

Abstract
Animal models of neurodegenerative diseases such as inherited peripheral neuropathies sometimes accurately recreate the pathophysiology of the human disease, and sometimes accurately recreate the genetic perturbations found in patients. Ideally, models achieve both, but this is not always possible; nonetheless, such models are informative. Here we describe two animal models of inherited peripheral neuropathy: mice with a mutation in tyrosyl tRNA‐synthetase, Yars E196K, modeling dominant intermediate Charcot‐Marie‐Tooth disease type C (diCMTC), and mice with a mutation in serine palmitoyltransferase long chain 1, Sptlc1 C133W, modeling hereditary sensory and autonomic neuropathy type 1 (HSAN1). Yars E196K mice develop disease‐relevant phenotypes including reduced motor performance and reduced nerve conduction velocities by 4 months of age. Peripheral motor axons are reduced in size, but there is no reduction in axon number and plasma neurofilament light chain levels are not increased. Unlike the dominant human mutations, the Yars E196K mice only show these phenotypes as homozygotes, or as compound heterozygotes with a null allele, and no phenotype is observed in E196K or null heterozygotes. The Sptlc1 C133W mice carry a knockin allele and show the anticipated increase in 1‐deoxysphingolipids in circulation and in a variety of tissues. They also have mild behavioral defects consistent with HSAN1, but do not show neurophysiological defects or axon loss in peripheral nerves or in the epidermis of the hind paw or tail. Thus, despite the biochemical phenotype, the Sptlc1 C133W mice do not show a strong neuropathy phenotype. Surprisingly, these mice were lethal as homozygotes, but the heterozygous genotype studied corresponds to the dominant genetics seen in humans. Thus, Yars E196K homozygous mice have a relevant phenotype, but imprecisely reproduce the human genetics, whereas the Sptlc1 C133W mice precisely reproduce the human genetics, but do not recreate the disease phenotype. Despite these shortcomings, both models are informative and will be useful for future research.
Keywords: axon degeneration, Charcot‐Marie‐Tooth, demyelination, HSN1, inherited peripheral neuropathy, sphingolipid synthesis, SPTLC2, tRNA synthetase, YARS
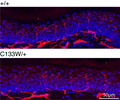
Intraepidermal nerve fiber density in wild type and Sptlc1‐C133W knockin mice, modeling hereditary sensory and autonomic neuropathy type 1.
1. INTRODUCTION
Inherited peripheral neuropathies are a heterogeneous collection of genetic disorders that result in the degeneration of peripheral motor and/or sensory axons, and in some cases autonomic neurons (Saporta & Shy, 2013). These diseases are typically Mendelian, monogenic disorders, and close to 100 loci in the human genome are associated with inherited peripheral neuropathies (Laura et al., 2019; Timmerman et al., 2014). Although genes remain to be identified, emphasis is shifting from gene discovery to understanding the pathophysiology and identifying and testing therapeutic strategies, although no curative treatments are yet approved for any form of these diseases. A key instrument in such mechanistic and preclinical research is an animal model that recapitulates the human disease. With the advent of CRISPR/Cas9 genome editing, creating “precision” models of human disease‐associated mutations has become increasingly feasible. However, these models can be precise in how well they recreate the human genetic lesion, or in how well they reproduce the human disease phenotype. Ideally, they will do both, but this is not always the reality and also not always necessary for the models to be useful (Tadenev & Burgess, 2019). Here we describe two mouse models of inherited peripheral neuropathy. The first is a knockin allele of Yars E196K, creating a model of dominant intermediate Charcot‐Marie‐Tooth disease type C (diCMTC) (Jordanova et al., 2006). These mice develop a relevant phenotype, but only when the mutation is homozygous, unlike the dominant human disease. The second model is a knockin allele of Sptlc1 C133W, creating a model of hereditary sensory and autonomic neuropathy type 1 (HSAN1) (Bejaoui et al., 2001; Dawkins et al., 2001). These mice precisely reproduce the human genetics, and develop a relevant biochemical phenotype, but despite modest behavioral changes, they do not show frank peripheral axon loss. Despite these deviations from the human conditions, both mouse models have been informative and will be useful for future preclinical and mechanistic research.
DiCMTC has both demyelinating and axonal clinical features. The “intermediate” designation denotes significantly reduced nerve conduction velocities (25–45 m/s), but less severely decreased than observed in most demyelinating neuropathies. DiCMTC patients typically present in the first two decades of life, but onset is variable. Distal leg and arm weakness is often the initial complaint. Motor symptoms and lower limb involvement predominate, but sensory deficits are also present. Sural nerve biopsies collected from a large pedigree showed clusters of regenerating fibers and age‐dependent reduction in fiber density and myelin thickness, but no onion bulb formation (Jordanova et al., 2003). The disease is slowly progressive (Pan et al., 2020).
The YARS gene encodes tyrosyl‐tRNA synthetase (a.k.a. TyrRS). The gene is ubiquitously expressed and the enzyme charges tyrosine onto its cognate tRNAs for translation. The pathogenic mechanism(s) by which dominant mutations in YARS cause peripheral neuropathy is unclear. Recessive mutations that are a mix of partial and complete loss‐of‐function alleles cause severe, multisystem syndromes that notably do not include peripheral neuropathy, and the parental carriers of these alleles are described as healthy (Nowaczyk et al., 2017; Tracewska‐Siemiatkowska et al., 2017; Williams et al., 2019). Therefore, the dominant neuropathy‐associated alleles of YARS are likely not simple haploinsufficiencies. Consistent with a possible gain‐of‐function (neomorph) activity, the mutant YARS protein adopts a stable conformation with novel exposed surfaces, allowing aberrant interactions with other proteins (Blocquel et al., 2017). Also, transgenic overexpression of DiCMTC alleles of YARS in Drosophila produces neuropathy‐relevant phenotypes (Niehues et al., 2015; Storkebaum et al., 2009). Importantly, the diCMTC‐associated allele we have modeled in mice, Yars E196K, is reported to have retained activity charging tyrosine onto tRNATyr in in vitro assays (Froelich & First, 2011; Jordanova et al., 2006).
The pathogenic mechanism by which dominant mutations in the serine‐palmitoyl transferase (SPT) cause HSAN1 is better understood (Bejaoui et al., 2001; Dawkins et al., 2001). HSAN1 patients show prominent loss of pain and temperature sensation with a typical onset in the lower limbs around 20 years‐of‐age (Fridman et al., 2015; Houlden et al., 2006). Electrophysiology and histopathology in skin and nerve biopsies are consistent with loss of sensory axons, but the disease is highly variable and motor weakness and signs of demyelination are also found in some patients. SPT is a multisubunit complex that typically conjugates palmitoyl‐CoA and L‐Serine to form a long chain base (LCB), which is the defining structure of all sphingolipids. The mammalian enzyme consists of three core subunits ‐ SPTLC1, SPTLC2 and SPTLC3, and forms tetrameric or even octameric complex (Hanada et al., 2000; Hornemann et al., 2006, 2009; Li et al., 2021; Wang et al., 2021). SPTLC2 and SPTLC3 carry a conserved PLP binding motif, which is required for catalytic activity. The SPT core structure is associated with additional protein such as ORMDL3 or ssSPTa and b, which are involved in regulating SPT activity and product spectrum to maintain cellular sphingolipid homeostasis (Li et al., 2021; Wang et al., 2021). Depending on its subunit composition and lipid substrate availability, SPT also metabolizes other fatty acyl‐CoAs, thereby forming long chain bases with different chain lengths and structures (Lone et al., 2020b). Moreover, SPT shows an alternative activity towards other amino acid substrates, particularly Alanine and Glycine, which produces an atypical category of 1‐deoxy‐sphingolipids (1‐deoxySL) (Penno et al., 2010; Zitomer et al., 2009). Metabolically, 1‐deoxySL are dead end metabolites (Bode et al., 2016; Lone et al., 2019; Penno et al., 2010), as the missing C1‐hydroxyl group precludes their conversion into complex SLs and also prevents their terminal degradation, as the catabolic intermediate sphingosine‐1‐P cannot be formed (Zitomer et al., 2009).
A common feature of all HSAN1‐SPT mutations is that this alternative activity with L‐Alanine and Glycine is greatly increased (Penno et al., 2010; Rotthier et al., 2010). As a result, elevated 1‐deoxySL are a hallmark for HSAN1, and they are found in blood and tissue of patients. The 1‐deoxySLs are toxic to axons and cause axon degeneration in a variety of systems, including when added to cultured dorsal root ganglion (DRG) sensory neurons (Penno et al., 2010). Thus, the mutations are effectively toxic gain‐of‐function alleles, producing atypical lipid intermediates that in turn cause neuropathy.
SPTLC1 mutations have been previously modeled in mice. Homozygous knockout animals die embryonically, and heterozygotes are normal (Hojjati et al., 2005). However, expression of the Sptlc1 C133W allele as a transgene caused both elevated 1‐deoxySL levels and neuropathy (Eichler et al., 2009; Penno et al., 2010; Rotthier et al., 2011). The mutant C133W protein was found to catalyze the formation of abnormal 1‐deoxySLs, but also to retain canonical activity in vitro (Bode et al., 2016). These results are consistent with the toxic gain‐of‐function mechanism, and the transgenic mice are good models of the human disease. However, they also involve caveats. The tissues producing the toxic 1‐deoxySLs are dependent on the transgene expression pattern and levels, which may differ from those of the endogenous gene. In addition, the mutant allele is expressed in the presence of normal levels of wild‐type Sptlc1, possibly masking concomitant loss‐of‐function effects. Finally, therapeutic testing can be confounded by results that silence the transgene, producing a false positive, and gene therapy approaches to silence the mutant allele are more complicated in the context of an artificial transgene system.
The mouse models of YARS/diCMTC and SPTLC1/HSAN1 we describe below successfully recreate some aspects of the human diseases, but are less accurate on others, and therefore also contain caveats. Their addition to the collection of available mouse models of inherited neuropathy will be of benefit, but their appropriateness for future mechanistic and preclinical studies will depend on the specific application.
2. METHODS
2.1. Mouse husbandry
All mice were bred and housed in the research animal facility at The Jackson Laboratory. Mice were maintained in a medium barrier pathogen‐free room and housed in PIV caging with a 12:12 light dark cycle. Mice were provided acidified water and 6% fat pelletized chow ad libitum. All mouse experiments were carried out following The Guide for the Care and Use of Laboratory Animals and procedures were reviewed and approved by the Institutional Animal Care and Use Committee of The Jackson Laboratory. The generation and use of specific mouse strains are described below, except the generation of Yars E196K mice, which has been described (Spaulding et al., 2021).
2.2. Generation of the Yars RRC100 allele
The RRC100 gene trap insertion (MGI:4127143) ES cell clone was obtained from Bay Genomics (Skarnes et al., 1995; Stryke et al., 2003). These cells (129P2/OlaHsd strain background) were expanded and microinjected into C57BL/6J blastocysts. Chimeric mice were bred for germline transmission. The insertion in Yars was identified by 5’ RACE performed by Bay Genomics. Subsequent analysis of the insertion site revealed that the gene trap construct disrupts the coding sequence of exon 11 of Yars. Mice are genotyped using the following primer combinations: FWD: 5’ CAG ATG CAG ATA GCC TTT ATG TG 3’ and REV: 5’ CTC CGC AAA CTC CTA TTT CTG AG 3’, where the forward primer is in exon 11 of Yars, and the reverse primer is in the gene trap construct.
2.3. Generation of the Sptlc1 C133W allele
The Sptlc1 C133W allele was generated by CRISPR/Cas9 genome editing. Two mutations were introduced, a T > G conversion changing codon 133 from TGT to TGG to accomplish the Cysteine to Tryptophan mutation, and also a silent mutation in the codon for Threonine 132, changing ACC to ACG to eliminate the PAM site of the CRISPR guide to prevent recutting. The sgRNA guide sequence used was AAGTACGGAGTGGGTACCTG, in which the TG of codon 133 sequence is at the 3’ end of the guide. A single stranded DNA oligo template was used to induce the mutations through strand invasion and homology directed repair. The donor oligo sequence was: ATTTGACACTTAAGAATACTTCTAAGACTCCTCAATGGAGTCTGACAAAATGCACTTACCAAATGTGCCATAGAACCCTCGAGGACC C CA C GTACCCACTCCGTACTTCTTTAAAGATGAAAAAGCT. This oligo is reverse strand sequence, the bold and underlined C introduces the C133W allele, and the italicized and underlined C introduces the silent change in T132. The sgRNA (50 ng/µl) and Cas9 mRNA (100 ngµl) were microinjected directly into C57BL/6J fertilized zygotes. A founder from this microinjection that carried the desired mutation was identified by PCR genotyping and backcrossed three generations to wild‐type C57BL/6J mice to eliminate mosaicism and possible off‐target events before establishing a research colony for the experiments described here. The primers used for genotyping by PCR amplification of genomic DNA and Sanger sequencing are: Forward: CATCCTCCAGGAAAGTCTGG, and Reverse: ACAGCCATGCACAACTGAAA. Genotyping can also be done using an endpoint assay to detect the nucleotide changes, the primers used are: Forward: GAGGGTGGTCTGCACTGTGT, Reverse: ACCAAATGTGCCATAGAACC, WT probe: TGGGTACCTGTGGTCCT and mutant probe AGTGGGTACGTGGGGTCCTCG.
2.4. Nerve conduction velocity
Nerve conduction velocity of motor axons of the sciatic nerve was measured. Mice were anaesthetized with 2% isoflurane and placed on a thermostatically regulated heating pad to maintain body temperature. Action potentials were produced by placing stimuli proximally at the sciatic notch or distally at the ankle. The latencies to elicit a compound muscle action potential (CMAP) recorded in the thenar muscles of the hind paw following proximal and distal stimuli was determined. NCV was calculated as [conduction distance/(proximal latency‐distal latency)].
2.5. Mouse behavioral testing
2.5.1. Wire hang test
The wire‐hang test (Gomez et al., 1997; Rafael et al., 2000) was used to assess motor function. Briefly, mice were placed on a 6” × 9” piece of wire mesh and then the mesh was inverted and held ~9” above a box filled with bedding. The latency (seconds) to a fall was timed and recorded, up to a maximum of 1 min. Each mouse performed three trials in a given session and a rest period of at least 30 s was given between individual trials. The results are reported as the average of 3 trials and as the best (maximum) time of the three trials.
2.5.2. Von Frey test
Mice were acclimated for 60 min on top of a wire mesh grid. One of a standard set of calibrated microfilaments (0.2–2 g) was applied to the plantar surface of the hind paw until it bowed and was maintained in place for 3 s or until the paw was withdrawn. A positive response was noted as a withdrawal from the microfilament and response to the next lower microfilament was tested, consistent with the “up‐down” method where if no response was observed, then the next highest microfilament in the series is applied. Both paws were tested consecutively, and paw withdrawal threshold (g) is defined as the minimum force applied resulting in withdrawal of the paw.
2.5.3. Rotarod test
An accelerating paradigm from 4 to 40 rpm was used for testing on the rotarod (Ugo‐Basile), with a maximum duration of 5 min per trial. After an acclimation period of 60 min in the testing room, mice were placed on the rotating rod and the latency to falling was recorded. This procedure was repeated on two consecutive days four times per day with an intertrial period of about 1 min, and the average of the last three trials on day 2 was used as a measure of locomotor ability.
2.5.4. Hot plate test
Mice were habituated to the testing room for 60 min. They were then placed inside a clear Plexiglas cylinder placed atop a hot plate (Harvard Apparatus) with the surface temperature maintained at 52 ± 2°C and observed for a maximum exposure time of 30 s. Latency to first instance of paw withdrawal, jumping, hind paw lick, or paw shake/flutter is recorded to the nearest 0.1 s. If no response is observed after 30 s, the subject is removed from the hot plate to avoid tissue damage. Subjects are placed in a holding cage until all mice within the home cage have been tested.
2.5.5. qRT‐PCR
Total RNA was prepared from snap‐frozen liver samples by homogenization in Trizol (ThermoFisher, #15596026). After addition of chloroform and isolation of the aqueous layer, 1.5 volumes of ethanol were added, and the solution transferred to an RNeasy Mini column (Qiagen, #74104). The rest of the kit protocol was followed for RNA purification with on‐column DNase I digestion (Qiagen, #79254). Purified RNA was then reverse‐transcribed using standard techniques (ThermoFisher, #18080400), including a control without reverse transcriptase. cDNA was then subjected to standard PCR and Sanger sequencing or to quantitative PCR using SYBR green reagents (ThermoFisher, #A46109) and performed on a ViiA7 machine (ThermoFisher). Primers for RT‐PCR and Sanger sequencing in Sptlc1 were as follows: Sptlc1 F: ACAAGTTGCAGGAGCGTTCT, Sptlc1 R: GGAATGGCACTGGCTATGGT. Primers for qPCR were as follows: Yars 5’ F: TGGAGAAGCTCAAGTTTATCAAAGG, Yars qPCR 5’ R: AGG GTGTTCCACCTGCTTCA, Yars 3’ F: TGAGCCGCCAGGTTGAAC, Yars 3’ R: AGTCGGCCTGCAGCTTCTC Sptlc1 qPCR A F: TGCCATTCCTGCGTACTCTAA, Sptlc1 qPCR A R: GCCTGTAATCCTTTCTGGATAGC, Sptlc1 qPCR B F: GCACGGTACTTGGACAAGGA, Sptlc1 qPCR B R: TAGCTCTTCCTCCGTCTGCT, beta actin qPCR F: GATCTGGCACCACACCTTCT, beta actin qPCR R: GGGGTGTTGAAGGTCTCAAA. Cycle threshold values as determined by the software (QuantStudio, ThermoFisher) were used to calculate arbitrary expression units, which are equal to 100‐times the fold change. Delta‐delta Cts were calculated using beta actin as the reference gene and the average of all WT samples as the reference group. p‐values were determined in Prism (GraphPad) using unpaired t‐test.
2.5.6. Immunoblot
Snap‐frozen spinal cords from n = 4 18‐month‐old mice per genotype were homogenized in RIPA buffer containing protease and phosphatase inhibitors (AG Scientific T‐2495) and centrifuged at 15k g for 15 min at 4°C to clear. Samples were boiled in Laemmli buffer and 15 µg of protein per sample was run on a 4–20% SDS‐PAGE gel (Bio‐Rad, #4561094) and transferred to PVDF using standard techniques. Blots were sequentially probed with anti‐SPTLC1 (mouse, 1:2000, BD, #611304) and anti‐GAPDH (rabbit, 1:2000, Cell Signaling Technology, #2118) antibodies; 0.02% sodium azide was included in the anti‐GAPDH primary solution to quench the first HRP reaction. Blots were visualized by incubation with HRP‐conjugated secondary antibody (1:50,000; Jackson ImmunoResearch, #711‐035–152 (anti‐rabbit), 715–035–150 (anti‐mouse)) followed by HRP substrate (Millipore, #WBLUR0100). Quantification of protein levels was performed by measuring SPTLC1 band intensity in Fiji and normalizing to GAPDH band intensity. p‐values were determined in Prism using unpaired t‐test.
2.5.7. Lipid analysis
Lipids were extracted from frozen tissues or plasma with 500 µl methanol containing 200 pmoles each of D7‐SA (d18:0) and D7 SO(d18:1) as internal standard on a shaker‐incubator at 37°C at 1400 rpm for 1 h. Lipids were hydrolyzed overnight at 65°C overnight and re‐extracted with CHCl3 as described (Zhakupova et al., 2016). Hydrolyzed lipids were dried under a stream of N2 and resuspended in 200 µl (tissues) or 70 µl (plasma) reconstitution buffer (70% methanol, 10 mM ammonium acetate, pH 8.5). LCBs were separated via a reverse phase C18 column (Uptispere 120 Å, 5 μm, 125 × 2 mm, Interchim, France) connected to a QTRAP 6500+ LC‐MS/MS System (Sciex) as described (Lone et al., 2020a). LCBs were normalized to internal standards and plasma volume or to protein amounts extracted for tissues. Tissue pellets in urea (8 M) were homogenized in a Precellys 24 tissue homogenizer (Bertin Technologies) post extraction. Protein concentration was determined using the Bradford assay.
2.5.8. Serine supplementation
Supplemental Serine was provided by mixing 1.5 g of L‐Serine (AjiPure pharmaceutical grade L‐serine) into a DietGel 76A food cup (ClearH2O), which was provided in place of standard chow and water. During this supplementation, the control diet was DietGel 76A without added Serine. DietGel is 76% water and 24% solid nutrients, of which 4.7% of the total is protein, although the specific content of Serine is not listed. A single DietGel cup is ~75g. Therefore, assuming Serine is ~5% of the total amino acid content, adding 1.5g of supplemental Serine constitutes an approximately 10‐fold increase.
2.5.9. Nerve histology and immunohistochemistry
Femoral nerve histology
The femoral nerves, which include both a motor and sensory branch (Scherer et al., 2005) were dissected free and fixed by immersion in 2% paraformaldehyde, 2% glutaraldehyde in 0.1 M cacodylate buffer for 12 h at 4°C. Nerves were then plastic embedded, sectioned at 1.0 µm thickness, and stained with Toluidine Blue. Images were collected at 40× magnification on a Nikon Eclipse 600 microscope with DIC‐Nomarski optics. Images were analyzed for axon number using an automated method in Fiji/ImageJ that was manually confirmed by visual inspection of images for mis‐identified axons. Axon diameter was measured in Fiji/ImageJ with the Measure & Label Plug‐in.
Caudal nerve histology
To examine the caudal nerves, a 1 cm cross‐section of tail was cut starting 2.5 cm from the base (proximal end) of the tail. The skin of the tail was opened at the ventral midline, and the ventral branches of the caudal nerve were exposed by removing overlying tendons. The proximal 0.5 cm of the caudal nerves were then dissected free, embedded and analyzed as described above for the femoral nerves.
Intraepidermal nerve fiber density
Intraepidermal nerve fiber density staining was performed as described by Su & Schwarz (Su & Schwarz, 2017). The skin of the tail (obtained from the section described above) or skin from a 3 mm punch biopsy of the plantar surface of the hind paw was fixed overnight at 4°C in Zamboni's fixative and embedded in OCT for cryostat sectioning. Cross‐sections of the skin 30 µm in thickness were mounted on charged glass slides and stained for sensory nerve endings in the skin using primary antibodies against the neuronal marker PGP9.5 (Proteintech, #14730‐1‐AP), and Alexa 555‐conjugated donkey anti‐rabbit antibodies for visualization. 40× confocal stacks through the entire 30 µm section were collected and viewed as maximum projections. The number of intraepidermal nerve fibers in each image (~300 µm of skin) were counted and normalized to the number of fibers per 250 µm. Two confocal stacks from each tissue sample (paw and tail) were analyzed and averaged together for each animal, with n = 10 animals per genotype.
2.5.10. Muscle weight to body weight ratio
To test for selective muscle atrophy, the triceps surae muscles (medial and lateral gastrocnemius, soleus, and plantaris) were dissected free from the Achilles tendon to the origin at the knee. Overlying vastus muscle was removed, and the muscle group was weighed and compared to total body weight. The triceps surae was chosen because it is a relatively distal muscle that atrophies in many peripheral neuropathy patients, and because the large size of the muscle group minimizes inconsistencies in dissection and weighing.
2.5.11. Circulating neurofilament light chain quantification
Neurofilament light chain (NEFL) was quantified in mouse plasma obtained by lancet puncture of the submandibular vein in mice prior to neurophysiological testing, or at least 24 h after such analysis. Approximately 50 µl of plasma was collected from each mouse. Samples were stored at −80°C and then shipped to Quanterix for Simoa quantification of neurofilament light chain levels. All samples, mutant and control, for each model were analyzed in a single batch.
2.5.12. Statistics
Statistical tests used are stated in figure legends. Differences were considered significant with a p value <0.05.
3. RESULTS
3.1. DiCMTC‐relevant phenotypes in Yars E196K mice
Mice carrying the human diCMTC‐associated allele Yars E196K were generated by homologous recombination as described (Spaulding et al., 2021). The E196K allele of YARS is strongly associated with diCMTC. It was identified in a large, multi‐generation pedigree, it segregated with disease in that family, and it was not found in ethnically matched healthy control subjects (Jordanova et al., 2003, 2006). To briefly describe the mouse allele, codon 196 falls near the end of exon 5, and a G > A mutation was introduced to change Glutamic acid (GAG) to Lysine (AAG) (Figure 1a). A loxP‐flanked Neomycin resistance cassette was also inserted 30 base pairs into intron 5, and was excised by mating to a ubiquitously expressed Cre transgenic strain so that only a loxP site and a small amount of vector sequence remains in the 1731 bp intron. Mice were confirmed to express the G > A mutation and to splice correctly from exon 5 to exon 6 by RT‐PCR analysis, and Yars transcript levels were not altered (Spaulding et al., 2021). Consistent with the retained tRNA charging activity observed in in vitro assays (Froelich & First, 2011; Jordanova et al., 2006), the Yars E196K mice are viable as homozygotes.
FIGURE 1.
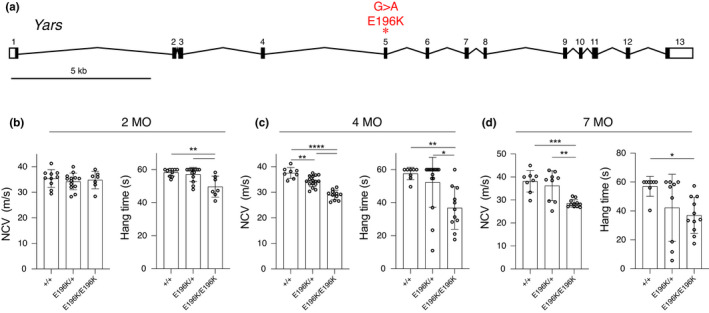
Yars E196K phenotype. (a) Diagram depicting the mouse Yars gene structure and the G > A point mutation made in exon 5. (b–d) Nerve conduction velocity (NCV) and wire hang experiments were conducted on WT, Yars E196K/+, and Yars E196K/E196K mice at 2, 4, and 7 months of age. 2‐month N = 10 WT, 13 Yars E196K/+, and 7 Yars E196K/E196K mice. 4‐month N = 8 WT, 16 Yars E196K/+, and 11 Yars E196K/E196K mice. 7‐month N = 8 WT, 9 Yars E196K/+, and 11 Yars E196K/E196K mice, pooled data from both sexes are shown. Bar graphs show individual data points and mean ± SD. For each experiment, a one‐way ANOVA with Tukey's multiple comparisons was used to determine significance. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 [Correction added on 16 February 2022, after first online publication: Figure 1 was originally published as Figure 3. The Figures have been corrected in this version]
Heterozygous Yars E196K/+ mice, recreating the dominant disease‐associated genotype, and homozygous Yars E196K/E196K mice were compared to wild‐type littermates in a series of clinically relevant assays. At 2 months of age, the Yars E196K/+ mice were largely unaffected, although homozygotes showed a modest but significant decrease in performance in a wire hang (inverted grid) test of grip strength and endurance (Figure 1b). By 4‐months‐of‐age, nerve conduction velocity in sciatic motor axons was significantly reduced in both heterozygotes and homozygotes, although the reduction in heterozygotes was less and only nominally below the normal range (37.58 ± 2.13, 34.34 ± 2.12, 28.96 ± 1.88 in WT, hets, and homs respectively). Performance in the wire hang test became increasingly variable in the heterozygotes, but was reduced to an even greater degree in the homozygotes (Figure 1c). Despite these indications of progressive neuromuscular dysfunction, neither males nor females showed reduced body weight, even in homozygotes (Figure 3c). At 7‐months‐of‐age, nerve conduction velocity was significantly reduced in homozygotes (38.11 ± 4.71 vs. 28.54 ± 1.66, p = 0.0004), although heterozygotes were variable and changes did not reach statistical significance. Performance in the wire hang test was similarly variable in heterozygotes, but was reduced in homozygotes (Figure 1d). Body weights remained unchanged compared to control littermates at 7‐months‐of‐age (not shown).
FIGURE 3.
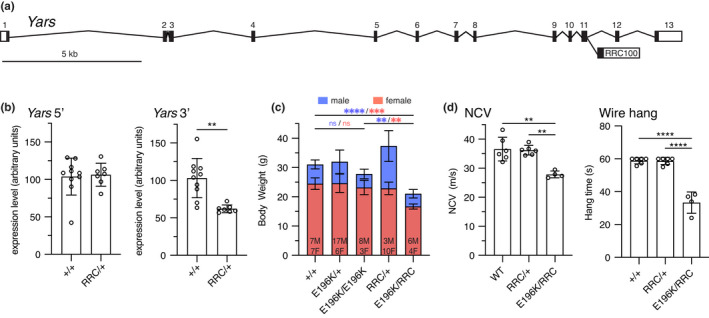
Yars E196K and null Yars RRC phenotype. (a) Schematic showing the RRC100 gene trap insertion in exon 11 of Yars. (b) Yars mRNA levels were measured in WT and Yars RRC/+ mice by qRT‐PCR using two different sets of primers, made to either the 5’ or 3’ end of the transcript. N = 10 WT and 7 Yars RRC/+ mice. (c) Body weight measurements of WT, Yars RRC/+, YarsE196K /+, Yars E196K/E196K and Yars RRC/E196K mice taken at 4 months of age. Data is presented as a superimposed bar graph of male and female body weight values. Ns for each sex are indicated, asterisks are color coded with sex. (d) NCV and wire hang experiments were conducted at 4‐months‐of‐age with WT, Yars RRC/+, and Yars RRC/E196K mice. N = 6 WT, 6 Yars RRC/+, 4 Yars RRC/E196K mice, pooled data from both sexes are shown. Bar graphs show individual data points and mean ± SD. Data were analyzed by unpaired, two‐tailed t‐test (b) or one‐way ANOVA with Tukey's multiple comparisons test (c, d). **p < 0.01, ***p < 0.001, ****p < 0.0001 [Correction added on 16 February 2022, after first online publication: Figure 3 was originally published as Figure 1. The Figures have been corrected in this version]
DiCMTC is an intermediate form of CMT, with moderately reduced nerve conduction velocities in patients, similar to the changes observed in mice (Figure 1c,d). To examine the basis for this reduced conduction velocity, we examined peripheral nerves by both light and electron microcopy. The femoral nerve consists of a predominantly motor branch that innervates the quadriceps muscle in the thigh, and a sensory branch that becomes the saphenous nerve more distally and innervates the skin of the lower leg. Cross sections of the motor branch of the femoral nerve at 4‐months‐of‐age showed decreased axon diameters in homozygous mutant mice, but not in heterozygotes (Figure 2a,b). The axon number was not changed (Figure 2c, 544.3 ± 12.39, 541.3 ± 9.87, and 538.0 ± 16.12 in WT, hets and homs, respectively), indicating that axons are smaller, but they do not degenerate. Consistent with this, plasma levels of neurofilament light chain, a biomarker of some forms of CMT (Sandelius et al., 2018), were not elevated (Figure 2d). Essentially the same changes were found at 7‐months‐of‐age (Figure S1), indicating little or no progression over this time period. The sensory branch of the femoral nerve did not reveal any notable histopathological changes (Figure S2). The smaller axons in homozygous mutant mice may contribute to the reduced motor nerve conduction velocity; however, we also examined myelination by electron microscopy. The G‐ratio (the relationship of the axon diameter to total nerve fiber diameter including myelin) in motor and sensory nerves was not changed in the homozygous mutant mice, indicating myelin thickness is preserved (Figure 2e). The packing of the myelin wraps was also examined and was also unchanged (Figure 2f). These results suggest that myelination is not altered in the Yars E196K mice, and this is not the explanation for the reduced conduction velocities.
FIGURE 2.
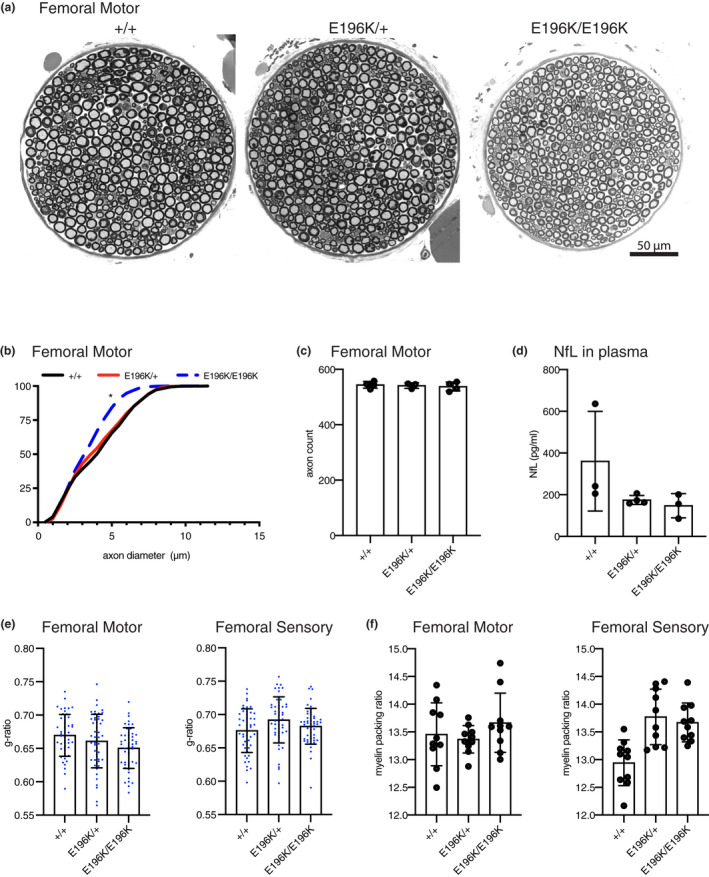
Femoral nerve histology of 4‐month‐old Yars E196K mice. (a) 40X images of toluidine blue stained sections of the motor branch of the femoral nerve taken from WT, Yars E196K/+, and Yars E196K/E196K mice at 4‐months‐of‐age. (b) Cumulative histogram of axon diameters found in femoral motor nerves of WT, Yars E196K/+, and Yars E196K/E196K. Homozygous mutant mice have significantly smaller axons compared to WT. (c) Axon number in the motor branch of the femoral nerve. (d) Levels of plasma neurofilament light chain (NfL) (e) The g‐ratio in the motor and sensory branches of the femoral nerve. (f) Myelin packing from the motor and sensory branches of the femoral nerve. Analyses in (e) and (f) were done using transmission electron micrographs. Measurements from nerve sections of at least three mice per genotype were taken for all experiments. Bar graphs show individual data points and mean ± SD. Data were analyzed by one‐way ANOVA (c and d) or nested one‐way ANOVA (b, e, and f) with Tukey's multiple comparisons test. * in (b) denotes comparison between WT and Yars E196K/E196K
3.2. Examination of a null allele of Yars
Human genetics suggests that diCMTC is not a simple haploinsufficiency, and the retained activity of the E196K allele of YARS argues against a simple loss‐of‐function mechanism. To explore this in mice, we used a gene trap allele of Yars, RRC100. Gene trap mutations intercept splicing and disrupt gene function at the mRNA level (Skarnes et al., 1995). The Yars RRC100 gene trap is inserted in exon 11 of Yars (Figure 3a). Analysis of transcript abundance using qRT‐PCR primers upstream of the insertion revealed no change in mRNA levels, but primers downstream of the insertion revealed ~50% levels of mRNA, consistent with the gene trap disrupting expression of downstream exons, leading to a truncated mRNA (Figure 3b). The Yars E196K allele was viable in combination with the RRC100 gene trap, which was homozygous lethal, as anticipated. Body weight was reduced in E196K/RRC100 compound heterozygotes, whereas it was not reduced in E196K homozygotes, or RRC100 heterozygotes (Figure 3c). The Yars RRC100 mice as heterozygotes did not have phenotype‐relevant phenotypes based on sciatic motor nerve conduction velocity or grip strength and endurance in the wire hang test. In E196K/RRC100 compound heterozygotes, the severity of the neuropathy at 4‐months‐of‐age was comparable to E196K as a homozygote (Figure 3d, NCV 36.58 ± 4.1, 36.19 ± 1.65, 27.84 ± 1.18 in WT, RRC100/+, and compound hets, respectively, and vs. 28.96 ± 1.88 in E196K homozygotes, Figure 1e). The viability of YarsRRC100/E196K compound heterozygotes again argues against a loss‐of‐function in the E196K allele, although the reduced body weight may indicate some reduction in function. Furthermore, the lack of phenotype in RRC100 heterozygotes indicates there is no haploinsufficiency in mice, consistent with humans (Nowaczyk et al., 2017; Tracewska‐Siemiatkowska et al., 2017; Williams et al., 2019).
3.3. Sptlc1 C133W knockin mice to model HSAN1
The C133W mutation in mouse Sptlc1 was created using CRISPR/Cas9 genome editing to introduce a T > G change in exon 5 of the gene, changing codon 133 from TGT (Cysteine) to TGG (Tryptophan). A second silent C > G mutation was also introduced in codon 132 (ACC to ACG, both encoding Threonine) to eliminate the PAM‐site and prevent recutting with Cas9 after successful editing (Figure 4a,b). Heterozygous mice were intercrossed in an attempt to create homozygotes to increase the gene dosage of the C133W allele. However, in 93 offspring recovered, no homozygous mutant mice were identified when genotyped at 1‐week‐of‐age, compared to 34 WT and 56 heterozygous littermates (genotyping was inconclusive on three pups), leading us to conclude that the C133W allele is homozygous lethal embryonically or perinatally (Chi‐square test p < 0.0001). To look for changes in gene expression or transcript stability that may exacerbate a partial loss‐of‐function, we performed qRT‐PCR on mRNA from heterozygous mice and wild‐type littermates using two different amplicons (Figure 4c,d). No change was detected in an amplicon in the 5’ end of the transcript, whereas a modest but significant decrease in transcript levels was found in an amplicon in the 3’ end of the transcript. This decrease is unlikely to be the basis for the lethality, since heterozygous null mice do not have a phenotype. Consistent with the qRT‐PCR data, WT and Sptlc1 C133W/+ mice display similar levels of SPTLC1 protein as assessed by western blot (Figure 4e).
FIGURE 4.
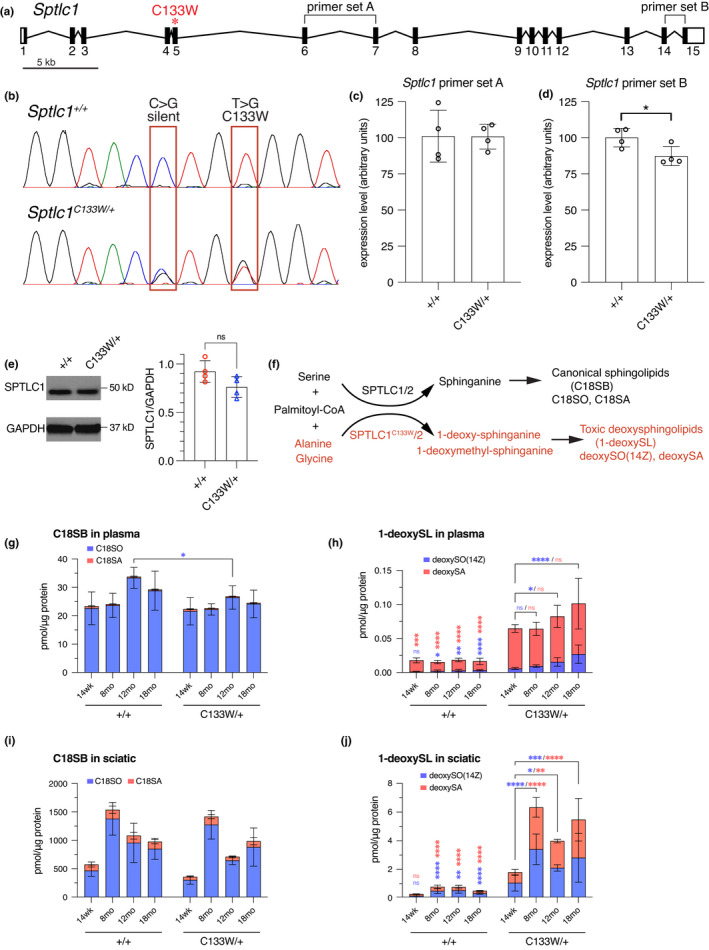
Generation of the Sptlc1C133W knockin allele. (a) Gene diagram of Sptlc1 indicating the location of the C133W mutation in exon 5. (b) Chromatograms of Sanger sequencing from RT‐PCR products obtained by amplifying a portion of Sptlc1 from cDNA prepared from Sptlc1 +/+ and Sptlc1 C133W /+ liver samples. A silent C > G mutation and the T > G transversion causing the change from Cysteine to Tryptophan at position 133 are indicated. (c, d) Expression of Sptlc1 in Sptlc1 +/+ and Sptlc1 C133W /+ as assessed by qRT‐PCR using liver cDNA samples. Two different primer sets (denoted in A) within Sptlc1 were used: primer set A (c) and B (d), and both were normalized to beta actin. Only primer set B showed a change, with an ~20% reduction in the Sptlc1 C133W /+. (e) Western blot reveals similar levels of SPTLC1 in Sptlc1 +/+ and Sptlc1 C133W /+ spinal cord (left). Lysates from n = 4 mice per genotype were analyzed, and levels of SPTLC1 were normalized to GAPDH (right). (f) Wild‐type SPTLC1, along with SPTLC2, are the catalytic components of the serine palmitoyltransferase complex (SPT), which catalyzes the condensation of palmitoyl‐CoA and Serine to generate sphinganine (black text). SPTLC1C133W has altered substrate specificity, and can use Alanine and Glycine in addition to Serine. However, these alternative amino acids generate novel, toxic, 1‐deoxy‐sphingolipid products (red text). (g) Plasma levels of canonical sphinoglipid C18SO and C18SA were essentially unchanged with genotype and age, only 12‐month‐old Sptlc1 C133W/+ mice showed a small decrease in C18SO from control values. (h) Plasma levels of the 1‐deoxy‐sphingolipids were elevated compared to controls and increased with age in Sptlc1 C133W /+ mice. Similar profiles of canonical (i) and 1‐deoxySLs (j) were observed in sciatic nerve. Data points in (c), (d) are individual mice. Data in (g–j) are presented as stacked bar graphs for different canonical (C18SO and C18SA) and 1‐deoxySLs (deoxySO(14Z) and deoxy SA), which are color coded as noted. Values are mean ± SD. Values were compared by unpaired, two‐tailed t‐test (c, d) or by ordinary two‐way ANOVA with Sidak's multiple comparisons test (g–j, independent analysis for each metabolite). ns: p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Vertical asterisks over WT data denote difference compared to C133W mutants at the same age. Brackets and asterisks over C133W data denote changes at different time points within that genotype. Asterisks are color coded to match each metabolite
3.4. Sptlc1 C133W mice develop a relevant biochemical phenotype
The SPTLC1 protein is in a multiprotein complex, where it typically conjugates L‐Serine and palmitoyl‐CoA to produce sphinganine and subsequently ceramide and complex sphingolipids (Figure 4f). Mutations in SPTLC1 alter the substrate preference of the enzyme also allowing the conjugation of Glycine or Alanine in addition to Serine, thereby forming the atypical metabolites 1‐deoxy‐ and 1‐deoxymethyl‐sphinganine (1‐deoxySLs). Levels of both canonical sphingolipids and toxic 1‐deoxySLs were analyzed by mass spectrometry in plasma, sciatic nerve, and other tissues from the Sptlc1 C133W/+ mice, and compared to levels in littermate controls (Figure 4g–j, and Figure S3). Although total levels of canonical sphingolipids (18SO, C18SA) were largely similar between mutant and control mice (Figure 4g,i), levels of the 1‐deoxySLs (deoxySO(14Z), deoxySA) were increased in the mutant mice (Figure 4h,j). This was also true in liver, cerebellum (for deoxySA), forebrain, and skeletal muscle (Figure S3). The levels of 1‐deoxySLs also gradually increased with age, with levels of 14 weeks being lower than 12‐ and 18‐month time points (Figure 4h,j and Figure S3). Thus, the knockin mice develop a relevant biochemical phenotype that is comparable to the previous transgenic mouse model and HSAN1 patients (Eichler et al., 2009).
Supplementation with Serine reduces 1‐deoxySL formation by competing with Alanine and Glycine for the active site of the SPT enzyme (Fridman et al., 2019; Garofalo et al., 2011). We showed this is also true in the Sptlc1 C133W/+ knockin mice (Figure 5a,b). At 20‐weeks‐of‐age, mice were shifted from normal chow to a L‐Serine supplemented diet for two weeks and were then shifted back to normal chow. A control cohort of both mutant and wild type littermates was given the same test diet, but without added Serine. Blood samples were collected at 20 weeks, immediately prior to the Serine supplementation, at 22 weeks, after 2 weeks on the high Serine diet, and at 24 weeks, 2 weeks after the mice had returned to normal chow. As anticipated, the Sptlc1 C133W/+ mice showed elevated plasma 1‐deoxySLs, but deoxySA levels were decreased after 2 weeks of Serine supplementation compared to mice on the same diet without added Serine and compared to baseline levels before Serine supplementation and after return to normal chow (Figure 5b). Therefore, the knockin allele is responding to Serine supplementation in a manner consistent with previous models.
FIGURE 5.
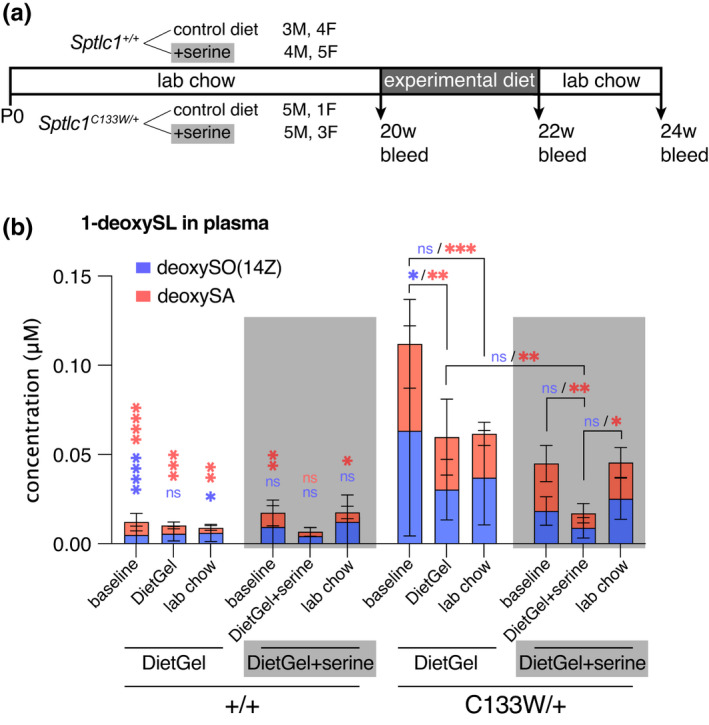
Serine supplementation reduces 1‐deoxySL levels in Sptlc1 C133W /+ mice. (a) Overview of serine supplementation experiment. Sptlc1 +/+ and Sptlc1 C133W /+ mice were randomly assigned to receive either serine supplemented DietGel 76A, or DietGel 76A alone for 2 weeks. Plasma was collected before the start of the diet manipulation, at the end of the 2‐week period, and again 2 weeks after returning to normal lab chow. (b) Levels of 1‐deoxySLs in plasma from the serine supplementation study. Mutant mice had elevated deoxySA levels, and these were reduced while mice were on a serine‐supplemented diet. N = 7 WT mice on control diet, 9 WT mice on serine supplemented diet, 6 Sptlc1 C133W/+ mice on control diet, and 8 Sptlc1 C133W/+ on serine supplemented diet, all groups were a mix of males and females. Data were analyzed within each metabolite by ordinary two‐way ANOVA with Sidak's multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Vertical asterisks over WT data denote difference compared to C133W mutants in the equivalent group. Brackets and asterisks over C133W data denote changes across groups within that genotype. Asterisks are color coded to match each metabolite. Stacked bar graphs show mean ± SD
3.5. Behavioral phenotypes in Sptlc1 C133W mice
The Sptlc1 C133W/+ mice were evaluated using a number of sensory and motor tests relevant to peripheral neuropathy. Both males and females were included in testing, but results for each sex were analyzed separately since several tests are known to have sex‐specific differences in outcomes. All mice were 12 months of age when tested. In the hot‐plate test of thermal nociception, male Sptlc1 C133W/+ mice had a greater latency to respond, suggesting decreased thermal sensitivity (Figure 6a). However, females showed no difference from wild‐type controls. Using von Frey filaments to test mechanical sensitivity, neither sex showed a phenotype (Figure 6b). In the accelerating rotarod, mutant females had a shorter latency to fall, consistent with impaired sensory/motor performance, but no effect was seen in males (Figure 6c). Notably, both wild type and mutant males performed poorly in this test, possibly due to obesity by 1 year of age, although body weight did not differ with genotype in either sex (Figure 6d). In tests of motor performance, no differences were seen in grip strength or endurance using the wire‐hang test when the best performance out of three trials was evaluated (Figure 6e). However, when the average of three trials was evaluated, mutant females actually outperformed their wild‐type littermates (Figure 6f). Again, all males performed rather poorly in this test, likely due to excessive body weight. Using clinical neurophysiology, no differences were seen in motor nerve conduction velocity in the sciatic nerve, or in compound muscle action potential amplitude in the muscles of the hind paw (Figure 6g,h). A decrease in the ratio of muscle weight (triceps surae) to total body weight (MW:BW) can be an indication of selective muscle atrophy. The MW:BW ratio was not different between mutant mice and controls (Figure 6i), and trended towards an increase in the mutant mice. This may be attributable to a trend towards decreased body weight in the mutant mice (Figure 6d), but is inconsistent with selective muscle atrophy. Overall, behavioral changes were very mild at 1‐year‐of‐age, with decreased sensitivity to thermal stimuli in males and decreased rotarod performance in females being the most clearly relevant changes observed. Behavioral phenotyping at 6 months of age did not reveal any significant differences in sensory or motor tests in younger, less obese and more compliant mice, and phenotyping at 18 months‐of‐age did not reveal any late‐onset phenotypes (Figure S4).
FIGURE 6.
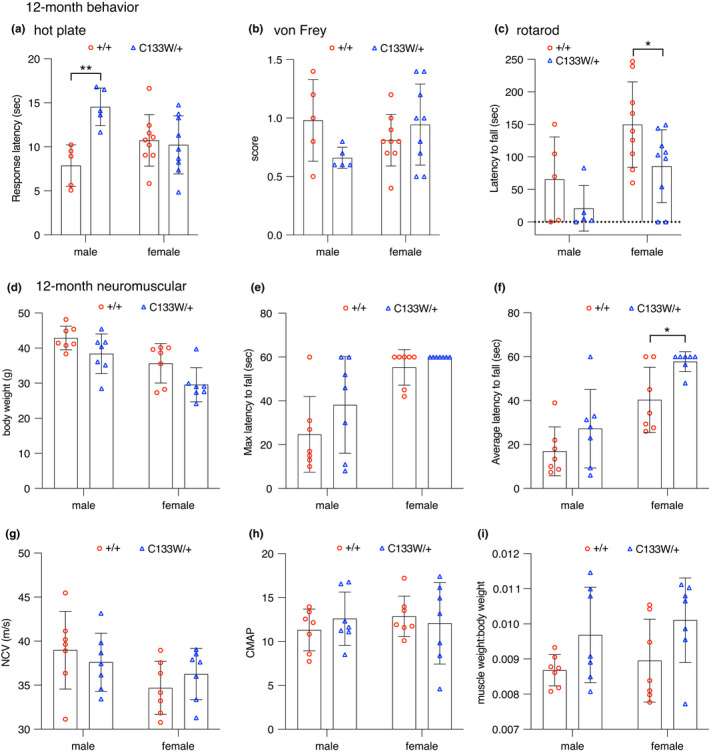
Sptlc1C133W /+ mice show mild impairment in tests of motor or sensory function at 12 months of age. (a–c) Female Sptlc1C133W /+ mice performed similarly to littermate controls on the hot plate, whereas males had an increased latency to response (a). No differences were observed on the von Frey test (B). Rotarod testing revealed a reduced latency to fall in female, but not male, Sptlc1 C133W /+ mice (c). (d) Body weight was unchanged in Sptlc1 C133W/+ mice of either sex. (e–f) Sptlc1 C133W /+ mice performed similarly to littermate controls on the wire hang test, with no change seen in the maximum latency to fall over the three‐trial test (e) and a modest increase in the average latency in female Sptlc1 C133W /+ mice (f). (g–h) No differences were found in sciatic motor nerve conduction velocity (g) or the compound muscle action potential amplitudes in the hind paw muscles, elicited from nerve stimulation at the ankle (h). There was no gross indication of muscle atrophy, as muscle weight to body weight ratio (MW:BW) (i) was changed in Sptlc1 C133W /+ mice compared to controls. Data points represent values from individual mice, bar graphs show mean ± SD, and were analyzed within each sex by unpaired, two‐tailed t‐test. *p < 0.05, **p < 0.01
3.6. Histopathology and Immunofluorescence evaluation of peripheral nerves
To more closely examine peripheral nerves for signs of neuropathy, we performed both histopathology and immunolabeling in 18‐month‐old Sptlc1 C133W/+ mice. We examined the motor and sensory branches of the femoral nerve (Figure 7a,b), and the ventral caudal nerve in the tail (Figure 7c). No differences in axon number were detected in any of these nerves (Figure 7d–f). Axon diameters were also similar (Figure 7g–i), although a small but significant decrease in femoral sensory axon caliber was found (Figure 7h). Consistent with the lack of axon loss or atrophy, plasma neurofilament light chain levels were unchanged between mutant and control mice (Figure 7j). To examine small, distal sensory nerve endings in the skin, we used a PGP9.5 antibody to measure intraepidermal nerve fiber density (IENFD) (Figure 7k). We took skin biopsies from the plantar surface of the hind paw, and from the tail sheath when collecting caudal nerve samples. No change in nerve fiber density between mutant and wild type mice was seen in either the skin of the hind paw or tail (Figure 7l). Therefore, there is no evidence for axon degeneration in the Sptlc1 C133W/+ mice in a variety of peripheral nerves, even at 18‐months‐of‐age. Previous studies in transgenic mice overexpressing the Sptlc1C133W allele as a transgene showed neuropathy relevant phenotypes (Eichler et al., 2009); however a comparison of 1‐deoxySL levels in serum or liver samples from the transgenic mice did not reveal significantly higher levels in the transgenic samples (Figure S5).
FIGURE 7.
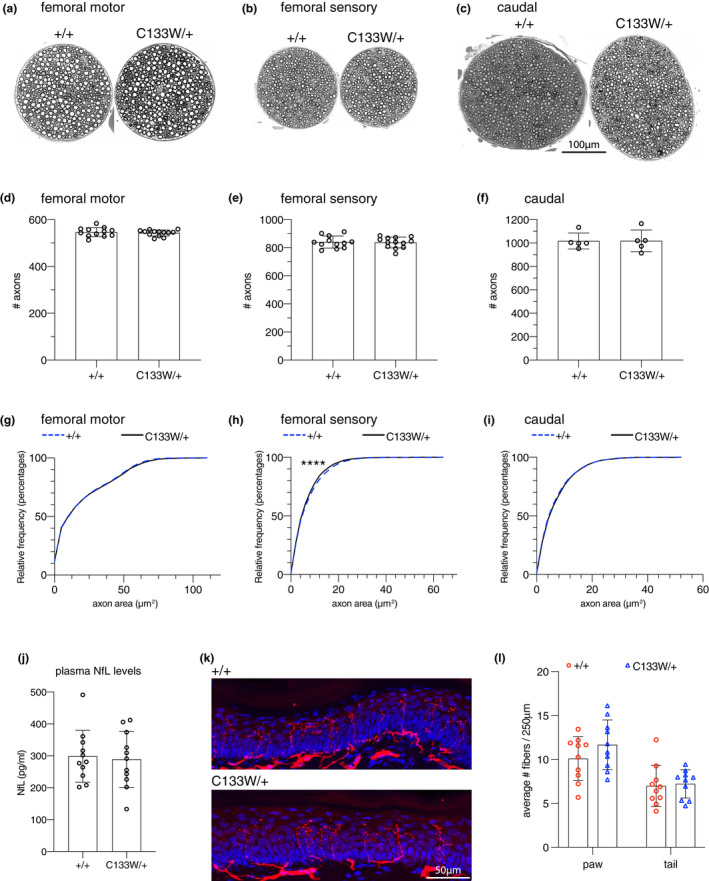
Histological analysis of Sptlc1 C133W /+ mice. (a–c) The femoral motor (a) or sensory (b) branches, and the caudal nerve (c) of 18‐month‐old Sptlc1 +/+ and Sptlc1 C133W /+ mice. (d–f) Counts of myelinated axons in these nerves, data points represent individual mice. (g–i) Cumulative histograms of axon cross‐sectional area in these nerves. The axons of the femoral sensory nerve showed a small but significant decrease in size in the Sptlc1 C133W/+ mice (h). (j) Plasma neurofilament light chain levels are unchanged in 20‐month‐old Sptlc1 C133W /+ mice, consistent with the preservation of axons. N = 11 animals per genotype. (k) Intra‐epidermal nerve fiber staining of sections of hind paw glabrous skin from 18‐month‐old Sptlc1 +/+ and Sptlc1C133W /+ mice. PGP9.5 signal is in red, and DAPI is in blue. (l) Quantification of intra‐epidermal nerve fiber density from hind paw and tail skin sections from 18‐month‐old Sptlc1 +/+ and Sptlc1 C133W /+ mice. N = 10 animals per genotype. Bar graphs show individual data points and mean ± SD. Data were analyzed by unpaired, two‐tailed t‐test (d–f, j, l) or by Kolmogorov–Smirnov and nested t‐test (g–i). ****p < 0.0001 by K–S test
4. DISCUSSION
We have generated and characterized two precision mouse models of inherited peripheral neuropathy. The first, Yars E196K, models diCMTC by introducing a disease‐associated mutation into the mouse Yars gene. These mice precisely recreate the human disease allele, but they only develop CMT‐relevant phenotypes as homozygotes, whereas diCMTC YARS mutations, including E196K, are dominant in humans. Nonetheless, this model has been informative and will be useful for future mechanistic and preclinical studies, as described below. The second model, Sptlc1 C133W, creates a knockin mouse for HSAN1. This model more accurately recreates the human genetics than previous transgenic models and was thus useful for demonstrating that this mutation is lethal when homozygous, and also for examining levels of abnormal 1‐deoxysphingolipids (1‐deoxySLs) in tissues that may not have had normal Sptlc1 expression levels with the transgene. However, while the Sptlc1 C133W knockin mice do show a relevant biochemical phenotype and mild behavioral deficits, they do not show dysfunction or axon loss in neurophysiological or histological assays. Presumably, this milder phenotype is because 1‐deoxySL production was lower than in the transgenic strain.
The Yars E196K mice have phenotypes that are relevant to the clinical features of diCMTC, including degenerative (not congenital) weakness, and reduced nerve conduction velocities. In this regard, they present a valid model of diCMTC. Although the recessive nature of the phenotype is a deviation from the human genetics, it is also informative. First, the fact that the E196K allele is viable as a homozygote and in combination with the RRC100 gene trap allele is consistent with the residual tRNA charging activity seen in vitro (Froelich & First, 2011; Jordanova et al., 2006). Related mouse models of CMT2D with mutations in Gars are not viable as homozygotes or as compound heterozygotes with a null allele, and may have both loss‐of‐function and neomorphic activities (Motley et al., 2011). Loss of function in vivo can also arise for other reasons such as protein instability or mislocalization, but the viability of E196K homozygotes and RRC100 compound heterozygotes argues that these are not significant issues. The lack of phenotype in the RRC100 heterozygotes is also consistent with the neuropathy not being due to haploinsufficiency. The finding that the E196K/RRC100 compound heterozygotes have reduced body weight, but very similar neuropathy phenotypes, does not fit neatly into a genetic mechanism. If partial loss of function in E196K was the cause, then combining it with a stronger (null) loss‐of‐function allele (RRC100) should make the phenotype more severe, and this may underlie the body weight differences between E196K homozygotes and E196K/RRC100 compound heterozygotes, but it is not true of NCV and grip strength. If instead the E196K allele was a neomorph, where the mutant allele takes on a new, toxic activity, then doubling the genetic dose of this toxic activity in E196K homozygotes should be worse than one copy in E196K/RRC100 compound heterozygotes.
Since YARS (TyrRS) is a dimerizing protein, a possibility is that the phenotype requires the presence of mutant homodimers. Given the dominant human genetics and the phenotype caused by overexpression of mutant transgenes in Drosophila, even in the presence of the endogenous fly Yars gene (Niehues et al., 2015; Storkebaum et al., 2009), one would have to speculate that sufficient levels of the mutant protein must be present to drive mutant‐homodimer formation. The levels of homodimer formation in mice (presumably 25% of the total, but this is untested) would be below the threshold required to induce neuropathy in this model organism. A previous in vivo mouse study used adenovirus to deliver both wild type and mutant FLAG‐tagged human YARS to peripheral neurons by direct injection into sciatic nerve (Lee et al., 2017). The wild type and E916K YARS proteins were similarly expressed in spinal cord and DRG cell bodies, whereas G41R YARS was not expressed well. Much less anti‐FLAG labeling was found in the peripheral nerves in the E196K mutant compared to wild type. Whether this was due to a trafficking or localization defect, or whether the transduced cells underwent rapid axon degeneration is not clear, and the pathogenicity of mutant YARS overexpression was not examined. However, the mutant YARS proteins did behave differently than wild‐type YARS in this system.
Two recent studies address the mechanisms underlying tRNA‐synthetase associated peripheral neuropathies (Spaulding et al., 2021; Zuko et al., 2021). The first (Zuko et al., 2021), identified a mechanism of tRNA sequestration by mutant GARS that leads to ribosome stalling. This mechanism is supported biochemically by a much slower off‐rate of the tRNAs from the mutant enzyme and an increased steady‐state level of tRNA bound to the mutant enzyme. It is also supported genetically by rescue of the neuropathy in both Drosophila and mouse models by overexpressing tRNAGly, whereas previous work has shown that overexpression of the wild type synthetase does not rescue (Motley et al., 2011). The second study (Spaulding et al., 2021), found that affected motor and sensory neurons activate the integrated stress response (ISR) through the kinase GCN2, a consistent consequence of stalled ribosomes. The chronic activation of the ISR contributes to the severity of the neuropathy, and genetic deletion or pharmacological inhibition of GCN2 greatly reduces the severity of the phenotype in Gars mutant mice. Importantly, the Yars E196K mice were also examined (Spaulding et al., 2021), and were shown to have activation of the ISR as homozygotes, with phospho‐eIF2α labeling in alpha motor neurons in the spinal cord and a stress‐response gene expression signature. However, there was very little ISR activation in heterozygous mice. The degree of tRNA sequestration may only be sufficient to robustly elicit the subsequent ISR activation in homozygous Yars E196K mice, although a partial‐loss‐of‐function mechanism may also contribute to ISR activation through GCN2 due to uncharged tRNAs. The data presented here indicate that ISR activation correlates well with the manifestation of clinically relevant phenotypes such as decreased grip strength, reduced nerve conduction velocity, and reduced axon size. The Yars E196K mice as homozygotes will be useful in preclinical studies testing whether GCN2 inhibitors are effective in treating these deficits, as they were in Gars/CMT2D models (Spaulding et al., 2021).
The Sptlc1 C133W knockin allele is also informative. While Sptlc1 knockout mice are known to be lethal (Hojjati et al., 2005), the C133W allele retains some normal activity, in addition to its neomorphic activity of conjugating Glycine or Alanine onto palmitoyl‐CoA (Bode et al., 2016). Therefore, it is surprising that the C133W allele is not viable when homozygous. It is formally possible that doubling the levels of the C133W allele results in the production of lethal levels of 1‐deoxySLs, but this would reflect a sharp threshold for transitioning from a very mild phenotype to a lethal phenotype. Alternatively, homeostatic control of sphingolipid synthesis may be irreparably perturbed in homozygotes, or there may be a closely linked off‐target mutations that account for this lethality. However, given that the Sptlc1 knockout is lethal, it seems likely that the retained activity is simply insufficient.
The Sptlc1 C133W knockin mice will be useful for testing gene therapy approaches or strategies designed to lower the levels of toxic deoxysphingoid bases, but not for testing therapies designed to protect axons from degeneration in the presence of 1‐deoxySLs. The latter can be tested in the previously published transgenic mice that do show neuropathy. That the 1‐deoxySL levels are not higher in the transgenic mice is surprising, but our comparison did not examine the most relevant tissues such as sciatic nerve, so a tissue‐specific difference may have been missed. Nonetheless, testing gene therapy approaches such as allele‐specific‐knockdown of the mutant Sptlc1 would be more challenging to execute and interpret using the transgenic model. First, the mutant Sptlc1 transgene is likely expressed at a higher level than the endogenous mouse gene, and the existing transgenic strain used a hamster Sptlc1 cDNA. Despite high similarity, this could complicate sequence‐dependent gene therapy approaches such as allele‐specific knockdown or knockdown‐and‐replacement with a wild‐type Sptlc1 sequence. Second, it is unclear that the neuropathy is a cell autonomous problem, and our results demonstrate that many tissues are producing the abnormal 1‐deoxySLs and they are present in the circulation. Therefore, determining which tissues to target with a gene therapy could produce misleading results with a transgene, which in this case used the chicken beta‐actin promoter with a CMV enhancer for ubiquitous high‐level expression. Our results indicate that 1‐deoxySLs can be found in many tissues, but not uniformly, and some tissues may be more important to target than others in producing the 1‐deoxySLs that cause axon loss.
SPTLC1 was also recently associated with a childhood form of amyotrophic lateral sclerosis (Mohassel et al., 2021). Unlike the mutations that lead to HSAN1, the mutations associated with childhood ALS increase enzymatic activity (hypermorphic alleles) and cause the over production of sphingolipids, and they do not per‐se cause an increase in deoxy sphingolipids. Therefore, the C133W allele is not an effective model for this disorder and other disease‐specific mutations need to be developed. However, the finding that SPTLC1 also underlies a motor neuron disease emphasizes the critical role of sphingolipid synthesis in neuronal homeostasis.
In summary, the mouse models described here recreate important aspects of their respective human diseases, but are also imperfect in other aspects. They will be valuable for future research provided these imperfections are considered. A single mouse model rarely captures all the complexity and variability of patients, necessitating a battery of models, which can reflect the specific salient features of the disease that are to be the focus of future research.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
TJH, IB, MGS, and CLH created and analyzed the Yars E196K mouse model. ALDT, CLH and KEM created and analyzed the Sptlc1 C133W mouse model. MAL performed quantification of lipids by mass spectroscopy. TH and RWB supervised experimental design and data interpretation. RWB wrote the manuscript with input from all authors.
Supporting information
Fig S1‐S5
ACKNOWLEDGEMENTS
We would like to thank the Scientific Services at The Jackson Laboratory for technical assistance, particularly Genome Engineering Technologies, the Center for Biometric Analysis, Electron Microscopy, Light Microscopy, and the Rare and Orphan Disease Center. We would also like to thank Drs. Florian Eichler and Yi Gong at Harvard, Mass. General Hospital for providing transgenic Sptlc1‐C133W transgenic mouse samples for analysis. This work was supported by grants from the National Institutes of Health (R37 NS054154, R24 NS098523, and R01 NS113583) to RWB, and the Swiss National Science Foundation (SNF 31003A_179371) and European Joint Programme on Rare Diseases (EJP RD+SNF 32ER30_187505) to TH. The Scientific Services at The Jackson Laboratory are supported in part by NIH grant CA34196.
Hines, T.J. , Tadenev, A.L.D. , Lone, M.A. , Hatton, C.L. , Bagasrawala, I. , Stum, M.G. , et al (2022) Precision mouse models of Yars/dominant intermediate Charcot‐Marie‐Tooth disease type C and Sptlc1/hereditary sensory and autonomic neuropathy type 1. Journal of Anatomy, 241(5), 1169–1185. Available from: 10.1111/joa.13605
Timothy J. Hines, Abigail L. D. Tadenev and Museer A. Lone contributed equally, co‐first authors.
DATA AVAILABILITY STATEMENT
All data relevant to this publication are included in the manuscript of supplemental materials.
REFERENCES
- Bejaoui, K. , Wu, C. , Scheffler, M.D. , Haan, G. , Ashby, P. , Wu, L. et al. (2001) SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nature Genetics, 27, 261–262. [DOI] [PubMed] [Google Scholar]
- Blocquel, D. , Li, S. , Wei, N. , Daub, H. , Sajish, M. , Erfurth, M.L. et al. (2017) Alternative stable conformation capable of protein misinteraction links tRNA synthetase to peripheral neuropathy. Nucleic Acids Research, 45, 8091–8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode, H. , Bourquin, F. , Suriyanarayanan, S. , Wei, Y. , Alecu, I. , Othman, A. et al. (2016) HSAN1 mutations in serine palmitoyltransferase reveal a close structure‐function‐phenotype relationship. Human Molecular Genetics, 25, 853–865. [DOI] [PubMed] [Google Scholar]
- Dawkins, J.L. , Hulme, D.J. , Brahmbhatt, S.B. , Auer‐Grumbach, M. & Nicholson, G.A. (2001) Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit‐1, cause hereditary sensory neuropathy type I. Nature Genetics, 27, 309–312. [DOI] [PubMed] [Google Scholar]
- Eichler, F.S. , Hornemann, T. , McCampbell, A. , Kuljis, D. , Penno, A. , Vardeh, D. et al. (2009) Overexpression of the wild‐type SPT1 subunit lowers desoxysphingolipid levels and rescues the phenotype of HSAN1. Journal of Neuroscience, 29, 14646–14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman, V. , Oaklander, A.L. , David, W.S. , Johnson, E.A. , Pan, J. , Novak, P. et al. (2015) Natural history and biomarkers in hereditary sensory neuropathy type 1. Muscle and Nerve, 51, 489–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman, V. , Suriyanarayanan, S. , Novak, P. , David, W. , Macklin, E.A. , McKenna‐Yasek, D. et al. (2019) Randomized trial of l‐serine in patients with hereditary sensory and autonomic neuropathy type 1. Neurology, 92, e359–e370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froelich, C.A. & First, E.A. (2011) Dominant Intermediate Charcot‐Marie‐Tooth disorder is not due to a catalytic defect in tyrosyl‐tRNA synthetase. Biochemistry, 50, 7132–7145. [DOI] [PubMed] [Google Scholar]
- Garofalo, K. , Penno, A. , Schmidt, B.P. , Lee, H.J. , Frosch, M.P. , von Eckardstein, A. et al. (2011) Oral L‐serine supplementation reduces production of neurotoxic deoxysphingolipids in mice and humans with hereditary sensory autonomic neuropathy type 1. Journal of Clinical Investigation, 121, 4735–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez, C.M. , Maselli, R. , Gundeck, J.E. , Chao, M. , Day, J.W. , Tamamizu, S. et al. (1997) Slow‐channel transgenic mice: a model of postsynaptic organellar degeneration at the neuromuscular junction. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience, 17, 4170–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada, K. , Nishijima, M. , Fujita, T. & Kobayashi, S. (2000) Specificity of inhibitors of serine palmitoyltransferase (SPT), a key enzyme in sphingolipid biosynthesis, in intact cells. A novel evaluation system using an SPT‐defective mammalian cell mutant. Biochemical Pharmacology, 59, 1211–1216. [DOI] [PubMed] [Google Scholar]
- Hojjati, M.R. , Li, Z. & Jiang, X.C. (2005) Serine palmitoyl‐CoA transferase (SPT) deficiency and sphingolipid levels in mice. Biochimica Et Biophysica Acta, 1737, 44–51. [DOI] [PubMed] [Google Scholar]
- Hornemann, T. , Penno, A. , Rutti, M.F. , Ernst, D. , Kivrak‐Pfiffner, F. , Rohrer, L. et al. (2009) The SPTLC3 subunit of serine palmitoyltransferase generates short chain sphingoid bases. Journal of Biological Chemistry, 284, 26322–26330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornemann, T. , Richard, S. , Rutti, M.F. , Wei, Y. & von Eckardstein, A. (2006) Cloning and initial characterization of a new subunit for mammalian serine‐palmitoyltransferase. Journal of Biological Chemistry, 281, 37275–37281. [DOI] [PubMed] [Google Scholar]
- Houlden, H. , King, R. , Blake, J. , Groves, M. , Love, S. , Woodward, C. et al. (2006) Clinical, pathological and genetic characterization of hereditary sensory and autonomic neuropathy type 1 (HSAN I). Brain, 129, 411–425. [DOI] [PubMed] [Google Scholar]
- Jordanova, A. , Irobi, J. , Thomas, F.P. , van Dijck, P. , Meerschaert, K. , Dewil, M. et al. (2006) Disrupted function and axonal distribution of mutant tyrosyl‐tRNA synthetase in dominant intermediate Charcot‐Marie‐Tooth neuropathy. Nature Genetics, 38, 197–202. [DOI] [PubMed] [Google Scholar]
- Jordanova, A. , Thomas, F.P. , Guergueltcheva, V. , Tournev, I. , Gondim, F.A. , Ishpekova, B. et al. (2003) Dominant intermediate Charcot‐Marie‐Tooth type C maps to chromosome 1p34‐p35. American Journal of Human Genetics, 73, 1423–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laura, M. , Pipis, M. , Rossor, A.M. & Reilly, M.M. (2019) Charcot‐Marie‐Tooth disease and related disorders: an evolving landscape. Current Opinion in Neurology, 32, 641–650. [DOI] [PubMed] [Google Scholar]
- Lee, S. , Panthi, S. , Jo, H.W. , Cho, J. , Kim, M.S. , Jeong, N.Y. et al. (2017) Anatomical distributional defects in mutant genes associated with dominant intermediate Charcot‐Marie‐Tooth disease type C in an adenovirus‐mediated mouse model. Neural Regeneration Research, 12, 486–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S. , Xie, T. , Liu, P. , Wang, L. & Gong, X. (2021) Structural insights into the assembly and substrate selectivity of human SPT‐ORMDL3 complex. Nature Structural and Molecular Biology, 28, 249–257. [DOI] [PubMed] [Google Scholar]
- Lone, M.A. , Hulsmeier, A.J. , Saied, E.M. , Karsai, G. , Arenz, C. , von Eckardstein, A. et al. (2020b) Subunit composition of the mammalian serine‐palmitoyltransferase defines the spectrum of straight and methyl‐branched long‐chain bases. Proceedings of the National Academy of Sciences of the United States of America, 117, 15591–15598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lone, M.A. , Hülsmeier, A.J. , Saied, E.M. , Karsai, G. , Arenz, C. , von Eckardstein, A. et al. (2020) Subunit composition of the mammalian serine‐palmitoyltransferase defines the spectrum of straight and methyl‐branched long‐chain bases. Proceedings of the National Academy of Sciences, 117(27), 15591–15598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lone, M.A. , Santos, T. , Alecu, I. , Silva, L.C. & Hornemann, T. (2019) 1‐Deoxysphingolipids. Biochimica Et Biophysica Acta ‐ Molecular and Cell Biology, 1864, 512–521. [DOI] [PubMed] [Google Scholar]
- Mohassel, P. , Donkervoort, S. , Lone, M.A. , Nalls, M. , Gable, K. , Gupta, S.D. et al. (2021) Childhood amyotrophic lateral sclerosis caused by excess sphingolipid synthesis. Nature Medicine, 27(7), 1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley, W.W. , Seburn, K.L. , Nawaz, M.H. , Miers, K.E. , Cheng, J. , Antonellis, A. et al. (2011) Charcot‐Marie‐Tooth‐linked mutant GARS is toxic to peripheral neurons independent of wild‐type GARS levels. PLoS Genetics, 7, e1002399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehues, S. , Bussmann, J. , Steffes, G. , Erdmann, I. , Kohrer, C. , Sun, L. et al. (2015) Impaired protein translation in Drosophila models for Charcot‐Marie‐Tooth neuropathy caused by mutant tRNA synthetases. Nature Communications, 6, 7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowaczyk, M.J. , Huang, L. , Tarnopolsky, M. , Schwartzentruber, J. , Majewski, J. , Bulman, D.E. et al. (2017) A novel multisystem disease associated with recessive mutations in the tyrosyl‐tRNA synthetase (YARS) gene. American Journal of Medical Genetics. Part A, 173, 126–134. [DOI] [PubMed] [Google Scholar]
- Pan, Y. , Kafaie, J. & Thomas, F.P. (2020) Longitudinal 16‐year study of dominant intermediate CMT type C neuropathy. Muscle and Nerve, 61, 111–115. [DOI] [PubMed] [Google Scholar]
- Penno, A. , Reilly, M.M. , Houlden, H. , Laura, M. , Rentsch, K. , Niederkofler, V. et al. (2010) Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. Journal of Biological Chemistry, 285, 11178–11187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafael, J.A. , Nitta, Y. , Peters, J. & Davies, K.E. (2000) Testing of SHIRPA, a mouse phenotypic assessment protocol, on Dmd(mdx) and Dmd(mdx3cv) dystrophin‐deficient mice. Mammalian Genome: Official Journal of the International Mammalian Genome Society, 11, 725–728. [DOI] [PubMed] [Google Scholar]
- Rotthier, A. , Auer‐Grumbach, M. , Janssens, K. , Baets, J. , Penno, A. , Almeida‐Souza, L. et al. (2010) Mutations in the SPTLC2 subunit of serine palmitoyltransferase cause hereditary sensory and autonomic neuropathy type I. American Journal of Human Genetics, 87, 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotthier, A. , Penno, A. , Rautenstrauss, B. , Auer‐Grumbach, M. , Stettner, G.M. , Asselbergh, B. et al. (2011) Characterization of two mutations in the SPTLC1 subunit of serine palmitoyltransferase associated with hereditary sensory and autonomic neuropathy type I. Human Mutation, 32, E2211–E2225. [DOI] [PubMed] [Google Scholar]
- Sandelius, A. , Zetterberg, H. , Blennow, K. , Adiutori, R. , Malaspina, A. , Laura, M. et al. (2018) Plasma neurofilament light chain concentration in the inherited peripheral neuropathies. Neurology, 90, e518–e524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saporta, M.A. & Shy, M.E. (2013) Inherited peripheral neuropathies. Neurologic Clinics, 31, 597–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer, S.S. , Xu, Y.T. , Messing, A. , Willecke, K. , Fischbeck, K.H. & Jeng, L.J. (2005) Transgenic expression of human connexin32 in myelinating Schwann cells prevents demyelination in connexin32‐null mice. Journal of Neuroscience, 25, 1550–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes, W.C. , Moss, J.E. , Hurtley, S.M. & Beddington, R.S. (1995) Capturing genes encoding membrane and secreted proteins important for mouse development. Proceedings of the National Academy of Sciences of the United States of America, 92, 6592–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding, E.L. , Hines, T.J. , Bais, P. , Tadenev, A.L.D. , Schneider, R. , Jewett, D. et al. (2021) The integrated stress response contributes to tRNA synthetase‐associated peripheral neuropathy. Science, 373, 1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storkebaum, E. , Leitao‐Goncalves, R. , Godenschwege, T. , Nangle, L. , Mejia, M. , Bosmans, I. et al. (2009) Dominant mutations in the tyrosyl‐tRNA synthetase gene recapitulate in Drosophila features of human Charcot‐Marie‐Tooth neuropathy. Proceedings of the National Academy of Sciences of the United States of America, 106, 11782–11787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryke, D. , Kawamoto, M. , Huang, C.C. , Johns, S.J. , King, L.A. , Harper, C.A. et al. (2003) BayGenomics: a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Research, 31, 278–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, C. & Schwarz, T.L. (2017) O‐GlcNAc transferase is essential for sensory neuron survival and maintenance. Journal of Neuroscience, 37, 2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadenev, A.L.D. & Burgess, R.W. (2019) Model validity for preclinical studies in precision medicine: precisely how precise do we need to be? Mammalian Genome, 30, 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman, V. , Strickland, A.V. & Zuchner, S. (2014) Genetics of Charcot‐Marie‐Tooth (CMT) disease within the frame of the human genome project success. Genes, 5, 13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracewska‐Siemiatkowska, A. , Haer‐Wigman, L. , Bosch, D.G.M. , Nickerson, D. , Bamshad, M.J. Van de Vorst, M. et al. (2017). An expanded multi‐organ disease phenotype associated with mutations in YARS. Genes, 8, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Niu, Y. , Zhang, Z. , Gable, K. , Gupta, S.D. , Somashekarappa, N. et al. (2021) Structural insights into the regulation of human serine palmitoyltransferase complexes. Nature Structural and Molecular Biology, 28, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, K.B. , Brigatti, K.W. , Puffenberger, E.G. , Gonzaga‐Jauregui, C. , Griffin, L.B. , Martinez, E.D. et al. (2019) Homozygosity for a mutation affecting the catalytic domain of tyrosyl‐tRNA synthetase (YARS) causes multisystem disease. Human Molecular Genetics, 28, 525–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhakupova, A. , Debeuf, N. , Krols, M. , Toussaint, W. , Vanhoutte, L. , Alecu, I. et al. (2016) ORMDL3 expression levels have no influence on the activity of serine palmitoyltransferase. The FASEB Journal, 30, 4289–4300. [DOI] [PubMed] [Google Scholar]
- Zitomer, N.C. , Mitchell, T. , Voss, K.A. , Bondy, G.S. , Pruett, S.T. , Garnier‐Amblard, E.C. et al. (2009) Ceramide synthase inhibition by fumonisin B1 causes accumulation of 1‐deoxy‐sphinganine: A novel category of bioactive 1‐deoxy‐sphingoid bases and 1‐deoxy‐dihydroceramides biosynthesized by mammalian cell lines and animals. Journal of Biological Chemistry, 284, 4786–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuko, A. , Mallik, M. , Thompson, R. , Spaulding, E.L. , Wienand, A.R. , Been, M. et al. (2021) tRNA overexpression rescues peripheral neuropathy caused by mutations in tRNA synthetase. Science, 373, 1161–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S5
Data Availability Statement
All data relevant to this publication are included in the manuscript of supplemental materials.