

1. NARRATIVE
1.1. Contextual background
As we age, our cognitive abilities deteriorate [1], without necessarily progressing to dementia. One of the earliest and most striking cognitive changes in the aging process is the alteration of memory. Episodic memory, our ability to remember recently acquired experiences gradually deteriorates from middle age to older age. Our ability to create and storage memories (encoding and storage) along with its retrieval [2] becomes less efficient, interfering with our daily activities.
Major research efforts have focused on trying to distinguish the memory decline attributable to normal aging from those that indicate pathological aging. Such studies shown that the effects of aging in our memory performance are very heterogeneous, with clear inter-individual vulnerabilities. Some people exhibit little change in their memory ability to extreme old age, while others experience a rapid and severe memory decline that might culminate in a clinical diagnosis of Alzheimer’s disease. Understanding the causal factors underlying over-time memory performance is increasingly important given the health care crisis of an aging world’s population. Psychological, health-related, environmental, education and genetics [3] factors have been reported as significant contributors to the variability observed in the trajectories of episodic performance across individuals.
Twin and family studies support the notion that episodic memory is under strong genetic influence in older persons in healthy and demented populations[4]. In recent years, different study designs and approaches have been used to genetically characterize episodic memory trajectories. The majority of the genetic studies on episodic memory have been cross-sectional either using genome-wide arrays [5–7] or candidate genes approaches [8–18]. Genetic studies based on longitudinal measures of episodic memory are few, and predominantly focused on candidate genes [19, 20]. Genome-wide association studies (GWAS) of cognitive abilities assessing the contribution of common variants [11, 17, 18, 21–29] have consistently reported modest genetic effects, partly due to limited sample sizes that compromise the statistical power to identify loci at a genome-wide significance level. As reported for other complex phenotypes [30, 31], such as autoimmune and cardiovascular diseases, genomic analysis including rare variants might reveal its unique roles in cognitive genetics.
In the present study, we integrated common and rare genetic variants and transcriptomics data for the identification of novel episodic memory loci.
1.2. Study design and main results
To guarantee a better understanding of the impacts of ageing, cohort differences and period effects in the trajectories of memory performance, we considered a longitudinal study design.
The identification of genetic risk/protective factors underlying memory function are commonly based on cross-sectional data and genetic studies based on longitudinal data are less frequently implemented. Contrary to cross-sectional designs in which a temporal sequence cannot be established, longitudinal methods are uniquely able to capture genetic variation associated with the rate of cognitive decline [32], allowing the separation of population trends (fixed effects) and individual differences about the trends (random effects). The availability of longitudinal measures of memory performance allow us to expand genetic analyses beyond the dichotomous case-control phenotype, typically resulting in loss of measurement information as well as effect size and statistical power.
To study trajectories of memory performance in elderly cohorts, we have used a previously described latent curve models approach (LCM) [33]. The resulting slopes of repeated measures of memory are used as quantitative phenotype for genetic analyses [32].
Since GWAS common variants explain a modest fraction of the genetic variance of cognitive abilities [25], low-frequency and rare genetic variants have been proposed as responsible for the uncharacterized genetic risk underlying cognitive traits [30]. A cost-efficient approach to characterize the contribution of rare variants to memory function is their genotype imputation, that is, statistically inference of untyped rare variants’ genotypes based on a reference panel of whole genome sequenced individuals [34]. The publically available Haplotype Reference Panel (HRC) reference panel contains over 39 million SNPs from 27,165 individuals, and reported high performance and accuracy for imputation for admixed populations such as African-Americans [35] and Caribbean Hispanics [36].
In addition to the traditional SNP-based approaches [37], we have also considered gene-based GWAS association tests. Gene-based analyses increases the statistical power of discovery by i) aggregating the disparate signals from multiple independent causal variants within the gene and ii) by reducing the multiple testing burden (~1,000,000 million SNPs versus ~20,000 genes). Moreover, since the impact of genetic heterogeneity due to underlying linkage disequilibrium patterns (different SNPs being linked to the causal variants) is reduced when considering the gene as the unit of analysis, it can alleviate limitations in replication leading to more consistent results [38].
In an attempt to improve our understanding of the genetic architecture of memory function, our study has included participants from ethnically diverse populations: Caribbean Hispanics and African-Americans. A disproportionate majority of participants in cognitive genetics research are of European descent. However, it is well established that the effect of genetic variants vary between populations based on the reported differences in the genetic architecture of populations [39]. Moreover, low-frequency and rare variants tend to be ethnic specific (i.e. exhibit little sharing among diverged populations) and enriched in admixed populations[40]. The inclusion of multi-ancestry cohorts in genetics studies are needed to fully characterize human genomic variation, bolster our understanding of disease etiology, and ensure that genetic testing is broadly accessible.
Results from APOE-stratified GWAS analyses and brain transcriptomics identified Doublecortin Domain Containing 2 gene (DCDC2) as a novel predictor of memory maintenance among non-carriers of APOE-ε4. DCDC2 brain expression appeared associated with episodic memory maintenance and lower burden of pathological Alzheimer’s hallmarks. Moreover, when AD cases were compared to cognitively healthy participants, DCDC2 expression was decreased across all brain areas.
1.3. Study conclusions, disease implications, and therapeutic opportunities
Our multi-omics data integrative approach using meta-analysis results from eight independent GWAS of episodic memory trajectories and brain transcriptomics for three independent cohorts identified DCDC2 as a putative gene for protection against episodic memory decline and a potential to reduce risk of dementia.
To our knowledge, this is the first study reporting DCDC2 association with longitudinal changes in episodic memory performance. Interestingly, the DCDC2 gene was previously reported as genome-wide significantly associated with general cognitive function (p < 5 × 10−8) in a sample of more than 300,000 subjects from three different European cohorts including UKBB [25].
The DCX domain-containing protein 2 (DCDC2) gene is one of the most conserved genes of the doublecortin (DCX) superfamily, a group of proteins that regulate filamentous actin structure in developing neurons. DCDC2 binds to tubulin and enhances microtubule polymerization [41, 42] influencing synaptic plasticity [43]. It is well documented that cytoskeleton dynamics in the adult brain affect fundamental processes, such as memory and learning, which are often compromised in neuro degenerative diseases [44, 45]. In fact, genetically modified mice studies showed that DCDC2 mutations resulted in persistent memory impairments [46, 47]. Multiple epidemiological genetic studies linked variants within DCDC2 gene to reading abilities including dyslexia [48–55]. A recent re-evaluation suggested that evidence in support of the DCDC2 deletion as a risk factor for dyslexia was statistically weak [56]. Our results in the Non-Hispanic White sample of the WHICAP cohort did not find significant association between DCDC2 and language trajectories.
Reinforcing its role in brain development, DCDC2 has also been found to interact with ciliary proteins. Ciliary proteins play an important role in neurogenesis, neuronal migration, and underlie a growing list of human disorders including developmental delays and cognitive deficits. Protein–protein interaction network analysis[57] revealed a link between cilia function, neuronal function, and neurological disorders such as Alzheimer’s disease. These results provide a novel therapeutic avenue in which drugs targeting proteins in the cilia interactome might be repurposed for treating neurological disorders.
The inverse association between brain expression levels and lower amyloid and tau pathology may selectively upregulate DCDC2 expression in the dorsolateral prefrontal cortex, conferring protection against Alzheimer’s pathology. Follow-up studies are needed to determine whether reserve mechanisms (brain reserve [58, 59], cognitive reserve [58, 59] and brain maintenance [59, 60]) might act as moderators.
Our results found differential brain expression of DCDC2 when AD cases and cognitively healthy participants were compared. Specifically, gene expression in AD cases appeared nominally downregulated for two brain areas, superior temporal gyrus (temporal lobe), and inferior frontal gyrus (prefrontal cortex). Future studies incorporating neuroimaging data will be needed to validate these results and gain a better understanding of its neuroanatomical correlates.
The identification of DCDC2 gene as a predictor of memory maintenance in older adulthood provides the possibility of identifying population subgroups at-risk of memory decline and dementia, paving the way for precision medicine intervention [32, 61–63]. Compared to the universal “one-size-fits-all” approach (generalized prevention strategies for all individuals), a precision medicine approach offers the opportunity to personalize interventions that hold the promise of advancing memory decline prevention strategies [64]. To be used as a diagnostic system and more efficient treatment of age-related memory impairment it will require i) to define groups of individuals for whom a cognitive intervention is warranted and ii) to develop and test novel treatments and interventions that can be applied with a degree of specificity to distinct subpopulations of individuals[65]. Finally, it is important to consider that relying solely on genetics may miss unknown underlying memory decline mechanisms. In addition to genetics, a precision medicine approach should also encompass recommendations to target lifestyle factors and medical comorbidities on an individual basis.
1.4. Limitations, unanswered questions, and future directions
Our study has some limitations. First, trajectories of episodic memory were modelled as a linear function of time, hence we did not consider potential nonlinear age effects. Second, we did not consider the contribution of additional protective or/and risk factors, socio-economic status, mental or behavioral health, and clinical comorbid conditions that may be associated with maintenance/decline of memory. Third, potential interactions between genetic variants and these risk/resilience additional factors may also contribute to set courses toward memory progression over time. Fourth, we cannot rule out the possibility that additional regulatory mechanisms might regulate DCDC2 expression variation.
Future translational studies will investigate the role of DCDC2 variants in cytoskeleton dynamics via generation of CRISPR-pluripotent cellular models expressing different variants of DCDC2 gene and differentiated into neurons (cortical or hippocampal). Cytoskeleton structure and organelle distribution can be assessed by confocal imaging using these cell models. Furthermore, expression of proteins involved in posttranslational modifications of microtubules, such as acetylation can be also investigated by western blot and qPCR analysis.
2. Consolidated description of methods and results
Using latent class models, we have estimated episodic memory trajectories in 35,245 ethnically diverse older adults representing eight independent cohorts. We conducted APOE-stratified GWAS analyses and combined individual cohorts ‘results via meta-analysis. Three independent transcriptomics datasets were used to further interpret GWAS signals.
We identified DCDC2 gene significantly associated with episodic memory (Pmeta=3.3 × 10−8) among non-carriers of APOE-ε4. Brain transcriptomics revealed an association between episodic memory maintenance and i) increased dorsolateral prefrontal cortex DCDC2 expression (p=3.8 × 10−4) and ii) lower burden of pathological Alzheimer’s hallmarks (PHF-tau p=0.003, and amyloid-beta load p=0.008). Additional transcriptomics results comparing Alzheimer’s disease and cognitively healthy brain samples showed a downregulation of DCDC2 levels in superior temporal gyrus (p=0.007) and inferior frontal gyrus (p=0.013).
3. Complete methods and results
3.1. Methods
Study Cohorts.
All study participants provided written informed consent and the study procedures were approved by the Institutional Review Boards within each of the corresponding institutions. All study procedures were performed in accordance with the World Medical Association Declaration of Helsinki ethical principles for medical research.
The present study includes eight independent study cohorts: 1) The Alzheimer’s Disease Genetics Consortium and National Alzheimer’s Coordinating Center (ADGC_NACC), 2) The National Institute on Aging Late-Onset Alzheimer Disease Family Based Study (NIA-LOAD), 3) The Chicago Health and Aging Project (CHAP), 4) The Religious Orders Study and Rush Memory and Aging Project (ROSMAP), 5) The Washington Heights-Inwood Columbia Aging Project (WHICAP), 6) The Long Life Family Study (LLFS), 7) The Alzheimer’s Disease Neuroimaging Initiative (ADNI) and the 8) United Kingdom Biobank (UKBB). Detailed characteristics and methodologies for study cohorts can be found elsewhere [33, 66–68].
Within each of the study cohorts, inclusion criteria for participants were based on the availability of longitudinal episodic memory scores (minimum of two visits to a maximum of 15), socio-demographic variables (sex, age, education and ethnic background), and imputed GWAS genotyped data using the Haplotype Reference Consortium (HRC v1.1).
An overview of the study design is summarized in Supplemental Figure 1.
Episodic memory.
In the WHICAP cohort, episodic memory was derived as the average of standardized measures for total immediate recall, delayed recall, and delayed recognition of the Selective Reminding Tests [69]. In the ADNI cohort, the Rey Auditory Verbal Learning Test (RAVLT) [27, 70] served as a measure of episodic memory. In the UKBB, as previously described [23], participants’ scores on the pairs matching test can be used as a measure of episodic visual memory. As previously described [33], in the rest of cohorts, episodic memory was quantified as the average of the standardized Wechsler Memory Scale tests.
Alzheimer’s disease.
In all study cohorts, except for LLFS and UKBB, participants were classified as dementia patients or non-cognitively impaired (NCI) participants using NINCDS-ADRDA) criteria [71]. In the LLFS cohort, dementia status was categorized based on a previously described diagnostic algorithm [72]. In the UKBB cohort, cognitive impairment was defined using a 1.5-SD cut-off below demographically adjusted episodic memory scores (age, education, and sex). UKBB study participants were classified as non-cognitive impaired (NCI) if their standardized adjusted memory scores were greater than 1.5 SD below the mean.
Statistical Analysis
Statistical analyses were performed using a dataset freeze from 2019 for which complete and accurate phenotypic and genomic information was available.
Episodic memory trajectories (EMTs).
As previously described [33], episodic memory trajectories were derived using Latent Class Mixed Models (LCMM). The LCMM estimated episodic memory slope was used as quantitative outcome.
Genome-wide genotype (GWAS) imputation.
Genome-wide genotyped data was imputed using the Haplotype Reference Consortium panel (HRC v1.1) through the Michigan Imputation online server [73].
Quality control metrics.
Samples were excluded for analyses purposes based on: cryptic relatedness (duplicates or first degree relatives) calculated as identity by descent estimates using PLINK [74] software, and genotype call missing rate greater than 10%. Only variants with high imputation quality (r2≥ 0.8) were retained for analyses purposes.
Population substructure.
To account for population stratification, principal component analysis was conducted using PLINK software [74] and the top three principal components were retained as covariates in regression models.
Gene-based association analyses.
Gene-based annotations were generated using ANNOVAR software [75] and were limited to intronic, exonic, 3’ and 5’ untranslated regions variants. Analyses were conducted only for genes with at least 10 annotated variants. Gene-based test were run using the optimal single-nucleotide polymorphism–set Sequence Kernel Association Test (SKAT-O) as implemented in EPACTS [76]. Covariates in the linear regression models included sex, age at last evaluation, education and the top three principal components. For LLFS cohort, further covariates adjustment included kinship correlation matrix. All analyses were conducted independently in three different APOE strata: no APOE stratification, APOE-ε4 carriers vs. non-carriers. Gene-level significance was established as p ≤ 2.7 × 10−6 after Bonferroni’s correction for multiple testing (an average of 20,000 genes annotated across all cohorts).
SNP-based and gene-based meta-analysis.
Meta-analysis of the gene-based and SNP-based association results was carried out using inverse variance–weighted model based on p-values/sample size and metrics to measure between-study heterogeneity (Cochran’s Q-test)[77] as implemented in METAL software [78]. Using Bonferroni for multiple testing correction, a conservative threshold for significance was set as p ≤ 2.5×10−6 and p≤ 1.6 × 10−4 for gene-based and SNP-based respectively).
DCDC2 SNP-based analyses in APOE-ε4 non-carriers.
Variants in DCDC2 gene were individually tested for its association with episodic memory using EPACTS software. Sex, age at last evaluation, education, principal components, and kinship matrix (only for the LLFS cohort) were included as covariates in the model. SNP-level significance was established as p ≤ 1.5 × 10−5 after Bonferroni’s correction for multiple testing based on the total number of SNPs tested in the meta-analysis.
SNP-based APOE interaction analyses.
The regression-based approach implemented in the epistasis module of PLINK [74] was used to run test pair-wise interactions between the strongest DCDC2 associated variant in the SNP-based meta-analysis (rs1340698) and APOE genotype, carriers and non-carries of APOE-ε4.
Brain transcriptomic analyses.
RNA sequencing data processed in the present study can be accessed on the Accelerating Medicines Partnership- Alzheimer’s Disease (AMP-AD) Synapse knowledge portal (https://www.synapse.org). The AMP-AD is a public-private partnership focused on the development of new drug targets to prevent or treat Alzheimer’s disease. The threshold for nominal significance was defined as P-values ≤0.05.
Brain transcriptomic analysis Religious Orders Study and Rush Memory and Aging Project (ROSMAP) study.
RNA sequencing (RNA-seq) data generated by ROSMAP [79–82] consisted of post-mortem dorsolateral prefrontal cortex (DLPFC) brain tissue from 624 participants (254 syndromic Alzheimer’s disease, 169 mild cognitive impairment and 201 no cognitive impairment).
Brain transcriptomic analysis in The Mount Sinai Brain Bank (MSBB) study.
The MSBB analyses included a total of 476 samples collected from four different brain areas: parahippocampal gyrus (PHG), inferior frontal gyrus (IFG), superior temporal gyrus (STG) and the frontal pole FP (n=476). Detailed specific sample characteristics and methodological pipeline can be found elsewhere [83].
Brain transcriptomic analysis in the Mayo clinic dataset.
The analyses of the Mayo RNA-seq dataset included samples harvested from temporal cortex and cerebellum. Detailed specific sample characteristics and methodological pipeline can be found elsewhere [84].
Summary data-based Mendelian Randomization (SMR).
We used a Mendelian Randomization approach to investigate whether DCDC2 variants associated with episodic memory performance could act through DCDC2 gene expression levels in brain. eQTLs analyses were performed using SMR software [85]. Because of the lack of publically available episodic memory GWAS summary statistics, we relied on SNP-based association results from the largest cohort in our study, UKBB cohort (DCDC2_noE4 strata, n= 14,874). Reference eQTL data were obtained from the Brain-eMeta dataset, which includes brain tissue eQTL data from GTEx v6, the CommonMind Consortium (CMC), ROS/MAP, and the Brain eQTL Almanac project (Braineac). The linkage disequilibrium (LD) estimation was based on the entire UKBB sample (n= 20,184). Software and reference database details can be accessed at https://cnsgenomics.com/software/smr/#SMR&HEIDIanalysis.
DCDC2 patterns of linkage disequilibrium (LD).
We investigated the linkage disequilibrium pattern between most significant associated SNPs in the Mendelian randomization analyses (topSMR) and DCDC2 topSNPs in the GWAS meta-analysis (noE4 SNP-based association strata). All LD analyses were performed using NIH web-based application LDlink (LD matrix module) (https://ldlink.nci.nih.gov/?tab=home) (Myers, 2020).
DCDC2 and APOE interaction.
Gene-gene interaction was tested using epistasis module of PLINK [74].
3.2. Results
The characteristics of the participants within each are summarized in Table 1. A higher percentage of women was observed across all cohorts. The average age (at baseline and at last evaluations) and education of the participants were 72 ± 8, 78 ± 8 and 14 ± 3, respectively. The majority of the participants across cohorts were non-carriers of the APOE-ε4 allele, and as expected, lower frequency of dementia when compared to APOE-ε4 carriers.
Table 1.
Characteristics of the study participants by cohort.
| N | women | ageBA | ageLE | educ | EMTStables | EMTDecliners | demBA | non-demBA | APOE_ε4 | APOE_nonε4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | % | n | % | n | % | n | % | n | % | n | % | n | % | |||||
| ADNI | 1,090 | 634 | 58 | 74±7 | 79±8 | 16±3 | 380 | 35 | 710 | 65 | 322 | 30 | 768 | 70 | 501 | 46 | 589 | 54 |
| CHAP | 696 | 431 | 62 | 72±5 | 82±6 | 15±3 | 362 | 52 | 334 | 48 | 10 | 1 | 686 | 99 | 165 | 24 | 531 | 76 |
| LLFS | 1,874 | 1,040 | 55 | 64±11 | 71±11 | 12±3 | 1,047 | 56 | 827 | 44 | 131 | 7 | 1,743 | 93 | 400 | 21 | 1,474 | 79 |
| NACC_ADGC | 6,774 | 3,845 | 57 | 74±9 | 78±9 | 16±3 | 4,014 | 59 | 2,760 | 41 | 3,016 | 44 | 3,758 | 55 | 2,731 | 40 | 4,043 | 60 |
| NIA-LOAD | 460 | 298 | 65 | 73±9 | 77±8 | 16±3 | 253 | 55 | 207 | 45 | 31 | 7 | 429 | 93 | 152 | 34 | 308 | 64 |
| ROSMAP | 1,265 | 883 | 70 | 79±8 | 87±7 | 16±4 | 651 | 51 | 614 | 49 | 952 | 75 | 313 | 25 | 317 | 25 | 948 | 75 |
| UKBB | 20,184 | 10,322 | 51 | 55±8 | 63±7 | 91% | 17,451 | 86 | 2,733 | 14 | 1,390 | 7 | 18,794 | 93 | 5,310 | 26 | 14,874 | 74 |
| WNHW | 619 | 370 | 60 | 76±7 | 80±8 | 13±4 | 597 | 93 | 22 | 7 | 45 | 7 | 574 | 93 | 121 | 19 | 498 | 81 |
| WAA | 736 | 532 | 72 | 75±6 | 79±7 | 12±4 | 712 | 97 | 24 | 3 | 37 | 5 | 699 | 95 | 244 | 33 | 492 | 67 |
| WCH | 1,547 | 1,093 | 71 | 76±6 | 81±7 | 7±4 | 972 | 61 | 614 | 39 | 561 | 35 | 1,025 | 65 | 402 | 25 | 1,184 | 75 |
EMTs: Episodic Memory Trajectories; ageBA: age at baseline evaluation; ageLE: age at last evaluation; demBA: dementia status at baseline evaluation; non-demBA: non-dementia status at baseline evaluation; WNHW: WHICAP Non-Hispanic Whites; WAA: WHICAP African-Americans; WCH: WHICAP Caribbean-Hispanics
Episodic memory trajectories.
Within study cohorts’ trajectories of episodic memory are shown in Supplemental Figure 2. Consistent with previous literature, the majority of the participants were aggregated into the EMTStables cluster (individuals exhibiting sustained or improved memory function over time). LCMM plots could not be generated for the LLFS cohort because, as described in the methods section, a different regression framework was used.
Meta-analysis of genomewide gene-based test of association.
The quantile-quantile plots for the gene-based association results within each of the cohorts stratify by APOE status are shown in Supplemental Figures 3–5. The average’s statistics for SNP allele frequencies (minimum, maximum, average and standard deviation) stratify by study’s cohort are shown in Supplemental Table 1. In the non-APOE stratified sample, the meta-analysis results (Table 2) revealed the doublecortin domain-containing family member (DCDC2) gene as the strongest association signal (Pmeta= 3.7 × 10−7). More interestingly, the DCDC2-EM association was significant stronger among non-APOE-ε4 study participants (Pmeta=3.3 × 10−8). Additional potential novel loci were observed in both APOE strata, however, none of the associations reached the same significance level as DCDC2. Secondary analyses excluding the UKBB cohort (Supplemental Table 2) corroborated that associations reported (Table 2) were not solely driven by the largest cohort in the study.
Table 2.
Top significant genes (p≤10−6) in the genome-wide gene-based meta-analysis stratify by APOE status.
| Meta-analysis | ADNI | CHAP | LLFS | NACC | NIA-LOAD | ROSMAP | UKBB | WNHW | WAA | WCH | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr_Gene | N | Pmeta | PHet | N | P | N | P | N | P | N | P | N | P | N | P | N | P | N | P | N | P | N | P | |
| noAPOE | 6_DCDC2 | 35,250 | 3.3E-07 | 0.204 | 1,090 | 0.377 | 696 | 0.741 | 1,874 | 0.030 | 6,774 | 0.473 | 460 | 0.536 | 1,265 | 0.006 | 20,184 | 6.7E-04 | 619 | 0.953 | 736 | 0.732 | 1,547 | 0.002 |
| 16_FBXL19 | 35,245 | 4.5E-06 | 0.208 | 1,090 | 0.384 | 696 | 0.278 | 1,874 | 0.667 | 6,774 | 0.048 | 460 | 0.029 | 1,265 | 0.093 | 20,184 | 0.018 | 619 | 0.280 | 736 | 0.506 | 1,547 | 3.5E-04 | |
| 15_ICE2 | 35,245 | 2.8E-06 | 0.262 | 1,090 | 0.780 | 696 | 0.294 | 1,874 | 0.854 | 6,774 | 0.006 | 460 | 0.703 | 1,265 | 0.016 | 20,184 | 0.006 | 619 | 0.318 | 736 | 0.001 | 1,547 | 0.434 | |
| 17_KRT37 | 35,245 | 9.0E-06 | 0.445 | 1,090 | 0.034 | 696 | 0.066 | 1,874 | 0.550 | 6,774 | 0.605 | 460 | 0.298 | 1,265 | 0.352 | 20,184 | 8.5E-04 | 619 | 1.000 | 736 | 0.567 | 1,547 | 0.275 | |
| 16_MTHFSD | 35,245 | 8.3E-06 | 0.769 | 1,090 | 0.504 | 696 | 0.188 | 1,874 | 0.299 | 6,774 | 0.850 | 460 | 0.308 | 1,265 | 0.430 | 20,184 | 1.7E-04 | 619 | 0.417 | 736 | 0.115 | 1,547 | 0.085 | |
| 16_NPRL3 | 35,245 | 9.5E-06 | 0.642 | 1,090 | 0.323 | 696 | 0.787 | 1,874 | 0.334 | 6,774 | 0.000 | 460 | 0.140 | 1,265 | 0.207 | 20,184 | 0.041 | 619 | 0.805 | 736 | 0.219 | 1,547 | 0.659 | |
| 11_OR4C45 | 35,245 | 6.6E-06 | 0.834 | 1,090 | 0.735 | 696 | 0.066 | 1,874 | 0.638 | 6,774 | 0.005 | 460 | 0.495 | 1,265 | 0.069 | 20,184 | 0.008 | 619 | 0.933 | 736 | 0.588 | 1,547 | 0.476 | |
| APOE_ε4 | 6_AKAP12 | 10,333 | 2.8E-06 | 0.815 | 502 | 0.003 | 165 | 0.445 | 400 | 0.110 | 2731 | 0.059 | 152 | 1.000 | 316 | 0.753 | 5,310 | 0.001 | 121 | 1.000 | 241 | 0.486 | 395 | 0.436 |
| 16_ANXA11 | 10,332 | 6.1E-06 | 0.421 | 501 | 0.043 | 165 | 0.548 | 400 | 0.589 | 2731 | 0.140 | 152 | 0.076 | 316 | 0.106 | 5,310 | 0.007 | 121 | 0.091 | 241 | 0.894 | 395 | 0.009 | |
| 15_FIBP | 10,332 | 8.6E-06 | 0.962 | 501 | 0.844 | 165 | 0.273 | 400 | 0.336 | 2731 | 0.122 | 152 | 0.326 | 316 | 0.077 | 5,310 | 0.000 | 121 | 0.994 | 241 | 0.336 | 395 | 0.374 | |
| 17_KBTBD12 | 10,332 | 5.6E-06 | 0.809 | 501 | 0.009 | 165 | 0.751 | 400 | 0.379 | 2731 | 0.003 | 152 | 0.561 | 316 | 0.793 | 5,310 | 0.019 | 121 | 0.740 | 241 | 0.859 | 395 | 0.077 | |
| 16_KIT | 10,332 | 2.3E-06 | 0.952 | 501 | 0.454 | 165 | 0.037 | 400 | 0.160 | 2731 | 0.046 | 152 | 0.459 | 316 | 0.481 | 5,310 | 0.001 | 121 | 0.355 | 241 | 0.194 | 395 | 0.378 | |
| 16_L3MBTL3 | 10,333 | 2.9E-06 | 0.946 | 502 | 0.193 | 165 | 0.432 | 400 | 0.179 | 2731 | 0.001 | 152 | 0.697 | 316 | 0.051 | 5,310 | 0.018 | 121 | 0.671 | 241 | 0.737 | 395 | 0.334 | |
| 11_MERTK | 10,332 | 3.9E-06 | 0.304 | 501 | 0.002 | 165 | 0.062 | 400 | 0.024 | 2731 | 0.246 | 152 | 0.138 | 316 | 0.293 | 5,310 | 0.006 | 121 | 0.347 | 241 | 0.584 | 395 | 0.392 | |
| 6_PADI4 | 10,332 | 9.6E-06 | 0.407 | 501 | 0.291 | 165 | 0.412 | 400 | 0.509 | 2731 | 0.026 | 152 | 0.158 | 316 | 0.042 | 5,310 | 0.050 | 121 | 0.591 | 241 | 0.011 | 395 | 0.024 | |
| 10_SUCLG1 | 10,332 | 6.5E-06 | 0.103 | 501 | 0.253 | 165 | 0.131 | 400 | 0.492 | 2731 | 1.000 | 152 | 0.249 | 316 | 0.370 | 5,310 | 3.0E-05 | 121 | 1.000 | 241 | 0.001 | 395 | 0.472 | |
| APOE_noε4 | 6_DCDC2 | 24,913 | 3.4E-08 | 0.284 | 593 | 0.087 | 531 | 0.351 | 1474 | 0.038 | 4043 | 0.307 | 308 | 0.400 | 948 | 0.010 | 14,874 | 5.3E-04 | 498 | 0.003 | 492 | 0.181 | 1152 | 0.132 |
| 16_MTHFSD | 24,909 | 7.8E-07 | 0.560 | 589 | 0.100 | 531 | 0.212 | 1474 | 0.046 | 4043 | 1.000 | 308 | 0.112 | 948 | 0.778 | 14,874 | 4.1E-05 | 498 | 0.248 | 492 | 0.330 | 1152 | 0.140 | |
| 15_ARSK | 24,909 | 2.5E-06 | 0.937 | 589 | 0.849 | 531 | 1.000 | 1474 | 0.270 | 4043 | 0.002 | 308 | 0.881 | 948 | 0.824 | 14,874 | 2.0E-04 | 498 | 0.897 | 492 | 0.261 | 1152 | 0.705 | |
| 17_RALGDS | 24,909 | 3.9E-06 | 0.986 | 589 | 0.415 | 531 | 0.286 | 1474 | 0.578 | 4043 | 0.061 | 308 | 0.816 | 948 | 0.629 | 14,874 | 3.1E-04 | 498 | 0.062 | 492 | 0.443 | 1152 | 0.444 | |
| 16_CYP2W1 | 24,909 | 6.9E-06 | 0.955 | 589 | 0.437 | 531 | 1.000 | 1474 | 0.087 | 4043 | 0.273 | 308 | 0.566 | 948 | 0.607 | 14,874 | 9.5E-05 | 498 | 0.245 | 492 | 0.216 | 1152 | 1.000 | |
| 16_TTC37 | 24,909 | 5.7E-07 | 0.956 | 589 | 0.676 | 531 | 0.727 | 1474 | 0.333 | 4043 | 1.000 | 308 | 0.169 | 948 | 0.780 | 14,874 | 1.6E-04 | 498 | 0.289 | 492 | 0.002 | 1152 | 0.669 | |
| 11_DHX36 | 24,909 | 8.4E-06 | 0.138 | 589 | 0.025 | 531 | 0.002 | 1474 | 0.141 | 4043 | 0.004 | 308 | 0.916 | 948 | 0.064 | 14,874 | 0.031 | 498 | 0.921 | 492 | 0.884 | 1152 | 0.800 | |
| 6_CASP3 | 24,909 | 9.2E-06 | 0.041 | 589 | 0.191 | 531 | 0.032 | 1474 | 0.009 | 4043 | 0.022 | 308 | 0.027 | 948 | 0.204 | 14,874 | 0.153 | 498 | 0.007 | 492 | 0.931 | 1152 | 0.085 | |
Meta-analysis of DCDC2 single-SNP association in the non-carriers of the APOE-ε4.
A total 1,144 variants in DCDC2 appeared to be present in all study cohorts. The results from the SNP-based meta-analysis are summarized in Table 3, and study’s regional association plots are shown in Figure 1. The strongest SNP-based association corresponded to intronic common SNP rs1340698 (Pmeta=1.3 × 10−7). As seen in Supplemental Figure 6, the strong regional LD block (r2 ≥0.6) included the top-associated SNP rs1340698. The top SNP is located in the vicinity of a weak neuronal enhancer that connects to one of the two DCDC2 promoters. However, nor the SNP or the LD block yielded significant eQTL effects in standard datasets (GTEx, GRASP).
Table 3.
Top significant SNPs (p≤10–7) in the meta-analysis of DCDC2 no-APOE _E4 strata
| common variants | rare+ultra-rare variants | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs | rs1340698 | rs114941574 | rs112854846 | rs6933291 | rs73394040 | rs73394022 | rs114540201 | rs75359737 | rs147661578 | rs116394689 | rs807711 | rs73727536 | rs150137064 | rs73727537 | |
| bp | 24256726 | 24310169 | 24236194 | 24346573 | 24245058 | 24225761 | 24315294 | 24350002 | 24318524 | 24335865 | 24294560 | 24357033 | 24191780 | 24366667 | |
| A1/A2 | A/G | T/C | A/G | C/G | A/G | A/C | A/G | A/G | A/C | A/G | T/C | C/G | A/G | T/C | |
| Metal | N | 24890 | 24897 | 24902 | 24856 | 24898 | 24903 | 24873 | 24829 | 24873 | 24877 | 24900 | 24883 | 24834 | 24883 |
| Pmeta | 1.3E-07 | 1.8E-07 | 1.9E-07 | 1.9E-07 | 2.2E-07 | 2.3E-07 | 2.4E-07 | 2.6E-07 | 0.002 | 0.004 | 0.012 | 0.017 | 0.032 | 0.041 | |
| Phet | 0.348 | 0.631 | 0.618 | 0.255 | 0.610 | 0.636 | 0.607 | 0.287 | 0.512 | 0.082 | 0.183 | 0.221 | 0.093 | 0.157 | |
| ADNI n=589 | MAF | 0.03 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.0084 | 0.0059 | 0.0051 | 0.0076 | 0.0067 | 0.0067 |
| B | −0.04 | 0.03 | 0.03 | 0.06 | 0.03 | 0.03 | 0.03 | 0.06 | 0.02 | 0.09 | 0.06 | 0.08 | −0.07 | 0.08 | |
| SE | 0.02 | 0.03 | 0.02 | 0.03 | 0.02 | 0.02 | 0.03 | 0.03 | 0.04 | 0.05 | 0.05 | 0.04 | 0.05 | 0.05 | |
| P | 0.111 | 0.358 | 0.253 | 0.029 | 0.253 | 0.253 | 0.358 | 0.029 | 0.675 | 0.073 | 0.222 | 0.074 | 0.140 | 0.070 | |
| CHAP n=531 | MAF | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.0066 | 0.0028 | 0.0028 | 0.0047 | 0.0066 | 0.0047 |
| B | 0.02 | 0.02 | 0.02 | 0.03 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | −0.02 | 0.03 | −0.02 | 0.00 | −0.02 | |
| SE | 0.01 | 0.02 | 0.01 | 0.01 | 0.01 | 0.01 | 0.02 | 0.01 | 0.02 | 0.04 | 0.04 | 0.03 | 0.02 | 0.03 | |
| P | 0.157 | 0.155 | 0.157 | 0.062 | 0.157 | 0.157 | 0.155 | 0.073 | 0.347 | 0.585 | 0.415 | 0.515 | 0.975 | 0.515 | |
| LLFS n=1,474 | MAF | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.0054 | 0.0017 | 0.0041 | 0.0024 | 0.0126 | 0.0024 |
| B | 0.00 | −0.01 | −0.01 | −0.01 | −0.01 | −0.01 | −0.01 | −0.01 | −0.01 | 0.09 | 0.03 | 0.05 | −0.03 | 0.05 | |
| SE | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.03 | 0.05 | 0.03 | 0.04 | 0.02 | 0.04 | |
| P | 0.788 | 0.498 | 0.676 | 0.628 | 0.676 | 0.676 | 0.498 | 0.691 | 0.679 | 0.094 | 0.458 | 0.286 | 0.179 | 0.286 | |
| NACC n=4,043 | MAF | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.0075 | 0.0043 | 0.0019 | 0.0047 | 0.0119 | 0.0046 |
| B | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | −0.02 | 0.00 | −0.02 | 0.02 | −0.01 | 0.01 | |
| SE | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.02 | 0.02 | 0.03 | 0.02 | 0.01 | 0.02 | |
| P | 0.364 | 0.224 | 0.278 | 0.241 | 0.278 | 0.278 | 0.244 | 0.183 | 0.178 | 0.845 | 0.474 | 0.458 | 0.512 | 0.526 | |
| NIA-LOAD n=308 | MAF | 0.02 | 0.02 | 0.02 | 0.03 | 0.02 | 0.02 | 0.02 | 0.03 | 0.0065 | 0.0065 | NA | 0.0065 | 0.0097 | 0.0065 |
| B | 0.01 | 0.01 | 0.02 | −0.01 | 0.01 | 0.02 | 0.01 | −0.01 | −0.12 | 0.05 | NA | 0.05 | 0.04 | 0.05 | |
| SE | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.06 | 0.06 | NA | 0.06 | 0.05 | 0.06 | |
| P | 0.775 | 0.673 | 0.540 | 0.782 | 0.673 | 0.540 | 0.673 | 0.774 | 0.038 | 0.370 | NA | 0.370 | 0.386 | 0.370 | |
| ROSMAP n=948 | MAF | 0.02 | 0.02 | 0.02 | 0.03 | 0.02 | 0.02 | 0.02 | 0.03 | 0.0053 | 0.0042 | 0.0016 | 0.0042 | 0.0127 | 0.0042 |
| B | −0.03 | −0.02 | −0.02 | −0.02 | −0.02 | −0.02 | −0.02 | −0.02 | 0.01 | 0.06 | −0.09 | 0.07 | −0.04 | 0.07 | |
| SE | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.03 | 0.03 | 0.05 | 0.03 | 0.02 | 0.03 | |
| P | 0.018 | 0.129 | 0.066 | 0.048 | 0.066 | 0.066 | 0.129 | 0.055 | 0.626 | 0.059 | 0.087 | 0.031 | 0.045 | 0.031 | |
| UKBB n=14,857 | MAF | 0.03 | 0.02 | 0.02 | 0.03 | 0.02 | 0.02 | 0.02 | 0.03 | 0.0049 | 0.0039 | 0.0025 | 0.0041 | 0.0100 | 0.0042 |
| B | 0.01 | 0.02 | 0.01 | 0.01 | 0.01 | 0.01 | 0.02 | 0.01 | −0.02 | −0.01 | 0.01 | −0.01 | 0.00 | 0.00 | |
| SE | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | |
| P | 1.4E-04 | 9.6E-05 | 1.6E-04 | 1.7E-04 | 1.7E-04 | 1.7E-04 | 8.3E-05 | 2.8E-04 | 0.057 | 0.291 | 0.479 | 0.560 | 0.603 | 0.850 | |
| WNHW n=498 | MAF | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.0191 | 0.0050 | 0.0070 | NA | NA | NA |
| B | −0.03 | −0.03 | −0.03 | −0.03 | −0.03 | −0.03 | −0.03 | −0.03 | 0.00 | 0.00 | −0.02 | NA | NA | NA | |
| SE | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | NA | NA | NA | |
| P | 0.002 | 0.010 | 0.002 | 0.014 | 0.002 | 0.000 | 0.010 | 0.014 | 0.653 | 0.787 | 0.233 | NA | NA | NA | |
| WAA n=492 | MAF | 0.16 | 0.05 | 0.15 | 0.06 | 0.15 | 0.15 | 0.05 | 0.06 | 0.0007 | 0.0641 | 0.1606 | 0.0385 | 0.0020 | 0.0385 |
| B | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | −0.01 | 0.00 | 0.00 | 0.00 | 0.01 | NA | |
| SE | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 | NA | |
| P | 0.153 | 0.052 | 0.151 | 0.058 | 0.152 | 0.155 | 0.052 | 0.038 | 0.032 | 0.057 | 0.166 | 0.038 | 0.003 | NA | |
| WCH N=1,151 | MAF | 0.09 | 0.03 | 0.08 | 0.04 | 0.08 | 0.09 | 0.03 | 0.04 | 0.0030 | 0.0196 | 0.0745 | 0.0192 | 0.0040 | 0.0192 |
| B | −0.01 | −0.01 | −0.01 | 0.00 | −0.01 | −0.01 | −0.01 | 0.00 | 0.02 | −0.01 | −0.01 | 0.01 | 0.00 | 0.01 | |
| SE | 0.00 | 0.01 | 0.00 | 0.01 | 0.00 | 0.00 | 0.01 | 0.01 | 0.02 | 0.01 | 0.01 | 0.01 | 0.02 | 0.01 | |
| P | 0.040 | 0.314 | 0.066 | 0.686 | 0.061 | 0.074 | 0.471 | 0.627 | 0.490 | 0.580 | 0.172 | 0.323 | 0.911 | 0.323 | |
Figure 1.
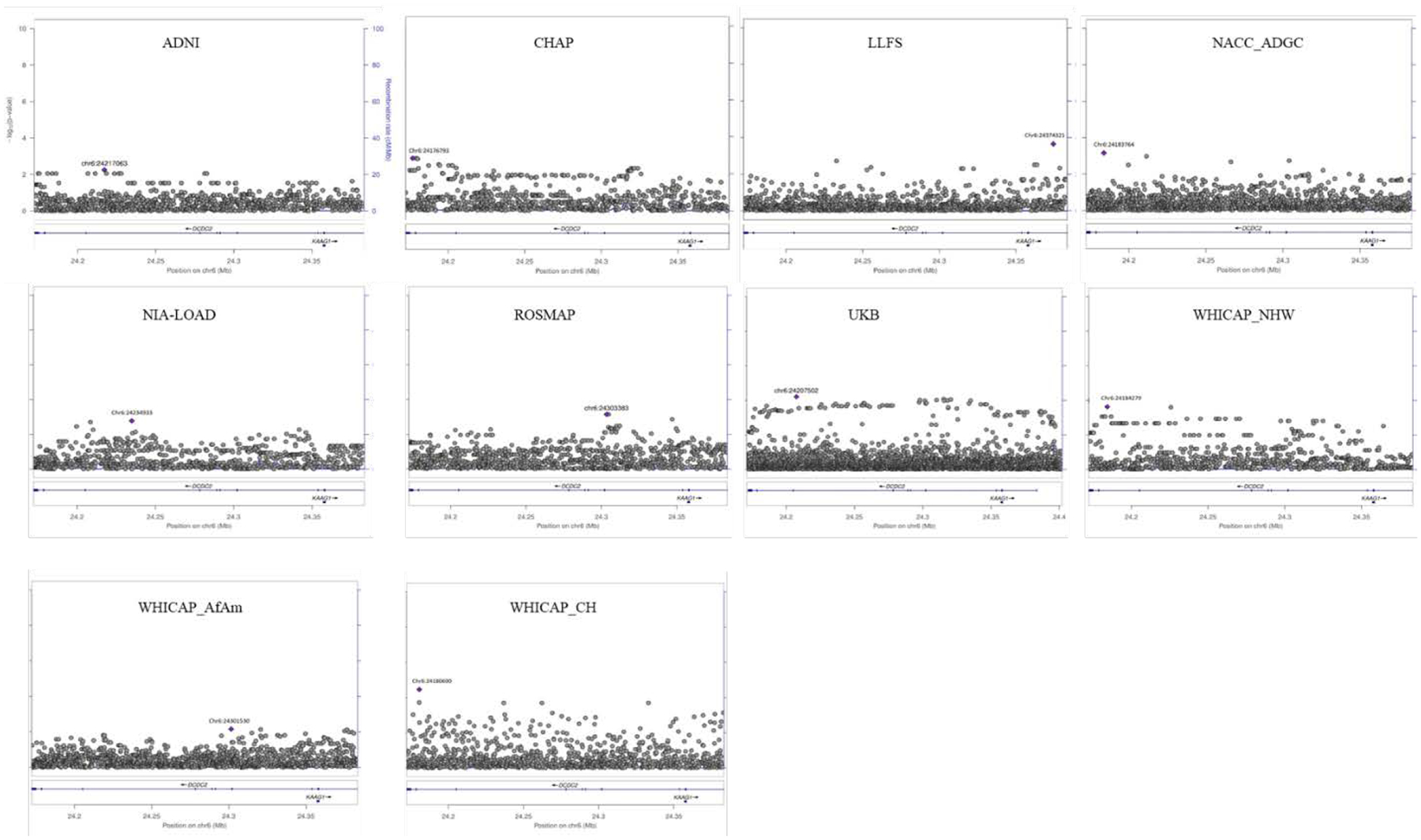
Regional association plots for SNP based DCDC2 analysis in the APOE-noE4 strata.
The X-axis represent the GRCh37/hg19 chromosomal position (Mb) of the tested SNP variant(s); the left Y-axis correspond to the statistical strength of the SNP association (log10 (p value)). The right y-axis displays the estimated recombination rates (cM/Mb) to reflect the local LD structure. NHW: Non-Hispanic Whites; AfAm: African-Americans; CH: Caribbean-Hispanics.
DCDC2 and APOE interaction.
The results from epistatic models (Table 4) revealed that there is no significant interaction between the strongest DCDC2 associated variant in the SNP-based meta-analysis (rs1340698) and APOE genotype.
Table 4.
Common SNP-based DCDC2-APOE epistasis models by study cohort.
| Cohort | TEST | rs1340698 | |||
|---|---|---|---|---|---|
| A1 | N | B | P | ||
| ADNI | SNP | G | 1,090 | 0.04 | 0.134 |
| E4 | G | 1,090 | −0.07 | 1.7E-15 | |
| SNP*ε4 | G | 1,090 | −0.07 | 0.078 | |
| CHAP | SNP | G | 696 | 0.00 | 0.919 |
| E4 | G | 696 | −0.02 | 0.002 | |
| SNP*ε4 | G | 696 | 0.00 | 0.908 | |
| LLFS | SNP | G | 1,874 | 0.01 | 0.731 |
| E4 | G | 1,874 | 0.00 | 0.671 | |
| SNP*ε4 | G | 1,874 | 0.03 | 0.514 | |
| NACC | SNP | G | 6,774 | 0.01 | 0.376 |
| E4 | G | 6,774 | −0.04 | 1.4E-25 | |
| SNP*ε4 | G | 6,774 | −0.01 | 0.382 | |
| NIA-LOAD | SNP | G | 482 | 0.01 | 0.877 |
| E4 | G | 482 | −0.03 | 0.007 | |
| SNP*ε4 | G | 482 | 0.04 | 0.393 | |
| ROSMAP | SNP | G | 1,265 | −0.03 | 0.022 |
| E4 | G | 1,265 | −0.03 | 8.6E-08 | |
| SNP*ε4 | G | 1,265 | −0.01 | 0.837 | |
| UKB | SNP | G | 20,174 | 0.01 | 9.8E-05 |
| E4 | G | 20,174 | 0.00 | 0.529 | |
| SNP*ε4 | G | 20,174 | −0.01 | 0.097 | |
| WHICAP_NHW | SNP | G | 619 | −0.03 | 3.6E-04 |
| E4 | G | 619 | 0.00 | 0.383 | |
| SNP*ε4 | G | 619 | 0.04 | 0.008 | |
| WHICAP_AfAm | SNP | G | 741 | 0.00 | 0.519 |
| E4 | G | 741 | 0.00 | 0.461 | |
| SNP*ε4 | G | 741 | 0.00 | 0.871 | |
| WHICAP_CH | SNP | G | 1,529 | 0.00 | 0.220 |
| E4 | G | 1,529 | −0.01 | 1.7E-05 | |
| SNP*ε4 | G | 1,529 | −0.01 | 0.452 | |
Brain transcriptome results.
ROSMAP results (Table 5) revealed FDR-adjusted association between episodic memory maintenance and increased DCDC2 expression in dorsolateral prefrontal cortex (p=3.8 × 10−4). When evaluating additional ROSMAP neuropathological traits, the increased DCDC2 expression levels were associated with: Tau protein (measured as the average cortical density of antibodies to abnormally phosphorylated Tau in eight brain regions, p=0.003), overall amyloid beta level (measured as the average of the percent area that is occupied by amyloid beta in eight different brain regions, p=0.008), neurofibrillary tangle burden (measured as the average of tangle count in silver-stained slides from 5 regions, p=0.009), neuritic plaque burden (measured as the average of neuritic plaque count in silver-stained slides from 5 regions, p=0.011) and global burden of Alzheimer’s disease pathology (measured as the average of counts in three pathologies: neurofibrillary tangles, neuritic and diffuse plaques in silver-stained slides from 5 regions, p=0.012).
Table 5.
Association of DCDC2 mRNA levels with cognitive and pathological phenotypes in the ROSMAP cohort.
| Trait | n | logFC | t | P | Padj | FDRPadj |
|---|---|---|---|---|---|---|
| Slope of global cognition | 661 | 1.10 | 4.73 | 2.8E-06 | 7.4E-05 | 0.002 |
| Slope of episodic memory | 660 | 0.97 | 4.31 | 1.9E-05 | 3.8E-04 | 0.004 |
| Neuronal neurofibrillary tangles | 691 | −0.06 | −3.70 | 2.3E-04 | 0.003 | 0.021 |
| Amyloid beta protein | 692 | −0.06 | −3.26 | 0.001 | 0.008 | 0.042 |
| Neurofibrillary tangle burden | 698 | −0.17 | −3.32 | 0.001 | 0.009 | 0.038 |
| Neuritic plaque burden | 698 | −0.13 | −3.17 | 0.002 | 0.011 | 0.039 |
| Pathological AD diagnosis | 698 | −0.11 | −3.46 | 0.001 | 0.012 | 0.036 |
| Global measure of pathology | 698 | −0.10 | −2.88 | 0.004 | 0.024 | 0.063 |
| Neuronal loss substantia nigra | 696 | −0.08 | −2.97 | 0.003 | 0.026 | 0.061 |
| Transactive response DNA binding protein | 640 | −0.05 | −2.50 | 0.013 | 0.138 | 0.290 |
| Pathologic diagnosis of Lewy body diseases | 674 | −0.04 | −2.07 | 0.039 | 0.332 | 0.634 |
| Diffuse plaque burden | 698 | −0.06 | −1.47 | 0.142 | 0.455 | 0.796 |
| Global Parkinsonian Summary Score | 696 | −0.03 | −1.81 | 0.071 | 0.482 | 0.779 |
| Arteriolosclerosis | 692 | −0.03 | −1.37 | 0.173 | 0.665 | 0.998 |
| Any distribution of α-synuclein | 674 | −0.06 | −1.78 | 0.075 | 0.668 | 0.935 |
| Gross cerebral infarctions | 698 | 0.03 | 0.93 | 0.354 | 0.798 | 1.047 |
| Micro cerebral infarctions | 698 | −0.03 | −1.03 | 0.303 | 0.821 | 1.014 |
| Cerebral amyloid angiopathy | 683 | −0.02 | −0.71 | 0.481 | 0.875 | 1.021 |
| Diagnosis of Parkinson | 695 | 0.03 | 0.48 | 0.630 | 0.891 | 0.985 |
| Hippocampal sclerosis | 694 | −0.04 | −0.80 | 0.423 | 0.898 | 0.943 |
| Cerebral Atherosclerosis | 695 | 0.00 | 0.17 | 0.863 | 0.964 | 0.964 |
Differential brain expression results from MSBB and Mayo datasets (Figure 2) revealed an overall decreased DCDC2 expression (across all brain areas when AD cases were compared to controls. DCDC2 downregulated expression achieved nominally statistical significance (~2-fold change, p<0.05) in two specific brain areas: superior temporal gyrus (p=0.007) and inferior frontal gyrus (p=0.013).
Figure 2.
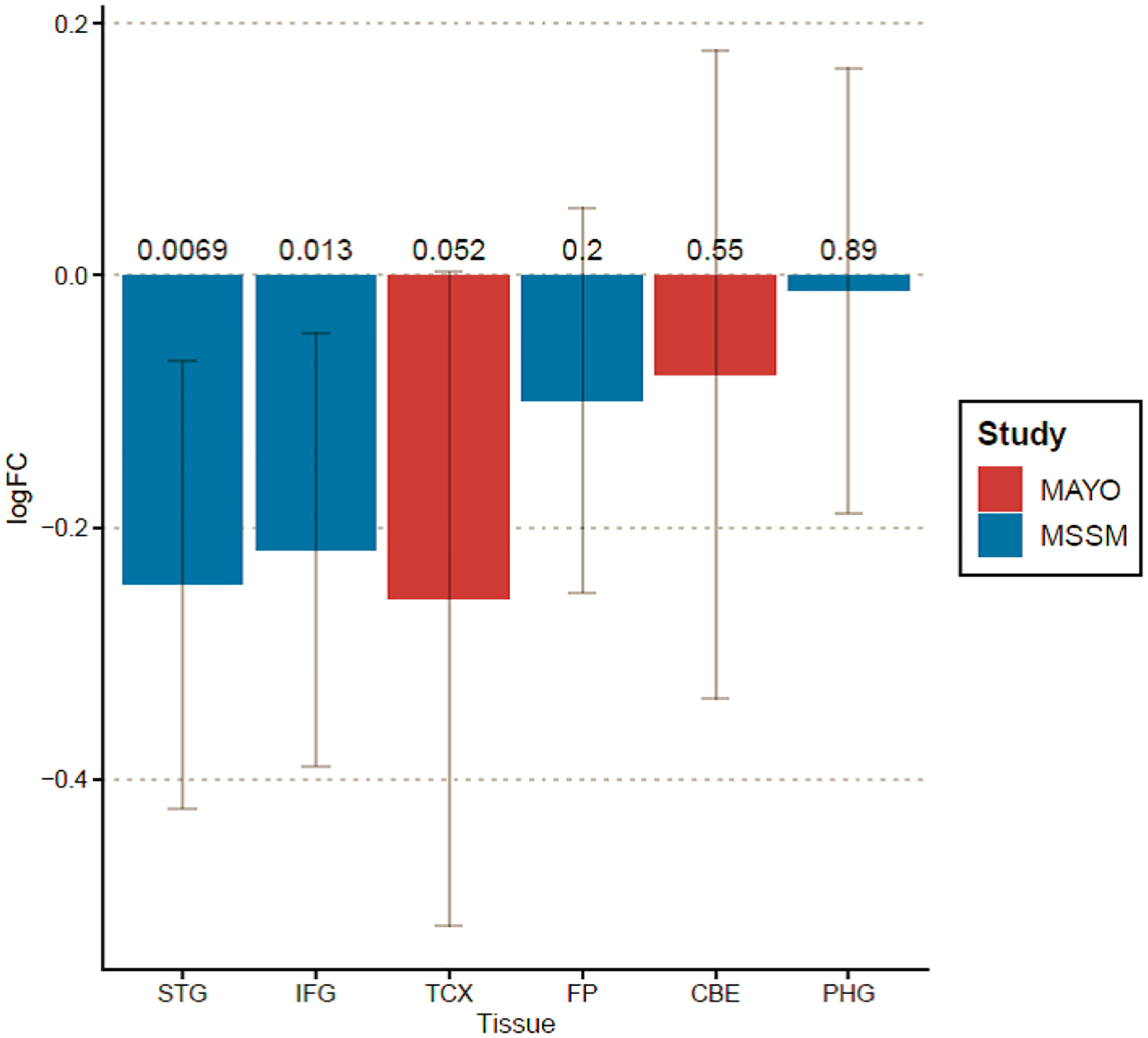
DCDC2 brain transcriptome results from Mount Sinai Brain Bank (MSBB) and Mayo Clinic datasets.
The X-axis represents the brain regions analyzed from each cohort: Mount Sinai Brain Bank: superior temporal gyrus (STG), inferior frontal gyrus (IFG), frontal pole (FP), and parahippocampal gyrus (PHG); Mayo Clinic: temporal cortex (TCX) and cerebellum (CBE). The Y-axis correspond to the estimated tissue-specific fold change in DCDC2 expression (in red upregulation, in blue downregulation) and the 95% confidence intervals.
Mendelian randomization results identified common variant rs12216513 as significant eQTL for DCDC2 expression (B=0.29, SE=0.04, p=1.1 × 10−11). This DCDC2 variant is in tight LD with meta-analysis topSNPs, common (rs1340698, D’=0.88) and rare (rs147661578, D’=0.84). However, the effect of DCDC2 variants on episodic memory performance over-time is not mediated by its brain expression (SMR p-value=0.950) (Supplemental Figure 7).
Because the widely reported association of DCDC2 with phonological awareness and phonemic decoding [86], secondary analyses in WHICAP tested the DCDC2 association with LCMM estimated trajectories of language [87]. The gene-based association results indicated no significant association between DCDC2 and decay of language in none of the APOE strata considered (Supplementary Figure 8).
Supplementary Material
Supplemental Figure 1. Overview of the study design.
Supplemental Figure 2. Episodic memory trajectories considering all subjects at baseline within each of the study cohorts.
NHW: Non-Hispanic Whites; AfAm: African-Americans; CH: Caribbean-Hispanics. The X-axis correspond to the time of follow-up in years (ranging from O to 15); the Y-axis correspond to the residual episodic memory score (ranging from −6 to 4) after being adjusted for sex, age, education, episodic memory scores at baseline and total years of follow-up (truncated to a maximum of 15 years.
Supplemental Figure 3. QQ-plots of genome-wide gene-based analysis in the non-stratified sample.
The x axis displays the expected p-values from a theoretical normal distribution; the y-axis represents the dataset observed p-values.
Supplemental Figure 4. qqplots of genome-wide gene-based analysis in APOE-E4 strata.
The x axis displays the expected p-values from a theoretical normal distribution; the y-axis represents the dataset observed p-values.
Supplemental Figure 5. qqplots of genome-wide gene-based analysis in APOE-noE4 strata.
The x axis displays the expected p-values from a theoretical normal distribution; the y-axis represents the dataset observed p-values.
Supplemental Figure 6. Matrix of linkage disequilibrium for topSNPs in the singleSNP based meta-analysis.
The dimensions of the square plot correspond to the number of SNP variants tested. SNPs are displayed based on their GRCh37 genomic coordinates. Measures of SNP pairwise linkage disequilibrium consisted of D’ (blue colored) and R2 (red colored) statistics.
Supplemental Figure 7. Mendelian randomization DCDC2-brain eQTLs.
The x-axis displays the position (in Mb) for each of the SNPs tested within chromosomal region 6p22.3. In the top panel, each colored circle represents the −log10 association GWAS p-values for each of the SNPs. The hollow diamonds show the p-values for probes considered in the analyses. The bottom panel displays the eQTLs p-values of the SNPs from Brain-eMeta dataset. The dotted line highlighted in red indicates SMR threshold of significance.
Supplemental Figure 8. Trajectories of language in WHICAP Non-Hispanic Whites study.
The X-axis correspond to the time of follow-up in years (ranging from 0 to 15); the Y-axis correspond to the residual episodic memory score (ranging from −6 to 4) after being adjusted for sex, age, education, episodic memory scores at baseline and total years of follow-up (truncated to a maximum of 15 years).
References
- 1.Vidal-Pineiro D, et al. , The Functional Foundations of Episodic Memory Remain Stable Throughout the Lifespan. Cereb Cortex, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glisky EL, Changes in Cognitive Function in Human Aging, in Brain Aging: Models, Methods, and Mechanisms, Riddle DR, Editor. 2007: Boca Raton (FL). [Google Scholar]
- 3.Bearden CE and Glahn DC, Cognitive genomics: Searching for the genetic roots of neuropsychological functioning. Neuropsychology, 2017. 31(8): p. 1003–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson RS, et al. , Heritability of different forms of memory in the Late Onset Alzheimer’s Disease Family Study. J Alzheimers Dis, 2011. 23(2): p. 249–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Papassotiropoulos A, et al. , A genome-wide survey and functional brain imaging study identify CTNNBL1 as a memory-related gene. Mol Psychiatry, 2013. 18(2): p. 255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pawlowski TL and Huentelman MJ, Identification of a common variant affecting human episodic memory performance using a pooled genome-wide association approach: a case study of disease gene identification. Methods Mol Biol, 2011. 700: p. 261–9. [DOI] [PubMed] [Google Scholar]
- 7.Andrews SJ, et al. , Association of AKAP6 and MIR2113 with cognitive performance in a population-based sample of older adults. Genes Brain Behav, 2017. 16(4): p. 472–478. [DOI] [PubMed] [Google Scholar]
- 8.Athanasiu L, et al. , A genetic association study of CSMD1 and CSMD2 with cognitive function. Brain Behav Immun, 2017. 61: p. 209–216. [DOI] [PubMed] [Google Scholar]
- 9.Barral S, et al. , Genotype patterns at PICALM, CR1, BIN1, CLU, and APOE genes are associated with episodic memory. Neurology, 2012. 78(19): p. 1464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Frias CM, et al. , COMT gene polymorphism is associated with declarative memory in adulthood and old age. Behav Genet, 2004. 34(5): p. 533–9. [DOI] [PubMed] [Google Scholar]
- 11.de Vries CF, et al. , Klotho gene polymorphism, brain structure and cognition in early-life development. Brain Imaging Behav, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dempster E, et al. , Association between BDNF val66 met genotype and episodic memory. Am J Med Genet B Neuropsychiatr Genet, 2005. 134B(1): p. 73–5. [DOI] [PubMed] [Google Scholar]
- 13.Huentelman MJ, et al. , Calmodulin-binding transcription activator 1 (CAMTA1) alleles predispose human episodic memory performance. Hum Mol Genet, 2007. 16(12): p. 1469–77. [DOI] [PubMed] [Google Scholar]
- 14.Milnik A, et al. , Association of KIBRA with episodic and working memory: a meta-analysis. Am J Med Genet B Neuropsychiatr Genet, 2012. 159B(8): p. 958–69. [DOI] [PubMed] [Google Scholar]
- 15.Papassotiropoulos A, et al. , Common Kibra alleles are associated with human memory performance. Science, 2006. 314(5798): p. 475–8. [DOI] [PubMed] [Google Scholar]
- 16.Sigmund JC, et al. , Fine-mapping at the HTR2A locus reveals multiple episodic memory-related variants. Biol Psychol, 2008. 79(2): p. 239–42. [DOI] [PubMed] [Google Scholar]
- 17.Ward DD, et al. , APOE and BDNF Val66Met polymorphisms combine to influence episodic memory function in older adults. Behav Brain Res, 2014. 271: p. 309–15. [DOI] [PubMed] [Google Scholar]
- 18.Papenberg G, et al. , Dopamine and glutamate receptor genes interactively influence episodic memory in old age. Neurobiol Aging, 2014. 35(5): p. 1213 e3–8. [DOI] [PubMed] [Google Scholar]
- 19.Josefsson M, et al. , Genetic and lifestyle predictors of 15-year longitudinal change in episodic memory. J Am Geriatr Soc, 2012. 60(12): p. 2308–12. [DOI] [PubMed] [Google Scholar]
- 20.Smith JA, et al. , Genetic effects and gene-by-education interactions on episodic memory performance and decline in an aging population. Soc Sci Med, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Albrecht MA, et al. , Longitudinal cognitive decline in the AIBL cohort: The role of APOE epsilon4 status. Neuropsychologia, 2015. 75: p. 411–9. [DOI] [PubMed] [Google Scholar]
- 22.Bertola L, et al. , Predictors of Episodic Memory Performance Across Educational Strata: Multiple-Group Comparisons. J Int Neuropsychol Soc, 2019. 25(9): p. 901–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cornelis MC, et al. , Age and cognitive decline in the UK Biobank. PLoS One, 2019. 14(3): p. e0213948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davies G, et al. , Genetic contributions to variation in general cognitive function: a meta-analysis of genome-wide association studies in the CHARGE consortium (N=53949). Mol Psychiatry, 2015. 20(2): p. 183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davies G, et al. , Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat Commun, 2018. 9(1): p. 2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davies G, et al. , Genome-wide association study of cognitive functions and educational attainment in UK Biobank (N=112 151). Mol Psychiatry, 2016. 21(6): p. 758–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding X, et al. , Evaluating trajectories of episodic memory in normal cognition and mild cognitive impairment: Results from ADNI. PLoS One, 2019. 14(2): p. e0212435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panizzon MS, et al. , Genetic and environmental architecture of changes in episodic memory from middle to late middle age. Psychol Aging, 2015. 30(2): p. 286–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tromp D, et al. , Episodic memory in normal aging and Alzheimer disease: Insights from imaging and behavioral studies. Ageing Res Rev, 2015. 24(Pt B): p. 232–62. [DOI] [PubMed] [Google Scholar]
- 30.Bomba L, Walter K, and Soranzo N, The impact of rare and low-frequency genetic variants in common disease. Genome Biol, 2017. 18(1): p. 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Momozawa Y and Mizukami K, Unique roles of rare variants in the genetics of complex diseases in humans. J Hum Genet, 2021. 66(1): p. 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kerner B, North KE, and Fallin MD, Use of longitudinal data in genetic studies in the genome-wide association studies era: summary of Group 14. Genet Epidemiol, 2009. 33 Suppl 1: p. S93–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee S, et al. , Episodic memory performance in a multi-ethnic longitudinal study of 13,037 elderly. PLoS One, 2018. 13(11): p. e0206803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitt M, et al. , Improved imputation accuracy of rare and low-frequency variants using population-specific high-coverage WGS-based imputation reference panel. Eur J Hum Genet, 2017. 25(7): p. 869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vergara C, et al. , Genotype imputation performance of three reference panels using African ancestry individuals. Human Genetics, 2018. 137(4): p. 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sariya S, et al. , Rare Variants Imputation in Admixed Populations: Comparison Across Reference Panels and Bioinformatics Tools. Front Genet, 2019. 10: p. 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang MH, et al. , COMBAT: A Combined Association Test for Genes Using Summary Statistics. Genetics, 2017. 207(3): p. 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li MX, et al. , GATES: A Rapid and Powerful Gene-Based Association Test Using Extended Simes Procedure. American Journal of Human Genetics, 2011. 88(3): p. 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sirugo G, Williams SM, and Tishkoff SA, The Missing Diversity in Human Genetic Studies. Cell, 2019. 177(4): p. 1080. [DOI] [PubMed] [Google Scholar]
- 40.Igartua C, et al. , Ethnic-specific associations of rare and low-frequency DNA sequence variants with asthma. Nat Commun, 2015. 6: p. 5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Girard M, et al. , DCDC2 Mutations Cause Neonatal Sclerosing Cholangitis. Hum Mutat, 2016. 37(10): p. 1025–9. [DOI] [PubMed] [Google Scholar]
- 42.Grammatikopoulos T, et al. , Mutations in DCDC2 (doublecortin domain containing protein 2) in neonatal sclerosing cholangitis. J Hepatol, 2016. 65(6): p. 1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaworski J, et al. , Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron, 2009. 61(1): p. 85–100. [DOI] [PubMed] [Google Scholar]
- 44.Dent EW, Of microtubules and memory: implications for microtubule dynamics in dendrites and spines. Mol Biol Cell, 2017. 28(1): p. 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Breuss MW, et al. , Tubulins and brain development - The origins of functional specification. Mol Cell Neurosci, 2017. 84: p. 58–67. [DOI] [PubMed] [Google Scholar]
- 46.Truong DT, et al. , Mutation of Dcdc2 in mice leads to impairments in auditory processing and memory ability. Genes Brain Behav, 2014. 13(8): p. 802–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gabel LA, et al. , Mutation of the dyslexia-associated gene Dcdc2 impairs LTM and visuo-spatial performance in mice. Genes Brain Behav, 2011. 10(8): p. 868–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Couto JM, et al. , Association of attention-deficit/hyperactivity disorder with a candidate region for reading disabilities on chromosome 6p. Biol Psychiatry, 2009. 66(4): p. 368–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meng H, et al. , DCDC2 is associated with reading disability and modulates neuronal development in the brain. Proc Natl Acad Sci U S A, 2005. 102(47): p. 17053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schumacher J, et al. , Strong genetic evidence of DCDC2 as a susceptibility gene for dyslexia. Am J Hum Genet, 2006. 78(1): p. 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilcke A, et al. , The role of gene DCDC2 in German dyslexics. Ann Dyslexia, 2009. 59(1): p. 1–11. [DOI] [PubMed] [Google Scholar]
- 52.Newbury DF, et al. , Investigation of dyslexia and SLI risk variants in reading- and language-impaired subjects. Behav Genet, 2011. 41(1): p. 90–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scerri TS, et al. , DCDC2, KIAA0319 and CMIP are associated with reading-related traits. Biol Psychiatry, 2011. 70(3): p. 237–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, et al. , Association of DCDC2 Polymorphisms with Normal Variations in Reading Abilities in a Chinese Population. PLoS One, 2016. 11(4): p. e0153603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marino C, et al. , DCDC2 genetic variants and susceptibility to developmental dyslexia. Psychiatric Genetics, 2012. 22(1): p. 25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scerri TS, et al. , The DCDC2 deletion is not a risk factor for dyslexia. Transl Psychiatry, 2017. 7(7): p. e1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karunakaran KB, et al. , Cilia interactome with predicted protein-protein interactions reveals connections to Alzheimer’s disease, aging and other neuropsychiatric processes. Sci Rep, 2020. 10(1): p. 15629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stern Y, Cognitive reserve. Neuropsychologia, 2009. 47(10): p. 2015–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perneczky R, et al. , Translational research on reserve against neurodegenerative disease: consensus report of the International Conference on Cognitive Reserve in the Dementias and the Alzheimer’s Association Reserve, Resilience and Protective Factors Professional Interest Area working groups. BMC Med, 2019. 17(1): p. 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nyberg L, et al. , Memory aging and brain maintenance. Trends Cogn Sci, 2012. 16(5): p. 292–305. [DOI] [PubMed] [Google Scholar]
- 61.Nelson MR, et al. , The support of human genetic evidence for approved drug indications. Nat Genet, 2015. 47(8): p. 856–60. [DOI] [PubMed] [Google Scholar]
- 62.King EA, Davis JW, and Degner JF, Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet, 2019. 15(12): p. e1008489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Small SA, Age-related memory decline: current concepts and future directions. Arch Neurol, 2001. 58(3): p. 360–4. [DOI] [PubMed] [Google Scholar]
- 64.Berkowitz CL, et al. , Precision Medicine for Alzheimer’s Disease Prevention. Healthcare (Basel), 2018. 6(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rees E and Owen MJ, Translating insights from neuropsychiatric genetics and genomics for precision psychiatry. Genome Med, 2020. 12(1): p. 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barral S, et al. , Cognitive function in families with exceptional survival. Neurobiol Aging, 2012. 33(3): p. 619 e1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bycroft C, et al. , The UK Biobank resource with deep phenotyping and genomic data. Nature, 2018. 562(7726): p. 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weiner MW, et al. , The Alzheimer’s disease neuroimaging initiative: progress report and future plans. Alzheimers Dement, 2010. 6(3): p. 202–11 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ruff RM, Light RH, and Quayhagen M, Selective Reminding Tests: a normative study of verbal learning in adults. J Clin Exp Neuropsychol, 1989. 11(4): p. 539–50. [DOI] [PubMed] [Google Scholar]
- 70.Moradi E, et al. , Rey’s Auditory Verbal Learning Test scores can be predicted from whole brain MRI in Alzheimer’s disease. Neuroimage Clin, 2017. 13: p. 415–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McKhann GM, et al. , The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement, 2011. 7(3): p. 263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cosentino S, et al. , Reduced prevalence of cognitive impairment in families with exceptional longevity. JAMA Neurol, 2013. 70(7): p. 867–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Das S, et al. , Next-generation genotype imputation service and methods. Nat Genet, 2016. 48(10): p. 1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Purcell S, et al. , PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet, 2007. 81(3): p. 559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang K, Li M, and Hakonarson H, ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res, 2010. 38(16): p. e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Center for Statistical Genetics, M.U. EPACTS Efficient and Parallelizable Association Container Toolbox. 2014; Available from: http://genome.sph.umich.edu/wiki/EPACTS.
- 77.Cochran WG, The combination of estimates from different experiments. Biometrics, 1954. 10: p. 101–129. [Google Scholar]
- 78.Willer CJ, Li Y, and Abecasis GR, METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 2010. 26(17): p. 2190–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bennett DA, et al. , Overview and findings from the religious orders study. Curr Alzheimer Res, 2012. 9(6): p. 628–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bennett DA, et al. , Overview and findings from the rush Memory and Aging Project. Curr Alzheimer Res, 2012. 9(6): p. 646–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.De Jager PL, et al. , A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research. Sci Data, 2018. 5: p. 180142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mostafavi S, et al. , A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat Neurosci, 2018. 21(6): p. 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang M, et al. , The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer’s disease. Sci Data, 2018. 5: p. 180185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Allen M, et al. , Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci Data, 2016. 3: p. 160089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhu Z, et al. , Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet, 2016. 48(5): p. 481–7. [DOI] [PubMed] [Google Scholar]
- 86.DeMille MMC, et al. , Worldwide distribution of the DCDC2 READ1 regulatory element and its relationship with phoneme variation across languages. Proc Natl Acad Sci U S A, 2018. 115(19): p. 4951–4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zahodne LB, et al. , Cognitive declines precede and predict functional declines in aging and Alzheimer’s disease. PLoS One, 2013. 8(9): p. e73645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Overview of the study design.
Supplemental Figure 2. Episodic memory trajectories considering all subjects at baseline within each of the study cohorts.
NHW: Non-Hispanic Whites; AfAm: African-Americans; CH: Caribbean-Hispanics. The X-axis correspond to the time of follow-up in years (ranging from O to 15); the Y-axis correspond to the residual episodic memory score (ranging from −6 to 4) after being adjusted for sex, age, education, episodic memory scores at baseline and total years of follow-up (truncated to a maximum of 15 years.
Supplemental Figure 3. QQ-plots of genome-wide gene-based analysis in the non-stratified sample.
The x axis displays the expected p-values from a theoretical normal distribution; the y-axis represents the dataset observed p-values.
Supplemental Figure 4. qqplots of genome-wide gene-based analysis in APOE-E4 strata.
The x axis displays the expected p-values from a theoretical normal distribution; the y-axis represents the dataset observed p-values.
Supplemental Figure 5. qqplots of genome-wide gene-based analysis in APOE-noE4 strata.
The x axis displays the expected p-values from a theoretical normal distribution; the y-axis represents the dataset observed p-values.
Supplemental Figure 6. Matrix of linkage disequilibrium for topSNPs in the singleSNP based meta-analysis.
The dimensions of the square plot correspond to the number of SNP variants tested. SNPs are displayed based on their GRCh37 genomic coordinates. Measures of SNP pairwise linkage disequilibrium consisted of D’ (blue colored) and R2 (red colored) statistics.
Supplemental Figure 7. Mendelian randomization DCDC2-brain eQTLs.
The x-axis displays the position (in Mb) for each of the SNPs tested within chromosomal region 6p22.3. In the top panel, each colored circle represents the −log10 association GWAS p-values for each of the SNPs. The hollow diamonds show the p-values for probes considered in the analyses. The bottom panel displays the eQTLs p-values of the SNPs from Brain-eMeta dataset. The dotted line highlighted in red indicates SMR threshold of significance.
Supplemental Figure 8. Trajectories of language in WHICAP Non-Hispanic Whites study.
The X-axis correspond to the time of follow-up in years (ranging from 0 to 15); the Y-axis correspond to the residual episodic memory score (ranging from −6 to 4) after being adjusted for sex, age, education, episodic memory scores at baseline and total years of follow-up (truncated to a maximum of 15 years).