

Abstract
BACKGROUND AND AIMS:
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), caused by autoimmune regulator (AIRE) mutations, manifests with chronic mucocutaneous candidiasis (CMC) and multisystem autoimmunity, most often hypoparathyroidism (HP) and adrenal insufficiency (AI). European cohorts previously reported a ~10% prevalence of APECED-associated hepatitis (APAH) with presentations ranging from asymptomatic laboratory derangements to fatal fulminant hepatic failure. Herein, we characterized APAH in a large APECED cohort from the Americas.
APPROACH AND RESULTS:
Forty-five consecutive patients with APECED were evaluated (2013-2015) at the National Institutes of Health (NIH; NCT01386437). Hepatology consultation assessed hepatic and autoimmune biomarkers and liver ultrasound in all patients. Liver biopsies evaluated autoimmune features and fibrosis. The 16S ribosomal RNA (rRNA) sequencing was performed in 35 patients’ stools (12 with and 23 without APAH). Among 43 evaluable patients, 18 (42%) had APAH; in 33.3% of those with APAH, APAH occurred before developing classic APECED diagnostic criteria. At APAH diagnosis, the median age was 7.8 years, and patients manifested with aminotransferase elevation and/or hyperbilirubinemia. All patients with APAH were in clinical remission during their NIH evaluation while receiving immunomodulatory treatment. We found no difference in age, sex, or prevalence of CMC, AI, or HP between patients with or without APAH. Autoantibody positivity against aromatic L-amino acid decarboxylase, cytochrome P450 family 1 subfamily A member 2, histidine decarboxylase (HDC), bactericidal/permeability-increasing fold-containing B1, tryptophan hydroxlase, and 21-h ydroxylase (21-OH), and the homozygous c.967_979del13 AIRE mutation were associated with APAH development. Classical serological biomarkers of autoimmune hepatitis (AIH) were only sporadically positive. AIH-like lymphoplasmacytic inflammation with mild fibrosis was the predominant histological feature. Stool microbiome analysis found Slackia and Acidaminococcus in greater abundance in patients with APAH.
CONCLUSIONS:
APAH is more common than previously described, may present early before classic APECED manifestations, and most often manifests with milder, treatment-responsive disease. Several APECED-associated autoantibodies, but not standard AIH-associated biomarkers, correlate with APAH.
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) is a monogenic autoimmune disorder caused by impaired central tolerance.(1) Biallelic mutations in the autoimmune regulator (AIRE) gene are required for diagnosis, or a patient must have ≥2 among the classic triad manifestations of chronic mucocutaneous candidiasis (CMC), hypoparathyroidism (HP), and adrenal insufficiency (AI).(2,3) Beyond the triad, several endocrine and nonendocrine manifestations develop with varying frequencies among patient cohorts, including hypothyroidism, hypogonadism, type 1 diabetes, urticarial eruption, vitiligo, alopecia, asplenia, Sjogren-like syndrome, pneumonitis, gastritis, pernicious anemia, intestinal dysfunction, nephritis, and hepatitis.(2,4–7)
APECED-associated hepatitis (APAH) has been reported to have a wide clinical presentation ranging from mild asymptomatic laboratory derangements to life-threatening fulminant hepatic failure requiring emergent liver transplantation. APAH has been reported in nearly all large APECED cohorts with an overall prevalence of ~16% but a reported prevalence as low as ~3% in certain populations.(2,8–11) APAH is the initial APECED manifestation in <2% of patients.(8–11) APAH is considered to share common features with autoimmune hepatitis (AIH) and was previously considered to fit into an AIH subtype. AIH is associated with extrahepatic organ-specific autoimmunity including several nontriad APECED manifestations, such as type 1 diabetes, thyroiditis, and pernicious anemia.(5,12) Because of this extrahepatic disease association, APECED is considered in the differential diagnosis for patients presenting with AIH. Indeed, autoimmunity is considered the prevailing pathological mechanism of APAH. However, other studies suggest that APAH may be a unique clinical variant separate from classical AIH. For instance, Vogel et al. showed that only 2 of 188 patients with AIH had AIRE mutations.(13) In addition, hallmark AIH-associated antibodies, such as anti–smooth muscle antibody (ASMA), antinuclear antibody (ANA), liver-kidney microsome (LKM), antimitochondrial antibody, and soluble liver antigen (SLA), are variably present in patients with APECED with low sensitivity or specificity for liver disease.(3,4,14,15) Moreover, sera from large cohorts of Italian and Finnish patients with APECED revealed no consistent pattern in autoantigens for AIH type 1, AIH type 2, or APAH.(16,17) Few hepatic autoantigens have been r eported in patients with APECED and APAH.(17–19) Autoantibodies targeting cytochrome P450 family 1 subfamily A member 2 (CYP1A2) and aromatic L-amino acid decarboxylase (AADC) are enriched in patients with APAH; however, CYP1A2 autoantibodies were reported to lack sensitivity, and AADC autoantibodies were reported to lack specificity for APAH.(16,19) No liver-specific autoantigen has been identified to date that reliably and specifically predicts all patients with APAH. Collectively, these data argue that APAH may be a distinct entity from classical AIH.
Conversely, APAH shares some clinical and histological features with AIH. Typically, APAH manifests in early life before the third decade. Predominant reported histological features include mixed inflammatory infiltrates with plasma cell and eosinophil predominance, which accompany interface hepatitis.(14) Biopsies show varying degrees of fibrosis, but most prior reports suggest advanced fibrosis to cirrhosis may occur at a young age. Clinically, APAH may be silent, detected as only mild aminotransferase elevations, whereas other patients may present with fulminant hepatic failure. Interestingly, this wide clinical variation can occur even among siblings.(3)
To date, the description of APAH is largely confined to case reports and small case series. Although attempts have been made to predict patients with APECED who would develop APAH, there has been no reliable correlative AIRE mutation or autoantibody pattern yet identified. A detailed description of APAH features and their potential correlation with other APECED manifestations is limited to predominantly European cohorts and is lacking in other patient populations, such as in the Americas. Moreover, limited data exist on APAH treatment responses.
We systematically characterized clinical, biochemical, serological, histological, and treatment data of APAH in a large APECED cohort predominantly from the Americas followed up at the NIH through unbiased enrollment of 43 consecutive patients who underwent uniform and extensive multidisciplinary evaluation, including hepatology consultation regardless of the presence of liver disease. We provide a set of data on APAH in American patients with APECED and data on the response of APAH to classical AIH-associated immunosuppressive treatments. We show that, contrary to previous reports, the clinical course of APAH is more indolent with less-advanced fibrosis and demonstrate that APAH can be controlled with immunosuppressive treatment. Moreover, the gut microbiome can modulate immune responses, and dysbiosis has been described in several autoimmune disorders including AIH and in patients with APECED with intestinal symptoms, although evaluation of the gut microbiome in patients with APAH is lacking.(20–22) For that, we employed 16S rRNA sequencing to characterize the gut microbiome in patients with APECED with or without APAH and found alterations in specific bacterial genera in patients with APAH.
Patients and Methods
PATIENTS
Patients were evaluated at the NIH in a National Institute of Allergy and Infectious Diseases institutional review board–approved prospective observational natural history study (NCT01386437). All patients, or if minors, their parents, provided informed written consent. The study was conducted in accordance with the Declaration of Helsinki.
Patients were predominantly recruited from North and South America. Inclusion criteria included the presence of biallelic AIRE mutations/deletions or a clinical APECED diagnosis based on having ≥2 of the classic triad manifestations (CMC, HP, AI). Diagnosis of CMC, HP, AI, and other endocrine or nonendocrine manifestations was made as described.(6) Patients were excluded if they were found not to have biallelic AIRE mutations/deletions and did not meet a classic APECED diagnostic dyad after protocol evaluation.
DIAGNOSIS OF APAH AND LABORATORY, HISTOLOGICAL, AUTOANTIBODY, AND MICROBIOMIC ANALYSES
All patients were seen by the Gastroenterology and Hepatology consult service at the NIH regardless of whether they had a history of or active gastrointestinal or hepatic manifestations of APECED at the time of NIH evaluation. The patients’ prior labs and liver biopsies, when available, were reviewed for the presence of AIH. Laboratory testing assessed standard measures of hepatic function such as aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), total and direct bilirubin, gamma-glutamyltransferase (GGT), primary and secondary bile acids; markers of hepatic synthetic function, namely international normalized ratio (INR) and platelet counts; and classical AIH-associated markers, such as ANA, immunoglobulins, ASMA, anti-LKM, and SLA. Other causes of chronic liver disease, including viral hepatitis, were excluded with serologic and/or PCR testing and biopsy assessment. As no patients were found to have positive AIH biomarkers at the time of NIH evaluation, the diagnosis of APAH relied on diagnostic liver biopsy in all cases where biopsies were available. Both transjugular and percutaneous liver biopsy specimens were considered adequate so long as there was sufficient tissue for histological analysis. If a liver biopsy was unavailable, diagnosis relied on improvement in liver-associated enzyme abnormalities after initiation of treatment for APAH or AIH as determined by review of clinical documentation. If patients had multiple liver biopsies, the earliest biopsy was used to determine the date of APAH diagnosis, as immunosuppressive treatment can affect subsequent hepatic histology.
Liver biopsies, whether performed at or outside the NIH, were reviewed by an expert hepatopathologist (D.K.) to establish or confirm the diagnosis. Liver specimens were evaluated for autoimmune features and scored based on the histological activity index and Ishak fibrosis (IF) scoring systems. Lymphocyte immunophenotyping was performed on selected cases based on material availability using antibodies to CD3, CD4, CD8, and CD20.
Sera from patients with APECED were subjected to autoantibody analysis by radioligand binding assay, and stool from patients with APECED was subjected to microbiomic analyses by 16S rRNA sequencing, as described in the Supporting Information.
STATISTICAL ANALYSES
Statistical analyses were performed using SAS, Prism, and R. P < 0.05 was considered significant. To compare dichotomous variables, Pearson’s chi-square test and Fisher’s exact test were employed. Kaplan–Meier curves and log rank tests were used to analyze time to development to APAH.
Results
APAH IS A COMMON MANIFESTATION OF AMERICAN PATIENTS WITH APECED AND FEATURES AN INDOLENT CLINICAL COURSE
Forty-five consecutive patients were referred for evaluation at the NIH (May 2013-July 2015), through self-referral or referral from their primary or subspecialty physicians. Among those patients, 2 were excluded from analysis; one did not meet clinical APECED criteria or carry AIRE mutations/deletions on genetic testing. Another patient with APECED had undergone hematopoietic stem cell transplantation 17 years before NIH evaluation and was excluded to avoid the confounding effects of this intervention. Therefore, 43 patients were included in our study. Table 1 presents their clinical and laboratory data at the time of the NIH evaluation. Twenty-seven (62.8%) were female. Forty (93%) were from the Americas (37 from the United States; one each from Canada, Argentina, and Colombia); the remainder were from Germany, the United Kingdom, and the United Arab Emirates. Forty-two patients were Caucasian, amongst which five coidentified as Hispanic, and one patient was Asian. The most common AIRE mutant alleles identified were c.967_979del13 (p.L323SfsX51), followed by c.769C>T (R257X) (Supporting Table S1). Among the classic triad manifestations, 37 (86%) had AI, 38 (88%) had HP, and 37 (86%) had CMC.
TABLE 1.
Demographic, Clinical, and Laboratory Features of Patients With APECED and With and Without APAH at the Time of NIH Evaluation
| Characteristic | All Patients | Patients With APAH | Patients Without APAH | P Value |
|---|---|---|---|---|
| Number of patients | 43 | 18 | 25 | N/A |
| Median age at NIH evaluation (years) | 20 (16-32) | 19 (14-23) | 24 (15-38) | 0.5 |
| Median age at diagnosis of hepatitis (years) | 7.8 (3.5-14) | 7.8 (3.5-14) | N/A | N/A |
| Female sex | 27 (63) | 13 (72) | 14 (56) | 0.35 |
| Ethnicity: | ||||
| Caucasian | 42 (98) | 18 (100) | 24 (96) | |
| Hispanic | 5 (12) | 3 (17) | 2 (8) | |
| Asian | 1 (2) | 0 (0) | 1 (4) | |
| Classic triad manifestations | ||||
| CMC | 37 (86) | 15 (83) | 22 (88) | 0.68 |
| HP | 38 (88) | 15 (83) | 23 (92) | 0.63 |
| AI | 37 (86) | 14 (83) | 23 (92) | 0.22 |
| Other nontriad manifestations | ||||
| Enamel hypoplasia | 37 (86) | 17 (94) | 20 (80) | 0.37 |
| Hypothyroidism | 12 (28) | 4 (22) | 8 (32) | 0.73 |
| Growth hormone deficiency | 7 (16) | 4 (22) | 3 (12) | 0.43 |
| DM type 1 | 6 (14) | 4 (22) | 2 (8) | 0.22 |
| Hypogonadism | 17 (40) | 6 (33) | 11 (44) | 0.54 |
| Gastritis | 20 (47) | 10 (56) | 10 (40) | 0.37 |
| Pernicious anemia | 12 (28) | 5 (28) | 7 (28) | >0.99 |
| Intestinal dysfunction | 33 (77) | 17 (94) | 16 (64) | 0.03 |
| Keratoconjunctivitis | 15 (35) | 6 (33) | 9 (36) | >0.99 |
| APECED rash | 28 (65) | 16 (89) | 12 (48) | 0.01 |
| Vitiligo | 17 (40) | 9 (50) | 8 (32) | 0.34 |
| Alopecia | 11 (26) | 4 (22) | 7 (28) | 0.74 |
| Nail dystrophy | 13 (30) | 5 (28) | 8 (32) | >0.99 |
| Pneumonitis | 17 (40) | 11 (61) | 6 (24) | 0.03 |
| Sjogren-like syndrome | 20 (47) | 9 (50) | 11 (44) | 0.76 |
| TIN | 2 (5) | 0 (0) | 2 (8) | 0.50 |
| HTN | 10 (23) | 2 (11) | 8 (32) | 0.15 |
| Asplenia | 4 (9) | 3 (17) | 1 (4) | 0.29 |
| Laboratory values | ||||
| ALT (U/L) | 22 (17-30) | 22 (18-33) | 25 (17-26) | 0.58 |
| AST (U/L) | 23 (17-30) | 23 (16-36) | 23 (19-25) | 0.53 |
| ALP (U/L) | 109 (75-181) | 141 (78-227) | 106 (70-166) | 0.11 |
| ALP% (age and sex adjusted normal) | 64 (53-89) | 80 (64-120) | 57 (48-73) | 0.03 |
| GGT (U/L) | 15 (12-26) | 15 (13-32) | 15 (11-24) | 0.48 |
| Total bilirubin (mg/dL) | 0.3 (0.2-0.5) | 0.3 (0.2-0.5) | 0.3 (0.2-0.5) | 0.88 |
| Direct bilirubin (mg/dL) | 0.2 (0.1-0.2) | 0.2 (0.2-0.2) | 0.2 (0.1-0.2) | 0.48 |
| Albumin (g/dL) | 4.1 (3.8-4.3) | 4.0 (3.8-4.2) | 4.2 (4.0-4.4) | 0.06 |
| ESR (mm/hour) | 12 (7-18) | 14 (10-18) | 10 (5-16) | 0.06 |
| CRP (mg/L) | 2.15 (0.7-7.27) | 0.6 (0.5-5.6) | 5.1 (0.8-7.3) | 0.24 |
| IgG (g/dL) | 1,070 (894-1,313) | 1,250 (902-1,610) | 1,050 (892-1,170) | 0.10 |
| Platelets (K/μL) | 266 (202-339) | 261 (225-376) | 274 (202-330) | 0.65 |
| INR | 1.04 (0.99-1.11) | 1.07 (0.99-1.12) | 1.04 (0.98-1.11) | 0.36 |
| Ferritin (μg/L) | 33 (14-72) | 38 (14-65) | 29 (13-107) | 1.0 |
| Autoimmune biomarkers | ||||
| ANA (+) | 7 (16.3) | 2 (11.1) | 5 (20) | 0.68 |
| ASMA (+) | 0 (0) | 0 (0) | 0 (0) | N/A |
| Anti-LKM (+) | 0 (0) | 0 (0) | 0 (0) | N/A |
| AMA (+) | 0 (0) | 0 (0) | 0 (0) | N/A |
Data are presented as n (%) or median (IQR).
Abbreviations: AMA, antimitochondrial antibody; DM, diabetes mellitus; HTN, hypertension; IgG, immunoglobulin G; IQR, interquartile range; N/A, not applicable; TIN, tubulointerstitial nephritis.
Eighteen patients (41.9%) had a history of APAH; the diagnosis was confirmed through histologic examination of prior liver biopsies at the NIH in 16 of them, whereas in the remaining 2 patients in whom the prior liver biopsy was unavailable for review at NIH, the diagnosis was made by clinical history and response to immunosuppressive treatment. No previously unrecognized cases of APAH were diagnosed during the NIH evaluation. The median age of diagnosis of APAH was 7.8 years (mean, 9.7 years; range, 2-31 years; interquartile range, 3.5-14 years). In 6 (33%) of the 18 patients with APAH, APAH developed before meeting a classic diagnostic dyad of APECED; the mean age of APAH diagnosis in these patients was 3.8 years (range, 2-9 years). The mean interval between onset of APAH and development of a classic diagnostic dyad in 4 of these 6 patients was 0.625 years (range, 0.5-1.5 years). The remaining 2 patients developed APAH at 6 years (14 years old at the time of the study) and 8 years (12 years old at the time of the study) and have had a genetic diagnosis of APECED but have not yet developed a classic diagnostic dyad. The remaining 12 patients developed APAH after already having had a diagnosis of APECED, with APAH developing at a mean age of 12.1 years (range, 2.5-31.9 years).
The mean age of patients with APAH at the time of their NIH evaluation was 20.2 years (range, 7-31.9 years) relative to a mean age of 28.1 years (range, 7.3-67.8 years) of those without APAH. Female patients were more commonly affected by APAH (13 patients, 72.2%) than male patients (5 patients, 27.8%). Fifteen were from the United States, and one each was from Colombia, Argentina, and the United Kingdom. Fourteen (77.8%) had AI, 15 (83.3%) had HP, and 15 (83.3%) had CMC. Patient age and the prevalence of CMC, HP, or AI were not statistically different between those with and those without APAH. The prevalence of intestinal dysfunction (94% versus 64%; P = 0.03), APECED rash (89% versus 48%; P = 0.01), and pneumonitis (61% versus 24%; P = 0.03) was significantly greater among patients with APAH versus those without APAH (Table 1).
At the time of APAH diagnosis, 9 (50%) patients had overt signs or symptoms consistent with liver derangement, including 7 (38.8%) patients with jaundice and/or scleral icterus and 2 (11.1%) patients with pruritus without accompanying jaundice, which resolved with APAH-directed treatment. Because of coexistent intestinal dysfunction in the vast majority of patients with APAH (Table 1), presence of abdominal pain, nausea, or vomiting could not be reliably attributed to APAH. Similarly, fatigue, another symptom of liver dysfunction, could not be readily attributed to APAH given other potential causes, such as AI. Notably, no patient presented with acute liver failure. At the time of NIH evaluation, all patients were asymptomatic from an APAH standpoint.
A BIOCHEMICAL PATTERN OF HEPATOCELLULAR INJURY AND SPORADIC POSITIVITY OF SEROLOGIC BIOMARKERS OF CLASSICAL AIH UNDERLIE APAH
At the time of NIH evaluation, all patients with APAH had the APAH medically controlled. As a result, levels of ALT, AST, albumin, bilirubin, INR, and immunoglobulin G as well as platelet counts were not statistically different between patients with and without APAH (Table 1). ALP trended higher in those with APAH at the time of NIH evaluation, although this did not reach statistical significance (P = 0.11). When ALP was transformed as a percentage of upper limit of normal-age-sex-adjusted ALP (ALP%), ALP% was significantly greater in patients with APAH (P = 0.03), and this difference remained significant when adjusted for the presence of HP (P = 0.02). Among inflammatory markers, erythrocyte sedimentation rate (ESR; P = 0.06), but not C-reactive protein (CRP), was greater in patients with APAH at the time of NIH evaluation (Table 1). Levels of primary and secondary bile acids in serum were not different between patients with and without APAH at the time of NIH evaluation (data not shown).
Laboratory data could be obtained from 6 patients at the time of APAH diagnosis, all before the NIH evaluation. These patients had a predominantly hepatocellular injury pattern with elevated AST and ALT, with aminotransferase levels exceeding 1,000 IU/L in 2 out of 6 evaluable patients (33.3%); conversely, ALP, GGT, and bilirubin were variably elevated (Table 2).
TABLE 2.
APAH Features a Hepatocellular Injury Pattern With Only Sporadic Positivity of Serologic Biomarkers of Classic AIH
| Patient # | AST (U/L) | ALT (U/L) | ALP (U/L) | Total Bilirubin (mg/dL) | Direct Bilirubin (mg/dL) | GGT (U/L) | Classical AIH Markers |
||
|---|---|---|---|---|---|---|---|---|---|
| ANA | ASMA | Anti-LKM | |||||||
| 1 | 241 | 324 | 199 | 0.4 | N/A | 35 | − | + | + |
| 2 | 88 | 134 | 230 | 0.5 | 0.5 | 42 | − | N/A | − |
| 3 | 930 | 1,992 | 164 | 2.3 | 0.7 | 525 | − | − | − |
| 4 | 93 | 105 | N/A | N/A | N/A | 525 | N/A | − | − |
| 5 | 2,603 | 1,587 | 618 | 7.2 | 4.3 | N/A | N/A | − | +* |
| 6 | 98 | 75 | 133 | 0.41 | N/A | 81 | − | − | − |
Weak positive at titer 1:20.
Abbreviation: N/A, not available.
Interestingly, at the time of APAH diagnosis, only a minority of patients had positive serologic biomarkers of classical AIH. Specifically, 2 out of 6 evaluable patients were positive for anti-LKM and 1 out of 5 evaluable patients was positive for ASMA (also positive for anti-LKM), whereas none of the 4 evaluable patients were positive for ANA (Table 2). At the time of NIH evaluation, during APAH remission, none of the 18 patients with APAH had positive classical serologic biomarkers of AIH, namely anti-LKM, ASMA, or SLA (Table 1). Two out of 18 patients with APAH were positive for ANA at the time of NIH evaluation; however, the frequency of ANA positivity was not statistically different between patients with and without APAH (2 of 18 versus 5 of 25, respectively) (Table 1).
As part of the NIH evaluation, all patients underwent liver imaging with ultrasound, which did not reveal consistent abnormalities in those with APAH. Specifically, hepatomegaly was seen in 4 patients without APAH but in no patients with APAH. A coarsened echotexture on ultrasound was observed in 6 patients with APAH (33.3%) compared with 3 patients without APAH (12%; P = 0.09). Increased echogenicity was seen in 4 patients with APAH (graded as mild to moderate in all), as opposed to 6 patients without APAH (graded as mild in 5 and severe in 1 patient). One of the patients without APAH who had mildly increased echogenicity underwent liver biopsy, which revealed fatty infiltration.
THE HOMOZYGOUS AIRE c.967_979del13 MUTATION AND SEVERAL AUTOANTIBODIES CORRELATE WITH THE PRESENCE OF APAH
We examined whether there is a genotype-phenotype correlation between specific AIRE mutations and the presence of APAH. We found an association between carrying the AIRE c.967_979del13 mutation in homozygosity, but not in heterozygosity, and development of APAH; however, no such correlation was evident for the AIRE c.769C>T mutation or other AIRE mutations (Supporting Table S1, Supporting Fig. S1).
We then investigated whether certain serum autoantibodies might distinguish patients with and without APAH. A significantly higher prevalence of autoantibodies against AADC, CYP1A2, HDC, bactericidal/permeability-increasing fold-containing B1 (BPIFB1), tryptophan hydroxylase (TPH), and 21-OH were identified in patients with APAH relative to those without APAH (Table 3). Concordantly, these six autoantibodies were significantly associated with the time to development of APAH (Fig. 1). Conversely, we found no correlation between autoantibodies against transglutaminase 4, nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing 5, tyrosine phosphatase-related islet antigen 2, glutamic acid decarboxylase 65, sex determining region Y-box 10, 17-OH, potassium channel regulator, side-chain cleavage enzyme, interleukin-22, interferon (IFN)-α4, or IFN-ω and the time to development of APAH (Supporting Figs. S2 and S3). Autoantibodies against AADC and TPH had the highest sensitivity for APAH (~95%) but lacked specificity (Table 3). Conversely, among all autoantibodies examined, only autoantibodies against CYP1A2 exhibited 100% specificity for APAH, although with low sensitivity (~30%) (Table 3).
TABLE 3.
Summary of Autoantibody Positivity in Patients With APECED and With or Without APAH
| Autoantibodies | All Patients (n = 42) | Patients With APAH (n = 17) | Patients Without APAH (n = 25) | HR (95% CI) | P Value |
|---|---|---|---|---|---|
| AADC | 32 (76.2) | 16 (94.1) | 16 (64) | 8.39 (1.1-63.8) | 0.03 |
| CYP1A2 | 5 (11.9) | 5 (29.4) | 0 (0) | 7.89 (2.54-24.49) | 0.004 |
| HDC | 22 (52.3) | 13 (76.5) | 9 (36) | 4.38 (1.4-13.73) | 0.01 |
| TPH | 32 (76.2) | 16 (94.1) | 16 (64) | 6.3 (0.83-47.53) | 0.03 |
| BPIFB1 | 14 (33) | 10 (58.8) | 4 (16) | 3.85 (1.46-10.18) | 0.004 |
| 21-OH | 19 (45.2) | 11 (64.7) | 8 (32) | 2.94 (1.06-8.17) | 0.04 |
| GAD65 | 17 (40.4) | 8 (47.1) | 9 (36) | 1.27 (0.49-3.3) | 0.47 |
| IA-2 | 1 (2.3) | 0 (0) | 1 (4) | 0 (0-inf) | 0.41 |
| IFN-α4 | 41 (97.6) | 17 (100) | 24 (96) | (0-inf) | 0.40 |
| IFN-ω | 40 (95.2) | 16 (94.1) | 24 (96) | 0.84 (0.11-6.36) | 0.78 |
| IL-22 | 38 (90.4) | 16 (94.1) | 22 (88) | 2.18 (0.29-16.44) | 0.51 |
| KCNRG | 6 (14.3) | 3 (17.7) | 3 (12) | 1.05 (0.3-3.68) | 0.61 |
| NALP5 | 24 (57.1) | 12 (70.5) | 12 (48) | 1.93 (0.68-5.5) | 0.15 |
| SCC | 27 (64.2) | 11 (64.7) | 16 (64) | 1.16 (0.42-3.16) | 0.96 |
| SOX10 | 13 (30.1) | 6 (35.3) | 7 (28) | 1.21 (0.45-3.28) | 0.62 |
| TGM4 | 9 (21.4) | 2 (11.8) | 7 (28) | 0.35 (0.08-1.56) | 0.21 |
| 17-OH | 21 (50) | 10 (58.8) | 11 (44) | 1.56 (0.59-4.14) | 0.35 |
Data are presented as n (%) or median (IQR).
Abbreviations: CI, confidence interval; GAD65, glutamic acid decarboxylase 65; HR, hazard ratio; IA-2, tyrosine phosphatase-related islet antigen 2; IL-22, interleukin-22; inf, infinity; KCNRG, potassium channel regulator; NALP5, nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing 5; SCC, side-chain cleavage enzyme; SOX10, sex determining region Y-box 10; TGM4, transglutaminase 4.
FIG. 1.
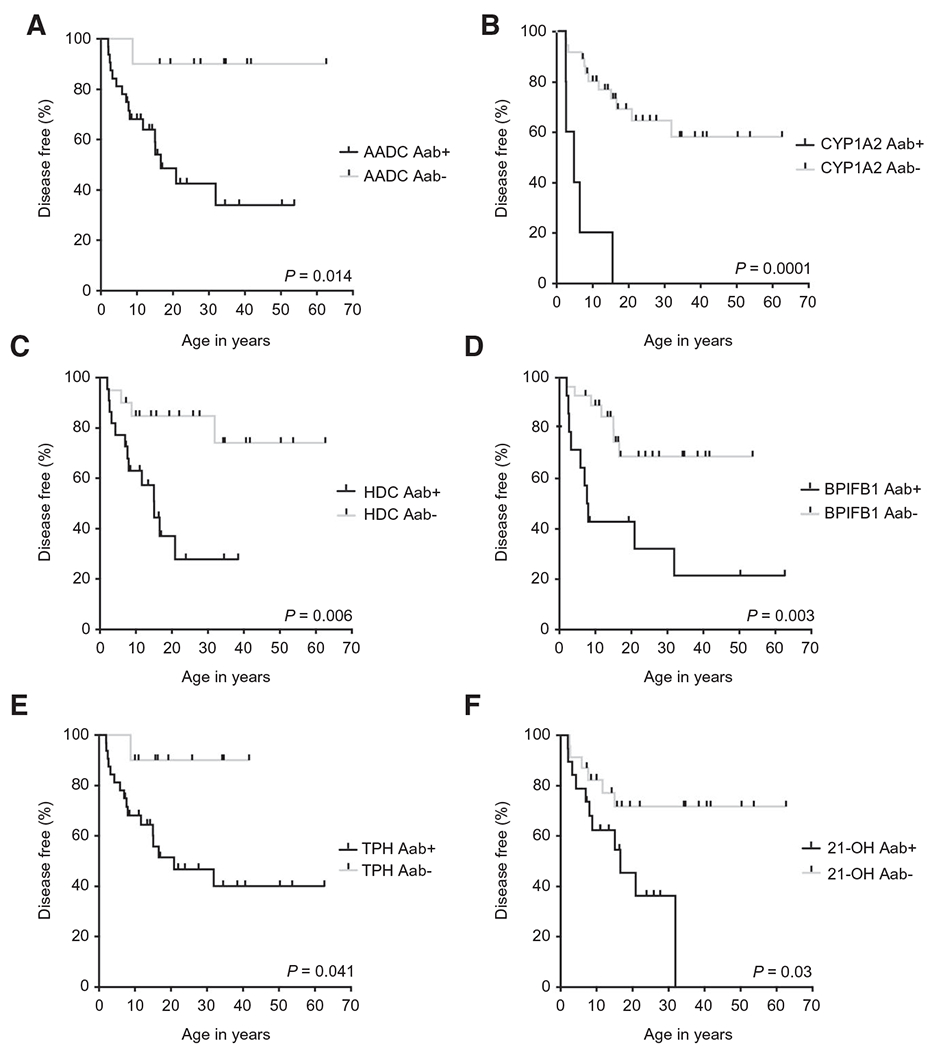
A variety of tissue-specific autoantibodies correlate with the time to development of APAH. Shown are Kaplan-Meier curves illustrating the correlation between the time to development of APAH with the presence or absence of autoantibodies against (A) AADC, (B) CYP1A2, (C) HDC, (D) BPIFB1, (E) TPH, and (F) 21-OH. P values were generated using the log-rank test for significance (n = 42). Abbreviations: APAH, APECED-associated hepatitis; KM, Kaplan-Meier; AADC, aromatic L-amino acid decarboxylase; BPIFB1, bactericidal/permeability-increasing fold-containing B1; CYP1A2, steroid 21-hydroylase; HDC, histidine decarboxylase; TPH, tryptophan hydroxylase; 21-OH, 21-hydroxylase.
APAH PRESENTS WITH AIH-LIKE LYMPHOPLASMACYTIC INFLAMMATION AND MILD FIBROSIS
Biopsies from 16 patients with APAH were available for review (Fig. 2, Supporting Figs. S4 and S5, Table 4). All but one biopsy showed either no fibrosis or just portal fibrotic expansion (IF stage of 1-2). Specifically, 11 biopsies (69%) showed no or very mild fibrosis with IF stage of 0-1 and 4 (25%) had an IF stage of 2. One biopsy (6.25%) showed bridging fibrosis with an IF stage of 3, and none were cirrhotic. Most of the biopsies (12/16, 75%) showed a chronic hepatitis pattern of inflammation with moderate-to-marked portal inflammation and interface hepatitis with only mild-to-moderate parenchymal inflammation. Two cases (12.5%) showed relatively more parenchymal inflammation resembling acute hepatitis, and in two cases (12.5%), inflammation was minimal. The overall degree of inflammatory activity was moderate to severe, with 10 cases (62.5%) showing either confluent perivenular or bridging necrosis. Increased numbers of plasma cells were seen in half of the cases, wherein the cells were mainly distributed at the edges of portal areas and with interface hepatitis was present, as is classically seen in idiopathic AIH. Eosinophils were noted in five cases (31.3%), mainly scattered within portal areas. The mean total inflammation score was 10 (median, 11; range, 1-16) (Table 4). The correlation between the histological activity index score and the degree of fibrosis trended toward significance (Supporting Fig. S6). Mild steatosis was seen in only 2 (12.5%) patients.
FIG. 2.
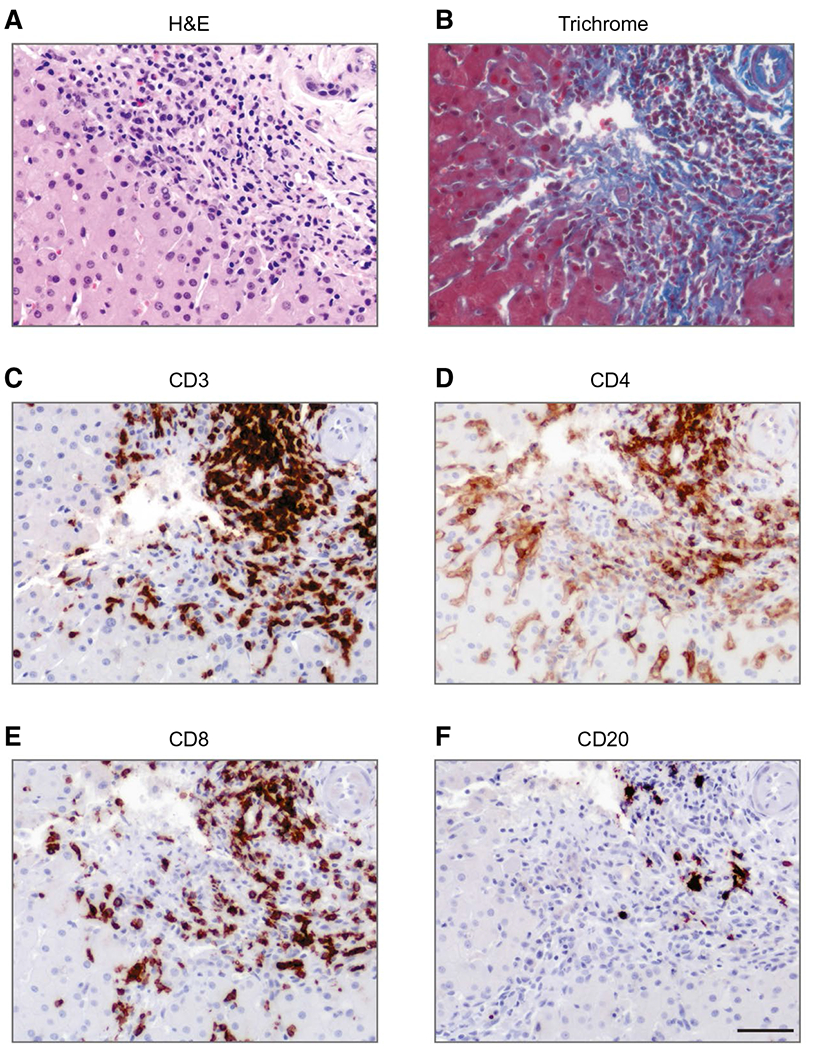
APAH features AIH-like lymphoplasmacytic inflammation with mild fibrosis. Shown are serial sections of a liver biopsy from a patient with APAH stained with (A) H&E, (B) trichrome, and immunostains for (C) CD3, (D) CD4, (E) CD8, and (F) CD20. Scale bar: 50 μm. Abbreviations: AIH, autoimmune hepatitis; APAH, APECED-associated hepatitis; H&E, hematoxylin and eosin.
TABLE 4.
Histological Characteristics of Liver Biopsies in patients With APECED and APAH
| Patient # | Age at Liver Biopsy (Years) | Sex | Histological Activity Index Score |
IF Stage |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Periportal Necrosis | Lobular Inflamm | Portal Inflamm | Inflamm (Total) | Fibrosis (Total) | Piecemeal Necrosis | Lobular Inflamm | Portal Inflamm | Confluent Necrosis | Inflamm (Total) | Fibrosis (Total) | Plasma Cells | Eosinophils | Histological Pattern | |||
| 1 | 7.7 | F | 4 | 3 | 3 | 11 | 1 | 4 | 2 | 3 | 2 | 11 | 2 | Increased | Increased | Chronic hepatitis |
| 2 | 2.0 | M | 3 | 4 | 1 | 8 | 0 | 3 | 3 | 2 | 0 | 8 | 0 | Increased | Not increased | Chronic hepatitis |
| 3 | 31.9 | F | 6 | 4 | 4 | 14 | 3 | 4 | 3 | 3 | 4 | 14 | 3 | Increased | Increased | Chronic hepatitis |
| 4 | 15.1 | F | 3 | 1 | 1 | 5 | 1 | 3 | 1 | 2 | 0 | 6 | 1 | Increased | Not increased | Chronic hepatitis |
| 5 | 8.8 | F | 0 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | Not increased | Not increased | Nonspecific changes |
| 6 | 3.2 | F | 1 | 1 | 1 | 3 | 1 | 1 | 1 | 1 | 0 | 3 | 1 | Not increased | Not increased | Nonspecific changes |
| 7 | 5.9 | F | 6 | 4 | 3 | 13 | 0 | 4 | 4 | 3 | 5 | 16 | 0 | Increased | Not increased | Acute hepatitis |
| 8 | 20.9 | F | 5 | 3 | 3 | 11 | 1 | 3 | 2 | 2 | 4 | 11 | 2 | Increased | Not increased | Chronic hepatitis |
| 9 | 2.7 | F | 3 | 3 | 1 | 7 | 1 | 3 | 2 | 2 | 0 | 7 | 2 | Not increased | Not increased | Chronic hepatitis |
| 10 | 4.3 | F | 5 | 4 | 1 | 10 | 0 | 2 | 3 | 2 | 5 | 12 | 0 | Not increased | Not increased | Acute hepatitis |
| 11 | 9.8 | M | 4 | 3 | 4 | 11 | 1 | 3 | 2 | 3 | 0 | 8 | 2 | Increased | increased | Chronic hepatitis |
| 12* | 2.1 | M | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| 13 | 15.0 | F | 3 | 4 | 1 | 8 | 1 | 3 | 4 | 2 | 3 | 12 | 1 | Increased | Not increased | Chronic hepatitis |
| 14 | 7.0 | M | 6 | 4 | 3 | 13 | 1 | 3 | 3 | 3 | 4 | 13 | 1 | Increased | Not increased | Chronic hepatitis |
| 15 | 2.1 | M | 6 | 4 | 3 | 3 | 0 | 4 | 4 | 3 | 5 | 16 | 0 | Not increased | Increased | Chronic hepatitis |
| 16 | 16.6 | F | 4 | 4 | 3 | 11 | 1 | 4 | 2 | 3 | 2 | 11 | 1 | Increased | Not increased | Chronic hepatitis |
| 17 | 11.7 | F | 4 | 4 | 3 | 11 | 1 | 4 | 2 | 2 | 3 | 11 | 1 | Increased | Increased | Chronic hepatitis |
| 18* | 8.0 | F | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
In 2 of the 18 patients with APAH, we could not obtain the prior liver biopsies for review at NIH.
Abbreviations: F, female; Inflamm, inflammation; M, male; N/A, not available.
We performed immunohistochemical stains for CD3, CD4, CD8, and CD20 in 6 cases and found that CD3+ T-lymphocytes were more prevalent than CD20+ B-lymphocytes. Among T-lymphocytes, CD8+ T-cells predominated over CD4+ T-cells in the majority of cases (5/6, 83.3%; Fig. 2 and Supporting Fig. S5). Taken together, our cases of APAH showed moderate-to-severe inflammatory activity, early-stage fibrosis, and features of plasma cell infiltrates and confluent/bridging necrosis, which are seen more often in AIH than chronic viral hepatitis.
NOT ALL AMINOTRANSFERASE ELEVATIONS IN PATIENTS WITH APECED ARE DUE TO APAH
Of the 25 patients with APECED without APAH in our study, 3 underwent diagnostic liver biopsy to elucidate the underlying etiology of observed aminotransferase elevations. One patient had nonspecific chronic portal inflammatory changes not felt to meet criteria for APAH or AIH. The second patient had sinusoidal dilation and minimal chronic inflammation without fibrosis, also inconsistent with a diagnosis of APAH or AIH. That patient was on oral contraceptives at the time of liver biopsy, which was considered the possible explanation for the transaminitis, which resolved after the liver biopsy. The third patient was diagnosed with fatty liver disease on a biopsy performed in an outside hospital that was unavailable for review at NIH. Therefore, aminotransferase elevations in patients with APECED may be caused by etiologies other than APAH.
APAH IS RESPONSIVE TO IMMUNOMODULATORY THERAPY
At the time of the NIH evaluation, all 18 patients with APAH had their disease in clinical and biochemical remission while they were receiving immunosuppressive treatment. The immunomodulatory treatment regimens used most often included standard-of-care therapy for AIH with a goal to decrease hepatic inflammation and fibrosis progression. As such, azathioprine or 6-mercaptopurine with or without corticosteroids was used in the majority of patients (n = 10, 55.6%); less often, mycophenolate mofetil (as monotherapy or in combination with rituximab, n = 3, 16.7%), prednisone monotherapy, cyclosporine, or tacrolimus were used (Table 5). Notably, 16 (88.9%) patients with APAH responded to the initial immunomodulatory regimen used to control APAH; few of these regimens are atypical for AIH but were chosen by primary providers for accompanying autoimmune manifestations (Table 5). The remainder 2 (11.1%) patients with APAH required augmentation of initially introduced immunosuppression to achieve APAH control; specifically, one patient required addition of rituximab to mycophenolate mofetil and another patient required addition of cyclosporine to 6-mercaptopurine on prednisone weaning.
TABLE 5.
Treatment Regimens Used for the Management of APAH in Our APECED Cohort at the Time of the NIH Evaluation
| Treatment Regimen | Number of Patients (% of Total Patients with APAH) |
|---|---|
| Combination of prednisone with azathioprine | 6 (33.3) |
| 6-mercaptopurine or azathioprine monotherapy | 4 (22.2) |
| Prednisone monotherapy | 2 (11.1) |
| Combination of mycophenolate mofetil and rituximab | 2 (11.1) |
| Cyclosporine monotherapy | 1 (5.6) |
| Mycophenolate mofetil monotherapy | 1 (5.6) |
| Combination of prednisone, mycophenolate mofetil, and tacrolimus* | 1 (5.6) |
| Combination of 6-mercaptopurine and cyclosporine† | 1 (5.6) |
Combination immunosuppression was prescribed for limbal stem cell transplantation.
Combination immunosuppression was prescribed because of initial difficulty to control APAH on 6-mercaptopurine monotherapy on prednisone weaning. Since then, the patient has been on azathioprine monotherapy with complete control of APAH.
PATIENTS WITH APAH HAVE ALTERED ABUNDANCE OF CERTAIN OTUS IN STOOL
Because dysbiosis has been implicated in the pathogenesis of autoimmune disorders and AIH,(20,21) we asked whether patients with APECED and APAH have an altered microbiomic signature in stool relative to patients with APECED without APAH. We performed microbiome analysis through 16S rRNA sequencing from 35 patient stool samples, 12 with APAH and 23 without APAH. We found no significant difference in alpha diversity (Chao1 and Shannon indices), beta diversity (by UniFrac distances), or bacterial taxa at the phylum and order levels between patients with and without APAH (Fig. 3). However, when using the linear discriminant analysis (LDA) effect size method to examine differential abundance of OTUs as possible biomarkers, the genus Slackia (LDA = 4.06, P = 0.014) and the genus Acidaminococcus (LDA = 4.91, P = 0.047) showed greater abundance in patients with APAH, whereas several members from the Bacilli class were more abundant in the stool of patients without APAH (Fig. 3).
FIG. 3.
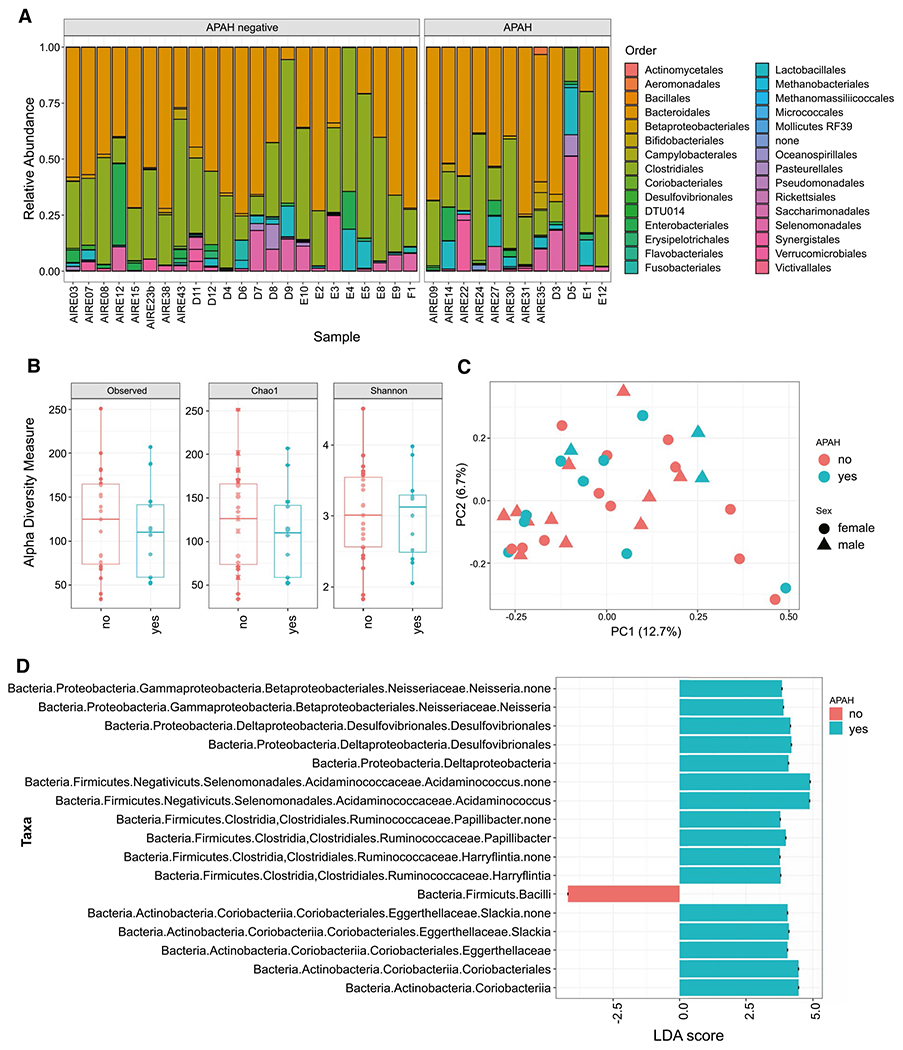
Taxonomy, diversity and biomarker microbiome analysis in stool samples from APECED patients with and without APAH. (A) Relative abundance of taxa at order taxonomy rank. (B) comparison of alpha diversity calculations using Chao1 species richness and Shannon diversity indices. (C) Principle Coordinates Analysis based on unweighted UniFrac. In addition to using colors to separate patients with or without APAH, the plot uses circles and triangles to indicate the sex of patients. (D) Results of biomarker discovery using LEfSe algorithms; the taxa with higher abundance in APAH samples and significant LDA score are shown in green color. Abbreviations: APECED, Autoimmune polyendocrinopathy-Candidiasis-Ectodermal dystrophy; APAH, APECED-associated hepatitis; LDA, linear discriminant analysis; PC, principle coordinates.
Discussion
APAH is among the most important APECED manifestations given the potential risk for fulminant hepatic failure and death. In our comprehensive analysis of APAH from a large, predominantly American APECED cohort, APAH onset mirrored that of European cohorts, typically before the third decade of life, with a third of APAH cases occurring before developing a classic APECED diagnostic dyad. Therefore, APECED should be considered by hepatologists in children who develop hepatitis in the setting of CMC or endocrine or nonendocrine autoimmune manifestations even if a classic APECED diagnostic dyad is absent (Supporting Fig. S7). A finding in our study is the 42% prevalence of APAH, which is 2-fold to 4-fold greater than described.(4)
Patients with APECED, to date, have been described as developing AIH type 2 or, previously, AIH type 3. Based on our and other published work, we would use restraint in diagnosing AIH in APECED, as patients with APAH do not consistently display classical AIH biomarkers. Although seronegative AIH can occur, it represents a small overall percentage of AIH.(23,24) Our results suggest one of two possibilities: patients with APECED may have a novel clinical presentation of AIH, or they may have a unique disease entity that has been misclassified as AIH. Drug-induced liver injury from hepatotoxic medications, microbiome alterations, or a hepatitis due to antigens reaching the liver that would normally not be present, would be cleared, or would be nonreactive could all account for hepatic decompensation. Further study to determine the etiology of hepatic inflammation in APECED is warranted. Our data demonstrate that besides APAH, patients with APECED may also be at risk for other common liver diseases, including steatohepatitis, from chronic steroid exposure and drug-induced liver injury from hepatotoxic medications (e.g., triazoles).
APAH has previously been considered an AIH subset given variably present classical AIH biomarkers in affected patients with APECED. However, no patient among our APECED cohort had classical AIH biomarkers at the time of NIH evaluation regardless of the presence or absence of abnormal liver-associated enzymes or whether there was histological APAH diagnosis. This finding suggests that currently available serological biomarkers of AIH are not universally useful in detecting hepatic disease in APECED but retain utility in ruling out classical AIH. However, the possibility of waning antibody titers while on immunosuppressive treatment cannot be ignored as a possible explanation. For that, we examined the presence of serological biomarkers of AIH at the time of diagnostic biopsy, which in all cases was made before patient evaluation at NIH. We were able to retrieve serologic profiles from 6 out of 18 patients at the time of diagnosis and found that only 2 of those 6 patients had positive classical AIH biomarkers; notably, the pattern of biomarker positivity was nonspecific, with one patient having positive anti-LKM and another having both positive anti-ASMA and anti-LKM.
Although there were no differences in alpha or beta diversity between patients with and without APAH, the genera Slackia and Acidaminococcus were more abundant in patients with APAH, whereas several members from the Bacilli class were found more often in patients without APAH. Interestingly, the Slackia species, which has not been previously directly linked to AIH, belongs to the family Coriobacteriaceae, which carries out important functions such as the conversion of bile salts and steroids.(25) Hence, they may influence host homeostasis by modulating bile acid and lipid metabolism. In fact, Coriobacteriaceae have been linked to several autoimmune disorders, and alterations in bile salts have been associated with gut dysbiosis and liver cirrhosis progression.(26–28) In addition, Acidaminococcus was recently reported to be enriched in the stool of patients with rheumatoid arthritis, ankylosing spondylitis, and ulcerative colitis.(29,30) Although Acidaminococcus has not been previously associated with AIH, prior studies have shown that Veillonella, another member of the Negativicutes class, was enriched in patients with classical AIH and associated with disease activity.(21) In our cohort, there was no significant difference in serum primary and secondary bile acid profiles between patients with or without APAH. However, we did not examine the abundance and relative composition of bile acids in duodenal aspirates or stool from patients with or without APAH. Therefore, the interplay between bile acids, microbiome, and APAH requires further investigation. In addition, given the enrichment of intestinal dysfunction (a reported frequent APECED manifestation that features chronic diarrhea, constipation, or an alternating pattern of both(6)) in patients with APAH in our cohort, future studies will be required to (1) examine the composition of mucosa-associated and stool microbiota in patients with APECED with intestinal dysfunction and (2) determine whether Slackia and Acidaminococcus promote a leaky gut in patients with APECED with resultant microbial translocation to the liver.
At present, it is unclear what drives the hepatic inflammation, which we have termed APAH. As classical AIH biomarkers are unreliably present, we do not believe there is sufficient evidence to categorize APAH within the current standard classifications of AIH. APAH could represent a variant of AIH. Given that the liver is critically involved in regulating immune functions, APAH may represent a reactive inflammatory state with subsequent hepatic injury as the liver attempts to modulate the immune dysregulation inherent to APECED pathogenesis.
Although prior case reports have described fulminant hepatitic failure and emergent liver transplantation, the liver disease in our cohort was more indolent. However, 2 patients had markedly elevated aminotransferase levels ranging into the thousands at the time of initial diagnosis. Currently published biopsy data suggest that patients present with advanced fibrosis and cirrhosis even at a young age. Our findings argue against this clinical presentation, as our patients did not feature advanced liver disease, and actually, only 5 patients had an IF stage >2, with only one patient having more advanced fibrosis with an IF of 3. No patients had cirrhosis or portal hypertension-associated complications. One possibility to explain the relative absence of severe liver disease in our cohort may be that our patients were extensively screened for multiorgan autoimmune disorders, including APAH, once diagnosed with APECED, bringing them to clinical attention sooner than other patients. It is also possible that patients had been started on immunosuppressive therapy for other reasons before the diagnosis of APAH, attenuating the APAH once it developed. Alternatively, publication bias of more severe cases may have occurred in prior reports; in our study design, all patients were evaluated in a systematic manner that may have allowed for earlier detection of disease stages in a larger proportion of patients. More studies in additional American and European patients with APECED will be required to define the relative frequency of early-onset severe liver disease in APECED and determine whether that reflects population-specific differences or delays in therapeutic intervention. Given the reports of fulminant hepatic failure and the significantly elevated aminotransferase levels in some of our patients, physicians should screen patients with APECED with aminotransferase elevation for APAH to ensure early diagnosis and initiation of treatment to avoid potentially severe complications, including hepatic failure (Supporting Fig. S7).
ALP% was significantly greater in our patients with APAH, but there was no significant difference in GGT, ESR, or CRP between patients with or without APAH. A possible explanation is that APAH does indeed represent a unique disease entity separate from AIH. This hypothesis is supported by the fact that elevated ALP, outside coexistent overlap syndromes, is considered a negative predictor for AIH. However, we are cautious about this conclusion. We attempted to remove the confounders of age and HP, which could cause elevations in bone-derived ALP, but again saw significantly increased ALP% in patients with APAH. Another possible ALP source could be intestinal sites, and we found more prevalent intestinal dysfunction in patients with APECED with APAH. However, to date, there has been no clear connection between APECED-associated enteropathy and APAH, and we found no clear association between APAH and TPH autoantibody reactivity, which is associated with APECED-associated enteropathy(12); indeed, although the prevalence of TPH-targeted autoantibodies in patients with APAH was >90%, the same autoantibodies were also detected in ~65% of those without APAH. In addition, there were no major differences in alpha or beta diversity of the microbiome of patients with APECED with and without APAH. Although it is noteworthy that abundance of certain bacterial genera was significantly different between stool of patients with APECED with and without APAH, it is unclear whether microbiome variations alone drive APAH; further study and longer follow-up are needed. A final alternate explanation of our ALP% findings could be that they are subject to type 1 error given our overall small sample size. We thus tentatively suggest age-adjusted ALP as a possible biomarker for APAH, but further study is warranted.
Several organ-specific autoantibodies are enriched in various APECED clinical manifestations. Hence, APECED serves as a unique model for studying organ-specific autoimmunity because it is monogenic and many molecular targets associated with its autoimmune processes are known. Furthermore, given that hepatitis may occur in up to ~40% of patients with APECED and could have significant clinical consequences, identification of liver-targeted autoantibodies may facilitate the diagnosis of APAH and identification of patients at risk to develop APAH. Indeed, we show that autoantibody profiles differ between patients with and without APAH. Consistent with reported data, CYP1A2-targeted autoantibodies were associated with APAH.(12,16) In our cohort, they were present in less than a third of patients with APAH but had 100% specificity for APAH, as no patient without APAH tested positive for CYP1A2. Autoantibodies against AADC, previously associated with vitiligo, and TPH, previously associated with enteropathy, were also more frequently found in patients with APAH. However, as opposed to CYP1A2, they had low specificity for APAH. These results are in line with Finnish data wherein AADC-targeted autoantibodies were present in 92% of patients with APAH but were also frequent in patients with APECED without signs of hepatitis.(19) The association of APAH with autoantibodies against BPIFB1, previously associated with pneumonitis, and autoantibodies against 21-OH, previously associated with AI, is intriguing and requires confirmation in future data sets.
Our study has several limitations. Although we evaluated a relatively large APECED cohort, we still have examined an overall small patient population. Referral bias could explain the higher APAH prevalence in our cohort. However, we did not enroll patients with APECED at NIH based on the presence or absence of APAH (or any other manifestation). Although we cannot control for referral bias from outside institutions, all referred patients were evaluated in a consecutive consistent manner with only 2 patients excluded, as one did not meet the diagnostic criteria for APECED and another presented after transplantation with curative intent. Additionally, all patients regardless of symptoms underwent a systematic clinical and research evaluation guided by hepatology consultation. A limiting factor in our analysis is that the laboratory parameters analyzed were taken at the time of first visit to NIH, which in no instance coincided with the liver biopsy or clinical determination used to diagnose APAH. Despite extensive attempts, we were unable to confirm serologic assessment for classical AIH at the time of APAH diagnosis for several patients. However, we obtained the vast majority of liver biopsies for review by an expert hepatopathologist at NIH. Despite the small sample size, data from our cohort suggest that APAH is characterized by robust hepatic inflammation with a rise in aminotransferases typically in a hepatocellular pattern consistent with an AIH-like hepatitis. Although we were unable to clearly define the serologic presentation at the time of diagnosis with our limited sample size, our data suggest that disease control is possible with immunosuppressive therapy, as all patients at the time of NIH evaluation had their disease under clinical and biochemical control, as evidenced by normal or near-normal aminotransferases. Importantly, our findings suggest that early recognition and control of APAH may prevent the development of fibrosis and complications of end-stage liver disease even after a severe initial clinical presentation.
In conclusion, we found APAH to be more prevalent in American patients with APECED than previously described in European cohorts, to feature less severity, and to be responsive to immunomodulation. Although classical AIH biomarkers lack sensitivity and specificity for APAH, several APECED-related autoantibodies, including AADC, CYP1A2, and HDC, were associated with APAH with variable sensitivity and specificity. The potential contribution of specific bacterial genera in the pathogenesis of APAH deserves investigation in future studies. Collectively, further investigation is warranted before defining APAH as a variant of AIH based on currently accepted classifications.
Supplementary Material
Potential conflict of interest:
Dr. Kampe received grants from Shire/Takeda.
Supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases and the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health. This project was also funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract no. HHSN261200800001E.
Abbreviations
- AADC
aromatic l-amino acid decarboxylase
- AI
adrenal insufficiency
- AIH
autoimmune hepatitis
- AIRE
autoimmune regulator
- ALP
alkaline phosphatase
- ALP%
percentage of upper limit of normal-age-sex-adjusted alkaline phosphatase
- ALT
alanine aminotransferase
- ANA
antinuclear antibody
- APAH
autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy–associated hepatitis
- APECED
autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy
- ASM
anti–smooth muscle antibody
- AST
aspartate aminotransferase
- BPIFB1
bactericidal/permeability-increasing fold-containing B1
- CMC
chronic mucocutaneous candidiasis
- CRP
C-reactive protein
- CYP1A2
cytochrome P450 family 1 subfamily A member 2
- ESR
erythrocyte sedimentation rate
- GGT
gamma-glutamyltransferase
- HDC
histidine decarboxylase
- HP
hypoparathyroidism
- IF
Ishak fibrosis
- IFN
interferon
- INR
international normalized ratio
- LDA
linear discriminant analysis
- LKM
liver-kidney microsome
- NIH
National Institutes of Health
- OH
hydroxylase
- rRNA
ribosomal RNA
- SLA
soluble liver antigen
- TPH
tryptophan hydroxylase
Footnotes
Supporting Information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.31421/suppinfo.
REFERENCES
- 1).Constantine GM, Lionakis MS. Lessons from primary immunodeficiencies: autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol Rev 2019;287:103–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Wolff AS, Erichsen MM, Meager A, Magitta NF, Myhre AG, Bollerslev J, et al. Autoimmune polyendocrine syndrome type 1 in Norway: phenotypic variation, autoantibodies, and novel mutations in the autoimmune regulator gene. J Clin Endocrinol Metab 2007;92:595–603. [DOI] [PubMed] [Google Scholar]
- 3).Michele TM, Fleckenstein J, Sgrignoli AR, Thuluvath PJ. Chronic active hepatitis in the type I polyglandular autoimmune syndrome. Postgrad Med J 1994;70:128–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Betterle C, Greggio NA, Volpato M. Clinical review 93: autoimmune polyglandular syndrome type 1. J Clin Endocrinol Metab 1998;83:1049–1055. [DOI] [PubMed] [Google Scholar]
- 5).Kluger N, Jokinen M, Krohn K, Ranki A. Gastrointestinal manifestations in APECED syndrome. J Clin Gastroenterol 2013;47:112–120. [DOI] [PubMed] [Google Scholar]
- 6).Ferre EM, Rose SR, Rosenzweig SD, Burbelo PD, Romito KR, Niemela JE, et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight 2016;1:e88782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Ferre EMN, Break TJ, Burbelo PD, Allgäuer M, Kleiner DE, Jin D, et al. Lymphocyte-driven regional immunopathology in pneumonitis caused by impaired central immune tolerance. Sci Transl Med 2019;11:eaav5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Bruserud O, Oftedal BE, Landegren N, Erichsen MM, Bratland E, Lima K, et al. A longitudinal follow-up of autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab 2016;101:2975–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Meloni A, Willcox N, Meager A, Atzeni M, Wolff ASB, Husebye ES, et al. Autoimmune polyendocrine syndrome type 1: an extensive longitudinal study in Sardinian patients. J Clin Endocrinol Metab 2012;97:1114–1124. [DOI] [PubMed] [Google Scholar]
- 10).Orlova EM, Sozaeva LS, Kareva MA, Oftedal BE, Wolff ASB, Breivik L, et al. Expanding the phenotypic and genotypic landscape of autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab 2017;102:3546–3556. [DOI] [PubMed] [Google Scholar]
- 11).Dominguez M, Crushell E, Ilmarinen T, McGovern E, Collins S, Chang B, et al. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in the Irish population. J Pediatr Endocrinol Metab 2006;19:1343–1352. [DOI] [PubMed] [Google Scholar]
- 12).Lankisch TO, Jaeckel E, Strassburg CP. The autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy or autoimmune polyglandular syndrome type 1. Semin Liver Dis 2009;29:307–314. [DOI] [PubMed] [Google Scholar]
- 13).Vogel A, Liermann H, Harms A, Strassburg CP, Manns MP, Obermayer-Straub P. Autoimmune regulator AIRE: evidence for genetic differences between autoimmune hepatitis and hepatitis as part of the autoimmune polyglandular syndrome type 1. Hepatology 2001;33:1047–1052. [DOI] [PubMed] [Google Scholar]
- 14).Goldstein NS, Rosenthal P, Sinatra F, Dehner LP. Liver disease in polyglandular autoimmune disease type one: clinicopathologic study of three patients and review of the literature. Pediatr Pathol Lab Med 1996;16:625–636. [PubMed] [Google Scholar]
- 15).Clemente MG, Obermayer-Straub P, Meloni A,Strassburg CP, Arangino V, Tukey RH, et al. Cytochrome P450 1A2 is a hepatic autoantigen in autoimmune polyglandular syndrome type 1. J Clin Endocrinol Metab 1997;82:1353–1361. [DOI] [PubMed] [Google Scholar]
- 16).Obermayer-Straub P, Perheentupa J, Braun S, Kayser A, Barut A, Loges S, et al. Hepatic autoantigens in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Gastroenterology 2001;121:668–677. [DOI] [PubMed] [Google Scholar]
- 17).Clemente MG, Meloni A, Obermayer-Straub P, Frau F, Manns MP, de Virgiliis S. Two cytochromes P450 are major hepatocellular autoantigens in autoimmune polyglandular syndrome type 1. Gastroenterology 1998;114:324–328. [DOI] [PubMed] [Google Scholar]
- 18).Gebre-Medhin G, Husebye ES, Gustafsson J, Winqvist O, Goksøyr A, Rorsman F, et al. Cytochrome P450IA2 and aromatic l-amino acid decarboxylase are hepatic autoantigens in autoimmune polyendocrine syndrome type I. FEBS Lett 1997;412:439–445. [DOI] [PubMed] [Google Scholar]
- 19).Husebye ES, Gebre-Medhin G, Tuomi T, Perheentupa J, Landin-Olsson M, Gustafsson J, et al. Autoantibodies against aromatic l-amino acid decarboxylase in autoimmune polyendocrine syndrome type I. J Clin Endocrinol Metab 1997;82:147–150. [DOI] [PubMed] [Google Scholar]
- 20).Czaja AJ. Factoring the intestinal microbiome into the pathogenesis of autoimmune hepatitis. World J Gastroenterol 2016;22:9257–9278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Wei Y, Li Y, Yan L, Sun C, Miao Q, Wang Q, et al. Alterations of gut microbiome in autoimmune hepatitis. Gut 2020;69:569–577. [DOI] [PubMed] [Google Scholar]
- 22).Naskali E, Dettmer K, Oefner PJ, Pereira PAB, Krohn K, Auvinen P, et al. Serotonin and tryptophan metabolites, autoantibodies and gut microbiome in APECED. Endocr Connect 2019;8:69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Czaja AJ. Autoantibody-negative autoimmune hepatitis. Dig Dis Sci 2012;57:610–624. [DOI] [PubMed] [Google Scholar]
- 24).Miyake Y, Iwasaki Y, Kobashi H, Yasunaka T, Ikeda F, Takaki A, et al. Clinical features of antinuclear antibodies-negative type 1 autoimmune hepatitis. Hepatol Res 2009;39:241–246. [DOI] [PubMed] [Google Scholar]
- 25).Wegner K, Just S, Gau L, Mueller H, Gérard P, Lepage P, et al. Rapid analysis of bile acids in different biological matrices using LC-ESI-MS/MS for the investigation of bile acid transformation by mammalian gut bacteria. Anal Bioanal Chem 2017;409:1231–1245. [DOI] [PubMed] [Google Scholar]
- 26).Bernstein CN, Forbes JD. Gut microbiome in inflammatory bowel disease and other chronic immune-mediated inflammatory diseases. Inflamm Intest Dis 2017;2:116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Jangi S, Gandhi R, Cox LM, Li N, von Glehn F, Yan R, et al. Alterations of the human gut microbiome in multiple sclerosis. Nat Commun 2016;7:12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Kakiyama G, Pandak WM, Gillevet PM, Hylemon PB, Heuman DM, Daita K, et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol 2013;58:949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Butera A, Di Paola M, Vitali F, De Nitto D, Covotta F, Borrini F, et al. IL-13 mRNA tissue content identifies two subsets of adult ulcerative colitis patients with different clinical and mucosa-associated microbiota profiles. J Crohns Colitis 2020;14:369–380. [DOI] [PubMed] [Google Scholar]
- 30).Lee JY, Mannaa M, Kim Y, Kim J, Kim GT, Seo YS. Comparative analysis of fecal microbiota composition between rheumatoid arthritis and osteoarthritis patients. Genes 2019;10:748. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.