

Abstract
While studies have elucidated many pathophysiological elements of COVID-19, little is known about immunological changes during COVID-19 resolution. We analyzed immune cells and phosphorylated signaling states at single-cell resolution from longitudinal blood samples of patients hospitalized with COVID-19, pneumonia and/or sepsis, and healthy individuals by mass cytometry. COVID-19 patients showed distinct immune compositions and an early, coordinated, and elevated immune cell signaling profile associated with early hospital discharge. Intra-patient longitudinal analysis revealed changes in myeloid and T cell frequencies and a reduction in immune cell signaling across cell types that accompanied disease resolution and discharge. These changes, together with increases in regulatory T cells and reduced signaling in basophils, also accompanied recovery from respiratory failure and were associated with better outcomes at time of admission. Therefore, although patients have heterogeneous immunological baselines and highly variable disease courses, a core immunological trajectory exists that defines recovery from severe SARS-CoV-2 infection.
Keywords: COVID-19, immune response, immune cell signaling, disease resolution, recovery
Graphical abstract
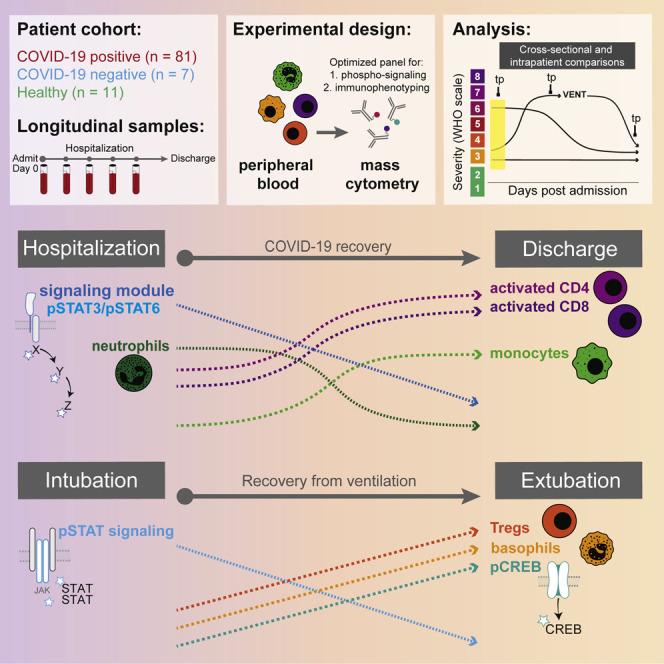
Immunological changes during COVID-19 resolution remain unknown. Burnett, Okholm, Tenvooren et al. analyze longitudinal blood samples from hospitalized COVID-19 patients by single-cell mass cytometry, identifying a conserved set of immunological processes and cell signaling states that uniquely accompany COVID-19 recovery and associate with better clinical outcomes at time of admission.
Introduction
SARS-CoV-2 and the resulting disease COVID-19 has resulted in over 517,000,000 infected individuals and more than 6,200,000 deaths globally as of May 15, 2022 (World Health Organization, 2021a). In a prospective study of adults confirmed with SARS-CoV-2, 91% of patients were asymptomatic or were outpatients with mild illness, while 9% required inpatient care (Logue et al., 2021). These patients can develop severe diseases, including pneumonia, acute respiratory distress syndrome (ARDS), or multiple organ failure, and often require supplemental oxygen support or, in the most critical cases, mechanical ventilation. Although a small percentage of all infected patients succumb to the disease (1.3%) (Centers for Disease Control and Prevention, 2021), the majority of hospitalized patients successfully combat and clear the infection. Many studies have focused on features defining the subset of patients who ultimately succumb to the disease; however, it is also essential to characterize successful resolution and identify conserved immune features during this interval.
The immunopathology of COVID-19 is broadly characterized by lymphopenia, lymphocyte dysfunction, abnormalities of innate immune cells, and increased cytokine production (Lucas et al., 2020; Mann et al., 2020; Mathew et al., 2020; Yang et al., 2020). Early observations of serum cytokine levels in COVID-19 patients revealed high levels of circulating IL-6, generating the hypothesis of an IL-6-driven cytokine storm and resulting immunopathology (Moore and June, 2020; Yang et al., 2020). However, other studies suggest that IL-6 levels do not differ between severe and moderate COVID-19 patients (Wilson et al., 2020) and may even be lower in severe COVID-19 than in other similar critical illnesses (Sinha et al., 2020). While a meta-analysis evaluating IL-6-neutralizing therapies concluded that they may provide some benefit, individual clinical trials report conflicting results, raising questions about when and for whom they should be used (Tsai et al., 2020; RECOVERY Collaborative Group, 2021; Rosas et al., 2021; WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group et al., 2021). Corticosteroid treatment is another strategy to modulate immune signaling and has been broadly adopted based on the results of prospective randomized clinical trials (Angus et al., 2020; RECOVERY Collaborative Group et al., 2021). However, their benefit may be modest (Wagner et al., 2021) and vary across different subsets of patients (Chen et al., 2021; Sinha et al., 2021; Wagner et al., 2021). Additionally, insufficient type I interferon (IFN) signaling and autoantibodies that inhibit type I IFN have been linked to a subset of severe cases of COVID-19, suggesting that type I immune responses and IFN signaling are likely protective (Zhang et al., 2020a; Asano et al., 2021; Chang et al., 2021; Combes et al., 2021; van der Wijst et al., 2021; Wang et al., 2021). High serum cytokine levels, along with observations of broad immunological misfiring, have been observed across patient subsets, indicating a delicate balance between productive and destructive immune responses and suggesting the importance of evaluating immune cell signaling. However, it remains unclear what, if any, immune cell signaling is protective and how immune cell signaling dynamics change over time in patients who resolve or fail to resolve COVID-19.
While many studies have made significant contributions to our understanding of the immune system and its relation to COVID-19, most analytical approaches are cross-sectional and describe the immunological differences between COVID-19 severity groups defined by clinical metrics, such as the WHO score. In comparison, longitudinal studies are uniquely capable of assessing changes in the immune response during disease progression or resolution over time. Elucidating the immunological events that accompany successful disease resolution is essential to informing the management of patient care and contextualizing the deviations from successful resolution that characterize the most severe disease cases. Because the infection timeline is highly variable, and human immunological responses are diverse, understanding immunological dynamics during this specific recovery period requires longitudinal monitoring and high-dimensional data from a large cohort of patients. Here, we investigated intra-patient immunological changes across clinically relevant time points to identify changes in immune responses that accompany effective COVID-19 resolution. We obtained 230 longitudinal peripheral blood samples from 81 hospitalized COVID-19 patients, 7 patients with pneumonia and/or sepsis unrelated to SARS-CoV-2 (COVID-19-negative patients), and 11 healthy individuals. To investigate changes in immune cell signaling states over time, we utilized mass cytometry with a panel of antibodies specific for immune cell phenotyping and for measuring phosphorylated cell signaling proteins. We identified distinct immune cell composition and signaling states in COVID-19 patients compared to COVID-19-negative patients and healthy individuals. Additionally, we discovered a conserved and coordinated immune response, including changes in myeloid and T cell abundance and phenotypes, as well as a reduction in pan-immune cell signaling, that accompanies COVID-19 resolution and hospital discharge. Furthermore, these and other features were relevant to resolution in the most severe mechanically ventilated patients, and these immune cell states correlated with better clinical outcomes at time of admission. Our findings indicate that, although patients have heterogeneous immunological baselines and highly variable disease courses, there exists a core immunological trajectory that defines recovery from severe SARS-CoV-2 infection. Our results provide a working model of a successful immune response trajectory among patients with COVID-19 requiring hospitalization, deviations from which are associated with extended hospitalization and mortality.
Results
Longitudinal peripheral blood analysis was performed in hospitalized COVID-19-positive and COVID-19-negative patients
To investigate the composition of circulating immune cells and the cell signaling states that characterize SARS-CoV-2 infections and distinguish it from other respiratory infections, we collected longitudinal peripheral blood (PB) samples from COVID-19 patients and COVID-19-negative patients with pneumonia and/or sepsis (PCR negative for SARS-CoV-2) admitted to UCSF Medical Center and Zuckerberg San Francisco General Hospital. PB samples and corresponding patient demographics and clinical parameters, e.g., World Health Organization (WHO) severity scores (World Health Organization, 2021b), ventilation duration, and hospital length of stay, were collected throughout inpatient care (Tables S1, S2, and S3). PB samples from 11 healthy individuals were obtained as controls (Table S4), though median age was younger than for hospitalized groups. Whole blood was fixed using Smart Tube proteomic stabilizer and stored at -80°C. All samples were processed, stained, and analyzed by mass cytometry to quantify the expression of 30 protein markers and 14 phosphorylated signaling molecules (Table S5). Samples that met quality control standards (methods) were normalized across batches (methods, Figure S1A) resulting in 205 samples from 81 COVID-19 patients, 14 samples from 7 COVID-19-negative patients, and single samples from each of 11 healthy individuals (Figure 1A and S1B, Table S6). COVID-19 patients were classified into COVID-19 severity groups based on their WHO score at day of sampling (3: mild, 4: moderate, 5–7: severe) (World Health Organization, 2021b). We manually gated 38 canonical immune cell populations (Figure S1C) and evaluated immune cell population frequencies, protein expression patterns, and immune cell signaling pathways specific to COVID-19 course escalation and resolution.
Figure 1.

COVID-19 immune phenotype and composition is highly divergent from healthy individuals and has distinct features compared to other severe respiratory infections
(A) Overview of cohort. Patients were admitted to the hospital and enrolled in the study at day 0. Peripheral blood samples were collected up until day 28 of hospitalization. Corresponding clinical parameters and WHO scores were documented. 205 samples from 81 COVID-19-positive patients were included in the final cohort. Additionally, 14 samples from 7 COVID-19-negative patients with other respiratory diseases and 11 healthy individuals were included in the study. On average, we obtained 2 (range of 1–7) usable blood samples per patient.
(B) t-SNE plot of all patient samples at day 0 (n = 83) using phenotypic markers colored by major immune cell populations. Upper right panel: t-SNE plot of healthy samples (n = 11); middle right panel: t-SNE plot of COVID-19-negative samples (n = 6); lower right panel: t-SNE plot of COVID-19-positive samples (n = 66).
(C) Immune cell population abundance at day 0 in COVID-19-positive (+), COVID-19-negative (-) patients, and healthy individuals (H). Nominal p values obtained by Wilcoxon Rank Sum Test, followed by Benjamini-Hochberg correction with FDR < 0.1.
(D) Correlation between cell population abundance at day 0 and clinical outcomes, e.g., ventilation duration (vent duration) and hospital length of stay (hosp los) for COVID-19-positive patients (n = 65, excluding the patient that is hospitalized for 260 days). Correlation estimates are obtained by Spearman correlation.
(E) Protein expression on neutrophils (F) in COVID-19-positive (COV+), COVID-19-negative (COV-) patients, and healthy controls at day 0 (Wilcoxon Rank Sum Test, Benjamini-Hochberg correction with FDR < 0.1).
(F) Frequency of monocyte subsets in COVID-19-positive (COV+), COVID-19=negative (COV-) patients, and healthy controls at day 0. Nominal p values obtained by Wilcoxon Rank Sum Test. See also Figure S1.
Immune cell compositions in COVID-19 patients, COVID-19-negative patients, and healthy individuals are distinct on day of admission
First, we characterized the immunological landscape of COVID-19 patients, COVID-19-negative patients (critically ill, mechanically ventilated controls with pneumonia and/or sepsis unrelated to SARS-CoV-2 infection), and healthy individuals to assess immunological signatures that may be specific to COVID-19 at day of admission (day 0). Dimensionality reduction by t-distributed stochastic neighbor embedding (t-SNE) using only phenotypic markers revealed distinct immune cell compositions between COVID-19-positive, COVID-19-negative, and healthy individuals (Figure 1B). Consistent with previous studies, COVID-19 patients exhibited a significantly different immune cell composition compared with healthy individuals, with significant frequency differences across almost all manually gated immune cell populations (FDR < 0.1, Figure 1C) (Mathew et al., 2020). To determine modules of immune changes, we evaluated whether distinct immune cell populations correlate with each other as well as with patient demographics or clinical parameters. We found a coordinated adaptive immune response in which several T cell subsets and B cell frequencies were positively correlated with one another (Figure 1D). In contrast, the innate arm demonstrated a dichotomous relationship, with an anti-correlation between neutrophil and monocyte frequencies. Additionally, monocyte frequencies at day 0 were positively correlated with T cell subsets and negatively correlated with ventilation duration (Figure 1D), suggesting there may be a coordinated immune response associated with better clinical outcome.
Monocyte and neutrophil composition reveal distinct compartmental shifts in the innate immune arm of COVID-19 infection
Large shifts in innate immune compartments were evident between COVID-19 patients, patients with other respiratory infections, and healthy controls (Figure 1B); therefore, we further investigated the composition of neutrophils and monocytes. While neutrophil frequency was not significantly different between COVID-19 patients and the healthy individuals (Figures 1C and S1D), we found that a variety of proteins were altered in their expression on neutrophils across groups. Neutrophils from COVID-19 patients exhibited significantly increased expression of CD11c, CD14, CD16, and PD-L1, suggesting a highly activated and inflammatory neutrophil phenotype in COVID-19 patients (FDR < 0.1, Figure 1E). Additionally, while the frequency of all monocytes was comparable between groups (FDR > 0.1, Figure 1C), composition of monocyte subsets (defined as classical, intermediate, and non-classical) was significantly different between patients with COVID-19 and other respiratory infections compared with healthy individuals (FDR < 0.1, Figure 1C). Patients exhibited a significant increase in the frequency of intermediate monocytes along with a relative decrease in classical monocytes (Figure 1F).
Cross-sectional analysis of COVID-19 severity groups reveals few immunological features that distinguish severity states
We next evaluated the immunological differences between COVID-19 severity groups across time (Figure S1E). We found no significant differences between severity groups at day 0 (FDR > 0.1, Figure S1F) and only few population differences at day 4 and day 7 (FDR < 0.1, Figure S1G). Within each severity group, comparisons across time showed that plasmablasts contract from day 0 to day 7 in the majority of severe COVID-19 patients (FDR < 0.1, Figure S1H), while activated CD4 T cells are upregulated from day 0 to day 7 in mild COVID-19 patients (FDR < 0.1, Figure S1I). The paucity of differences between severity groups suggested that significant variability may exist in the timing of disease escalation and resolution across individuals and therefore the immunological processes that mediate these changes over time.
Early, coordinated, and activated immune cell signaling is associated with early hospital discharge in COVID-19 patients
To gain insights into key immune cell signaling modules associated with COVID-19, we measured the phosphorylation state of 14 signaling molecules across all immune cell subsets (Figure 2A). First, we evaluated the median expression of phosphorylated signaling proteins across all CD45+ hematopoietic PB cells in COVID-19-positive, COVID-19-negative, and healthy individuals at day 0. Differential expression analysis revealed five signaling molecules (pSTAT1, pPLCγ2, pZAP70/pSyk, pCREB, and pSTAT3) that were upregulated in COVID-19 patients compared with healthy individuals (FDR < 0.1, Figure 2B). To determine whether a specific cell type was driving the higher signaling state in COVID-19 patients, we evaluated the median phosphorylation state of the respective signaling molecules within manually gated immune cell subsets. We found higher median signaling across the majority of cell subsets, showing that immune cell signaling states are coordinated across most cell types simultaneously and not driven by signaling within a specific cell type (Figure S2A). Consistent with our observations for immune cell populations, we observed no signaling differences within and across severity groups at day 0, day 4, and day 7 (FDR > 0.1, Figures S2B and S2C).
Figure 2.

Early, coordinated, and activated immune cell signaling is associated with early hospital discharge in COVID-19 patients
(A) Signaling schematic. Stars denote phosphorylated signaling molecules that are measured in the CyTOF panel.
(B) Expression of signaling molecules in CD45+ CD235a/b-negative peripheral blood immune cells at day 0 in COVID-19-positive (+), COVID-19-negative (-) patients, and healthy individuals (H). Nominal p values obtained by Wilcoxon Rank Sum Test, followed by Benjamini-Hochberg correction with FDR < 0.1.
(C and D) Correlation between signaling molecule expressions at day 0 for COVID-19+ patients (n = 66) (C) and COVID-19- patients (n = 6) (D). Correlation estimates are obtained by Spearman correlation.
(E) Differential expression analysis of signaling molecules at day 0 between COVID-19+ patients that are discharged early (≤ 30 days of admission, n = 59) and late (>30 days after admission, n = 7). Nominal p values obtained by Wilcoxon Rank Sum Test. The log2 fold changes (late versus early) are plotted against the negative log10 (nominal p values). Colors indicate whether signaling molecules are significantly higher in early discharged patients (blue) or late discharged patients (purple) or not differentially expressed (FALSE, gray) after Benjamini-Hochberg correction, FDR < 0.1.
(F) Correlation between pSTAT3, pERK, pS6, and pSTAT6 signaling at day 0 and ventilation duration for ventilated COVID-19+ patients (n = 16). Correlation estimates and nominal p values are obtained by Spearman correlation, followed by Benjamini-Hochberg correction. Blue lines and gray shadows represent the best-fitted smooth line and 95% confidence interval. See also Figure S2.
To investigate coordinated signaling modules in CD45+ cells, we evaluated correlations between the expression of signaling molecules at day 0. For COVID-19 patients, we observed a coordinated, positive signaling response (Figure 2C), which was absent in patients with other respiratory infections or sepsis (Figure 2D). To evaluate the relevance of this early, coordinated, and activated signaling signature, we examined associations between the expression of signaling molecules at day 0 and clinically relevant outcomes. By splitting the patients into two groups based on time until discharge, we found that the expression of 8 of the 14 signaling molecules were significantly higher at day 0 in patients that were discharged early (≤30 days, n = 59) compared with patients that were discharged late (> 30 days, n = 7) (FDR < 0.1, Figure 2E). No signaling molecules were higher at day 0 in patients who were discharged late (Figure 2E). We also observed an overall trend of negative correlations between median signaling in CD45+ cells and hospital length of stay as a continuous variable, though these did not reach statistical significance (FDR > 0.1, Figure S2D). Consistent with these findings, for patients on mechanical ventilation, we observed an overall trend of negative correlations between CD45+ signaling and ventilation duration, with the strongest correlations observed for pSTAT3, pERK, pS6 (nominal p value < 0.05, FDR = 0.1, Figure 2F), and pSTAT6 (nominal p value < 0.05, FDR = 0.12, Figure 2F). Median pSTAT3 expression in several cell subtypes was significantly correlated with ventilation duration (FDR < 0.1, Figure S2F), and other cell type-specific signaling features exhibited negative correlations as well, though these did not reach statistical significance (FDR > 0.1, Figure S2F). Correspondingly, signaling within cell subtypes demonstrated a broad positive correlation with each other (Figure S2G), indicating coordinated signaling states across cell populations. Taken together, our results show that coordinated high signaling at day of admission is associated with shorter length of hospitalization and mechanical ventilation.
Conserved immunological processes and changes in cell signaling states accompany disease resolution and discharge
Although cross-sectional analysis can provide insights into the immunological state of COVID-19 patients and severity groups, the natural heterogeneity of patient immune responses and significant differences in their disease time courses may obscure immunological processes that mediate recovery. Therefore, we aimed to identify conserved changes within patients, over time, that are tied to clinically relevant outcomes. Given that the majority of our patients successfully recovered from the infection, albeit after differing lengths of hospitalization, we investigated immunological changes that occurred within patients from time of admission (tp1) to time of discharge (tp2) from the hospital (Figures 3A and S3A). For this analysis, we included patients who were discharged within 30 days of admission across all disease severity states at time of enrollment (n = 32), allowing us to identify conserved features among all COVID-19 patients who successfully recover. A variety of immune cell subsets significantly changed in frequency between tp1 and tp2 (FDR < 0.1, Figure 3B). Monocytes, as well as activated CD4 and CD8 T cells, significantly increased at the time of discharge (tp2) as patients resolved the infection (Figure 3C). Conversely, neutrophils and conventional type 1 dendritic cells (cDC1s) significantly decreased in frequency by time of discharge (Figure 3C). For most COVID-19 patients, the overall composition of immune cells became more like that of healthy individuals at the time of discharge compared with the time of enrollment (distance between PCA centroids: healthy versus tp1 = 3.2, healthy versus tp2 = 2.8, Figure 3D). However, some immune cell populations exhibited deviations away from healthy at the time of discharge, most notably activated CD4 and CD8 T cells (CD38+ HLA-DR+) as well as monocytes (Figure 3E). This indicates that the immune state at the time of discharge is characterized by the restoration of certain elements of the immune response that were perturbed early in infection alongside a continued immunological process, including an expansion of activated T cells, that proceeds past the time patients stabilize for discharge.
Figure 3.

Conserved immunological processes and changes in cell signaling states accompany disease resolution and discharge
(A) Illustration of intra-patient analysis from admission to discharge for patients who are successfully discharged from the hospital within 30 days of admission (n = 32).
(B) Paired differential abundance analysis of immune cell populations between the first (tp1) and second (tp2) timepoints illustrated in 3A (paired Wilcoxon Rank Sum Test). The log2 fold changes (tp2 versus tp1) are plotted against the negative log10 (nominal p values). Colors indicate if cell populations are significantly down- (blue) or upregulated (purple) from tp1 to tp2 or not differentially expressed (FALSE, gray) after Benjamini-Hochberg correction, FDR < 0.1.
(C) Frequency of monocytes, neutrophils, cDC1, and CD8 activated T cells at tp1 and tp2. Lines connect samples from the same patient. Nominal p values obtained by paired Wilcoxon Rank Sum Test. CD8 activated T cells and cDC1 cells are shown as a percentage of parent populations (e.g., CD8 T cells and dendritic cells, respectively), while monocytes and neutrophils are shown as a percentage of all cells.
(D) Principal component analysis of significant immune cell subsets in 3B for tp1, tp2, and healthy controls. Immune cell directionality and contribution to PCA space denoted on right (top). Summary ellipsoid of tp1, tp2, and healthy patients in PCA space on right (bottom).
(E) Population frequencies of significant immune cell subsets in 3B for tp1, tp2, and healthy controls. Stars indicate median value for each group. Cell populations are highlighted in green if tp2 is closer to healthy than tp1 and highlighted in yellow if tp2 is moving away from healthy.
(F and G) Protein expression on CD8- and CD4 activated T cells (F) and on monocyte subsets (G) at tp1 and tp2. Mean protein expression values have been log10 transformed, scaled, and centered on heatmap. Bars indicate mean protein expression across all samples. Only significant proteins are shown (Wilcoxon Rank Sum Test, Benjamini-Hochberg correction with FDR < 0.1).
(H) Scatter plots of CD11c and HLA-DR expression on non-classical monocytes in patient 1344 at day 0 (top) and day 7 (bottom).
(I) Expression of signaling molecules in significant immune cell subsets in 3B at tp1 and tp2. Median signaling expression values have been centered on heatmap. Only significant signaling molecules are shown (Wilcoxon Rank Sum Test, Benjamini-Hochberg correction with FDR < 0.1 within each cell type).
(J) Expression of pTBK1 in CD8 activated T cells, and pSTAT3 expression in CD8 activated T cells and classical monocytes at tp1 and tp2. Lines connect samples from the same patient. Nominal p values obtained by paired Wilcoxon Rank Sum Test.
(K) Expression of PD-L1 on non-classical monocytes at tp1 and tp2. Lines connect samples from the same patient. Nominal p values obtained by paired Wilcoxon Rank Sum Test. See also Figure S3.
Patients who successfully resolve COVID-19 have robust pan-hematopoietic signaling and cytotoxic activated T cells at day of admission
To obtain more granular insights into the immunological perturbations that accompany COVID-19 recovery, we evaluated phenotypic changes and signaling dynamics within immune cell populations that changed during disease resolution. We focused on cell populations whose frequencies move away from relative frequencies observed in healthy controls, indicating they continue to have a dynamic response during infection resolution. Activated CD4 and CD8 T cells exhibited a reduction in the expression of GranzymeB and CD45RA as patients transition from early infection to discharge (FDR < 0.1, Figures 3F and S3B), consistent with a transition from more activated effector cells to more of a memory phenotype. We also observed a significant change in the phenotype of circulating monocytes, which expressed high PD-L1 at time of admission but higher expression of CD4, CD11c, and HLA-DR at time of discharge (FDR < 0.1, Figures 3G, 3H, and S3B). Similarly, we observed a reduction in PD-L1 expression on neutrophils at time of discharge (FDR < 0.1, Figure S3B).
We then analyzed the median values of phosphorylated signaling molecules within the relevant immune cell subtypes to evaluate changes in cell signaling during this resolution phase. A variety of cell signaling proteins were significantly downregulated within the key immune cell populations at time of discharge (FDR < 0.1, Figure 3I). Several signaling molecules changed in a coordinated fashion across different immune cell types (e.g., pTBK1, pERK, and pSTAT3), with the broadest signaling changes observed in activated CD8 T cells and monocyte subsets (FDR < 0.1, Figures 3I and 3J). These observations are consistent with previous studies describing the relationship between IL-6 expression and pSTAT3 signaling and subsequent upregulation of PD-L1 in monocytes (FDR < 0.1, Figures 3F and 3K) (Zhang et al., 2020b). Although signaling trajectories trended in the same direction among most patients (Figure S3C), we did not observe a clear trend toward healthy individuals (Figure S3D), likely explained by the expression variability and difficulty of measuring signaling molecules in rare populations in healthy individuals, e.g., activated CD8 T cells (Figure 3E). Taken together, our results suggest that a coordinated set of changes in immune cell abundances and signaling states occur in patients who successfully resolve COVID-19.
Immune changes associated with COVID-19 resolution differ in patients who are hospitalized for more than 30 days or die from COVID-19
To determine if the immune features identified in the resolution phase are specific to patient recovery, we analyzed patients who had delayed disease resolution, i.e. who remained hospitalized for more than 30 days (“late discharge”; n = 6) or who died from COVID-19 (“ultimately deceased”; n = 5) (Figures 4A and S4A). First, we evaluated changes in immune cell population frequencies occurring within these patients but found no significant changing populations between tp1 and tp2 for either group (FDR > 0.1, Figure S4B). Focusing on cell populations that significantly changed between these time points in patients who were discharged in ≤30 days, patients with poor clinical outcomes exhibited different patterns over time as well as greater variability (Figures 4B and 4C, S4C, and S4D). Next, we evaluated signaling dynamics in late discharge and ultimately deceased patients to determine if the observed changes in early discharge patients were evident. In contrast to patients resolving COVID-19 in ≤30 days, which exhibited consistent changes from high to low signaling states over time, we observed no significant changes between tp1 and tp2 for late discharged and ultimately deceased patients (FDR > 0.1, Figures 4D, S4F, S4G, and S4H). Instead, these patients exhibited discoordinate signaling directionality in activated CD8 T cells (Figures 4E and S4I), a complete lack of pS6 signaling in cDC1s (Figure 4E), and marginal changes in monocyte signaling states over time (Figures 4F and S4I). When the late discharged patients were within 30 days of discharge, the trajectory of several immune resolution features, e.g., monocytes, neutrophils, and signaling molecules, did resemble the recovery trajectories in patients hospitalized ≤30 days, suggesting that the resolution phase engages in these patients as well before they are discharged (Figures 4G and S4J). Taken together, these results indicate that changes in cell signaling evident in early discharge patients over time were not evident in patients with poor clinical outcomes. Furthermore, these results suggest that the immune processes observed during resolution through discharge are specific to a successful response against COVID-19.
Figure 4.

Immune changes associated with COVID-19 resolution differ in patients who are hospitalized for more than 30 days or die from COVID-19
(A) Illustration of intra-patient analysis of patients who are hospitalized for >30 days (n = 6) and patients who die (n = 5).
(B) Median cell population frequencies at tp1 (red) and tp2 (blue) for patients who are discharged ≤ 30 days, >30 days, and deceased. Error bars represent standard errors.
(C) Representative scatter plots of activated CD8 T cells (defined by CD38 and HLA-DR expression), at tp1 (left) and tp2 (right) for patients who are discharged ≤ 30 days, >30 days, and deceased.
(D) Magnitude of change illustrated by log2FC∗-log10 (p value) of signaling molecules (identified in Figure 3I) for patients who are discharged within 30 days (≤30 days, green), discharged after 30 days (>30 days, blue), and die (red). Nominal p values obtained by paired Wilcoxon Rank Sum Test.
(E and F) Median signaling molecule expression at tp1 (red) and tp2 (blue) for patients who are discharged ≤30 days, >30 days, and deceased. Error bars represent standard errors.
(G) Monocyte frequencies (left plots) and CD8 activated pERK expressions (right plots) relative to time to discharge in all samples from patients who are discharged ≤30 days (n = 142 samples) or >30 days (n = 30 samples). Black lines connect samples from the same patient. Blue lines and gray shadows represent the best-fitted smooth line and 95% confidence interval. Dotted lines intersect the x-axis at day 30. See also Figure S4.
Core immune resolution features characterize COVID-19 patients recovering from ventilation
Having established immune features that accompany COVID-19 resolution among our entire patient cohort, we next examined the immunological changes within only the most severe patients who required mechanical ventilation (Figure S5A). We analyzed immunological changes between three key time points: the first time point after a patient was intubated (tp1), the last time point before they were extubated (tp2), and the first time point after a patient was successfully extubated (tp3) (Figure 5A). This allowed us to evaluate the immunological dynamics that occur during ventilation (tp1 versus tp2) and during successful recovery from intubation (tp1 versus tp3). First, we analyzed the within-patient immune cell frequency changes between tp1 and tp3 (n = 9, Figure S5B). Consistent with patients resolving COVID-19, monocytes and activated CD4 and CD8 T cells significantly increased in frequency, while neutrophil frequency decreased during ventilation resolution (FDR < 0.1, Figures 5B and 5C). Additionally, ventilation resolution was characterized by an increase in CD4 regulatory T cells (Tregs) and basophils at time of recovery (FDR < 0.1, Figure 5B). These changes were collectively associated with a coordinated trajectory of recovery from tp1 to tp3 (Figure 5D). Despite these coordinated changes, patients did not return to an immune composition comparable to healthy donors, indicating that the time of extubation remains an active immunological phase of disease resolution from the most severe form of COVID-19. Some key immune cell populations that remain different from healthy controls included both activated CD4 and CD8 T cells as well as Tregs (Figures 5E and S5C). Of these changes, only the observed increase in activated CD8 T cells was apparent within patients during intubation (tp1 versus tp2; n = 11), suggesting that additional dynamic changes are specific to the resolution of severe COVID-19 (FDR < 0.1, Figures S5D and S5E).
Figure 5.

Recovery from severe COVID-19 requires core immune resolution features and additional regulatory T cell and basophil upregulation
(A) Illustration of intra-patient analysis of ventilated patients. Three timepoints are considered: tp1 (first sample after a patient has been put on a ventilator), tp2 (last sample before the patient is removed from a ventilator), and tp3 (first sample after a patient is successfully removed from ventilation support).
(B) Paired differential abundance analysis of immune cell populations between the first (tp1) and third (tp3) timepoints illustrated in 5A (paired Wilcoxon Rank Sum Test). The log2 fold changes (tp3 versus tp1) are plotted against the negative log10 (nominal p values). Colors indicate whether cell populations are significantly down- (blue) or upregulated (purple) from tp1 to tp3 or not differentially expressed (FALSE, gray) after Benjamini-Hochberg correction, FDR < 0.1.
(C) Frequency of monocytes, neutrophils, CD4 Treg, and CD8 activated T cells at tp1 and tp3. Lines connect samples from the same patient. Nominal p values obtained by paired Wilcoxon Rank Sum Test. CD8 activated T cells are shown as a percentage of parent population (e.g., CD8 T cells), while monocytes, neutrophils, and CD4 Tregs are shown as a percentage of all cells.
(D) Principal component analysis of significant immune cell subsets in 5B for tp1, tp3, and healthy controls. Immune cell directionality and contribution to PCA space denoted on the right.
(E) Population frequencies of significant immune cell subsets in 3B for tp1, tp3, and healthy controls. Stars indicate median value for each group. Cell populations are highlighted in green if tp3 is closer to healthy than tp1 and highlighted in yellow if tp3 is moving away from healthy.
(F and G) Protein expression on CD8 activated T cells (F) and on monocyte subsets (G) at tp1 and tp3. Mean protein expression values have been log10 transformed, scaled, and centered on heatmap. Bars indicate mean protein expression across all samples. Only significant proteins are shown (Wilcoxon Rank Sum Test, Benjamini-Hochberg correction with FDR < 0.1).
(H) Expression of PD-L1 on non-classical monocytes at tp1 and tp3. Lines connect samples from the same patient. Nominal p values obtained by paired Wilcoxon Rank Sum Test.
(I) Left: Scatter plots of CD11c and HLA-DR expression on non-classical monocytes in patient 1276 at day 0 (tp1, top) and day 28 (tp3, bottom). Right: Expression of CD64 on non-classical monocytes for patient 1279 from day 0 (tp1) to day 28 (tp3).
(J) Expression of signaling molecules in significant immune cell subsets in 5B at tp1 and tp3. Median signaling expression values have been centered on heatmap. Only significant signaling molecules are shown (Wilcoxon Rank Sum Test, Benjamini-Hochberg correction with FDR < 0.1 within each cell type).
(K) Expression of pSTAT1 (left) and pCREB (right) in CD4 Tregs at tp1 (blue) and tp3 (orange) for representative patients.
(L) Expression of pSTAT1 and pCREB in CD4 Tregs at tp1 and tp3. Lines connect samples from the same patient. Nominal p values obtained by paired Wilcoxon Rank Sum Test.
(M) Principal component analysis of significant signaling molecules in 5I for tp1, tp3, and healthy controls. Immune cell directionality and contribution to PCA space denoted on the right. See also Figure S5.
COVID-19 ventilation recovery is associated with T cell and monocyte phenotypic changes and a transition from pSTAT to pCREB dominated signaling
Next, we further analyzed changes in immune cell activation and cell signaling dynamics that accompany ventilation resolution. Consistent with recovery trajectories in patients resolving COVID-19, activated CD8 T cells expressed higher HLA-DR and lower CCR7 at the time of extubation (FDR < 0.1, Figure 5F), while neutrophils expressed lower PD-L1 (FDR < 0.1, Figure S5F). Additionally, while there was no difference in monocyte subset frequencies (FDR > 0.1, Figure S5G), non-classical (CD16+) monocytes exhibited a shift from a CD64+ PD-L1+ phenotype during ventilation to a CD4+ CD11c+ HLA-DR+ activated monocyte phenotype at the time of extubation (FDR < 0.1, Figures 5G, 5H, and 5I). CD64+ expression on non-classical monocytes incrementally decreased between tp1 and tp3 (Figure 5I).
Cell signaling states also changed markedly from the time of intubation to the time of extubation. During early time points of mechanical ventilation (tp1), higher expression of pSTAT1, pSTAT3, and pSTAT5 signaling was evident in CD4 Tregs, basophils, and activated CD8 T cells (FDR < 0.1, Figures 5J, 5K, 5L, and S5H). Conversely, pCREB signaling was significantly increased after extubation (tp3) in CD4 Tregs and non-classical monocytes (FDR < 0.1, Figures 5J, 5K, 5L, and S5H), suggesting there is a transition from inflammatory cytokine signaling response to pro-survival signaling within these cells, specifically. Visualizing these signaling trajectories in PCA space revealed a coordinated trajectory of immune cell signaling that accompanies extubation across patients (Figure 5M), though signaling states remained distinct from those in healthy individuals (Figure S5I). Taken together, our analyses identify a conserved set of immunological processes that are consistent among patients who recovered from mechanical ventilation as a result of COVID-19, elucidating an additional layer of immunological changes, e.g., increases in CD4 Tregs, basophils, and pCREB signaling, that are specific to these patients compared to recovery in patients who did not require mechanical ventilation.
Core immune resolution features define patients with better clinical outcomes at time of admission
Having identified a signature of immune remodeling during COVID-19 recovery, we next investigated if the early presence of these features was associated with better patient outcomes. We evaluated the immune composition of severe COVID-19 patients before or on the day they were ventilated (vent, n = 13) and compared it to the immunological state at time of admission (day 0) for patients who never required ventilation (no vent, n = 50) (Figures 6A and S6A). Differential abundance analysis of immune cell frequencies revealed higher frequencies of monocytes and CD4 Tregs, as well as decreased neutrophil frequencies, in patients who never required ventilation (FDR < 0.1, Figures 6B and 6C). Similar results were obtained when exclusively analyzing samples collected prior to ventilation (vent, n = 8) (FDR < 0.1, Figures S6B and S6C). Patients who never required ventilation exhibited an immune state more like those of the healthy controls (Figure S6D). While monocytes were significantly downregulated at time of admission in patients who required ventilation, we observed a consistent increase from time of intubation to time of discharge with the highest incline occurring right after time of extubation (Figure 6D). The opposite directionality was observed for neutrophils (Figure 6D). CD4 Tregs, which are known to play a role in ARDS resolution and pulmonary recovery, demonstrate a gradual increase in frequency during patient intubation followed by the steepest increase after extubation (Garibaldi et al., 2013; Mock et al., 2014) (Figure S6E). Additionally, the phenotype of monocytes in patients who never require ventilation resembles the activated monocyte subset identified during discharge and ventilation recovery, expressing significantly higher CD4 and CD11c (FDR < 0.1, Figures S6F and S6G). Furthermore, basophil and CD4 Treg signaling states that were identified during ventilation resolution were already significantly higher in patients who required ventilation at time of admission (p < 0.05, Figures 6E and S6H) and consistently decreased during ventilation (Figure 6F).
Figure 6.

Core immune resolution features define patients with better clinical outcomes at time of admission
(A) Illustration of inter-patient analysis of ventilated patients (vent, n = 13) versus patients who are never ventilated (no vent, n = 50). For ventilated patients, the latest sample before the patient is put on a ventilator is used. For non-ventilated patients, day 0 is used.
(B) Differential abundance analysis of immune cell populations between ventilated and non-ventilated patients illustrated in 6A (Wilcoxon Rank Sum Test). The log2 fold changes (vent versus no vent) are plotted against the negative log10 (nominal p values). Colors indicate if cell populations are significantly down- (blue) or upregulated (purple) for vent versus no vent or not differentially expressed (FALSE, gray) after Benjamini-Hochberg correction, FDR < 0.1.
(C) Frequency of monocytes, neutrophils, CD4 Tregs, and CD8 EM3 T cells in vent and no vent patients. Nominal p values obtained by Wilcoxon Rank Sum Test. CD8 EM3 T cells parent population (e.g., CD8 T cells), while monocytes, neutrophils, and CD4 Tregs are shown as a percentage of all cells.
(D) Monocyte (left plots) and neutrophil (right plots) frequencies relative to intubation/extubation in all samples from ventilated patients. Black lines connect samples from the same patient. Blue lines and gray shadows represent the best-fitted smooth line and 95% confidence interval. Dotted lines intersect the x axis at day of intubation or extubation.
(E) Expression of pSTAT3 and pPLCg2 in basophils in non-ventilated and ventilated patients as well as healthy individuals. Nominal p values obtained by Wilcoxon Rank Sum Test.
(F) Expression of pSTAT5 in CD4 Tregs relative to intubation or extubation in all samples from ventilated patients.
G) Graphical summary depicting the trajectories of key immune features involved in COVID-19 resolution and ventilation recovery. See also Figure S6.
In conclusion, we identified a set of conserved core immune features that accompany disease resolution, including changes in myeloid and T cell abundances as well as reduction in pan-immune cell activation, with additional features that identify patients who recover from ventilation, e.g., an increase of CD4 Tregs and basophils (Figure 6G). These ventilation-specific features are significantly different at time of admission between patients who will require mechanical ventilation and those that never require ventilation, and thus associate with poorer clinical outcomes (Figure S6I).
Discussion
Human immunology studies are inherently challenging because of the variability across individuals. The urgency to understand and respond to COVID-19 provided an opportunity to recruit, study, and analyze a large number of individuals responding to the same infection over a finite period of time (April 2020–April 2021). Since individuals recover from infection across a variable amount of time, these studies highlight the benefit of longitudinal analysis anchored on key clinical events in the disease process. This analytical approach revealed the unifying trends among patients that define clinically relevant events, such as discharge from the hospital or extubation after mechanical ventilation, regardless of initial disease severity or time to recovery.
Our findings are consistent with several recent reports of immune responses to COVID-19 while contributing an understanding of the processes that accompany disease recovery, including changes in immune cell signaling states. Although some studies have suggested that early intervention to modulate immune hyperactivation may be beneficial in severe COVID-19 (Lucas et al., 2020), our data indicate that early immune cell signaling is associated with shorter hospitalization and ventilation duration. This indicates that an early robust immune response, driven by pSTAT signaling, and subsequent contraction during recovery may be beneficial to resolving COVID-19. Baseline differences in this signaling state across patients at the time of hospital admission and the dynamic regulation of signaling over time within individual patients during recovery may, at least in part, explain conflicting reports from studies targeting immune cell signaling pathways with IL-6 inhibitors or steroids. In patients who require mechanical ventilation, additional immunological changes, including increased Tregs and basophils and reduced cell signaling in basophils, also accompany recovery in addition to the core recovery trajectory observed in patients who did not require ventilation. In our analysis, the STAT1 pathway downstream of type I IFN signaling was not differentially activated between patients with different disease severities. Instead, our study identified that many signaling pathways are activated simultaneously at the time of hospitalization, consistent with a recent report of concordant production of cytokines associated with type 1, 2, and 3 immune responses in patients with severe COVID-19 (Lucas et al., 2020). Despite the importance of B cells to generate SARS-CoV-2-neutralizing antibodies (Lucas et al., 2021), we did not identify changes in circulating B cells associated with the recovery trajectory. This aligns with the clinical observation that B cell-deficient patients or patients with agammaglobulinemia can recover from COVID-19 (Soresina et al., 2020; Bange et al., 2021) and suggests that B cells may play a role in contributing to immunological memory as compared to the resolution of severe COVID-19. Our work identified regulatory T cells as significantly changing only in patients who require ventilation, starting at significantly lower frequencies than in patients who never require ventilation support but steeply increasing after extubation. These findings are consistent with their critical role in pulmonary repair and ARDS recovery (Garibaldi et al., 2013; Mock et al., 2014).
Overall, our study identifies core immunological changes that accompany disease recovery from severe COVID-19 and provides a foundational model of a successful anti-SARS-CoV-2 immune response to contextualize divergent immune processes during poor disease outcomes in immunosuppressed or immunocompromised patients, long-haul COVID-19 patients, pediatric patients with MIS-C, or response to new variants. By elucidating a conserved trajectory of successful recovery, this study also nominates key immunological processes that could be targeted to enable recovery of severe disease in COVID-19 patients and perhaps other acute respiratory infections.
Limitations of the study
Variability across patients limited the number of significant conclusions drawn from cross-sectional analyses. In addition, healthy controls were on average younger than the hospitalized patients, though ranges overlapped. Samples were only collected during the hospitalization period, precluding analysis of later convalescent time points. Additionally, the majority of our patients recovered successfully. The limited sample size and variable immune states of patients with long-term hospital stays and deceased patients made it challenging to fully understand the defining immune characteristics of patients with the worst outcomes.
Consortia
The members of the UCSF Comet Consortium are Ravi Patel, Yumiko Abe-Jones, Saurabh Asthana, Alexander Beagle, Sharvari Bhide, Cathy Cai, Maria Calvo, Sidney A. Carrillo, Suzanna Chak, Zachary Collins, Spyros Darmanis, Gabriela K. Fragiadakis, Rajani Ghale, Jeremy Giberson, Pat Glenn, Ana Gonzalez, Kamir Hiam-Galvez, Alejandra Jauregui, Serena Ke, Tasha Lea, Deanna Lee, Raphael Lota, Leonard Lupin-Jimenez, Viet Nguyen, Nishita Nigam, Logan Pierce, Priya Prasad, Arjun Rao, Sadeed Rashid, Nicklaus Rodriguez, Bushra Samad, Cole Shaw, Austin Sigman, Pratik Sinha, Kevin Tang, Luz Torres Altamirano, Erden Tumurbaatar, Vaibhav Upadhyay, Alyssa Ward, Kristine Wong, Chun Jimmie Ye, Kimberly Yee, and Mingyue Zhou.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mass cytometry antibodies are found in Table S5 | This paper | N/A |
| Biological samples | ||
| Blood sample | UCSF hospital under the IMPACC study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Benzonase | Sigma-Aldrich | Cat# E8263-25KU; RRID: N/A |
| Calibration beads | EQTM Four Element Fluidigm | Cat#201078; RRID: N/A |
| TrueStain FcX (anti-mouse CD16/32 antibody (clone 93) | BioLegend | Cat#101320 |
| Cell Acquisition Solution | Fluidigm | Cat#201240 |
| Critical commercial assays | ||
| MaxPar Antibody Conjugation Kit | Fluidigm | Cat#201300 |
| Deposited data | ||
| Mass cytometry data | This paper | Mendeley data: https://doi.org/10.17632/pmjrc8kw9x.2 |
| Software and algorithms | ||
| Cytobank analysis software | Cytobank, Inc | https://cytobank.org RRID: SCR_014043 |
| Cellengine analysis software | Primitybio | https://primitybio.com/cellengine.html RRID: N/A |
| Normalizer/Debarcoding | Parker Institute for Cancer Immunotherapy | https://github.com/ParkerICI/premessa RRID: N/A |
| R environment | R Development Core Team |
https://www.r-project.org/ RRID:SCR_001905 |
| Other | ||
| Helios mass cytometer | Fluidigm | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Matthew Spitzer (matthew.spitzer@ucsf.edu).
Materials availability
This study did not generate new unique materials. Information regarding antibody conjugates is presented in Table S5.
Subject details
Human subjects
Patients, or a designated surrogate, provided informed consent to participate in the study. The study is approved by the UCSF Institutional Review Board: IRB 20-30497.Clinical study was designed and implemented according to the IMPACC study ((Null et al., 2021)). Patients were recruited from UCSF hospital system and Zuckerberg San Francisco General Hospital and they, or a designated surrogate, provided informed consent to participate in the study. Patients with presumed COVID-19 were enrolled within three days of hospital admission and peripheral blood samples were collected under a protocol approved by the UCSF Institutional Review Board (IRB 20-30497). Patients with confirmed positive SARS-CoV-2 polymerase chain reaction (PCR) were designated as COVID-19 positive cohort (n = 81) and patients without confirmed SARS-CoV-2 PCR were designated COVID-19 negative (n = 7). Healthy donors (n = 11) were recruited (IRB 19-27147) for a single peripheral blood time point and consisted of unexposed patients in a similar age range as the hospitalized cohort. Clinical data and peripheral blood samples were collected at time of enrollment and throughout hospitalization (mainly on days 4, 7, 14, 21, and 28). If escalation of care was required, samples were collected within 24 and 96 h of care escalation. All COVID-19 patients in this study were admitted into the UCSF hospital system and remained there for the duration of our study. By definition, all in-patients reflect a World Health Organization (WHO) COVID-19 severity score of 3 or greater. Patient severity was determined by the clinical team to reflect the WHO COVID-19 severity scoring at each clinical time point throughout in-patient treatment. Based on WHO stratifications (World Health Organization, 2021b) and consulting with the treating physician teams, our study combined WHO score 5, 6, and 7 into the most severe clinical group. WHO scores of 3 and 4 correspond to Mild and Moderate groups, respectively. Participant age, gender, and additional demographic details are provided in Table S2.
Method details
Peripheral blood sample collection and processing
Blood samples were collected in one EDTA tube and processed within 6 h of collection. Whole blood was divided in 540 μL aliquots then fixed by addition of 756 μL of SmartTube Stabilizer from SmartTube Inc (Fisher Sci. Cat# 501351692). After gentle mixing at room temperature for 10 min, the samples were transferred to labeled cryovials and immediately carried to −80°C for long term storage.
Sample thawing and filtering
Samples were subsequently thawed after being placed 10 min into a 4°C refrigerator then incubated for 15 min in a room temperature water bath. After filtering with 70μm Cell Strainer (Celltreat, Cat# 229483) and washing in 45 mL Milli-Q H2O, samples were counted and barcoded.
Antibodies and staining procedure
The source for all mass cytometry antibodies can be found in Table S5. Antibodies were conjugated to their associated metals with MaxPar X8 labeling reagent kits (Fluidigm) according to manufacturer instructions, diluted with Candor PBS Antibody Stabilization solution (Candor Bioscience, CAT#130 050) supplemented with 0.02% sodium azide, and filtered through an UltrafreeMC 0.1-mm centrifugation filter (Millipore) before storage at 4°C. To reduce tube-to-tube pipetting variations, part of the signaling antibody panel came from lyophilized antibody cocktail, made at Stanford University as previously described ((Han et al., 2018)). Surface and intracellular master antibody cocktails were made and kept at −80°C in order to stain up to 600 samples.
Mass-tag cellular barcoding
Prior to antibody staining, mass tag cellular barcoding of prepared samples was performed by incubating cells with distinct combinations of isotopically-purified palladium ions chelated by isothiocyanobenzyl-EDTA as previously described ((Zunder et al., 2015)). After counting, 1∗106 cells from aliquot were barcoded with distinct combinations of stable Pd isotopes for 15 min at room temperature on a shaker in Maxpar Barcode Perm Buffer (Fluidigm, cat#201057). Cells were washed twice with cell staining media (PBS with 0.5% BSA and 0.02% NaN3), and pooled into a single 15 mL tube.
Mass cytometry staining
Barcoded cells were stained with Fc Receptor Blocking Solution (BioLegend, Cat#422302) at 20 mg/mL for 5 min at RT on a shaker. Surface antibody cocktail is then added with a 500 ul final reaction volume for 30 min at RT on a shaker. Following staining, cells were washed twice with cell staining media. Before intracellular staining, cells were permeabilized for 10 min with methanol at 4°C. Methanol is then removed by washing the cells 2 times with cells staining media. Intracellular cocktail is then added to the cells in 500 uL final reaction volume for 1 h at RT on a shaker. Cells were washed twice in cell staining media to remove antibodies excess and then stained with 1mL of 1:4000 191/193Ir Iridium intercalator solution (Fluidigm,Cat#201192B) diluted in PBS with 4% PFA overnight. Before mass cytometry run, cells were washed once with cell staining media, and twice with Cell Acquisition Solution (Fluidigm, Cat# 201240).
Mass cytometry
Mass cytometry samples were diluted in Cell Acquisition Solution containing bead standards (Fluidigm, Cat#201078) to approximately 106 cells/mL and then analyzed on a Helios mass cytometer (Fluidigm) equilibrated with Cell Acquisition Solution. Approximately 0.5x106 cell events were collected for each sample at an even rate of 400–500 events/second.
Data normalization and de-barcoding
Bead standard data normalization and de-barcoding of the pooled samples into their respective conditions was performed using the R package from the PICI institute available at https://github.com/ParkerICI/premessa.
Quality control inclusion and exclusion criteria
In order to ensure high quality sample collection, processing, and staining across the cohort we developed a set of inclusion criteria required for each sample to be used in our data analysis. We processed and ran CyTOF on 498 peripheral blood samples. After debarcoding and normalization, samples were uploaded to Cell Engine to assess adequate staining and cell number. Each barcode plate was run with a healthy PB control sample aliquoted from two healthy donors to validate staining and for normalization between barcode plates. If the control PB sample failed to stain the major immune cell populations (T cell, B cell, granulocytes, monocytes), no samples from that barcode plate were included. Individual samples were then assessed for CD45+ composition (>50% CD45+ staining required), cell abundance (>5,000 cells per sample required), and representation of the major immune cell populations (T cell, B cell, granulocytes, monocytes). 230 samples passed QC and were used in the batch normalization.
Quantification and statistical analysis
Batch normalization
All manually gated immune cells (CD45+) from samples meeting our inclusion criteria (n = 230) were downloaded as FCS files from cellEngine. Premessa (Gherardini, 2021) (https://github.com/ParkerICI/premessa) and cytofCore (Bruggner et al., 2021) (https://github.com/nolanlab/cytofCore) were used to harmonize panels between runs, and CytoNorm (Van Gassen, 2021) (https://github.com/saeyslab/CytoNorm) were utilized to correct for batch effect. All markers were used for batch effect normalization, except for Ki-67, which failed for several CyTOF runs and were excluded in the final data. Samples were separately normalized to control 1 and 2, and subsequently combined into one final data set of normalized FCS files.
Manual gating
Batch effect normalized FCS files were uploaded to Cell Engine for manual gating. Major immune cell populations were identified based on prior gating strategy (Allen et al., 2020). T cell subsets were further identified based on phenotypic markers specified in prior publication that suggested these specific subtypes could play a role in COVID-19 severity (Mathew et al., 2020).
t-SNE visualization
The multiparameter dimensionality reduction method t-distributed stochastic neighbor embedding (t-SNE) was employed to visualize major shifts in immune distribution between COVID-19 positive, COVID-19 negative, and healthy individuals. CD45+ immune cells from healthy peripheral blood samples were compared to day 0 (D0) peripheral blood samples from COVID-9 positive and negative individuals and respective groups were concatenated into a single FSC file which was then used in the t-SNE algorithm on Cell Engine (cellengine.com). Only phenotypic markers were used as analysis channels and no phospho-signaling channels were input into the t-SNE visualization. The default settings for t-SNE plot were utilized and a default of 90 nearest neighbors (k) was used. Manually gated immune cell populations were used to color the t-SNE plot to identify representative immune populations on the plot.
Defining groups and samples
For intra-patient resolution analyses, we defined three different groups; patients who were discharged within 30 days of enrollment in the study (≤30 days), patients who were discharged after 30 days of enrollment in the study (>30 days), and patients who died. For patients who were discharged ≤30 days, the last sample (tp2) had to be obtained within 7 days of discharge. For patients who were discharged >30 days and patients who died, the last sample (tp2) had to be obtained within 50 days of discharge. For all groups, the first sample (tp1) had to be obtained within 14 days of enrollment. For intra-patient ventilation recovery analysis, samples had to be obtained within 7 days of the point of interest, e.g. going on a ventilator or coming off a ventilator. For all comparisons; if multiple samples fulfilled the requirements, we used the sample closest to the event of interest. The number of patients and specific sampling timepoints used for each analysis are illustrated in the supplementary figures.
Statistical analysis
All statistical tests were performed in R (Team and Others, 2013; RStudio Team, 2016). The non-parametric Wilcoxon rank sum test was utilized to compare immune population frequencies, median protein expression values, and median signaling molecule values between groups of interest. For intra-patient analysis, we used the paired Wilcoxon rank sum test. For multiple testing corrections, we applied Benjamini-Hochberg correction and statistical differences were declared significant at FDR <0.1. Most of the plots were produced with the R package ggplot2 (Wickham, 2016).
Acknowledgments
This work was supported by grants from The Carlsberg Foundation (T.L.H.O.); COVID-19 Fast Grants (M.H.S.); NCI 1F31CA260938-01 (C.E.B.); NSF GRFP (C.E.B.); UCSF Discovery Fellowship (C.E.B.); NIH NIAID Immunophenotyping Assessment in a COVID-19 Cohort (IMPACC) Network (3U19AI077439-13S1 and 3U19AI077439-13S2); Howard Hughes Medical Institute James H. Gilliam Fellowship (P.M.S.); NHLBI R35 HL140026 (C.S.C.); funding from NHLBI and NIAID to C.R.L.; NHLBI K23 and a DOD grants to C.M.H.; grant 2019-202665 from the Chan Zuckerberg Foundation and TSK-020586 from Genentech; and NIH Grants P30DK063720, S10OD018040, S10OD018040, and S10OD021822 to the UCSF Flow Cytometry CoLab. M.H.S. is an investigator of the Chan Zuckerberg Biohub and the Parker Institute for Cancer Immunotherapy.
Author contributions
C.E.B., I.T., P.M.S., K.M.A., D.J.E., and M.H.S. conceived the project and designed CyTOF experiments. C.E.B., T.L.H.O., and M.H.S. conceptualized the study. C.E.B., I.T., D.M.M., and S.T. performed CyTOF experiments. T.L.H.O. and C.E.B. performed data analysis and generated all figures. A.W. provided clinical information. C.E.B., T.L.H.O., and M.H.S. wrote and revised the manuscript. M.H.S. supervised the study. C.S.C., C.M.H., C.R.L., M.F.K., P.G.W., and D.J.E., founded and led the COMET Consortium. All authors read and approved the final manuscript.
Declaration of interests
M.H.S. is a board member and equity holder in Teiko.bio and has received research support from Roche/Genentech, Bristol Myers Squibb, Pfizer, and Valitor. C.S.C. has received funding from NHLBI, FDA, DOD, Genentech, and Quantum Leap Healthcare Collaborative and is on consulting/advisory boards for Vasomune, Gen1e Life Sciences, Janssen, and Cellenkos. C.M.H. has been consulting for Spring Discovery. P.G.W. has a contract from Genentech to study COVID-19.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper received support from a program designed to increase minority representation in science. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Published: July 12, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2022.06.004.
Contributor Information
The UCSF COMET Consortium:
Ravi Patel, Yumiko Abe-Jones, Saurabh Asthana, Alexander Beagle, Sharvari Bhide, Cathy Cai, Maria Calvo, Sidney A. Carrillo, Suzanna Chak, Zachary Collins, Spyros Darmanis, Gabriela K. Fragiadakis, Rajani Ghale, Jeremy Giberson, Pat Glenn, Ana Gonzalez, Kamir Hiam-Galvez, Alejandra Jauregui, Serena Ke, Tasha Lea, Deanna Lee, Raphael Lota, Leonard Lupin-Jimenez, Viet Nguyen, Nishita Nigam, Logan Pierce, Priya Prasad, Arjun Rao, Sadeed Rashid, Nicklaus Rodriguez, Bushra Samad, Cole Shaw, Austin Sigman, Pratik Sinha, Kevin Tang, Luz Torres Altamirano, Erden Tumurbaatar, Vaibhav Upadhyay, Alyssa Ward, Kristine Wong, Chun Jimmie Ye, Kimberly Yee, and Mingyue Zhou
Supplemental information
A) Summary table of COVID-19-positive patient demographics and clinical parameters, e.g., WHO score at time of sampling, max WHO score, hospital length of stay, ventilation duration, etc. B) Summary table of COVID-19-negative patient demographics and clinical parameters, e.g., WHO score at time of sampling, max WHO score, hospital length of stay, ventilation duration, etc.
A) Per-patient table of COVID-19- positive patient demographics and clinical parameters, e.g., WHO score at time of sampling, max WHO score, hospital length of stay, ventilation duration, etc. B) Per-patient table of COVID-19-negative patient demographics and clinical parameters, e.g., WHO score at time of sampling, max WHO score, hospital length of stay, ventilation duration, etc.
Data and code availability
-
•
Mass cytometry data are publicly available from Mendeley Data at https://doi.org/10.17632/pmjrc8kw9x.2.
-
•
No new code or algorithms were developed during this study. All code used will be provided upon request without limitations.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Allen B.M., Hiam K.J., Burnett C.E., Venida A., DeBarge R., Tenvooren I., Marquez D.M., Cho N.W., Carmi Y., Spitzer M.H. Systemic dysfunction and plasticity of the immune macroenvironment in cancer models. Nat. Med. 2020;26:1125–1134. doi: 10.1038/s41591-020-0892-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angus D.C., Derde L., Al-Beidh F., Annane D., Arabi Y., Beane A., van Bentum-Puijk W., Berry L., Bhimani Z., Bonten M., et al. Effect of hydrocortisone on mortality and organ support in patients with severe COVID-19: the REMAP-CAP COVID-19 corticosteroid domain randomized clinical trial. JAMA: the journal of the American Medical Association. 2020;324:1317–1329. doi: 10.1001/jama.2020.17022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano T., Boisson B., Onodi F., Matuozzo D., Moncada-Velez M., Maglorius Renkilaraj M.R.L., Zhang P., Meertens L., Bolze A., Materna M., et al. X-linked recessive TLR7 deficiency in ∼1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 2021;6:eabl4348. doi: 10.1126/sciimmunol.abl4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bange E.M., Han N.A., Wileyto P., Kim J.Y., Gouma S., Robinson J., Greenplate A.R., Hwee M.A., Porterfield F., Owoyemi O., et al. CD8+ T cells contribute to survival in patients with COVID-19 and hematologic cancer. Nat. Med. 2021;27:1280–1289. doi: 10.1038/s41591-021-01386-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention CDC COVID data tracker, trends in number of COVID-19 cases and deaths in the US reported to CDC, by State/Territory. Available at: 2021. https://covid.cdc.gov/covid-data-tracker/#trends_dailydeaths
- Chang S.E., Feng A., Meng W., Apostolidis S.A., Mack E., Artandi M., Barman L., Bennett K., Chakraborty S., Chang I., et al. New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat. Commun. 2021;12:5417. doi: 10.1038/s41467-021-25509-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Xie J., Su N., Wang J., Sun Q., Li S., Jin J., Zhou J., Mo M., Wei Y., et al. Corticosteroid therapy is associated with improved outcome in critically ill patients with COVID-19 with hyperinflammatory phenotype. Chest. 2021;159:1793–1802. doi: 10.1016/j.chest.2020.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combes A.J., Courau T., Kuhn N.F., Hu K.H., Ray A., Chen W.S., Chew N.W., Cleary S.J., Kushnoor D., Reeder G.C., et al. Global absence and targeting of protective immune states in severe COVID-19. Nature. 2021;591:124. doi: 10.1038/s41586-021-03234-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garibaldi B.T., D'Alessio F.R., Mock J.R., Files D.C., Chau E., Eto Y., Drummond M.B., Aggarwal N.R., Sidhaye V., King L.S. Regulatory T cells reduce acute lung injury fibroproliferation by decreasing fibrocyte recruitment. Am. J. Respir. Cell Mol. Biol. 2013;48:35–43. doi: 10.1165/rcmb.2012-0198OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherardini P.F. 2021. Premessa: R Package for Pre-processing of Flow and Mass Cytometry Data. [Google Scholar]
- Han G., Spitzer M.H., Bendall S.C., Fantl W.J., Nolan G.P. Metal-isotope-tagged monoclonal antibodies for high-dimensional mass cytometry. Nat. Protoc. 2018;13:2121–2148. doi: 10.1038/s41596-018-0016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue J.K., et al. JAMA Network Open; 2021. Sequelae in Adults at 6 Months after COVID-19 Infection; p. e210830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas C., Wong P., Klein J., Castro T.B.R., Silva J., Sundaram M., Ellingson M.K., Mao T., Oh J.E., Israelow B., et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584:463–469. doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas C., Klein J., Sundaram M.E., Liu F., Wong P., Silva J., Mao T., Oh J.E., Mohanty S., Huang J., et al. Delayed production of neutralizing antibodies correlates with fatal COVID-19. Nat. Med. 2021;27:1178–1186. doi: 10.1038/s41591-021-01355-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann E.R., Menon M., Knight S.B., Konkel J.E., Jagger C., Shaw T.N., Krishnan S., Rattray M., Ustianowski A., Bakerly N.D., et al. Longitudinal immune profiling reveals key myeloid signatures associated with COVID-19. Sci. Immunol. 2020;5 doi: 10.1126/sciimmunol.abd6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew D., Giles J.R., Baxter A.E., Oldridge D.A., Greenplate A.R., Wu J.E., Alanio C., Kuri-Cervantes L., Pampena M.B., D'Andrea K., et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369 doi: 10.1126/science.abc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mock J.R., Garibaldi B.T., Aggarwal N.R., Jenkins J., Limjunyawong N., Singer B.D., Chau E., Rabold R., Files D.C., Sidhaye V., et al. Foxp3+ regulatory T cells promote lung epithelial proliferation. Mucosal Immunol. 2014;7:1440–1451. doi: 10.1038/mi.2014.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore J.B., June C.H. Cytokine release syndrome in severe COVID-19. Science. 2020;368:473–474. doi: 10.1126/science.abb8925. [DOI] [PubMed] [Google Scholar]
- Null N., et al. Immunophenotyping assessment in a COVID-19 cohort (IMPACC): a prospective longitudinal study. Science Immunol. 2021;6:eabf3733. doi: 10.1126/sciimmunol.abf3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggner R., Linderman M., Finck R. 2021. cytofCore: cytofCore: Analysis Tools for CyToF Mass Cytometer Data. [Google Scholar]
- RECOVERY Collaborative Group, et al. Dexamethasone in hospitalized patients with Covid-19. N. Engl. J. Med. 2021;384:693–704. doi: 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RECOVERY Collaborative Group Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet. 2021;397:1637–1645. doi: 10.1016/S0140-6736(21)00676-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas I.O., Bräu N., Waters M., Go R.C., Hunter B.D., Bhagani S., Skiest D., Aziz M.S., Cooper N., Douglas I.S., et al. Tocilizumab in hospitalized patients with severe Covid-19 pneumonia. N. Engl. J. Med. 2021;384:1503–1516. doi: 10.1056/NEJMoa2028700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RStudio Team RStudio: Integrated Development Environment for R. Boston, MA: RStudio, Inc. 2016. http://www.rstudio.com/
- Sinha P., Furfaro D., Cummings M.J., Abrams D., Delucchi K., Maddali M.V., He J., Thompson A., Murn M., Fountain J., et al. Latent Class Analysis reveals COVID-19-related acute respiratory distress syndrome Subgroups with differential responses to corticosteroids. Am. J. Respir. Crit. Care Med. 2021;204:1274–1285. doi: 10.1164/rccm.202105-1302OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha P., Matthay M.A., Calfee C.S. Is a “cytokine storm” relevant to COVID-19? JAMA Intern. Med. 2020;180:1152–1154. doi: 10.1001/jamainternmed.2020.3313. [DOI] [PubMed] [Google Scholar]
- Soresina A., et al. Two X-linked agammaglobulinemia patients develop pneumonia as COVID-19 manifestation but recover. Pediatr. Allergy Immunol.: official publication of the European Society of. 2020;31:565–569. doi: 10.1111/pai.13263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team R.C., et al. R: a language and environment for statistical computing. 2013. http://r.meteo.uni.wroc.pl/web/packages/dplR/vignettes/intro-dplR.pdf
- Tsai A., Diawara O., Nahass R.G., Brunetti L. Impact of tocilizumab administration on mortality in severe COVID-19. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-76187-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gassen S. 2021. CytoNorm: Normalisation of Cytometry Data Measured across Multiple Batches. [Google Scholar]
- Wagner C., Griesel M., Mikolajewska A., Mueller A., Nothacker M., Kley K., Metzendorf M.I., Fischer A.L., Kopp M., Stegemann M., et al. Systemic corticosteroids for the treatment of COVID-19. Cochrane Database Syst. Rev. 2021;8:CD014963. doi: 10.1002/14651858.CD014963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.Y., Mao T., Klein J., Dai Y., Huck J.D., Jaycox J.R., Liu F., Zhou T., Israelow B., Wong P., et al. Diverse functional autoantibodies in patients with COVID-19. Nature. 2021;595:283–288. doi: 10.1038/s41586-021-03631-y. [DOI] [PubMed] [Google Scholar]
- WHO Rapid Evidence Appraisal for COVID-19 Therapies REACT Working Group. Vale C.L., Godolphin P.J., Fisher D., Higgins J.P.T., Spiga F., Savovic J., Tierney J., Baron G., Benbenishty J.S., et al. Association between administration of IL-6 Antagonists and mortality among patients hospitalized for COVID-19: a meta-analysis. JAMA. 2021;326:499–518. doi: 10.1001/jama.2021.11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. Springer; 2016. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- van der Wijst M.G.P., Vazquez S.E., Hartoularos G.C., Bastard P., Grant T., Bueno R., Lee D.S., Greenland J.R., Sun Y., Perez R., et al. Type I interferon autoantibodies are associated with systemic immune alterations in patients with COVID-19. Sci. Transl. Med. 2021;13:eabh2624. doi: 10.1126/scitranslmed.abh2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J.G., Simpson L.J., Ferreira A.M., Rustagi A., Roque J., Asuni A., Ranganath T., Grant P.M., Subramanian A., Rosenberg-Hasson Y., et al. Cytokine profile in plasma of severe COVID-19 does not differ from ARDS and sepsis. JCI insight. 2020;5 doi: 10.1172/jci.insight.140289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization WHO Coronavirus (COVID-19) Dashboard, WHO Coronavirus (COVID-19) Dashboard. 2021. https://covid19.who.int/
- World Health Organization WHO R&D Blueprint novel Coronavirus COVID-19 therapeutic trial Synopsis. 2021. https://www.who.int/blueprint/priority-diseases/key-action/COVID-19_Treatment_Trial_Design_Master_Protocol_synopsis_Final_18022020.pdf
- Yang L., Liu S., Liu J., Zhang Z., Wan X., Huang B., Chen Y., Zhang Y. COVID-19: immunopathogenesis and Immunotherapeutics. Signal Transduct. Target. Ther. 2020;5:128. doi: 10.1038/s41392-020-00243-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Bastard P., Liu Z., Le Pen J., Moncada-Velez M., Chen J., Ogishi M., Sabli I.K.D., Hodeib S., Korol C., et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370 doi: 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Liu Y., Yan Z., Yang H., Sun W., Yao Y., Chen Y., Jiang R. IL-6 promotes PD-L1 expression in monocytes and macrophages by decreasing protein tyrosine phosphatase receptor type O expression in human hepatocellular carcinoma. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2019-000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zunder E.R., Finck R., Behbehani G.K., Amir E.A.D., Krishnaswamy S., Gonzalez V.D., Lorang C.G., Bjornson Z., Spitzer M.H., Bodenmiller B., et al. Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat. Protoc. 2015;10:316–333. doi: 10.1038/nprot.2015.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A) Summary table of COVID-19-positive patient demographics and clinical parameters, e.g., WHO score at time of sampling, max WHO score, hospital length of stay, ventilation duration, etc. B) Summary table of COVID-19-negative patient demographics and clinical parameters, e.g., WHO score at time of sampling, max WHO score, hospital length of stay, ventilation duration, etc.
A) Per-patient table of COVID-19- positive patient demographics and clinical parameters, e.g., WHO score at time of sampling, max WHO score, hospital length of stay, ventilation duration, etc. B) Per-patient table of COVID-19-negative patient demographics and clinical parameters, e.g., WHO score at time of sampling, max WHO score, hospital length of stay, ventilation duration, etc.
Data Availability Statement
-
•
Mass cytometry data are publicly available from Mendeley Data at https://doi.org/10.17632/pmjrc8kw9x.2.
-
•
No new code or algorithms were developed during this study. All code used will be provided upon request without limitations.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.