

Abstract
SOX2 variants and deletions are a common cause of anophthalmia and microphthalmia (A/M). This article presents data from a cohort of patients with SOX2 variants, some of whom have been followed for 20+ years. Medical records from patients enrolled in the A/M Research Registry and carrying SOX2 variants were reviewed. Thirty-seven patients were identified, ranging in age from infant to 30 years old. Eye anomalies were bilateral in 30 patients (81.1%), unilateral in 5 (13.5%), and absent in 2 (5.4%). Intellectual disability was present in all with data available and ranged from mild to profound. Seizures were noted in 18 of 27 (66.6%) patients, usually with abnormal brain MRIs (10/15, 66.7%). Growth issues were reported in 14 of 21 patients (66.7%) and 14 of 19 (73.7%) had gonadotropin deficiency. Genitourinary anomalies were seen in 15 of 19 (78.9%) male patients and 5 of 15 (33.3%) female patients. Patients with SOX2 nucleotide variants, whole gene deletions or translocations are typically affected with bilateral or unilateral microphthalmia and anophthalmia. Other associated features include intellectual disability, seizures, brain anomalies, growth hormone deficiency, gonadotropin deficiency, and genitourinary anomalies. Recommendations for newly diagnosed patients with SOX2 variants include eye exams, MRI of the brain and orbits, endocrine and neurology examinations. Since the clinical spectrum associated with SOX2 alleles has expanded beyond the originally reported phenotypes, we propose a broader term, SOX2-associated disorder, for this condition.
Keywords: anophthalmia, growth, intellectual disability, microphthalmia, SOX2
1 |. INTRODUCTION
Anophthalmia and microphthalmia (A/M) are defects in eye development that lead to absence of the globe or reduction in globe size, unilaterally or bilaterally. A/M was first described in the medical literature in 1917 (Davies, 1917). SOX2 heterozygous pathogenic variants are identified in 15–20% of patients with A/M (Williamson & FitzPatrick, 2014), making SOX2 pathogenic variants the most frequent known genetic cause of A/M (Williamson & FitzPatrick, 2014). SOX2-related A/M is an autosomal dominant condition (Williamson & FitzPatrick, 2014) discovered in 2003 (Fantes et al., 2003) when the gene was found to be disrupted in a patient with a t(3;11) translocation and bilateral anophthalmia (Driggers et al., 1999). This condition has previously been named “Anophthalmia-Esophageal-Genital syndrome (AEG)”, as suggested by Shah et al. (1997) and supported by other publications (Williamson et al., 2006). Over time, other features associated with SOX2 pathogenic variants were described, including intellectual disability, developmental delays, brain, and endocrine anomalies, while the initially identified features were noted to be variable (Driggers et al., 1999; Ragge et al., 2005; Schneider et al., 2008; Zhou et al., 2008). Thus, AEG syndrome now seems to be too narrow of a description for this condition.
Many patients have been described in case reports, but this is the first longitudinal report of a large cohort of patients with SOX2 pathogenic variants or whole gene deletions, compared with previously reported patients with SOX2 pathogenic variants and deletions.
This article presents 37 patients with SOX2 variants or deletions from the A/M Research Registry and aims to provide clinical guidance for providers.
2 |. METHODS
Editorial Policies and Ethical Considerations: This study was approved by the ethics committee from the Institutional Review Boards of Einstein Medical Center, Philadelphia and of Children’s Wisconsin. Informed consent from all participants was obtained.
The Anophthalmia/Microphthalmia (A/M) Research Registry was established in 1994 at the Albert Einstein Medical Center, Philadelphia to assemble an international database of individuals with A/M to determine the incidence of A/M and to provide deep phenotyping of the registry population. In 1999, the registry began offering a gene testing protocol. Eligibility criteria for enrollment in the registry included a diagnosis of unilateral or bilateral microphthalmia, anophthalmia, or coloboma in enrollees themselves or in a family member, or the presence of a pathogenic variant in a known A/M gene. The registry gathers longitudinal phenotypic data by collecting medical records and regular communication with families.
Thirty-seven patients had SOX2 pathogenic variants or deletions. Figure 1 details the location of each variant within the SOX2 gene. Information regarding each patient’s phenotype is included in Tables 1, S2 and S3. Table S5 describes the ACMG classifications of the variants based on ACMG classification guidelines (Richards et al., 2015). Tables 2–6 describe SOX2 variant type and presence or absence of various health issues, including eye anomalies, seizures, brain anomalies, growth hormone and gonadotropin deficiency and genitourinary anomalies. Brain anomalies were generally detected on standard brain MRI without particular focus on specific brain regions such as hippo-campus or peri-hippocampal region. Thirty-five patients have bilateral or unilateral microphthalmia or anophthalmia. Patients were classified as having unilateral microphthalmia or anophthalmia after a thorough ocular examination of their other eye for accurate phenotyping. Patients 22 and 28, who did not have A/M, enrolled after whole exome sequencing identified SOX2 pathogenic variants. Patients enrolled after being seen either in the Albert Einstein Medical Center Genetics Clinic, at the clinic offered during the biennial family support group meetings hosted by ICAN, (International Children’s Anophthalmia Network), or by referral to the Registry by ICAN or their physician.
FIGURE 1.
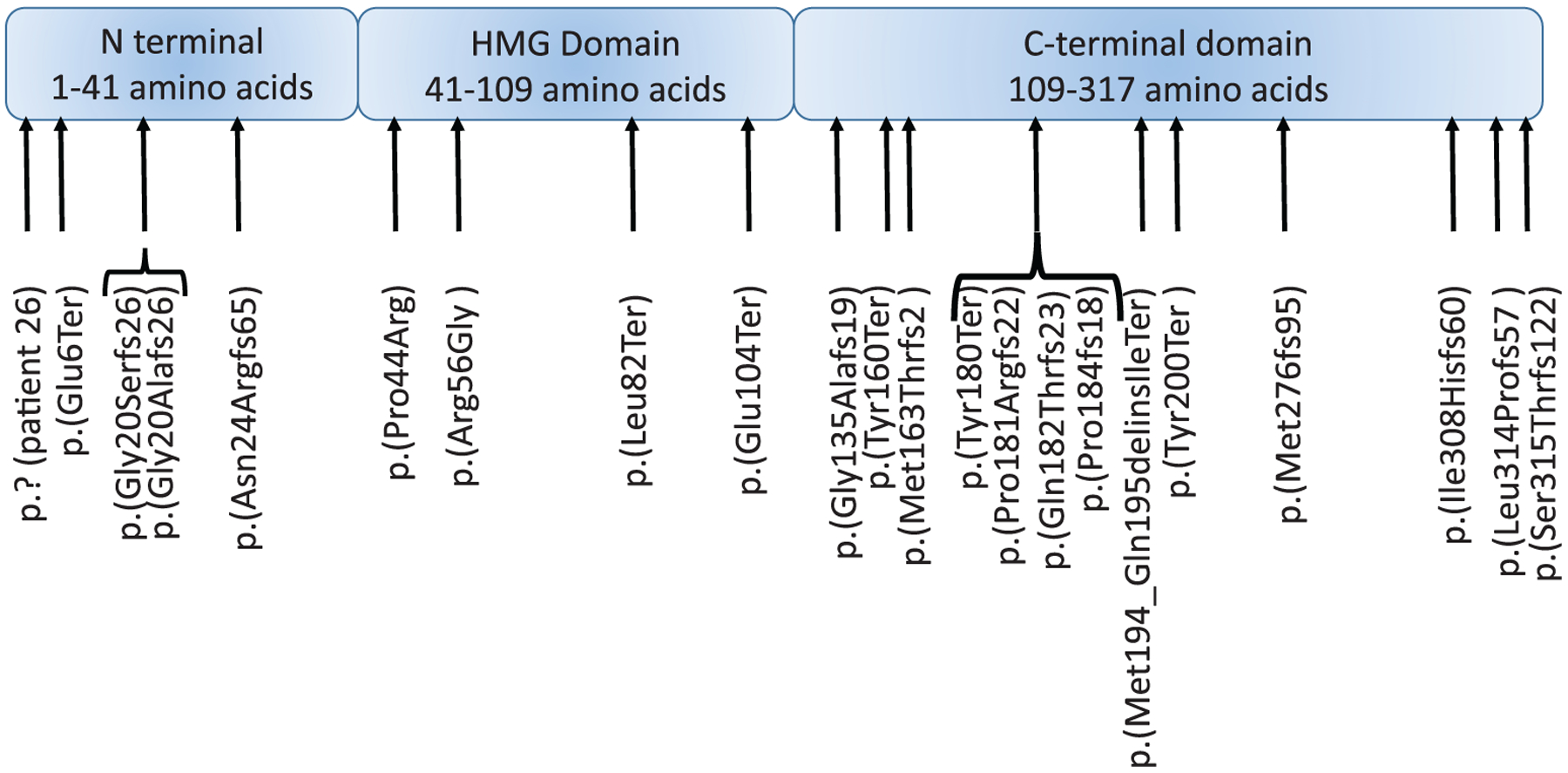
Schematic diagram of the SOX2 gene and locations of variants identified in the 37 patients reported. Amino acid locations of each domain adapted from Weina and Utika, (2014)
TABLE 1.
37 patients with SOX2 variants or deletions and their gender, age at time of last evaluation, presence of absence of microphthalmia or anophthalmia, brain anomalies, seizures, cognitive delay, gait abnormalities, autism, growth hormone and gonadotropin deficiencies and genitourinary anomalies
| p | Sex | Age | SOX2 (NM_003106.3) variant | Protein effect | Type of variant/test type | Eye anom. | Brain ano | Sz | Cognitive delay | Gait abnl | Autism | GHD | GD | GU | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 20y | 46 XX t(3;11)(q27;p11.2) | Unk | Translocation/karyotype | BA | + | F | mild | + | − | + | + | + | Driggers et al., 1999; Fantes et al., 2003; Ragge et al., 2005 |
| 2 | F | 18y | arr[GRCh37] 3q26.33 (180102701-181991155)x1 | Loss | Deletion/array (C) | BA | − | − | severe-profound | + | + | + | + | − | |
| 3 | M | 20y | arr[GRCh37] 3q26.33q27.2 (181171210-184706091)x1 | Loss | Deletion/array (C) | BA | Unk | Unk | profound | +, | − | + | + | + | |
| 4 | F | 6y | arr[GRCh37] 3q26.33q26.33 (182902731-182945128)x1 | Loss | Deletion/array (C) | BA | + | + | moderate-severe | Unk | − | NL | NL | − | |
| 5 | F | 26y | arr[GRCh37] 3q26.33q26.33 (182871341-182987855)x1 | Loss | Deletion/array (R) | BM | + | − | severe-profound | + | − | Unk | + | + | Schilter et al., 2013, (pt 4) |
| 6 | F | 14y | 3q26.33 microdeletion (breakpoints Unk) | Loss | Deletion/array (C) | BA | + | + | Unk | Unk | − | Unk | Unk | − | |
| 7 | M | 19y | arr[GRCh37] 3q26.33 (180913778-181432287)x1 | Loss | Deletion/array (R) | BA | + | Unk | severe | Unk | − | + | + | + | Schilter et al., 2013, (pt 1) |
| 8 | F | 12y | arr[GRCh37] 3q26.33q26.33 (180834336-183551661)x1 | Loss | Deletion/array (C) | BA | + | + | severe | + | − | Unk | Unk | − | |
| 9 | M | 6y | c.70_89del | p.(Asn24Argfs*65) | Frameshift/Sanger (C) | BA | Unk | + | Unk | Unk | − | Unk | Unk | − | |
| 10 | M | 6y | c.70_89del | p.(Asn24Argfs*65) | Frameshift/Sanger (C) | BA | + | + | severe-profound | + | + | NL | + | + | |
| 11 | F | 11y | c.70_89del | p.(Asn24Argfs*65) | Frameshift/Sanger (R) | BM | + | Unk | mild-moderate | + | − | + | Unk | + | Schneider et al., 2009, Pt 2 |
| 12 | M | 18y | c.70_89del | p.(Asn24Argfs*65) | Frameshift/Sanger (R) | BM | − | F | severe | + | − | + | + | + | Schneider et al., 2009, pt 4 |
| 13 | M | 19y | c.70_89del | p.(Asn24Argfs*65) | Frameshift/Sanger (R) | BM | Unk | Unk | Unk | + | + | Unk | Unk | + | Schneider et al., 2009, pt 3 |
| 14 | F | 2y | c.70_89del | p.(Asn24Argfs*65) | Frameshift/Sanger (C) | BA | + | − | Unk | + | − | NL | Unk | − | |
| 15 | F | 13y | c.70_89del | p.(Asn24Argfs*65) | Frameshift/Sanger (C) | BA | Unk | − | moderate | + | − | + | + | − | |
| 16 | M | 29y | c.921_930del | p.(lle308Hisfs*60) | Frameshift/Sanger (R) | BA | − | F | mild to moderate | Unk | − | NL | NL | + | |
| 17a | F | 19y | c.551delC | p.(Pro184fs*18) | Frameshift/Sanger (R) | RALM | + | + | severe | + | − | + | + | + | Schneider et al., 2008, Zhou et al., 2008 |
| 18a | F | 12y | c.551delC | p.(Pro184fs*18) | Frameshift/Sanger (R) | BM | + | Unk | Unk | + | − | + | Unk | − | Schneider et al., 2008, Zhou et al., 2008 |
| 19 | M | 12y | c.131C>G | p.(Pro44Arg) | Missense/Sanger (R) | LM | + | − | mild | + | − | Unk | Unk | + | Schneider et al., 2009, pt 9, |
| 20 | M | 18y | c.310G>T | p.(Glu104Ter) | Nonsense/Sanger(R) | BA | + | + | moderate-severe | + | − | + | + | − | Zhou et al., 2008, pt 40A |
| 21 | M | 19y | c.828delG | p.(Met276llefs*95) | Frameshift/Sanger (C) | BA | − | + | moderate- | + | + | Unk | + | + | |
| 22 | F | 7y | c.582_583delGCinsTT | p.(Met194_Gln195delinslleTer) | Nonsense/WES (C) | − | + | + | moderate-severe | + | − | NL | NL | − | |
| 23 | M | 15y | c.486_487dup | p.(Met163Thrfs*2) | Frameshift/Sanger (R) | BA | Unk | + | severe-profound | Unk | − | Unk | Unk | + | Schneider et al., 2009, pt 5 |
| 24 | F | 4y | c.-13_43del | p.? | Initiation codon del/Sanger (C) | RALM | − | − | mild | Unk | − | + | NL | − | |
| 25 | M | 30y | c.166C>G | p.(Arg56Gly) | Missense/Sanger (C) | BA | + | Unk | mild | + | − | + | + | + | |
| 26 | M | 23y | c.941delT | p.(Leu314Profs*57) | Frameshift/Sanger (R) | BA | − | + | mild | + | − | NL | + | − | Schneider et al., 2009, pt 7 |
| 27 | M | 1y | c.600C>G | p.(Tyr200Ter) | Nonsense/NGS (C) | UM | − | − | Unk | Unk | − | Unk | Unk | + | |
| 28 | M | 6y | c.245 T>A | p.(Leu82Ter) | Nonsense/WES (C) | − | + | − | severe-profound | + | + | NL | NL | + | |
| 29 | M | 4y | c.402delC | p.(Gly135Alafs*19) | Frameshift/Sanger (R) | RM | + | Unk | Unk | Unk | − | Unk | Unk | + | |
| 30 | F | 13y | c.16G>T | p.(Glu6Ter) | Nonsense/Sanger(R) | RALM | Unk | Unk | Unk | + | − | Unk | Unk | + | Schneider et al., 2009, (pt 1) |
| 31 | M | 8y | c.542delC | p.(Pro181Argfs*22) | Frameshift/Sanger (C) | LM | − | − | mild | Unk | − | + | Unk | + | |
| 32 | M | 3 mos | c.538_542dupTACCC | p.(Gln182Thrfs*23) | Frameshift/Sanger (C) | BA | − | Unk | Unk | Unk | − | Unk | Unk | − | |
| 33 | F | 12y | c.59delG | p.(Gly20Alafs*26) | Frameshift/Sanger (R) | BM | + | + | mild-moderate | Unk | − | Unk | Unk | − | |
| 34 | M | 13y | c.540C>G | p.(Tyr180Ter) | Nonsense/Research described in Schneider et al., 2009 | RMLA | + | + | Unk | Unk | − | + | Unk | + | Schneider et al., 2009, (pt 6) |
| 35 | F | 25y | c.58_59delinsT | p.(Gly20Serfs*26) | Frameshift/Sanger (C) | BA | − | + | mild | + | − | Unk | + | + | |
| 36 | F | 10y | c.943_944del | p.(Ser315Thrfs*122) | Frameshift/Sanger (C) | LA | Unk | Unk | Unk | + | Unk | Unk | Unk | Unk | |
| 37 | F | 8y | c.480C>G | p.(Tyr160Ter) | Nonsense/WES (C) | BM | + | + | profound | + | − | + | Unk | − | Gorman et al., 2016; Martinez et al., 2019 |
Abbreviations: +, present; −, absent; Anom.: anomalies; BA, bilateral anophthalmia; BM, bilateral microphathlmia; C, clinical; COL, coloboma; DD, developmental delay; F, febrile; GD, Gonadotropin Deficiency; GHD, Growth Hormone Deficiency; GU, Genitourinary; LA, left microphthalmia; LM, left microphthalmia; mo, months; NGS next generation sequencing; NL, normal; Pt, Patient; RA, right anophthalmia; RALM, right anophthalmia left microphthalmia; R, research; Ref., Reference; RM, right microphthalmia, RMLA, right microphthalmia left anophthalmia; SZ, Seizures; UM, unilateral microphthalmia; Unk, unknown; WES whole exome sequencing; y, years.
Siblings.
TABLE 2.
SOX2 variant types and presence or absence of anophthalmia and microphthalmia
| Patients in this study | Bilateral eye anomalies | Unilateral eye anomalies | Normal | |
|---|---|---|---|---|
| Total | 37 | 31 | 0 | 0 |
| Deletion/translocation | 8 | 8 | 0 | 0 |
| c.70_89del | 7 | 7 | 0 | 0 |
| Frameshift | 13 | 10 | 3 | 0 |
| Missense | 2 | 1 | 1 | 0 |
| Nonsense | 7 | 4 | 1 | 2 |
TABLE 6.
SOX2 variant types and presence or absence of genitourinary (GU) anomalies
| Female GU anomalies | Female normal GU | Male GU anomalies | Male normal GU | |
|---|---|---|---|---|
| Total | 5 | 14 | 15 | 5 |
| Deletion/translocation | 1 | 5 | 2 | 0 |
| c.70_89del | 1 | 2 | 3 | 1 |
| Frameshift | 2 | 4 | 5 | 2 |
| Missense | 0 | 0 | 2 | 0 |
| Nonsense | 1 | 2 | 3 | 1 |
| In-frame | 0 | 0 | 0 | 1 |
SOX2 variants were identified through research-based Sanger sequencing or array CGH, as previously described (Schneider et al., 2008; Schneider et al., 2009; Zhou et al., 2008) or through clinical testing (Table 1 and Figure 1). Intragenic variants are named using transcript NM_003106.3. Fifteen patients have been previously published (Driggers et al., 1999; Fantes et al., 2003; Gorman et al., 2016; Martinez & Madsen, 2019; Ragge et al., 2005; Schilter et al., 2013; Schneider et al., 2008, 2009; Zhou et al., 2008) (Table 1). All variants are available through ClinVar or the SOX2 LOVD database (https://databases.lovd.nl/shared/genes/SOX2).
3 |. RESULTS
The 37 patients with SOX2 variants ranged in age from 3 months to 30 years at the time of review with 62.1% under the age of 18 and 37.9% over 18 (Table 1). There were 18 females (48.7%) and 19 males (51.3%).
3.1 |. Molecular findings
A range of SOX2 alterations was identified (Table 1 and Figure 1). The 3q26 genomic region was disrupted in 8 patients. Seven patients carried a deletion of 3q26 (7/37, 18.9%) and another had a chromosome 3:11 translocation, which included the SOX2 gene. Deletions of this area have been previously reported (Alatzoglou et al., 2011; Bakrania et al., 2007; Chassaing et al., 2007; Chassaing et al., 2014; Dennert et al., 2017; Gerth-Kahlert et al., 2013; Male et al., 2002; Mulvihill et al., 2017; Sisodiya et al., 2006; Suzuki et al., 2014).
The most common pathogenic variant was c.70_89del p. (Asn24Argfs*65) in seven patients (7/37, 18.9%). This frameshift variant, which causes a premature stop codon, has been previously reported (Bakrania et al., 2007; Chassaing et al., 2007; Chassaing et al., 2014; Errichiello et al., 2018; Gerth-Kahlert et al., 2013; Kelberman et al., 2006; Kelberman et al., 2008; Reis et al., 2010; Suzuki et al., 2014; Zenteno et al., 2005; Zenteno et al., 2006). In total, 20 patients had a frameshift pathogenic variant, including the seven patients with c.70_89del; the other 13 patients had private frameshift variants.
The remaining patients’ variants are unique and never previously reported. Seven patients had nonsense variants, leading to truncation of the normal protein, one of which was a complex indel. Missense variants were identified in three patients. One in-frame indel was also identified in Patient 24 that is predicted to be pathogenic.
We identified two families with proven mosaicism of a SOX2 variant. Patients 18 and 19 are siblings and have been described previously (Schneider et al., 2008; Zhou et al., 2008). They both had bilateral eye anomalies and the c.551del pathogenic variant in SOX2. Their mother was mosaic for the same variant in her blood sample and was completely unaffected. Patient 10 and her sister (not enrolled) both had severe bilateral microphthalmia and the c.70_89del pathogenic variant. Their unaffected mother was found to be mosaic for this variant.
3.2 |. Anophthalmia/microphthalmia
Nineteen patients (19/37, 51.4%) had bilateral anophthalmia (Table 1). Four patients had unilateral anophthalmia with microphthalmia of the contralateral eye (4/37, 10.8%). All seven patients (7/37, 18.9%) with bilateral microphthalmia clinically resembled bilateral anophthalmia due to the severity of disease (Table 1). Among five patients with unilateral eye anomalies (5/37, 13.5%), four had unilateral microphthalmia and one had unilateral anophthalmia. Table 2 describes the variant types associated with bilateral and unilateral eye disease.
In total, 30 patients (30/37, 81.1%) had bilateral eye disease and five had unilateral eye disease. We identified two patients without A/M despite the presence of SOX2 pathogenic variants without mosaicism (2/37, 5.4%); both patients had nonsense variants. They presented with severe neurologic symptoms that led to exome sequencing after prior nondiagnostic routine testing. Eye findings in Patient 28 showed right eye esotropia and strabismus requiring patching while Patient 22 had a normal eye exam. Neither patient’s variant had been previously reported in an affected individual nor in the general population through gnomAD.
3.3 |. Development and cognition
Previous studies (Table S4) have shown that patients with pathogenic variants in SOX2 have developmental delay and intellectual disability (ID). Therefore, the Registry collected data from participants concerning development and intellectual disability (Tables 1, 3 and S2). Twenty-six patients had adequate records to classify their cognitive development. Eleven patients had insufficient records and could not be classified; they are therefore excluded from this part of the analysis. The classifications of intellectual disability severity that are used come from Table 9–1 of Mental Disorders and Disabilities among Low-Income Children (Committee to Evaluate the Supplemental Security Income Disability Program for Children with Mental Disorders, 2015) (Table S1). In our subgroup of 26 patients, seven patients are classified with mild ID (7/26, 26.9%) and three with mild to moderate ID (3/26, 11.5%). Five patients had moderate to severe ID (5/26, 19.2%). Four patients had severe ID (4/26, 15.4%) and five had severe to profound ID (5/26, 19.2%) (Table 1). Individuals who are nonverbal, markedly delayed, need significant support and often cannot complete activities of daily living independently are diagnosed with severe or severe to profound ID. Two patients were classified with profound ID (2/26, 7.7%) (Tables 1 and 3). These two patients were in wheelchairs and had significant health problems outside of their anophthalmia. Genotype–phenotype correlations are described in Table 3.
TABLE 3.
SOX2 variant types and intellectual disability severity
| Normal | Mild ID | Mild to moderate ID | Moderate ID | Moderate to severe ID | Severe ID | Severe to profound ID | Profound ID | Total | |
|---|---|---|---|---|---|---|---|---|---|
| Total | 1 | 6 | 3 | 1 | 5 | 4 | 5 | 2 | 28 |
| Deletion/translocation | 0 | 1 | 0 | 0 | 1 | 2 | 2 | 1 | 7 |
| c.70_89del | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 3 |
| Frameshift | 0 | 4 | 3 | 0 | 1 | 1 | 1 | 0 | 10 |
| Missense | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
| Nonsense | 0 | 0 | 0 | 0 | 2 | 0 | 1 | 1 | 4 |
A diagnosis of autism had been made in five patients (5/26, 19.2%), all of whom had moderate to profound ID. Many of these individuals had frameshift variants (four patients); one had a deletion of the SOX2 gene.
No patients with a nonsense variant or a deletion of the SOX2 gene had mild or mild to moderate ID; this could indicate that these variants predispose to more significant ID. Patients with frameshift variants were represented in multiple levels of ID, suggesting that these patients could have any level of ID.
Although patients with SOX2 pathogenic variants/deletions do vary in their intellectual ability, as outlined above, our longitudinal analysis of this cohort suggests that these patients do not appear to worsen with age. The distribution of cognitive abilities appears similar among adults compared to those under 18, suggesting that the prevalence of intellectual disability severity may remain stable when these individuals become young adults. However, follow-up is recommended since adult-onset neurodegeneration in patients with SOX2 syndrome has been reported elsewhere (Ragge et al., 2013).
3.4 |. Brain MRIs and seizures
We received results from brain MRIs performed on 30 of 39 patients; 20 patients had an abnormal brain MRI (20/30, 66.6%) (Tables 1 and 4). Brain MRIs were of standard resolution and were not focused on any specific region. Brain MRIs were available for 16 of 18 patients with seizures (Tables 1 and 4); of these 5 were normal and 11 abnormal. Eight patients with brain MRIs available reported never having seizures (4 abnormal) and seizure status was unknown for the remaining 6 (5 abnormal). Overall, the data suggests that while a person with a SOX2 pathogenic variant who has an abnormal brain MRI is more likely to develop seizures, a normal brain MRI does not rule out development of seizures nor does an abnormal MRI guarantee development of seizures.
TABLE 4.
SOX2 variant type and presence or absence of seizures and brain anomalies
| Seizures | No seizures | Unknown seizure | Brain anomalies | Normal brain MRI | Unknown brain MRI | Seizures and brain anomalies | Brain anomalies, no seizures | Brain anomalies, seizures unknown | Seizures, no brain anomalies | Seizures, unknown brain |
|
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | 20 | 8 | 11 | 20 | 10 | 9 | 11 | 4 | 4 | 5 | 3 |
| Deletion/translocation | 4 | 2 | 2 | 6 | 1 | 1 | 4 | 1 | 1 | 0 | 0 |
| c.70_89del | 4 | 1 | 2 | 3 | 1 | 3 | 1 | 1 | 1 | 1 | 1 |
| Frameshift | 7 | 2 | 4 | 4 | 7 | 2 | 2 | 0 | 2 | 4 | 1 |
| Missense | 0 | 0 | 2 | 2 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| Nonsense | 4 | 2 | 1 | 5 | 1 | 1 | 4 | 1 | 0 | 0 | 0 |
Patients with frameshift variants appeared to be more likely to report seizures than not (10 patients with seizures and 4 without). This trend was also observed, though to a lesser extent in patients with deletions of the SOX2 gene or nonsense variants (4 patients with seizures and 2 without in each group).
3.5 |. Gait
Twenty-three individuals were noted to have an abnormal wide-based, ataxic gait. Twelve individuals did not have records indicating their gait, and two individuals were noted to be hypotonic but did not have records regarding their gait.
3.6 |. Growth and endocrine
Growth parameters and/or endocrine evaluations were available for 21 patients (Tables 1, 4 and S3). Two patients were noted to have growth hormone issues but did not provide their endocrine records. Fifteen patients (15/21, 71.4%) had growth delays, of which nine had a diagnosis of growth hormone deficiency and six had short stature which may be caused by growth hormone deficiency. Seven patients reported normal growth hormone levels (Table 1).
Fourteen patients had a diagnosis of gonadotropin deficiency (LH, FSH, testosterone deficiency, hypogonadism, hypogonadotropic hypogonadism) (14/19, 73.7%, 5 had normal gonadotropins) (Table 1). Nine patients also had growth hormone deficiency or short stature in addition to gonadotropin deficiency. Eighteen patients did not have medical records available for their endocrine evaluations or had never had an endocrine evaluation. Several patients did not provide the Registry with endocrine evaluations but had genitourinary anomalies or growth hormone deficiencies noted in other medical records.
Out of 19 male patients with information regarding genitourinary anomalies, 15 patients (15/19, 78.9%) had at least one genitourinary anomaly, including micropenis, undescended testicles, shawl scrotum, and other testicular anomalies (Tables 1 and 6). Four males had reportedly normal genitourinary systems. Six female patients reported a female genitourinary anomaly such as infantile uterus, labial adhesions, or hypoplastic labia (6/17, 35.3%). Eleven reported normal genitourinary tracts and one was unknown.
4 |. DISCUSSION
Heterozygous pathogenic variants in SOX2 were first reported as a cause of anophthalmia in 2003 in Patient 1 with bilateral anophthalmia and absent optic nerves, delayed walking with ataxic gait, ongoing weakness of her legs, delayed puberty, short stature, developmental delay and a single febrile seizure (Driggers et al., 1999; Fantes et al., 2003; Ragge et al., 2005). Review of this large cohort of patients with SOX2 variants provides a better description of the natural history of this disorder.
4.1 |. Anophthalmia prevalence
Of the 37 patients in the registry with SOX2 variants, 51.3% had bilateral anophthalmia, 18.9% had bilateral severe microphthalmia, and 10.8% had unilateral anophthalmia and contralateral severe microphthalmia. Unilateral eye anomalies and one normal eye were present in 13.5% and 2 patients had normal eye size (Patients 24 and 30). In total, 30/37 (81.1%) patients with SOX2 variants or deletions had bilateral eye defects.
One hundred and forty-four individuals have been described previously with SOX2 pathogenic variants (Table S4). In total, bilateral eye disease was identified in 106 of 144 (73.6%), consistent with the rate in our patients (81.1%). There are now 18 patients (including our two) reported with normal eyes and SOX2 pathogenic variants. However, this number is expected to rise as more patients with neurological disorders are analyzed with whole exome sequencing. Historically, SOX2 was analyzed in patients with A/M, so most patients with SOX2 variants are affected with A/M, a classic case of ascertainment bias.
4.2 |. Variant specific ocular findings
The recurrent c.70_89del frameshift variant, reported in 12 of 144 (8.3%) previously reported patients (Table S4) was also present in 7/37 (18.9%) patients reported here (Table 1), further supporting that this is a hot-spot mutation. While all patients with this variant reported here have bilateral anophthalmia or microphthalmia, 6 of 12 (50%) of the previously reported cases had unilateral anophthalmia or had normal eyes (Errichiello et al., 2018; Gerth-Kahlert et al., 2013; Zenteno et al., 2006), suggesting that phenotypic variability does not correlate fully with genotype. Among all patients with frameshift variants in the Registry, 3 of 19 (15.8%) had unilateral microphthalmia or anophthalmia and 16 of 19 (84.2%) had bilateral anophthalmia, similar to the distribution in the literature, with 38 of 54 (70.4%) patients affected with bilateral A/M.
Among the seven Registry patients with nonsense variants, four (57.1%) have bilateral A/M. In contrast, among patients in the literature, 26 of 30 (86.7%) had bilateral A/M; three other patients in the literature had normal eyes and nonsense variants.
The Registry includes seven individuals with a deletion of the whole SOX2 gene (Table 1) and one patient with an initiation codon deletion, which would be expected to result in a similar complete loss of protein product, all of whom demonstrated bilateral A/M (100%). Twenty-eight previously reported patients have had whole gene deletions with bilateral A/M seen in 22 of 28 (78.6%) (Table S4). Three of the six without had normal eye size with minor concerns such as strabismus, retinal folds or unilateral coloboma.
Two individuals from the Registry had a missense variant: one with bilateral anophthalmia, and one with unilateral microphthalmia. In contrast, 32 patients have been previously reported in the literature with missense variants (Table S4); of these, 15 of 31 (48.4%) had bilateral A/M, 4 of 31 (12.9%) had unilateral A/M, 4 of 31 (12.9%) had other anomalies (coloboma, microcornea), and 8 of 31 (25.8%) had normal eyes.
Overall, the occurrence of bilateral A/M in our cases was higher in almost every group compared to the literature, consistent with the fact that these patients were part of an A/M registry. The type of variant appears to have some effect on phenotype with missense variants having a lower penetrance for bilateral A/M phenotypes compared to loss-of-function (17/34 [50%] vs. 116/148 [78.4%], respectively).
4.3 |. Mosaicism
Two cases of mosaicism were previously reported in addition to the two new cases of mosaicism reported here: in a family with two affected children, maternal germline mosaicism was identified for the c.70_86del variant (Chassaing et al., 2007) and in a Moroccan family with two of six pregnancies affected, a pathogenic missense variant, c.138T>G, p.(Asn46Lys), was identified in both affected pregnancies and present at a lower level in the mother’s sample (Faivre et al., 2006). In one additional family, two children with ocular anomalies were born to a mother with idiopathic hypogonadotropic hypogonadism and a c.837del p.(Gly280Alafs*91) pathogenic variant was identified in all three individuals, with varying levels in the mother’s blood compared to saliva, suggesting possible mosaicism, though a sequencing artifact could not be ruled out (Stark et al., 2011). While the mother has been the carrier in all reported familial cases with mosaicism, the possibility of paternal mosaicism cannot be ruled out. With five known cases of A/M patients with SOX2 variants inherited from a parent with normal eyes, we recommend that all parents of children with SOX2 pathogenic variants should be tested to rule out mosaicism. Additionally, different tissues should be tested, and next generation sequencing should be considered as opposed to Sanger sequencing to avoid allele dropout. The identification of mosaicism is important for determining the recurrence risk for the patient’s parents.
4.4 |. Patients without A/M
While many of the first cases in the literature with normal eyes and SOX2 variants were identified because other family members were found to have pathogenic variants (Chassaing et al., 2007; Mihelec et al., 2009; Stark et al., 2011; Zenteno et al., 2006), several were the first in their families, presenting with overlapping syndromic features including brain anomalies, hypogonadotropic hypogonadism (HH), developmental delay/intellectual disability (ID), seizures, or genital anomalies (Blackburn et al., 2018; Dennert et al., 2017; Errichiello et al., 2018; Kelberman et al., 2006; Pilz et al., 2019; Shima et al., 2017; Takagi et al., 2013). Some of these patients, on further examination, had slight ocular anomalies, such as narrowed palpebral fissure (Zenteno et al., 2006), mild retinal anomalies (Errichiello et al., 2018; Mihelec et al., 2009; Pilz et al., 2019; Shima et al., 2017; Takagi et al., 2013), subtle anterior segment anomalies (Dennert et al., 2017, Mihelec et al., 2009), ocular motility disorders (Errichiello et al., 2018; Pilz et al., 2019), or optic nerve hypoplasia (Kelberman et al., 2008). Some patients were normal intellectually while others had severe cognitive impairment, and some were too young to evaluate. As more patients with normal eyes undergo whole exome sequencing, we expect that SOX2 pathogenic variants will continue to be identified in patients without A/M. Unbiased screening through whole exome sequencing will enable better determination of the true incidence of A/M in patients with a SOX2 pathogenic variant.
4.5 |. Brain anomalies and seizures
Individuals with SOX2 pathogenic variants have an increased risk for brain anomalies and seizures. Among patients with data available from our cohort and the literature combined, 87 patients out of 114 in total (76.3%, Tables 1, 4, S2 and S4) had brain anomalies and 41 of 63 individuals (65%) had seizures. SOX2 is a developmental regulator transcription factor that is expressed in the brain and eyes (Sisodiya et al., 2006), which may explain the high rate of brain anomalies and seizures. Seizures were seen in the majority of patients with frameshift variants (10/15 patients), gene deletions (4/6 patients), and nonsense variants (4/6 patients) (Tables 1 and 4), similar to rates in the literature with nonsense variants (10/15), deletions (4/6) and missense variants (2/2) (Table S4). However, among previously reported patients with frameshift variants, only 5 of 11 had seizures (Table S4). A large proportion of previously reported patients did not mention seizures (Table S4), and it is unclear if these patients were seizure-free or never had a neurologic evaluation. A strength of this article is documentation of seizure-free patients within our cohort.
Brain MRI was performed on 30 patients in this cohort, with abnormalities identified in 20including 6 of 7 with gene deletion, 7 of 15 with frameshift, 5 of 6 with nonsense, and 2 of 2 with missense variants. While this data suggests a lower rate of anomalies for patients with frameshift variants, the sample size is very small. Within the literature, abnormal brain MRIs were reported more often than normal brain MRIs for patients with all types of variants (Table 4 and S4), possibly because abnormal brain MRI results are more likely to be included in a publication compared to normal results. In total, 89 patients had an abnormal brain MRI (Tables 1, 4 and S4), indicating that brain MRI abnormalities are common among patients with any variants in the SOX2 gene. Very few patients in the literature have been described as having normal brain MRIs, so this may be under-reported and should be investigated further.
The cohort reported here also provided insight on patients with seizures and the presence or absence of a brain anomaly. In this cohort, brain MRI results were available for 16 of 18 patients with seizures, of which 11 were abnormal; an abnormal brain MRI was also reported in 5 of 9 seizure-free patients (Table 4). However, in the literature, there are far more patients reported with seizures without information about brain anomalies, indicating that brain MRIs are not always performed after seizures are noted (Table S4). Given the previous association of mesial temporal malformations in patients with SOX2-related seizure disorders (Sisodiya et al., 2006), a broader study of brain MRIs from patients with SOX2 variants, including detailed evaluation of the peri-hippocampal region and correlation with seizure status would be of interest.
4.6 |. Intellectual disability and developmental delay
Many previous publications have described developmental delays and/or intellectual disability in patients with SOX2 variants (Table S4). However, this publication is the first to describe a cohort of older individuals with SOX2 pathogenic variants and to report on their cognition. Twenty-six patients had information available regarding their cognitive ability (Tables 1 and 3). Among the 13 characterized patients over the age of 18, five of those patients have mild to moderate intellectual disability, and eight are moderately to profoundly affected. There did not appear to be a correlation between variant type and severity of intellectual disability in our cohort. Table 3 describes ID severity based on SOX2 variant type. Eight patients in the literature had reportedly normal intelligence (Table S4). However, over 90 patients in the literature (Table S4) and 11 patients in our cohort did not have any information specified about their intelligence, so it is possible that there is a bias and that there are additional patients with normal intelligence and SOX2 variants.
4.7 |. Abnormal gait
An abnormal gait has been noted anecdotally for many patients with SOX2 variants. In total, 55 individuals, including 24 individuals from this study, were noted to have a wide-based, ataxic gait. While there were no reports in the literature of normal gait, in most cases gait is not described at all. Therefore, further investigation is needed to determine the prevalence of the wide-based ataxic gait. The gait anomalies seem to be associated with muscle weakness and to improve with physical therapy, but most individuals are somewhat unsteady as they get older and tire easily. Data from this registry suggests that physical therapy is important for management of individuals with SOX2 pathogenic variants and deletions.
4.8 |. Growth hormone and gonadotropin deficiency
Another significant issue for these patients is growth hormone and gonadotropin deficiency. Both growth hormone deficiency and short stature have been reported in patients with SOX2 pathogenic variants (Table S4) and SOX2 has been shown to be involved in the development and function of hypothalamo-pituitary and reproductive axes (Kelberman et al., 2006). Mice with SOX2 heterozygous variants show a reduction in size and poor fertility (Kelberman et al., 2006). There is evidence that SOX2 is part of the pituitary transcription pathway, causing growth hormone deficiency and hypopituitarism when it is deleted or mutated (Kelberman et al., 2006). Detailed phenotyping of our cohort supports this association, with 10 of our patients having documented growth hormone deficiency, and an additional 6 reporting short stature (14/37, 37.8%, Tables 1 and 5). Seven reportedly had normal height and 15 with unknown height. Within the literature, a similar frequency was noted with 46 of 145 (32%) reported to have growth hormone deficiency or short stature (Table S4). Patients with SOX2 pathogenic variants should see an endocrinologist early and have height and growth hormone levels closely monitored.
TABLE 5.
SOX2 variant types and growth hormone and gonadotropin deficiency presence or absence
| Growth hormone deficiency | No growth hormone deficiency | Unknown | Gonadotropin deficiency | Normal gonadotropin | Unknown | Short stature | |
|---|---|---|---|---|---|---|---|
| Total | 16 | 7 | 16 | 14 | 5 | 20 | 21 |
| Deletion/translocation | 4 | 1 | 3 | 5 | 1 | 2 | 5 |
| c.70_89del | 3 | 1 | 2 | 3 | 0 | 4 | 4 |
| Frameshift | 4 | 3 | 7 | 4 | 2 | 7 | 7 |
| Missense | 1 | 1 | 1 | 1 | 0 | 1 | 1 |
| Nonsense | 3 | 0 | 2 | 1 | 2 | 4 | 3 |
Estrogen and testosterone elevations lead to the onset of puberty and development of secondary sex characteristics. Delayed puberty or hypogonadotropic hypogonadism is marked by lack of sexual development and/or low gonadotropin levels (Dye et al., 2018), also known as secondary hypogonadism. Male patients with hypogonadotropic hypogonadism may also have congenital genitourinary anomalies, such as micropenis, cryptorchidism, and undescended testes (Richard-Eaglin, 2018). Patients with SOX2 pathogenic variants have been reported to have delayed puberty, genitourinary anomalies, hypogonadism and/or hypogonadotropic hypogonadism (Table S4) Gonadotropin deficiencies were documented in 49 out of 58 in our cohort and in the literature (84.5%, Tables 1 and 5, Tables S3 and S4). However, this may be artificially elevated, as there were only three patients in the literature reported with normal assessment of gonadotropin function.
Between our cohort and the literature, 61 individuals out of 94 with data available (64.9%) had genitourinary anomalies (Tables 1 and 6, S3 and S4). Within our cohort, five female patients had a genitourinary anomaly (13 females had normal GU tract) and 15 male patients had at least one genitourinary anomaly (4 had normal GU tract) (Tables 1 and 6). Therefore, there appears to be a lower prevalence of genitourinary anomalies in female patients (28% vs. 79%). It is not clear how many individuals with SOX2 pathogenic variants have normal gonadotropin levels, but this should also be monitored in affected patients, particularly if the patient has delayed puberty or a genitourinary anomaly.
5 |. RECOMMENDATIONS
Patients with variants or deletions of the SOX2 gene are at risk for multiple health concerns. Upon diagnosis, patients should undergo a thorough evaluation by an ophthalmologist for ocular anomalies, with potential consideration of prosthesis or surgery if needed, and physical examination to determine if they have any genitourinary anomalies. Other common syndromic features noted in the literature in patients with SOX2 deficiency, not specifically tracked in this population but important to consider, include esophageal anomalies and hearing loss. Imaging at the time of diagnosis should include brain MRI which may require special pituitary protocols to visualize the pituitary region. If seizures are noted or suspected, patients should undergo an EEG and referral to a neurologist should be considered. Seizures can occur at any age and may occur even if a brain MRI is normal. Due to the risk for intellectual disability and learning disabilities, patients’ milestones and development should be followed closely with a low threshold for referral to early intervention and/or a developmental pediatrician. Patients with SOX2 pathogenic variants or deletions are at risk of failure to thrive (particularly with esophageal anomalies) and short stature, so growth and pubertal development should be closely followed with a low threshold for referral to the appropriate specialist, with particular attention to the high risk of growth hormone and gonadotropin deficiencies. As patients reach adulthood, they should continue to follow with an ophthalmologist and a primary care provider along with any other specialists needed for co-morbidities. Depending on their cognitive ability and family support, they may either live at home or in a group setting and may need support with activities of daily living or employment.
6 |. CONCLUSION
Most patients with SOX2 pathogenic variants in our cohort of 37 individuals have structural eye anomalies, usually bilaterally, however, patients without A/M have also been identified. Other features include neurological anomalies with abnormal gait, intellectual disability, seizures, endocrine and esophageal anomalies. We expect that the phenotypic range of SOX2 will continue to expand as more patients with developmental anomalies undergo exome sequencing. Since the phenotypic spectrum of individuals with SOX2 pathogenic variants has widened beyond anophthalmia, esophageal anomalies and genital anomalies, we propose using the name SOX2-associated disorder instead of AEG.
Supplementary Material
ACKNOWLEDGMENTS
Funding for this project provided by grants from The Albert Einstein Society (AES) of the Einstein Healthcare Network as well as the National Institutes of Health award R01EY025718.
We would like to thank ICAN (International Children’s Anophthalmia Network) for their constant support and sharing of information about this project with new families who contact them. Our deep appreciation to all the families for their willingness to share information about their family member with SOX2 syndrome.
Funding information
Albert Einstein Society; National Institutes of Health, Grant/Award Number: R01EY025718
Footnotes
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author, LAW, upon reasonable request.
REFERENCES
- Alatzoglou KS, Andoniadou CL, Kelberman D, Buchanan CR, Crolla J, Arriazu MC, Roubicek M, Moncet D, Martinez-Barbera JP, & Dattani MT (2011). SOX2 haploinsufficiency is associated with slow progressing Hypothalamo-pituitary tumours. Human Mutation, 32, 1376–1380. 10.1002/humu.21606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakrania P, Robinson DO, Bunyan DJ, Salt A, Martin A, Crolla JA, Wyatt A, Fielder A, Ainsworth J, Moore A, Read S, Uddin J, Laws D, Pascuel-Salcedo D, Ayuso C, Allen L, Collin JRO, & Ragge NK (2007). SOX2 anophthalmia syndrome: 12 new cases demonstrating broader phenotype and high frequency of large gene deletions. The British Journal of Ophthalmology, 91, 1471–1476. 10.1136/bjo.2007.117929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn PR, Chacon-Camacho OF, Ortiz-Gonzalez XR, Reyes M, Lopez-Uriarte GA, Zarei S, Bhoj EJ, Perez-Solorzano S, Vaubel RA, Murphree MI, Nava J, Cortes-Gonzalez V, Parisi JE, Villanueva-Mendoza C, Tirado-Torres IG, Li D, Klee EW, Pichurin PN, & Zenteno JC (2018). Extension of the mutational and clinical spectrum of SOX2 related disorders: Description of six new cases and a novel association with suprasellar teratoma. American Journal of Medical Genetics, 176(12), 2710–2719. 10.1002/ajmg.a.40644 [DOI] [PubMed] [Google Scholar]
- Chassaing N, Causse A, Vigouroux A, Delahaye A, Alessandri JL, Boespflug-Tanguy O, Boute-Benejean O, Dollfus H, Duban-Bedu B, Gilbert-Dussardier B, Giuliano F, Gonzales M, Holder-Espinasse M, Isidor B, Jacquemont ML, Lacombe D, Martin-Coignard D, Mathieu-Dramard M, Odent S, … Calvas P (2014). Molecular findings and clinical data in a cohort of 150 patients with anophthalmia/microphthalmia. Clinical Genetics, 86, 326–334. 10.1111/cge.12275 [DOI] [PubMed] [Google Scholar]
- Chassaing N, Gilbert-Dussardier B, Nicot F, Fermeaux V, Encha-Razavi F, Fiorenza M, Toutain A, & Calvas P (2007). Germinal mosaicism and familial recurrence of a SOX2 mutation with highly variable phenotypic expression extending from AEG syndrome to absence of ocular involvement. American Journal of Medical Genetics, 143A, 289–291. 10.1002/ajmg.a.31524 [DOI] [PubMed] [Google Scholar]
- Committee to Evaluate the Supplemental Security Income Disability Program for Children with Mental Disorders, Board on the Health of Select Populations, Board on Children, Youth, and Families, Institute of Medicine, Division of Behavioral and Social Sciences and Education, The National Academies of Sciences, Engineering, and Medicine, Boat TF, & Wu JT (2015). Clinical characteristics of intellectual disabilities. In: Mental disorders and disabilities among low-income children. National Academies Press (US). [PubMed] [Google Scholar]
- Davies DL (1917). Anophthalmia and microphthalmia. British Journal of Ophthalmology, 1(7), 415–423. 10.1136/bjo.1.7.415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennert N, Engels H, Cremer K, Becker J, Wohlleber E, Albrecht B, Ehret JK, Ludecke H-J, Suri M, Carignani G, Renieri A, Kukuk GM, Wieland T, Andrieux J, Strom TM, Wieczorek D, Dieux-Coesslier A, & Zink AM (2017). De novo microdeletions and point mutations affecting SOX2 in three individuals with intellectual disability but without major eye malformations. American Journal of Medical Genetics Part A, 173A, 435–443. 10.1002/ajmg.a.38034 [DOI] [PubMed] [Google Scholar]
- Driggers RW, Macri CJ, Greenwald J, Carpenter D, Avallone J, Howard-Peebles PN, & Levin SW (1999). Isolated bilateral Anophthalmia in a girl with an apparently balanced de novo translocation: 46,XX,t(3;11)(q27;p11.2). American Journal of Medical Genetics. Part A, 87, 201–202. [DOI] [PubMed] [Google Scholar]
- Dye AM, Nelson GB, & Diaz-Thomas A (2018). Delayed puberty. Pediatric Annals, 47(1), e16–e22. 10.3928/19382359-20171215-01 [DOI] [PubMed] [Google Scholar]
- Errichiello E, Gorone C, Giuliano L, Iadarola B, Cosentino E, Rossato M, Kurtas NE, Delledonne M, Mattina T, & Zuffardi O (2018). SOX2: Not always eye malformations. Severe genital but no major ocular anomalies in a female patient with the recurrent c.70del20 variant. European Journal of Medical Genetics, 61, 335–340. 10.1016/j.ejmg.2018.01.011 [DOI] [PubMed] [Google Scholar]
- Faivre L, Williamson KA, Faber V, Laurent N, Grimaldi M, Thauvin-Robinet C, Durand C, Mugneret F, Gouyon J-B, Bron A, Huet F, Hayward C, van Heyningen V, & FitzPatrick DR (2006). Recurrence of SOX2 anophthalmia syndrome with gonosomal mosaicism in a phenotypically normal mother. American Journal of Medical Genetics. Part A, 140A, 636–639. 10.1002/ajmg.a.31114 [DOI] [PubMed] [Google Scholar]
- Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JRO, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van Heyningen V, & FitzPatrick DR (2003). Mutations in Sox2 cause anophthalmia. Nature Genetics, 33(4), 461–463. 10.1038/ng1120 [DOI] [PubMed] [Google Scholar]
- Gerth-Kahlert C, Williamson K, Ansari M, Rainger JK, Hingst V, Zimmermann T, Tech S, Guthoff RF, van Heyningen V, & FitzPatrick DR (2013). Clinical and mutation analysis of 51 probands with anophthalmia and/or severe microphthalmia from a single center. Molecular Genetics & Genomic Medicine, 1(1), 15–31. 10.1002/mgg3.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman KM, Lynch SA, Schneider A, Grange DK, Williamson KA, FitzPatrick DR, & King MD (2016). Status dystonicus in two patients with SOX2-anopthalmia syndrome and nonsense mutations. American Journal of Medical Genetics. Part A, 170(11), 3048–3050. 10.1002/ajmg.a.37849 [DOI] [PubMed] [Google Scholar]
- Kelberman D, de Castro SCP, Huang S, Crolla JA, Palmer R, Gregory JW, Taylor D, Cavallo L, Faienza MF, Fischetto R, Achermann JC, Martinez-Barbera JP, Rizzoti K, Lovell-Badge R, Robinson ICAF, Gerrelli D, & Dattan MT (2008). SOX2 plays a critical role in the pituitary, forebrain, and eye during human embryonic development. The Journal of Clinical Endocrinology and Metabolism, 93(5), 1865–1873. 10.1210/jc.2007-2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, Chong WK, Kirk JMW, Achermann JC, Ross R, Carmignac D, Lovell-Badge R, Robinson ICAF, & Dattani MT (2006). Mutations within SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. Journal of Clinical Investigation, 116, 2442–2455. 10.1172/JCI28658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Male A, Davies A, Bergbaum A, Keeling J, FitzPatrick D, Ogilvie CM, & Berg J (2002). Delineation of an estimated 6.7 Mb candidate interval for an anophthalmia gene at 3q26.33-q28 and description of the syndrome associated with visible chromosome deletions of this region. European Journal of Human Genetics, 10, 807–812. 10.1038/sj.ejhg.5200890 [DOI] [PubMed] [Google Scholar]
- Martinez E, & Madsen EC (2019). Status dystonicus, hyperpyrexia, and acute kidney injury in a patient with SOX2-anophthalmia syndrome. American Journal of Medical Genetics, 179A, 1395–1397. 10.1002/ajmg.a.61144 [DOI] [PubMed] [Google Scholar]
- Mihelec M, Abraham P, Gibson K, Krowka R, Susman R, Storen R, Chen Y, Donald J, Tam PPL, Grigg JR, Flaherty M, Gole GA, & Jamieson RV (2009). Novel SOX2 partner-factor domain mutation in a four-generation family. European Journal of Human Genetics, 17, 1417–1422. 10.1038/ejhg.2009.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvihill J, Sarkar A, & Dixit A (2017). Seeing the diagnosis on karyotype—SOX2 and eye development. Ophthalmic Genetics, 38(6), 580–583. 10.1080/13816810.2017.1290119 [DOI] [PubMed] [Google Scholar]
- Pilz RA, Korenke GC, Steeb R, Strom TM, Felbor U, & Rath M (2019). Exome sequencing identifies a recurrent SOX2 deletion in a patient with gait ataxia and dystonia lacking major ocular malformations. Journal of the Neurological Sciences, 401, 34–36. 10.1016/j.jns.2019.04.007 [DOI] [PubMed] [Google Scholar]
- Ragge NK, Lorenz B, Schneider A, Bushby K, de Sanctis L, de Sanctis U, Salt A, Collin JRO, Vivian AJ, Free SL, Thompson P, Williamson KA, Sisodiya SM, van Heyningen V, & FitzPatrick DR (2005). SOX2 anophthalmia syndrome. American Journal of Medical Genetics. Part A, 135A, 1–7. 10.1002/ajmg.a.30642 [DOI] [PubMed] [Google Scholar]
- Ragge NK, Quaghebeur G, & Stewart H (2013). SOX2 anophthalmia syndrome in adulthood—A neurodegenerative picture? Clinical Genetics, 83, 482–484. 10.1111/j.1399-0004.2012.01922.x [DOI] [PubMed] [Google Scholar]
- Reis LM, Tyler RC, Schneider A, Bardakjian T, & Semina EV (2010). Examination of SOX2 in variable ocular conditions identifies a recurrent deletion in microphthalmia and lack of mutations in other phenotypes. Molecular Vision, 16, 768–773. [PMC free article] [PubMed] [Google Scholar]
- Richard-Eaglin A (2018). Male and female hypogonadism. The Nursing Clinics of North America, 53(3), 395–405. 10.1016/j.cnur.2018.04.006 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, & Rehm HL (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–423. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilter KF, Reis LM, Schneider A, Bardakjian TM, Abdul-Rahman O, Kozel BA, Zimmerman HH, Broeckel U, & Semina EV (2013). Whole-genome copy number variation analysis in anophthalmia and microphthalmia. Clinical Genetics, 84(5), 473–481. 10.1111/cge.12202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Bardakjian T, Reis LM, Tyler RC, & Semina EV (2009). Novel SOX2 mutations and genotype–phenotype correlation in anophthalmia and microphthalmia. American Journal of Medical Genetics. Part A, 149A, 2706–2715. 10.1002/ajmg.a.33098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Bardakjian TM, Zhou J, Hughes N, Keep R, Dorsainville D, Kherani F, Katowitz J, Schimmenti LA, Hummel M, FitzPatrick DR, & Young TL (2008). Familial recurrence of SOX2 anophthalmia syndrome: Phenotypically normal mother with two affected daughters. American Journal of Medical Genetics. Part A, 149A(12), 2794–2798. 10.1002/ajmg.a.32384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah D, Jones R, Porter H, & Turnpenny P (1997). Bilateral microphthalmia, esophageal atresia, and cryptorchidism: The anophthalmia-esophageal-genital syndrome. American Journal of Medical Genetics, 70(2), 171–173. [DOI] [PubMed] [Google Scholar]
- Shima H, Ishii A, Wada Y, Kizawa J, Yokoi T, Azuma N, Matsubara Y, Suzuki E, Nakamura A, Narumi S, & Fukami M (2017). SOX2 nonsense mutation in a patient clinically diagnosed with non-Syndromic hypogonadotropic hypogonadism. Endocrine Journal, 64(8), 813–817. 10.1507/endocrj.ej17-0078 [DOI] [PubMed] [Google Scholar]
- Sisodiya SM, Ragge NK, Cavalleri GL, Hever A, Lorenz B, Schneider A, Williamson KA, Stevens JM, Free SL, Thompson PJ, van Heyningen V, & FitzPatrick DR (2006). Role of SOX2 mutations in human hippocampal malformations and epilepsy. Epilepsia, 47(3), 534–542. 10.1111/j.1528-1167.2006.00464.x [DOI] [PubMed] [Google Scholar]
- Stark Z, Storen R, Bennetts B, Savarirayan R, & Jamieson RV (2011). Isolated Hypogonadotropic hypogonadism with SOX2 mutation and anophthalmia/microphthalmia in offspring. European Journal of Human Genetics, 19, 753–756. 10.1038/ejhg.2011.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J, Azuma N, Dateki S, Soneda S, Muroya K, Yamamoto Y, Saito R, Sano S, Nagai T, Wada H, Endo A, Urakami T, Ogata T, & Fukami M (2014). Mutation spectrum and phenotypic variation in nine patients with SOX2 abnormalities. Journal of Human Genetics, 59, 353–356. 10.1038/jhg.2014.34 [DOI] [PubMed] [Google Scholar]
- Takagi M, Narumi S, Asakura Y, Muroya K, Hasegawa Y, Adachi M, & Hasegawa T (2013). A novel mutation in SOX2 causes Hypogonadotropic Hypogonadism with mild ocular malformations. Hormone Research in Pædiatrics, 81(2), 133–138. 10.1159/000355279 [DOI] [PubMed] [Google Scholar]
- Weina K, & Utika J (2014). SOX2 and cancer: Current research and its implications in the clinic. Clinical and Translational Medicine, 3, 19. 10.1186/2001-1326-3-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson KA, & FitzPatrick DR (2014). The genetic architecture of microphthalmia, anophthalmia and coloboma. European Journal of Medical Genetics, 57, 369–380. 10.1016/j.ejmg.2014.05.002 [DOI] [PubMed] [Google Scholar]
- Williamson KA, Hever AM, Rainger J, Rogers RC, Magee A, Fiedler Z, Keng WT, Sharkey FH, McGill N, Hill CJ, Schneider A, Messina M, Turnpenny PD, Fantes JA, van Heyningen V, & FitzPatrick DR (2006). Mutations in SOX2 cause anophthalmia-esophageal-genital (AEG) syndrome. Human Molecular Genetics, 15(9), 1413–1422. [DOI] [PubMed] [Google Scholar]
- Zenteno JC, Gascon-Guzman G, & Tovilla-Canales JL (2005). Bilateral anophthalmia and brain malformations caused by a 20-bp deletion in the SOX2 gene. Clinical Genetics, 68, 564–566. 10.1111/j.1399-0004.2005.00518.x [DOI] [PubMed] [Google Scholar]
- Zenteno JC, Perez-Cano HJ, & Aguinaga M (2006). Anophthalmia-esophageal atresia syndrome caused by an SOX2 gene deletion in monozygotic twin brothers with markedly discordant phenotypes. American Journal of Medical Genetics. Part A, 1401A, 1899–1903. 10.1002/ajmg.a.31384 [DOI] [PubMed] [Google Scholar]
- Zhou J, Kherani F, Bardakjian TM, Katowitz J, Hughes N, Schimmenti LA, Schneider A, & Young TL (2008). Identification of novel mutations and sequence variants in the SOX2 and CHX10 genes in patients with anophthalmia/microphthalmia. Molecular Vision, 14, 583–592. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, LAW, upon reasonable request.