while left ventricular ejection fraction was markedly depressed after the initial episode of pressure overload (blue), it completely recovered after 24 h (*P < 0.05, PE vs. baseline). After 2 wk of RPO (red), the development of interstitial fibrosis reduced stretch-induced myocyte injury. There was no longer a deterioration in EF during or after pressure overload. In addition, as illustrated in D, the development of interstitial fibrosis with RPO caused a marked attenuation of troponin I release when LVEDP was transiently elevated after 2 wk. These data indicate that the development of reduced LV compliance limits LV filling yet prevents myocyte injury from excessive diastolic myocyte strain. *P < 0.05, PE vs. corresponding baseline. Values are means ± SE. Modified from Weil et al. (74) and republished with permission of the American College of Cardiology.
IS ACCENTUATED MYOCYTE STRAIN THE CAUSE OF TROPONIN I RELEASE IN THE ABSENCE OF PROLONGED ISCHEMIA OR INFARCTION?
The data presented thus far supports the notion that diastolic myocyte strain induces calpain activation, myocyte apoptosis, and troponin I degradation and release in the absence of ischemia. The factors responsible for inducing apoptosis and troponin release during brief ischemia of a duration compatible with angina pectoris are less clear. The development of troponin elevation after 10 min of ischemia in swine (43) and <90 s of ischemia in humans (46) is too short to produce a critical level of myocyte energy depletion. Furthermore, the delayed troponin release after reperfusion is restored is not typical of the rapid release characteristic of reperfused myocardial infarction where sarcolemmal disruption and myocardial necrosis predominate. An alternative possibility is that troponin I release arises from abnormalities in contraction that cause myofibrillar strain from elongation throughout all or part of systole during ischemia. The rapid development of contractile dysfunction that quickly culminates in dyskinesis within minutes of the onset of ischemia was originally described in experimental studies of Tenant and Wiggers (Fig. 8) and published in the American Journal of Physiology over 85 years ago (81). Tyberg and colleagues (82) subsequently showed that the strength of contraction of isolated cardiac muscle could influence deformation and lead to elongation of weakly contracting myocardium produced by hypoxia in vitro. In subsequent studies, they demonstrated that late systolic elongation developed during less severe ischemia in vivo with systolic elongation of the weak ischemic segment determined by the normally contracting adjacent myocardium (83). Indeed, other investigators have suggested that the extent of systolic dyskinesis during ischemia is a major determinant of the extent of systolic dysfunction and temporal course of recovery after reperfusion in stunned myocardium (84–86). It is also possible that abnormal myocyte strain during ischemia is amplified by disruption of the interstitial collagen matrix in stunned myocardium (30, 87). Like diastolic strain (53), brief ischemia of a duration of 2–15 min has been associated with free radical-mediated injury that contributes to contractile dysfunction in stunning and can also be attenuated by free radical scavengers (70, 88–90). Thus, the sensitivity of myocytes to strain-induced injury from dyskinesis may be the basis for many forms of myocardial injury and troponin elevations that arise in the absence of the more prolonged ischemia required for necrosis that is associated with an acute coronary syndrome.
Figure 8.

Tenant and Wiggers’ (81) original description of the effects of brief ischemia on cardiac contraction. Left: innovative optical myograph used to assess cardiac contraction (strain) during an acute occlusion of the left anterior descending coronary artery. Middle: original recordings of aortic pressure and dynamic myocardial strain throughout that cardiac cycle on a beat-to-beat basis during a coronary occlusion. Vertical bars indicate 1) end-diastole, 2) onset of ejection, 3) end-systole or aortic valve closure, and 4) onset of diastole and mitral valve opening. Systolic shortening at rest (C) transitions to lengthening or dyskinesis during brief ischemia (D). Right: left ventricular pressure below which are strain measurements demonstrating the rapid and progressive reductions in systolic shortening on a beat-to-beat basis during 125 s of ischemia. Dyskinesis and systolic lengthening consistently developed by 1 min of ischemia. Adapted from Tennant and Wiggers (81) and reproduced with permission of the American Physiological Society.
DOES A DIFFERENTIAL RESPONSE TO STRAIN-INDUCED MYOCARDIAL INJURY DETERMINE THE HEART FAILURE PHENOTYPE?
Based upon the previous discussion, it appears plausible that strain-induced myocyte injury and myocyte apoptosis may be responsible for the acute and chronic troponin elevations found in many of the pathophysiological states not associated with an acute coronary syndrome. The myocyte loss that results from chronic injury is also consonant with the prognostic ability of circulating troponin to predict the subsequent development of heart failure (91). Nevertheless, it remains unclear why fairly similar frequencies of myocyte injury and myocyte apoptosis in experimental studies of preload elevation (54) and brief regional ischemia (43) result in divergent chronic effects on myocardial fibrosis. This differential response to injury may underlie the structural phenotypes of HFpEF and HFrEF (Fig. 9) (16, 59, 72, 74). One possibility is that it simply relates to the magnitude of strain or differences between diastolic strain from preload elevation versus systolic dyssynergy from repetitive ischemia. Alternatively, there may be differences in the inflammatory response to myocyte injury from preload elevation versus brief ischemia (92, 93). Finally, it is possible that repetitive diastolic myocardial strain, while insufficient to produce ventricular hypertrophy, differentially activates myofibroblasts to stimulate interstitial collagen and myocardial matrix formation (94–96). Although maladaptive in terms of the effects of reduced LV compliance on ventricular performance, the fibrosis prevents hypertrophy as well as chronic myocyte loss from preload-induced myocyte apoptosis.
Figure 9.
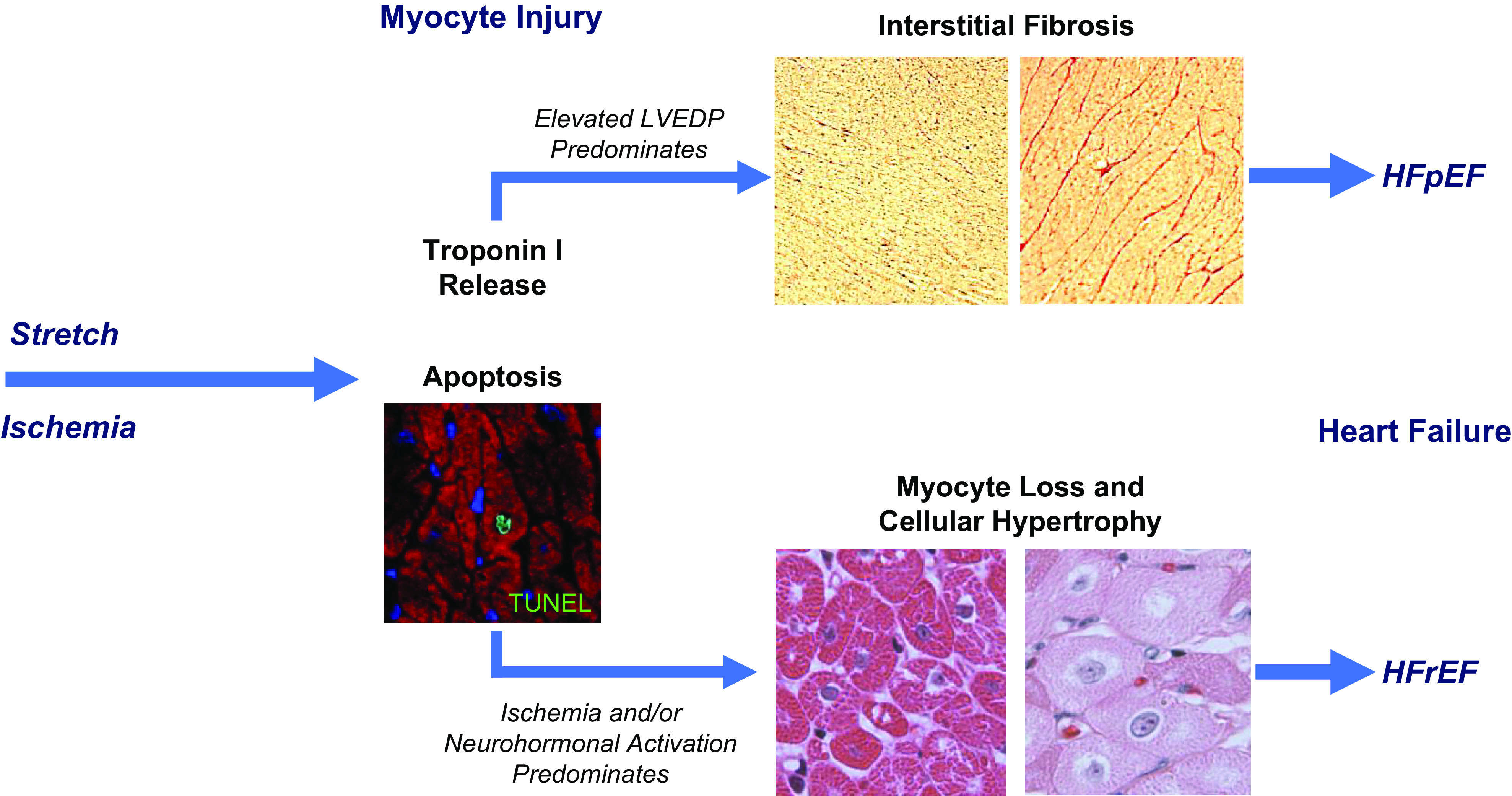
Differential effects of ischemia and preload-induced myocardial injury on the functional phenotype of heart failure. Preload elevation and ischemia both lead to troponin release in association with myocyte apoptosis by TUNEL staining. While single episodes are reversible, repetitive injury from either stress leads to myocyte loss. In the case of preload elevation, the diastolic strain also results in marked interstitial fibrosis. This may arise as a consequence of an increased inflammatory response to myocyte injury or activation of myofibroblasts in response to mechanical stimuli. The development of fibrosis prevents progressive myocyte loss but results in a physiological phenotype of HFpEF with concentric left ventricular (LV) remodeling and reduced LV compliance. Over long periods of time, labile systolic hypertension, reduced aortic compliance, and aging likely contribute to episodic preload elevation in humans. In contrast, the development of apoptosis in response to repetitive nontransmural ischemia appears to be insufficient to elicit a robust fibrotic response. As a result, myocyte loss persists and cellular hypertrophy predominates. When this impacts a large amount of viable myocardium or is associated with irreversible injury from a myocardial infarction, global LV function deteriorates and neurohormonal activation leads to further myocyte injury and loss throughout the heart. Over time, this leads to left ventricular dilatation and ischemic cardiomyopathy with a phenotype of HFrEF. HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction.
This oversimplified paradigm emphasizes the central role of transient strain-induced myocyte injury in the development of heart failure and how this can develop without proinflammatory risk factors, uncontrolled hypertension, or postinfarction left ventricular remodeling. This may explain why some patients develop ischemic cardiomyopathy and HFrEF from chronic coronary artery disease without having a large myocardial infarction. It may also explain how HFpEF develops in the subset of patients without sustained hypertension or obesity which is particularly germane to the phenotype in aging subsets. It is not meant to minimize the known importance of proinflammatory risk factors on the development of heart failure. Indeed, the same risk factors negatively impact the outcome of both HFpEF and HFrEF. Thus, it is likely that when myocyte injury from reversible ischemia or preload elevation occurs in the setting of proinflammatory risk factors such as obesity, diabetes, and endothelial dysfunction, the development and progression of both the HFrEF and HFpEF phenotypes becomes accelerated.
In conclusion, there is now a large body of evidence to indicate that troponin elevation due to myocyte injury in the absence of myocardial infarction adversely impacts prognosis. This appears to reflect a low rate of myocyte death that can lead to a significant loss of myocytes when the injury is chronic or recurrent. While preventing myocyte apoptosis from neurohormonal activation favorably impacts the prognosis of patients with HFrEF (97), the role of attenuating strain-induced myocyte injury in heart failure remains unproven and further studies are needed. Interventions to prevent strain-induced inflammation from myocyte injury and strain-induced myofibroblast activation may be particularly germane to HFpEF since implementing approaches to block neurohormonal activation have largely been unsuccessful.
GRANTS
The work was funded by Department of Veterans Affairs Grant 1IO1BX002659; National Heart, Lung, and Blood Institute Grants HL-055324 and HL-061610; National Center for Advancing Translational Sciences Grant UL1TR001412; and the Albert and Elizabeth Rekate Fund in Cardiovascular Medicine.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author.
AUTHOR CONTRIBUTIONS
J.M.C.Jr. interpreted results of experiments, prepared figures, drafted manuscript, edited and revised manuscript, and approved final version of manuscript.
ACKNOWLEDGMENTS
This work was presented in part as the 2021 Carl Wiggers Lecture at the 2021 Experimental Biology Meeting.
REFERENCES
- 1.Beltrami CA, Finato N, Rocco M, Feruglio GA, Puricelli C, Cigola E, Quaini F, Sonnenblick EH, Olivetti G, Anversa P. Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circulation 89: 151–163, 1994. doi: 10.1161/01.cir.89.1.151. [DOI] [PubMed] [Google Scholar]
- 2.Olivetti G, Quaini F, Sala R, Lagrasta C, Corradi D, Bonacina E, Gambert SR, Cigola E, Anversa P. Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J Mol Cell Cardiol 28: 2005–2016, 1996. doi: 10.1006/jmcc.1996.0193. [DOI] [PubMed] [Google Scholar]
- 3.Anversa P, Kajstura J. Myocyte cell death in the diseased heart. Circ Res 82: 1231–1233, 1998. doi: 10.1161/01.res.82.11.1231. [DOI] [PubMed] [Google Scholar]
- 4.Jennings RB, Reimer KA, Hill ML, Mayer SE. Total ischemia in dog hearts, in vitro 1. Comparison of high energy phosphate production, utilization, and depletion, and of adenine nucleotide catabolism in total ischemia in vitro vs. severe ischemia in vivo. Circ Res 49: 892–900, 1981. doi: 10.1161/01.res.49.4.892. [DOI] [PubMed] [Google Scholar]
- 5.Reimer KA, Jennings RB, Hill ML. Total ischemia in dog hearts, in vitro 2. High energy phosphate depletion and associated defects in energy metabolism, cell volume regulation, and sarcolemmal integrity. Circ Res 49: 901–911, 1981. doi: 10.1161/01.res.49.4.901. [DOI] [PubMed] [Google Scholar]
- 6.Reimer KA, Jennings RB. The “wavefront phenomenon” of myocardial ischemic cell death. II. Transmural progression of necrosis within the framework of ischemic bed size (myocardium at risk) and collateral flow. Lab Invest 40: 633–644, 1979. [PubMed] [Google Scholar]
- 7.Jennings RB, Schaper J, Hill ML, Steenbergen C Jr, Reimer KA. Effect of reperfusion late in the phase of reversible ischemic injury. Changes in cell volume, electrolytes, metabolites, and ultrastructure. Circ Res 56: 262–278, 1985. doi: 10.1161/01.res.56.2.262. [DOI] [PubMed] [Google Scholar]
- 8.Reichlin T, Hochholzer W, Bassetti S, Steuer S, Stelzig C, Hartwiger S, Biedert S, Schaub N, Buerge C, Potocki M, Noveanu M, Breidthardt T, Twerenbold R, Winkler K, Bingisser R, Mueller C. Early diagnosis of myocardial infarction with sensitive cardiac troponin assays. N Engl J Med 361: 858–867, 2009. doi: 10.1056/NEJMoa0900428. [DOI] [PubMed] [Google Scholar]
- 9.Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, White HD; ESC Scientific Document Group. Fourth universal definition of myocardial infarction. Eur Heart J 40: 237–269, 2019. doi: 10.1093/eurheartj/ehy462. [DOI] [PubMed] [Google Scholar]
- 10.Giannitsis E, Katus HA. Cardiac troponin level elevations not related to acute coronary syndromes. Nat Rev Cardiol 10: 623–634, 2013. doi: 10.1038/nrcardio.2013.129. [DOI] [PubMed] [Google Scholar]
- 11.Thielmann M, Sharma V, Al-Attar N, Bulluck H, Bisleri G, Bunge JJ, Czerny M, Ferdinandy P, Frey UH, Heusch G, Holfeld J, Kleinbongard P, Kunst G, Lang I, Lentini S, Madonna R, Meybohm P, Muneretto C, Obadia J-F, Perrino C, Prunier F, Sluijter JPG, Van Laake LW, Sousa-Uva M, Hausenloy DJ. ESC Joint Working Groups on Cardiovascular Surgery and the Cellular Biology of the Heart Position Paper: peri-operative myocardial injury and infarction in patients undergoing coronary artery bypass graft surgery. Eur Heart J 38: 2392–2407, 2017. doi: 10.1093/eurheartj/ehx383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruetzler K, Smilowitz NR, Berger JS, Devereaux PJ, Maron BA, Newby LK, De Jesus Perez V, Sessler DI, Wijeysundera DN. Diagnosis and management of patients with myocardial injury after noncardiac surgery: a scientific statement from the American Heart Association. Circulation 144: e287–e305, 2021. doi: 10.1161/CIR.0000000000001024. [DOI] [PubMed] [Google Scholar]
- 13.Kleinbongard P, Heusch G. A fresh look at coronary microembolization. Nat Rev Cardiol 19: 265–280, 2022. doi: 10.1038/s41569-021-00632-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chapman AR, Adamson PD, Shah ASV, Anand A, Strachan FE, Ferry AV, Ken Lee K, Berry C, Findlay I, Cruikshank A, Reid A, Gray A, Collinson PO, Apple F, McAllister DA, Maguire D, Fox KAA, Vallejos CA, Keerie C, Weir CJ, Newby DE, Mills NL, High SI; High-STEACS Investigators. High-sensitivity cardiac troponin and the universal definition of myocardial infarction. Circulation 141: 161–171, 2020. doi: 10.1161/CIRCULATIONAHA.119.042960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mallat Z, Fornes P, Costagliola R, Esposito B, Belmin J, Lecomte D, Tedgui A. Age and gender effects on cardiomyocyte apoptosis in the normal human heart. J Gerontol A Biol Sci Med Sci 56: M719–M723, 2001. doi: 10.1093/gerona/56.11.M719. [DOI] [PubMed] [Google Scholar]
- 16.Lim H, Fallavollita JA, Hard R, Kerr CW, Canty JM Jr.. Profound apoptosis-mediated regional myocyte loss and compensatory hypertrophy in pigs with hibernating myocardium. Circulation 100: 2380–2386, 1999. doi: 10.1161/01.CIR.100.23.2380. [DOI] [PubMed] [Google Scholar]
- 17.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Evidence for cardiomyocyte renewal in humans. Science 324: 98–102, 2009. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saraste A, Pulkki K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc Res 45: 528–537, 2000. doi: 10.1016/s0008-6363(99)00384-3. [DOI] [PubMed] [Google Scholar]
- 19.Olivetti G, Giordano G, Corradi D, Melissari M, Lagrasta C, Gambert SR, Anversa P. Gender differences and aging: effects on the human heart. J Am Coll Cardiol 26: 1068–1079, 1995. doi: 10.1016/0735-1097(95)00282-8. [DOI] [PubMed] [Google Scholar]
- 20.Olivetti G, Melissari M, Capasso JM, Anversa P. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ Res 68: 1560–1568, 1991. doi: 10.1161/01.res.68.6.1560. [DOI] [PubMed] [Google Scholar]
- 21.Kimenai DM, Shah ASV, McAllister DA, Lee KK, Tsanas A, Meex SJR, Porteous DJ, Hayward C, Campbell A, Sattar N, Mills NL, Welsh P. Sex differences in cardiac troponin I and T and the prediction of cardiovascular events in the general population. Clin Chem 67: 1351–1360, 2021. doi: 10.1093/clinchem/hvab109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhatia PM, Daniels LB. Highly sensitive cardiac troponins: the evidence behind sex-specific cutoffs. J Am Heart Assoc 9: e015272, 2020. doi: 10.1161/JAHA.119.015272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia X, Sun W, Hoogeveen RC, Nambi V, Matsushita K, Folsom AR, Heiss G, Couper DJ, Solomon SD, Boerwinkle E, Shah A, Selvin E, de Lemos JA, Ballantyne CM. High-sensitivity troponin I and incident coronary events, stroke, heart failure hospitalization, and mortality in the ARIC study. Circulation 139: 2642–2653, 2019. doi: 10.1161/CIRCULATIONAHA.118.038772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karlén J, Karlsson M, Eliasson H, Bonamy A-KE, Halvorsen CP. Cardiac Troponin T in Healthy Full-Term Infants. Pediatr Cardiol 40: 1645–1654, 2019. doi: 10.1007/s00246-019-02199-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kajstura J, Mansukhani M, Cheng W, Reiss K, Krajewski S, Reed JC, Quaini F, Sonnenblick EH, Anversa P. Programmed cell death and expression of the protooncogene bcl-2 in myocytes during postnatal maturation of the heart. Exp Cell Res 219: 110–121, 1995. doi: 10.1006/excr.1995.1211. [DOI] [PubMed] [Google Scholar]
- 26.Barbosky L, Lawrence DK, Karunamuni G, Wikenheiser JC, Doughman Y-Q, Visconti RP, Burch JBE, Watanabe M. Apoptosis in the developing mouse heart. Dev Dyn 235: 2592–2602, 2006. doi: 10.1002/dvdy.20885. [DOI] [PubMed] [Google Scholar]
- 27.Martínez-Lagunas K, Yamaguchi Y, Becker C, Geisen C, Deruiter MC, Miura M, Fleischmann BK, Hesse M. In vivo detection of programmed cell death during mouse heart development. Cell Death Differ 27: 1398–1414, 2020. doi: 10.1038/s41418-019-0426-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Canty JM Jr, Suzuki G. Myocardial perfusion and contraction in acute ischemia and chronic ischemic heart disease. J Mol Cell Cardiol 52: 822–831, 2012. doi: 10.1016/j.yjmcc.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heusch G. Myocardial stunning and hibernation revisited. Nat Rev Cardiol 18: 522–536, 2021. doi: 10.1038/s41569-021-00506-7. [DOI] [PubMed] [Google Scholar]
- 30.Zhao MJ, Zhang H, Robinson TF, Factor SM, Sonnenblick EH, Eng C. Profound structural alterations of the extracellular collagen matrix in postischemic dysfunctional (“stunned”) but viable myocardium. J Am Coll Cardiol 10: 1322–1334, 1987. doi: 10.1016/S0735-1097(87)80137-7. [DOI] [PubMed] [Google Scholar]
- 31.Gao WD, Atar D, Liu Y, Perez NG, Murphy AM, Marban E. Role of troponin I proteolysis in the pathogenesis of stunned myocardium. Circ Res 80: 393–399, 1997. [PubMed] [Google Scholar]
- 32.Thomas SA, Fallavollita JA, Lee TC, Feng J, Canty JM Jr.. Absence of troponin I degradation or altered sarcoplasmic reticulum uptake protein expression after reversible ischemia in swine. Circ Res 85: 446–456, 1999. doi: 10.1161/01.res.85.5.446. [DOI] [PubMed] [Google Scholar]
- 33.Marston SB, Redwood CS. Modulation of thin filament activation by breakdown or isoform switching of thin filament proteins: physiological and pathological implications. Circ Res 93: 1170–1178, 2003. doi: 10.1161/01.RES.0000105088.06696.17. [DOI] [PubMed] [Google Scholar]
- 34.Feng J, Schaus BJ, Fallavollita JA, Lee TC, Canty JM. Jr.. Preload induces troponin I degradation independently of myocardial ischemia. Circulation 103: 2035–2037, 2001. doi: 10.1161/01.CIR.103.16.2035. [DOI] [PubMed] [Google Scholar]
- 35.Smith MA, Schnellmann RG. Calpains, mitochondria, and apoptosis. Cardiovasc Res 96: 32–37, 2012. doi: 10.1093/cvr/cvs163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Labugger R, Organ L, Collier C, Atar D, Van Eyk JE. Extensive troponin I and T modification detected in serum from patients with acute myocardial infarction. Circulation 102: 1221–1226, 2000. doi: 10.1161/01.cir.102.11.1221. [DOI] [PubMed] [Google Scholar]
- 37.McDonough JL, Labugger R, Pickett W, Tse MY, MacKenzie S, Pang SC, Atar D, Ropchan G, Van Eyk JE. Cardiac troponin I is modified in the myocardium of bypass patients. Circulation 103: 58–64, 2001. doi: 10.1161/01.cir.103.1.58. [DOI] [PubMed] [Google Scholar]
- 38.Turer AT, Addo TA, Martin JL, Sabatine MS, Lewis GD, Gerszten RE, Keeley EC, Cigarroa JE, Lange RA, Hillis LD, de Lemos JA. Myocardial ischemia induced by rapid atrial pacing causes troponin T release detectable by a highly sensitive assay: insights from a coronary sinus sampling study. J Am Coll Cardiol 57: 2398–2405, 2011. doi: 10.1016/j.jacc.2010.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sabatine MS, Morrow DA, de Lemos JA, Jarolim P, Braunwald E. Detection of acute changes in circulating troponin in the setting of transient stress test-induced myocardial ischaemia using an ultrasensitive assay: results from TIMI 35. Eur Heart J 30: 162–169, 2009. doi: 10.1093/eurheartj/ehn504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siegel AJ, Sholar M, Yang J, Dhanak E, Lewandrowski KB. Elevated serum cardiac markers in asymptomatic marathon runners after competition: is the myocardium stunned? Cardiology 88: 487–491, 1997. doi: 10.1159/000177396. [DOI] [PubMed] [Google Scholar]
- 41.Shave R, Baggish A, George K, Wood M, Scharhag J, Whyte G, Gaze D, Thompson PD. Exercise-induced cardiac troponin elevation: evidence, mechanisms, and implications. J Am Coll Cardiol 56: 169–176, 2010. doi: 10.1016/j.jacc.2010.03.037. [DOI] [PubMed] [Google Scholar]
- 42.Balmain BN, Sabapathy S, Yamada A, Shiino K, Chan J, Haseler LJ, Kavanagh JJ, Morris NR, Stewart GM. Cardiac perturbations after high-intensity exercise are attenuated in middle-aged compared with young endurance athletes: diminished stress or depleted stimuli? Am J Physiol Heart Circ Physiol 320: H159–H168, 2021. doi: 10.1152/ajpheart.00427.2020. [DOI] [PubMed] [Google Scholar]
- 43.Weil BR, Young RF, Shen X, Suzuki G, Qu J, Malhotra S, Canty JM Jr.. Brief myocardial ischemia produces cardiac troponin I release and focal myocyte apoptosis in the absence of pathological infarction in swine. JACC Basic Transl Sci 2: 105–114, 2017. doi: 10.1016/j.jacbts.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishikawa Y, Saffitz JE, Mealman TL, Grace AM, Roberts R. Reversible myocardial ischemic injury is not associated with increased creatine kinase activity in plasma. Clin Chem 43: 467–475, 1997. doi: 10.1093/clinchem/43.3.467. [DOI] [PubMed] [Google Scholar]
- 45.Hoole SP, Heck PM, White PA, Read PA, Khan SN, West NEJ, O'Sullivan M, Dutka DP. Stunning and cumulative left ventricular dysfunction occurs late after coronary balloon occlusion in humans. JACC Cardiovasc Interv 3: 412–418, 2010. doi: 10.1016/j.jcin.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 46.Árnadóttir Á, Pedersen S, Bo Hasselbalch R, Goetze JP, Friis-Hansen LJ, Bloch-Münster A-M, Skov Jensen J, Bundgaard H, Iversen K. Temporal release of high-sensitivity cardiac troponin T and I and copeptin after brief induced coronary artery balloon occlusion in humans. Circulation 143: 1095–1104, 2021. doi: 10.1161/CIRCULATIONAHA.120.046574. [DOI] [PubMed] [Google Scholar]
- 47.deFilippi CR, Mills NL. Rapid cardiac troponin release after transient ischemia. Circulation 143: 1105–1108, 2021. doi: 10.1161/CIRCULATIONAHA.120.052649. [DOI] [PubMed] [Google Scholar]
- 48.Amgalan D, Pekson R, Kitsis RN. Troponin release following brief myocardial ischemia: apoptosis versus necrosis. JACC Basic Transl Sci 2: 118–121, 2017. doi: 10.1016/j.jacbts.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev 99: 1765–1817, 2019. doi: 10.1152/physrev.00022.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heusch G. Coronary blood flow in heart failure: cause, consequence and bystander. Basic Res Cardiol 117: 1, 2022. doi: 10.1007/s00395-022-00909-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P. Apoptosis in the failing human heart. N Engl J Med 336: 1131–1141, 1997. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 52.Shizukuda Y, Buttrick PM, Geenen DL, Borczuk AC, Kitsis RN, Sonnenblick EH. beta-adrenergic stimulation causes cardiocyte apoptosis: influence of tachycardia and hypertrophy. Am J Physiol Heart Circ Physiol 275: H961–H968, 1998. doi: 10.1152/ajpheart.1998.275.3.h961. [DOI] [PubMed] [Google Scholar]
- 53.Cheng W, Li B, Kajstura J, Li P, Wolin MS, Sonnenblick EH, Hintze TH, Olivetti G, Anversa P. Stretch-induced programmed myocyte cell death. J Clin Invest 96: 2247–2259, 1995. doi: 10.1172/JCI118280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weil BR, Suzuki G, Young RF, Iyer V, Canty JM Jr.. Troponin release and reversible left ventricular dysfunction after transient pressure overload. J Am Coll Cardiol 71: 2906–2916, 2018. doi: 10.1016/j.jacc.2018.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schulz R, Oudiz RJ, Guth BD, Heusch G. Minimal alpha 1- and alpha 2-adrenoceptor-mediated coronary vasoconstriction in the anaesthetized swine. Naunyn Schmiedebergs Arch Pharmacol 342: 422–428, 1990. doi: 10.1007/BF00169459. [DOI] [PubMed] [Google Scholar]
- 56.Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest 111: 1497–1504, 2003. doi: 10.1172/JCI17664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen C, Ma L, Linfert DR, Lai T, Fallon JT, Gillam LD, Waters DD, Tsongalis GJ. Myocardial cell death and apoptosis in hibernating myocardium. J Am Coll Cardiol 30: 1407–1412, 1997. doi: 10.1016/S0735-1097(97)00309-4. [DOI] [PubMed] [Google Scholar]
- 58.Depre C, Kim S-J, John AS, Huang Y, Rimoldi OE, Pepper JR, Dreyfus GD, Gaussin V, Pennell DJ, Vatner DE, Camici PG, Vatner SF. Program of cell survival underlying human and experimental hibernating myocardium. Circ Res 95: 433–440, 2004. doi: 10.1161/01.RES.0000138301.42713.18. [DOI] [PubMed] [Google Scholar]
- 59.Fallavollita JA, Perry BJ, Canty JM Jr.. 18F-2-deoxyglucose deposition and regional flow in pigs with chronically dysfunctional myocardium: Evidence for transmural variations in chronic hibernating myocardium. Circulation 95: 1900–1909, 1997. doi: 10.1161/01.cir.95.7.1900. [DOI] [PubMed] [Google Scholar]
- 60.Fallavollita JA, Canty JM Jr.. Differential 18F-2-deoxyglucose uptake in viable dysfunctional myocardium with normal resting perfusion: Evidence for chronic stunning in pigs. Circulation 99: 2798–2805, 1999. doi: 10.1161/01.cir.99.21.2798. [DOI] [PubMed] [Google Scholar]
- 61.Valeti U, Fallavollita JA, Canty JM Jr.. Assessing contractile reserve with β-adrenergic stimulation selectively releases troponin I in hibernating myocardium (Abstract). Circulation 104: Suppl. II–673, 2001. [Google Scholar]
- 62.Fallavollita JA, Lim H, Canty JM Jr.. Myocyte apoptosis and reduced SR gene expression precede the transition from chronically stunned to hibernating myocardium. J Mol Cell Cardiol 33: 1937–1944, 2001. doi: 10.1006/jmcc.2001.1457. [DOI] [PubMed] [Google Scholar]
- 63.Elsässer A, Schlepper M, Klövekorn WP, Cai WJ, Zimmermann R, Müller KD, Strasser R, Kostin S, Gagel C, Münkel B, Schaper W, Schaper J. Hibernating myocardium: an incomplete adaptation to ischemia. Circulation 96: 2920–2931, 1997. doi: 10.1161/01.cir.96.9.2920. [DOI] [PubMed] [Google Scholar]
- 64.Zelt JGE, Liu PP, Erthal F, deKemp RA, Wells G, O'Meara E, Garrard L, Beanlands RSB, Mielniczuk LM. N-terminal pro B-type natriuretic peptide and high-sensitivity cardiac troponin T levels are related to the extent of hibernating myocardium in patients with ischemic heart failure. Can J Cardiol 33: 1478–1488, 2017. doi: 10.1016/j.cjca.2017.06.012. [DOI] [PubMed] [Google Scholar]
- 65.Depre C, Tomlinson JE, Kudej RK, Gaussin V, Thompson E, Kim SJ, Vatner DE, Topper JN, Vatner SF. Gene program for cardiac cell survival induced by transient ischemia in conscious pigs. Proc Natl Acad Sci USA 98: 9336–9341, 2001. doi: 10.1073/pnas.171297498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fallavollita JA, Malm BJ, Canty JM Jr.. Hibernating myocardium retains metabolic and contractile reserve despite regional reductions in flow, function, and oxygen consumption at rest. Circ Res 92: 48–55, 2003. doi: 10.1161/01.RES.0000049104.57549.03. [DOI] [PubMed] [Google Scholar]
- 67.Hu Q, Suzuki G, Young RF, Page BJ, Fallavollita JA, Canty JM Jr.. Reductions in mitochondrial O(2) consumption and preservation of high-energy phosphate levels after simulated ischemia in chronic hibernating myocardium. Am J Physiol Heart Circ Physiol 297: H223–H232, 2009. doi: 10.1152/ajpheart.00992.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Page BJ, Young RF, Suzuki G, Fallavollita JA, Canty JM Jr.. The physiological significance of a coronary stenosis differentially affects contractility and mitochondrial function in viable chronically dysfunctional myocardium. Basic Res Cardiol 108: 354, 2013. doi: 10.1007/s00395-013-0354-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dewald O, Frangogiannis NG, Zoerlein M, Duerr GD, Klemm C, Knuefermann P, Taffet G, Michael LH, Crapo JD, Welz A, Entman ML. Development of murine ischemic cardiomyopathy is associated with a transient inflammatory reaction and depends on reactive oxygen species. Proc Natl Acad Sci USA 100: 2700–2705, 2003. doi: 10.1073/pnas.0438035100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bolli R, Jeroudi MO, Patel B, DuBose CM, Lai EK, Roberts R, McCay PB. Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proc Natl Acad Sci USA 86: 4695–4699, 1989. doi: 10.1073/pnas.86.12.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bolli R, Zughaib M, Li XY, Tang XL, Sun JZ, Triana JF, McCay PB. Recurrent ischemia in the canine heart causes recurrent bursts of free radical production that have a cumulative effect on contractile function. A pathophysiological basis for chronic myocardial “stunning”. J Clin Invest 96: 1066–1084, 1995. doi: 10.1172/JCI118093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fallavollita JA, Canty JM Jr.. Ischemic cardiomyopathy in pigs with two-vessel occlusion and viable, chronically dysfunctional myocardium. Am J Physiol Heart Circ Physiol 282: H1370–H1379, 2002. doi: 10.1152/ajpheart.00138.2001. [DOI] [PubMed] [Google Scholar]
- 73.Perrino C, Naga Prasad SV, Mao L, Noma T, Yan Z, Kim H-S, Smithies O, Rockman HA. Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J Clin Invest 116: 1547–1560, 2006. doi: 10.1172/JCI25397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Weil BR, Techiryan G, Suzuki G, Konecny F, Canty JM Jr.. Adaptive reductions in left ventricular diastolic compliance protect the heart from stretch-induced stunning. JACC Basic Transl Sci 4: 527–541, 2019. doi: 10.1016/j.jacbts.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dewald O, Frangogiannis NG, Zoerlein MP, Duerr GD, Taffet G, Michael LH, Welz A, Entman ML. A murine model of ischemic cardiomyopathy induced by repetitive ischemia and reperfusion. Thorac Cardiovasc Surg 52: 305–311, 2004. doi: 10.1055/s-2004-821153. [DOI] [PubMed] [Google Scholar]
- 76.Weil BR, Smith C, Konecny F, Techiryan G, Zimmer H, Canty J. Persistent left ventricular diastolic stiffening despite cessation of intermittent myocardial stretch in swine (Abstract). FASEB J 32, 2018. doi: 10.1096/fasebj.2018.32.1_supplement.848.9. [DOI] [Google Scholar]
- 77.Patel B, Ismahil MA, Hamid T, Bansal SS, Prabhu SD. Mononuclear phagocytes are dispensable for cardiac remodeling in established pressure-overload heart failure. PLoS One 12: e0170781, 2017. doi: 10.1371/journal.pone.0170781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M, Prabhu SD. CCR2+ monocyte-derived infiltrating macrophages are required for adverse cardiac remodeling during pressure overload. JACC Basic Transl Sci 3: 230–244, 2018. doi: 10.1016/j.jacbts.2017.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Samson R, Jaiswal A, Ennezat PV, Cassidy M, Le Jemtel TH. Clinical phenotypes in heart failure with preserved ejection fraction. J Am Heart Assoc 5: e002477, 2016. doi: 10.1161/JAHA.115.002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cohen JB, Schrauben SJ, Zhao L, Basso MD, Cvijic ME, Li Z, Yarde M, Wang Z, Bhattacharya PT, Chirinos DA, Prenner S, Zamani P, Seiffert DA, Car BD, Gordon DA, Margulies K, Cappola T, Chirinos JA. Clinical phenogroups in heart failure with preserved ejection fraction detailed phenotypes, prognosis, and response to spironolactone. JACC Heart Fail 8: 172–184, 2020. doi: 10.1016/j.jchf.2019.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tennant R, Wiggers CJ. The effects of coronary occlusion on myocardial contraction. Am J Physiol 112: 351–361, 1935. doi: 10.1152/ajplegacy.1935.112.2.351. [DOI] [Google Scholar]
- 82.Tyberg JV, Parmley WW, Sonnenblick EH. In-vitro studies of myocardial asynchrony and regional hypoxia. Circ Res 25: 569–579, 1969. doi: 10.1161/01.res.25.5.569. [DOI] [PubMed] [Google Scholar]
- 83.Tyberg JV, Forrester JS, Wyatt HL, Goldner SJ, Parmley WW, Swan HJ. An analysis of segmental ischemic dysfunction utilizing the pressure-length loop. Circulation 49: 748–754, 1974. doi: 10.1161/01.cir.49.4.748. [DOI] [PubMed] [Google Scholar]
- 84.Heusch G, Guth BD, Widmann T, Peterson KL, Ross J Jr.. Ischemic myocardial dysfunction assessed by temporal Fourier transform of regional myocardial wall thickening. Am Heart J 113: 116–124, 1987. doi: 10.1016/0002-8703(87)90018-4. [DOI] [PubMed] [Google Scholar]
- 85.Przyklenk K, Kloner RA. What factors predict recovery of contractile function in the canine model of the stunned myocardium? Am J Cardiol 64: 18F–26F, 1989. doi: 10.1016/0002-9149(89)90741-8. [DOI] [PubMed] [Google Scholar]
- 86.Mazhari R, Omens JH, Pavelec RS, Covell JW, McCulloch AD. Transmural distribution of three-dimensional systolic strains in stunned myocardium. Circulation 104: 336–341, 2001. doi: 10.1161/01.CIR.104.3.336. [DOI] [PubMed] [Google Scholar]
- 87.Charney RH, Takahashi S, Zhao M, Sonnenblick EH, Eng C. Collagen loss in the stunned myocardium. Circulation 85: 1483–1490, 1992. doi: 10.1161/01.cir.85.4.1483. [DOI] [PubMed] [Google Scholar]
- 88.Sekili S, McCay PB, Li XY, Zughaib M, Sun JZ, Tang L, Thornby JI, Bolli R. Direct evidence that the hydroxyl radical plays a pathogenetic role in myocardial “stunning” in the conscious dog and demonstration that stunning can be markedly attenuated without subequent adverse effects. Circ Res 73: 705–723, 1993. doi: 10.1161/01.res.73.4.705. [DOI] [PubMed] [Google Scholar]
- 89.Li XY, McCay PB, Zughaib M, Jeroudi MO, Triana JF, Bolli R. Demonstration of free radical generation in the “stunned” myocardium in the conscious dog and identification of major differences between conscious and open-chest dogs. J Clin Invest 92: 1025–1041, 1993. doi: 10.1172/JCI116608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sun JZ, Tang XL, Park SW, Qiu Y, Turrens JF, Bolli R. Evidence for an essential role of reactive oxygen species in the genesis of late preconditioning against myocardial stunning in conscious pigs. J Clin Invest 97: 562–576, 1996. doi: 10.1172/JCI118449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Myhre PL, Claggett B, Ballantyne CM, Selvin E, Rosjo H, Omland T, Solomon SD, Skali H, Shah AM. Association between circulating troponin concentrations, left ventricular systolic and diastolic functions, and incident heart failure in older adults. JAMA Cardiol 4: 997–1006, 2019. doi: 10.1001/jamacardio.2019.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Paulus WJ, Zile MR. From systemic inflammation to myocardial fibrosis: the heart failure with preserved ejection fraction paradigm revisited. Circ Res 128: 1451–1467, 2021. doi: 10.1161/CIRCRESAHA.121.318159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62: 263–271, 2013. doi: 10.1016/j.jacc.2013.02.092. [DOI] [PubMed] [Google Scholar]
- 94.MacKenna D, Summerour SR, Villarreal FJ. Role of mechanical factors in modulating cardiac fibroblast function and extracellular matrix synthesis. Cardiovasc Res 46: 257–263, 2000. doi: 10.1016/s0008-6363(00)00030-4. [DOI] [PubMed] [Google Scholar]
- 95.Tschumperlin DJ, Ligresti G, Hilscher MB, Shah VH. Mechanosensing and fibrosis. J Clin Invest 128: 74–84, 2018. doi: 10.1172/JCI93561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Frangogiannis NG. Cardiac fibrosis. Cardiovasc Res 117: 1450–1488, 2021. doi: 10.1093/cvr/cvaa324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hartupee J, Mann DL. Neurohormonal activation in heart failure with reduced ejection fraction. Nat Rev Cardiol 14: 30–38, 2017. doi: 10.1038/nrcardio.2016.163. [DOI] [PMC free article] [PubMed] [Google Scholar]