

Abstract
Methyl groups are ubiquitous in biologically active molecules. Thus, new tactics to introduce this alkyl fragment into polyfunctional structures are of significant interest. With this goal in mind, a direct method for the Markovnikov hydromethylation of alkenes is reported. This method exploits the degenerate metathesis reaction between the titanium methylidene unveiled from Cp2Ti(μ-Cl)(μ-CH2)AlMe2 (Tebbe’s reagent) and unactivated alkenes. Protonolysis of the resulting titanacyclobutanes in situ effects hydromethylation in a chemo-, regio-, and site-selective manner. The broad utility of this method is demonstrated across a series of mono- and di-substituted alkenes containing pendant alcohols, ethers, amides, carbamates, and basic amines.
Keywords: hydromethylation, polyfunctional structures, site-specificity, synthetic methods, titanacyclobutanes
Graphical Abstract
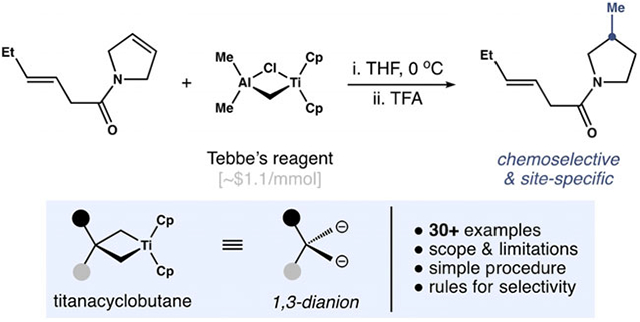
A method for the direct Markovnikov hydromethylation of unactivated alkenes by protonolysis of titanacyclobutanes has been developed. This approach enables site-specific incorporation of a methyl group into complex, polyfunctional molecules and has been demonstrated with a series of mono- and di-substituted alkenes containing pendant alcohols, ethers, amides, carbamates, and basic amines.
Site-specific methylation is a valuable strategy to optimize the pharmacology of bioactive small molecules.[1] This “magic methyl” effect has inspired new strategies for selective C–H bond methylation that facilitate the late-stage diversification of complex structures.[2,3] The addition of methane across a C–C π-system provides an appealing and complementary approach to small-molecule methylation. Nevertheless, despite advances in catalytic alkene hydrofunctionalization,[4] there are few direct methods for regioselective hydromethylation.[5–7] A procedure involving cyclopropanation and reductive C–C bond cleavage provides an indirect approach to this problem (Figure 1).[8] In contrast, the Baran group developed a more direct, branch-selective hydromethylation procedure using Fe-mediated hydrogen-atom transfer.[9,10] Herein, we describe the utility of Cp2Ti(μ-Cl)(μ-CH2)AlMe2 (1; Cp = C5H5) as a hydromethylation reagent.[11] This method facilitates site-specific incorporation of methyl substituents into polyfunctional structures and circumvents several of the intrinsic limitations of existing hydromethylation tactics.
Figure 1.
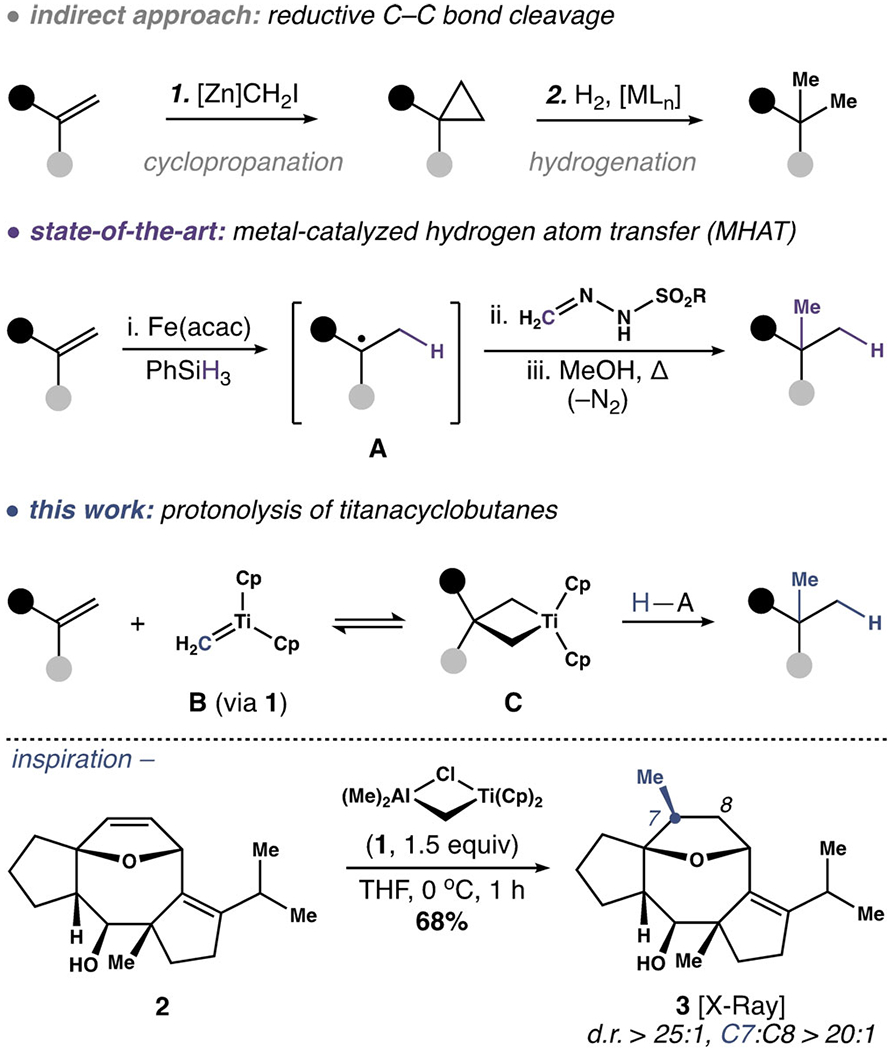
Regioselective hydromethylation of unactivated alkenes explored in the context of structure 2.
Our interest in hydromethylation strategies emerged from a program to synthesize fusicoccane diterpenes.[12] An early route intercepted structure 2, from which the regioselective addition of methane to the C7–C8 alkene became an attractive option to install the C7 methyl group of this terpene family. In practice, the poor reactivity profile of 2 made this task challenging. For example, exposure of 2, or protected variants, to combinations of electrophilic metals (e.g. Pd,[13] Fe,[14] Cu[15]) and nucleophilic methyl surrogates (e.g. ZnMe2) returned only starting material. Cyclopropanation was also intractable, requiring 10 equiv (TFA)ZnCH2I to achieve modest conversion.[16] Conversely, the Mukaiyama-type hydromethylation reported by Baran yielded only traces of target structure 3 alongside significant quantities of the corresponding net hydrogenation product arising from competitive reduction of radical species A.[17]
In search of a solution, we revisited the pioneering work of Tebbe et al.[18] and Grubbs and co-workers[19] concerning reagent 1, which serves as a progenitor to titanium methylidene B. While best known as an intermediate for carbonyl methylenation, B also participates in a degenerate metathesis reaction with unactivated alkenes.[20] In select cases, the resultant titanacyclobutanes (i.e. C) have been isolated and reacted with acid to give formal hydromethylation products.[20] However, these examples are largely constrained to simple hydrocarbons lacking other functional groups.[21,22] Thus, with some experimentation, we were pleased to find that the reaction of 1.5 equiv 1 with 2 afforded 3[23] in 68% yield after addition of SiO2 to the reaction.[24] The enhanced reactivity and regioselectivity (C7/C8 = 21:1) achieved with reagent 1 in this complex setting compelled us to explore further. Our efforts to transform this chemistry into a general method for alkene hydromethylation are summarized below.
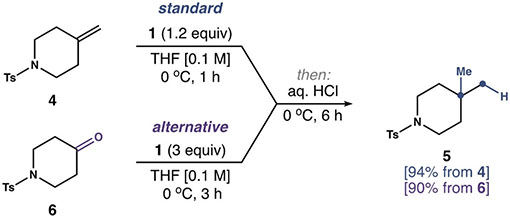
Our study began with a detailed investigation of reaction conditions (Table 1). Reaction parameters were explored using piperidine 4, which reacted with a solution of 1 (0.3–0.4 m in PhMe)[25] and 4-dimethylaminopyridine (DMAP) at 0°C.[19c] After 6 h, addition of HCl furnished an inseparable mixture of net Markovnikov hydromethylation product 5 (16% conversion) and 4 (entry 1). We observed no reaction in the absence of a Lewis base (entry 2). In contrast, the reaction was improved using THF as the Lewis base (entry 3), and the best results (94% yield) were obtained using THF as the solvent (entry 4). We found that HCl could be replaced by trifluoroacetic acid (TFA) without impacting the reaction efficiency (entry 5). Moreover, the reaction was executed on a gram scale to give 5 in 89 % yield (entry 6). Importantly, commercial solutions of 1 (0.5 m in PhMe) did not give comparable results (entry 7). The concentration of 1 was accurate; however, commercial 1 was darker than solutions of 1 freshly prepared from Cp2TiCl2 and AlMe3.[26] The same problem was encountered with prepared solutions of 1 after about 120 h, which suggests the formation of impurities upon storage.[27] With this practical issue noted, we found that ketone 6 was converted into 5 in 90 % yield with 3 equiv 1 using otherwise identical conditions. As such, this method also allows the direct geminal dimethylation of ketones.[28]
Table 1:
| |||
|---|---|---|---|
|
| |||
| Entry | Deviation from standard procedure | t [h] | Yield 5 [%] |
| 1 | PhMe, 1 equiv DMAP | 6 | 16[d] |
| 2 | PhMe | 6 | 0 |
| 3 | PhMe/THF (2:1) | 2 | 80 |
| 4 | none | 1 | 94 |
| 5 | TFA as proton source[e] | 1 | 92 |
| 6 | none, gram scale | 1 | 89 |
| 7 | commercial solution of 1[f] | 6 | 70[d] |
Yields are based on isolated 5.
Reactions were carried out on a 0.2 mmol scale.
Reagent 1 was prepared directly before use.[25]
Reflects conversion of 4 into 5 as judged by 1H NMR spectroscopic analysis of the unpurified reaction mixture.
The reaction was treated with TFA at −78°C and warmed to rt.
Commercial 1 at 0.52 m (in PhMe) was used as received.
A series of control reactions shed light on the properties of the titanacyclobutane 7 formed by the reaction of 1 and 4 (Scheme 1). It was unnecessary to isolate this transient species, which we observed in 1H NMR spectra of unpurified reaction mixtures before protonolysis.[25] Instead, the thermal stability of 7 was established by forming the metallacycle in situ at 0°C, then warming the reaction for 1 h before the addition of HCl. This analysis revealed significant cyclo-reversion to 4 after 1 h at 35°C (50% by 1H NMR spectroscopy).[29] Conversely, 7 persisted for 10 h at 0°C when precautions were taken to exclude air. The introduction of oxygen (1 atm) resulted in the rapid formation of by-products, the most significant being aldehyde 8. However, when handled as described, 7 functions as a useful 1,3-dianion equivalent. This feature was showcased by the reaction of DCl with 7 at 0°C to furnish isotopically labeled 5-d2 in 90% yield and with ≥ 90 % deuterium incorporation.
Scheme 1.
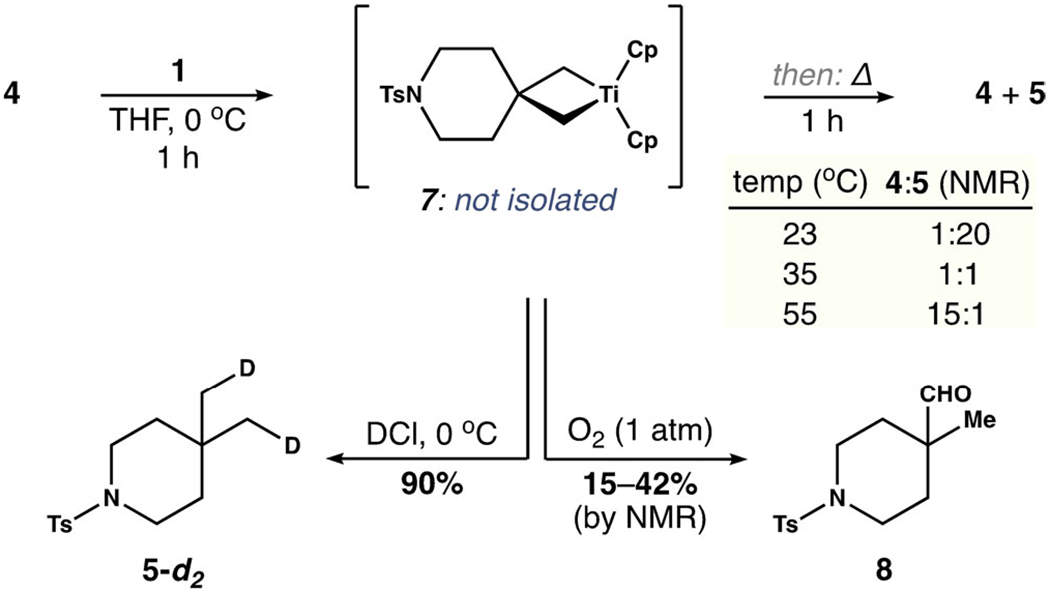
Reactivity of titanacyclobutane 7.
As shown in Scheme 2, the regioselectivity of this method was highlighted using α-olefins (9). In principle, both branched (b) and linear (l) hydromethylation isomers of 10 are accessible by protonolysis of titanacycles I and II, respectively. However, using the standard procedure, 4-phenyl-1-butene (9a) gave branched alkane 10a (72% yield, > 25:1 b/l), indicating the selective generation of the primary alkyl titanacycle I. In comparison, pyridine congener 9b afforded 10b in 71 % yield, but with reduced regioselectivity (3:1 b/l). In this case, the secondary alkyl titanacycle II is stabilized by coordination to the pendant nitrogen atom through a six-membered chelate. Increasing the reaction time to 3 h before the addition of acid improved the branched selectivity (10b, 3:1→6:1 b/l), which suggests that intermediates I and II equilibrate under the reaction conditions. A directing effect was not observed using homoallylic ether 9c, as shown by the regioselective formation of 10c after 1 h (52% yield, > 25:1 b/l). In contrast, modest branched selectivity was observed after 1 h with allyl arene derivatives 9d and 9e. As expected, the regioselectivity in both cases was enhanced to >25:1 by extending the reaction time. This modification allowed 10d and 10e to be isolated in yields of 78% and 85%, respectively.
Scheme 2.
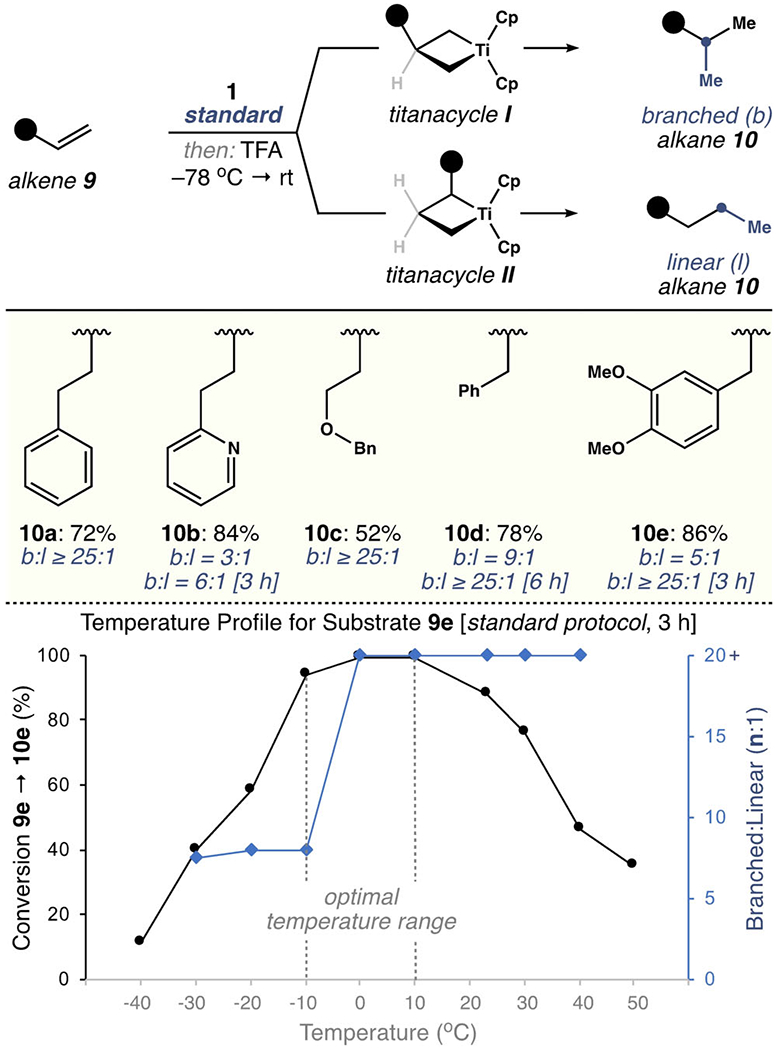
Regioselective hydromethylation of α-olefins.
To interrogate the role of temperature on the formation and equilibration of titanacyclobutanes I and II, we studied the conversion of methyl eugenol (9e) into products 10e using 1H NMR spectroscopy. Thus, 9e was reacted with 1.2 equiv 1 in THF (0.1m) for 3 h at various temperatures, then treated with TFA at −78°C. These experiments revealed a temperature window of −10°C to 10°C to achieve a high conversion into 10e (≥95%). Using these conditions, the regioselectivity (b/l ratio) improved from 8:1 at Ȓ10°C to >25:1 at 0°C. Taken together, these data demonstrate the importance of time and temperature as variables for reaction optimization.
With these considerations in mind, the hydromethylation of substituted alkenes was explored. As highlighted in Scheme 3, alkenes 11 were divided into four groups (I–IV) based on the structure of the titanacyclobutane formed during the reaction. Group I included exocyclic 1,1-disubstituted alkenes derived from nitrogen heterocycles (11a–g). Substrates of this type reacted at 0°C to afford branched products exclusively. Pendent carbamates (12a) and amides (12b) were tolerated, as were small- (12d–f) and medium-sized (12g) ring systems. In contrast, hydroquinoline 11c reacted slowly under the standard conditions, presumably because the resultant titanacyclobutane is more sterically hindered.
Scheme 3.
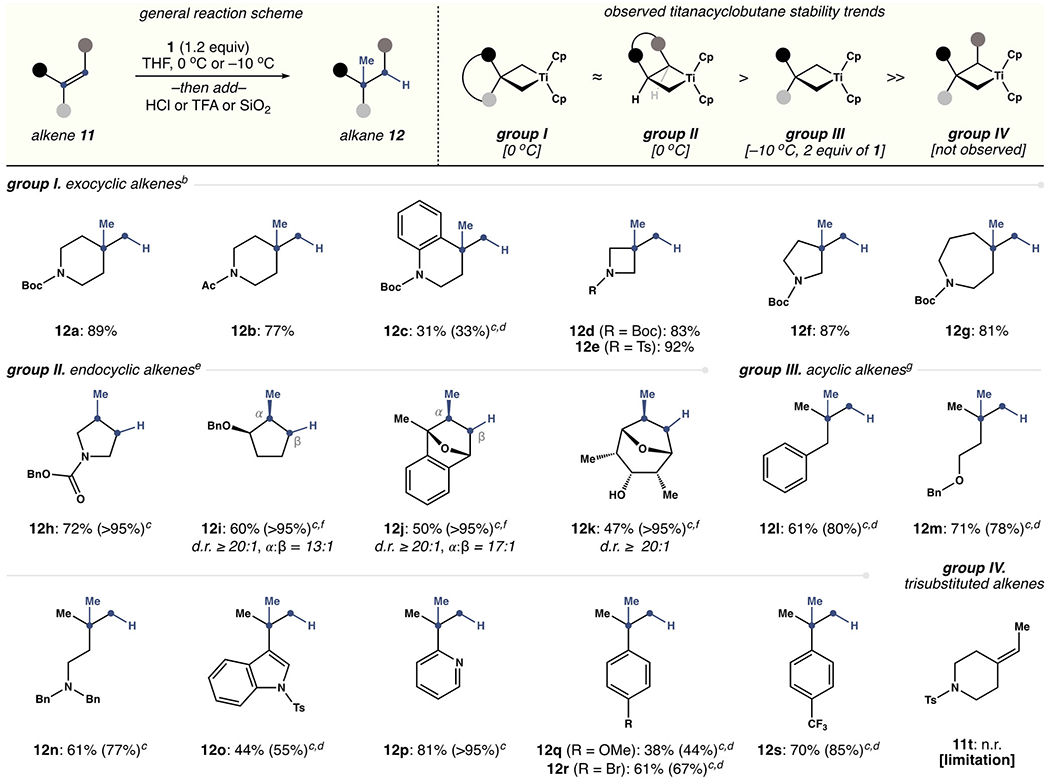
Scope and limitations of the titanium-mediated hydromethylation of alkenes.[a] [a] Yields are based on isolated 12. [b] Conditions: 1.2 equiv 1, 1 h; 3m aq HCl, 0°C, 6 h. [c] Conversion of 11 into 12 as judged by 1H NMR spectroscopic analysis of the unpurified reaction mixture. [d] Unreacted 11 was removed by treating the mixture of 11 and 12 with Meta-chloroperoxybenzoic acid (m-CPBA).[25] [e] Conditions: 1.2 equiv 1, 3 h; TFA, −78°C→rt, 6 h. [f] SiO2 in EtOAc used in place of TFA. [g] Conditions: 2 equiv 1, THF (0.1 m), −10°C, 3 h; TFA, −78°C→rt, 6 h.
Group II consisted of endocyclic 1,2-disubstituted alkenes (11h–k), which reacted at 0°C in an identical fashion to Group I. Similarly, heterocyclic carbamates (12h), ethers, and alcohols (12i–k) were tolerated.[30] We observed that the methyl group was selectively delivered to the more congested position (α) within 12i and 12j. This outcome is consistent with a requirement to place the titanium atom in the least sterically encumbered position. In comparison, branched acyclic alkenes in Group III (11l–s) were less reactive, requiring an excess of 1 (2 equiv) and longer reaction times (3 h, −10°C) to obtain useful results. Nevertheless, 1,1-disubstituted alkyl (11l–n) and (hetero)aryl (11o–s) alkenes were transformed to branched alkanes 12l–s in reasonable yield. Alkanes 12q–s derived from α-methylstyrene derivatives are noteworthy, as this alkene class is a limitation of the Baran hydromethylation.[9] On the other hand, trisubstituted alkenes (e.g. 11t) were unreactive. In addition, reactions of substrates bearing pendent aldehydes, ketones, or esters were complicated by competitive carbonyl methylenation.
With these limitations established, we set out to exploit the noted differences in alkene reactivity to achieve site-specific hydromethylation (Scheme 4). Thus, we found that the acyclic alkene in 13 reacted selectively to give 14 exclusively. We also observed complete selectivity for the α-olefin in 15 to afford 16 in 84% yield. Likewise, the cyclic alkene of 17 was functionalized, leaving the branched acyclic alkene untouched en route to amide 18. In contrast, the α-olefin of 19 reacted preferentially to give pyrrole 20 in 72% yield following oxidation of the pyrroline ring during purification.[31] Taken together, these competition experiments established the following order of alkene reactivity: α-olefins > cyclic alkenes > acyclic branched alkenes ⪢ trisubstituted alkenes.
Scheme 4.
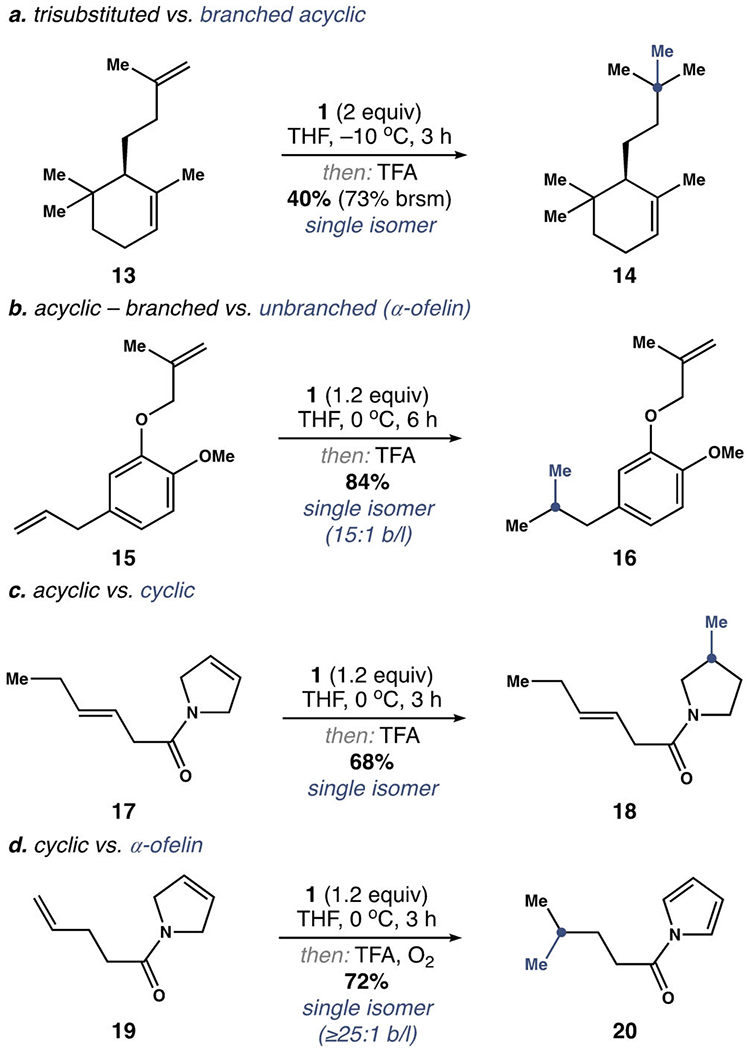
Site-specific hydromethylation.
In summary, a method for the direct hydromethylation of alkenes has been developed. This approach harnesses Tebbe’s reagent (1) to generate titanacyclobutanes from alkenes. These transient 1,3-dianion equivalents react in situ with exogenous acid to furnish net hydromethylation products with excellent regioselectivity. In defining the scope and limitations of this method, we established a clear hierarchy for alkene reactivity that allows site-specific hydromethylation within complex, polyfunctional molecules. This feature is especially useful for natural product synthesis and the late-stage diversification of bioactive small molecules.
Supplementary Material
Acknowledgements
This work was supported by the NIGMS branch of the National Institutes of Health (NIH) under award number R01GM125926. We thank Dr. Xinsong Lin (FSU) for assistance with X-ray crystallography and mass spectrometry.
Footnotes
Supporting information and the ORCID identification number for one of the authors of this article can be found under: https://doi.org/10.1002/anie.202103278.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Schönherr H, Cernak T, Angew. Chem. Int. Ed 2013, 52, 12256–12267; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2013, 125, 12480–12492. [Google Scholar]
- [2].Feng K, Quevedo RE, Kohrt JT, Oderinde MS, Reilly U, White MC, Nature 2020, 580, 621–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Friis SD, Johansson MJ, Ackermann L, Nat. Chem 2020, 12, 511–519. [DOI] [PubMed] [Google Scholar]
- [4].For select reviews, see; (a) Beller M, Seayd J, Tillack A, Jiao H, Angew. Chem. Int. Ed 2004, 43, 3368–3398; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2004, 116, 3448–3479; [Google Scholar]; (b) Shenvi RA, Matos JLM, Green SA, Org. React 2019, 383–470; [Google Scholar]; (c) Beletskaya IP, Najera C, Yus M, Russ. Chem. Rev 2020, 89, 250–274. [Google Scholar]
- [5].For Markovnikov hydromethylation methods, see; (a) Parnes ZN, Bolestova GI, Akhrem IS, Kursanov DN, J. Chem. Soc. Chem. Commun 1980, 748; [Google Scholar]; (b) Terao J, Watanabe T, Saito K, Kambe N, Sonoda N, Tetrahedron Lett. 1998, 39, 9201–9204; [Google Scholar]; (c) Fontaine F-G, Tilley TD, Organometallics 2005, 24, 4340–4342. [Google Scholar]
- [6].For anti-Markovnikov hydromethylation methods, see; (a) Clausen F, Kischkewitz M, Bergander K, Studer A, Chem. Sci 2019, 10, 6210–6214; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhu Q, Nocera DG, J. Am. Chem. Soc 2020, 142, 17913–17918. [DOI] [PubMed] [Google Scholar]
- [7].Zirconium-catalyzed carboalumination (ZACA) reactions also provide an entry point to formal alkene hydromethylation, see; (a) Negishi E, Dalton Trans. 2004, 827–848; [DOI] [PubMed] [Google Scholar]; (b) Van Horn DE, Valente LF, Idacavage MJ, Negishi E, J. Organomet. Chem 1978, 156, C20–C24. [Google Scholar]
- [8].For examples in natural product total synthesis, see; (a) McMurry JE, Erion MD, J. Am. Chem. Soc 1985, 107, 2712–2720; [Google Scholar]; (b) Pattenden G, Roberts L, Blake A, J. Chem. Soc. Perkin Trans. 1 1998, 863–868; [Google Scholar]; (c) Deng J, Ning Y, Tian H, Gui J, J. Am. Chem. Soc 2020, 142, 4690–4695. [DOI] [PubMed] [Google Scholar]
- [9].Dao HT, Li C, Michaudel Q, Maxwell BD, Baran PS, J. Am. Chem. Soc 2015, 137, 8046–8049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].For a related catalytic MHAT hydroalkylation method, see; Green SA, Huffman TR, McCourt RO, van der Puyl V, Shenvi RA, J. Am. Chem. Soc 2019, 141, 7709–7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pine SH, Org. React 1993, 43, 1–90. [Google Scholar]
- [12].Salvati AE, Law JA, Liriano J, Frederich JH, Chem. Sci 2018, 9, 5389–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lautens M, Hiebert S, J. Am. Chem. Soc 2004, 126, 1437–1447. [DOI] [PubMed] [Google Scholar]
- [14].Nakamura M, Matsuo K, Inoue T, Nakamura E, Org. Lett 2003, 5, 1373–1375. [DOI] [PubMed] [Google Scholar]
- [15].(a) Bertozzi F, Pineschi M, Macchia F, Arnold LA, Minnaard AJ, Feringa BL, Org. Lett 2002, 4, 2703–2705; [DOI] [PubMed] [Google Scholar]; (b) Zhang W, Wang L, Shi W, Zhou Q, J. Org. Chem 2005, 70, 3734–3736. [DOI] [PubMed] [Google Scholar]
- [16].Lorenz JC, Long J, Yang Z, Xue S, Xie Y, Shi Y, J. Org. Chem 2004, 69, 327–334. [DOI] [PubMed] [Google Scholar]
- [17].Competitive reduction of nucleophilic radical A was reported as a problem using sterically hindered substrates. See Ref. [9].
- [18].(a) Tebbe FN, Parshall GW, Reddy GS, J. Am. Chem. Soc 1978, 100, 3611–3613; [Google Scholar]; (b) Tebbe FN, Harlow RL, J. Am. Chem. Soc 1980, 102, 6149–6151. [Google Scholar]
- [19].(a) Pine SH, Zahler R, Evans DA, Grubbs RH, J. Am. Chem. Soc 1980, 102, 3270–3272; [Google Scholar]; (b) Ott KC, Grubbs RH, J. Am. Chem. Soc 1981, 103, 5922–5923; [Google Scholar]; (c) Buchwald SL, Grubbs RH, J. Am. Chem. Soc 1983, 105, 5490–5491; [Google Scholar]; (d) Ho SCH, Straus DA, Grubbs RH, J. Am. Chem. Soc 1984, 106, 1533–1534. [Google Scholar]
- [20].For seminal reports, see; (a) Howard TR, Lee JB, Grubbs RH, J. Am. Chem. Soc 1980, 102, 6876–6878; [Google Scholar]; (b) Krusic PJ, Tebbe FN, Inorg. Chem 1982, 21, 2900–2902; [Google Scholar]; (c) Lee JB, Ott KC, Grubbs RH, J. Am. Chem. Soc 1982, 104, 7491–7496. [Google Scholar]
- [21].For examples, see; Brown-Wensley KA, Buchwlad SL, Cannizzo L, Clawson L, Ho S, Meinhardt D, Stille JR, Straus D, Grubbs RH, Pure Appl. Chem 1983, 55, 1733–1744, [Google Scholar]; and references therein.
- [22].Hydromethylation was noted as a by-product competing reaction? in carbonyl methylenation reactions employing excess 1, see; Harrowven DC, Lucas MC, Howes PD, Tetrahedron 2001, 57, 791–804. [Google Scholar]
- [23].The utility of 1 in carbonyl-olefin metathesis using highly functionalized substrates has been demonstrated, see; (a) Stille JR, Grubbs RH, J. Am. Chem. Soc 1986, 108, 855–856; [Google Scholar]; (b) Nicolaou KC, Postema MHD, Claiborne CF, J. Am. Chem. Soc 1996, 118, 1565–1566. [Google Scholar]
- [24].Deposition Number 2059774 (for 3) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
- [25].See the Supporting Information for additional details.
- [26].Reagent 1 was prepared from Cp2TiCl2 and AlMe3 (2.0m in PhMe). Based upon current prices for these reagents, the cost of 1 was ca. $1.1mmol−1 when prepared as described in the Supporting Information. In our hands, the titration of 1 with 2-tert-butylcyclohexanone was difficult to interpret. An alternative titration using p-anisaldehyde was developed. These procedures were adapted from an earlier report:; Cannizzo LF, Grubbs RH, J. Org. Chem 1985, 50, 2386–2387. [Google Scholar]
- [27].Krusic and Tebbe studied the degradation of 1 in PhMe. Several titanium species are generated, including (Cp2TiCl)2. See Ref. [20b].
- [28].For an overview of methods relating to ketone gem-dialkylation, see; Seebach D, Angew. Chem. Int. Ed 2011, 50, 96–101; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2011, 123, 99–105. [Google Scholar]
- [29].The thermal cycloreversion of titanacyclobutanes to alkenes and Cp2TiCH2 has been described. See Refs. [19] and [20].
- [30].The relative stereochemistry of 12i–12k was assigned by comparison to previously reported data for these structures.
- [31].Structures 17 and 19 were intrinsically sensitive to oxidation and slowly oxidized to the corresponding acyl pyrroles under ambient conditions.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.