

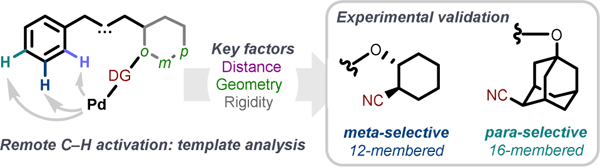
Abstract
The ability to differentiate and selectively activate remote C–H bonds represents a perennial challenge in the field of C–H activation. Since its first report in 2012, a now-established “directing template” (DT) approach remains demonstrably effective for the functionalization of remote C–H bonds. As selectivity is hypothesized to be principally determined by the optimal positioning of the reactive catalyst to a target C–H bond, a DT’s spatial factors are particularly important towards achieving high selectivity, though a systematic study on its requisite factors remain unelucidated. Through an in-depth analysis of 119 structurally unique published remote-DTs, this report summarizes the key factors that are central towards achieving high selectivity at defined aryl positions, which are experimentally corroborated through the development of new aliphatic meta and para-selective DTs for electronically unbiased arenes. These empirical rules—which summarize key distance and geometric factors—are expected to be useful tools for the future development of site-selective arene C–H activation, as well as other reactions that rely on covalent/non-covalent DT-mediated remote regioselection.
Graphical Abstract
INTRODUCTION
Remote C–H bonds, which often possess similar steric and electronic properties, can pose a considerable challenge for catalysts to differentiate and selectively target in C–H activation reactions.1 Within a biological context, the selective transformation of remote bonds can be achieved through organizational and directing effects conferred by weak interactions in an enzymatic scaffold. However, translating these specialized effects in organometallic catalysis remains difficult.1b One strategy to circumvent this obstacle involves utilizing a directing template (DT) bearing a Lewis basic directing group (DG) to position the reactive catalyst close to the target C–H bond,2–5 which is proposed to favor reaction at the desired position by preorganizing the macrocyclophane-like C–H cleavage transition state.5,6 Since its initial disclosure in 2012,7 such a DT strategy based off a macrocyclophane (MCP) structure has stood out an eminent approach for the selective distal functionalization of arene rings. In this strategy, the selectivity of C–H activation was hypothesized to be principally determined by the spatial factors encoded by the terms “distance” and “geometry” (Figure 1).8 In the ensuing decade, this strategy has been comprehensively validated in over 50 reports that targets a range of remote C–H bonds.
Figure 1.

Overview of the key factors hypothesized to confer remote selectivity in directing template (DT)-mediated arene C–H functionalization. DG = directing group.
The success of leveraging a MCP structure to control remote regioselectivity alludes to its potential generality, such that this strategy could be employed to impart regioselection at a range of positions for diverse substrate classes. Despite the seemingly straightforward proposed factors that determine remote regioselection, there has been no systematic study reported on the role played by each of the spatial factors (“distance” and “geometry”) in giving the observed site-selectivity in DT-mediated processes. Thus, the development of successful DTs tends to rely on a combination of opaque chemical intuition and serendipitous screening despite over 10 years of fervent research to date in the area. Given the potential generalizability of this approach for remote regioselection, the development of clear guidelines for DT design would greatly facilitate the future rational design of new directing motifs.
To examine our longstanding hypothesis that simple spatial factors—i.e. template distance and geometry—are sufficient to tune catalyst positioning, and therefore able to capture the selectivity of a remote-DT, we undertook a systematic analysis of 119 structurally unique published ortho,9–12 meta7,9,13–54 and para-DTs55–59 published between 2012–2021. We report herein, in this hybrid literature analysis/research article, the key factors that were found to be particularly determinant for selectivity in DT-mediated remote arene C–H activation. These quantitatively driven findings were validated through experimental and ab initio analyses, leading to the development user-friendly guidelines for future DT design. Using these guidelines, we successfully developed novel aliphatic meta- and para-selective DTs, and project that these guidelines can be broadly applicable for the future development a range of DT-mediated site-selective reactions of diverse arene scaffolds.
RESULTS AND DISCUSSION
Quantitative Analysis of Published DTs.
Our analysis commenced through the tabulation of all structurally unique substrate-DT combinations, which include a range of covalent and non-covalent DTs across a range of metals and catalytic cycles. These structures were subdivided into two components: the “substrate” (the mono arene component where C–H activation occurs) and “template” (the remainder of the macrocycle). These structures were further categorized into their constituent site-selectivity (ortho, meta and para) and an indication of the magnitude of selectivity (good: >80:20, medium: 67:33 to 80:20, poor: <67:33).60 Through this initial categorization, we collated 119 structurally unique macrocycles published between 2012–2021, 8 of which were ortho-directing, 98 meta-directing and 13 para-directing. Within this set, 85 exhibited good selectivity, 8 medium and 26 poor (Figure 2A). Pleasingly, the spread of data for DTs exhibiting good site-selectivity could be cross-validated against ones with poorer regioselectivity to test the robustness of our findings (see the SI for further discussion).
Figure 2.

Key findings from published DTs (2012–2021; total DTs = 119) for MCP size and DT geometry. General trends illustrated using the subset of DTs with “good” selectivity (r.r. > 80:20; total DTs = 85). r.r.: regioisomeric ratio; SD = standard deviation; X denotes average values on box and whisker graphs, while a horizontal line within the interquartile range (IQR) denotes the median.
After encoding each unique structure into their constituent chemoinformatic parameters (see the SI for a detailed analysis and key assumptions), a series of univariate comparisons were made with the goal of uncovering parameters that led to tight and differentiable clustering of data for each class of DT. Through this, we found two structural features to be particularly predictive for site selectivity: 1) MCP size (“distance”; or the number of atoms within a macro-cyclometallated species — see Figure 1 and the SI); specifically, each farther carbon targeted on the arene ring requires approximately three more carbons in the DT to achieve selectivity for the more distal carbon (ortho: 8.4 ± 1.4, n = 8; meta: 12.5 ± 1.6, n = 71; para: 16.0 ± 0.6, n = 6; Figure 2B), and 2) geometric features of DT backbone; specifically ortho DTs do not contain meta, linear or para components, and meta DTs do not contain para components (Figure 2C). Both findings can be intuitively understood as physical requirements to facilitate macrocycle closure in a thermodynamically favorable manner. In terms of MCP size, DTs required a certain size to be able to readily access the C–H cleavage pretransition state at a given position. In terms of MCP geometry for a given MCP size, DTs cannot contain too many “rigid spacers”, which disfavors ring closure. This can be understood as the presence of E double bonds (in meta and para motifs) as well as linear (triple bonds) disfavoring ring closure in smaller macrocycles such as in the ortho position, or for the meta position, the presence of a para component to disfavors final ring closure (vide infra for further discussion).63–66
Given the diversity and complexity across published DTs—which contain a range of DGs, as well as a range of backbone motifs that connect the arene substrate to the DG—we investigated whether a method of scoring individual geometric components could be developed to allow for the rapid assessment of the suitability of a given DT for the activation of a particular remote C–H bond. To this end, we found that a weighted linear sum of the para, meta, ortho and linear template components gave appreciable clustering of DT data according to selectivity. Through consideration of i) sign/directionality, ii) displacement and iii) numerical simplicity through simple integer terms, we captured the relative ranks of each geometric component and identified a relationship represented by a “DT score” (5npara+3nmeta+2nlinear–northo; n = number of a given component) that gave tight clustering and adequate demarcation for the majority of surveyed DTs (see the SI for a detailed discussion). Such DT score captured the intuitive understanding that a higher score corresponds to more “linear” spacers, wherein a number is reduced when the template is forced to “bend back” onto itself. Through this, we found that a score of –1 and 0 best represented ortho-selective templates, 1 and 2 represented meta, and a score around 6 represented para (Figure 2D). Finally, preliminary investigations into the role of DT rigidity (number/number of contiguous/fraction of rotatable/unrotatable bonds – see the SI) gave inconclusive evidence on their role in remote regioselection prompting further experimental investigation (vide infra).
With these preliminary “distance” and “geometric” factors quantified based on DTs exhibiting good selectivity, we cross-validated our findings through the evaluation of medium and poorly selective templates. We found that the mean MCP size and DT score for the set of poor-medium selectivity meta DTs were higher than the ideal cases (MCP size: 14.4 ± 1.6, average DT score = 4, interquartile range (IQR) = 1–7, n = 27, Figure 3A), while the DT score for poorly selective para DTs tended to score higher and across a wider range than in the ideal case (average DT score = 8, IQR = 6–9, n = 7, Figure 3B, see the SI for discussion of apparent exceptions); the latter case however, is confounded by the modest sample size arising from sparse literature on para-selective directed C–H activation of electronically unbiased substrates,27,57,59 Altogether, these observed trends indicate that simple tools (MCP size and DT score) could provide a rapid guideline to determine the suitability of a given DT for targeting a desired C–H bond. These findings also provide a starting point to experimentally validate and investigate the underlying molecular features which may contribute to the observed trends.
Figure 3.

Cross validation of MCP size and DT score against a set of moderate and poorly selective DTs (Total DTs = 34). Both sets of data indicate that good regioselection for a desired position fall within a narrow window for both template distance and geometric factors.
Experimental Validation through the Development of Aliphatic meta-Selective DTs.
Building from our analyses, we next embarked on a series of experimental studies to investigate the validity of the obtained observations. Given the relative ease in which reliable ortho activation could occur through a canonical small (five or six-membered) ring cyclometallation regimen, we focused our experimental validation on verifying the importance of these identified factors on the more challenging nature of meta and para regioselection.
We commenced our experimental study by identifying several boundary case scenarios. We first hypothesized that rigidity played a minimal role for meta regioselection based on preliminary analyses and previous literature reports, where flexible linker/MCP structures did not appear to adversely affect meta selectivity.21,29 To evaluate this hypothesis, we elected to test a series of simple, flexible alkyl nitriles substrates of variable linker lengths (Scheme 1A)—the minimum possible component predicted to enable meta directivity across a range of MCP size (DT score = 2).
Scheme 1.

MCP Size vs. meta-Selectivity for Linear Alkyl Nitriles and 3-Pyridyl Motifs
Interestingly, the combination of even this simple predicted meta-directing motif and the optimal MCP size for meta-selectivity (2b, MCP = 12) allowed for high reactivity and moderate meta selectivity (up to 77:23 m:p, no ortho product observed). These results constitute the only example where nitriles with unfunctionalized linkers are capable of directing to the meta position. Homologation to the 13- and 14-membered MCP (2c-2e) resulted in an erosion in meta selectivity (though reactivity remained unaffected), most notably revealing a modest restoration of ortho reactivity. For the 16-membered substrate (2d, predicted optimal para distance), selectivity for the para position was not seen, affording a similar selectivity profile to product 2c. Likewise, the truncated 11 membered nitrile (2a) registered a drop off in reactivity, with selectivity corresponding to non-directed reactivity (ortho, meta and para products seen, see the SI for further details).
While 3-pyridyl/5-pyrimidyl motifs have been featured in several template designs,14,18,20 initial inspection using our scoring heuristic indicated that these motifs, by themselves, may not be ideal for meta selection, falling just above the predicted ideal range (DT score = 3, c.f. meta range: 1 and 2, Scheme 1A).18,19,30 To this end, a series of simple 3-pyridyl-containing DTs were investigated (Scheme 1B). Indeed, the bare 2-fluoro-3-pyridyl motif uniformly provided poor meta selectivity and modest reactivity, giving a mixture of meta and para products in 4c-4f (MCP = 12 to 16) with no ortho products seen. Reactivity reduction was again observed upon moving from the 12-membered (4c) to the 11-membered MCP (4a), highlighting that the correct MCP size has a clear effect on overall reaction efficacy despite differences in DT geometry.
While these results preliminarily affirm the contributions of both overall MCP size (distance) and DT geometry towards the reactivity and selectivity of a particular scaffold, the comparatively modest selectivity (m:p:o = 77:23:0, up to 80:20:0 at 125 °C, see the SI), and the presence of rigid motifs proximate to the DG in published DTs indicated that—contrary to our initial hypothesis—a greater degree of rigidification might be necessary to obtain high meta selectivities. Thus, we generated a simple trans-2-hydroxycyclohexanecarbonitrile DT (T1, DT score = 1) motif and investigated its meta selectivity across a range of anchoring motifs and linker lengths (Scheme 2), with a notable benefit in that the alicyclic DT-linker scaffold of T1 enables facile regioisomeric product discrimination without DT signals (from canonical aryl-containing DTs) obfuscating selectivity analyses.
Scheme 2.

MCP Size vs. meta-Selectivity Study with an Alicyclic trans-2-Hydroxycyclohexanecarbonitrile DT (T1) with Various Carboxylic Acids and Alcohols
DG-adjacent conformational restriction markedly improved selectivity for substrate class 5 relative to their non-rigid analogs 1. For carboxylic acid substrates, optimal meta selectivity was observed for the substrate forming a 12-membered MCP (6c, m:p > 95:5). In addition, high selectivities were observed with 11- and 13-membered MCP (6b and 6d), with yield and selectivity sharply dropping off for the benzoic acid derivative (6a, MCP = 10). A similar distance dependence was observed with alcohol-containing substrates, where 12-membered MCP (6j) gave the highest meta selectivity. Similarly high selectivities were also observed with the 11- and 13-membered congeners (6i and 6k), once again with reactivity and selectivity precipitously dropping below this length (6g and 6h). Extending beyond the optimal MCP size, product selectivity, but not yield, was again observed to noticeably drop (6e, MCP = 14, m:o:p = 51:29:20; 6f, MCP = 16, m:o:p = 46:35:19); an observation that contradicted a single previous report.21 Again, the erosion of selectivity appears to be driven by the appearance of the ortho product.
In conjunction with data presented in Scheme 1, these results revealed four useful insights for DT-mediated meta regioselection. First, a clear minimum MCP size was required to obtain reactivity regardless of DT geometry—substrates that formed MCP sizes smaller than the required 12-membered MCP threshold typically gave poor reactivity and meta-selectivity (2a, 4a, 6a, 6g, and 6h). In particular, substrates forming smaller MCPs (MCP < 11) showed yield and product distributions broadly consistent with intermolecular control experiments in which the DG and arene core are unlinked, suggesting that non-directed, acyclic mechanisms may dominate with these substrates. This indicated a strong minimum distance-dependence for the DT to enable intramolecular directed reactivity to occur.
Second, extending MCP size beyond the optimum meta-range (12) towards the predicted optimal para range (16) did not result a switch from meta to para regioselection (2d and 4e). In concert with the above point, this indicated that MCP size (distance) was a necessary, but by itself, an insufficient factor dictating meta regioselection.
Third, extending MCP size beyond the optimum meta range (12) registered a drop in meta selectivity (but not reactivity), with the decrease in meta selectivity largely driven by an increase in ortho functionalized product for rigid nitrile motifs (2c-d, 6e-f). This observation points to the fact that larger MCPs can allay the incurred strain by the rigid nitrile DG motif/DT scaffolds and enable functionalization at the more proximate ortho position. Conversely, it also indicates that high meta-selectivity arises in part due to the DT disfavoring ortho functionalization through the higher strain incurred by the smaller MCP required for ortho C–H activation. Notably, our data show the combination of optimal MCP size and minimal DT geometry are by themselves sufficient to prevent ortho functionalization (2b and 4b).
The “strain” hypotheses were rationalized through a series of qualitative DFT analyses modelling ring strain on simplified macrocyclic carbocycles (Figure 4, also see the SI for further detail).65 This highlights that for a given MCP size for meta C–H activation, shifting the macrocyclic connection towards the more proximal ortho position (MCP size –1) incurs prohibitive amounts of ring strain to disfavor its functionalization. It also highlights that increasing MCP size globally reduces overall strain upon ring closure, rationalizing the reduction in selectivity and appearance of ortho functionalized products observed for larger ring sizes.62,66 This observation implies that a combination of factors remote regioselection results from a cumulation of favorable catalyst positioning for the target site, in conjunction with disfavoring reactivity at alternative sites.
Figure 4.

Quantitative assessment of the computed energy difference between open and closed chain forms of simplified carbocycles bearing a linear motif across chain length and arene position. These data highlight that as macrocycle size increases, both the overall MCP strain and differential strain between the o-, m- and p-MCPs decrease.
Fourth, in addition to preventing ortho functionalization (point three, vide supra), our data show that optimal MCP size (distance) and DT geometry, while sufficient to achieve broad meta-selectivity, were by themselves insufficient to achieving high meta selectivity. Increasing DT rigidity was also required to suppress undesired reactivity at the more distal para position (c.f. 2b vs. 6c and 6j).30
To further investigate our revised hypothesis in that high selectivity is a composite function of disfavoring the ortho site while positioning the catalyst near the targeted meta-C–H bond, we modeled substrates 5a, 5c, and 5e using density functional theory (DFT) methods, which form 10-, 12-, and 14-membered MCP intermediates respectively. We were particularly interested in the position and distance of the coordinating nitrile DG relative to the ortho, meta, and para C–H bonds.40 Given the conformational flexibility of these substrates, we focused on relevant conformers that place the DG in proximity to the C–H bonds of interest; i.e. capable of intramolecular directed reactivity. For each set of relevant conformers, we then obtained the Boltzmann-averaged distances (Figure 5A) of the nitrile DG to the nearest ortho, meta, and para C–H bonds.
Figure 5.
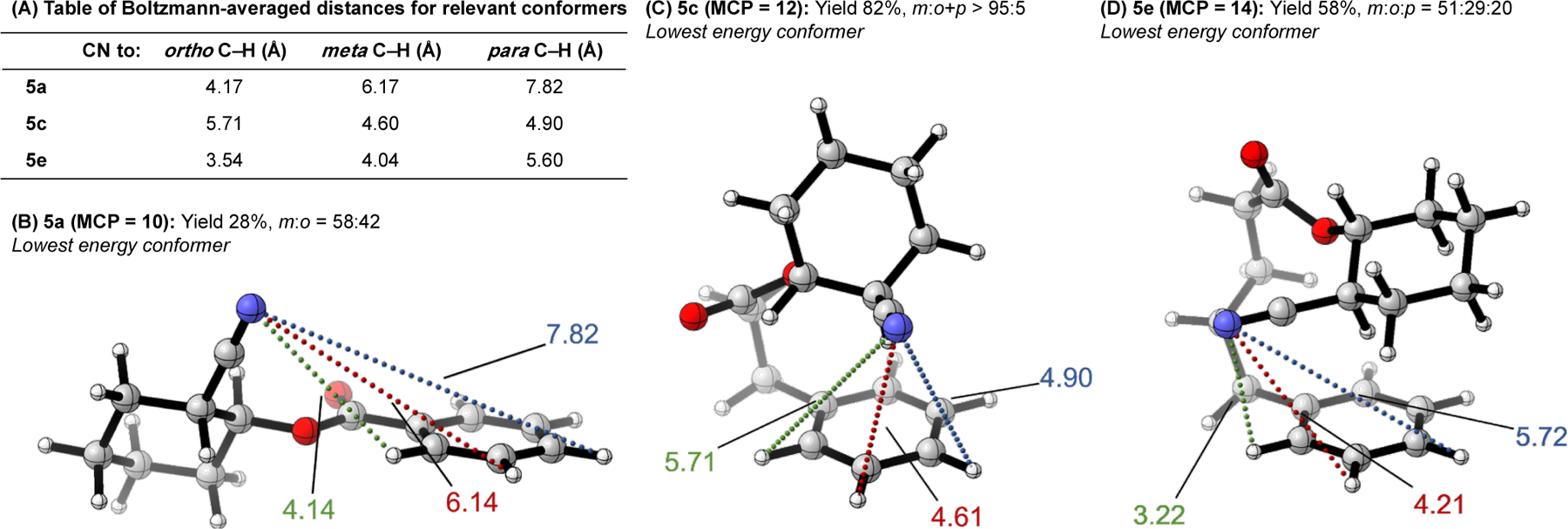
Investigation of DG position, and its relative distance and geometry to target arene C–H bonds for conformers relevant to intramolecular DT-mediated processes through density functional theory (DFT) analysis. Geometries and energies calculated at the ωB97X-D/def2-SVP level of theory.
Unsurprisingly, 5a (MCP size = 10, Figure 5B), showed nitrile–H distances that are likely too great to direct Pd in an intramolecular fashion (o: 4.17 Å, m: 6.17 Å, p: 7.82 Å). The results for 5c (MCP size = 12, Figure 5C), which provides high experimental meta selectivity, indicate the nitrile DG is in closest proximity to the meta C–H bond (o: 5.71 Å, m: 4.60 Å, p: 4.90 Å). Inspection of the relevant conformers indicates that the overall structure would have to undergo considerable distortion to reach the ortho C–H bond, supporting the argument that selectivity also arises from disfavoring ortho activation. Furthermore, the para C–H bond, while relatively close to the nitrile DG, is not in the correct alignment (geometry) for a Pd C–H activation event. Finally, the results for 5e (MCP size = 14, Figure 5D), which exhibits low experimental meta selectivity, indicates the nitrile DG could be near any of the C–H bonds (o: 3.54 Å, m: 4.04 Å, p: 5.60 Å). Notably, the ortho C–H bond is now accessible, which is consistent with experimental observations and fully supports our revised hypothesis—that MCP sizes above the optimal value results in increased conformational flexibility/decreased ring strain, rendering the DT unable to prohibit accessibility to the undesired ortho site.
Rational Development of Aliphatic para-Selective DTs.
A notable absence in the remote DT literature is comprehensive coverage of effective para selective DTs. To date, 13 unique substrate-DT scaffolds has been reported—only 6 of which bearing good selectivity. Of note, studies towards the DT-mediated functionalization of electronically unbiased substrates are especially scant.27,57,59 Analysis of published DTs—caveated by their small sample size—indicated that para-selective DTs possessed a higher geometric score (DT ~ 6) and optimal MCP distance (MCP ≥ 16). In addition, we observed that all published para DTs tended to possess at least two aromatic rings in the linker (which accounts for least six atoms within the MCP) for optimal DG positioning.55,57,59 Due to structural constraints imposed by these linker designs, only a limited class of phenyl substrates—specifically benzyl and phenethyl-containing scaffolds—have been successfully employed for para functionalization. Given the low sample size and high degree of structural similarity between most reported para-selective DTs, we wanted to experimentally validate the factors that drive para regioselection and explore the generality of our obtained insights. Using our experimental data, we aimed towards the simplification/rational development of a “minimum para-selective unit” capable of extending para-selective reactivity to higher order phenylalkyl-containing compounds beyond current benzyl- and phenethyl-containing substrates.
Our investigation began by exploring DT-substrate combinations that corresponded to the predicted optimal 16-membered MCP, specifically focusing on the para-functionalization of 1-phenylbutanoate-containing substrates (Scheme 3A). First, the use of a simple para-cyanophenol motif (DT score = 7, predicted to be over the optimum DT score for para direction) was shown to be ineffective (8a). Based on this four carbon DT scaffold, we pondered whether selectivity could be improved by “reducing” the DT score (or in geometric terms, incorporating a motif that enables the DT to “bend” the DG towards the substrate) in line with other successful para templates.55–58 We noted that cis-disposed 1,4-substituted cyclohexyl motifs possess intrinsic concavity, which we hypothesize could act as an ortho-embedded para spacer without resorting to the addition of an extra ortho phenyl motif (see Scheme 3A and the SI for an empirical rationalization). Our hypothesis that introducing a pucker to the scaffold would improve para selectivity was verified using cis-disposed motif, which provided a preference for the para position (8b, p:m:o = 70:30:0) with a simple alicyclic scaffold; the use of which again enabled rapid visual spectroscopic inspection of the resulting product selectivity without confounding aryl signals typical of other para-selective DTs. Notably, the corresponding trans isomer 7c, which contains a similar geometry to 7a (DT score ~ 7), gave poor selectivity profiles ascribed to intermolecular (non-directed) reactivity.
Scheme 3.
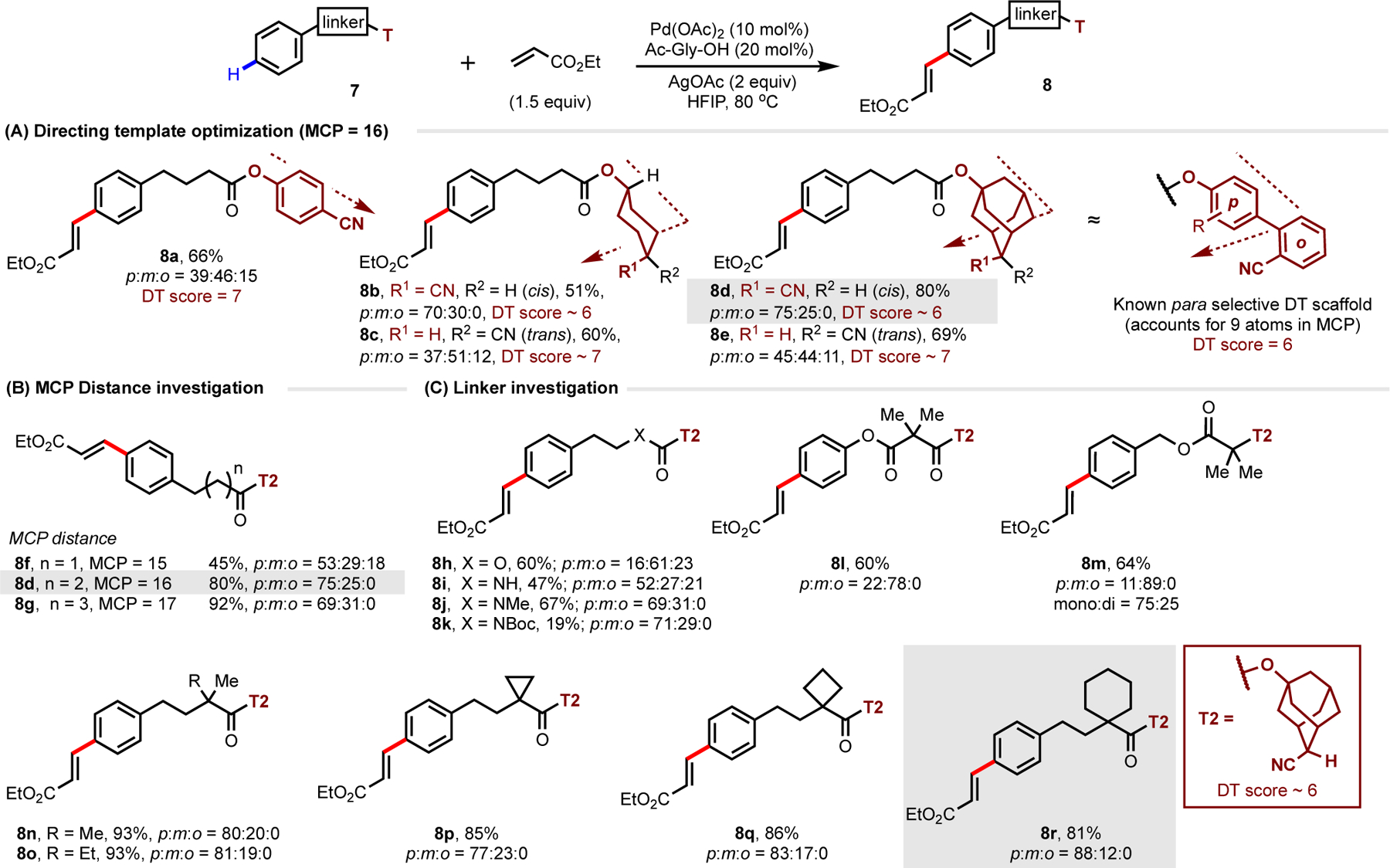
Empirical Findings Guide the Development of a New para-Selective Alicyclic DT (T2) Based on MCP Size, DT Geometry and Rigidity
Taking note that that improved selectivity can be obtained from DG-adjacent scaffold rigidification (vide supra), we surmised that the flexibility of substrate 7b might preclude more selective functionalization. As such, we rigidified the core structure of the DT while preserving the overall geometry through a locked cis-disposed adamantyl scaffold T2, which markedly improved yield and conferred modest improvements in para selectivity for the cis isomer (8d). Again, the use of the alternative trans-disposed template (8e) gave similarly poor selectivity profiles similar to 8a and 8c. The resulting reactivity and selectivity improvement provided confirmation of both 1) the DT geometry required to reach the para position (DT score ~ 6), and 2) the requirement of DT rigidity to reduce undesired adjacent meta functionalization.
So far, our para-selective DT optimization campaign revealed broad similarities for the factors required for meta and para regioselection. We were next curious about whether a similar MCP size dependence on para selectivity might be operative. Indeed, modulation of linker distance outside of the optimal 16-membered MCP adversely affected reaction efficacy (Scheme 3B), in complete accordance with data obtained for meta-selective DTs. Linker truncation (8f, MCP = 15) registered a large drop in yield and selectivity analogous to meta-selective DTs, while linker homologation (8g, MCP = 17) resulted in a modest decrease in selectivity with no detrimental effect on reactivity, again echoing the data obtained for meta-selective DTs. These cumulative results again affirm the importance of a combination of optimum MCP size and DT geometry for broad para regioselection. Once again however, these results suggested that while the necessary MCP size and DT geometric factors have been fulfilled for para selectivity, they themselves were not fully sufficient to enable the exceptionally high selectivity to be achieved even after DT rigidification. Noting that all previously reported para templates possessed a high degree of overall MCP structural rigidity relative to 7d, we surmised that increasing linker rigidity might allow our novel DT to disfavor the more proximate meta functionalization.
Investigation into the linker chemical space revealed a sensitivity of its composition to selectivity (Scheme 3C); in some cases, causing a reversal in selectivity understood to arise from adverse linker conformational effects (8l-8m). We found that the introduction of a quaternary center alpha to the carbonyl improved selectivity (8n-8r), attributed to conformational restriction via the Thorpe-Ingold effect. 8r was given up to 88:12:0 p:m:o selectivity, affording comparable selectivities to previously published para-selective templates for unbiased arenes substrates. In all cases, no ortho reactivity was observed despite the larger MCP size, contrasting observations arising from analogous meta-directing DTs at similar MCP sizes. DFT optimized geometries of 7d and 7r indicated the nitrile could not access the ortho C–H bonds despite the larger MCP size (MCP = 16), thus disfavoring any directed ortho C–H activation (see SI for details). This differs with the calculated structures for substrates possessing meta DT with longer linkers (5e; MCP = 14, Figure 5D), which distinctly show the capability of the nitrile DG to access the ortho site. These computational results affirm both our experimental observations and literature analysis indicating a properly tuned combination of and MCP size DT geometry works synergistically to position the catalyst, and is critical to disfavoring undesirable ortho products, wherein without both factors high selectivity cannot be achieved.
The resulting selectivity achieved with template T2 indicated that the rational development of a minimal para-selective DT from our literature-based findings was possible. In addition to MCP size and DT geometry, it also highlighted the paramount importance of DT and overall MCP rigidity through conformational restriction to ensure high DT-mediated para selectivity. In doing so, this enabled the development of a fully alicyclic DT capable of para-selective functionalization of phenylbutyl-containing substrates—the most flexible substrate to date amenable to para C–H activation—inaccessible to prior published DTs.59, 61
Guidelines for Future DT Design.
Following a quantitative analysis of 119 published DTs over 2012–2021, experimental corroboration affirms the empirical hypothesis that MCP size (“distance”) and DT “geometry” serve as the vital factors for controlling DT-mediated remote site-selection in a broad sense, as well as revealing the importance of DT rigidity to improve regioselection for a target C–H bond. Careful consideration of these factors then enabled the rational development of new alicyclic meta and para selective DTs, which confirm the possibility of using these guidelines for rational DT design. In doing so, these alicyclic DTs enable facile spectroscopic determination of selectivity as well as expanding the types of substrates amenable to remote functionalization. Taking all our experimental data, we explain the importance of three key factors important for DT-mediated meta and para-selective remote regioselection.67–72
1. MCP size (distance):
At the minimum end, MCP size is disproportionately important to switching on reactivity regardless of DT geometry. It is a necessary but, by itself, an insufficient requirement for remote selectivity, with the optimal MCP distance clustering around 12 for meta, and 16 for para regioselection. Finally, overly large MCP sizes erode selectivity for a target position typically through reactivity restoration for more proximate positions (see point 3: DT/MCP rigidity below)
2. DT geometry:
We observed that there is a qualitative ranking of geometric components for remote regioselection. This observation can most usefully be approximated in a scoring system given by 5npara+3nmeta+2nlinear–northo; where n is the number of a given component. In short—for templates of the optimum MCP size—ortho templates (score: –1 and 0) should not contain meta, linear or para motifs, meta templates (score: 1 and 2) should not contain para motifs, while para templates (score: 6) can contain all components. Intuitively, this observation can be understood as a need to avoid too many rigid spacers, or trans containing motifs, so as to enable the DT to be able to close onto the target C–H bond with minimal energetic penalty.
3. DT/MCP Rigidity:
Experimental data also highlight that DT selectivity is principally determined through an interplay between a) catalyst positioning to favor target bond activation, and b) engineering MCP strain to disfavor undesired bond activation. While optimal MCP size and DT geometry can enable broad selectivity, DT rigidification, which could be achieved by incorporating unrotatable bonds or leveraging the Thorpe-Ingold effect, is necessary to obtain higher selectivities. In the case of meta regioselection, DG-adjacent rigidity is sufficient and helps abate undesired para C–H activation. Rigidity is particularly important for para regioselection, where both DG-adjacent and increased overall MCP rigidity is required to disfavor proximate meta functionalization by imbuing a higher strain penalty for the DT to direct macrocyclometallation onto the meta position. These considerations are particularly important in the future development of new aliphatic DTs, as aliphatic motifs lack the rigidity conferred by rigid phenyl units typically utilized in established DTs.
Exemplified through the development of alicyclic meta and para-selective DTs, these three factors are noted to be particularly important for remote regioselection. From this, we additionally summarize key factors that we project ensures the greatest chance of achieving the desired meta and para-regioselection through a DT strategy shown in Table 1 and Figure 7.
Table 1.
Guidelines for the Development of meta- and para-Selective Templates
| MCP Size | Available Template Componentsa | DT scoreb | Rigidity Required | |
|---|---|---|---|---|
| Meta | 12 | ortho, linear, meta | 1, 2 | DG adjacent |
| Para | 16 | ortho, linear, meta, para | 6 | DG adjacent + Overall MCP |
See also Figure 7
DT score = 5npara + 3nmeta + 2nlinear – northo
Figure 7.

Graphical summary of distance and geometry factors and their importance for imparting remote regioselection.
CONCLUSIONS
In summary, the analysis of published DTs revealed important factors (distance, geometry and rigidity) that appeared to deterministically guide regioselection in remote arene C–H activation. Two types of novel aliphatic meta and para-selective templates were also successfully developed under the guidance of these rules. We anticipate that these guidelines will aid future efforts in the rational design of DTs, which could address the challenge of achieving remote regioselectivity both within and outside of the realm of C–H activation. Indeed, our approach and guidelines could serve as a framework for the future development of other directing templates, as well as catalytic manifolds, to impart remote regioselection.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully thank the Scripps Research Institute and the NIH (National Institute of General Medical Sciences grant R01 GM102265) for financial support. H.S.P. thank the Korea Foundation for Advanced Studies for a predoctoral fellowship. We thank Dr. Jason Chen, Brittany Sanchez, and Emily Sturgell (Scripps Automated Synthesis Facility) for assistance with HRMS. We thank Dr. Jake Bailey and the UCSD Crystallography Facility for X-ray crystallographic analysis.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Detailed analysis, experimental and computational procedures, full characterization of compounds including 1H and 13C NMR spectra, mass spectrometry data and crystallographic data CCDC 2098006, CCDC 2105518 (PDF)
The authors declare no competing financial interests.
REFERENCES
- (1).(a) Dutta U; Maiti S; Bhattacharya T; Maiti D Arene Diversification through Distal C(sp2)-H Functionalization. Science 2021, 372. DOI 10.1126/science.abd5992. [DOI] [PubMed] [Google Scholar]; (b) Whitehouse CJC; Bell SG; Wong LL P450BM3 (CYP102A1): Connecting the Dots. Chem. Soc. Rev 2012, 41, 1218–1260. [DOI] [PubMed] [Google Scholar]
- (2).Breslow R; Winnik MA Remote Oxidation of Unactivated Methylene Groups. J. Am. Chem. Soc 1969, 91, 3083–3084. [Google Scholar]
- (3).Das S; Incarvito CD; Crabtree RH; Brudvig GW Molecular Recognition in the Selective Oxygenation of Saturated C-H Bonds by a Dimanganese Catalyst. Science 2006, 312, 1941–1943. [DOI] [PubMed] [Google Scholar]
- (4).Moreira RF; Wehn PM; Sames D Highly Regioselective Oxygenation of C−H Bonds: Diamidomanganese Constructs with Attached Substrates as Catalyst Models. Angew. Chem. Int. Ed 2000, 39, 121. [Google Scholar]
- (5).Whisler MC; MacNeil S; Snieckus V; Beak P Beyond Thermodynamic Acidity: A Perspective on the Complex-Induced Proximity Effect (CIPE) in Deprotonation Reactions. Angew. Chem. Int. Ed 2004, 43, 2206–2225. [Google Scholar]
- (6).Yang Y-F; Cheng G-J; Liu P; Leow D; Sun T-Y; Chen P; Zhang X; Yu J-Q; Wu Y-D; Houk KN Palladium-Catalyzed Meta-Selective C–H Bond Activation with a Nitrile-Containing Template: Computational Study on Mechanism and Origins of Selectivity. J. Am. Chem. Soc 2014, 136, 344–355. [DOI] [PubMed] [Google Scholar]
- (7).Leow D; Li G; Mei TS; Yu J-Q Activation of Remote Meta-C-H Bonds Assisted by an End-on Template. Nature 2012, 486, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Meng G; Lam NYS; Lucas EL; Saint-Denis TG; Verma P; Chekshin N; Yu J-Q Achieving Site-Selectivity for C-H Activation Processes Based on Distance and Geometry: A Carpenter’s Approach. J. Am. Chem. Soc 2020, 142, 10571–10591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Li S; Ji H; Cai L; Li G Pd(II)-Catalyzed Remote Regiodivergent Ortho- and Meta-C-H Functionalizations of Phenylethylamines. Chem. Sci 2015, 6, 5595–5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Li Q; Knight BJ; Ferreira EM Palladium(II)-Catalyzed Ortho-Arylation of Aromatic Alcohols with a Readily Attachable and Cleavable Molecular Scaffold. Chem. Eur. J 2016, 22, 13054–13058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Knight BJ; Rothbaum JO; Ferreira EM The Design of a Readily Attachable and Cleavable Molecular Scaffold for Ortho-Selective C–H Alkenylation of Arene Alcohols. Chem. Sci 2016, 7, 1982–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Baccalini A; Vergura S; Dolui P; Maiti S; Dutta S; Maity S; Khan FF; Lahiri GK; Zanoni G; Maiti D Cobalt-Catalyzed C(Sp2)–H Allylation of Biphenyl Amines with Unbiased Terminal Olefins. Org. Lett 2019, 21, 8842–8846. [DOI] [PubMed] [Google Scholar]
- (13).Bera M, M.; Maji A; Sahoo SK; Maiti D Palladium(II)-Catalyzed Meta-C–H Olefination: Constructing Multisubstituted Arenes through Homo-Diolefination and Sequential Hetero-Diolefination. Angew. Chem. Int. Ed 2015, 54, 8515–8519 [Google Scholar]
- (14).Chu L; Shang M; Tanaka K; Chen Q; Pissarnitski N; Streckfuss E; Yu J-Q Remote Meta-C-H Activation Using a Pyridine-Based Template: Achieving Site-Selectivity via the Recognition of Distance and Geometry. ACS Cent. Sci 2015, 1, 394–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Modak A; Mondal A; Watile R; Mukherjee S; Maiti D Remote Meta C–H Bond Functionalization of 2-Phenethylsulphonic Acid and 3-Phenylpropanoic Acid Derivatives. Chem. Commun 2016, 52, 13916–13919. [Google Scholar]
- (16).Maji A; Bhaskararao B; Singha S; Sunoj RB; Maiti D Directing Group Assisted: Meta-Hydroxylation by C–H Activation. Chem. Sci 2016, 7, 3147–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Li S; Cai L; Ji H; Yang L; Li G Pd(II)-Catalysed Meta-C-H Functionalizations of Benzoic Acid Derivatives. Nat. Commun 2016, 7 DOI 10.1038/ncomms10443. [DOI] [Google Scholar]
- (18).Davis HJ; Mihai MT; Phipps RJ Ion Pair-Directed Regiocontrol in Transition-Metal Catalysis: A Meta-Selective C–H Borylation of Aromatic Quaternary Ammonium Salts. J. Am. Chem. Soc 2016, 138, 12759–12762. [DOI] [PubMed] [Google Scholar]
- (19).Davis HJ; Genov GR; Phipps RJ Meta-Selective C−H Borylation of Benzylamine-, Phenethylamine-, and Phenylpropylamine-Derived Amides Enabled by a Single Anionic Ligand. Angew. Chem. Int. Ed 2017, 56, 13351–13355. [Google Scholar]
- (20).Bag S; Jayarajan R; Dutta U; Chowdhury R; Mondal R; Maiti D Remote Meta-C–H Cyanation of Arenes Enabled by a Pyrimidine-Based Auxiliary. Angew. Chem. Int. Ed 2017, 56, 12538–12542. [Google Scholar]
- (21).Zhang L; Zhao C; Liu Y; Xu J; Xu X; Jin Z Activation of Remote Meta-C−H Bonds in Arenes with Tethered Alcohols: A Salicylonitrile Template. Angew. Chem. Int. Ed 2017, 56, 12245–12249. [Google Scholar]
- (22).Dutta U; Modak A; Bhaskararao B; Bera M; Bag S; Mondal A; Lupton DW; Sunoj RB; Maiti D Catalytic Arene Meta-C-H Functionalization Exploiting a Quinoline-Based Template. ACS Catal 2017, 7, 3162–3168. [Google Scholar]
- (23).Wan L; Dastbaravardeh N; Li G; Yu J-Q Cross-Coupling of Remote Meta-C–H Bonds Directed by a U-Shaped Template. J. Am. Chem. Soc 2013, 135, 18056–18059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhang Z; Tanaka K; Yu J-Q Remote Site-Selective C–H Activation Directed by a Catalytic Bifunctional Template. Nature 2017, 543, 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Achar TK; Ramakrishna K; Pal T; Porey S; Dolui P; Biswas JP; Maiti D Regiocontrolled Remote C−H Olefination of Small Heterocycles. Chem.– A Eur. J 2018, 24, 17906–17910. [Google Scholar]
- (26).Fang L; Saint-Denis TG; Taylor BLH; Ahlquist S; Hong K; Liu S; Han L; Houk KN; Yu J-Q Experimental and Computational Development of a Conformationally Flexible Template for the Meta-C–H Functionalization of Benzoic Acids. J. Am. Chem. Soc 2017, 139, 10702–10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hoque ME; Bisht R; Haldar C; Chattopadhyay B Noncovalent Interactions in Ir-Catalyzed C–H Activation: L-Shaped Ligand for Para-Selective Borylation of Aromatic Esters. J. Am. Chem. Soc 2017, 139, 7745–7748. [DOI] [PubMed] [Google Scholar]
- (28).Modak A; Patra T; Chowdhury R; Raul S; Maiti D Palladium-Catalyzed Remote Meta-Selective C–H Bond Silylation and Germanylation. Organometallics 2017, 36, 2418–2423. [Google Scholar]
- (29).Jayarajan R; Das J; Bag S; Chowdhury R; Maiti D Diverse Meta-C−H Functionalization of Arenes across Different Linker Lengths. Angew. Chem. Int. Ed 2018, 57, 7659–7663. [Google Scholar]
- (30).(a) Mihai MT; Davis HJ; Genov GR; Phipps RJ Ion Pair-Directed C-H Activation on Flexible Ammonium Salts: Meta-Selective Borylation of Quaternized Phenethylamines and Phenylpropylamines. ACS Catal 2018, 8, 3764–3769. [Google Scholar]; (b) The only examples where a simple 3-pyridyl motif was used in meta-regioselection are borylation processes, which by itself possess appreciable intrinsic non-directed meta selectivity. These examples also employ non-covalent anchoring strategies, which also may behave differently to more tightly associated DT-substrate structures.
- (31).Yang G; Zhu D; Wang P; Tang RY; Yu J-Q Remote C−H Activation of Various N-Heterocycles Using a Single Template. Chem. Eur. J 2018, 24, 3434–3438. [DOI] [PubMed] [Google Scholar]
- (32).Jin Z; Chu L; Chen YQ; Yu J-Q Pd-Catalyzed Remote Meta-C–H Functionalization of Phenylacetic Acids Using a Pyridine Template. Org. Lett 2018, 20, 425–428. [DOI] [PubMed] [Google Scholar]
- (33).Achar TK; Zhang X; Mondal R; Shanavas MS; Maiti S; Maity S; Pal N; Paton RS; Maiti D Palladium-Catalyzed Directed Meta-Selective C−H Allylation of Arenes: Unactivated Internal Olefins as Allyl Surrogates. Angew. Chem. Int. Ed 2019, 58, 10353–10360. [Google Scholar]
- (34).Dai H-X; Li G; Zhang XG; Stepan AF; Yu J-Q Pd(II)-Catalyzed Ortho- Or Meta-C–H Olefination of Phenol Derivatives. J. Am. Chem. Soc 2013, 135, 7567–7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Li S; Wang H; Weng Y; Li G Carboxy Group as a Remote and Selective Chelating Group for C−H Activation of Arenes. Angew. Chem. Int. Ed 2019, 131, 18673–18678.. [Google Scholar]
- (36).Bag S; Petzold M; Sur A; Bhowmick S; Werz DB; Maiti D Palladium-Catalyzed Selective Meta-C−H Deuteration of Arenes: Reaction Design and Applications. Chem. Eur. J 2019, 25, 9433–9437. [DOI] [PubMed] [Google Scholar]
- (37).Brochetta M; Borsari T; Bag S; Jana S; Maiti S; Porta A; Werz DB; Zanoni G; Maiti D Direct Meta-C−H Perfluoroalkenylation of Arenes Enabled by a Cleavable Pyrimidine-Based Template. Chem. Eur. J 2019, 25, 10323–10327. [DOI] [PubMed] [Google Scholar]
- (38).Xu J; Chen J; Gao F; Xie S; Xu X; Jin Z; Yu J-Q Sequential Functionalization of Meta-C-H and Ipso-C-O Bonds of Phenols. J. Am. Chem. Soc 2019, 141, 1903–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Xu H; Liu M; Li LJ; Cao YF; Yu J-Q; Dai H-X Palladium-Catalyzed Remote Meta-C–H Bond Deuteration of Arenes Using a Pyridine Template. Org. Lett 2019, 21, 4887–4891. [DOI] [PubMed] [Google Scholar]
- (40). Fan Z; Bay KL; Chen X; Zhuang Z; Park HS; Yeung K-S; Houk KN; Yu J-Q Rational Development of Remote C−H Functionalization of Biphenyl: Experimental and Computational Studies. Angew. Chem. Int. Ed 2020, 59, 4770–4777. This is a notable outlier case (MCP = 8), in which invoking the (hetero)bimetallic speciation MCP leads to an MCP size of 12 in full accordance with our guidelines.
- (41).Gholap A; Bag S; Pradhan S; Kapdi AR; Maiti D Diverse Meta-C–H Functionalization of Amides. ACS Catal 2020, 10, 5347–5352. [Google Scholar]
- (42).Casali E; Kalra P; Brochetta M; Borsari T; Gandini A; Patra T; Zanoni G; Maiti D Overriding Ortho Selectivity by Template Assisted Meta-C–H Activation of Benzophenones. Chem. Commun 2020, 56, 7281–7284. [Google Scholar]
- (43).Bag S; K, S.; Mondal A; Jayarajan R; Dutta U; Porey S; Sunoj RB; Maiti D Palladium-Catalyzed Meta-C–H Allylation of Arenes: A Unique Combination of a Pyrimidine-Based Template and Hexafluoroisopropanol. J. Am. Chem. Soc 2020, 142, 12453–12466. [DOI] [PubMed] [Google Scholar]
- (44).Porey S; Zhang X; Bhowmick S; Kumar Singh V; Guin S; Paton RS; Maiti D; Singh VK; Guin S; Paton RS; Maiti D Alkyne Linchpin Strategy for Drug:Pharmacophore Conjugation: Experimental and Computational Realization of a Meta-Selective Inverse Sonogashira Coupling. J. Am. Chem. Soc 2020, 142, 3762–3774. [DOI] [PubMed] [Google Scholar]
- (45).Lee S; Lee H; Tan KL Meta-Selective C–H Functionalization Using a Nitrile-Based Directing Group and Cleavable Si-Tether. J. Am. Chem. Soc 2013, 135, 18778–18781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Cai L; Li S; Zhou C; Li G Carboxyl-Assisted Meta-Selective C–H Functionalizations of Benzylsulfonamides. Org. Lett 2020, 22, 7791–7796. [DOI] [PubMed] [Google Scholar]
- (47).Bag S; Jana S; Pradhan S; Bhowmick S; Goswami N; Sinha SK; Maiti D Imine as a Linchpin Approach for Meta-C–H Functionalization. Nat. Commun 2021, 12, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Ramesh P; Sreenivasulu C; Gorantla KR; Mallik BS; Satyanarayana G A Simple Removable Aliphatic Nitrile Template 2-Cyano-2,2-Di-Isobutyl Acetic Acid for Remote Meta-Selective C–H Functionalization. Org. Chem. Front 2021, 8, 1959–1969. [Google Scholar]
- (49).Li S; Zhang C; Fu L; Wang H; Cai L; Chen X; Wang X; Li G Arene C–H Iodination Using Aryl Iodides. CCS Chem 2021, 8, 2360–2371. [Google Scholar]
- (50).Yang G; Lindovska P; Zhu D; Kim J; Wang P; Tang RY; Movassaghi M; Yu J-Q Pd(II)-Catalyzed Meta-C-H Olefination, Arylation, and Acetoxylation of Indolines Using a U-Shaped Template. J. Am. Chem. Soc 2014, 136, 10807–10813. [DOI] [PubMed] [Google Scholar]
- (51).Tang RY; Li G; Yu J-Q Conformation-Induced Remote Meta-C-H Activation of Amines. Nature 2014, 507, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Bera M; Modak A; Patra T; Maji A; Maiti D Meta-Selective Arene C–H Bond Olefination of Arylacetic Acid Using a Nitrile-Based Directing Group. Org. Lett 2014, 16, 5760–5763. [DOI] [PubMed] [Google Scholar]
- (53).Kuninobu Y; Ida H; Nishi M; Kanai M A Meta-Selective C–H Borylation Directed by a Secondary Interaction between Ligand and Substrate. Nat. Chem 2015, 7, 712–717. [DOI] [PubMed] [Google Scholar]
- (54).Deng Y; Yu J-Q Remote Meta-C–H Olefination of Phenylacetic Acids Directed by a Versatile U-Shaped Template. Angew. Chem. Int. Ed 2015, 54, 888–891. [Google Scholar]
- (55).Bag S; Patra T; Modak A; Deb A; Maity S; Dutta U; Dey A; Kancherla R; Maji A; Hazra A; Bera M; Maiti D Remote Para-C–H Functionalization of Arenes by a D-Shaped Biphenyl Template-Based Assembly. J. Am. Chem. Soc 2015, 137, 11888–11891. [DOI] [PubMed] [Google Scholar]
- (56).Patra T; Bag S; Kancherla R; Mondal A; Dey A; Pimparkar S; Agasti S; Modak A; Maiti D Palladium-Catalyzed Directed Para C−H Functionalization of Phenols. Angew. Chem. Int. Ed 2016, 55, 7751–7755. [Google Scholar]
- (57).Li M; Shang M; Xu H; Wang X; Dai H-X; Yu J-Q Remote Para-C–H Acetoxylation of Electron-Deficient Arenes. Org. Lett 2019, 21, 540–544. [DOI] [PubMed] [Google Scholar]
- (58).Pimparkar S; Bhattacharya T; Maji A; Saha A; Jayarajan R; Dutta U; Lu G; Lupton DW; Maiti D Para-Selective Cyanation of Arenes by H-Bonded Template. Chem. Eur. J 2020, 26, 11558–11564. [DOI] [PubMed] [Google Scholar]
- (59).Chen X; Fan S; Zhang M; Gao Y; Li S; Li G Palladium-Catalyzed Remote Para-C–H Activation of Arenes Assisted by a Recyclable Pyridine-Based Template. Chem. Sci 2021, 12, 4126–4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Control experiments show that non-directed selectivity roughly gives 0.5:1:1 selectivity between ortho:meta:para positions, which affirms a 67:33 cut off for poor selectivity. The cut off for good selectivity was empirically determined where the desired product accounts for 80%+ of the product mixture, as well as generating a cut off that gives a large enough sample size for ortho, meta and para-directing DTs to generate meaningful conclusions from.
- (61).We assessed “flexibility” based on quantifying i) the number of contiguous rotatable bonds, ii) the number of contiguous sp3 centers. See attached spreadsheet and supporting information for more information of other parameters assessed. In terms of number of contiguous sp3 centers, 8r contains three, while the next most flexible template from reference 59 contains two.
- (62).Squillacote ME; DeFellipis J; Shu Q How Stable Is Trans-Cycloheptene? J. Am. Chem. Soc 2005, 127, 15983–15988. [DOI] [PubMed] [Google Scholar]
- (63).Allinger NL; Sprague JT Conformational Analysis. LXXXIV. A Study of the Structures and Energies of Some Alkenes and Cycloalkenes by the Force Field Method. J. Am. Chem. Soc 1972, 94, 5734–5747. [Google Scholar]
- (64).Sicher J; Svoboda M; Závada J; Turner RB; Goebel P Stereochemical Studies—Xxxvi : An Approach to Conformational Analysis of Medium Ring Compounds. Unsaturated Ten-Membered Ring Derivatives. Tetrahedron 1966, 22, 659–671. [Google Scholar]
- (65).Dudev T; Lim C Ring Strain Energies from Ab Initio Calculations. J. Am. Chem. Soc 1998, 120, 4450–4458. [Google Scholar]
- (66).Wiberg KB The Concept of Strain in Organic Chemistry. Angew. Chem. Int. Ed 1986, 25, 312–322. [Google Scholar]
- (67).Several other directing ligand/template-mediated processes outside of the surveyed C–H activation literature appear to conform with the data presented in this manuscript. The references cited below (references 68–72) are representative papers that indicate the potential applicability of this approach.
- (68).Golding WA; Pearce-Higgins R; Phipps RJ Site-Selective Cross-Coupling of Remote Chlorides Enabled by Electrostatically Directed Palladium Catalysis. J. Am. Chem. Soc 2018, 140, 13570–13574. [DOI] [PubMed] [Google Scholar]
- (69).Golding WA; Phipps RJ Electrostatically-Directed Pd-Catalysis in Combination with C–H Activation: Site-Selective Coupling of Remote Chlorides with Fluoroarenes and Fluoroheteroarenes. Chem. Sci 2020, 11, 3022–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Yang L; Uemura N; Nakao Y Meta-Selective C–H Borylation of Benzamides and Pyridines by an Iridium–Lewis Acid Bifunctional Catalyst. J. Am. Chem. Soc 2019, 141, 7972–7979. [DOI] [PubMed] [Google Scholar]
- (71).Trouvé J; Zardi P; Al-Shehimy S; Roisnel T; Gramage-Doria R Enzyme-like Supramolecular Iridium Catalysis Enabling C−H Bond Borylation of Pyridines with Meta-Selectivity. Angew. Chem. Int. Ed 2021, 60, 18006–18013. [Google Scholar]
- (72).Zhang T; Luan YX; Lam NYS; Li JF; Li Y; Ye M; Yu J-Q A Directive Ni Catalyst Overrides Conventional Site Selectivity in Pyridine C–H Alkenylation. Nat. Chem 2021, 13, 1207–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.